Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(13):3732-3753. doi:10.7150/thno.34377 This issue Cite
Research Paper
A Multifunctional Nanotherapy for Targeted Treatment of Colon Cancer by Simultaneously Regulating Tumor Microenvironment
Qixiong Zhang1*, Fuzhong Zhang1*, Shanshan Li2*, Renfeng Liu1,3, Taotao Jin1,3, Yin Dou1, Zhenhua Zhou3 ![]() , Jianxiang Zhang1
, Jianxiang Zhang1 ![]()
1. Department of Pharmaceutics, College of Pharmacy, Third Military Medical University, Chongqing 400038, China
2. Department of Pharmaceutical Engineering, College of Pharmaceutical Sciences, Southwest University, Chongqing 400715, China
3. Department of Neurology, Southwest Hospital, Third Military Medical University, Chongqing 400038, China
* These authors contributed equally to this work.
Received 2019-2-24; Accepted 2019-4-22; Published 2019-5-31
Abstract

Colitis-associated colon cancer (CAC) is a widely recognized cancer, while treatment with the existing chemotherapeutic drugs affords limited clinical benefits. Herein we proposed a site-specific, combination nanotherapy strategy for targeted treatment of CAC by the oral route.
Methods: A reactive oxygen species (ROS)-responsive and hydrogen peroxide-eliminating material OCD was synthesized, which was further produced into a functional nanoparticle (OCD NP). The antioxidative stress and anti-inflammatory effects of OCD NP were examined by in vitro and in vivo experiments. By packaging an anticancer drug camptothecin-11 (CPT-11) into OCD NP, a ROS-responsive nanotherapy CPT-11/OCD NP was obtained, and its antitumor activity was evaluated by both in vitro and in vivo studies. Preliminary safety studies were also performed for CPT-11/OCD NP in mice.
Results: OCD NP significantly attenuated oxidative stress and inhibited inflammatory response in different cells and mice with induced colitis. CPT-11/OCD NP could selectively release drug molecules under intestinal pH conditions and at high levels of ROS. In C26 murine colon carcinoma cells, this nanotherapy showed significantly higher antitumor activity compared to free CPT-11 and a non-responsive CPT-11 nanotherapy. Correspondingly, oral delivery of CPT-11/OCD NP notably inhibited tumorigenesis and tumor growth in mice with induced CAC. By combination therapy with the nanovehicle OCD NP in the inflammatory phase, more desirable therapeutic effects were achieved. Furthermore, CPT-11/OCD NP displayed excellent safety profile for oral administration at a dose that is 87.3-fold higher than that employed in therapeutic studies.
Conclusions: Anticancer nanotherapies derived from intrinsic anti-inflammatory nanocarriers are promising for targeted combination treatment of inflammation-associated tumors by simultaneously shaping pro-inflammatory microenvironment toward a relatively normal niche sensitive to chemotherapy.
Keywords: colitis-associated colon cancer, anti-inflammation, reactive oxygen species, nanoparticle, camptothecin-11, site-specific therapy
Introduction
Colorectal cancer (CRC) is one of the commonly diagnosed cancers worldwide, with poor prognosis after metastasis to lymph nodes or distant organs, and therefore causing high mortality [1-3]. In addition to surgery, chemotherapeutic drugs, such as oxaliplatin, 5-fluorouracil, and irinotecan, are frequently used in first-line treatment of CRC [4]. However, systemic administration of chemotherapeutic drugs generally leads to dose-limiting toxicity in non-target tissues and organs [5, 6], while treatment by the most preferable and acceptable route of oral delivery more often results in unsatisfactory bioavailability and serious side effects, such as mucositis, edema, and diarrhea [7-9].
Recently, increasing evidence has demonstrated that the nanoparticle (NP)-based targeting strategy is effective and promising for molecular imaging and therapy of CRC [10-13]. A diverse array of nanocarriers, including liposomes, lipid NPs, polymeric NPs, nanogels, high density lipoprotein, and organic/inorganic hybrid nanomaterials [10, 14, 15], have been investigated for targeted treatment of CRC, by loading different drugs such as CPT-11 [16], doxorubicin [17], oxaliplatin [18], curcumin [19], 5-aminolevulinic acid [20], and nucleic acids [21-23]. The targeting capability of these NPs can be further enhanced, by peripheral decoration with antibodies [24], fragments of antibodies [25], peptides [26], aptamers [27], and other small molecules [20]. In other cases, the magnetic- and/or photo-responsive nanoplatforms were used for targeted delivery of therapeutics to the CRC sites [28-30]. Nevertheless, these nanotherapies have been largely investigated for CRC treatment by intravenous injection [12], and only a few of them are administered by oral delivery [31, 32]. Whereas the majority of these nanotherapies are under preclinical development, several liposome-based nanomedicines (i.e. CPX-1, LE-SN38, and Thermodox) are currently in clinical trials [11]. Unfortunately, only limited therapeutic outcome was afforded in some cases. For instance, patients (with metastatic CRC) treated with LE-SN38 did not show slowed cancer progression [11]. Consequently, other innovative strategies are necessary for effective and safe therapy of CRC. In particular, site-specific therapy of CRC by the patient-friendly oral delivery route is desperately required.
As well documented, the risk of CRC development is high in patients with long-term chronic gastrointestinal disorders, such as inflammatory bowel disease (IBD) [33-35]. Crohn's disease and ulcerative colitis are two major types of IBD. It was reported that 18-20% patients suffering Crohn's disease and 8% patients with ulcerative colitis would develop colitis-associated colon cancer (CAC) after 30 years [36]. Chronic intestinal inflammation is a key factor in the onset of carcinogenesis in IBD patients, and it can further promote tumor growth and progression [37]. The molecular mediators and cellular effectors of inflammation are important components of the tumor microenvironment, exhibiting many tumor-promoting effects, such as increasing the proliferation and survival of malignant cells, facilitating angiogenesis and metastasis, attenuating adaptive immune responses, and impairing responses to therapy [38-40]. Consequently, normalization of the tumor inflammatory microenvironment represents a new regimen for CAC treatment. On the other hand, long-term oxidative stress caused by overproduced reactive oxygen species (ROS) considerably contributes to the inflammation-cancer transformation [39, 40].
In view of the above unaddressed problems and based on our previous findings on ROS-scavenging anti-inflammatory materials [27], herein we hypothesize that ROS-triggered site-specific delivery of chemotherapeutic drugs, in combination with anti-inflammatory therapy via the functional nanocarrier itself, can serve as a new regimen for effective treatment of CAC by the oral route (Figure 1). As a proof of concept, a ROS-responsive and hydrogen peroxide-eliminating material (defined as OCD) was synthesized based on a cyclic polysaccharide. Both in vitro and in vivo experiments were performed to demonstrate anti-inflammatory activity of OCD NPs. Subsequently, a ROS-responsive nanotherapy was produced using irinotecan (i.e. camptothecin 11, abbreviated as CPT-11) as a model drug, and its in vitro release profiles were examined under the gastrointestinal conditions simulating oxidative stress. In addition to in vitro antitumor effects, in vivo targeting and efficacy of the ROS-responsive CPT-11 nanotherapy were investigated in a mouse model of CAC. The therapeutic benefits of normalizing pro-inflammatory microenvironment by the nanocarrier were also demonstrated.
Study design of targeted treatment of colitis-associated colon cancer (CAC) by normalizing inflammatory microenvironment using a functional nanovehicle. An antitumor drug CPT-11 is loaded into a nanoparticle (NP) derived from a ROS-responsive β-cyclodextrin material OCD. On the one hand, OCD NP can target diseased sites of CAC and selectively release the loaded CPT-11 molecules; while on the other hand, OCD is able to normalize inflammatory and oxidative microenvironment by eliminating over-produced H2O2. Desirable antitumor efficacy can be achieved by the ROS-responsive nanotherapy CPT-11/OCD NP, in combination with anti-inflammatory therapy with the nanocarrier OCD NP at the inflammatory phase.
Materials and methods
Materials
4-(Aminomethyl)phenylboronic acid pinacol ester (AM-PBAP), N,N'-carbonyldiimidazole (CDI), 4-dimethylaminopyridine (DMAP), α-amylase, and pepsin were purchased from Sigma-Aldrich (U.S.A.). Anhydrous dichloromethane (DCM) and anhydrous N,N'-dimethylformamide (DMF) were obtained from J&K Scientific Ltd. (China). Triethylamine (TEA) and lecithin were purchased from TCI (Tokyo, Japan). β-Cyclodextrin (β-CD) was supplied by Zhiyuan Biotechnology Co., Ltd. (China). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[(carbonyl-methoxy polyethylene glycol-2000) (DSPE-PEG) was obtained from Corden Pharma (Switzerland). Poly(lactide-co-glycolide) (PLGA, 75:25, with intrinsic viscosity of 0.50-0.65) was purchased from Polysciences Inc. (U.S.A.). Dulbecco's Modified Eagle's medium (DMEM), fetal bovine serum (FBS), and trypsin were purchased from Gibco (U.S.A.). Cyanine5 NHS ester (Cy5) and Cyanine7.5 NHS ester (Cy7.5) were obtained from Lumiprobe (U.S.A.). Nocodazole, amiloride, chlorpromazine, genistein, irinotecan (CPT-11), and Z-VAD-FMK were supplied by MedChemExpress (U.S.A.). Pyridinium iodide (PI) and FITC Annexin V Apoptosis Detection Kits containing 7-aminoactinomycin D (7-AAD) were purchased from Biolegend (U.S.A.). Alexa Fluor 488-labeled rabbit anti-mouse topoisomerase I monoclonal antibodies (#ab197505) and anti-E-cadherin antibodies (#ab76055) were supplied by Abcam (U.S.A.). PE-labeled rabbit anti-mouse cleaved caspase-3 monoclonal antibodies (#9978) and PE-labeled rabbit anti-mouse cleaved poly(ADP-ribose) polymerase (PARP) monoclonal antibodies (#67495) were purchased from Cell Signaling Technology, Inc. (U.S.A.). Cytofix/Cytoperm Plus Fixation/Permeabilization Kit (with BD GolgiStop) was purchased from BD Biosciences (U.S.A.). Azoxymethane (AOM), lipopolysaccharides (LPS) from E. coli, and phorbol-12-myristate-13-acetate (PMA) were obtained from Sigma-Aldrich (U.S.A.). Dextran sulfate sodium (DSS, Mw = 36-50 kDa) was purchased from MP Biomedicals (U.S.A.). Mouse lipocalin-2 ELISA kit was purchased from Boster Biological Technology Co. Ltd. (China), while other ELISA kits were obtained from Neobioscience (China). Kits for quantification of molecules related to oxidative stress were purchased from BioAssay Systems (U.S.A.). ROS Assay Kit based on 2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA) was obtained from Beyotime Biotechnology Co. Ltd. (China). All the other reagents are commercially available and used as received.
Synthesis and characterization of a H2O2-eliminating material based on β-CD
A H2O2-eliminating β-CD material (OCD) was synthesized according to our previously established method [27]. Briefly, β-CD (0.25 g, 0.22 mmol) and CDI (0.249 g, 1.54 mmol) were dissolved in anhydrous DMF at room temperature. After constant magnetic stirring for 90 min, the reaction mixture was added to anhydrous diethyl ether. Then the solution was filtered and thoroughly rinsed with ether to obtain CDI-activated β-CD (CDI-CD). Subsequently, AM-PBAP (0.359 g, 1.54 mmol) and CDI-CD were dissolved in DMF, into which 0.25 mL of TEA was added. After stirring overnight, the final product OCD was precipitated from ether, collected by centrifugation at 5752g for 5 min, and dried to give a white powder.
Materials characterization
1H NMR spectra were acquired using an Agilent DD2 600 MHz NMR spectrometer. Fourier-transform infrared (FT-IR) spectroscopy was performed on a PerkinElmer spectrometer (100S, U.S.A.). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) was conducted on a MALDI-7090 TOF-TOF mass spectrometer (Shimadzu).
Characterization of inclusion interaction between CPT-11 and OCD
To characterize inclusion formation of OCD and CPT-11, they were co-dissolved in DMSO-d6 at a molar ratio of 1:1. Then 2D 1H-1H NOESY spectra of OCD/CPT-11 were acquired on an Agilent NMR spectrometer with a mixing time of 300 ms.
Fabrication of various NPs by a nanoprecipitation/self-assembly method
All NPs based on different materials were prepared by a modified nanoprecipitation/self-assembly method. Briefly, 50 mg of the carrier material (OCD or PLGA) was dissolved in 2 mL of methanol (for OCD) or acetonitrile (in the case of PLGA). The obtained solution was added dropwise into 10 mL of deionized water containing 6 mg DSPE-PEG and 4 mg lecithin that was preheated at 65°C. The suspension was stirred at room temperature for 2 h. Then the organic solvent and water were removed by vacuum evaporation at 60°C and concentrated to a volume of 8 mL. Finally, NPs were obtained by freeze-drying. NPs containing different payloads (Cy5, Cy7.5, or CPT-11) were fabricated by following the similar procedures. In this case, free payload was removed by dialysis (MWCO: 3500 Da) against deionized water for 24 h, and payload containing NPs were collected after lyophilization.
Characterization of NPs
Dynamic light scattering and ζ-potential measurements were performed on a Malvern Zetasizer Nano ZS instrument at 25°C. Transmission electron microscopy (TEM) observation was carried out using a JEM-1400 microscope (JEOL, Japan). Differential scanning calorimetry (DSC) was conducted on a Q2000 instrument (TA, U.S.A.). To quantify the drug content in NPs, 5 mg lyophilized drug-loaded NPs was dispersed in ethanol for extraction. The drug concentration was quantified by UV-Vis spectrophotometry (TU-1901, Persee Co., Ltd., Beijing). The detection wavelength for CPT-11, Cy5, and Cy7.5 was 372, 646, and 788 nm, respectively.
In vitro H2O2-eliminating capability of OCD or OCD NP
Accurately weighed samples of OCD or OCD NP were incubated in 4 mL of PBS containing 1.0 mM H2O2 at 37°C for 24 h. Using the QuantiChrom peroxide assay kit (BioAssay Systems, U.S.A.), the concentration of remaining H2O2 was quantified by measuring the absorbance at 585 nm, and the H2O2-eliminating capacity was calculated. Similarly, we examined the effects of buffer pH values on the H2O2-scavenging capability of OCD or OCD NP.
In vitro hydrolysis of OCD NP in different solutions
The hydrolysis behavior of OCD NP (1.0 mg/mL) was examined in 4 mL of PBS (10 mM, pH 7.4) containing various concentrations of H2O2 at 37°C. Quantitative experiments were conducted by measuring the absorbance of NP-containing aqueous solutions at 500 nm after incubation for various periods of time (0, 5, 10, 15, 20, 25, 30, 40, 60, 80, 120, 180, 240, and 300 min). Similarly, hydrolysis of OCD NP in buffer solutions with or without 1.0 mM H2O2 was studied at different pH values (varying from pH 1.2, 4.8, 7.4, 9.0, to 11.0). In addition, hydrolysis profiles of OCD NP were tested in the presence of different digestive enzymes including α-amylase, pepsin, and trypsin.
In vitro drug release tests of CPT-11/OCD NP
Briefly, 10 mg CPT-11/OCD NP was incubated in 6 mL of fresh medium. At predetermined time points, 3 mL of release medium was withdrawn. The concentration of CPT-11 was measured by UV-Vis spectrometry at 372 nm. Through the similar procedures, release behaviors were tested in the presence of different concentrations of H2O2. In addition, simulated gastric fluid (50 mL of PBS containing 0.16 g pepsin, 0.1 g NaCl, 0.35 mL of 38% HCl solution) and simulated intestinal fluid (50 mL of PBS with 0.5 g trypsin, 0.34 g KH2PO4, and 76 mg NaOH) were prepared. After NPs were incubated with these two simulated gastrointestinal fluids for 8 h, the concentration of CPT-11 in release medium was detected. Also, size distribution and ζ-potential of CPT-11/OCD NP in PBS or two simulated gastrointestinal fluids were determined. In a separate study, changes of average hydrodynamic diameter and polydispersity index (PDI) of CPT-11/OCD NP during 8 h of incubation were determined.
Cytotoxicity evaluation by MTT assay
C26 mouse colon carcinoma cells and Caco-2 human intestinal epithelial cells were cultured in DMEM supplemented with 10% (v/v) FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin in a 5% CO2 humidified environment at 37°C. For the methyl thiazolyl tetrazolium (MTT) assay, cells were planted at 1 × 104 cells/well in 96-well plates for 24 h before OCD NP was added. Subsequently, cells were treated with the medium containing NPs at various concentrations for 12 or 24 h. The cell viability was quantified by the MTT assay.
Intracellular uptake of OCD NP by C26 cells
C26 cells were seeded in 12-well plates at a density of 2 × 105 cells per well in 1 mL of growth medium. After 24 h, the culture medium was replaced with 1 mL of fresh medium containing 5 μg/mL Cy5-loaded OCD NP and incubated for various periods of time (0.5, 1, 2, 4, and 8 h). Then, the cells were digested and fluorescence intensity was determined using a flow cytometer (BD Accuri C6). Through similar procedures, dose-dependent (1.25, 2.5, 5, 10, and 20 μg/mL Cy5/OCD NP) internalization profiles were examined after 2 h of incubation. In order to confirm the specific pathways dominating cellular uptake of OCD NP in C26 cells, cells were pretreated with different endocytic inhibitors (including nocodazole, amiloride, chlorpromazine, genistein, and sodium azide), and then co-incubated with Cy5/OCD NP (at 5 μg/mL) for 2 h. Quantitative analysis was performed by flow cytometry.
In vitro anti-inflammatory activity of OCD NP
Firstly, we examined in vitro anti-inflammatory effects of OCD NP in typical inflammatory cells. Specifically, murine macrophage RAW264.7 cells (2 × 105 cells/well) were cultured in 12-well plates with culture medium (RPMI-1640, 10 wt% FBS, 1 wt% penicillin-streptomycin solution) for 12 h. Then fresh medium was changed and cultured in the presence of 100 ng/mL PMA for 1 h, followed by incubation with 100 μg/mL OCD NP or PLGA NP for 6 h. Subsequently, cells were washed three times with Hank's balanced salt solution (HBSS) and treated with 10 μM DCFH-DA in the dark at 37°C for 40 min. Collected cells were washed three times with HBSS, and fluorescent intensities were measured by a flow cytometer (Accuri C6, BD). After culture media were collected, the levels of inflammatory cytokines including tumor necrosis factor (TNF)-α and interleukin (IL)-6 were determined by ELISA kits (Neobioscience) according to the manufacturer's instructions. Through the similar procedures, anti-inflammatory effects of OCD NP were investigated in neutrophils that were collected from the peritoneal cavity of BALB/c mice after stimulation with PMA.
In addition, anti-inflammatory activity of OCD NP was interrogated in intestinal epithelial cells. In brief, Caco-2 human epithelial cells (2 × 105 cells/well) were cultured in 12-well plates with RPMI-1640 for 12 h. Then cells were incubated in fresh medium containing 20 μg/mL LPS for 1 h, followed by incubation with 100 μg/mL OCD NP or PLGA NP for 6 h. Subsequently, the levels of TNF-α and IL-1β were quantified by ELISA kits.
Effects of CPT-11/OCD NP treatment on the cell cycle of C26 cells
The effects of CPT-11/OCD NP treatment on the cell cycle of C26 cells were evaluated by quantifying the number of G2/M cells. After treatment with CPT-11/OCD NP (at 30 μg/mL of CPT-11) for 6 h. Cells were collected and washed with PBS, and then resuspended in PBS containing 0.03% Triton X-100, 200 mg/mL RNase A, and 50 μg/mL PI. After further incubation at room temperature in the dark for 15 min, cell cycle analysis was performed by flow cytometry of 10000 cells in each group (BD Accuri C6). Data were analyzed using the FlowJo V10 software. The percentage of cells in the G2/M phase was calculated to assess proliferation of C26 cells.
In vitro anti-apoptotic activity of CPT-11/OCD NP in C26 cells
Analysis of apoptosis was conducted using FITC Annexin V Apoptosis Detection Kit with 7-AAD according to the manufacture's protocols. Briefly, C26 cells were seeded in a 6-well plate at 4 × 105 cells/well and incubated overnight. The medium was then replaced with fresh culture medium containing CPT-11, CPT-11/PLGA NP, or CPT-11/OCD NP at 30 μg/mL CPT-11 for all formulations. After 6 h of incubation, cells were washed with BioLegend's cell staining buffer, digested with 0.25 wt% trypsin, and collected by centrifugation. After the cells were resuspended in 100 μL of annexin V binding buffer with 2.5 μL of annexin V and 5 μL of 7-AAD viability staining solution at 1 × 105 cells/mL, they were vortexed gently and incubated in the dark for 15 min. Finally, 400 μL of Annexin V binding buffer was added for analysis by flow cytometry.
Effects of CPT-11/OCD NP on the expression of topoisomerase I, cleaved caspase-3, and cleaved PARP in C26 cells
C26 cells were seeded in 12-well plates at 2 × 105 cells/well and incubated with growth medium overnight. Then cells were treated with CPT-11, CPT-11/PLGA NP, or CPT-11/OCD NP at 30 μg/mL of CPT-11. After 6 h, cells were washed, digested, collected, and then resuspended in 1 mL of PBS containing 250 μL of fixation/permeabilization solution at 4°C for 20 min. Subsequently, cells were washed two times with 1 mL of BD Perm/Wash buffer, and resuspended in 200 μL of BD Perm/Wash buffer. After cells were stained with Alexa Fluor 488-labeled topoisomerase I antibody at 22°C for 30 min, flow cytometry analysis was performed. Similarly, after the cells were incubated with CPT-11/OCD NP and 10 μM Z-VAD-FMK, the expression levels of cleaved caspase-3 and PARP were quantified by flow cytometry.
Animals
All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals proposed by the National Institutes of Health. All procedures and protocols were approved by the Animal Ethics Committee at Third Military Medical University. BALB/c mice (18-20 g) were obtained from the Animal Center at the Third Military Medical University. Animals were housed in standard mouse cages under conditions of optimum light, temperature, and humidity, with ad libitum access to water and food. Before further experiments were performed, all mice were acclimatized for at least 7 days.
Induction of colitis-associated cancer by AOM/DSS in mice
Colitis-associated cancer (CAC) was induced according to the previously described protocols [41], with some modifications. Briefly, mice were intraperitoneally injected with AOM at 10 mg/kg body weight. Then mice were subjected to three cycles of DSS treatment, each consisting of 2% DSS for 7 days followed by a 14-day recovery period with regular water.
Study on the localization of orally delivered OCD NP in diseased mice by ex vivo imaging
Mice with CAC were induced with AOM/DSS as mentioned above. At day 22, Cy7.5-labeled OCD NP or PLGA NP was orally administered at a dose of 0.5 mg Cy7.5 in each mouse, while free Cy7.5 was used as a control. At 6 h after administration, whole colon tissues were harvested and imaged simultaneously using an IVIS Spectrum system (Ex/Em, 745/820 nm; exposure time, 2 s; binning, 8; F/Stop, 1). Fluorescence intensity of the colon tissue was then analyzed by Living Image Software.
Evaluation on the degree of CAC
At the end of the experiment, colon tissues were isolated. Colonic tumors were counted and measured using a dissecting microscope. According to the oncology evaluation criteria, the number of tumors per mouse, the average size of tumors, and the size distribution of tumors were determined. In addition, the tumor burden was calculated based on the tumor number and tumor size to assess the degree of CAC.
Quantification of MPO, pro-inflammatory cytokines, and oxidative mediators in the colonic tissues
The colonic tissues were isolated from mice after different treatments, and they were homogenized in ice-cold potassium phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethylammonium bromide. The homogenates were sonicated, freeze-thawed 3 times, and centrifuged at 10,621g at 4°C for 15 min. The levels of myeloperoxidase (MPO), TNF-α, IL-1β, and IL-6 in the supernatant were determined using the corresponding detection kits.
To quantify the level of H2O2, 100 mL of supernatant from the tissue homogenate was mixed with 1 mL of aqueous solution of molybdic acid (2.4 mmol/mL) that was preheated at 37°C. After the mixture was incubated for 1 min, 1 mL of the termination reagent was added. Then the complex formed by H2O2 and molybdic acid was measured at 405 nm, and the concentration of H2O2 was calculated. As for malondialdehyde (MDA), its level in the supernatant of homogenate was quantified by using QuantiChrom TBARS assay kit.
Immunofluorescence analysis of TNF-α in the colon
After different treatments, colonic tissues were isolated and embedded in Tissue-Tek O.C.T. Compound immediately. Histological sections were then prepared and incubated with primary rabbit anti-TNF-α antibody overnight at 4°C. Subsequently, the slides were incubated with the secondary antibody of Alexa Fluor 647-conjugated goat anti-rabbit IgG at room temperature for 1 h. After washing with PBS, sections were stained with DAPI. Fluorescence images were acquired using a Zeiss LSM confocal system.
Histological and immunohistochemical assessments
After tissues were fixed in 10% formalin, they were embedded in paraffin. Tissue sections (6-μm thickness) were stained with hematoxylin and eosin (H&E). For staining with E-cadherin, paraffin-embedded colon sections of 6-μm thickness were deparaffinized in xylene, incubated in 3% H2O2 in PBS for 30 min, and treated with 10 mM sodium citrate buffer (pH 6.0) containing 0.05% Tween 20, followed by heating in a pressure cooker for 10 min (antigen retrieval). Sections were blocked with goat serum at 37°C for 45 min, and then incubated with E-cadherin antibody (Abcam) at 37°C for 1 h. After washing with PBS containing 0.01% Tween 20, sections were incubated with biotinylated secondary antibody at 37°C for 30 min, and then treated with reagents from the Vectastain ABC kit (Vector Laboratories) to allow color development. After counterstaining with hematoxylin, sections were observed by fluorescence microscopy (Olympus, Japan).
TUNEL assay
Apoptotic cells were detected by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay using the in situ cell death detection kit (Roche Diagnostics, U.S.A.). Images were acquired using an Olympus fluorescence microscope equipped with a Hamamatsu ORCA03G digital camera.
Inhibition of colonic inflammation in CAC mice by OCD NP
To evaluate the anti-inflammatory effect of OCD NP, mice with CAC were induced by AOM/DSS as aforementioned, which were then randomly assigned to 3 groups. In the diseased control group, mice were untreated during days 7-21. For the OCD NP and PLGA NP groups, mice were orally administered with OCD NP or PLGA NP at 30 mg/kg every day during days 7-21, respectively. In addition, healthy mice without AOM/DSS treatment were used in the normal control. Body weight, feces, and physical activity were monitored daily. The levels of lipocalin-2 (Lcn-2) in feces were determined every two days. At day 22, all mice were euthanized, and colons were harvested. After homogenization, the homogenate supernatant was used to determine the levels of MPO, TNF-α, IL-1β, and IL-6. The H2O2 and MDA levels in colonic tissues were measured as aforementioned. In addition, the major organs and colon tissues were paraffin-embedded and sectioned for H&E or E-cadherin immunohistochemical staining.
In vivo pharmacokinetic study
Mice were intraperitoneally injected with AOM at 10 mg/kg, followed by treatment with 2% DSS for 7 days. Then mice were orally administered with different CPT-11 formulations at 5 mg/kg of CPT-11. At predefined time points (15, 30, 60, 120, 180, 240, and 480 min), blood samples and colonic tissues were collected. The whole blood samples were immediately centrifuged at 12,000g for 2 min to obtain plasma. Then 800 μL of ice-cold methanol was added into 200 μL of plasma to precipitate protein. After centrifugation at 12,000g for 2 min, the supernatant was purged with nitrogen, reconstituted with methanol, and detected by high-performance liquid chromatography (HPLC) [42]. The mobile phase was consisted of acetonitrile and ammonium acetate buffer (75 mM, pH 6.4) containing 5 mM tetrabutylammonium phosphate at 22:78 (v/v). Reversed-phase C18 columns were used, and detection wavelength was 370 nm. To determine drug distribution in colonic tissues, they were homogenized and centrifuged. CPT-11 in the supernatant was extracted with acetonitrile. Quantification was also performed by HPLC. Typical pharmacokinetic parameters such as the maximum plasma/colon concentration (Cmax), time to reach Cmax (Tmax), and the area under the plasma/colon drug concentration-time curve (AUC) were calculated.
In vivo efficacies of CPT-11/OCD NP in CAC mice
According to the aforementioned procedures, the mouse model of CAC was established by induction with AOM/DSS during days 1-7, days 22-27, and days 43-48. Then mice were divided into five groups: the AOM/DSS group, AOM/DSS-induced mice without treatment during the periods of days 7-21, days 28-42, and days 49-63; the CPT-11 group, AOM/DSS-induced mice without treatment during days 7-21, and treated with 5 mg/kg CPT-11 during days 28-42 and days 49-63; the T1 group, AOM/DSS-induced mice without treatment during days 7-21, and treated with CPT-11/OCD NP during days 28-42 and days 49-63; the T2 group, AOM/DSS-induced mice treated with OCD NP during days 7-21, and treated with CPT-11/OCD NP during days 28-42 and days 49-63; the TP group, AOM/DSS-induced mice treated with PLGA NP during days 7-21, and treated with CPT-11/PLGA NP during days 28-42 and days 49-63. For all CPT-11 nanotherapies, their dose was 5 mg/kg of CPT-11. In addition, healthy mice without treatment with AOM/DSS were used in the control group.
The changes of body weight were measured once a week, while Lcn-2 levels in feces were determined by kits at day 21, 42 and 63, respectively. At the end of experiment at day 64, colonic tumors were counted and measured using a dissecting microscope. Major organs and colon tissues were collected, and the related organ index was calculated. The indicators of oxidative stress and pro-inflammatory factors were determined by commercially available kits. Paraffin sections of colon tissues were subjected to H&E staining or E-cadherin immunohistochemical analysis. In addition, the colon cryosections were used to TUNEL assay.
Preliminary safety evaluation of CPT-11/OCD NP
Male BALB/c mice were randomly assigned into three groups (n = 10). CPT-11/OCD NP in saline was orally administered at 1.5 or 3.0 g/kg. In the control group, mice were orally administered with the same volume of saline. After administration, the body weight of mice and their behaviors were monitored for any signs of illness each day. After two weeks, mice were euthanized. Blood samples were collected for hematological analysis and quantification of biochemical markers relevant to liver/kidney functions. Major organs were resected and weighed, and their organ index was calculated. Histopathological sections were prepared and stained with H&E.
Statistical analysis
Data are expressed as mean ± standard deviation (SD). Statistical analysis was assessed using one-way ANOVA test. A value of p < 0.05 was considered statistically significant.
Results and Discussion
Synthesis and characterization of a H2O2-eliminating material OCD
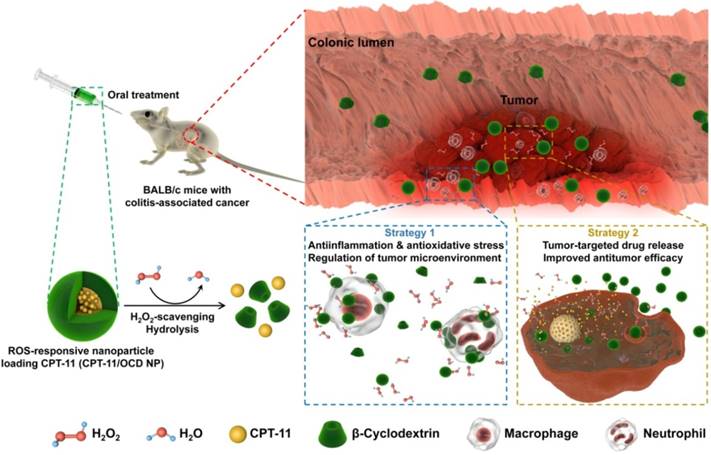
According to our previously established methods [27], a H2O2-eliminating material OCD was synthesized by chemical functionalization of β-CD with an oxidation-labile moiety of AM-PBAP (Figure 2A-B). β-CD was used as a scaffold molecule due to its multiple advantages, such as low cost, well-defined structure, good in vivo safety, and inclusion capability with various compounds for further functionalization or therapeutic loading, which are very important for translation studies [43]. Of note, CDs and their derivatives have already been widely used in different drug formulations [44]. The chemical structure of synthesized OCD was confirmed by 1H NMR, FT-IR, and MALDI-TOF mass spectrometry (Figure S1A-C). Calculation based on the 1H NMR spectrum revealed approximately 5 AM-PBAP units were conjugated onto each β-CD molecule. According to our previous findings on the hydrolysis mechanisms of PBAP-based β-CD materials [27], in the presence of H2O2, the PBAP moiety may be oxidized to a phenolic compound that undergoes a quinone methide rearrangement (Figure S2A). Through a series of electron transfer processes, OCD can be rapidly hydrolyzed into water-soluble products, including β-CD, 4-hydroxybenzylamine, pinacol, and boric acid (Figure S2B-C). Concomitant with hydrolysis in PBS at pH 7.4, OCD was able to efficiently eliminate H2O2, in a dose-dependent manner (Figure 2C). Of note, the H2O2-eliminating capability of OCD was closely associated with pH of buffer solutions, which was remarkably enhanced with the increased pH value when it was below pH 7.4 (Figure 2D). This pH-dependent H2O2-scavenging capacity is related to the pH-sensitive hydrolysis performance of OCD. As demonstrated in our previous study [27], hydrolysis of OCD was considerably inhibited in buffers with low pH. This can be attributed to the fact that hydroxyl ion is generally required for hydrolysis of pinacol ester, while low pH may suppress the removal of pinacol from AM-PBAP, thereby impairing hydrolysis of OCD NP [45]. Collectively, these results demonstrated that the ROS-responsive material OCD can effectively eliminate H2O2.
Preparation and physicochemical characterization of a H2O2-eliminating nanovehicle. A-B, Schematic illustration of the synthesis of an oxidation-responsive material OCD and engineering of a H2O2-eliminating nanocarrier OCD NP. C, Dose-dependent elimination of H2O2 in PBS by OCD at pH 7.4. D, The effect of pH values of buffer solutions on H2O2-eliminating capability of OCD. E-F, TEM image (E) and size distribution profile (F) of OCD NP. G-H, Hydrolysis profiles of OCD NP in buffers with various concentrations of H2O2 (G) or at different pH values with 1 mM H2O2 (H). I, OCD NP dose-dependent elimination of H2O2 in PBS at pH 7.4. J, H2O2-eliminating capability of OCD NP at different pH values. All data are presented as mean ± SD (n = 3).
Preparation and characterization of a H2O2-eliminating nanocarrier
By a modified nanoprecipitation/self-assembly method [46], OCD NP was prepared in the presence of a small amount of lecithin and DSPE-PEG (Figure 2B). Lecithin was used to introduce an amphiphilic layer around the hydrophobic core of OCD, in which the DSPE-PEG component can be effectively anchored to provide resulting nanovehicles with peripheral PEG chains, thereby affording good colloidal stability. Characterization by TEM suggested that OCD NP exhibited a well-defined spherical shape (Figure 2E), with relatively narrow size distribution (Figure 2F). The mean hydrodynamic diameter of OCD NP was 103 ± 4 nm, and the ζ-potential was -32 mV. In addition, NPs based on a biodegradable polymer PLGA that has been approved by U.S. Food and Drug Administration were prepared via the same method and used as a control without H2O2-scavenging capability (Figure S3A). The mean hydrodynamic diameter of PLGA NP was 95 ± 1 nm.
Hydrolysis behavior and H2O2-eliminating capability of OCD NP
The hydrolysis profiles of OCD NP were first tested in buffers containing various concentrations of H2O2 (Figure 2G). As expected, only slight hydrolysis occurred after OCD NP was incubated in PBS (pH 7.4) without H2O2 for 5 h. By contrast, remarkably accelerated hydrolysis profiles were observed with increase in the H2O2 concentration. In PBS (pH 7.4) containing 1.0 mM of H2O2, a hydrolysis percentage of 80% was found at 180 min after incubation. Accordingly, hydrolysis of OCD NP was highly sensitive to the level of H2O2. To interrogate the hydrolysis of OCD NP under various pH conditions, which is intimately related to oral drug delivery, experiments were conducted in buffers with 1.0 mM H2O2 at different pH values. In the entire pH spectrum corresponding to the gastrointestinal tract, OCD NP displayed notably rapid hydrolysis at pH 7.4 compared to that at other pH values (Figure 2H). At pH 1.2, even with H2O2, this low pH considerably inhibited hydrolysis of OCD NP.
Considering that α-amylase is capable of hydrolyzing CDs [47], we also investigated the stability of OCD NP in aqueous solution of α-amylase. It was found that OCD NP was relatively stable for at least 24 h in the presence of α-amylase (Figure S4A). On the one hand, the conjugated PBAP groups may inhibit contact of α-amylase to the β-CD skeleton by the steric hindrance effects. Meanwhile, the hydrophobic core of OCD NP is surrounded by a hydrophilic PEG shell, which may further reduce the binding of α-amylase to OCD. These effects collectively accounted for the stability of OCD NP in the presence of α-amylase. Also, our results showed that OCD NP was stable upon incubation in aqueous solutions containing other digestive enzymes such as pepsin and trypsin (Figure S4B-C). Consequently, these results demonstrated that OCD NP can protect the loaded drugs from hydrolysis by digestive enzymes in the gastrointestinal tract.
Similar to that of OCD, the H2O2-eliminating capability of OCD NP was also dependent on its dose (Figure 2I). The amount of eliminated H2O2 was almost linearly proportional to the quantity of OCD NP. The calculated H2O2-eliminating efficiency of OCD NP was approximately 0.33 μmol/mg. Also, in line with the hydrolysis profile, low pH reduced the H2O2-eliminating ability of OCD NP (Figure 2J). Together, these data substantiated that OCD NP can be rapidly hydrolyzed in response to high levels of H2O2, concomitant with elimination of H2O2 in dose and pH-dependent patterns.
Internalization of OCD NP in C26 murine colon carcinoma cells
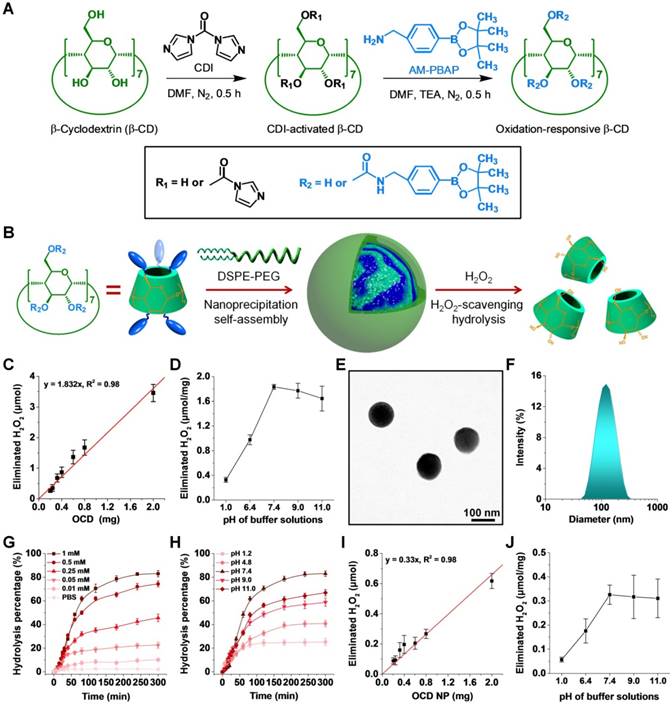
It is well known that CPT-11 achieves its antitumor activity by inhibiting type I topoisomerase, which is expressed in the nucleus [48]. Accordingly, cellular internalization is a prerequisite for CTP-11 nanotherapies to perform their therapeutic effects. First, we demonstrated that the blank nanocarrier OCD NP showed negligible cytotoxicity in C26 murine colon carcinoma cells and human epithelial colorectal adenocarcinoma Caco-2 cells after 12 h of incubation at the examined doses (Figure S5). Even at a dose as high as 1000 μg/mL, high viability was detected for both cells. Then, we examined cellular uptake profiles of OCD NP by flow cytometry, using Cy5-labeled OCD NP. With prolonged incubation, internalized Cy5/OCD NP notably increased (Figure 3A-B). In dose-dependent experiments, flow cytometry analysis showed enhanced endocytosis of Cy5/OCD NP in C26 cells with increase in its dose (Figure 3C-D).
In vitro intracellular uptake of OCD NP by C26 murine colon carcinoma cells. A-B, Representative flow cytometry curves (A) and quantitative data (B) illustrating time-dependent internalization of 5 μg/mL Cy5/OCD NP in C26 cells. C-D, Flow cytometry profiles (C) and quantitative results (D) of cellular uptake of Cy5/OCD NP in a dose-dependent manner. In this case, the incubation time was 2 h. E-F, Flow cytometry profiles (E) and quantitative results (F) indicating the effects of different endocytosis inhibitors on internalization of Cy5/OCD NP in C26 cells. In all cases, the dose of Cy5/OCD NP was 5 μg/mL, while the incubation time was 2 h. All data are presented as mean ± SD (n = 3). **p < 0.01, ***p < 0.001; ns, no significance.
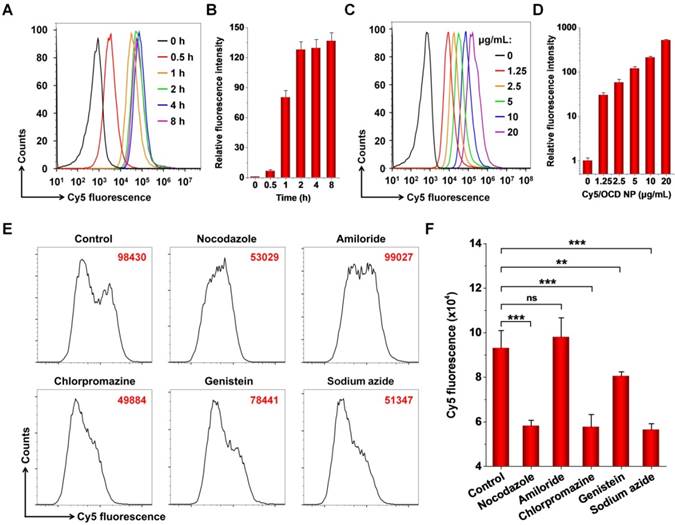
To address the pathways responsible for endocytosis of OCD NP by C26 cells, different inhibitors were used. Among them, nocodazole can inhibit the polymerization of F-actin and induce microtubule depolymerization [49], while amiloride is an inhibitor of micropinocytosis [50]. Chlorpromazine may suppress clathrin-mediated endocytosis [51], and genistein is capable of attenuating caveolae-mediated endocytosis [52]. On the other hand, sodium azide can deplete cellular ATP [53], while it is required for energy-dependent internalization. Flow cytometric analysis showed that cellular uptake of Cy5/OCD NP was significantly inhibited when C26 cells were pre-incubated with nocodazole, chlorpromazine, genistein, or sodium azide, but amiloride had no significant effect (Figure 3E-F). These results suggested that endocytosis of OCD NP in C26 cells can be realized by multiple pathways, including caveolae- and clathrin-mediated ATP-dependent processes. As well documented, NPs internalized by clathrin-mediated endocytosis will eventually be transported to endolysosomes [54]. Our previous studies showed that endocytosed NPs based on ROS-responsive β-CD materials were co-localized with LysoTracker-labeled endolysosomes in different cells [46, 55, 56]. Nevertheless, endolysosomal escape was also observed after prolonged incubation. Accordingly, the endolysosomal pathways are mainly responsible for intracellular trafficking of OCD NP, agreeing with previous studies on other NPs [57]. Furthermore, compared to RAW264.7 macrophages in the resting state, C26 cells exhibited a relatively high level of intracellular ROS (Figure S6), which can promote ROS-responsive hydrolysis of OCD NP and triggerable drug release in C26 cells.
In vitro anti-inflammatory activity of OCD NP in different cells
Based on the H2O2-eliminating capacity and cellular internalization performance of OCD NP, we examined its anti-inflammatory effects in different cells, including RAW264.7 macrophages, peritoneal neutrophils, and Caco-2 epithelial cells. Whereas the level of intracellular ROS in RAW264.7 cells significantly increased by stimulation with PMA, treatment with OCD NP significantly reduced ROS generation (Figure 4A-B). Meanwhile, OCD NP significantly attenuated the expression of TNF-α and IL-6 in PMA-induced RAW264.7 cells (Figure 4C-D). In both cases, PLGA NP had no significant effects. Of note, both OCD NP and PLGA NP did not increase the levels of ROS, TNF-α, and IL-6. Similarly, we found that OCD NP effectively inhibited oxidative stress and inflammatory response in PMA-stimulated peritoneal neutrophils (Figure 4E-H). In this case, treatment with PLGA NP even potentiated PMA-mediated oxidative stress (Figure 4E-F). These results demonstrated that OCD NP displays anti-oxidative stress and anti-inflammatory effects in inflammatory cells.
In vitro anti-oxidative stress and anti-inflammatory activity of OCD NP. A-B, Typical flow cytometry profiles (A) and quantitative analysis (B) of relative ROS levels in PMA-stimulated RAW264.7 macrophages. C-D, Relative levels of TNF-α (C) and IL-6 (D) in RAW264.7 cells. E-F, Flow cytometric curves (E) and quantitative data (F) of relative levels of ROS in peritoneal neutrophils stimulated with PMA. G-H, Relative levels of TNF-α (G) and IL-6 (H) in neutrophils. In these cases, RAW264.7 cells or peritoneal neutrophils were first exposed to 100 ng/mL PMA for 1 h, followed by incubation with NPs at 100 μg/mL for 6 h. Cells were incubated with 10 μM DCFH-DA for analysis of intracellular ROS. In another parallel experiment, TNF-α and IL-6 in cell lysates were measured by ELISA. The same experiment was performed in mouse peritoneal neutrophils (E-H). All data are presented as mean ± SD (n = 5). **p < 0.01, ***p < 0.001; ns, no significance.
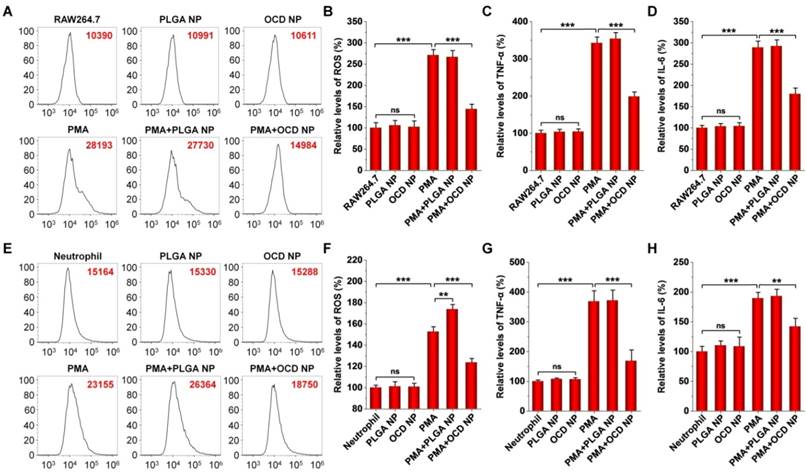
On the other hand, inflammatory response in intestinal epithelial cells induced by endotoxins is an important cause of colitis and colon cancer [58]. We found that OCD NP reduced the levels of TNF-α and IL-1β in epithelial-like Caco-2 cells stimulated by LPS (also known as endotoxins) (Figure S7). Collectively, these results demonstrated that OCD NP can effectively inhibit oxidative stress and attenuate inflammatory response in both inflammatory cells and epithelial cells.
Accumulation of OCD NP in the colon of mice with induced CAC
A common complication of ulcerative colitis is CAC [59]. The exacerbated activation and maintenance of inflammation, without inadequate resolution, can promote malignant progression and eventually leads to tumor development [36]. The selective accumulation of NPs in the colon is the first step toward effective oral therapy of CAC. Therefore we first examined the oral targeting capability of OCD NP in mice with AOM/DSS-induced CAC that recapitulates the aberrant crypt foci-adenoma-carcinoma sequence (Figure 5A) [60]. In this aspect, Cy7.5-labeled OCD NP was prepared (Figure S3B), and another Cy7.5-labeled nanovehicle based on PLGA was used as the control (Figure S3C). At 6 h after oral administration of different Cy7.5 formulations, ex vivo imaging showed a considerably high fluorescent signal in the colon isolated from CAC mice treated with Cy7.5/OCD NP, compared to that of mice administered with free Cy7.5 or Cy7.5/PLGA NP (Figure 5B). Further quantitative analysis confirmed the highest accumulation of Cy7.5/OCD NP, although Cy7.5/PLGA NP also exhibited stronger fluorescence than Cy7.5 (Figure 5C).
Accumulation of orally administered OCD NP in the colon of mice with AOM/DSS-induced colitis-associated colon cancer (CAC). A, Schematic of establishment of CAC in mice by AOM/DSS induction and treatment with different Cy7.5 formulations by oral administration. B-C, Representative ex vivo images (B) and quantitative analysis (C) illustrating distribution of Cy7.5 fluorescence signals in colon tissues at 6 h after oral administration. All data are presented as mean ± SD (n = 3). ***p < 0.001.
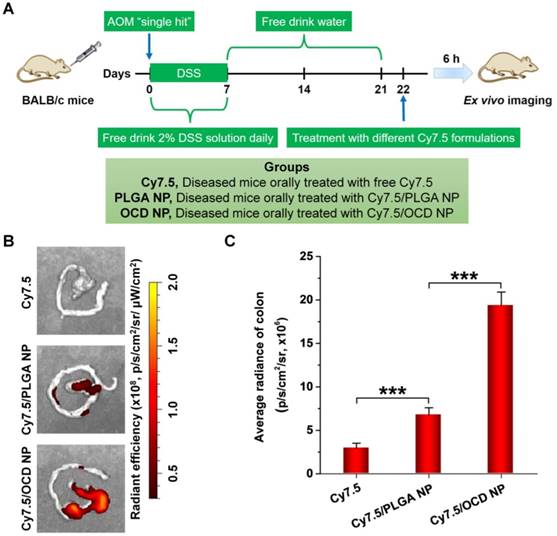
Taken together, these results demonstrated that orally administered OCD NP can selectively accumulate in the inflamed colon of CAC mice. On the one hand, this is mainly attributed to inflammation-mediated opening of the tight junctions in epithelial cells that can notably increase colonic mucosal permeability [61]. On the other hand, the stability of OCD NP in the upper gastrointestinal tract guarantees its successful transport to the colon, which is a prerequisite for colon targeting. Notably, the targeting efficiency of OCD NP was significantly higher than that of control PLGA NP, which should be attributed to the multiple binding interactions of OCD with biological molecules upon triggering by the inflammatory microenvironment [55, 62]. In addition, positive charges on the intestinal epithelium during inflammation can facilitate their electrostatic interactions with OCD NP that displays more negative charges after ROS-triggered hydrolysis [62, 63]. This finding also suggested that, by packaging into OCD NP, the loaded antitumor molecules can be preferentially targeted to the colitis/tumor tissue, thereby effectively maximizing their bioavailability at diseased sites and decreasing their indiscriminate distribution in healthy organs.
Oral treatment with OCD NP inhibited the progression of inflammation in CAC mice
Based on the above promising result of OCD NP targeting at diseased colon tissues in CAC mice, in combination with its excellent H2O2-eliminating capability, we further examined in vivo anti-inflammatory activity of OCD NP in CAC mice (Figure 6A). Lcn-2 is a well-recognized sensitive biomarker for intestinal inflammation, and the level of fecal Lcn-2 can reflect the degree of inflammation in the colon of mice [64]. Compared to the normal control, mice induced with AOM/DSS showed significantly higher levels of fecal Lcn-2 (Figure 6B). Oral administration of OCD NP effectively reduced Lcn-2 levels. By contrast, PLGA NP had no therapeutic effects at all examined time points. In addition, CAC mice displayed significantly shortened colon length compared to normal mice, while OCD NP treatment maintained the colon length (Figure 6C). Further, we quantified different mediators relevant to oxidative stress and inflammatory response in colon tissues. For CAC mice, the levels of H2O2 and MDA significantly increased (Figure 6D-E). Treatment with the control nanovehicle PLGA NP showed no therapeutic effects, consistent with its inert and inactive properties. By contrast, OCD NP effectively decreased both H2O2 and MDA in colon tissues. Correspondingly, treatment with OCD NP dramatically reduced the expression of typical inflammatory mediators including MPO, TNF-α, IL-1β, and IL-6 (Figure 6F-I), while PLGA NP displayed no significant benefits. Also, immunofluorescence staining confirmed that the expression of TNF-α in colon sections remarkably decreased by oral treatment with OCD NP (Figure S8).
OCD NP attenuates the progression of inflammation in CAC mice. A, Protocols for induction of CAC by AOM/DSS and treatment regimens. B, Lcn-2 levels in feces during the inflammation stage. C, Representative digital photos (left) and quantitative data (right) showing colon length of mice at day 21 after different treatments. D-E, The relative levels of H2O2 (D) and MDA (E) in colon tissues isolated from mice at day 21 after receiving treatment in the inflammatory phase. F-I, The levels of MPO (F), TNF-α (G), IL-1β (H), and IL-6 (I) in colon tissues. J-K, Representative microscopic images of H&E-stained sections (J) and immunohistochemistry analysis of E-cadherin in sections (K) of colon tissues after treatment with different formulations. Scale bars, 100 μm. All data are presented as mean ± SD (n = 10). *p < 0.05, **p < 0.01, ***p < 0.001; ns, no significance.
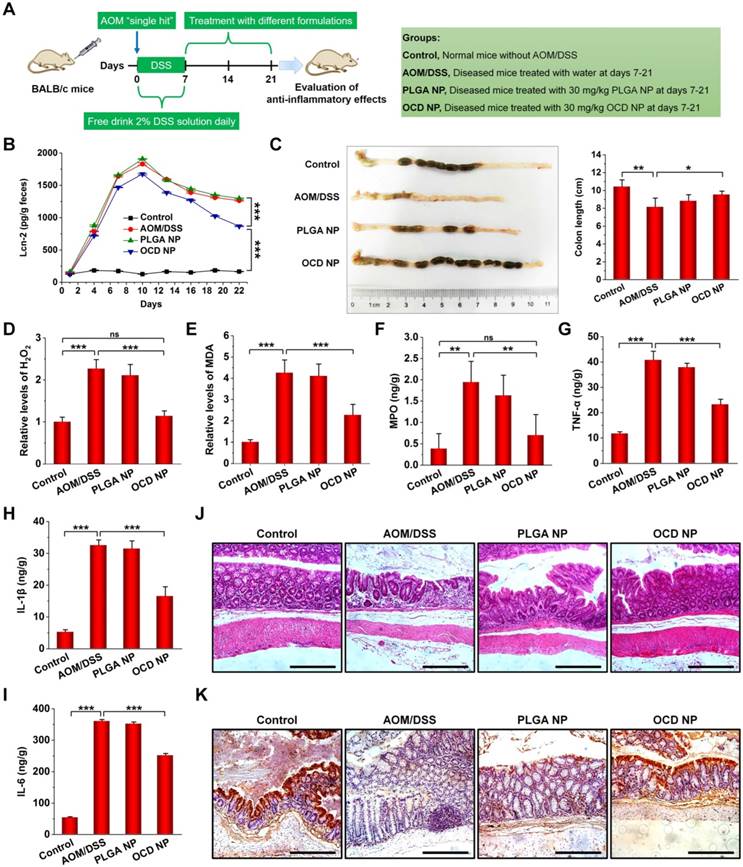
Examination on the histological sections of colon tissues revealed serious necrocytosis, significantly damaged mucous membrane, destruction of crypt structure, and considerable infiltration of inflammatory cells for AOM/DSS-induced mice (Figure 6J). The PLGA NP group also showed certain degrees of injuries in the colon tissues. Comparatively, nearly normal histological microstructure was observed for the OCD NP group, which was even comparable to that of the normal control. Previous studies have demonstrated that the expression of E-cadherin (a typical cell adhesion molecule) on the surface of intestine epithelial cells (IECs) is important for the formation of adherens junction to bind cells with each other [65]. We determined E-cadherin expression on the surface of IECs in the inflamed colon by immunohistochemistry (Figure 6K). Compared to the normal colon, the AOM/DSS-induced inflammatory colon exhibited markedly decreased E-cadherin expression. Whereas treatment with PLGA NP did not increased E-cadherin positive cells, OCD NP-treated mice showed considerably high expression of E-cadherin. On the other hand, treatment with OCD NP showed negligible effects on typical major organs, as illustrated by H&E-stained sections of heart, liver, spleen, lung, and kidney (Figure S9).
Collectively, these results demonstrated that oral administration of OCD NP can effectively attenuate oxidative stress and inhibit inflammation in the colon of mice with induced CAC, thereby protecting intestinal tissues from injury.
Fabrication and characterization of CPT-11-loaded NPs
Since the above results revealed H2O2-scavenging capability, preferential accumulation in the inflamed colon, and anti-inflammatory activity of orally administered OCD NP, we reasonably speculate that OCD NP can serve as a multifunctional nanocarrier for targeted treatment of CAC in mice. Among different anti-cancer drugs, CPT-11 has been widely used to treat lung, esophageal, gastric, and colorectal cancers [66]. CPT-11 can inhibit cellular DNA topoisomerase I, causing apoptosis and death of cancer cells. Unfortunately, CPT-11 has severe gastrointestinal toxicity [67], due to its direct damage to DNA. Moreover, efficacy of CPT-11 can be strongly suppressed by the cancer microenvironment. In particular, oxidative stress in the tumor microenvironment and activation of NF-κB increase cancer cell resistance to CPT-11 treatment [68, 69]. In view of these issues, CPT-11 was employed as a candidate drug in this conceptual proof study.
Through a similar nanoprecipitation technique as aforementioned, CPT-11 was physically entrapped into OCD NP, offering a spherical nanotherapy CPT-11/OCD NP (Figure 7A). The loading content of CPT-11 in CPT-11/OCD NP was 14.5% (Table S1). Measurement by DSC suggested that the loaded CPT-11 molecules in CPT-11/OCD NP were molecularly dispersed in the OCD nanocarrier other than in a crystal form (Figure S10), which is advantageous for rapid release of CPT-11 upon triggering by high levels of ROS. To investigate the possible inclusion interaction between CPT-11 and OCD, both 1H NMR and 2D 1H-1H NOESY spectra were acquired for different samples in DMSO-d6 (Figure S11 and Figure S12A). Of note, two groups of intermolecular NOE cross-peaks were observed in the 2D NOESY spectrum of CPT-11/OCD. One correlation signal at 5.78 and 6.56 ppm revealed interaction between -OH groups on the wider ring of OCD with H14' of CPT-11 (Figure S11 and Figure S12B). Another cross peak at 3.65 and 6.56 ppm indicated correlation between H3/H5 of OCD and H14' of CPT-11 (Figure S12C). These results suggested that the D- and E-ring of the camptothecin unit in CPT-11 was included in the hydrophobic cavity of OCD through the wider rim. Our result agrees with a previous study on CPT-11 and water-soluble negatively charged CD derivatives [70]. Consequently, both inclusion complexation and hydrophobic interactions may have contributed to entrapment of CPT-11 in OCD NP. In addition, PLGA NP containing CPT-11 was fabricated and used as a control nanotherapy (Figure S3D and Table S1). At the same drug feeding, the loading content of CPT-11/OCD NP was significantly higher than that of CPT-11/PLGA NP, which should be related to the inclusion-facilitated drug entrapment in the OCD core in the former case.
Physicochemical characterization, in vitro drug release, and in vitro antitumor activity of CPT-11-loaded OCD NP. A, TEM image of CPT-11/OCD NP. B, Particle size distribution and ζ-potential of CPT-11/OCD NP in different solutions. C, CPT-11 release behaviors in buffers simulating gastrointestinal conditions without H2O2. D-F, In vitro release profiles of CPT-11/OCD NP at different pH values with or without 1 mM H2O2. G, In vitro release of CPT-11 at various concentrations of H2O2. H-K, Relative cell viability of C26 cells (H, J) and calculated IC50 values (I, K) after incubation with different concentrations of CPT-11 or CTP-11/OCD NP for 12 h (H, I) or 24 h (J, K). Data are presented as mean ± SD (B-G, n = 3; H-K, n = 6). ***p < 0.001.
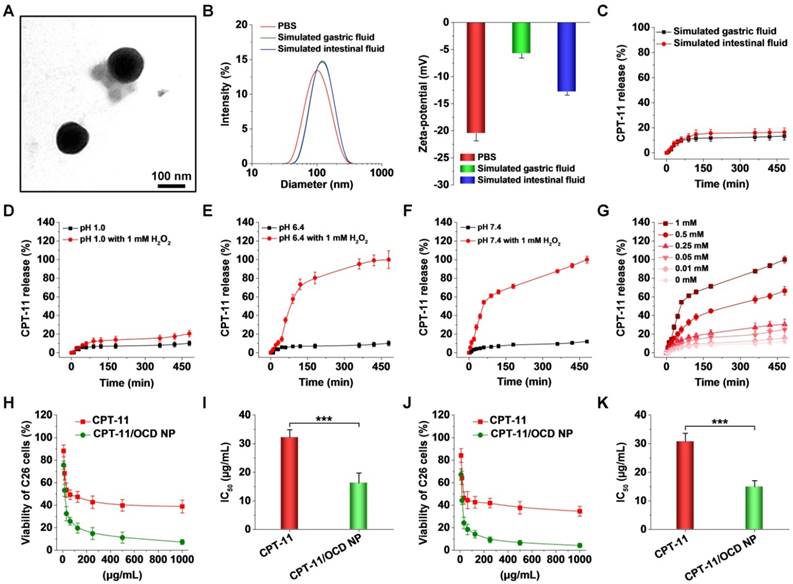
In 0.01 M PBS at pH 7.4, CPT-11/OCD NP exhibited relatively narrow size distribution, with mean hydrodynamic diameter of 121 nm and ζ-potential of -20.5 mV (Figure 7B). We also examined the stability of CPT-11/OCD NP in the simulated gastric fluid (0.32% pepsin, pH 1.2) and simulated intestinal fluid (1.0% trypsin, pH 7.4). After incubation with the simulated gastrointestinal fluid, the size distribution profile of CPT-11/OCD NP only slightly changed (Figure 7B, the left panel), with average size of 118 and 114 nm, respectively. Also, CPT-11/OCD NP had negative ζ-potential in the two simulated fluids (Figure 7B, the right panel). During 8 h of incubation in different solutions, only slight changes were observed for both average hydrodynamic diameter and polydispersity index (Figure S13). Accordingly, CPT-11/OCD NP was stable in the stimulated gastric/intestinal fluids, despite the presence of proteases and notable change in pH. On the one hand, this is due to the inherent hydrophobicity and stability of the carrier material OCD in the absence of H2O2. On the other hand, peripheral coating of DSPE-PEG can improve the stability of NPs in the simulated gastrointestinal environment, which is consistent with previous findings [71, 72].
In vitro release profiles of CPT-11/OCD NP
In simulated gastrointestinal fluids without H2O2, less than 20% of total CPT-11 was released even after 8 h of incubation (Figure 7C). Consistent with the hydrolysis profile of OCD NP, CPT-11 release at pH 1.2 was significantly inhibited, even in the presence of 1 mM H2O2 (Figure 7D). In PBS at either pH 6.4 or pH 7.4 with 1 mM H2O2, rapid release of CPT-11 was observed (Figure 7E-F). In both cases, above 65% of CPT-11 was released after 2 h of incubation. Also, release of CPT-11 was clearly dependent on the H2O2 concentration in 0.01 M PBS at pH 7.4 (Figure 7G). These results indicated that OCD NP can effectively prevent loaded CPT-11 from leaking in the gastric environment, while rapid release of CPT-11 under the intestinal pH conditions may be efficiently triggered by H2O2.
It has been demonstrated that CPT-11 is easy to open the lactone ring and transfer to a carboxylate form in alkaline environment, thereby leading to significantly decreased pharmacological activity [73]. Our tests by HPLC further substantiated that CPT-11 released from CPT-11/OCD NP retained its original structure (Figure S14).
In vitro antitumor evaluations of CPT-11/OCD NP in C26 cells
We then tested the antitumor activity of CPT-11/OCD NP in C26 cells. First, cytotoxicity of free CPT-11 and CPT-11/OCD NP was evaluated after 12 h of incubation. In both cases, significant dose-dependent cytotoxicity was observed for C26 cells. The calculated half maximal inhibitory concentration (IC50) value was 32.3 ± 2.6 and 16.3 ± 3.5 μg/mL for CPT-11 and CPT-11/OCD NP, respectively (Figure 7H-I). After 24 h of incubation, IC50 of CPT-11/OCD NP was also significantly lower than that of free CPT-11 (14.9 ± 2.1 versus 30.8 ± 2.9 μg/mL, Figure 7J-K). Consequently, packaging CPT-11 into OCD NP significantly potentiated its antitumor activity. In view of the slight effect of OCD NP alone on C26 cell viability (Figure S5B), the desirable antitumor activity observed herein should be largely attributed to enhanced cellular uptake and ROS-triggered release of CPT-11 in C26 cells.
It is well known that camptothecins mainly affect the spatial structure of cellular DNA topoisomerase I, thereby causing irreversible cell cycle arrest at the G2/M phase [74-76]. Accordingly, the effect of CPT-11/OCD NP on the cell cycle of C26 cells was examined by flow cytometry (Figure 8A). Treatment with CPT-11 at 30 μg/mL for 12 h caused approximately 33% of cells to stay in the G2/M phase, with an increase of nearly 10% compared to cells under normal conditions (Figure 8B). The effect of CPT-11/PLGA NP was comparable to that of free CPT-11 at the same dose of CPT-11. By contrast, incubation with CPT-11/OCD NP increased the G2/M cell ratio to around 40%, which is significantly higher than that of CPT-11.
In vitro antitumor activities of CPT-11/OCD NP in C26 cells. A-B, Typical flow cytometry profiles (A) and quantitative data (B) showing the percentage of G2/M cells. The cell cycle of C26 cells was measured by PI staining after treatment with different formulations for 6 h. C-D, Flow cytometric analysis of cell apoptosis of C26 cells. Apoptosis was determined by staining with Annexin V and 7-AAD at 6 h after different treatments. E-F, Flow cytometry quantification of topoisomerase I expression. After 6 h of incubation, intracellular topoisomerase I was stained with Alexa Fluor 488-labeled rabbit anti-mouse topoisomerase I monoclonal antibodies. G-H, Flow cytometric profiles (left) and quantitative analysis (right) illustrating the levels of cleaved caspase-3 (G) and cleaved PARP (H) in C26 cells. All data are presented as mean ± SD (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001; ns, no significance.
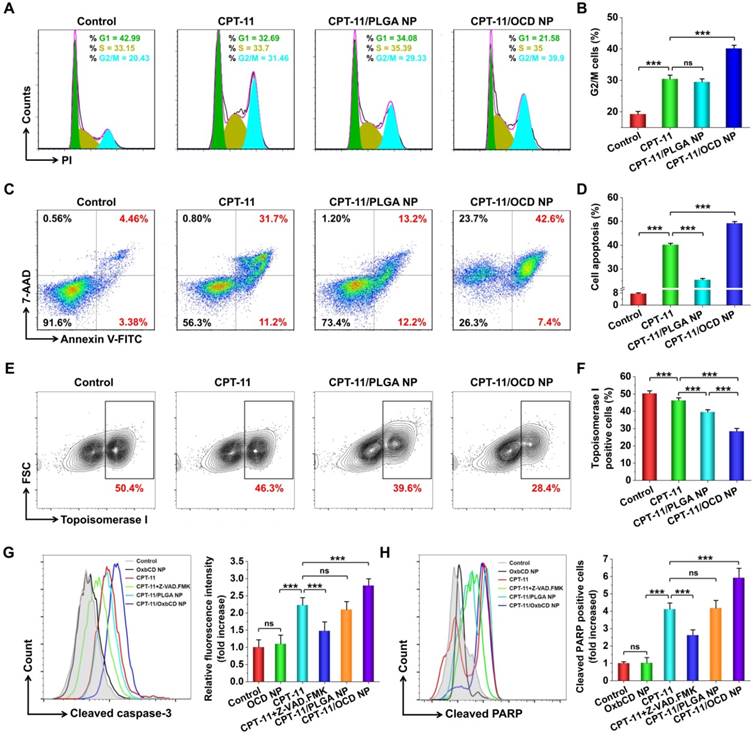
In line with the inhibited formation of DNA spatial structure, treatment with CPT-11 resulted in significant apoptosis of C26 cells (Figure 8C-D). Of note, cells incubated with CPT-11/OCD NP showed significantly high apoptosis, compared to those treated with CPT-11. By contrast, CPT-11/PLGA NP only induced slight apoptosis in C26 cells. As aforementioned, efficacy of CPT-11 in OCD NP can be enhanced by increasing intracellular delivery and triggered release in subcellular organelles, while activity of CPT-11 in PLGA NP may be compromised due to its delayed release. Further, we quantified the expression of topoisomerase I in C26 cells by flow cytometry (Figure 8E). As expected, treatment with CPT-11/OCD NP most effectively inhibited topoisomerase I expression in C26 cells, among all the examined CPT-11 formulations (Figure 8F). Consequently, this result demonstrated that CPT-11/OCD NP also exerted its antitumor activity by inhibiting topoisomerase I.
As well demonstrated, caspase-3 plays a central role in CPT-11-induced cell apoptosis, and high expression of cleaved caspase-3 indicates a high level of apoptosis [77]. During cell apoptosis, caspase-3 is also responsible for the cleavage of PARP-1 [78]. Both cleaved caspase-3 and PARP-1 have been used as important indicators of cell apoptosis. Compared to normal cells, CPT-11 treated C26 cells showed 2.2 and 4.1 times higher expression of cleaved caspase-3 and PARP-1, respectively (Figure 8G-H). When cells were simultaneously incubated with CPT-11 and an inhibitor of caspase-3 (i.e. Z-VAD-FMK), the cleavage of both caspase-3 and PARP-1 significantly decreased. Whereas treatment with the blank nanocarrier OCD NP showed no significant changes in these two apoptosis-related proteins, CPT-11/OCD NP increased the levels of cleaved caspase-3 and PARP-1 to 2.8 and 5.9 times higher than those of normal cells, respectively. Moreover, significant differences were observed between the free CPT-11 and CPT-11/OCD NP groups.
Together, these results demonstrated that antitumor activity of CPT-11 can be considerably potentiated by packing it into a ROS-responsive nanocarrier OCD NP. Cytotoxicity of the responsive nanotherapy CPT-11/OCD NP in C26 cells was mainly mediated by activation of caspase-3 and PARP-1 via inhibiting topoisomerase I, while the nanocarrier itself showed negligible effects. Notably, the responsive nanotherapy displayed significantly higher efficacy compared to the nonresponsive control nanotherapy CPT-11/PLGA NP.
In vivo pharmacokinetic and tissue distribution studies
Subsequently, we compared pharmacokinetic and colon distribution profiles of CPT-11, CPT-11/PLGA NP, and CPT-11/OCD NP in mice with AOM/DSS-induced colitis. At predetermined time points, both colon and blood samples were collected. For three formulations, CPT-11 levels in the colon tissues rapidly increased within the first 4 h, which were then decreased (Figure S15A). Notably, the CPT-11/OCD NP group showed remarkably higher CPT-11 levels at most time points examined, compared to those of the CPT-11 and CPT-11/PLGA NP groups. Furthermore, analysis of typical pharmacokinetic parameters suggested that Cmax and AUC of the CPT-11/OCD NP group were significantly higher than those of CPT-11 and CPT-11/PLGA NP groups (Figure S15B-C). In particular, the AUC value of CPT-11/OCD NP was 1.88 times of that corresponding to free CPT-11. These results are well consistent with the desirable targeting capability of OCD NP (Figure 5).
As for plasma levels of CPT-11, similar changing profiles were also observed for three different formulations. In this case, the lowest plasma CPT-11 levels were detected after treatment with CPT-11/OCD NP, while the highest levels were found for free CPT-11 (Figure S15D). Correspondingly, Cmax and AUC values of free drug were dramatically larger than those of nanotherapies (Figure S15E-F). Consequently, the ROS-responsive nanotherapy CPT-11/OCD NP can effectively deliver CPT-11 to the diseased colon tissue, increase its oral bioavailability, and simultaneously decrease its systemic exposure that is beneficial for reducing side effects.
In vivo efficacy of CPT-11/OCD NP in mice with AOM/DSS-induced CAC
Antitumor effects of CPT-11/OCD NP were investigated in CAC mice (Figure 9A). Normal mice were treated with saline in the control group, while the diseased mice received oral administration of free CPT-11 in the AOM/DSS group. In order to verify the advantages of combination therapy with OCD NP and CPT-11/OCD NP, two groups were examined. In one group (i.e. T1), diseased mice were treated with CPT-11/OCD NP during days 28-42 and days 49-63. In another group (i.e. T2), mice were subjected to treatment with OCD NP during days 7-21, followed by treatment with CPT-11/OCD NP during days 28-42 and days 49-63. A similar combination therapy with PLGA NP and CPT-11/PLGA NP was performed in diseased mice for the TP group.
In vivo efficacies of CPT-11/OCD NP for treatment of CAC in mice. A, Establishment of AOM/DSS-induced CAC in mice and treatment regimens. B, Representative digital photos show colon tumors in CAC mice after treatment with different formulations. C, The number of colon tumors in each mouse, tumor burden (calculated based on the number of tumors and the average tumor size), and the distribution of tumor size quantified after different treatments. D, The Lcn-2 levels in colon tissues of mice after treatment for 21, 42, and 63 days. E, Changes in the body weight of mice during 63 days of treatment. F, The organ index of livers isolated from mice subjected to different treatments. G-H, The relative levels of H2O2 (G) and MPO (H) in colon tissues. I-K, The levels of TNF-α (I), IL-1β (J), and IL-6 (K) in colon tissues quantified by ELISA. Control, Health mice without AOM/DSS induction; AOM/DSS, AOM/DSS-induced mice without treatment during the periods of days 7-21, days 28-42, and days 49-63; CPT-11, CAC mice without treatment during days 7-21, and treated with CPT-11 during days 28-42 and days 49-63; T1, CAC mice without treatment during days 7-21, and treated with CPT-11/OCD NP during days 28-42 and days 49-63; T2, CAC mice treated with OCD NP during days 7-21, followed by treatment with CPT-11/OCD NP during days 28-42 and days 49-63; TP, CAC mice treated with PLGA NP during days 7-21, followed by treatment with CPT-11/PLGA NP during days 28-42 and days 49-63. For all CPT-11-containing formulations, the dose was 5 mg/kg of CPT-11. All data are presented as mean ± SD (n = 10). *p < 0.05, **p < 0.01, ***p < 0.001; ns, no significance.
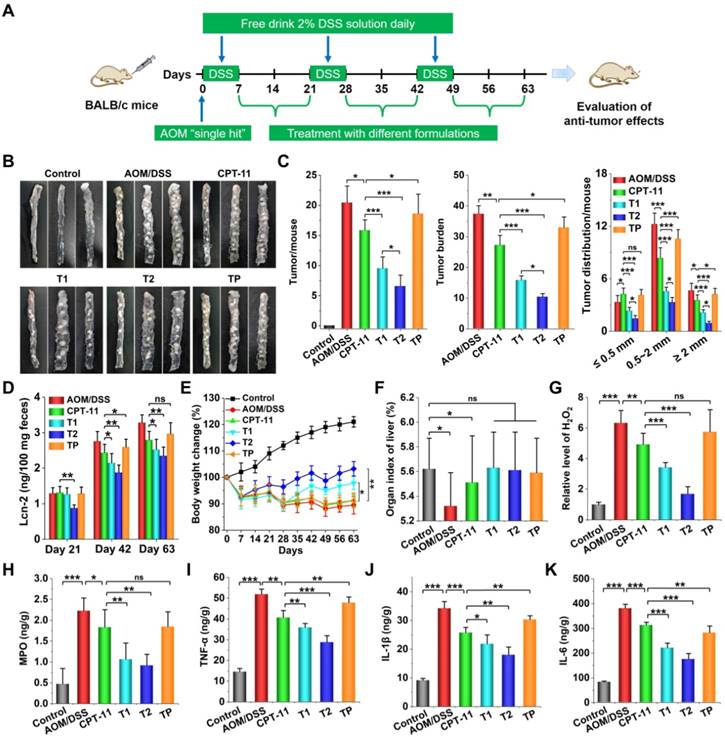
After 63 days, mice were euthanized and colon tissues were collected for evaluation of the degree of CAC. For the model group treated with AOM/DSS alone, the originally smooth colonic wall was accompanied by a large number of tumor lesions, and tumors developed from the middle to the distal portion of the colon. As for mice in the CPT-11 group orally treated with free CPT-11, the number of colon tumors decreased to a certain degree. Mice in the T1 group showed considerably reduced tumors. Of note, when mice were pre-treated with OCD NP during days 7-21, followed by treatment with CPT-11/OCD NP during days 28-42 and days 49-63 (i.e. the T2 group), less carcinogenesis was observed. By contrast, the therapeutic effect in the TP group was extremely limited (Figure 9B). Further quantitative analysis revealed significant efficacy in the T1 and T2 groups, as implicated by the significantly reduced tumor number per mouse and tumor burdens (Figure 9C), particularly the T2 group. Moreover, sequential treatment with OCD NP and CPT-11/OCD NP notably decreased the number of tumors. It is worth noting that both T1 and T2 groups showed more desirable results compared to the CPT and TP groups.
Importantly, CAC mice in the T2 groups exhibited the lowest level of Lcn-2 in the feces at all examined days (Figure 9D), although the content of Lcn-2 in the CPT-11 or T1 group also significantly decreased at days 42 and 63. In addition, mice in the T2 group displayed significantly restored body weight loss (Figure 9E), while no significant effects were observed in the CPT-11 and TP groups. In particular, after 7 or 28 days of treatment, the body weight of mice in the T1 or T2 group showed an increasing trend, while the mice treated with CPT-11 or CPT-11/PLGA NP decreased gradually. Meanwhile, the organ index of the liver of CAC mice was reversed to a normal level after different treatments (Figure 9F). Whereas the levels of H2O2 and MPO in the colon tissues of CAC mice were significantly higher than those of normal mice, treatment with CPT-11/OCD NP significantly reduced their levels (Figure 9G-H). Also, treatment with CPT-11 or CPT-11/OCD NP notably decreased the expression of typical pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) in colon tissues, particularly after combination therapy with OCD NP and CPT-11/OCD NP (Figure 9I-K). These results demonstrated that local oxidative stress and inflammation can be effectively attenuated by OCD NP.
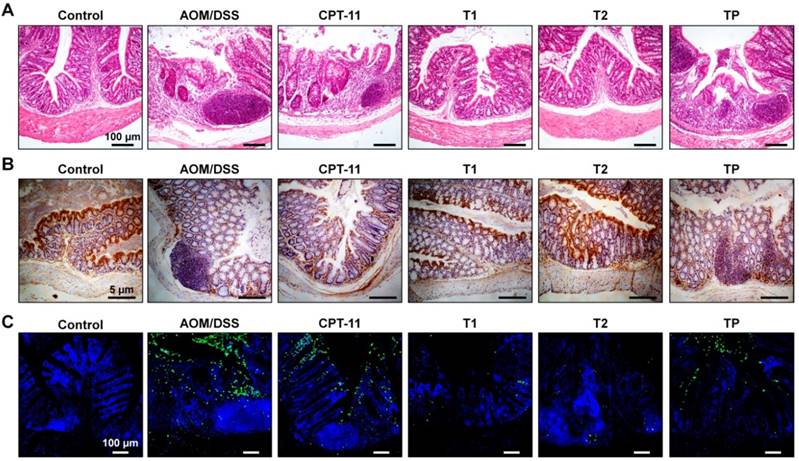
In line with the above findings, examination on H&E-stained sections of colon tissues showed obvious inflammatory cell infiltration and crypt disappearance for mice in the model group (Figure 10A). In addition, a high level of adenocarcinoma was observed in the colon section of CAC mice. Whereas treatment with different CPT-11 formulations mitigated carcinogenesis, more desirable outcomes were achieved by CPT-11/OCD NP or CPT-11/OCD NP in combination with OCD NP. The absence of E-cadherin, an adhesion molecule, is associated with the development, invasion, and metastasis of various cancers [79]. Downregulation of E-cadherin reduces the strength of cell adhesion within tissues, thereby allowing cancer cells to invade the surrounding tissue through the basement membrane. In addition, impaired expression of E-cadherin is linked to disturbed intestinal barrier function and homeostasis [79]. Immunohistochemistry analysis of colonic sections revealed dramatically higher expression of E-cadherin on colonic epithelial cells of mice in the T1 and T2 groups, compared to the untreated CAC mice and mice treated with CPT-11 or CPT-11/PLGA NP (Figure 10B). The E-cadherin level of the T2 group was even comparable to that of the normal control. This result is in accordance with that observed in anti-inflammatory studies in vivo (Figure 6K). Also, agreeing with well-maintained E-cadherin, TUNEL assay of colonic tissues near colon neoplasms showed that oral administration of CPT-11/OCD NP remarkably suppressed apoptosis in intestinal epithelial cells (Figure 10C).
Histological evaluations of therapeutic effects of CPT-11/OCD NP in CAC mice. A, H&E-stained sections of colon tissues excised from mice subjected to different treatments. B, Immunohistochemistry analysis of E-cadherin in the colon sections. C, Analysis of cell apoptosis by TUNEL assay. The green fluorescence indicates apoptotic intestinal epithelial cells, while the blue fluorescence shows DAPI-stained nuclei.
Collectively, these results demonstrated that CPT-11/OCD NP can suppress colorectal tumorigenesis, particularly by combination therapy with a H2O2-eliminating nanocarrier OCD NP. On the one hand, OCD NP serves as a ROS-responsive nanovehicle for targeting CAC and selectively releasing CPT-11 in response to a high level of ROS at diseased sites. On the other hand, it may function as a pharmacologically active nanocarrier capable of attenuating colitis-associated oxidative stress and inflammation. The multiple functions of OCD NP synergistically contribute to its antitumor activity in CAC mice.
In vivo safety evaluations of CPT-11/OCD NP for oral administration
Since the above findings evidenced that combination therapy with OCD NP and CPT-11/OCD NP is promising for targeted treatment of CAC, we performed additional animal studies to interrogate in vivo safety of CPT-11/OCD NP. To this end, CPT-11/OCD NP was orally administered in mice at 1.5 or 3.0 g/kg in BALB/c mice. Post administration, all animals showed normal breath, autonomic movements, and responses to various stimuli. No abnormalities such as convulsions, vomiting, and diarrhea were observed for CPT-11/OCD NP-treated mice. No animal death occurred at all examined doses. Mice in all groups displayed gradually increased body weight, and no statistical significance was found between the saline and CPT-11/OCD NP groups (Figure S16A).
At day 14, mice were euthanized. Calculation on the organ index of typical major organs revealed no abnormal changes in all CPT-11/OCD NP groups (Figure S16B). Quantification of typical hematological parameters, such as white blood cells (WBC), red blood cells (RBC), platelets (PLT), and hemoglobin (HGB), indicated no significant differences between the saline and CPT-11/OCD NP groups (Figure S17A-D). Clinical biochemistry analysis of biomarkers relevant to liver (ALT, alanine aminotransferase; AST, aspartate aminotransferase) and kidney (UREA, blood urea; CREA, creatinine) functions revealed no abnormal variations in the CPT-11/OCD NP groups (Figure S17E-H). Furthermore, H&E stained histological sections suggested that there were no distinguishable injuries or pathological changes in the heart, liver, spleen, lung, and kidney of mice treated with 1.5 or 3.0 g/kg CPT-11/OCD NP (Figure S18).
Taken together, CPT-11/OCD NP showed excellent safety after oral administration even at a dose 87.3-fold higher than that used for in vivo therapeutic evaluations. According to recent studies, the side effects of camptothecins are mainly due to double-strand DNA that can be produced by the destruction of spatial structure of DNA, causing increased oxidative stress and inflammatory response in the body [80]. OCD NP can alleviate the adverse reactions of CPT-11 by effectively eliminating overproduced H2O2.
Conclusions
In summary, we have developed a combination nanotherapy strategy for targeted treatment of colorectal cancer by an oral route. A ROS-responsive and H2O2-eliminating material OCD was synthesized, which can be easily produced into a functional nanocarrier OCD NP capable of scavenging H2O2. Both in vitro and in vivo studies demonstrated that OCD NP can effectively attenuate oxidative stress and inflammatory response. By packaging a candidate chemotherapeutic drug CPT-11 into OCD NP, a ROS-responsive antitumor nanotherapy was obtained, which can selectively release the loaded drug molecules under intestinal pH conditions with high levels of ROS. Compared to free CPT-11 and a non-responsive CPT-11 nanotherapy, the responsive nanotherapy CPT-11/OCD NP exhibited more efficacious in vitro antitumor effects. Consistently, oral delivery of CPT-11/OCD NP significantly inhibited tumorigenesis and tumor growth in mice with induced CAC. In combination with OCD NP in the inflammatory phase, more desirable antitumor effects were achieved, mainly resulting from reduced oxidative stress and inflammation in the diseased colons. Moreover, preliminary in vivo tests revealed excellent safety profile of CPT-11/OCD NP for oral administration. Collectively, our results substantiated that anticancer nanotherapies derived from intrinsic anti-inflammatory nanocarriers are promising for targeted treatment of inflammation-associated tumors by simultaneously normalizing inflammatory microenvironment.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Nos. 81701832 and 81471774), the Graduate Student Research Innovation Project of Chongqing, and the Program for Distinguished Young Scholars of TMMU.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gu MJ, Huang QC, Bao CZ, Li YJ, Li XQ, Ye D. et al. Attributable causes of colorectal cancer in China. BMC Cancer. 2018;18:38
2. Ouakrim DA, Pizot C, Boniol M, Malvezzi M, Boniol M, Negri E. et al. Trends in colorectal cancer mortality in Europe: retrospective analysis of the WHO mortality database. Brit Med J. 2015;351:h4970
3. Levin B, Lieberman DA, McFarland B, Smith RA, Brooks D, Andrews KS. et al. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: A joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA-Cancer J Clin. 2008;58:130-60
4. Shukla RK. Recent advances in oral anticancer agents for colon cancer. Future Oncol. 2013;9:1893-908
5. Staff NP, Grisold A, Grisold W, Windebank AJ. Chemotherapy-induced peripheral neuropathy: A current review. Ann Neurol. 2017;81:772-81
6. Payne AS, James WD, Weiss RB. Dermatologic toxicity of chemotherapeutic agents. Semin Oncol. 2006;33:86-97
7. Hugenholtz-Wamsteker W, Robbeson C, Nijs J, Hoelen W, Meeus M. The effect of docetaxel on developing oedema in patients with breast cancer: a systematic review. Eur J Cancer Care. 2016;25:269-79
8. Andreyev J, Ross P, Donnellan C, Lennan E, Leonard P, Waters C. et al. Guidance on the management of diarrhoea during cancer chemotherapy. Lancet Oncol. 2014;15:e447-60
9. Alterio D, Jereczek-Fossa BA, Fiore MR, Piperno G, Ansarin M, Orecchia R. Cancer treatment-induced oral mucositis. Anticancer Res. 2007;27:1105-25
10. Dong Z, Cui MY, Peng Z, Li Y, Wang X, Fang Z. et al. Nanoparticles for colorectal cancer targeted drug delivery and mr imaging: Current situation and perspectives. Curr Cancer Drug Targets. 2016;16:536-50
11. Cisterna BA, Kamaly N, Choi WI, Tavakkoli A, Farokhzad OC, Vilos C. Targeted nanoparticles for colorectal cancer. Nanomedicine. 2016;11:2443-56
12. You X, Kang Y, Hollett G, Chen X, Zhao W, Gu Z. et al. Polymeric nanoparticles for colon cancer therapy: overview and perspectives. J Mater Chem B. 2016;4:7779-92
13. Satterlee AB, Huang L. Current and future theranostic applications of the lipid-calcium-phosphate nanoparticle platform. Theranostics. 2016;6:918-29
14. Zhang M, Xu C, Wen L, Han MK, Xiao B, Zhou J. et al. A hyaluronidase-responsive nanoparticle-based drug delivery system for targeting colon cancer cells. Cancer Res. 2016;76:7208-18
15. Zhang M, Xiao B, Wang H, Han MK, Zhang Z, Viennois E. et al. Edible ginger-derived nano-lipids loaded with doxorubicin as a novel drug-delivery approach for colon cancer therapy. Mol Ther. 2016;24:1783-96
16. Lei S, Chien PY, Sheikh S, Zhang A, Ali S, Ahmad I. Enhanced therapeutic efficacy of a novel liposome-based formulation of SN-38 against human tumor models in SCID mice. Anti-Cancer Drugs. 2004;15:773-8
17. Chen KJ, Chaung EY, Wey SP, Lin KJ, Cheng F, Lin CC. et al. Hyperthermia-mediated local drug delivery by a bubble-generating liposomal system for tumor-specific chemotherapy. ACS Nano. 2014;8:5105-15
18. Yang C, Liu HZ, Fu ZX, Lu WD. Oxaliplatin long-circulating liposomes improved therapeutic index of colorectal carcinoma. BMC Biotechnol. 2011;11:21
19. Li L, Ahmed B, Mehta K, Kurzrock R. Liposomal curcumin with and without oxaliplatin: effects on cell growth, apoptosis, and angiogenesis in colorectal cancer. Mol Cancer Ther. 2007;6:1276-82
20. Yang SJ, Lin FH, Tsai KC, Wei MF, Tsai HM, Wong JM. et al. Folic acid-conjugated chitosan nanoparticles enhanced protoporphyrin IX accumulation in colorectal cancer cells. Bioconjug Chem. 2010;21:679-89
21. Xiao B, Viennois E, Chen Q, Wang L, Han MK, Zhang Y. et al. Silencing of intestinal glycoprotein cd98 by orally targeted nanoparticles enhances chemosensitization of colon cancer. ACS Nano. 2018;12:5253-65
22. Liang G, Zhu Y, Jing A, Wang J, Hu F, Feng W. et al. Cationic microRNA-delivering nanocarriers for efficient treatment of colon carcinoma in xenograft model. Gene Ther. 2016;23:829-38
23. Liu X, Li Y, Sun X, Muftuoglu Y, Wang B, Yu T. et al. Powerful anti-colon cancer effect of modified nanoparticle-mediated IL-15 immunogene therapy through activation of the host immune system. Theranostics. 2018;8:3490-503
24. Fay F, McLaughlin KM, Small DM, Fennell DA, Johnston PG, Longley DB. et al. Conatumumab (AMG 655) coated nanoparticles for targeted pro-apoptotic drug delivery. Biomaterials. 2011;32:8645-53
25. Vigor KL, Kyrtatos PG, Minogue S, Al-Jamal KT, Kogelberg H, Tolner B. et al. Nanoparticles functionalized with recombinant single chain Fv antibody fragments (scFv) for the magnetic resonance imaging of cancer cells. Biomaterials. 2010;31:1307-15
26. Kopansky E, Shamay Y, David A. Peptide-directed HPMA copolymer-doxorubicin conjugates as targeted therapeutics for colorectal cancer. J Drug Target. 2011;19:933-43
27. Zhang Q, Zhang F, Chen Y, Dou Y, Tao H, Zhang D. et al. Structure-property correlations of reactive oxygen species-responsive and hydrogen peroxide-eliminating materials with anti-oxidant and anti-inflammatory activities. Chem Mater. 2017:8221-38
28. Grifantini R, Taranta M, Gherardini L, Naldi I, Parri M, Grandi A. et al. Magnetically driven drug delivery systems improving targeted immunotherapy for colon-rectal cancer. J Control Release. 2018;280:76-86
29. Yu Q, Zhang Y-M, Liu Y-H, Xu X, Liu Y. Magnetism and photo dual-controlled supramolecular assembly for suppression of tumor invasion and metastasis. Sci Adv. 2018;4:eaat2297
30. Yu Q, Zhang Y-M, Liu Y-H, Liu Y. Magnetic supramolecular nanofibers of gold nanorods for photothermal therapy. Adv Therap. 2019;2:1800137
31. Vong LB, Yoshitomi T, Matsui H, Nagasaki Y. Development of an oral nanotherapeutics using redox nanoparticles for treatment of colitis-associated colon cancer. Biomaterials. 2015;55:54-63
32. Tummala S, Kuppusamy G, Satish Kumar MN, Praveen TK, Wadhwani A. 5-Fluorouracil enteric-coated nanoparticles for improved apoptotic activity and therapeutic index in treating colorectal cancer. Drug Deliv. 2016;23:2902-10
33. Stidham RW, Higgins PDR. Colorectal cancer in inflammatory bowel disease. Clin Colon Rectal Surg. 2018;31:168-78
34. Wang ZH, Fang JY. Colorectal Cancer in Inflammatory Bowel Disease: Epidemiology, Pathogenesis and Surveillance. Gastrointest Tumors. 2014;1:146-54
35. Kim ER, Chang DK. Colorectal cancer in inflammatory bowel disease: the risk, pathogenesis, prevention and diagnosis. World J Gastroenterol. 2014;20:9872-81
36. Grivennikov SI. Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol. 2013;35:229-44
37. Rogler G. Chronic ulcerative colitis and colorectal cancer. Cancer Lett. 2014;345:235-41
38. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883-99
39. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436-44
40. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860-7
41. Wirtz S, Popp V, Kindermann M, Gerlach K, Weigmann B, Fichtner-Feigl S. et al. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat Protoc. 2017;12:1295-309
42. Rivory LP, Robert J. Reversed-phase high-performance liquid chromatographic method for the simultaneous quantitation of the carboxylate and lactone forms of the camptothecin derivative irinotecan, CPT-11, and its metabolite SN-38 in plasma. J Chrom B Biomed Appl. 1994;661:133-41
43. Zhang JX, Ma PX. Cyclodextrin-based supramolecular systems for drug delivery: recent progress and future perspective. Adv Drug Deliv Rev. 2013;65:1215-33
44. Davis ME, Brewster ME. Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev Drug Discov. 2004;3:1023-35
45. Achilli C, Ciana A, Fagnoni M, Balduini C, Minetti G. Susceptibility to hydrolysis of phenylboronic pinacol esters at physiological pH. Cent Eur J Chem. 2013;11:137-9
46. Zhang D, Wei Y, Chen K, Zhang X, Xu X, Shi Q. et al. Biocompatible reactive oxygen species (ROS)-responsive nanoparticles as superior drug delivery vehicles. Adv Healthc Mater. 2015;4:69-76
47. Jodái I, Kandra L, Harangi J, Nánási P, Debrecen, Szejtli J. Hydrolysis of cyclodextrin by aspergillus oryzae α-amylase. Starch. 1984;36:140-3
48. Pommier Y. DNA Topoisomerase I inhibitors: chemistry, biology and interfacial inhibition. Chem Rev. 2009;109:2894-902
49. Blajeski AL, Phan VA, Kottke TJ, Kaufmann SH. G(1) and G(2) cell-cycle arrest following microtubule depolymerization in human breast cancer cells. J Clin Invest. 2002;110:91-9
50. Koivusalo M, Welch C, Hayashi H, Scott CC, Kim M, Alexander T. et al. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J Cell Biol. 2010;188:547-63
51. Chen F, Zhu L, Zhang Y, Kumar D, Cao G, Hu X. et al. Clathrin-mediated endocytosis is a candidate entry sorting mechanism for Bombyx mori cypovirus. Sci Rep. 2018;8:7268-78
52. Lee J, Twomey M, Machado C, Gomez G, Doshi M, Gesquiere AJ. et al. Caveolae-mediated endocytosis of conjugated polymer nanoparticles. Macromol Biosci. 2013;13:913-20
53. Muneyuki E, Makino M, Kamata H, Kagawa Y, Yoshida M, Hirata H. Inhibitory effect of NaN3 on the F0F1 ATPase of submitochondrial particles as related to nucleotide binding. Biochim Biophys Acta. 1993;1144:62-8
54. Iversen T-G, Skotland T, Sandvig K. Endocytosis and intracellular transport of nanoparticles: Present knowledge and need for future studies. Nano Today. 2011;6:176-85
55. Feng S, Hu Y, Peng S, Han S, Tao H, Zhang Q. et al. Nanoparticles responsive to the inflammatory microenvironment for targeted treatment of arterial restenosis. Biomaterials. 2016;105:167-84
56. Dou Y, Chen Y, Zhang X, Xu X, Chen Y, Guo J. et al. Non-proinflammatory and responsive nanoplatforms for targeted treatment of atherosclerosis. Biomaterials. 2017;143:93-108
57. Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145:182-95
58. Dickson I. Colorectal cancer: Bacterial biofilms and toxins prompt a perfect storm for colon cancer. Nat Rev Gastroenterol Hepatol. 2018;15:129
59. Nowacki TM, Bruckner M, Eveslage M, Tepasse P, Pott F, Thoennissen NH. et al. The risk of colorectal cancer in patients with ulcerative colitis. Dig Dis Sci. 2015;60:492-501
60. Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KS, McIntire CR, LeBlanc PM. et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity. 2010;32:367-78
61. Kitajima S, Takuma S, Morimoto M. Changes in colonic mucosal permeability in mouse colitis induced with dextran sulfate sodium. Exp Anim. 1999;48:137-43
62. Zhang Q, Tao H, Lin Y, Hu Y, An H, Zhang D. et al. A superoxide dismutase/catalase mimetic nanomedicine for targeted therapy of inflammatory bowel disease. Biomaterials. 2016;105:206-21
63. Zhang S, Ermann J, Succi MD, Zhou A, Hamilton MJ, Cao B. et al. An inflammation-targeting hydrogel for local drug delivery in inflammatory bowel disease. Sci Transl Med. 2015;7:300ra128
64. Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, Vijay-Kumar M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS One. 2012;7:e44328
65. Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799-809
66. Fujita K, Kubota Y, Ishida H, Sasaki Y. Irinotecan, a key chemotherapeutic drug for metastatic colorectal cancer. World J Gastroenterol. 2015;21:12234-48
67. Mayo BJ, Stringer AM, Bowen JM, Bateman EH, Keefe DM. Irinotecan-induced mucositis: the interactions and potential role of GLP-2 analogues. Cancer Chemother Pharmacol. 2017;79:233-49
68. Xu Y, Villalona-Calero MA. Irinotecan: mechanisms of tumor resistance and novel strategies for modulating its activity. Ann Oncol. 2002;13:1841-51
69. Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J Clin Invest. 2001;107:241-6
70. Cheng J-G, Yu H-J, Chen Y, Liu Y. Selective binding and controlled release of anticancer drugs by polyanionic cyclodextrins. Bioorg Med Chem. 2018;26:2287-90
71. Crater JS, Carrier RL. Barrier properties of gastrointestinal mucus to nanoparticle transport. Macromol Biosci. 2010;10:1473-83
72. Behzadi S, Serpooshan V, Tao W, Hamaly MA, Alkawareek MY, Dreaden EC. et al. Cellular uptake of nanoparticles: journey inside the cell. Chem Soc Rev. 2017;46:4218-44
73. Zhou JJ, Liu J, Xu B. Relationship between lactone ring forms of HCPT and their antitumor activities. Acta Pharmacol Sin. 2001;22:827-30
74. Goldwasser F, Shimizu T, Jackman J, Hoki Y, O'Connor PM, Kohn KW. et al. Correlations between S and G2 arrest and the cytotoxicity of camptothecin in human colon carcinoma cells. Cancer Res. 1996;56:4430-7
75. Prasad Tharanga Jayasooriya RG, Dilshara MG, Neelaka Molagoda IM, Park C, Park SR, Lee S. et al. Camptothecin induces G2/M phase arrest through the ATM-Chk2-Cdc25C axis as a result of autophagy-induced cytoprotection: Implications of reactive oxygen species. Oncotarget. 2018;9:21744-57
76. Li F, Jiang T, Li Q, Ling X. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am J Cancer Res. 2017;7:2350-94
77. Pang WJ, Xiong Y, Wang Y, Tong Q, Yang GS. Sirt1 attenuates camptothecin-induced apoptosis through caspase-3 pathway in porcine preadipocytes. Exp Cell Res. 2013;319:670-83
78. Soldani C, Scovassi AI. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis. 2002;7:321-8
79. van Roy F. Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nat Rev Cancer. 2014;14:121-34
80. Lian Q, Xu J, Yan S, Huang M, Ding H, Sun X. et al. Chemotherapy-induced intestinal inflammatory responses are mediated by exosome secretion of double-strand DNA via AIM2 inflammasome activation. Cell Res. 2017;27:784-800
Author contact
![]() Corresponding authors: Jianxiang Zhang, PhD, Prof. Department of Pharmaceutics, Third Military Medical University, 30 Gaotanyan Main St., Chongqing 400038, China. E-mail: jxzhang1980com. Zhenhua Zhou, PhD, Prof. Department of Neurology, Southwest Hospital. Third Military Medical University, 30 Gaotanyan Main St., Chongqing 400038, China E-mail: exploiter001com.
Corresponding authors: Jianxiang Zhang, PhD, Prof. Department of Pharmaceutics, Third Military Medical University, 30 Gaotanyan Main St., Chongqing 400038, China. E-mail: jxzhang1980com. Zhenhua Zhou, PhD, Prof. Department of Neurology, Southwest Hospital. Third Military Medical University, 30 Gaotanyan Main St., Chongqing 400038, China E-mail: exploiter001com.