Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2019; 9(8):2380-2394. doi:10.7150/thno.29724 This issue Cite
Research Paper
SIRT6, a novel direct transcriptional target of FoxO3a, mediates colon cancer therapy
Yingjie Zhang1,2,3,4, Liming Nie5, Keqian Xu1,2, Yang Fu6, Juchang Zhong3, Kongzhen Gu1,2, Lingling Zhang1,2 ![]()
1. Department of Laboratory Medicine, The Third Xiangya Hospital, Central South University, Changsha 410013, P.R. China
2. Department of Laboratory Medicine, Xiangya School of Medicine, Central South University, Changsha 410013, P.R. China
3. College of Biology, Hunan University, Changsha 410082, P.R. China
4. Shenzhen Institute, Hunan University, Shenzhen 518000, P.R. China
5. State Key Laboratory of Molecular Vaccinology and Molecular Diagnosis & Center for Molecular Imaging and Translational Medicine, School of Public Health, Xiamen University, Xiamen 361102, P.R. China
6. Department of Surgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, P.R. China
Received 2018-9-5; Accepted 2019-2-25; Published 2019-4-13
Abstract

SIRT6, NAD+-dependent deacetylase sirtuin 6, has recently shown to suppress tumor growth in several types of cancer. Colon cancer is a challenging carcinoma associated with high morbidity and death. However, whether SIRT6 play a direct role in colon tumorigenesis and the underlying mechanism are not understood.
Methods: To investigate the role of SIRT6 in colon cancer, we firstly analyzed the specimens from 50 colorectal cancer (CRC) patients. We generated shSIRT6 LoVo cells and xenograft mouse to reveal the essential role of SIRT6 in cell apoptosis and tumor growth. To explore the underlying mechanism of SIRT6 regulation, we performed FRET and real-time fluorescence imaging in living cells, real-time PCR, immunoprecipitaion, immunohistochemistry, flow cytometry and luciferase reporter assay.
Results: The expression level of SIRT6 in patients' specimens is lower than that of normal controls, and patients with higher SIRT6 level have a better prognosis. Here, we identified that transcriptional factor FoxO3a is a direct up-stream of SIRT6 and positively regulated SIRT6 expression, which in turn, promotes apoptosis by activating Bax and mitochondrial pathway. Functional studies reveal that Akt inactivation increases FoxO3a activity and augment its binding to SIRT6 promoter, leading to elevated SIRT6 expression. Knocking down SIRT6 abolished apoptotic responses and conferred resistance to the treatment of BKM120. Combinational therapies with conventional drugs showed synergistic chemosensitization, which was SIRT6-dependent both in vitro and in vivo.
Conclusion: The results uncover SIRT6 as a new potential biomarker for colon cancer, and its unappreciated mechanism about transcription and expression via Akt/FoxO3a pathway.
Keywords: SIRT6, FoxO3a, Akt, Colon cancer, Apoptosis
Introduction
SIRT6 belongs to the evolutionarily conserved histone deacetylases sirtuin family. It is widely expressed in mammalian cells and plays crucial roles in various biological processes, including metabolism, aging, DNA stability and repair, proliferation and differentiation [1-3]. SIRT6 is recently shown to be a tumor suppressor for evident in many areas [2, 4, 5]. Reduced SIRT6 level has been observed in various human cancers, such as liver, colon, ovarian cancer and hepatocellular carcinoma [2, 6, 7]. Decreased expression of SIRT6 was detected in the early stage of human colon cancer, which maintained during tumor progression [2], indicating the potential role of SIRT6 in both initiation and development of colon cancer.
Using DNA microarrays and Ingenuity Pathway Analysis (IPA), our previous studies show that except for PUMA [8-10], SIRT6 is another pro-apoptotic protein that decreased a lot in colon cancer (data not shown). Furthermore, relative low level of SIRT6 was detected in the tissue of colon cancer compared to that of normal colon from total 50 patients. Several studies have shown that the effect of SIRT6 on tumor suppression is probably mediated by the regulation of metabolism [3, 4]. While recent evidence give us some new insights that SIRT6 serves as a tumor suppressor by inhibiting proliferation and promoting apoptosis in a variety of cancer cells, such as breast cancer, cervical carcinoma and fibrosarcoma cells [11-14]. Therefore, how does SIRT6 exert its anti-tumor effects and whether SIRT6 could induce apoptosis are uncertain.
More importantly, the reason of SIRT6 heterogeneous expression in normal and colon cancer tissue, and the mechanism of SIRT6 regulation are poorly understood. There exist different factors that involved in SIRT6 regulation. RUNX2 repressed the expression of SIRT6 in MCF7 cells [15]. E2F1 bound to SIRT6 promoter directly to down-regulate its transcription, in prostate and bladder cancer cells [16]. SIRT6 expression was mediated by c-Fos in mouse model of liver cancer [17]. Importantly, recent studies reported a mechanism involving Akt signaling dependent SIRT6 regulation. Study in breast cancer cells showed that activated Akt bound to SIRT6 directly, leading the ubiquitin-mediated degradation of SIRT6 [18]. Besides, the transcription factors FoxO3a, a direct downstream and negative responsor of Akt, is also shown to be involved in SIRT6 regulation in liver cells [19]. However, the exact regulation mechanism behind is unknown.
Colon cancer is a challenging carcinoma associated with high morbidity and death [20, 21]. It has been known that intracellular PI3K/Akt signaling is essential in many cancers, including colon cancer [9, 22-24]. Death from colon cancer is frequently due to metastases that are resistant to the conventional drugs [25]. Our former study demonstrated that 5-FU-mediated Akt activation may be the potential mechanism for drug resistance [8]. So inhibition of this pathway has been an attractive therapy due to its high activation in cancers. BKM120, a novel oral pan PI3K inhibitor, targets all of the PI3K isoforms and potently inhibits Akt activity [26]. It exerts anti-tumor effects both in several cancer cell models with PI3KCA mutations and in xenograft models [27, 28]. BKM120 is well tolerated in Phase I clinical trials of several solid tumors [29].
In this study, we analyzed, for the first time, the regulation mechanism of SIRT6 in colon cancer, and we found that FoxO3a directly binds to SIRT6 promoter and activates its transcription, following dephosphorylated by inactivated Akt. Therefore, inhibiting Akt by BKM120 induced apoptosis in several colon cancer cell lines, which was dependent on SIRT6 activation. Furthermore, BKM120 synergized with Cisplatin or Regorafenib, the common chemotherapeutic agent of colon cancer, and exerted potent efficacy through the Akt/FoxO3a/SIRT6 cascade in vitro and in vivo. Together, our study uncovered a novel cross-talk between FoxO3a and SIRT6 and identified SIRT6 as a new potential biomarker in colon cancer, which may provide novel insights into colon cancer therapy.
Methods
Cell lines and tissue treatment
Human colorectal cancer cell lines (LoVo, HCT-116, SW48, HT-29, DLD1, SW480) were ordered from ATCC. HCT-116 p53-/- cell was a gift from Dr Bert Vogelstein (Johns Hopkins University). Cells were cultured in McCoy's5A modified media (InvitrogenTM), containing 10% FBS in 37 °C incubator with 5% CO2. For the cell treatment, various concentrations of BKM120 (0, 0.5, 1, 2.5, or 5μM) were mixed into the culture medium before analysis.
Surgical specimens of colon cancer were divided into two groups: 1) Specimens were transferred after excision into DMEM with 20% FCS, which were then transported to the laboratory. After three times PBS washing, each fresh tumor was minced into pieces of approximately 10 mg. These tumor pieces were then cultured in the presence of drugs dissolved with DMEM medium containing 20% FCS at 37°C and 5% CO2. RNA was extract after incubation for 3 h in PCR analysis, and protein level was analysed by Western bloting after incubation for the indicated time. 2) Another group of specimens were immediately put in liquid nitrogen after excision for other analyses.
Real time imaging of caspase-3 cleavage
A genetic caspase-3 activity reporter SCAT3, which composes CFP (donor), YFP (acceptor Venus) and a sequence of caspase-3 cleavage (DEVD), was designed to detect the activation of caspase-3 [30]. When caspase-3 is activated, the DEVD sequence was cleaved, leading the decrease of FRET (reflected by Ratio=YFP (FRET)/CFP). That is, the descending FRET ratio means caspase-3 activation.
FRET was explored on the LSM510 confocal microscopy (Zeiss). CFP was excited by the 458 nm Ar-Ion Laser. Emission fluorescence was recorded by CFP channel (470-500 nm bandpass) as well as a YFP channel (530 nm longpass) [9].
Plasmids and adenoviral vector
Mammalian expression vectors, pcDNA3.1-GFP, Flag-SIRT1, Flag-FoxO3a, WT-Akt, Myr-Akt, HA-Akt DN (K179M), and SIRT6 were purchased from Addgene. The shAkt and shFoxO3a vectors were described previously [8, 9]. An empty vector was used as control. The shRNA reagents for SIRT1 and SIRT6 were purchased from Dharmacon.
The constitutively active FoxO3a (Ad-Tm-FoxO3a) is mutated at three phosphorylation sites, so that it cannot be phosphorylated. The dominant-negative FoxO3a (Ad-DN-FoxO3a) lacks the C-terminus transactivation domain [31]. For adenoviral expression, Ad-Tm-FoxO3a and DN-FoxO3a were cloned into Kpn I-BamH I of pShuttle-CMV-EGFP, followed by linearizing and co-transforming into DH5α with the adenoviral virus pAdeno (Invitrogen). The reconstituted adenoviral DNA containing FoxO3a cDNA was then transfected into 293 cells.
GFP-FoxO3a nuclear translocation
For real-time imaging in living cell, LoVo cells transiently transfected with GFP-FoxO3a. The time-course of GFP-FoxO3a distribution pattern in the cells with/without BKM120 treatment was recorded in single living LoVo cell. GFP fluorescence was recorded which represents the localization of FoxO3a in the cell as previously described [32].
Luciferase reporter assay
SIRT6 promoter region about 2000bp was isolated from human genomic DNA. Then, we cloned it into the basic vector pGL3 (LUC) at SacI/Hind III site to create a pGL3-SIRT6-LUC (forward: 5'-TATGAGCTCTCAGGGTACCTGGGTAGCTC-3'; reverse: 5'-ATAAGCTTAAGTT-TCCCTTGTTGAGGCC-3'). After adenoviral infection with plasmids of interest, cells were incubated with reporter lysis buffer. Then, luciferase substrate was added into cell lysates. Reporter assay was done according to the dual-luciferase reporter system (Promega).
Antibodies and reagents
Major primary antibodies were from cell signaling technology (CST) (Table S1). HRP-conjugated secondary antibodies and the HRP substrate kit were ordered from the GE Healthcare. BKM120 and MK-2206 were bought from Selleck. Akt inhibitor VIII was purchased from Axon Medchem. Lipofectamine™ was bought from Invitrogen. Others were all from Sigma unless otherwise indicated.
Cell viability, survival and apoptosis assays
Cells were grown in 96-well plate at 5 × 103 cells/well. After different treatments, the viability of cell was determined by CCK-8 assay, as described previously [9].
Apoptotic cells were firstly detected by calculating cells with condensed chromatin after nuclear staining with Hoechst 33258 [9]. The activity of caspase-3/9 was determined by Western blot using both cleaved- and full-length of caspase9/3 antibodies. For analysis of apoptosis by flow cytometry, Annexin V-FITC and PI were used to stain cells after treatment, which were then detected by flow cytometry as described before [33].
Long-term cell survival was detected by colony formation assay [34]. Briefly, equal numbers of cells were seeded into 6-well plates. After two weeks incubation, we stained the cells with crystal violet before calculating colonies numbers.
Real-time quantitative PCR
RNA was isolated with Tri-Reagent according to our previous report [35]. Total 3μg/RT reaction of RNA from the colon cancer cells or tissues was usded. Real-time quantitative PCR was done as described [9, 35]. Primers for Sirt6 are as follows: Sense: 5'- CGTGGATGAGGTGATGTG-3' and antisense: 5'-GGCTTATAGGAACCATTGAGA-3'; Transcripts of β-actin were used for normalization.
Subcellular Fraction analysis
To detect FoxO3a nuclear translocaion or mitochondrial cyto c release, nuclear, cytosol and mitochondria fractions were separated by Cell Compartment Kit (Qproteome), followed by Western blot analysis as described [9, 10].
Chromatin immunoprecipitation
ChIP assay was carried out by ChIP Assay kit (Millipore) with FoxO3a antibody as described [9]. The chromatin precipitates were detected with semi-quantitative PCR as describe above using sirt6 primers: forward: 5'- AACTCTGCGTGGCATTCAAA -3', reverse: 5'- AAATGCGGGACACAGGCTAT -3'.
Xenograft studies
shVector (Empty vector) and shSIRT6 xenografts were established and measured as described [9]. Six-week-old female nu/nu mice (Vital River, China) were inoculated with 1 × 106 cells (shVector and shSIRT6 LoVo cells) for each flank. When the tumnor was measurable, mice were administered by oral gavage with 40mg/kg/d BKM120 or vehicle (0.5% methyl cellulose) for two weeks. The tumors were measured every other day. Immunohistochemistry analysis was performed on paraffin sections, as described [33].
Colorectal cancer (CRC) Patient Sample
30 and 20 surgically excised CRC patient samples with available tissue blocks were respectively selected from the First Affiliated Hospital of Zhengzhou University (Zhengzhou, China) and from the Third Xiangya Hospital of Central South University (Changsha, China). Normal healthy colon samples were also provided by these two hospitals. Samples were embedded in paraffin for IHC staining, or collected by quickly freezing in liquid nitrogen for other analysis.
Statistical analysis
Data are given in mean ± SEM. Student's t test was applied to analyze the differences between various treatments and control, and P <0.05 was thought to be significant. Kaplan-Meier analysis was used to show the survival time of the CRC patients.
Results
Clinical significance of SIRT6 expression in human colorectal cancer
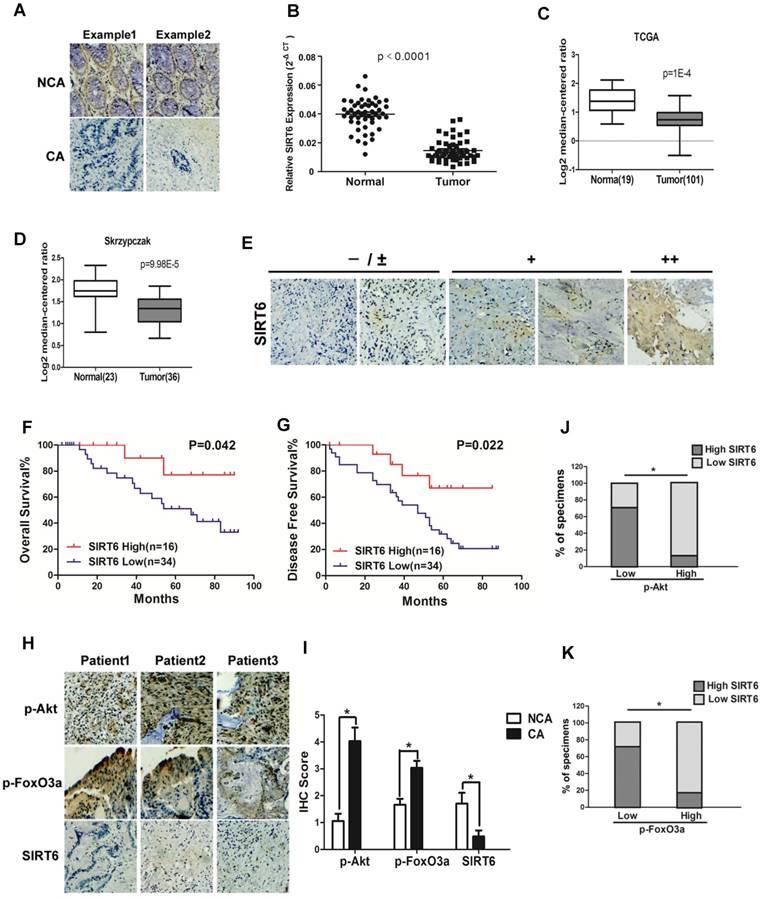
To investigate the role of SIRT6 in colorectal cancer, SIRT6 expression was firstly determined in 50 CRC (colorectal cancer) patient specimens. As shown in Figure 1A, there was high expression of SIRT6 in normal colon tissues. However, most colon cancer tissue (34 among 50 samples) expressed low level of SIRT6 or not expressed at all (72%, staining in <10% of tumor cells) (Table 1). Western analysis also showed decreased protein levels of SIRT6 in colon cancer tissue than that in adjacent non-cancerous tissue or in normal colon from healthy individuals (Figure 1A, S1A and S1B), consistent with the results from colon cancer cell lines and normal colon cells (Figure S1C). Furthermore, SIRT6 mRNA level is reduced in tissues of colon cancer compared to that of normal colon (Figure 1B). These are consistent with the Skrzypczak and TCGA data available from Oncomine (Figure 1C and 1D). Interestingly, although 68% of colon cancer tissue samples expressed extremely low (-) or low (±) level of SIRT6, there are still some cancer tissues showed high stain of SIRT6 in the IHC analysis (Figure 1E). Thus, we next wonder to know whether SIRT6 expression affect patients' clinical progression. Kaplan-Meier analysis showed that CRC patients with relative lower SIRT6 levels had shorter overall survival or disease-free survival times (Figure 1F and G).
Clinical significance of SIRT6 signaling in human colorectal cancer. (A-D) SIRT6 is down-regulated in colorectal cancer. (A) Immunohistochemical of SIRT6 level in colotectal cancer tissues (CA) and paired non-cancerous tissues (NCA). (100×). (B-D) Down-regulation of SIRT6 in patient samples from our hospital and from Oncomine database (TCGA and Skrzypczak). (B) The logarithmic scale 2-ΔΔCt was carried out to show the relative SIRT6 expression of patient samples. (C and D) Box plot of SIRT6 mRNA Log2 expression levels was evaluated in Oncomine database. (E-G) Low SIRT6 levels in CRC patients correlates with poor prognosis. (E) Representative immunohistochemical staining in colon cancer tissues with different degree of SIRT6 expression. Number of positive cells: (-) <10%; (±) 10-30%; (+) 30-60%; (++)>60%. (F and G) Kaplan-Meier curves of CRC patients with high versus low expression of SIRT6 (n=50; p< 0.05, log-rank test). (H-K) Inverse correlation between SIRT6 and p-Akt or p-FoxO3a expression in human colon cancer. (H) Immunohistochemical stainings of SIRT6, p-Akt and p-FoxO3a in colon cancer tissue. Three representative cases are shown. (I) Total IHC score of SIRT6, p-FoxO3a and p-Akt in colon cancer tissue and non-cancerous tissue (n=50). *P<0.05. (J and K) The percentages of CRC specimens with low or high p-FoxO3a or p-Akt levels and their relationship to SIRT6 expression.
Summary of SIRT6 expression in human colon cancer tissues by immunohistochemical analysis.
| Specimens | Case (n) | SIRT6 expression | SIRT6 positive (%) | Number(%) of cases expressing SIRT6 | ||||
|---|---|---|---|---|---|---|---|---|
| -(n) | +(n) | <10% | 49-10 | 75-50% | >75% | |||
| Cancer | 50 | 34 | 16 | 32 | 34(68) | 9(18) | 5(10) | 2(4) |
To study the correlation of SIRT6 and p-Akt or p-FoxO3a in patients, SIRT6 expression was analyzed in 50 cases of CRC specimens. Representative IHC staining showed marginally detectable SIRT6, but high level of p-Akt and p-FoxO3a in CRC samples (Figure 1H and 1I). Statistical analyses also demonstrated that SIRT6 level was positively correlated with FoxO3a activity, but inversely correlated with Akt activation (Figure 1J and 1K, P<0.05), supporting that aberrant expression of SIRT6 in colon cancer is regulated by Akt/FoxO3a.
Akt inhibition induced SIRT6 expression and apoptosis
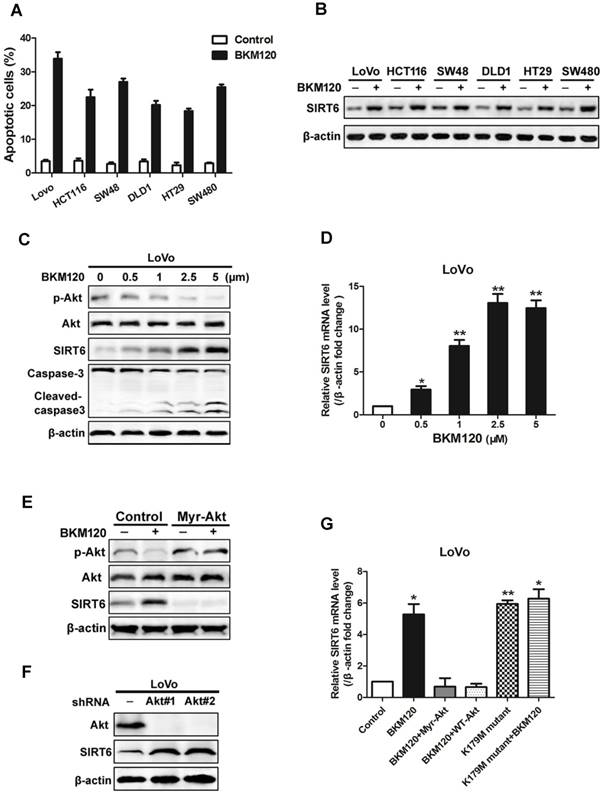
BKM120, a novel inhibitor of PI3K, is undergoing Phase I/II human clinical trials for solid tumors treatment. Here, analysis of 6 colon cancer cell lines revealed induction of apoptosis by BKM120 (Figure 2A, Table 2). SIRT6 was up-regulated by BKM120 in all these colon cancer cell lines (Figure 2B). Furthermore, BKM120 induced dose- and time-dependent increase of SIRT6 in mRNA and protein level (Figure 2C, 2D and S2A, S2B). We then investigated the mechanism of BKM120-induced SIRT6 expression in colon cancer cells. Constitutively active Akt (Myr-Akt) abolished SIRT6 expression induced by BKM120 (Figure 2E and S2C). In contrast, knockdown of Akt by shRNA led to increased SIRT6 expression in the absence of BKM120 (Figure 2F and S2D). Additionally, similar to BKM120, dominant negative Akt (K179M) led to positive regulation of SIRT6 mRNA (Figure 2G). These suggested that SIRT6 induction by BKM120 due to Akt inhibition.
Akt inhibition induced SIRT6 expression and cell apoptosis. (A-D) SIRT6 was induced by BKM120. (A and B) Various colorectal cancer cell lines were stimulated with 24 h of 5μM BKM120. (A) Apoptosis analysis was detected by calculating cells with condensed nucleus after Hoechst 33342 staining. (B) Western blotting analysis of SIRT6 expression. (C and D) LoVo cells with different doses of BKM120 for 24 h. (C) Western blot analysis of SIRT6, p-Akt (Ser473), Akt, c-caspase-3 and full-length caspase-3 expression. (D) Real-time PCR analysis of SIRT6 mRNA induction. *P<0.05, **P<0.01 vs. control. (E-G) SIRT6 was activated by Akt inhibition in LoVo cells. (E and F) Western blot analysis of p-Akt (Ser473), Akt or SIRT6 expression in LoVo cells transfected with control vector or (E) a constitutively active Akt (Myr-Akt) or (F) shAkt#1 or shAkt#2 in the presence/absence of 5μM BKM120. (G) SIRT6 mRNA was detected in LoVo cells transfected with Myr-Akt, WT-Akt, K179M mutant Akt in the presence/absence of BKM120.
The information of different colon cancer cell lines that used in our experiments.
| Cell lines Mutation | LoVo | HCT-116 | SW48 | DLD1 | HT29 | SW480 |
|---|---|---|---|---|---|---|
| p53 | - | - | - | + | + | + |
| PIK3CA | - | + | - | + | - | - |
| KRAS | + | + | - | + | - | + |
Recent study revealed that p53 transcriptionally activates expression of SIRT6 [36]. Here, our data showed that p53 level has no change in the presence of BKM120 (Figure S2E). Deletion of p53 had no effect on SIRT6 induction (Figure S2F), indicating p53 may be not involved in this process. Together, these suggested that Akt inhibition led to SIRT6 activation and apoptosis, which is a p53-independent event.
SIRT6 activation is mediated by FoxO3a
Foxo3a deficiency led to reduced SIRT6 expression in lipid metabolism of colon cancer cell and in liver cells [19, 37]. To explore the exact role of FoxO3a on SIRT6 induction, we firstly detected the activity of FoxO3a. BKM120 decreased phorphor-FoxO3a level in LoVo and DLD1 cells (Figure 3A). Moreover, nuclear translocation of FoxO3a was observed in cells treated with BKM120 (Figure 3B), which was confirmed by exogenous GFP-FoxO3a real-time imaging in living cell (Figure 3C), suggesting the activation of FoxO3a. The level of phosphor-FoxO3a (S315, T32 and S253) decreased with SIRT6 induction (Figure 3D). Similar results were obtained in fresh human tumor tissues (Figure 3E, 3F and S3A, S3B), indicating FoxO3a dephosphorylated and activated with SIRT6 induction.
Activation of FoxO3a by dephosphorylation mediated SIRT6 induction. (A-D) FoxO3a was dephosphorylated and activated by BKM120 in colon cancer cells. (A) Western blot showing p-FoxO3a (S253) after 8 or 24 h of BKM120 treatment in LoVo or in DLD1 cells. (B and C) FoxO3a translocated from cytosol to nucleus after BKM120 treatment in LoVo cells. (B) FoxO3a expression in both nuclear and cytoplamic fractions. LaminA/C and GAPDH were served as the marker of nucleus and cytosol. (C) Dynamics of GFP-FoxO3a nuclear translocation after BKM120 stimulation in single living cell after 5 μM BKM120 treatment. (D) The effect of BKM120 on FoxO3a phosphorylation at different sites. Western blot analysis of p-Akt and SIRT6 level as well as p-FoxO3a at T32, S253 and S315 sites in LoVo, DLD1 or HCT-116 cells after BKM120 treatment. (E and F) Expression of (E) SIRT6 mRNA or (F) p-FoxO3a (S253), FoxO3a and SIRT6 protein in colon tissue from normal person and in tumor tissue or adjacent tissue from colorectal cancer (CRC) patients in response to BKM120 treatment. (G) Effects of over-expression of FoxO3a or SIRT1 on SIRT6 protein level. Western blot analysis of SIRT6 level of LoVo cells after transfection of WT, Ad-DN-FoxO3a Ad-Tm-FoxO3a, or SIRT1. (H) Co-immunoprecipitation (Co-IP) analysis of the interaction between FoxO3a and Akt, Ac-K or SIRT1 in LoVo cells after 0, 12 or 24 h BKM120 treatment.
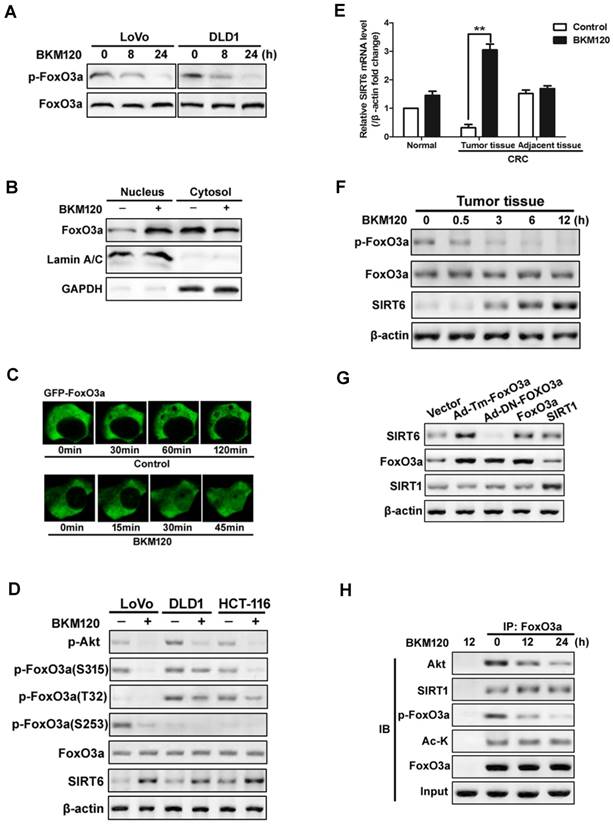
To further determine the relationship of FoxO3a and SIRT6, various FoxO3a mutants or inhibitors were used. Transfecting with constitutive active FoxO3a (unphosphorylatable FoxO3a, Ad-Tm-FoxO3a), or WT FoxO3a extremely increase the level of SIRT6 expression (Figure 3G). Whereas ectopic expression of Myr-Akt or WT-Akt increased the level of phosphor-FoxO3a, and blocked SIRT6 induction (Figure S3C and S3D). Impairment of Akt activity by Akt inhibitor activated FoxO3a and SIRT6 induction (Figure S3D and S3E). Interesting, the acetylation status of FoxO3a did not change with SIRT6 induction (Figure 3H). These suggested that dephosphorylation, but not deacetylation of FoxO3a upon Akt inhibition contributes to SIRT6 induction in colon cancer cells.
FoxO3a binds to SIRT6 promoter directly and activates its transcription
Kim et al. reported that SIRT1 binds and deacetylates FoxO3a, leading the formation of SFN (SIRT1-FoxO3a-NRF1) complex on the SIRT6 promoter, which facilitates SIRT6 transcription and expression [12]. Herein, our data showed that deletion of FoxO3a completely abolished SIRT6 induction by BKM120 (Figure 4A and 4B), indicating the requirement of FoxO3a for SIRT6 activation. However, knockdown of SIRT1 or NRF1 did not influence either SIRT6 promoter activity or its mRNA level (Figure 4B, 4C and S4A-S4D). Consistently, transfection of WT or Ad-Tm-FoxO3a improved the activity of SIRT6 promoter and SIRT6 mRNA level (Figure 4D). Also, ectopic expression of SIRT1 or NRF1 had no significant effect on SIRT6 promoter activity (Figure 4E and S4E).
Effects of FoxO3a on SIRT6 expression. (A-C) Effect of FoxO3a knockdown on SIRT6 protein expression. (A) Western blot analysis of SIRT6 and FoxO3a expression after 5μM BKM120 treatment for 24 h in LoVo cells with or without FoxO3a knockdown. (B and C) Effect of shRNA-mediated knockdown of FoxO3a, Akt and SIRT1 on (B) SIRT6 promoter reporter activity or (C) on SIRT6 mRNA level. (D and E) Effects of overexpression of Ad-Tm-FoxO3a (constitutively active FoxO3a) or WT-FoxO3a on (D) SIRT6 mRNA level or on (E) SIRT6 promoter reporter activity. (F) Schematic representation of human SIRT6 promoter in fragment A (between -295 to -276) (Fig.S4F) of the SIRT6 promoter, which contained FoxO3a binding sites that is the same as predicted sites. Three mutations (MT1, MT2, MT3) were produced by mutating the asterisks nucleotides. (G) LoVo cells were transiently transfected with luciferase reporters with WT or indicated mutant fragments (MT1, MT2, MT3) and then treated with BKM120 for 24 h. (H) Binding of FoxO3a to the SIRT6 promoter after 0, 8 or 12 h BKM120 treatment as revealed by ChIP assay. Primers were designed to amplify regions -345 to -226 for ChIP analysis. (I) The absence of FoxO3a blocked induction of SIRT6 mRNA by BKM120. shVector and shFoxO3a cells were incubated with/without BKM120 for 24 h. SIRT6 mRNA level was detected by Semi-Quantitative PCR. (J) Interaction of FoxO3a and SIRT1 in cultured cells with/without BKM120 treatment as revealed by immunoprecipitation (IP). Flag pulls down both phoshor-FoxO3a and acetylated-FoxO3a. (K) Binding of FoxO3a to the SIRT6 promoter after transfection of WT, Ad-DN-FoxO3a, Ad-Tm-FoxO3a or SIRT1 as revealed by ChIP assay.
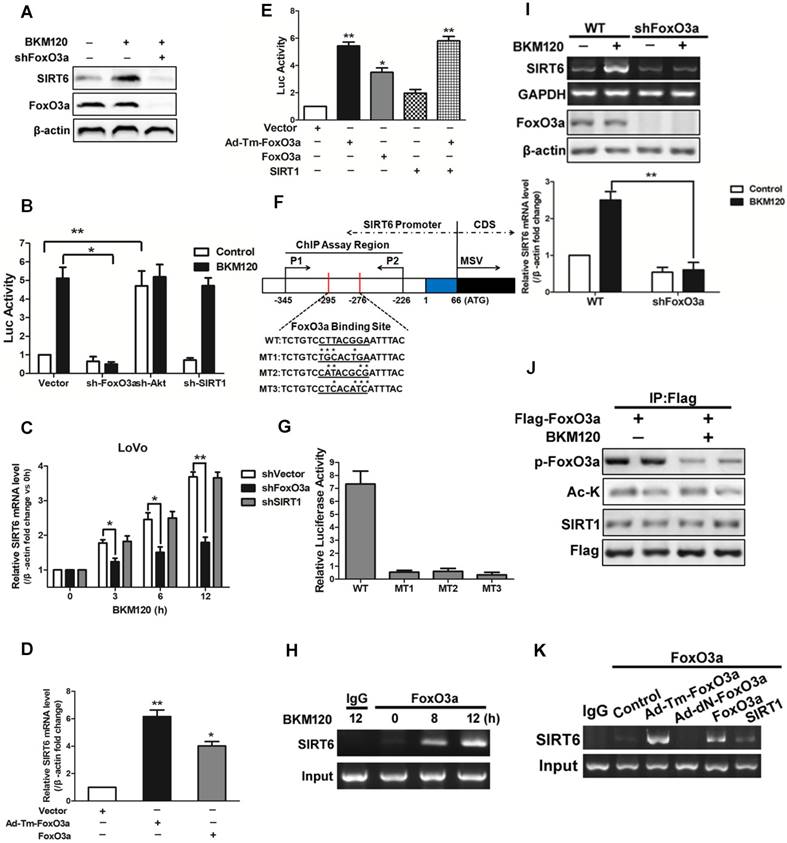
To determine whether FoxO3a can bind to SIRT6 promoter and figure out the exact binding sites, we firstly did a prediction by using bioinformatics analysis and obtained five potential binding sites (Figure S4F). Then we detected the activity of different region of SIRT6 promoter. As a result, BKM120 treatment strongly activated the proximal -295bp region of the SIRT6 promoter (fragments A in Figure S4F). We identified a FoxO3a binding site in this region (Figure 4F). SIRT6 promoter activation was completely inhibited when this FoxO3a binding site is mutated (Figure 4G), suggesting this region is indispensable. Furthermore, FoxO3a, but not SIRT1, bound to the SIRT6 promoter containing the FoxO3a binding site by ChIP assay (Figure 4H, 4I and S4G-S4I). FoxO3a was activated by dephorsphorylation, but not deacetylation, upon BKM120 stimulation (Figure 4J). Thus in the absence of BKM120, only Ad-Tm-FoxO3a (unphosphorylatable) bound to SIRT6 promoter obviously (Figure 4K). The fact that no binding of FoxO3a to SIRT1 protein (Figure S4J) excluded the possibility of SFN (SIRT1-FoxO3a-NRF1) complex on SIRT6 promoter. Taken together, these data indicated that FoxO3a binds to the proximal SIRT6 promoter region directly to promote its transcription in stimulation of BKM120.
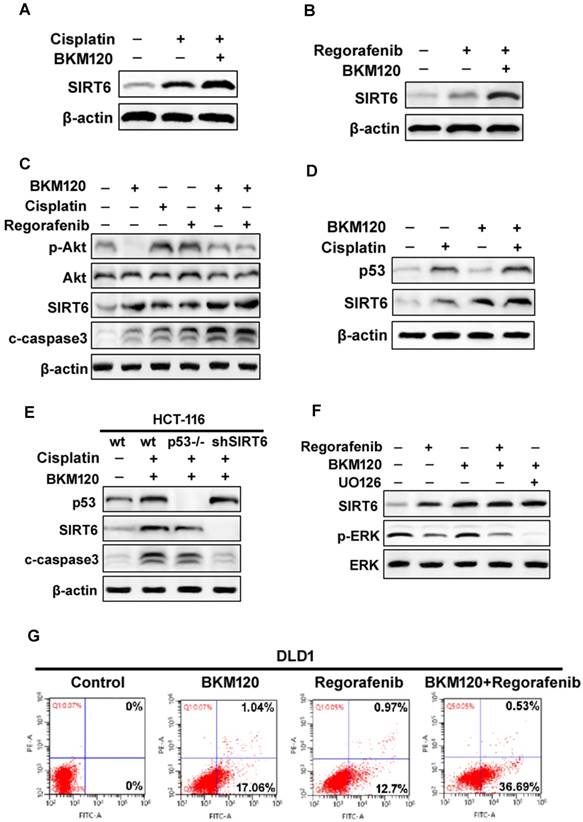
SIRT6 induction contributes to the synergetic effect of combinational therapies
The aberrant activation of Akt contributes to some classical drug assistant (Figure S5A) [8]. BKM120, a selective PI3K inhibitor, could be combined with other chemotherapy drug for solid tumor treatment [38]. We found that BKM120 combined with cisplatin or regorafenib expressed relative higher SIRT6 level, than that with single drug (Fig. 5A and 5B). Although cisplatin or regorafenib treatment alone caused slight increase in p-Akt level, they enhanced SIRT6 activation and cell apoptosis (Figure 5C). It seems that these comninational treatment mediated SIRT6 expression through different pathways. SIRT6 induction was via p53-dependent and -independent mechanisms after Cisplatin and BKM120 stimulation, respectively (Figure 5D and 5E). However, ERK inactivation was found after UO126 (ERK inhibitor) or regorafenib treatment (Figure 5F). Furthermore, ERK inhibition by UO126 led to FoxO3a translocation/dephosphorlation and SIRT6 induction (Figure S5B and S5C), whereas lack of FoxO3a abolished this SIRT6 induction (Figure S5D). Therefore, ERK may be involved in FoxO3a-dependent SIRT6 up-regulation induced by regorafenib. Importantly, the synergistic apoptosis by combinational treatment (Figure 5G) was highly suppressed in SIRT6 knockdown (shSIRT6) cells (Figure S5E and S5F), indicating the enhanced apoptosis was SIRT6-dependent.
SIRT6 augment the chemosensitization of BKM120. (A and B) Western blot analysis of SIRT6 level in the presence of 2.5 μM BKM120 combined with (A) 40 uM Cisplatin or (B) 20 mM Regorafenib for 24 h in LoVo cells. (C) Akt inhibition was involved in SIRT6 induction by combinational therapy. The level of SIRT6, p-Akt, cleaved-caspase3 was performed by western blot following 20 mM regorafenib, 40 μM cisplatin single treatment or combined with 2.5μM BKM120 for 24 h in LoVo cells. (D and E) p53 may mediate SIRT6 expression induced by Cisplatin or combined treatment. (D) LoVo cells were treated with 2.5 μM BKM120, 40 uM Cisplatin, or their combination. The expression levels of p53 and SIRT6 were detected by Western blot analysis. (E) The expression of p53, c-caspase-3 and SIRT6 were determined in wild-type, p53-/- or shSIRT6 (SIRT6 knockdown) HCT-116 cells. (F) The role of ERK inhibition in SIRT6 induction after regorafenib or its combination treatment with BKM120. LoVo cells were treated with either 20 mM Regorafenib, 20 mM UO126 or the combined treatment with 2.5 μM BKM120. Western blotting showing the level of SIRT6, p-ERK (Thr202/Tyr204) and total ERK. (G) Apoptosis in DLD1 cells treated with either 2.5 μM BKM120, 20mM Regorafenib or their combination therapy was detected by flow cytometry. Percentages of early apoptosis cells were shown in two right quadrants.
BKM120 induced SIRT6-dependent apoptosis
To explore the function of SIRT6, shSIRT6 cells were used. Apoptosis caused by BKM120 was inhibited in shSIRT6 cells in comparison with shVector cells. Flow cytometry result also showed less apoptosis in shSIRT6 cells after BKM120 treatment (Figure 6B). Lack of SIRT6 abolished BKM120-induced mitochondrial apoptotic events, such as cyto-c release and caspase-9/3 activation (Figure 6C-G, S6A and S6B). Apoptosis was abrogated in Bax-/- HCT-116 cell (Figure S6C), further confirming the mitochondrial pathway of apoptosis by BKM120. Furthermore, shSIRT6 cells had improved cell viability (Figure S6D and S6E) and long-term survival compared with shVector cells after BKM120 treatment (Figure 6H). These indicated SIRT6 is indispensible for BKM120-induced mitochondrial apoptosis and colon cancer cell growth suppression.
SIRT6 is indispensible for BKM120-induced apoptosis. (A and B) shVector or shSIRT6 LoVo cells were stimulated with BKM120 for 24 h. (A) Nuclei of cells were stained with Hoechst 33342. upper, representative pictures with arrows indicating fragmented nuclei (400×); lower, quantification analysis of apoptosis cells with fragmented nuclei. (B) Apoptosis analysis detected by flow cytometry. (C-E) Real-time detection of the activation of caspase-3 in single living cell. (C) Representative fluorescence images of CFP, YFP, Ratio as well as DIC image in LoVo cells transfected with SCAT-3 reporter. (D) The fluorescence images from Ratio channel at different time points after BKM120 treatments in shVector or shSIRT6 SCAT-3 LoVo cells. Scale bar: 10μm. (E) The fluorescence intensity of the Ratio (FRET/CFP) according to the images in D. (F) The expression of cleaved caspase-3, -9 and SIRT6 after 5 μM BKM120 treatment for 24 h in LoVo cells with or without SIRT6 deletion. (G) Western blot analysis showing cyto-c release from mitochondria to cytosol. The cytosolic and mitochondrial fractions were extracted from shVector and shSIRT6 cells after 24 h of BKM120 treatment. CoxIV and α-tublin were served as the mitochondrial and cytosolic marker. (H) Colony formation of shVector and shSIRT6 LoVo cells cultured 14 days after crystal violet staining in the presence/absence of BKM120. upper, representative photos of colonies; lower, quantification analysis of colony numbers. (I and J) SIRT6 bound to the promoter of survivin, leading the deacetylation at its H3K9 site. (I) Western blot analysis of AcH3K9, H3 and SIRT6 expression of nuclear fraction prepared from LoVo cells with or without SIRT6 deletion. (J) ChIP assay of the acetylation levels at H3K9 in the 5'-UTR of the survivin gene in samples extracted from shVector or shSIRT6 LoVo cells.
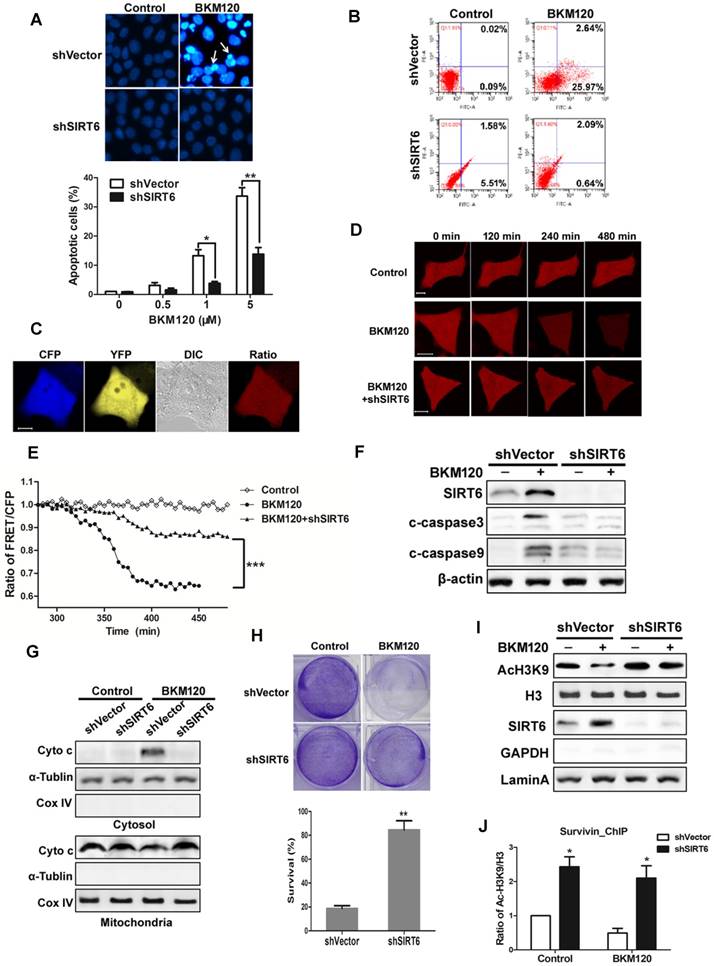
SIRT6 is an NAD-dependent histone deacetylase and a tumor suppresser [1-3], thus it might be recruited to the anti-apoptotic factor like survivin gene promoter to serve as a transcriptional factor. To assess this possibility, histone acetylation and survivin expression were detected. SIRT6 overexpression remarkably reduced acetylation of H3K9 (Figure S6F) and survivin expression (Figure S6G). In addition, decreased mRNA level of survivin was observed in SIRT6 overexpressed cells or in BKM120 treated cells (Figure S6H and S6I). Conversely, SIRT6 deficiency led to an increase of acetylation of H3K9 (Figure 6I) and high affinity to the 5'-UTR of survivin gene (Figure 6J). Therefore, SIRT6 promoted apoptosis probably through inhibiting survivin transcription by deacetylation of histone at H3K9 site in the promoter region.
SIRT6 shows potent anti-tumor effect in vivo
To determine the antitumor effect of SIRT6 in vivo, shVector or shSIRT6 LoVo cells were subcutaneously injected into each flank of nude mice to established xenograft tumors. The results showed that BKM120 therapy suppressed the growth of shVector tumors by ~75%. In contrast, shSIRT6 tumors were almost insensitive to BKM120 treatment compared with shVector tumors (Figure 7A-C). Moreover, Kaplan-Meier analyses showed that shSIRT6 mice had shorter survival than shVector mice in BKM120 treated groups (Figure 7D). These indicated that lack of SIRT6 abolished the antitumor activity of BKM120. Consistent with our in vitro data, decreased phospho-Akt and enhanced SIRT6 expression were observed after BKM120 treatment (Figure 7E and 7F). Comparing to shVector tumors, shSIRT6 tumors showed augment of Ki67 but reduced cleaved-caspase3 expression (Figure S7A and S7B), which confirmed pronounced increased proliferation and reduced apoptosis in response to BKM120. Collectively, our results demonstrated that BKM120 suppressed tumorigenesis through SIRT6-dependent manner.
SIRT6 mediates in vivo antitumor effects of BKM120. (A-F) Nude mice were used to established xengraft model with shVector or shSIRT6 tumor. Mice were subjected to 40 mg/kg/d BKM120 or vehicle for two weeks. (A) Tumors pictures in this test. (B) Tumor volume or (C) weight was measured. *P<0.05. (D) Survival curve of (Kaplan-Meier analysis of) shVector and shSIRT6 mice with or without BKM120 treatment (6 mice / group). (E) p-Akt (S473), SIRT6, ki67, cleaved-caspase3 and full-length caspase-3 levels were detected by Western blot in shVector or shSIRT6 tumors with/without BKM120 treatment. (F) Immunohistochemistry of ki67, p-Akt and cleaved-caspase-3 in shVector or shSIRT6 tumor tissues. (G and H) The regulation of SIRT6 by FoxO3a after BKM120 treatment in shVector tumors. (G) Western blotting analysis of p-FoxO3a, SIRT6 and p-Akt expression in tumors. (H) Chromatin immunoprecipitation (ChIP) was performed by using FoxO3a to pull down. Then PCR was done to detect sirt6 promote region that bound to FoxO3a. (I) Schematic representation of Akt inhibition-mediated SIRT6 induction and apoptotic signaling pathway.
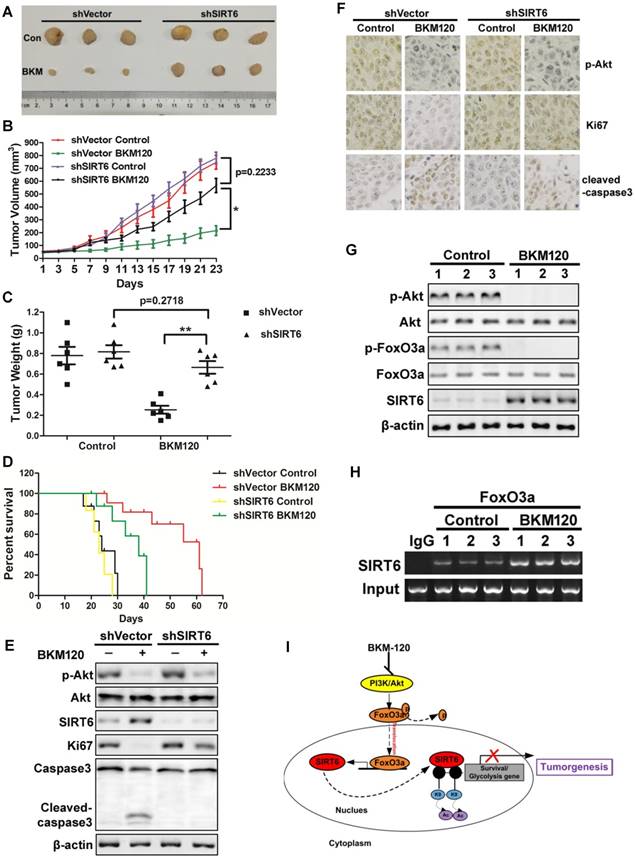
Together, to explore the regulation mechanism of SIRT6, we studied the relation among Akt, FoxO3a and SIRT6 in tumors from patients and got similar results as that in xenograft model. BKM120 activated FoxO3a by dephosphorlyation upon Akt inhibition (Figure 7G). Subsequently, FoxO3a bound to the promoter of SIRT6 (Figure 7H) to transcriptionally up-regulate SIRT6 expression (Figure S7C), which inhibited the expression of downstream survival protein survivin by deacetylated the histone at H3K9 site in the promoter region (Figure 7I).
Discussion
Our previous studies show that Akt inhibition is the key factor to overcome chemoresistance for colon cancer therapy. BKM120, a novel pan-class I (α, β, γ, δ) PI3K inhibitor, is now in Phase I/II clinical trial. In this study, we identified SIRT6 as a brand new transcriptional target of FoxO3a for the first time, and demonstrated Akt/FoxO3a/SIRT6 axis plays a crucial role in pro-apoptisis and colon cancer therapy by BKM120 both in vitro and in vivo. Lack of SIRT6 caused the resistance of colon cancer to BKM120 and combinational therapy. Mechanically, Akt is inhibited by BKM120, then separates and dephosphorlates FoxO3a, leading increased level of FoxO3a on SIRT6 promoter that triggered SIRT6 transcription. The activated SIRT6 deacetylated histone at H3K9 of some survival gene like survivin and down-regulates its transcription, finally initiates mitochondrial apoptosis. Our results indicated an unappreciated, but critical, role for Akt inactivation and SIRT6 induction as a novel target for the therapy or combinational therapy of colon cancer.
The function of SIRT6 in tumorigenesis is controversial. SIRT6 is downregulated in various human cancers, which is related with poor survival prognosis [2, 6, 7], like in ovarian cancer, pancreatic cancer, colorectal cancer, hepato-cellular carcinoma. However, SIRT6 is highly expressed and serves as an oncogene in some other cancer types, like prostate cancer, skin cancer and breast cancer [13, 39]. Even in the same cancer type, like non-small cell lung cancer, opposite roles of SIRT6 were reported [12, 40]. Here, 50 specimens of colorectal cancer tissues or paired normal colon tissues from CRC patients were analyzed and similar results were observed. Most of the colon cancer tissues expressed low or extremely low level of SIRT6, accompanied by high level of Akt activation or FoxO3a inhibition (Figure 1 and S1, Table 1). Meantime, patients with lower SIRT6 levels had shorter overall survival or disease-free survival times (Figure 1F and G). Therefore, SIRT6 may serves as a tumor suppressor in colorectal cancer.
Besides Akt, p53 has also been reported to regulate SIRT6, which could transcriptionally activate the expression of SIRT6 [36]. Our results showed that BKM120 induced apoptosis with increased SIRT6 expression but reduced Akt phosphorylation, in WT, p53-/- or p53 mutant cells (Figure S2E and S2F), indicating p53-independent but Akt inhibition-mediated SIRT6 induction and apoptosis. Some studies show that BKM120 induced pro-apoptotic effects and showed an increased response only in PI3KCA mutated or PI3K activating cells [8, 20]. Our results indicated that, after BKM120 treatment, SIRT6 expression and apoptosis increased in both wide-type (LoVo, SW48, HT29, SW480) and PIK3CA mutant (HCT-116, DLD1) colon cancer cells (Figure 2A, 2B, S2B-2E, Table 2). Moreover, BKM120 inhibited Akt activation with no difference between LoVo cell (PIK3CA wild type) and HCT-116 cells (PIK3CA mutant) (Figure 2C and S2F).
Since Akt aberrant activation is responsible for the resistant of many classic chemotherapy drugs like 5-FU, inhibiting Akt activity by combinational treatment come to our consideration. BKM120 showed clinical activity in patients with solid tumors, either as a single drug or in combination with other agent [38]. Our data indicated that the synergistic effect of cisplatin and BKM120 can be mediated by concurrent SIRT6 expression in both p53-dependent and -independent manners (Figure 5A and 5C-E). Regorafenib, another novel drug, induced FoxO3a-dependent SIRT6 expression by inactivating ERK (Figure 5B, 5C, 5F, S5B and S5C). Together, the chemosensitization effect of BKM120 is dependent on SIRT6 (Figure 5 and Figure S5B-5C), and involves concurrent SIRT6 induction through different pathways.
There are several regulators that might be involved in SIRT6 expression in different models [17-19]. RUNX2 was found to suppress both SIRT6 transcription and translation. E2F1 could directly bind to SIRT6 promoter and inhibit its promoter activity. In addition, c-Fos induced SIRT6 transcription to mediate liver cancer cell apoptosis. While in this study, our data from patients and colon cancer cell lines suggested that Akt inhibition led to FoxO3a activation and SIRT6 induction (Figure 2C-2G and 3A-3D), and we hypothesized that FoxO3a up-regulated SIRT6 expression in our model. Kim et al. found that SIRT1 binds and deacetylates FoxO3a, leading the increased level of SFN (SIRT1-FoxO3a-NRF1) complex on SIRT6 promoter to facilitate its transcription [12]. There also exist other different opinions, regarding that SIRT6 regulates FoxO3a by deacetylation [13, 41] or dephosphorylation [42].
To explore the mechanism of SIRT6 regulation, firstly, we revealed that only FoxO3a but not SIRT1, or NRF1 is involved in SIRT6 induction. Knockdown of FoxO3a blocked SIRT6 promoter activity, mRNA level and protein expression in the stimulation of BKM120 (Figure 4A-4C and S4A-S4B). Whereas, knockdown of SIRT1 or NRF1 did not affect SIRT6 induction (Figure 4B-4C and S4A-S4B, S4D). Furthermore, the level of phosphor-FoxO3a decreased (Figure 3A, 3D and 3F), while the level of FoxO3a acetylation did not change after Akt inhibition (Figure 3H and 4J), indicating the activation of FoxO3a by dephosphorylation, but not by deacetylation. Next, we further investigate the exact mechanism of SIRT6 regulation by FoxO3a. Five FoxO3a binding sites were predicted on the promoter of SIRT6 (Figure S4F), and ChIP assay results show that one of these binding sites directly interacts with activated FoxO3a (Figure 4F). This FoxO3a binding site is different from NRF1 binding site that Kim et al. reported [14]. CoIP result further confimed that SIRT1 did not interact with FoxO3a after Akt inhibition (Figure S4J), excluding the complex system of SIRT1, FoxO3a and NRF1 during SIRT6 induction. Together, we uncover a new mechanism of SIRT6 transcriptional regulation by FoxO3a. FoxO3a was dephosphorylated upon Akt inhibition, and this activated FoxO3a translocated from cytosol to nucleus to bind with SIRT6 promoter, leading to transcriptional expression of sirt6. This activated FoxO3a is indispensable and sufficient to induce SIRT6 expression.
Supplementary Material
Supplementary figures and table.
Acknowledgements
We would like to thank the support of the National Natural Science Foundation of China (NSFC) (No. 31701132, No. 31801140), the Basic Research Program of Shenzhen Municipal Science and Technology Innovation Committee (No. JCYJ20160530192802733), the Fundamental Research Funds for the Central Universities (No. 531107040909, No. 14700-502044001), the start funds from Department of Laboratory Medicine, Xiangya School of Medicine, Central South University, and the start funds from College of Biology, Hunan University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhong L, D'Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD. et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140:280-93
2. Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L. et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151:1185-99
3. Masri S, Rigor P, Cervantes M, Ceglia N, Sebastian C, Xiao C. et al. Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell. 2014;158:659-72
4. Lyssiotis CA, Cantley LC. SIRT6 puts cancer metabolism in the driver's seat. Cell. 2012;151:1155-6
5. Lerrer B, Cohen HY. The guardian: metabolic and tumour-suppressive effects of SIRT6. EMBO J. 2013;32:7-8
6. Marquardt JU, Fischer K, Baus K, Kashyap A, Ma S, Krupp M. et al. Sirtuin-6-dependent genetic and epigenetic alterations are associated with poor clinical outcome in hepatocellular carcinoma patients. Hepatology. 2013;58:1054-64
7. Lai CC, Lin PM, Lin SF, Hsu CH, Lin HC, Hu ML. et al. Altered expression of SIRT gene family in head and neck squamous cell carcinoma. Tumour biol. 2013;34:1847-54
8. Wang H, Zhang L, Yang X, Jin Y, Pei S, Zhang D. et al. PUMA mediates the combinational therapy of 5-FU and NVP-BEZ235 in colon cancer. Oncotarget. 2015;6:14385-98
9. Zhang L, Wang H, Li W, Zhong J, Yu R, Huang X. et al. Pazopanib, a novel multi-kinase inhibitor, shows potent antitumor activity in colon cancer through PUMA-mediated apoptosis. Oncotarget. 2017;8:3289-303
10. Zhang Y, Xing D, Liu L. PUMA promotes Bax translocation by both directly interacting with Bax and by competitive binding to Bcl-X L during UV-induced apoptosis. Mol Biol Cell. 2009;20:3077-87
11. Ran LK, Chen Y, Zhang ZZ, Tao NN, Ren JH, Zhou L. et al. SIRT6 Overexpression Potentiates Apoptosis Evasion in Hepatocellular Carcinoma via BCL2-Associated X Protein-Dependent Apoptotic Pathway. Clin Cancer Res. 2016;22:3372-82
12. Han Z, Liu L, Liu Y, Li S. Sirtuin SIRT6 suppresses cell proliferation through inhibition of Twist1 expression in non-small cell lung cancer. Int J Clin Exp Pathol. 2014;7:4774-81
13. Khongkow M, Olmos Y, Gong C, Gomes AR, Monteiro LJ, Yague E. et al. SIRT6 modulates paclitaxel and epirubicin resistance and survival in breast cancer. Carcinogenesis. 2013;34:1476-86
14. Van Meter M, Gorbunova V, Seluanov A. SIRT6: a promising target for cancer prevention and therapy. Adv Exp Med Biol. 2014;818:181-96
15. Choe M, Brusgard JL, Chumsri S, Bhandary L, Zhao XF, Lu S. et al. The RUNX2 Transcription Factor Negatively Regulates SIRT6 Expression to Alter Glucose Metabolism in Breast Cancer Cells. J Cell Biochem. 2015;116:2210-26
16. Wu M, Seto E, Zhang J. E2F1 enhances glycolysis through suppressing Sirt6 transcription in cancer cells. Oncotarget. 2015;6:11252-63
17. Min L, Ji Y, Bakiri L, Qiu Z, Cen J, Chen X. et al. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat Cell Biol. 2012;14:1203-11
18. Thirumurthi U, Shen J, Xia W, LaBaff AM, Wei Y, Li CW. et al. MDM2-mediated degradation of SIRT6 phosphorylated by AKT1 promotes tumorigenesis and trastuzumab resistance in breast cancer. Sci Signal. 2014;7:ra71
19. Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A. et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010;12:224-36
20. Li X, Yang T, Li CS, Song Y, Lou H, Guan D. et al. Surface Enhanced Raman Spectroscopy (SERS) for the Multiplex Detection of Braf, Kras, and Pik3ca Mutations in Plasma of Colorectal Cancer Patients. Theranostics. 2018;8:1678-89
21. Ren J, Ding L, Zhang D, Shi G, Xu Q, Shen S. et al. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics. 2018;8:3932-48
22. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
23. Briest F, Grabowski P. PI3K-AKT-mTOR-signaling and beyond: the complex network in gastroenteropancreatic neuroendocrine neoplasms. Theranostics. 2014;4:336-65
24. Xu YC, Wang X, Chen Y, Chen SM, Yang XY, Sun YM. et al. Integration of Receptor Tyrosine Kinases Determines Sensitivity to PI3Kalpha-selective Inhibitors in Breast Cancer. Theranostics. 2017;7:974-86
25. Green DR, Evan GI. A matter of life and death. Cancer cell. 2002;1:19-30
26. Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D. et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11:317-28
27. Lattanzio L, Tonissi F, Monteverde M, Vivenza D, Russi E, Milano G. et al. Treatment effect of buparlisib, cetuximab and irradiation in wild-type or PI3KCA-mutated head and neck cancer cell lines. Invest New Drugs. 2015;33:310-20
28. Estevez LG, Garcia E, Hidalgo M. Inhibiting the PI3K signaling pathway: buparlisib as a new targeted option in breast carcinoma. Clin Transl Oncol. 2016;18:541-9
29. Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D. et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30:282-90
30. Zhang L, Zhang Y, Xing D. LPLI inhibits apoptosis upstream of Bax translocation via a GSK-3beta-inactivation mechanism. J Cell Physiol. 2010;224:218-28
31. Skurk C, Maatz H, Kim HS, Yang J, Abid MR, Aird WC. et al. The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J Biol Chem. 2004;279:1513-25
32. Zhang L, Xing D, Gao X, Wu S. Low-power laser irradiation promotes cell proliferation by activating PI3K/Akt pathway. J Cell Physiol. 2009;219:553-62
33. Chen J, Zhong J, Liu Y, Huang Y, Luo F, Zhou Y. et al. Purified vitexin compound 1, a new neolignan isolated compound, promotes PUMA-dependent apoptosis in colorectal cancer. Cancer Med. 2018;7:6158-69
34. Sun L, Huang Y, Liu Y, Zhao Y, He X, Zhang L. et al. Ipatasertib, a novel Akt inhibitor, induces transcription factor FoxO3a and NF-kappaB directly regulates PUMA-dependent apoptosis. Cell death Dis. 2018;9:911
35. Zhang Z, Lau SW, Zhang L, Ge W. Disruption of Zebrafish Follicle-Stimulating Hormone Receptor (fshr) But Not Luteinizing Hormone Receptor (lhcgr) Gene by TALEN Leads to Failed Follicle Activation in Females Followed by Sexual Reversal to Males. Endocrinology. 2015;156:3747-62
36. Zhang P, Tu B, Wang H, Cao Z, Tang M, Zhang C. et al. Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proc Natl Acad Sci U S A. 2014;111:10684-9
37. Qi W, Fitchev PS, Cornwell ML, Greenberg J, Cabe M, Weber CR. et al. FOXO3 growth inhibition of colonic cells is dependent on intraepithelial lipid droplet density. J Biol Chem. 2013;288:16274-81
38. Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz-Ares L, Vansteenkiste J. et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res. 2015;21:730-8
39. Liu Y, Xie QR, Wang B, Shao J, Zhang T, Liu T. et al. Inhibition of SIRT6 in prostate cancer reduces cell viability and increases sensitivity to chemotherapeutics. Protein Cell. 2013;4:702-10
40. Azuma Y, Yokobori T, Mogi A, Altan B, Yajima T, Kosaka T. et al. SIRT6 expression is associated with poor prognosis and chemosensitivity in patients with non-small cell lung cancer. J Surg Oncol. 2015;112:231-7
41. Lee JJ, Lee HJ, Son BH, Kim SB, Ahn JH, Ahn SD. et al. Expression of FOXM1 and related proteins in breast cancer molecular subtypes. Int J Exp Pathol. 2016;97:170-7
42. Wang XX, Wang XL, Tong MM, Gan L, Chen H, Wu SS. et al. SIRT6 protects cardiomyocytes against ischemia/reperfusion injury by augmenting FoxO3alpha-dependent antioxidant defense mechanisms. Basic Res Cardiol. 2016;111:13
Author contact
![]() Corresponding author: Lingling Zhang, Ph.D., email: zhll0807edu.cn; Zhanglingling206com
Corresponding author: Lingling Zhang, Ph.D., email: zhll0807edu.cn; Zhanglingling206com