Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(8):2235-2251. doi:10.7150/thno.30678 This issue Cite
Research Paper
Sonic hedgehog pathway activation is associated with cetuximab resistance and EPHB3 receptor induction in colorectal cancer
Seong Hye Park1, Min Jee Jo1, Bo Ram Kim2, Yoon A Jeong1, Yoo Jin Na1, Jung Lim Kim2, Soyeon Jeong2, Hye Kyeong Yun1, Dae Yeong Kim1, Bu Gyeom Kim1, Sang Hee Kang3, Sang Cheul Oh1,2 ![]() , Dae-Hee Lee1,2
, Dae-Hee Lee1,2 ![]()
1. Graduate School of Medicine, Korea University College of Medicine, Seoul, 152-703, Republic of Korea.
2. Department of Oncology, Korea University Guro Hospital, Seoul, Republic of Korea.
3. Department of Surgery, Korea University Guro Hospital, Korea University College of Medicine, Seoul, 152-703, Republic of Korea.
Received 2018-10-15; Accepted 2019-2-17; Published 2019-4-12
Abstract

A major problem of colorectal cancer (CRC) targeted therapies is relapse caused by drug resistance. In most cases of CRC, patients develop resistance to anticancer drugs. Cetuximab does not show many of the side effects of other anticancer drugs and improves the survival of patients with metastatic CRC. However, the molecular mechanism of cetuximab resistance is not fully understood.
Methods: EPHB3-mediated cetuximab resistance was confirmed by in vitro western blotting, colony-forming assays, WST-1 colorimetric assay, and in vivo xenograft models (n = 7 per group). RNA-seq analysis and receptor tyrosine kinase assays were performed to identify the cetuximab resistance mechanism of EPHB3. All statistical tests were two-sided.
Results: The expression of EFNB3, which upregulates the EPHB3 receptor, was shown to be increased via microarray analysis. When resistance to cetuximab was acquired, EPHB3 protein levels increased. Hedgehog signaling, cancer stemness, and epithelial-mesenchymal transition signaling proteins were also increased in the cetuximab-resistant human colon cancer cell line SW48R. Despite cells acquiring resistance to cetuximab, STAT3 was still responsive to EGF and cetuximab treatment. Moreover, inhibition of EPHB3 was associated with decreased STAT3 activity. Co-immunoprecipitation confirmed that EGFR and EPHB3 bind to each other and this binding increases upon resistance acquisition, suggesting that STAT3 is activated by the binding between EGFR and EPHB3. Protein levels of GLI-1, SOX2, and Vimentin, which are affected by STAT3, also increased. Similar results were obtained in samples from patients with CRC.
Conclusion: EPHB3 expression is associated with anticancer drug resistance.
Keywords: colorectal cancer, Cetuximab Resistance, EPHB3, GLI-1
Introduction
Colorectal cancer (CRC) is the third leading cause of cancer worldwide. Metastatic CRC (mCRC) is one of the most common causes of cancer-linked death, and the anti-epidermal growth factor receptor (EGFR) antibodies cetuximab and panitumumab can be used to treat Kirsten rat sarcoma (KRAS) wild-type (WT) mCRC [1]. However, cetuximab is ineffective in CRC patients harboring KRAS mutations [2]. Moreover, almost all patients who initially respond to cetuximab become refractory, bearing evidence that acquired resistance to cetuximab is an important clinical problem. CRC cells usually overexpress EGFR, but most patients are resistant to cetuximab as a monotherapy or in combination with chemotherapeutic agents (5-FU, oxaliplatin, and irinotecan) [3-6]. Although mutation of KRAS and other genes, BRAF and NRAF, are found in primary and secondary resistance to cetuximab monotherapy, the pathways of tolerance are unclear. Cetuximab interacts strongly and competitively with EGFR, blocking the binding of its natural ligand EGF and transforming growth factor alpha (TGFα), causing receptor internalization [7, 8]. Combination therapy of cetuximab with anticancer drugs has been clinically approved for colorectal, head and neck, lung cancers [9-13]. Some of the proposed treatment strategies to overcome the resistance induced by downstream pathway reactivation are being examined in clinical trials combining anti-EGFR drugs with other targeted therapies. Preclinical research has reported that combined targeted treatments that lead to vertical interference of the EGFR pathway are a reasonable approach [13, 14]. However, further identification of tolerance pathways to anti-EGFR monotherapy is essential to develop productive therapies for CRC. Ultimately, understanding the molecular basis of the clinical feedback to cetuximab could lead to the identification of a subpopulation of patients who might benefit from cetuximab and avoid needless costs and drug toxicity.
Ephrin-related receptor tyrosine kinases (RTKs) have been involved in intercellular communication and signaling during embryonic development. Ephrin type-B receptor 3 (EPHB3), one of EPH transmembrane tyrosine kinase receptors (TKRs), has a critical function in tumor progression or regression in various cancers [15-20]. Chiu et al. [21] demonstrated that overexpression of EPHB3 suppressed tumor growth by enhancing cell-cell contact in CRC. In contrast, Zhang et al. [15] identified ephrinB3 as a negative regulator of cell proliferation and a positive regulator of cell survival in CRC cell lines. Consequently, the functions of EPH receptors in CRC remain obscure and controversial, and further study on this subject is warranted.
EGFR and hedgehog (HH) signaling have been described as key factors involved in the survival and proliferation of tumor cells [22-25]. HH signaling may also play an important role in the development of cancer-initiating (stem) cells (CSC) and drug resistance [26, 27]. EGFR and HH signaling synergize upstream of the GLI family zinc finger 1 (GLI-1) via extracellular signal-regulated kinase (ERK) / mitogen-activated protein kinase (MAPK) signaling [28]. EGF promotes the expression of GLI-1 and target genes PTCH1 and BCL2 in advanced gastric cancer [29], and the HH ligand sonic hedgehog (SHH) signals via phosphoinositide 3-kinase (PI3K) and MAPK to enhance expression of HH-specific targets in renal cancer [30, 31].
In the present study, we found a novel cetuximab resistance mechanism in CRC. Elevated expression of the EPHB3 receptor leads to the activation of the phosphorylation EGFR pathway and the STAT3 signaling cascade via HH signaling and confers resistance to cetuximab in CRC. The results gathered in this study will increase our understanding of the role of EPHs/HH in drug resistance in cancer biology and contribute to the development of a feasible therapeutic option for CRC treatment.
Methods
Cell culture and generation of resistant cells
Human colon carcinoma cell lines SW48, DLD-1, HT29, HCT116, and Colo205 were obtained from the American Type Culture Collection (ATCC) and maintained according to the ATCC's instructions. SW48R cells were kindly provided by the MOGAM Institute. Cetuximab-resistant cells (HT29, DLD-1, and HCT116) were obtained by increasing the cetuximab dosage stepwise from 1 μg/mL to 10 μg/mL over 5 months. Oxaliplatin-resistant cells (DLD-1 and Colo205) were obtained by increasing the oxaliplatin dosage stepwise from 0.05 μg/mL to 5 μg/mL over 1 year.
Reagents and antibodies
Erbitux (cetuximab) was purchased from Merck Serono (Burlington, Massachusetts, USA). The EPHB3 inhibitor (LDN-211904) was purchased from Merck Millipore. GANT61 was purchased from Selleckchem (Houston, TX, USA). The FGFR2 inhibitor (AZD4547) was purchased from Astrazeneca. The PDGFR Tyrosine Kinase inhibitor (imatinib) and VEGFR inhibitor (bevacizumab) were purchased from Calbiochem. Ephrin-B3 Fc chimera biotinylated protein (EFNB3 protein) was purchased from R&D Systems. Drug treatments were accomplished by aspirating the medium and replacing it with new medium containing the drugs. Anti-GLI-3 (1:1000) antibody was purchased from Bethyl. Anti-SOX2 (1:1000), anti-N-Cadherin (1:1000), and anti-E-Cadherin (1:1000) antibodies were purchased from BD Biosciences. Protein G PLUS-Agarose and anti-SHH (1:500), anti-Smoothened (1:1000), anti-EpCAM (1:1000), anti-Snail (1:1000), anti-EFNB3 (1:1000), and anti-HHIP (1:1000) antibodies were purchased from Santa Cruz Biotechnology. Anti-CD133 (1:1000) antibody was purchased from MACS. Anti-Vimentin (1:1000) antibody was purchased from Dako. Anti-Nanog (1:1000), and anti-EPHB3 (1:500) antibodies were purchased from Abcam. Anti-GLI-1 (1:500), anti-GLI-2 (1:1000), anti-patched (1:1000), anti-p-STAT3 (1:500), anti-STAT3 (1:1000), anti-cleaved PARP-1 (1:1000), anti-OCT4 (1:1000), anti-EGFR (1:1000), anti-p-ERK (1:1000), anti-ERK (1:1000), anti-p-mTOR (1:1000), anti-mTOR (1:1000), anti-p-AKT (1:1000), anti-AKT (1:1000), anti-p-JNK (1:1000), anti-JNK (1:1000), anti-p-EGFR (Y1045, 1:1000), anti-p-EGFR (Y992, 1:1000), anti-p-EGFR (Y1068, 1:1000), anti-VEGFR2 (1:500), and anti-HER2 (1:1000) antibodies were purchased from Cell Signaling. Anti-actin (1:10000) antibody was purchased from Sigma. For the secondary antibodies, anti-mouse-IgG-horseradish peroxidase (HRP, 1:200) and anti-rabbit-IgG-HRP (1:200) were purchased from Cell Signaling.
Patients and tissue specimens
Tissues from four cetuximab-resistant patients with colon cancer were collected from Korea University Guro Hospital tissue bank between 2009 and 2016. Four tissue samples before cetuximab treatment and another four which had developed resistance after cetuximab treatment were derived from colon cancer patients. This protocol was reviewed and permitted by the Institutional Review Board of Guro Hospital (KUGH16275-001).
Apoptosis assay (flow cytometry)
The translocation of phosphatidylserine, one of the markers of apoptosis, was detected by the binding of allophycocyanin (APC)-conjugated annexin V. Briefly, SW48 and SW48R cells, untreated or treated with the EPHB3 inhibitor siSTAT3, cetuximab, or a combination of the two agents, were resuspended for 24 h in the binding buffer provided in the Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit (BioBud). Cells were mixed with 1.25 μL of Annexin V-FITC reagent and incubated for 15 min at room temperature in the dark. The staining was terminated and cells were immediately analyzed by flow cytometry.
Co-immunoprecipitation
A total of 100Ø plates were washed with ice-cold PBS and incubated on ice for 5 min with 500 μL of lysis buffer (Cell Signaling, Cat. No. 9803) (1 mM phenylmethylsulfonyl fluoride (PMSF), protease inhibitor, phosphatase inhibitor). The cells were scrape-harvested, cellular debris was removed by centrifugation for 5 min at 15,000 × g at 4 °C, and the protein concentration was determined using a BCA kit (Thermo Scientific). Cell supernatants were incubated with a primary antibody overnight at 4 °C, followed by the addition of 50 μL protein G agarose beads (50% slurry) for 1 h at 4 °C. Beads were washed 5 times with ice-cold lysis buffer, separated by centrifugation for 30 s at 15,000 × g at 4 °C, and then heated for 10 min with 2× sample buffer (Biosesang, Korea) to release the immunoprecipitated proteins for subsequent electrophoresis and western blotting analysis.
EGF enzyme-linked immunosorbent assay (ELISA)
Cells (3 × 105/well) were seeded in 6-well plates and incubated overnight before treatment. After EGF treatment, cell culture medium was removed and stored at -80 °C. Levels of EGF protein in the medium were determined by ELISA using a commercial kit (RayBiotech) according to manufacturer's instructions [32]. All experiments were performed in triplicate.
Phospho-RTK assay
Levels of tyrosine phosphorylation of human receptor tyrosine kinases (RTKs) were detected by X-ray film using a commercial kit (R&D Systems) according to manufacturer's instructions and our previous report [33]. Each membrane contained kinase-specific antibodies spotted in duplicate.
Microarray and pathway analysis
Total RNA was isolated from second-passage cells using Trizol (Invitrogen) and purified using RNeasy columns (Qiagen, Valencia, CA, USA) according to the manufacturers' instructions. A microarray was used to evaluate alterations of mRNA expression between the cetuximab-resistant group and the control group. Three-samples from each of the two groups were subjected to microarray analysis on the Illumina Human HT-12 v4 Expression BeadChip (Illumina, Inc., San Diego, CA) platform. After exporting the array data, quantile normalization was performed using the Limma package [6] in R software version 3.3.2 (R Foundation for Statistical Computing, Vienna, Austria) [7]. To identify pathway changes, hallmark gene sets were applied to the microarray data from the Molecular Signatures Database using gene set enrichment analysis (GSEA) [8]. Gene ontology (GO) analysis was also conducted.
Animal experiments
All animal procedures were carried out in accordance with animal care guidelines approved by the Korea University Institutional Animal Care and Use Committee (IACUC). Four-week-old female BALB/c nude mice were obtained from Orient Bio (Orient Bio, Korea) and housed in a pathogen-free environment. The animals were acclimated for 1 week before the study and had free access to food and water. All surgeries were performed under Zoletil® 50/xylazine anesthesia, and animal suffering was minimized. SW48 and SW48R cells (1×106) in 100 μL of culture medium were mixed with 100 μL of Matrigel and implanted subcutaneously in 5-week-old BALB/c nude female mice. When the tumor volume reached approximately 100 mm3, mice were randomized into four drug treatment groups: control (n = 7), cetuximab (n = 7), EPHB3 inhibitor (n = 7), and cetuximab + EPHB3 inhibitor (n = 7). The mice were intraperitoneally (i.p.) administered 10 mg/kg cetuximab and/or 0.1 mg/kg EPHB3 inhibitor three times a week for 21 days. Tumor volume was calculated every 3 days for 34 days according to the following equation: tumor volume (mm3) = π/6 × length × (width) 2. Maximum tumor area and its corresponding section were calculated using MetaMorph software (Molecular Devices). Sections of formalin-fixed, paraffin-embedded tumor specimens were deparaffinized in xylene, rehydrated in graded alcohol and then subjected to immunohistochemical staining.
TUNEL assay
DNA fragmentation was visualized by the Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, as described by the manufacturer's protocol (Roche Applied Science). Fluorescent images were obtained using a Carl Zeiss confocal microscope (Weimar, Germany).
Immunofluorescence staining
Cells grown on glass coverslips were fixed with 3.7% formaldehyde for 15 min, followed by permeabilization with 0.5% Triton X-100 for 15 min at room temperature. Cells were then blocked 1 h with 3% bovine serum albumin. Primary antibodies were applied overnight at 4 °C, followed by incubation with secondary Alexa fluor-594-conjugated antibodies (Molecular Probes, Eugene, OR, USA) or FITC-conjugated secondary antibody (Sigma). The nuclei were counterstained with 4, 6-diamidino-2-phenylindole (DAPI). Images were acquired using a Carl Zeiss confocal microscope (Weimar, Germany). Quantification of the colocalization was performed from the captured fluorescence images and represented by a defined value provided by the ZEN 2011 software.
Colony formation assay
Cells were seeded into 60Ø plates, treated with the cetuximab and incubated at 37 ° C for 24 h. The cultured cells were seeded again into 6-well plates (500 cells/well) and then cultured at 37 °C. Culture medium was changed every three days. After two weeks, the cells were washed with PBS, fixed with 4% paraformaldehyde for 30 min, and then stained with crystal violet for 30 min for visualization and counting. Cells dyed with crystal violet were photographed using a mobile phone, and the number of colonies was counted to draw a graph.
Immunoblotting
Cells were lysed in Radioimmunoprecipitation assay buffer (RIPA buffer) (50 mM Tris, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, and 1% Na-deoxycholate [pH 7.4]) with protease inhibitor and phosphatase inhibitor cocktails, and then subjected to SDS-PAGE. The proteins were transferred onto nitrocellulose membranes (GE Healthcare life science), blocked with Tris-buffered saline (TBS) containing 0.2% Tween 20 and 5% skim milk, incubated with primary antibody, then incubated with horseradish peroxidase-labeled secondary antibody. Signals were detected using X-ray films.
Small interfering RNA (siRNA) and short hairpin RNA (shRNA)
GLI-1 siRNA (siGLI-1), STAT3 siRNA (siSTAT3), EPHB3 shRNA (h) lentiviral particles, and negative control siRNAs were obtained from SantaCruz Biotechnology. Cells were transfected with siRNA oligonucleotides using Lipofectamine RNAi Max reagents (Invitrogen) according to the manufacturer's introductions (200 nM).
Quantitative real-time PCR
Total RNA was extracted by using TRIzol reagent (Life Technologies). Amplification of transcripts was performed using a reverse transcriptase polymerase chain reaction kit (Life Technologies). Real-time PCR was performed using the QuantStudio 6 Flex system. Taqman probes were as follows: STAT3 (Hs00374280_m1), GLI-1 (Hs01110766_m1), EPHB3 (Hs00177903_m1) and GAPDH (Hs99999905_m1). GAPDH was used for the normalization of gene expression.
Survival assay (WST-1)
10,000 cells were grown in tissue culture-treated 96-well plates and treated as described in the Results. Cells were then treated with EZ-Cytox (WST-1 assay, DOGEN, Korea) for 3 h at 37 °C in an atmosphere of 5% CO2. Absorbance at 450 nm was determined using an enzyme-linked immunosorbent assay plate reader.
Immunohistochemical staining
The collected patients' tissues were cut into 5-μm sections, dewaxed in xylene and rehydrated in graded alcohol. Antigen retrieval was performed using microwave oven irradiation for 20 min in 0.01 M citrate buffer (pH 6.0). Endogenous peroxidase was blocked using 3% hydrogen peroxide in PBS for 12 min. The specimens were incubated with a protein-blocking solution consisting of PBS with 5% normal donkey serum for 30 min at room temperature. The sections were incubated with primary antibodies at 4 °C overnight. Primary antibodies for the following proteins were used: GLI-1 (1:30), p-STAT3 (1:30), EFNB3 (1:30), and EPHB3 (1:50). The levels of GLI-1, p-STAT3, EFNB3, and EPHB3 in the tumor cells was judged by an independent pathologist (Baek-Hui Kim) according to methods previously described (1). Briefly, the percentage of immunoreactive cells was used to divide the samples into five grades (percentage scores), as follows: 0 (<10%), 1 (10-25%), 2 (26-50%), 3 (51-75%), and 4 (> 75%). The staining intensity was divided into four grades (intensity scores) as follows: 0 (no staining), 1 (light brown), 2 (brown), and 3 (dark brown). Positive staining was determined using the following formula: overall score = percentage score × intensity score. Expression was classified into two groups (positive and negative) with a cut-off value based on the median value of the respective overall score.
Statistical analysis
The statistical significance of differences between two groups was analyzed with the unpaired Student's t-test using GraphPad Prism (version 7.0; GraphPad Software, Inc., La Jolla, CA). For multiple group comparisons and repeated measures, analysis of variance (ANOVA) and repeated-measures ANOVA (RM ANOVA), followed by post hoc least significant difference (LSD) test, were used. All P values were two-sided. P values less than 0.05 were considered statistically significant. Detailed information regarding the other methods is available in the Supplementary Materials and Methods.
Results
Cetuximab-resistant cells are associated with hedgehog, stemness, and EMT pathways
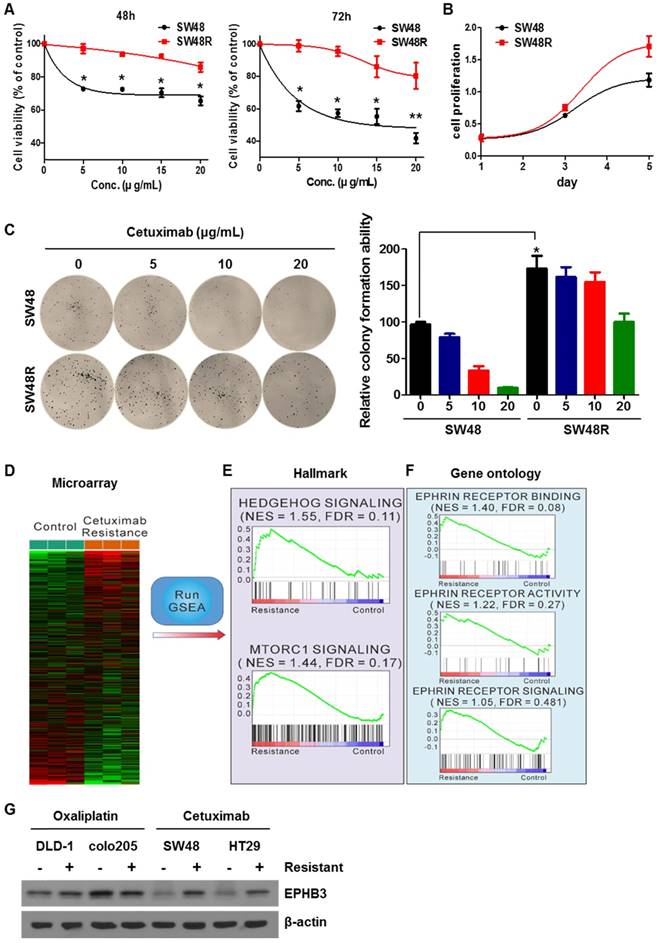
To study the difference between SW48 parent (SW48P) cells and SW48 cetuximab-resistant (SW48R) cells in colon cancer, we first confirmed the viability of both cells with or without cetuximab (Figure 1A). SW48R cells had relatively high cell viability and colony formation ability compared with SW48P cells (Figures 1B and 1C). mRNA expression in both groups was measured using a microarray (Figure 1D). Gene set enrichment analysis (GSEA) was performed to confirm pathway changes between the two groups. The GSEA of hallmark gene sets showed the highest increase for hedgehog signaling (normalized enrichment score (NES): 1.55; false discovery rate (FDR): 0.11) and the third highest increase for the mechanistic target of rapamycin (mTOR) signaling (NES 1.44, FDR 0.17) (Figure 1E, Supplementary Figures S2C and S2D). To evaluate the activity of ephrin at the mRNA level, gene ontology (GO) data sets were applied again to the microarray data with GSEA. Interestingly, all ephrin-related ontologies were found to be increased in the cetuximab-resistant group (Figure 1F). Especially, ephrin receptor binding increased beyond statistical significance (NES 1.40, FDR 0.08). We confirmed that cetuximab specifically induced EPHB3 activation in CRC cells and only activated the EPHB3 receptor. There was an increase in EPHB3 protein levels when cetuximab resistance was obtained (Figure 1G and Supplementary Figure S2E). The expression of the EPHB3 protein was not increased in oxaliplatin-resistant cells derived from other cell lines.
Effects of cetuximab on human CRC cell lines with acquired resistance to cetuximab, including ephrin-EPHB3 signaling and mTOR activation. (A) SW48 and SW48R cells were treated with increasing concentrations of cetuximab (5, 10, 15, 20 μg/mL) for 48 h and 72 h after overnight 2% FBS starvation. Cell viability was determined using a WST-1 assay. (B) SW48R cells showed improved cell proliferation for 5 days compared with SW48 cells. (C) SW48 and SW48R cells were treated with cetuximab (5, 10, 20 μg/mL). After two weeks, cells were stained with crystal violet and photographed. (D) Heat map showing the results of gene set enrichment analysis of genes significantly modified by cetuximab (10 μg/mL) in SW48 cells for 3 months. Plots of the mean gene expression values of leading-edge genes for each gene set. Lower levels of expression are displayed in green and higher levels in red. Gene sets representing differentially enriched pathways are grouped. (E and F) Enrichment plots of representative EPHB3 and mTOR gene sets. The relative gene positions of the gene sets are indicated by vertical lines below the graphs, which present the enrichment scores of individual genes. Lines clustered to the left represent higher-ranked genes in the list. Bottom plot shows the rank matrix of these genes. The position of leading-edge genes suggests a positive correlation between cetuximab treatment and the EGFR pathway. (G) Protein was collected and fractionated by SDS-PAGE, followed by immunoblotting for the indicated proteins. β-actin was used as a loading control. Data are expressed as the means of three independent experiments. **P < 0.01, *P < 0.05.
Cetuximab activates stemness, hedgehog signaling, and EMT in CRC cells
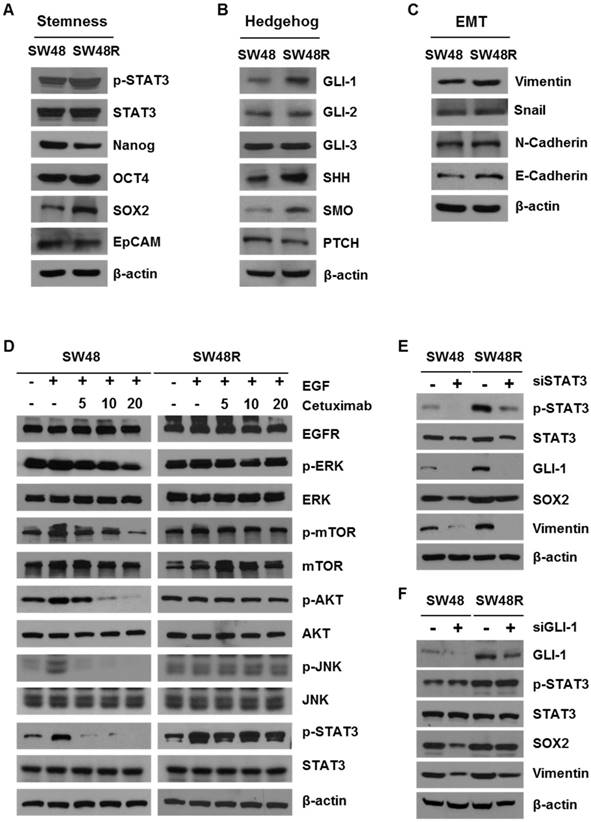
SW48 cetuximab-resistant cells showed increased levels of hedgehog (GLI-1, SHH, and SMO), stemness (p-STAT3 and SOX2) and epithelial-mesenchymal transition (Vimentin) pathway proteins (Figure 2A-C). On the other hand, we confirmed that protein expression of Nanog, OCT4, EpCAM, GLI-2, GLI-3, PTCH, Snail, and N-Cadherin did not change. Importantly, E-Cadherin levels increase in SW48R. These results suggest that binding of EPHB3 and its interacting factors is increased when resistance to cetuximab is acquired (addressed in the Discussion). To examine which proteins in SW48 cetuximab-resistant cells were affected, we confirmed the levels of EGFR downstream signaling proteins with and without cetuximab in the presence of EGF (Figure 2D). As a result, despite cetuximab resistance, p-STAT3 expression was affected by EGF. In addition, we confirmed that the increase was maintained with and without cetuximab. STAT3, which is already known to be associated with cancer, was thus selected as a target. In order to examine whether cetuximab resistance affected STAT3, we determined the phosphorylation of STAT3 associated with cetuximab resistance. Phosphorylation of STAT3 and GLI-1 are known to be correlated [34]. We knocked down the expression of STAT3 and GLI-1 using siRNAs (Figure 2E and 2F), which confirmed that GLI-1 was regulated according to the level of p-STAT3. The efficiency of the transfection with siGLI-1 and siSTAT3 is shown in Supplementary Figure S3G.
Cetuximab induced stemness, hedgehog signaling, and EMT in CRC cells. (A) Western blotting was performed to detect levels of stemness markers p-STAT3, Nanog, OCT4, SOX2, and EpCAM in SW48 and SW48R cells. β-actin was used as a loading control. (B) Western blotting was performed to detect levels of hedgehog markers GLI-1, GLI-2, GLI-3, SHH, SMO, and PTCH in SW48 and SW48R cells. (C) Western blotting was performed to detect levels of EMT markers E-Cadherin, N-Cadherin, Vimentin, and Snail in SW48 and SW48R cells. (D) SW48 and SW48R cells were treated with increasing concentrations of cetuximab (5, 10, and 20 μg/mL) for 24h, with EGF, after overnight 2% FBS starvation. Protein was collected and fractionated by SDS-PAGE, followed by immunoblotting for the indicated proteins. β-actin was used as a loading control. Data are expressed as the means of three independent experiments. (E) STAT3 was silenced in SW48R cells with STAT3 siRNA. The levels of GLI-1, p- STAT3, STAT3, SOX2, and Vimentin were detected by western blotting. (F) GLI-1 was silenced in SW48R cells with GLI-1 siRNA. The levels of GLI-1, p- STAT3, STAT3, SOX2, and Vimentin were detected by western blotting. β-actin was used as a loading control. Data are expressed as the means of three independent experiments. FBS: fetal bovine serum; EMT: epithelial-mesenchymal transition; siRNA: short interfering RNA.
Effect of EPHB3 on EFNB3 in models of acquired resistance to cetuximab
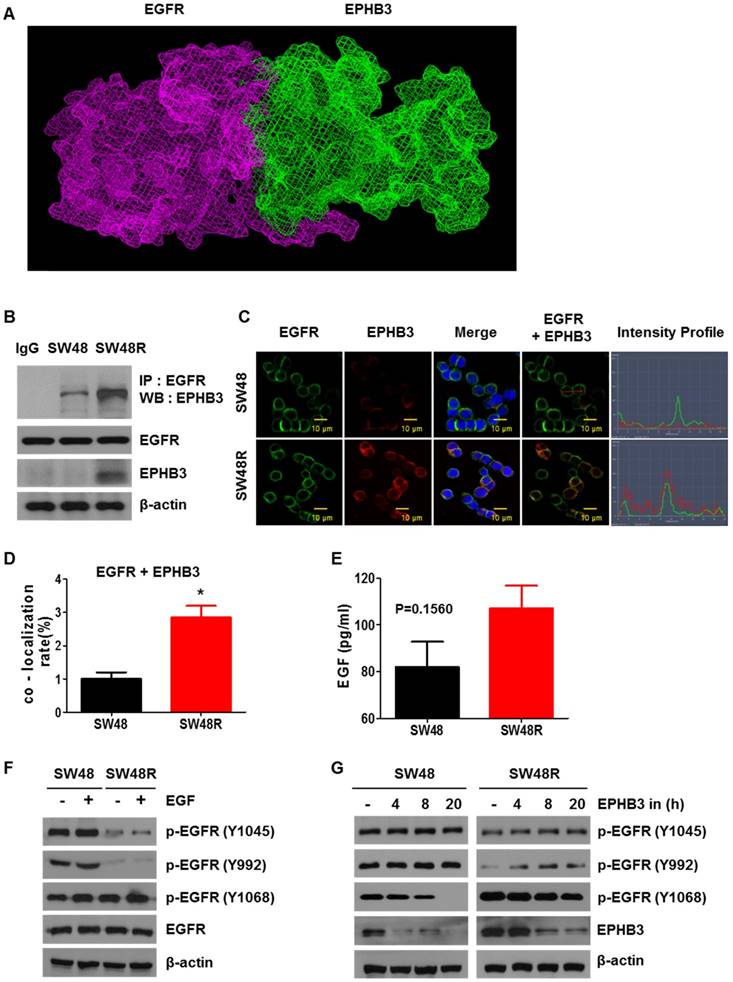
To assess the potential relevance of cetuximab resistance on colon cancer RNA levels, we conducted a microarray analysis for comparing RNA levels on samples from SW48P cells and cells with acquired resistance to cetuximab (Figure 3A). The results are shown in Table S1 as SW48 Resistance (SW48R)/SW48P fold-change of gene expression. EFNB3, which is known to be associated with cancer, was selected as a target [35]. We confirmed that the expression of EPHB3, the receptor of EFNB3, increased as the EFNB3 protein level increased. Based on the results shown in Figure 1D-G, we confirmed that the expression of the EphB3 receptor increased in the SW48 resistant cells as the EFNB3 protein level increased (Figure 3B). We confirmed the cell viability of both cells types in response to treatment with the EFNB3 protein (Figure 3C). The EFNB3- -treated group and SW48R cells (Figure 1A) showed similar viabilities. Therefore, EPHB3 expression is increased by EFNB3 when cetuximab resistance is acquired.
Ephrin-EPHB3 receptor signaling is upregulated in cetuximab-resistant cells. (A) Schematic representation of the proposed model related to cetuximab-induced EPHB3 and hedgehog activation. (B) Characterization of the expression of EPHB3 family members in EFNB3-treated SW48 parent cells. Proteins were collected and fractionated by SDS-PAGE followed by immunoblotting for the indicated proteins. β-actin was used as a loading control. (C) SW48 cells were treated with increasing concentrations of cetuximab (5, 10, 15, and 20 μg/mL) for 24 h and 48 h, with EFNB3, after overnight 2% FBS starvation. Cell viability was determined by the WST-1 assay. (D) SW48 and SW48R cells were treated with increasing concentrations of cetuximab (5, 10, and 20 μg/mL) for 24 h with EGF after overnight 2% FBS starvation. Stimulation was with EGF (10 ng/mL) for 30 min. Proteins were collected and fractionated by SDS-PAGE followed by immunoblotting for the indicated proteins. β-actin was used as a loading control. Data are expressed as the means of three independent experiments. (E) The immunofluorescence of EPHB3 was detected by confocal laser-scanning microscopy (original magnification, 40×). Scale bar: 10 µm. (F) Cetuximab treatment in cetuximab-sensitive cell line (KRAS wild-type (WT)) and cetuximab-resistant cell line (KRAS mutant-type (MT)) for 5 months. Proteins were collected and fractionated by SDS-PAGE, followed by immunoblotting for the indicated proteins. β-actin was used as a loading control. Data are expressed as the means of three independent experiments. (G) SW48 cells were treated with various anti-cancer drugs (cetuximab (EGFR), AZD4547 (FGFR), imatinib (PDGFR), and bevacizumab (VEGFR)) for 3 months. Proteins were collected and fractionated by SDS‑PAGE followed by immunoblotting for the indicated proteins. β-actin was used as a loading control. Data are expressed as the means of three independent experiments. FBS: fetal bovine serum. *P < 0.05.
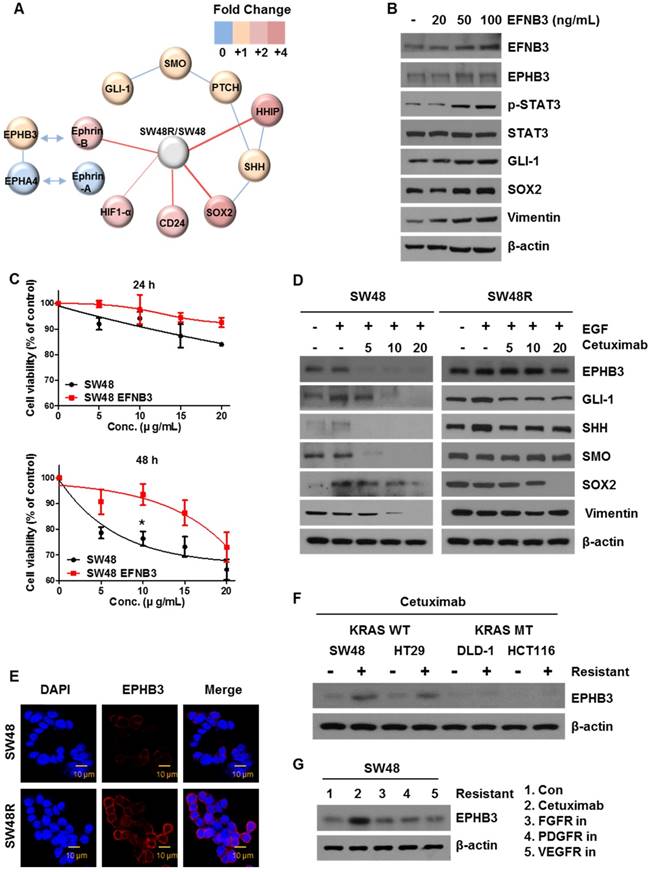
To explore the effect of EPHB3, we examined the protein levels of both cell types with or without cetuximab. We confirmed that key proteins (EPHB3, GLI-1, SOX2, Vimentin) were significantly decreased in SW48P cells compared with SW48R cells (Figure 3D), and cetuximab significantly reduced the levels of these proteins in the SW48P cells. Similar results were obtained for HT29 cells, with increased expression of EPHB3 upon resistance to cetuximab. (Figure 1G and Supplementary Figure S2G). This suggested that the EPHB3 protein specifically reacts to cetuximab. In agreement with these results, immunofluorescence studies demonstrated that EPHB3 levels were significantly increased in SW48R cells, as compared with SW48P cells (Figure 3E). Cetuximab is effective in CRC patients that have wild‑type KRAS. We compared wild-type KRAS with mutant-type KRAS cell lines to confirm EPHB3 protein expression. EPHB3 levels were increased when cetuximab resistance was acquired only in wild-type KRAS cell lines (Figure 3F). In addition, we investigated the protein levels of EPHB3 in cells resistant to antibodies for another receptor (not cetuximab). The increase in EPHB3 was only dependent on cetuximab resistance (Figure 3G). Before acquiring the inhibitor resistance of each receptor, EPHB3 protein expression in cells treated with various inhibitors was reduced by cetuximab in the SW48P cells (Supplementary Figure S2F).
STAT3 reduction through EPHB3 inhibition overcomes cetuximab resistance
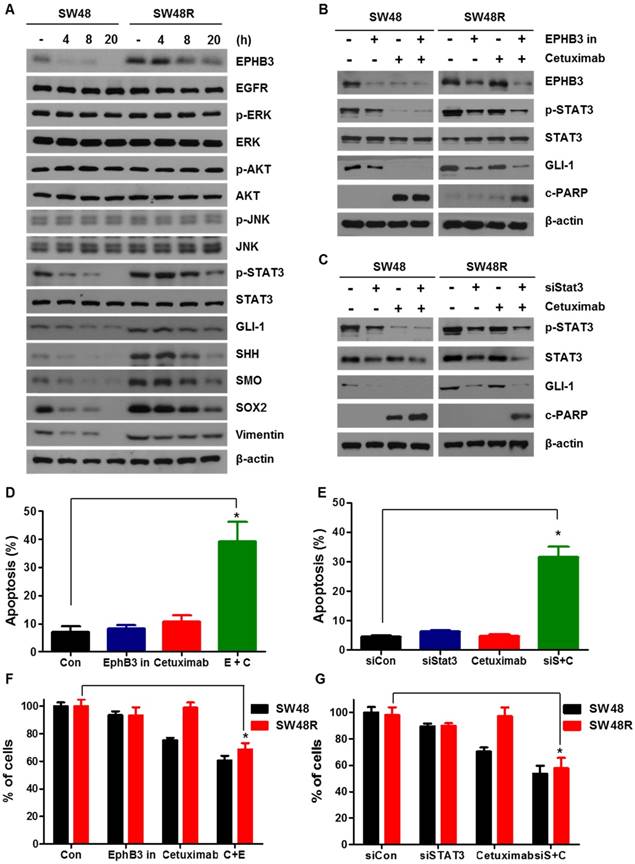
To study the different effects of EPHB3 in SW48P cells and SW48 cetuximab-resistant cells, we assessed the levels of STAT3 in both cell types, with or without EPHB3 (Figure 4A and Supplementary Figure S3D). Results showed that the levels of p-STAT3, GLI-1, SOX2 and Vimentin in SW48 resistant cells were decreased by EPHB3. We showed that cetuximab and the inhibition of EPHB3 in SW48R cells affects p-STAT3 and GLI-1 and induces apoptosis by increasing c-PARP. In addition, cetuximab and the inhibition of p-STAT3 modulated GLI-1 and increased c-PARP levels. These results suggest that EPHB3 affects EGFR and modulates EGFR down-signaling of p-STAT3 and GLI-1 (Figure 4B and 4C). Next, using GANT61, we confirmed that GLI-1 is the key gene in our hypothesis. The combination of cetuximab and GLI-1 inhibition in SW48R cells was shown to increase c-PARP (Supplementary Figure S2H). We also confirmed by flow cytometry that EPHB3 inhibition with siSTAT3 in combination with cetuximab increased apoptosis in SW48R cells (Figures 4D and 4E). This indicated that cetuximab resistance had been overcome. The same results were obtained using shEPHB3 (Supplementary figures S3E and S3F). The efficiency of the transfection with shEPHB3 is shown in Supplementary Figure S3G. We assessed cell viability in response to a combination of cetuximab with the EPHB3 inhibitor and a siRNA targeting STAT3 (siSTAT3) (Figure 4F and 4G). EPHB3 inhibition using siSTAT3 in SW48R cells decreased cell viability in combination with cetuximab.
Blockade of EPHB3 could effectively inhibit the proliferation and induction of apoptosis of SW48R cells. (A) The levels of EPHB3 signaling, hedgehog signaling, and cell growth signaling were examined using western blotting after treatment with an EPHB3 inhibitor (20 μM) for the indicated times. Total cell protein extracts were subjected to immunoblotting with the indicated antibodies, as described in the Materials and Methods. β-actin was used as a loading control. (B) Combinatorial treatment with cetuximab and the EPHB3 inhibitor led to loss of EPHB3 expression in SW48 and SW48R cells. The levels of c-PARP, STAT3, p- STAT3 and GLI-1 were detected by western blotting. (C) Knockdown of STAT3 cells treated with cetuximab led to loss of STAT3 expression in SW48 and SW48R cells. The levels of c-PARP, STAT3, p- STAT3 and GLI-1 were detected by western blotting. (D) SW48R cells treated with the EPHB3 inhibitor were stained with annexin V and propidium iodide (PI), and examined by FACS analysis. (E) SW48R cells transfected with control siRNA or STAT3 siRNA were stained with annexin V and PI, and examined by FACS analysis. (F) SW48 and SW48R cells were treated with cetuximab or the EPHB3 inhibitor for 24 h. The WST-1 assay was used to evaluate the effects of the EPHB3 inhibitor on proliferation. (G) SW48 and SW48R cells transfected with control or STAT3 siRNA were treated with cetuximab for 24 h. The WST-1 assay was used to evaluate the effects of STAT3 expression on proliferation. *P < 0.05. siRNA: short interfering RNA; PI: propidium iodide; FACS: fluorescence activated cell sorting.
Cetuximab resistance is caused by increased binding of EGFR and EPHB3
We observed an increase in EPHB3 levels in SW48R, but not in p-EPHB3. These results suggest that the increase in p-STAT3 expression is due to the activation of EGFR rather than activation of EPHB3 (Supplementary figures S3A and S3B). To study how EPHB3 affects EGFR and STAT3 in CRC, we first confirmed that there is an interaction between EGFR and EPHB3. We confirmed the possibility of indirect binding of EGFR and EPHB3 through structural modeling and found that though they are both large in size, they can structurally bind (Figure 5A). Co-immunoprecipitation and immunofluorescence confirmed the increase in interaction and co-localization caused by the binding of EGFR and EPHB3 in SW48R cells (Figure 5B-D), whereas EFNB3 and HHIP did not bind to EGFR (Supplementary Figure S3H and S3I). Next, we examined the effect of EGF using ELISA. The concentration of EGF in the supernatant was higher in SW48R cells than in SW48P cells, indicating that the binding of EGF to its receptor was low in SW48R cells (Figure 5E). Thus, we assumed that EGFR was affected by its binding to EPHB3. Finally, western blotting confirmed activation only of the p-EGFR (Y1068) site, known as a STAT3-activating site, when resistance to cetuximab was obtained [36, 37]. Also, p-EGFR (Y1068) reacted with EGF and its activity was reduced by the EPHB3 inhibitor (Figure 5F-G and Supplementary Figure S3C). In conclusion, the binding of EGFR to EPHB3 in SW48R is increased, resulting in the activation of the p-EGFR (Y1068) site, which in turn, results in increased phosphorylation of STAT3.
EGF treatment of SW48 cells renders these cells resistant to cetuximab, and EGF induces the EGFR-EPHB3 interaction. (A) EGFR domain (Red) and EPHB3 domain (green). (B) EGFR displayed increased association with EPHB3 in SW48R cells compared with SW48 cells. Cells were harvested and EGFR or EPHB3 were immunoprecipitated with anti-rabbit EGFR antibodies or anti-rabbit EPHB3 antibodies. The immunoprecipitated complexes were fractionated by SDS-PAGE and immunoblotting was performed on the indicated proteins. (C) SW48 and SW48R cells were immunostained for EGFR (green) or EPHB3 (red) and examined using confocal microscopy. Scale bar, 10 μm. (D) Quantification of the co-localization in fig C. Error bars represent the mean ± SEM from each cell (*P < 0.05). (E) Level of extracellular EGF protein expression by EPHB3 in SW48R cells compared with that in SW48 cells was measured using ELISA. Results are represented by the mean values of EGF concentrations, and error bars represent the SEM from three separate experiments. (F) Western blotting analysis of protein expression in SW48 and SW48R cells treated with EGF (10 ng/mL) for 30 min. The proteins were collected and fractionated by SDS-PAGE and immunoblotting was performed for the indicated proteins. (G) The protein levels of p-EGFR family members were evaluated using western blotting after treatment with the EPHB3 inhibitor (20 μM) at the indicated times. β-actin was used as a loading control. Data are expressed as the means of three independent experiments. ELISA: enzyme-linked immunosorbent assay.
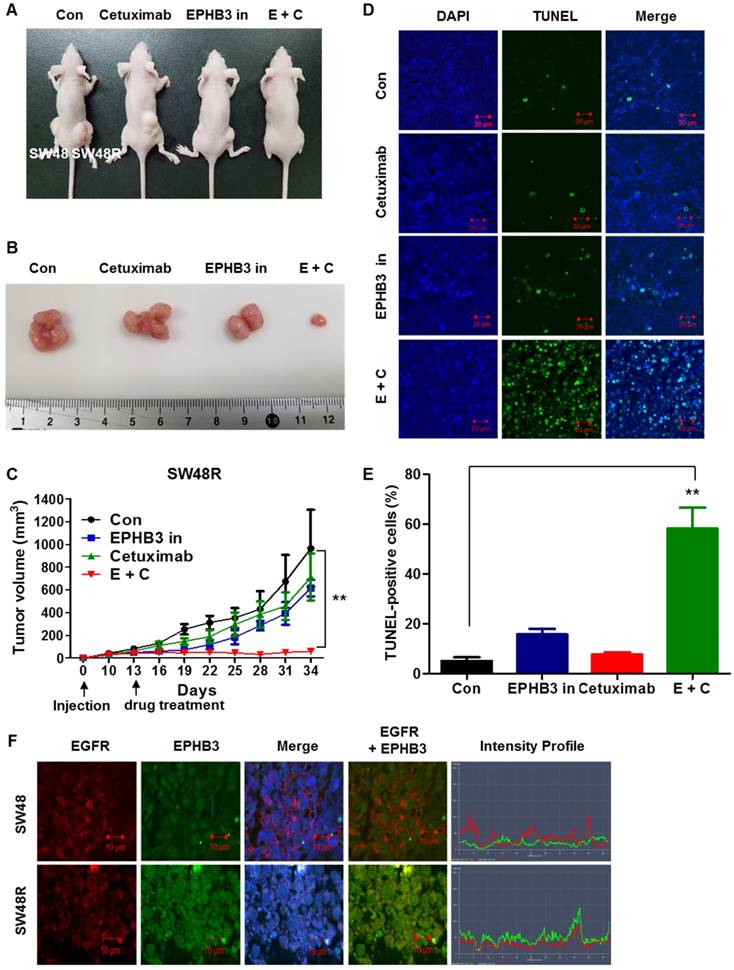
EPHB3 inhibits and overcomes cetuximab resistance in vivo
Finally, we investigated whether EPHB3 could inhibit colorectal tumorigenicity and resistance in vivo. SW48 and SW48R cells were subcutaneously injected into nude mice. After 3 weeks of treatment, well-formed tumors in the SW48R cetuximab and EPHB3 inhibitor combination group were much smaller than those in the control group (Figure 6A-C). A terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling (TUNEL) assay was performed to assess the apoptosis of SW48 and SW48R. The SW48R cetuximab and EPHB3 inhibitor combination group induced marked apoptosis, whereas only weakly positive staining could be observed in the control group (Figure 6D and 6E). Immunofluorescence staining was then performed to measure the expression of EGFR and EPHB3 in tumor tissues. Consistent with previous observations, we confirmed co-localization because of the increased binding of both factors in SW48R cells (Figure 6F). Taken together, these results suggest that EPHB3 could regulate cetuximab resistance in CRC in vivo.
Combination of cetuximab and EPHB3 inhibitor treatment of SW48R tumor cells leads to growth delay in vivo. (A and B) SW48R cells were implanted subcutaneously into nude mice, and then tumor growth was examined by measuring the tumor volume after 3 weeks of treatment with cetuximab (10 mg/kg), the EPHB3 inhibitor (0.1 mg/kg), or the combination of cetuximab and the EPHB3 inhibitor (every 2 days; n = 7). (C) Line graph showing the tumor volume (mm3) in SW48R cell tumor-bearing mice treated with PBS alone, cetuximab alone, the EPHB3 inhibitor alone, or a combination of cetuximab and the EPHB3 inhibitor, from day 0 to day 34. Error bars represent the mean ± SD from 5 mice. For statistical analysis, Student's t‐test (two‐sided, paired) was used. **P < 0.01. (D) Tumors were examined using the TUNEL assay, and DAPI was used to visualize the nuclei. (E) The percentage of TUNEL-positive cells was determined and plotted as a histogram. **P < 0.01. (F) The inhibition of EGFR and EPHB3 expression in SW48R tumors after combinatorial treatment is consistent with reduced proliferation and increased apoptosis. SW48R tumor samples after treatment with cetuximab, the EPHB3 inhibitor, or the combination of cetuximab and EPHB3 inhibitor in vivo were prepared and analyzed for EGFR and EPHB3 by Immunofluorescence staining. Images were measured by taking the average staining intensity quantified from three tumors per treatment group (three images/tumor, n = 7). Magnification 100×. PBS: phosphate buffered saline; TUNEL: terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling; DAPI: 4, 6-diamidino-2-phenylindole.
We then examined the expression of proteins (EPHB3, EFNB3, GLI-1, and p-STAT3) in both pre-cetuximab-treatment and cetuximab-resistant patient-derived samples, based on previous western blotting results. First, immunohistochemical analysis was performed to confirm EPHB3, EFNB3, GLI-1, and p-STAT3 expression. As shown in supplementary Figure S1A-D, EPHB3 was significantly highly expressed in cetuximab-resistant patient samples compared with pre-cetuximab treatment patient samples. Expression of EFNB3, GLI-1, and p-STAT3 was also higher in cetuximab-resistant samples compared with pre-cetuximab treatment samples, but no significance was noted. Collectively, these data show that EPHB3 affects cell sensitivity to cetuximab.
Discussion
Cetuximab plays a role by inhibiting EGF ligand binding to the extracellular domain of EGFR, thus blocking ligand-mediated EGFR signaling. Moreover, cetuximab elevates receptor internalization and degradation, and enhances antibody-dependent cell-mediated cytotoxicity. Cetuximab was the first FDA approved anti-EGFR antibody for CRC therapy, guided for combination with standard chemotherapy or radiotherapy in locally advanced, metastatic, and recurrent CRC [38, 39]. Despite promising advances in regional limit cure of CRC, further innovations of treatment are demanded. Despite the clinical success of cetuximab and panitumumab, the efficacy of these drugs is challenged by the development of resistance, which is achieved by activation of canonical and non-canonical signal transduction pathways [40]. There are many mechanisms of resistance to CRC treatment. Furthermore, CRC treatment via canonical and non-canonical Wnt signaling, crosstalk of pathways (e.g., NOTCH, mTOR, AKT/PI3K, NFκB) has also been reported to induce β-catenin activation. Therefore, in the present study, we focused on a microarray analysis and discovered previously unknown targets involved in overcoming intrinsic resistance to cetuximab.
The goals of this study were to identify whether resistance to cetuximab in CRC cells involved EPHB3 signaling, to disclose the molecular events underlying such changes, and to examine its role in tumor suppression. Finally, we used siRNAs and chemical inhibitors to validate the function of activated kinases in the proliferation of CRC cells and suggested several kinases that can overcome cetuximab resistance. Our results led us to suggest novel therapeutic targets against cetuximab resistance in regard to the kinome network derived from the results of phosphoproteomic analyses. We also found that alterations in cell behavior, morphology (Supplementary Figure S2A and S2B), and signaling molecules, resulting from EPHB3 signaling, contributed to both EMT and tumor suppression. Most importantly, we found that the EPHB3-ephrin-B interaction stimulated the junctional adhesion of E-Cadherin molecules, which are representative of tight junctions (Figure 2C).
EPH receptors constitute the largest subfamily of transmembrane TKRs, including 14 members of EPHAs and EPHBs [41]. Previous reports showed that EPHB3 is related to apoptotic signaling and acts as a tumor suppressor gene [17]. Zhang et al. found reduced expression of EPHB3 in human CRC tissues is related to poor survival and advanced recurrence [15], suggesting that EPHB3 might act as a tumor suppressor in CRC. However, we and another group found that EPHB3 has oncogenic functions in CRC patients treated with cetuximab [42]. In addition, recent studies have begun to report the interesting functions of EPH receptors in tumorigenesis. Overexpression of EPHs has been reported in numerous cancers, including breast, prostate, melanoma, and glioma [42-44]. For instance, EPHA2 is upregulated in breast cancer and activates tumor growth and invasion via increased Ras/MAPK signaling [45, 46]. However, due to the complicated functions of the EPH/ephrin system, recent reports show that the EPH/ephrin system may contribute as a tumor suppressor in certain cancer types and contexts [47]. A role in tumor suppression by the EPH/ephrin system is seen in CRC, in which EPHB signaling inhibits the transition of malignancy during CRC development by compartmentalizing the spread of cancer cells [48]. EPHB3 also interacts with integrin β1 to increase both tumor invasion and progress, raising the possibility that targeting EPHB3 may modulate integrin β1 signaling in CRC. Moreover, integrin β1 can trigger ligand-independent SRC-AKT signaling, inducing resistance to erlotinib in lung cancer [49]. However, the association of EPHB3 with integrin β1-SRC-AKT signaling, including its contribution to EGFR therapy resistance, remains uncertain in CRCs.
When activated by ephrin, the EPH receptor can transduce intracellular pathways that are associated with a variety of biological functions [50]. For instance, EPHB2 can promote cell proliferation via Abl-mediated increase in Cyclin-D1 expression and Src-mediated increase in STAT3 phosphorylation. In the present work, we found that EPHB3 can inhibit cancer cell growth in preclinical cancer models, including xenografts, in the absence of an EPHB3 inhibitor. Combination of an EPHB3 inhibitor and an EGFR inhibitor (cetuximab) led to increased anti-tumor effects in CRC cells. Furthermore, we confirmed the anti-tumor effects of EPHB3 inhibition using an EPHB3 inhibitor in mouse models as well as by resistance to cetuximab in tumor models that were not characterized by activating EGFR mutations (Figure 6A). Moreover, expression of EPHB3 in human CRC tissues that presented recurrence following cetuximab therapy demonstrated elevated EPHB3 levels compared with those from pretreatment CRC tissues. These observations suggest that inhibiting EPHB3 might improve the anti-tumor effects of EGFR inhibitors.
These findings reveal an important role for EPHB3 in cetuximab resistance of CRC cells. Our results showed that EPHB3 was a direct target of STAT3, and cell progression, including cell growth, invasion, cell cycle, and apoptosis, were regulated by increasing or decreasing EPHB3 expression. To explain whether the EPHB3 pathway was the main target in CRC in vivo, EPHB3 was suppressed by the EPHB3 inhibitor, which blocked tumor formation.
We also found increased expression of GLI-1 after EGFR suppression in EGFR-dependent SW48 cells. GLI-1 is a molecular factor for metastasis and cancer cell growth in various cancers [51]. In the present study, we discovered that “cross-talk” between EGFR and EPHB3 occurs through the HH/STAT3 cascade, but that HH inhibition might make cells more EGFR-dependent. However, the roles and underlying pathways of HH in drug-resistant cancer cells remain to be researched. EGFR and HH signaling synergize through the MAPK signaling in various tumor types [28, 52]. EGF stimulates the expression of GLI-1 and its target genes PTCH1and BCL2 in gastric cancer [53], and activates the HH ligand sonic hedgehog (SHH) pathway through PI3K and MAPK to increase the expression of HH specific targets [54]. Taken together, these results represent a powerful rationale for the development of combination therapy in xenograft models.
STAT3 is activated in various tumors [55]. STAT3 can be phosphorylated by the stimulation of upstream receptors and/or nonreceptor kinases including EGFR [56], IL-6/GP130 and Janus kinases (JAKs) [57], and SRC family kinases [58]. Continuous activation of STAT3 has been implicated in the development of resistance to conventional therapies in certain cancers. Additionally, overexpression of STAT3 has been observed in anticancer drug-resistant tumor cells [59]. For example, the activation of STAT3 has been reported in the tolerance to EGFR inhibitors in head and neck squamous cell carcinoma (HNSCC) and glioma [60, 61] and tolerance to EGFR inhibitors in the therapy of HNSCC patients was associated with increased expression of STAT3 [62]. These data suggest that activation of STAT3 induces tolerance to EGFR inhibitors in CRC, and a combination with STAT3 targeting may provide an effective therapeutic strategy.
Signaling of MEK/ERK and PI3K/AKT plays an important role in cetuximab resistance in CRC [63]. Our previous data and the present work found activation of ERK in HH-overexpressing gastric cancer cells [64]; however, this signaling was unchanged by manipulation of HH signaling in KRAS-WT CRC cells. Surprisingly, PI3K/AKT signaling can be activated in both KRAS-WT and KRAS-mutated CRC cells. The ERK signaling activated by SHH was responsible for PI3K/AKT activation in KRAS-mutated CRC cells. Our data demonstrated that targeting STAT3 using a STAT3 inhibitor in cetuximab-resistant cells sensitizes the cells to the EGFR inhibitor therapy.
We also performed an animal study using a combination of an EPHB3 inhibitor (LDN-211904) and cetuximab that could overcome cetuximab resistance and inhibit tumor growth (Supplementary Figure S4). Targeting the EPHB3 pathway has been shown to overcome cetuximab resistance in KRAS-WT CRC and our findings in cell and animal models showed that the EPHB3 pathway may be responsible for cetuximab resistance in CRC cells.
Conclusion
In summary, our results indicate that HH may play a role as an oncogene in cetuximab-resistant CRC cells by targeting EPHB3. We have provided a scheme of the working model of cetuximab resistance-induced activation of EPHB3 and hedgehog signaling via a graphic abstract. Collectively, our results using cells, mice, and patients strongly suggest the hypothesis that STAT3 could serve as a reliable biomarker for drug resistance, tumor recurrence, and survival prediction in CRC. Furthermore, the combination of an EPHB3 inhibitor with cetuximab could be effective in inhibiting STAT3-activated CSC stemness and cetuximab resistance in CRC.
Abbreviations
CRC: colorectal cancer; SMO: smoothened; GLI-1: Glioma-Associated Oncogene Homolog 1; PARP: poly (ADP-ribose) polymerase; PBS: phosphate-buffered saline solution; PUMA: p53 upregulated modulator of apoptosis; MDM: Mouse double minute 2 homolog; PI: propidium iodide; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; PMSF: phenylmethylsulfonyl fluoride; TBS: Tris-buffered saline; TUNEL: terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling; DAPI: 4,6-diamidino-2-phenylindole; ELISA: enzyme-linked immunosorbent assay; FBS: fetal bovine serum; EMT: epithelial-mesenchymal transition; siRNA: short interfering RNA; HRP: horseradish peroxidase; RIPA: radioimmunoprecipitation assay; TBS: Tris-buffered saline; APC: allophycocyanin; FITC: fluorescein isothiocyanate; PMSF: phenylmethylsulfonyl fluoride; GSEA: gene set enrichment analysis; GO: gene ontology; NES: normalized enrichment score; FDR: false discovery rate; HNSCC: head and neck squamous cell carcinoma.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
SW48 cetuximab-resistant cells were kindly provided by the MOGAM Institute. We thank MOGAM Institute for its support of the cell line. This research is based on data used in Seong Hye Park's doctoral dissertation (Korea University).
Funding
This work was supported by grant from the National Research Foundation (NRF) of Korea and funded by the Korean government (MSIP) (NRF-2017R1A2B2011684, NRF-2018M3A9G1075561).
Availability of data and materials
The data sets supporting the conclusions of this article are included within this article and the Supplementary Data.
Ethics approval and consent to participate
All experiments were approved by the Ethics Committee of Korea University.
Contributions
The study funders had no role in the design of the study; the collection, analysis, or interpretation of the data; the writing of the manuscript; or the decision to submit the manuscript for publication.
SHP conceived and designed the study, collected and assembled the data, analyzed and interpreted the data, and wrote the manuscript. MJJ and YAJ provided study materials. YJN, HKY, DYK, and BGK conceived and designed the study and analyzed and interpreted the data. SHK and BRK conceived and designed the study. JLK and SYJ collected and assembled the data, and analyzed and interpreted the data. SCO and DHL conceived and designed the study, provided financial support, collected and assembled the data, analyzed and interpreted the data, wrote the manuscript, and provided final approval of the manuscript. All authors discussed the results and commented on the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Abrams TA, Meyer G, Schrag D, Meyerhardt JA, Moloney J, Fuchs CS. Chemotherapy usage patterns in a US-wide cohort of patients with metastatic colorectal cancer. J Natl Cancer Inst. 2014;106:djt371
2. Migliardi G, Sassi F, Torti D, Galimi F, Zanella ER, Buscarino M. et al. Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin Cancer Res. 2012;18:2515-25
3. Liu G, Tu D, Lewis M, Cheng D, Sullivan LA, Chen Z. et al. Fc-gamma Receptor Polymorphisms, Cetuximab Therapy, and Survival in the NCIC CTG CO.17 Trial of Colorectal Cancer. Clin Cancer Res. 2016;22:2435-44
4. Segelov E, Thavaneswaran S, Waring PM, Desai J, Robledo KP, Gebski VJ. et al. Response to Cetuximab With or Without Irinotecan in Patients With Refractory Metastatic Colorectal Cancer Harboring the KRAS G13D Mutation: Australasian Gastro-Intestinal Trials Group ICECREAM Study. J Clin Oncol. 2016;34:2258-64
5. Saridaki Z, Weidhaas JB, Lenz HJ, Laurent-Puig P, Jacobs B, De Schutter J. et al. A let-7 microRNA-binding site polymorphism in KRAS predicts improved outcome in patients with metastatic colorectal cancer treated with salvage cetuximab/panitumumab monotherapy. Clin Cancer Res. 2014;20:4499-510
6. Wasan H, Meade AM, Adams R, Wilson R, Pugh C, Fisher D. et al. Intermittent chemotherapy plus either intermittent or continuous cetuximab for first-line treatment of patients with KRAS wild-type advanced colorectal cancer (COIN-B): a randomised phase 2 trial. Lancet Oncol. 2014;15:631-9
7. Hobor S, Van Emburgh BO, Crowley E, Misale S, Di Nicolantonio F, Bardelli A. TGFalpha and amphiregulin paracrine network promotes resistance to EGFR blockade in colorectal cancer cells. Clin Cancer Res. 2014;20:6429-38
8. Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301-11
9. Xu H, Yu Y, Marciniak D, Rishi AK, Sarkar FH, Kucuk O. et al. Epidermal growth factor receptor (EGFR)-related protein inhibits multiple members of the EGFR family in colon and breast cancer cells. Mol Cancer Ther. 2005;4:435-42
10. Troiani T, Martinelli E, Napolitano S, Vitagliano D, Ciuffreda LP, Costantino S. et al. Increased TGF-alpha as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res. 2013;19:6751-65
11. Loupakis F, Cremolini C, Fioravanti A, Orlandi P, Salvatore L, Masi G. et al. EGFR ligands as pharmacodynamic biomarkers in metastatic colorectal cancer patients treated with cetuximab and irinotecan. Target Oncol. 2014;9:205-14
12. Nukaga S, Yasuda H, Tsuchihara K, Hamamoto J, Masuzawa K, Kawada I. et al. Amplification of EGFR Wild-Type Alleles in Non-Small Cell Lung Cancer Cells Confers Acquired Resistance to Mutation-Selective EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2017;77:2078-89
13. Francis DM, Huang S, Armstrong EA, Werner LR, Hullett C, Li C. et al. Pan-HER Inhibitor Augments Radiation Response in Human Lung and Head and Neck Cancer Models. Clin Cancer Res. 2016;22:633-43
14. Yao YM, Donoho GP, Iversen PW, Zhang Y, Van Horn RD, Forest A. et al. Mouse PDX Trial Suggests Synergy of Concurrent Inhibition of RAF and EGFR in Colorectal Cancer with BRAF or KRAS Mutations. Clin Cancer Res. 2017;23:5547-60
15. Zhang G, Liu X, Li Y, Wang Y, Liang H, Li K. et al. EphB3-targeted regulation of miR-149 in the migration and invasion of human colonic carcinoma HCT116 and SW620 cells. Cancer Sci. 2017;108:408-18
16. Murphy M, Chatterjee SS, Jain S, Katari M, DasGupta R. TCF7L1 Modulates Colorectal Cancer Growth by Inhibiting Expression of the Tumor-Suppressor Gene EPHB3. Sci Rep. 2016;6:28299
17. Jagle S, Ronsch K, Timme S, Andrlova H, Bertrand M, Jager M. et al. Silencing of the EPHB3 tumor-suppressor gene in human colorectal cancer through decommissioning of a transcriptional enhancer. Proc Natl Acad Sci U S A. 2014;111:4886-91
18. Ji XD, Li G, Feng YX, Zhao JS, Li JJ, Sun ZJ. et al. EphB3 is overexpressed in non-small-cell lung cancer and promotes tumor metastasis by enhancing cell survival and migration. Cancer Res. 2011;71:1156-66
19. Holmberg J, Genander M, Halford MM, Anneren C, Sondell M, Chumley MJ. et al. EphB receptors coordinate migration and proliferation in the intestinal stem cell niche. Cell. 2006;125:1151-63
20. Li G, Ji XD, Gao H, Zhao JS, Xu JF, Sun ZJ. et al. EphB3 suppresses non-small-cell lung cancer metastasis via a PP2A/RACK1/Akt signalling complex. Nat Commun. 2012;3:667
21. Chiu ST, Chang KJ, Ting CH, Shen HC, Li H, Hsieh FJ. Over-expression of EphB3 enhances cell-cell contacts and suppresses tumor growth in HT-29 human colon cancer cells. Carcinogenesis. 2009;30:1475-86
22. Liebig H, Gunther G, Kolb M, Mozet C, Boehm A, Dietz A. et al. Reduced proliferation and colony formation of head and neck squamous cell carcinoma (HNSCC) after dual targeting of EGFR and hedgehog pathways. Cancer Chemother Pharmacol. 2017;79:411-20
23. Frohlich H, Bahamondez G, Gotschel F, Korf U. Dynamic Bayesian Network Modeling of the Interplay between EGFR and Hedgehog Signaling. PLoS One. 2015;10:e0142646
24. Della Corte CM, Bellevicine C, Vicidomini G, Vitagliano D, Malapelle U, Accardo M. et al. SMO Gene Amplification and Activation of the Hedgehog Pathway as Novel Mechanisms of Resistance to Anti-Epidermal Growth Factor Receptor Drugs in Human Lung Cancer. Clin Cancer Res. 2015;21:4686-97
25. Qin CF, Hao K, Tian XD, Xie XH, Yang YM. Combined effects of EGFR and Hedgehog signaling pathway inhibition on the proliferation and apoptosis of pancreatic cancer cells. Oncol Rep. 2012;28:519-26
26. Guo Y, Zhang K, Cheng C, Ji Z, Wang X, Wang M. et al. Numb-/low Enriches a Castration-Resistant Prostate Cancer Cell Subpopulation Associated with Enhanced Notch and Hedgehog Signaling. Clin Cancer Res. 2017;23:6744-56
27. Zhou M, Hou Y, Yang G, Zhang H, Tu G, Du YE. et al. LncRNA-Hh Strengthen Cancer Stem Cells Generation in Twist-Positive Breast Cancer via Activation of Hedgehog Signaling Pathway. Stem Cells. 2016;34:55-66
28. Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C. et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009;69:1284-92
29. Seto M, Ohta M, Asaoka Y, Ikenoue T, Tada M, Miyabayashi K. et al. Regulation of the hedgehog signaling by the mitogen-activated protein kinase cascade in gastric cancer. Mol Carcinog. 2009;48:703-12
30. Zhou J, Zhu G, Huang J, Li L, Du Y, Gao Y. et al. Non-canonical GLI1/2 activation by PI3K/AKT signaling in renal cell carcinoma: A novel potential therapeutic target. Cancer Lett. 2016;370:313-23
31. Dormoy V, Danilin S, Lindner V, Thomas L, Rothhut S, Coquard C. et al. The sonic hedgehog signaling pathway is reactivated in human renal cell carcinoma and plays orchestral role in tumor growth. Mol Cancer. 2009;8:123
32. Lim Y, Yoo J, Kim M-S, Hur M, Lee EH, Hur H-S. et al. GC1118, an anti-EGFR antibody with a distinct binding epitope and superior inhibitory activity against high-affinity EGFR ligands. Molecular cancer therapeutics. 2016;15:251-63
33. Lee S-y, Na YJ, Jeong YA, Kim JL, Oh SC, Lee D-H. Upregulation of EphB3 in gastric cancer with acquired resistance to a FGFR inhibitor. The international journal of biochemistry & cell biology. 2018;102:128-37
34. Dong W, Cui J, Tian X, He L, Wang Z, Zhang P. et al. Aberrant sonic hedgehog signaling pathway and STAT3 activation in papillary thyroid cancer. Int J Clin Exp Med. 2014;7:1786-93
35. Nakada M, Drake KL, Nakada S, Niska JA, Berens ME. Ephrin-B3 ligand promotes glioma invasion through activation of Rac1. Cancer Res. 2006;66:8492-500
36. Apicella M, Migliore C, Capeloa T, Menegon S, Cargnelutti M, Degiuli M. et al. Dual MET/EGFR therapy leads to complete response and resistance prevention in a MET-amplified gastroesophageal xenopatient cohort. Oncogene. 2017;36:1200-10
37. Katsila T, Juliachs M, Gregori J, Macarulla T, Villarreal L, Bardelli A. et al. Circulating pEGFR is a candidate response biomarker of cetuximab therapy in colorectal cancer. Clin Cancer Res. 2014;20:6346-56
38. Saltz LB, Meropol NJ, Loehrer PJ Sr, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201-8
39. Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A. et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337-45
40. Giampieri R, Scartozzi M, Del Prete M, Maccaroni E, Bittoni A, Faloppi L. et al. Molecular biomarkers of resistance to anti-EGFR treatment in metastatic colorectal cancer, from classical to innovation. Crit Rev Oncol Hematol. 2013;88:272-83
41. Shiuan E, Chen J. Eph Receptor Tyrosine Kinases in Tumor Immunity. Cancer Res. 2016;76:6452-7
42. Dominguez-Brauer C, Hao Z, Elia AJ, Fortin JM, Nechanitzky R, Brauer PM. et al. Mule Regulates the Intestinal Stem Cell Niche via the Wnt Pathway and Targets EphB3 for Proteasomal and Lysosomal Degradation. Cell Stem Cell. 2016;19:205-16
43. Astin JW, Batson J, Kadir S, Charlet J, Persad RA, Gillatt D. et al. Competition amongst Eph receptors regulates contact inhibition of locomotion and invasiveness in prostate cancer cells. Nat Cell Biol. 2010;12:1194-204
44. Amero P, Esposito CL, Rienzo A, Moscato F, Catuogno S, de Franciscis V. Identification of an Interfering Ligand Aptamer for EphB2/3 Receptors. Nucleic Acid Ther. 2016;26:102-10
45. Dunne PD, Dasgupta S, Blayney JK, McArt DG, Redmond KL, Weir JA. et al. EphA2 Expression Is a Key Driver of Migration and Invasion and a Poor Prognostic Marker in Colorectal Cancer. Clin Cancer Res. 2016;22:230-42
46. Brantley-Sieders DM, Caughron J, Hicks D, Pozzi A, Ruiz JC, Chen J. EphA2 receptor tyrosine kinase regulates endothelial cell migration and vascular assembly through phosphoinositide 3-kinase-mediated Rac1 GTPase activation. J Cell Sci. 2004;117:2037-49
47. Husa AM, Magic Z, Larsson M, Fornander T, Perez-Tenorio G. EPH/ephrin profile and EPHB2 expression predicts patient survival in breast cancer. Oncotarget. 2016;7:21362-80
48. Mathot L, Kundu S, Ljungstrom V, Svedlund J, Moens L, Adlerteg T. et al. Somatic Ephrin Receptor Mutations Are Associated with Metastasis in Primary Colorectal Cancer. Cancer Res. 2017;77:1730-40
49. Kanda R, Kawahara A, Watari K, Murakami Y, Sonoda K, Maeda M. et al. Erlotinib resistance in lung cancer cells mediated by integrin beta1/Src/Akt-driven bypass signaling. Cancer Res. 2013;73:6243-53
50. Marquardt T, Shirasaki R, Ghosh S, Andrews SE, Carter N, Hunter T. et al. Coexpressed EphA receptors and ephrin-A ligands mediate opposing actions on growth cone navigation from distinct membrane domains. Cell. 2005;121:127-39
51. Khatra H, Bose C, Sinha S. Discovery of Hedgehog Antagonists for Cancer Therapy. Curr Med Chem. 2017;24:2033-58
52. Keysar SB, Le PN, Anderson RT, Morton JJ, Bowles DW, Paylor JJ. et al. Hedgehog signaling alters reliance on EGF receptor signaling and mediates anti-EGFR therapeutic resistance in head and neck cancer. Cancer Res. 2013;73:3381-92
53. Kasper M, Schnidar H, Neill GW, Hanneder M, Klingler S, Blaas L. et al. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol Cell Biol. 2006;26:6283-98
54. Elia D, Madhala D, Ardon E, Reshef R, Halevy O. Sonic hedgehog promotes proliferation and differentiation of adult muscle cells: Involvement of MAPK/ERK and PI3K/Akt pathways. Biochim Biophys Acta. 2007;1773:1438-46
55. Turkson J, Jove R. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene. 2000;19:6613-26
56. Wu J, Liu W, Williams JP, Ratner N. EGFR-Stat3 signalling in nerve glial cells modifies neurofibroma initiation. Oncogene. 2017;36:1669-77
57. Wu X, Cao Y, Xiao H, Li C, Lin J. Bazedoxifene as a Novel GP130 Inhibitor for Pancreatic Cancer Therapy. Mol Cancer Ther. 2016;15:2609-19
58. Resetca D, Haftchenary S, Gunning PT, Wilson DJ. Changes in signal transducer and activator of transcription 3 (STAT3) dynamics induced by complexation with pharmacological inhibitors of Src homology 2 (SH2) domain dimerization. J Biol Chem. 2014;289:32538-47
59. Kuo WY, Hwu L, Wu CY, Lee JS, Chang CW, Liu RS. STAT3/NF-kappaB-Regulated Lentiviral TK/GCV Suicide Gene Therapy for Cisplatin-Resistant Triple-Negative Breast Cancer. Theranostics. 2017;7:647-63
60. Lo HW, Cao X, Zhu H, Ali-Osman F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin Cancer Res. 2008;14:6042-54
61. Sriuranpong V, Park JI, Amornphimoltham P, Patel V, Nelkin BD, Gutkind JS. Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res. 2003;63:2948-56
62. Tanizaki J, Okamoto I, Okabe T, Sakai K, Tanaka K, Hayashi H. et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clin Cancer Res. 2012;18:6219-26
63. Brand TM, Iida M, Wheeler DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther. 2011;11:777-92
64. Yoo YA, Kang MH, Lee HJ, Kim BH, Park JK, Kim HK. et al. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res. 2011;71:7061-70
Author contact
![]() Corresponding authors: Sang Cheul Oh, MD, PhD, Division of Oncology/Hematology, Department of Internal Medicine, Korea University Guro, Hospital, 148 Gurodong-gil, Guro-gu, Seoul 152-703, Korea; Phone: +82 2 2626 3060; E-mail: sachohac.kr. Dae-Hee Lee, PhD, Division of Oncology/Hematology, Department of Internal Medicine, Korea University Guro, Hospital, 148 Gurodong-gil, Guro-gu, Seoul 152-703, Korea; Phone: +82 2 2626 1998; E-mail: neogeneac.kr
Corresponding authors: Sang Cheul Oh, MD, PhD, Division of Oncology/Hematology, Department of Internal Medicine, Korea University Guro, Hospital, 148 Gurodong-gil, Guro-gu, Seoul 152-703, Korea; Phone: +82 2 2626 3060; E-mail: sachohac.kr. Dae-Hee Lee, PhD, Division of Oncology/Hematology, Department of Internal Medicine, Korea University Guro, Hospital, 148 Gurodong-gil, Guro-gu, Seoul 152-703, Korea; Phone: +82 2 2626 1998; E-mail: neogeneac.kr