Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Material and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(13):3940-3951. doi:10.7150/thno.31647 This issue Cite
Research Paper
Clathrin-mediated Endocytosis of Alpha-1 Antitrypsin is Essential for its Protective Function in Islet Cell Survival
Jingjing Wang1*, Wenyu Gou1*, Do-sung Kim1, Charlie Strange2#, Hongjun Wang1,3# ![]()
1. Department of Surgery, Medical University of South Carolina, Charleston, 29425
2. Department of Medicine, Medical University of South Carolina, Charleston, 29425
3. Department of Regenerative Medicine and Cell Biology, Medical University of South Carolina, Charleston, 29425
*JW and WG contributed equally and should be considered as co-first authors.
#CS and HW share senior authorship.
Received 2018-11-19; Accepted 2019-4-1; Published 2019-5-31
Abstract

Cytokine-induced pancreatic β cell death plays a pivotal role in both type 1 and type 2 diabetes. Our previous study showed that alpha-1 antitrypsin (AAT) inhibits β cell death through the suppression of cytokine-induced c-Jun N-terminal kinase (JNK) activation in an islet transplantation model. The aim of this study was to further understand how AAT impacts β cells by studying AAT endocytosis in human islets and a βTC3 murine insulinoma cell line.
Methods: In vitro, human islets and βTC3 cells were stimulated with cytokines in the presence or absence of chlorpromazine (CPZ), a drug that disrupts clathrin-mediated endocytosis. Western blot, real-time PCR and cell death ELISA were performed to investigate β cell death. The oxygen consumption rate (OCR) was measured on human islets. In vivo, islets were harvested from C57BL/6 donor mice treated with saline or human AAT and transplanted into the livers of syngeneic mice that had been rendered diabetic by streptozotocin (STZ). Islet graft survival and function were analyzed.
Results: AAT was internalized by β cells in a time- and dose-dependent manner. AAT internalization was mediated by clathrin as treatment with CPZ, profoundly decreased AAT internalization, cytokine-induced JNK activation and the downstream upregulation of c-Jun mRNA expression. Similarly, addition of CPZ attenuated cytokine-induced caspase 9 cleavage (c-casp 9) and DNA fragmentation, which was suppressed by AAT. Treatment of donor mice with AAT produced AAT internalization in islets, and resulted in a higher percentage of recipients reaching normoglycemia after syngeneic intraportal islet transplantation.
Conclusion: Our results suggest that AAT is internalized by β cells through clathrin-mediated endocytosis that leads to the suppression of caspase 9 activation. This process is required for the protective function of AAT in islets when challenged with proinflammatory cytokines or after islet transplantation.
Keywords: Alpha-1 antitrypsin, clathrin-mediated endocytosis, islet transplantation, graft survival, donor treatment
Introduction
Allogeneic islet transplantation is a promising treatment modality for patients with type 1 diabetes (T1D) [1]. Autologous islet transplantation is used to prevent pancreatogenic diabetes in chronic pancreatitis patients undergoing total pancreatectomy [2]. However, the outcomes of both forms of islet transplantation remain suboptimal. One of the major hurdles that limits clinical usage of islet transplantation is early loss of islet grafts mostly caused by an instant blood-mediated inflammatory reaction (IBMIR). These stresses induce islet cell death post transplantation in up to 60% of cells [3]. Similar to the process of islet damage in transplantation, the destructive effects toward β cells in T1D patients seem to overlap with several innate immune responses, including leukocyte infiltration and cytokine secretion [4]. These antigen-independent, inflammation-dependent events precede, as well as determine, the degree of antigen-specific immune responses in both T1D and allogeneic islet transplantation [5-7]. Indeed, pancreatic β cells are particularly prone to injury during inflammatory conditions, responding profoundly to cytokines such as interleukin-1 β (IL-1β) [8-10], which is further potentiated by tumor necrosis factor α (TNF-α) and interferon γ (IFN-γ) [11-13]. Therefore, drugs that can protect islets from inflammation could improve therapeutic outcomes for T1D with or without islet transplantation.
AAT is a major endogenous serine-protease inhibitor that suppresses the enzymatic activity of neutrophil elastase, cathepsin G, proteinase 3, and other proteinases [14]. Plasma AAT concentration can rise up to 4-fold in response to inflammatory stimuli [15, 16]. Purified plasma-derived AAT has been used in the treatment of emphysema in AAT-deficient individuals [17-19]. The importance of AAT in improving islet cell survival after transplantation was demonstrated in syngeneic, allogeneic islet transplantation models in rodents and cynomolgus models [20, 21]. We have shown that AAT protects islets from IBMIR in a syngeneic intraportal murine islet transplantation model [22]. Yet, the mechanism of action of AAT on islets still remains largely unclear. In the present study, we determine the molecular mechanism of AAT endocytosis in human islets and βTC3 cells, which will help further understand the mechanism by which β cells are protected by AAT.
Results
1. Kinetics of the internalization of AAT by β cells
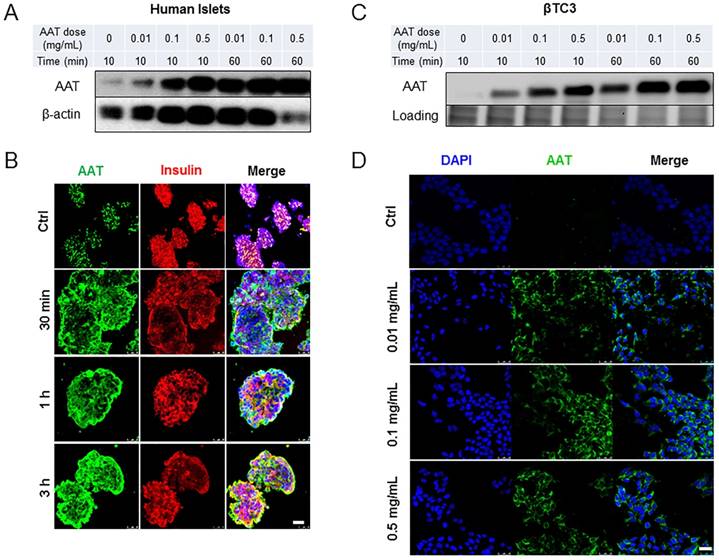
To investigate cellular uptake of AAT by β cells, human islets were incubated with increasing concentrations of AAT (0.01 to 0.5 mg/ml). A time-dependent AAT internalization, beginning as early as 10 min following incubation with AAT was observed. The internalization reached saturation at 1 hour (h) as observed by Western blot analysis (Figure 1A). Immunofluorescence confirmed these results and further showed that AAT was still presented in most islet cells 3 h after AAT treatment (Figure 1B). Co-staining with the anti-insulin or the anti-glucagon antibody showed that both β and α cells internalized AAT (Supplemental data, Figure 1A). A dose- and time-dependent internalization of AAT was also observed in βTC3 cells (Figure 1 C & D). AAT internalization was further confirmed by using a fluorescently labeled AAT in mouse islets and βTC3 cells (Supplemental data, Figure 1B & C).
Time course and dose response of AAT uptake by β cells. (A) Western blot on human islets cultured in the presence of AAT at indicated concentrations for indicated time periods. (B) Representative fluorescent micrographs show human islets stained for AAT (green), insulin (red) and nuclei (blue). Scale bar = 25 µm. (C) Western blot on βTC3 cells cultured in the presence of AAT at indicated concentrations for indicated time periods. (D) Representative fluorescent micrographs show human islets stained for AAT (green) and nuclei (blue). Scale bar = 25 µm. These experiments were repeated three times.
2. Mechanism of the internalization of AAT by β cells
Clathrin-mediated endocytosis plays an essential role in transporting molecules from the cell surface to cytosolic compartments. To explore the role of clathrin in the uptake of AAT in β cells, CPZ, a cationic amphiphilic inhibitor of clathrin-dependent endocytosis [23], was added to human islets and βTC3 cells 30 minutes (min) before the addition of AAT. CPZ prominently suppressed AAT internalization by human islets in the presence of either 0.1 mg/ml or 0.5 mg/ml AAT (Figure 2A), indicating that the endocytosis of a therapeutic dose of AAT (0.5 mg/ml) can be efficiently reduced by CPZ in human islets. Furthermore, among the two doses of CPZ used in this study, both doses (20 µM and 60 µM) were sufficient to inhibit AAT internalization (76% ± 16.8% vs. 62% ± 9.1% respectively). The 20 µM dose was selected for the following experiments. Immunofluorescent analysis showed strong inhibition of AAT presence in human islets following CPZ pretreatment (Figure 2B).
Effect of CPZ on AAT internalization in β cells. (A) CPZ inhibits AAT internalization in human islets. Top panel: representative immunoblot showing reduced AAT in CPZ pretreated human islets. Bottom panel: quantification of immunoblot result plotted as the mean fold-change in AAT internalization ± SEM from 3 independent experiments. * P < 0.05 by one-way ANOVA. (B) Human islets stained for AAT (green), insulin (red) and nuclei (blue). Scale bar = 25 µm. (C) CPZ inhibited AAT internalization in βTC3 cells. Top panel: representative immunoblots showing reduced AAT in cytosol fractions in CPZ-pretreated βTC3 cells. ATP1A1: ATPase Na+/K+ transporting subunit alpha1. Bottom panel: quantification of immunoblot result plotted as the mean fold-change in AAT internalization ± S.E. (D) βTC3 stained for AAT (green) and nuclei (blue). Scale bar = 10 µm.
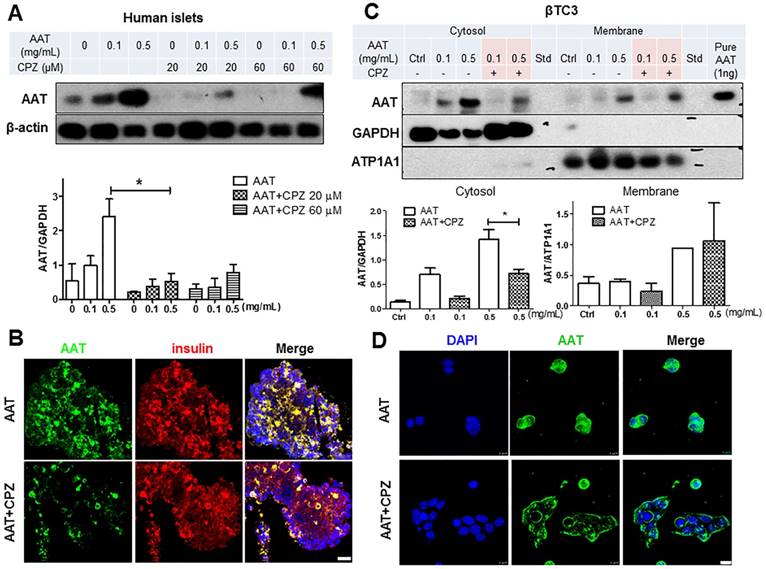
To confirm that CPZ block AAT endocytosis in β cells, βTC3 cells were incubated with AAT with or without CPZ pretreatment. Presence of AAT was observed in both the membrane and cytosol fractions of cells after incubation in a dose-dependent manner (Figure 2C). With the addition of CPZ, the cytosolic fraction of AAT was markedly reduced, indicating that AAT endocytosis was blocked. In contrast, membrane presence of AAT was not affected by CPZ in cells treated with 0.5 mg/ml AAT (Figure 2C). Consistent with these results, immunofluorescent analysis showed a diminished cytosol signal but strong membrane signal of AAT staining in βTC3 cells (Figure 2D). Similar results were also observed in mouse islets and βTC3 cells incubated with fluorescently labeled AAT (Supplemental data, Figure 1 B&C). Another major mechanism of endocytosis in cells is through the clathrin-independent pathway using caveoli [24, 25]. We therefore treated cells with the caveola inhibitor, filipin, to explore the possibility of AAT endocytosis through this clathrin-independent pathway. We found that pretreatment of human islets with filipin did not affect AAT uptake by islet cells, both at 0.1 mg/ml and 0.5 mg/ml AAT incubation (Supplemental data, Figure 2). These data demonstrated that AAT internalization by β cells is mostly mediated by clathrin-dependent endocytosis.
3. AAT endocytosis in cytokine-induced JNK pathway
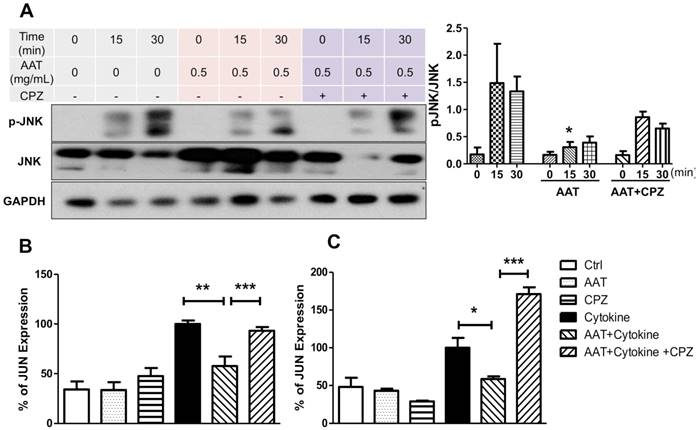
We reported that AAT suppressed cytokine-induced JNK phosphorylation in β cells [22]. To investigate if AAT endocytosis plays a role in this pathway, human islets were pretreated with CPZ and/or AAT followed by the stimulation of a cocktail of cytokines including IL-1β, INF-γ and TNF-α. AAT significantly decreased JNK phosphorylation at 15 min after cytokine treatment. This effect was reversed by the addition of CPZ (Figure 3A). To validate suppression of the cytokine-induced JNK signaling cascade by AAT, we quantified expression of the c-Jun gene, whose promoter transcriptional activity is positively regulated by JNK [26, 27]. A significant reduction by two-fold in c-Jun mRNA expression in human islet cells treated with cytokines for 6 h was observed (Figure 3B). Moreover, CPZ pre-treatment significantly blocked the inhibitory effect of AAT on cytokine-induced c-Jun mRNA expression (Figure 3B). Similar results were also observed in βTC3 cells (Figure 3C). These results demonstrated that clathrin-dependent endocytosis is required for the inhibitory effect of AAT on cytokine-induced JNK pathway activation.
Effect of AAT endocytosis on cytokine-induced JNK pathway. (A) Immunoblots and quantification of pJNK/JNK show cytokine-induced JNK phosphorylation in human islets was suppressed by AAT, and the addition of CPZ reduced this effect. The mRNA expression of c-JUN was measured in human islets (B) and βTC3 cells (C) after incubation with cytokines for 6 hours with or without AAT and/or CPZ pretreatments. Data are expressed as mean ± SEM from 3-5 independent experiments. * P < 0.05, ** P < 0.01 and *** P < 0.001 by one-way ANOVA.
4. AAT endocytosis in cytokine-induced mitochondrial dysfunction
To further elucidate the mechanistic impacts of AAT on cytokine-induced β cell death, the oxygen consumption rate (OCR) of human islets was measured since this correlates with islet viability and mitochondria function [28, 29]. OCR was significantly suppressed by cytokine treatment (68.2% ± 2.9% in cytokine-treated group vs non-treated controls) in human islets after incubation with cytokines for 24 h. Treatment with AAT significantly ameliorated this decrease (91.5% ± 6.1% of control group). Inhibition of clathrin moderately reversed the protective effect of AAT (74.8% ± 1.6% of control group) (Figure 4A). The levels of human islet OCR after each treatment are shown in Supplemental Table 1. Furthermore, cytokines significantly suppressed the expression of peroxisome proliferator-activated receptor γ coactivator 1 α (PGC1α), a key regulator of mitochondrial function and integrity [30, 31]. Treatment with AAT completely prevented the reduction of PGC1α. Addition of CPZ abolished the protective effect of AAT in respect of PGC1α mRNA expression (Figure 4B), suggesting that clathrin-mediated AAT endocytosis modulates mitochondrial function via regulation of PGC1α expression.
Effect of AAT on mitochondrial dysfunction. (A) AAT attenuated the inhibition of oxygen consumption rate (OCR) by cytokine treatment in human islets, and CPZ blocked human islets protective effect. The mean OCR/DNA (nmolO2/min·mg DNA) values were calculated and normalized to “fold change” against negative control. The mRNA expression of PGC1α (B), Bim (C) and NOS2 (D & E) was measured in human islets after incubation with cytokines for 6 or 24 h with or without AAT and/or CPZ pretreatments. Data are expressed as mean ± SEM from 3 independent experiments. * P < 0.05 by One-way ANOVA.
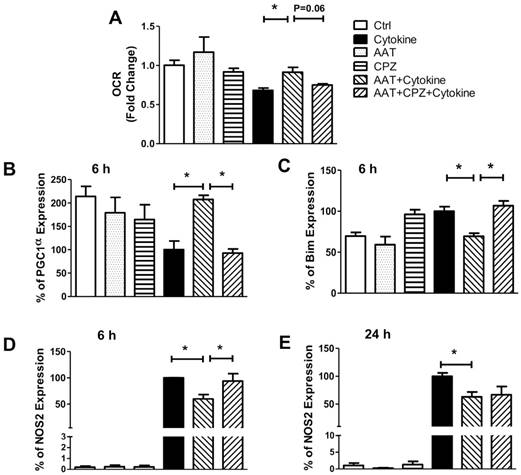
We measured the expression of BCL-2-like protein 11 (Bim) and nitric oxide synthase 2 (NOS2), both are associated with mitochondrial dysfunction and mitochondria-mediated apoptosis in cells. AAT significantly attenuated cytokine-induced upregulation of Bim mRNA expression (30%) in human islets 24 h after incubation in cytokines, which was reversed by CPZ pretreatment (Figure 4C). Furthermore, AAT significantly suppressed cytokine-induced NOS2 mRNA expressions as early as 6 h (40%) (Figure 4D) after cytokine stimulation, which lasted until 24 h post-treatment (37%) (Figure 4 E). Inhibition of clathrin diminished the protective effect of AAT, as evidenced by the upregulation of NOS2 gene expression in cells treated with CPZ, AAT and cytokines, to a similar level as the cells treated with cytokines only at 6 h after cytokine incubation. However, this reversal effect of CPZ was not observed after 24 h of cytokine treatment (Figure 4E).
5. AAT endocytosis in cytokine-induced β cell death
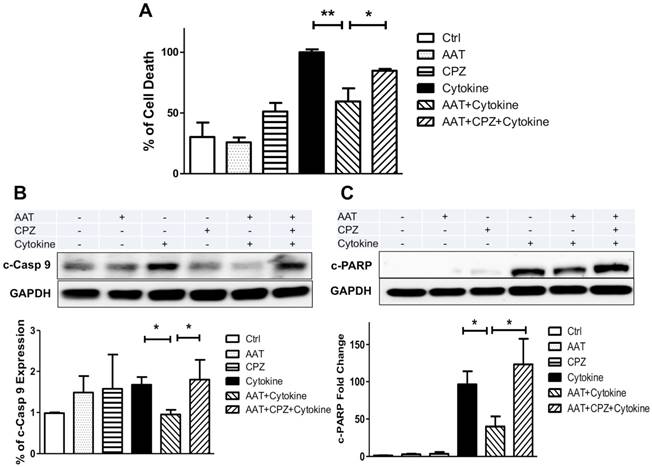
We next determined whether clathrin-mediated endocytosis is required for the anti-apoptotic effect of AAT. Human islets were cultured with cytokines for 96 h with or without AAT and/or CPZ pretreatment, and subjected to a cell death ELISA analysis that detects the level of DNA fragmentation as an indicator of apoptosis. As shown in Figure 5A, AAT significantly ameliorated cytokine-induced human islet cell death. CPZ partially reversed the beneficial effect of AAT after cytokine treatment on human islets. Studies showed that AAT improved cell survival by inhibition of cleaved-caspase 3 (c-casp3) [22, 32, 33]. However, above results suggested additional mechanisms might be responsible for the protective effects of AAT on β cells earlier than the activation of caspase 3 in the apoptosis pathway. To examine this possibility, we measured the expression level of cleaved caspase 9 (c-casp9) using Western blot analysis. Our data showed that cytokine-induced c-casp9 was significantly suppressed by AAT treatment on human islets, and the addition of CPZ diminished this protective effect (Figure 5B). In addition, cleaved Poly (ADP-ribose) polymerase (c-PARP) levels were significantly reduced in AAT-treated cytokine-stimulated islets, compared with cytokine only group. Again, the addition of CPZ markedly upregulated c-PARP levels regardless of the presence of AAT (Figure 5C). These observations indicate that clathrin-dependent AAT endocytosis promotes human islet survival through the inhibition of cytokine-induced c-casp9 activation, and results in reduced c-PARP and DNA fragmentation.
Effect of AAT endocytosis on cytokine-induced cell death. (A) Cell death measured in human islets pretreated with AAT and/or CPZ, followed by incubation with cytokines for 96 h using the Cell Death ELISA kit. Immunoblots show that AAT reduced (B) cytokine-induced cleaved caspase 9 (c-casp9) and (C) c-PARP expression in human islets, and CPZ reversed this effect. Data are expressed as mean ± SEM from 3 independent experiments. * P < 0.05 ** P < 0.01 by One-way ANOVA and Student's t test.
6. Islets from AAT-treated donors exhibited better outcome after transplantation
Next, we tested whether these findings can be translated into therapy to benefit islet survival after transplantation. We first assessed whether AAT can be internalized by islet cells when given to mice via intraperitoneal injection before islet isolation. C57BL/6 mice were treated with human AAT (80 mg/kg or 160 mg/kg i.p.) and islets were isolated 24 h after AAT injection. A robust level of human AAT was presented in islets harvested from AAT-treated mice as analyzed by Western blot and immunofluorescence analysis (Figure 6 A&B). AAT islets exhibited significantly higher OCR compared with those from vehicle-treated control mice (Figure 6C). Increased OCR was also confirmed in islets isolated from the Swiss mice treated with AAT compared to those treated with saline before islet isolation (Supplemental data, Figure 3B).
AAT donor-treatment enhances islet graft survival after transplantation. C57BL/6 mice were treated with saline (CTR) or human AAT at 80 mg/kg or 160 mg/kg body weight (AAT) for 24 h and islets were isolated and transplanted in diabetic C57BL/6 recipients. (A) Immunoblot and (B) immunofluorescence micrographs show the presence of human AAT in islets isolated from AAT-treated donors. Scale bar = 25 µm. (C) OCR of islets isolated from AAT-treated C57BL/6 mice donors. Donors treated with saline (CTR); Donors treated with 80 mg/kg AAT, and donors treated with 160 mg/kg AAT. Samples were measured in triplicates and mean was used for final calculation. (D) Islet graft survival curve shows that AAT donor-treatment leads to a higher percentage of recipients with normoglycemia. (E) Blood glucose levels during the IVGTT show at day 7 (E) and day 28 (F) that AAT islets function better in the recipients. Insets: area under the curve (AUC) of IPGTT glucose. *P < 0.05,
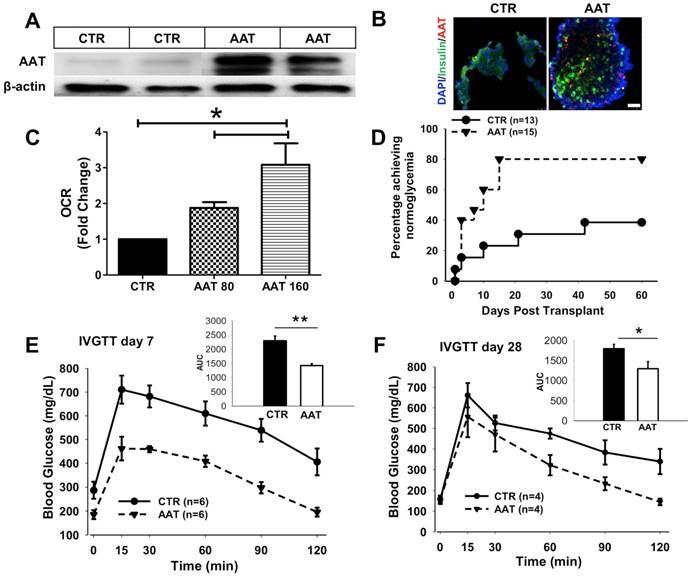
To further assess the survival advantage of islets isolated from mice treated with AAT, a marginal number of islets from C57BL/6 mice treated with saline (control) or AAT were transplanted into streptozotocin (STZ)-induced diabetic C57BL/6 mice. As shown in Figure 6D, Mice receiving islets from AAT-treated donors exhibited markedly improved islet graft survival. In the AAT treated group, 80% of recipients reached normoglycemia (n=15, P=0.02 vs. control by log-rank test) compared with 38.5% in the control group (n=13) at day 60 post-transplant. The average blood glucose levels in the AAT group were lower than control group (supplemental data, Figure 3A), suggesting that fewer islets had graft death after AAT exposure. At both time points, mice that received islets from AAT-treated donors showed a more rapid glucose clearance and had a significantly less area under the curve (AUC) after the glucose challenge than control mice during the intravenous glucose tolerance test (IVGTT) at day 7 and 28 post transplantation, respectively (Figure 6E). These data provided strong evidence that islets from AAT-treated donors internalized AAT, and exhibited enhanced mitochondria function and graft survival and function after islet transplantation.
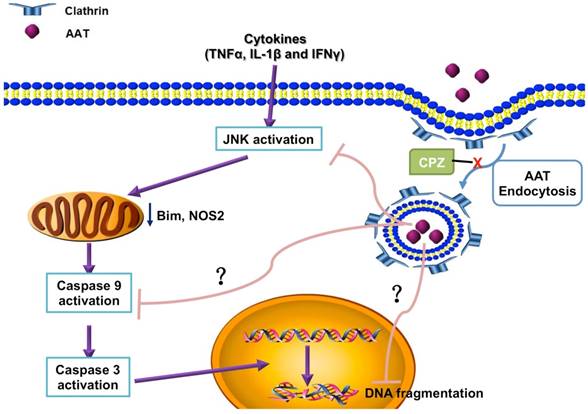
Simplified scheme showing the molecular mechanism of the protection effect of AAT on β cells. AAT enters β cells through clathrin-mediated endocytosis, resulted in the suppression of cytokine-induced JNK pathway activation, leading to the inhibition of mitochondrial dysfunction and caspase 9 activation. This protects β cells from cytokine-induced cell death.
Discussion
Pro-inflammatory cytokines play critical roles in the pathogenesis of T1D as well as in the inflammatory reactions associated with islet isolation and islet transplantation. AAT is an anti-inflammatory glycoprotein that exerts beneficial effects in islet transplantation and T1D. Despite growing evidence for the anti-inflammatory properties demonstrated for AAT, the mechanism of a direct effect of AAT on β cells remains to be defined. In this study, we found that AAT is internalized by β cells through clathrin-dependent endocytosis, leading to the suppression of the JNK pathway, inhibition of caspase 9 activation, and inhibition of mitochondria-mediated apoptosis. Furthermore, the benefit of AAT internalization was translated in vivo where we observed improved islet graft survival after syngeneic islet transplantation in mice receiving islets isolated from AAT treated donors. These results provide new insights into the mechanism(s) by which AAT achieves its protective effect on cytokine-induced islet damage.
The presence of AAT in the cytosol after incubation with exogenous AAT has been reported in lung endothelial cells, human monocyte-derived macrophages, primary rat alveolar macrophages, neutrophils, CD4+ T cells, mesenchymal stem cells, and Min6 cells [24, 33-36]. In this study, we show for the first time, that AAT was rapidly internalized by both human islets and βTC3 cells in a dose- and time-dependent manner, which is primarily regulated by clathrin-dependent endocytosis.
In lung endothelial cells, AAT is taken up predominantly by clathrin-mediated endocytosis. Cigarette smoke exposure decreased the ability of endothelial cells to internalize AAT both in vitro and in vivo, suggesting that internalization of AAT contributes to the maintenance of alveolar structure in addition to inhibition of extracellular neutrophil elastase [34]. Similarly, AAT internalized by CD4+ T cells resulted in the inhibition of HIV-1 replication by blocking the activation of the NF-κB pathway, which was abolished by CPZ, suggesting that clathrin-dependent endocytosis is required for the suppression of NF-κB pathway in CD4+ T cells [24]. In accordance with these results, we found that clathrin-dependent endocytosis plays a crucial role in the anti-apoptotic effects of AAT on β cells. In our previous study, we discovered that AAT reduced cytokine-induced JNK phosphorylation that served as a novel mechanism underling the beneficial effects of AAT on islets [22]. Importantly, in the current study, we further showed that clathrin-dependent AAT internalization is required for the suppressive effects of AAT on the JNK pathway previously reported. On the other hand, Bergin et al. demonstrated that surface AAT had the capacity of interrupting ligand-receptor interaction between TNF receptor 1/TNF receptor 2 and TNF-α and directly modulated TNF-α signaling in neutrophils [50]. This is consistent with our result that increased cell survival by AAT treatment was not completely abolished by the addition of CPZ. While we cannot irrefutably rule out the contribution of non-clathrin and non-caveoli-mediated endocytosis or macropinocytosis to the uptake of the protein, the observed effects using CPZ and filipino indicated that AAT was primarily internalized by clathrin-dependent endocytosis.
CPZ was reported to attenuate pancreatic β cell function through insulin receptor 2 degradation on mouse islets. However, the effective dose (50 µM) was more than two-fold higher than the dose that was used in the current study on AAT protective mechanisms (20 µM) [37]. Furthermore, CPZ treated islets showed no significant difference compared to non-treated islets in cell death-related parameters measured in this study (Figure 4 & 5), indicating the results observed in AAT plus CPZ plus cytokine groups are primarily caused by cytokine stimulation and the prevention of AAT endocytosis. Therefore, optimizing the uptake of AAT by human islets has the potential to become a target for therapy in T1D with or without islet transplantation for the purpose of improving islet survival under stress.
OCR is an indicator of islet cell viability and is used to predict the outcome of clinical islet autotransplantation [29, 38]. The effect of AAT on OCR seems to vary among different cells. Dendritic cells, regulatory T cells and natural killer cells from AAT-treated mice exhibited markedly higher OCR, whereas CD4+ and CD8+ effector T cells from the same animals showed lower OCR, compared to the control group [39]. We found that AAT slightly increased OCR in human islets. Furthermore, cytokine-suppressed OCR was ameliorated by the addition of AAT, strongly indicating that AAT promotes islet respiration. The upregulated OCR can also be attributed to the anti-apoptotic effect of AAT. Further, gene expression studies showed that AAT treatment influenced the expression levels of PGC1α, a key regulator of mitochondrial function and integrity, and Bim and NOS2, mitochondria-regulated apoptosis-related genes [40-43], implying that AAT reduced mitochondria-mediated apoptosis caused by cytokines in human islets.
In accordance with the suppression of JNK pathway as well as Bim and NOS2 expression, we found that expression of c-casp9, which is the initiator of the caspase cascade in the mitochondria-mediated apoptosis pathway that triggers the activation of c-casp3 [44], was also reduced by AAT treatment following cytokine stimulation. This is important because previously, studies have reported the direct inhibitory activity of AAT on c-casp3 in cell-free enzymatic activity assays and lung endothelial cell and other cells [32, 33, 45, 46]. Therefore, the anti-apoptotic effect of AAT was largely attributed to its inhibition of c-casp3. Our observation that AAT significantly reduced c-casp9 expression strongly indicated that AAT exerts protective effects on islets at stages earlier than the activation of c-casp3. A comprehensive analysis of AAT inhibitory ability on all caspases known to be involved in apoptosis showed that AAT only inhibited the effector caspases (-3, -6 and -7), but not the initiator caspases including c-casp9 [46]. Thus, the reduced c-casp9 by AAT in this study has most likely resulted from the suppression of the JNK pathway after cytokine stimulation. Our results proved that the anti-apoptotic effect of AAT on islets is not only caused by the direct inhibition of c-casp3 activity, but also the regulation of upstream factors of cytokine-induced apoptotic pathways, most likely beginning with the suppression of JNK phosphorylation. These novel findings shed light on the mechanisms underlying the protective effects of AAT on inflammation-induced islet cell death.
We show that a single injection of AAT to donor mice at a clinically relevant dose (160 mg/kg) significantly improved islet graft survival and function after syngeneic islet transplantation. Since transplant recipients did not receive any other treatment, the beneficial effect of AAT was likely explained by AAT uptake by islets and the attenuation of inflammatory response in islet cells. Importantly, this protection was sustained during the isolation process. These results not only confirmed that AAT endocytosis in islet cells occurs in vivo, but also provided a novel therapeutic strategy in islet transplantation. Our study offers evidence that it may be beneficial to give AAT before pancreatectomy in patients undergoing autologous islet transplantation.
To the best of our knowledge, this is the first report describing the use of AAT in the context of donor treatment in islet transplantation. In the context of allogeneic transplantation, islets suffer similar insults during the peri-transplant period. Our results of donor-treatment suggest that AAT could protect islets from stress, and has potential usage in allogeneic islet transplantation, by improving the outcome of islet isolation. This work extends the current paradigm of AAT as a potential anti-inflammatory therapy for recipients and reveals important additional functions of AAT in promoting islet survival through the process of islet procurement.
One limitation of this study is that it is difficult to inhibit AAT internalization by islet cells to confirm the precise role of AAT endocytosis in the improved islet graft function in vivo. β cell-specific clathrin knockout mice may be required to confirm this effect. In vitro culture of islets with AAT with or without CPZ before transplantation may also provide answers to this question. In fact, Abecassis and colleagues showed that AAT-pre-treated islet grafts exhibited increased IL-1 receptor antagonist, which were associated with significantly reduced levels of TNFα and IL-1β [47]. However, more studies on long-term graft survival and function are required to conclude the potential benefits of in vitro culture of islets with AAT and whether endocytosis plays a role in this.
In conclusion, this study demonstrated the mechanistic insight that AAT is internalized by β cells through clathrin-dependent endocytosis, leading to protection from cell death in vitro and after transplantation. Donor treatment with AAT may serve as an option to improve islet survival after transplantation.
Material and Methods
Mice
Male C57BL/6 and Swiss mice at 7-8 weeks of age were purchased from the Jackson Laboratory (Bar Harbor, ME) and the Taconic Biosciences (Germantown, NY). All experiments were approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina (protocol #AR170117).
Human AAT injection
Human AAT (Prolastin-C, Grifols) was dissolved in sterile water at 5 mg/ml and snap frozen in -80 ° C freezer before each use. Mice were injected with AAT (i.p.) or saline (control) 24 h before islets isolation.
Mouse islets isolation, diabetes induction, and islet transplantation
Islets were isolated by collagenase digestion as described previously [48]. C57BL/6 mice were rendered diabetic by a single dose streptozotocin (STZ) injection (225 mg/kg, i.p.; Sigma-Aldrich). At 5-7 days post STZ injection, mice with non-fasting blood glucose >300 mg/dL were used as recipients. For AAT donor treatment, C57BL/6 mice were treated with human AAT (80 mg/kg or 160 mg/kg, i.p.) and islets were isolated 24 h after injection. For each transplantation, 200-300 hand-picked mice islets (11 islets per gram of body weight) were transplanted into the recipients' liver as previously described [22]. Non-fasting blood glucose levels were measured every two or three days until 60 days post transplantation using a Freestyle Lite glucometer (Freestyle). Mice with blood glucose levels < 200 mg/dL were considered normoglycemic.
IVGTT
Recipient mice were fasted for 4 h and injected with glucose solution at 1g/kg via tail vein. Blood glucose levels were measured at 0, 15, 30, 60, 90, and 120 min after injection. Blood glucose area under the curve during the IVGTT was calculated using the Trapezoidal method [49].
Culture of human islets and βTC3 cells
Human islets were obtained from Georgetown University and cultured in CMRL 1066 (Corning) supplemented with 10% fetal bovine serum (FBS) (Gibco), 100 U/mL penicillin and 100 mg/mL streptomycin (Hyclone). βTC3 cells were cultured in DMEM with 10% FBS (Gibco), with penicillin-streptomycin (100 U/ml, Thermo Fisher).
Human islets and βTC3 treatment and Western blot
Human islets or βTC3 cells were incubated with AAT for 2 h before the addition of cytokines (50 U/ml TNFα + 50 U/ml IL-1β + 1000 U/ml INF-γ) for human islets, and 100 U/ml IL-1β and 1000 U/ml IFN-γ for βTC3 cells. For CPZ group, islets were incubated in CPZ for 1 h before AAT pretreatment. Samples were collected and lysed using RIPA Lysis and Extraction buffer (Thermo Fisher). Cytosolic or membrane fractions were extracted with the Plasma Membrane Protein Extraction Kit (Biovision), using the manufacturer's protocol. Lysates (10-30 µg) were separated by SDS-PAGE, transferred to PVDF membranes, and incubated with primary antibodies against phospho-JNK, total JNK, β-actin, ATPase Na+/K+ transporting subunit α 1 (ATP1A1), GAPDH (Cell Signaling Technology) and AAT (Sigma). Horseradish peroxidase-conjugated secondary antibodies were from Cell Signaling Technology. Signals were visualized using an ECL detection kit (Thermo Scientific). Relative protein expression of genes was quantified using ImageJ (NIH) or Image Lab™ Software (Bio-Rad).
AAT labeling and treatment
AAT was labeled using Alexa Fluor 488 Protein Labeling Kit (Invitrogen) following the manufacturer's instruction. Islets or βTC3 cells were cultured in the presence of labeled-AAT for 2 h, washed with cold PBS and immediately subjected to imaging using a Leica SP5 confocal microscope. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI).
Real-time PCR (qPCR) Analysis
RNA was extracted and reverse transcribed into cDNA using an RT-PCR kit (Bio-Rad). The Advanced Universal SYBR Green Supermix (Bio-Rad) was used for quantitative RT-PCR in a CFX96 Real-Time Thermalcycler (Bio-Rad). Fold changes in gene expression normalized to GAPDH or β-actin expression were plotted and compared between groups.
Immunofluorescence
Human islets were embedded in OCT compound (Sakura) and were cut into sections with a thickness of 5 μm. Sections were fixed in cold acetone for 5 min. βTC3 cells were washed and fixed in 2% paraformaldehyde for 15 min at room temperature. Sections were stained with anti-insulin (Invitrogen) and anti-AAT (Sigma) antibodies. Images were collected using a Leica SP5 confocal microscope.
Apoptosis ELISA Assay
Human islets were incubated in cytokine cocktails for 96 h. Apoptosis was measured using an In Situ Cell Death Detection Kit (Sigma) according to the manufacturer's recommendation.
Measurement of OCR
OCR was measured in islets using the MicroOxygen Uptake System, FO/SYSZ-P175 (Instech Laboratories, Plymouth Meeting, PA). DNA concentration of each sample was measured by fluoro spectrophotometry using the Quant-iTTM PicoGreen dsDNA Assay kit (Molecular Probes, Eugen, OR). Fluorescence was read by a microplate reader. The mean OCR/DNA (nmolO2/min·mg DNA) values were calculated and normalized to “fold change” against a negative control.
Statistical analysis
Percentages of mice reaching normoglycemia were plotted by Kaplan- Meier curves, and differences in graft survival were compared by the logrank test. Differences between groups were compared for statistical significance by ANOVA or Student's t test; p < 0.05 denoted significance. ANOVA was used for analyzing IVGTT results. Data are expressed as mean ± standard error of the mean (SEM).
Abbreviations
AAT: Alpha-1 antitrypsin; AUC: area under the curve; Bim: BCL-2-like protein 11; c-casp: cleaved caspase 9; CPZ: chlorpromazine; JNK: c-Jun N-terminal kinase; OCR: Oxygen consumption rate; IBMIR: instant blood-mediated inflammatory reaction; IL-1β: interleukin-1 β; NOS2: nitric oxide synthase 2; IFN-γ: interferon γ; PGC1α: peroxisome proliferator-activated receptor γ coactivator 1 α; STZ: streptozotocin; c-PARP: cleaved Poly (ADP-ribose) polymerase; T1D: Type 1 diabetes.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This study was supported in part by the National Institutes of Health (DK120394, DK105183, and DK099696 to HW). We thank the Reeves family for their generous donation.
Competing interests
CS consults for and participates in grants to MUSC by Grifols, the manufacturer of human AAT used in this study.
References
1. Bruni A, Gala-Lopez B, Pepper AR, Abualhassan NS, Shapiro AJ. Islet cell transplantation for the treatment of type 1 diabetes: recent advances and future challenges. Diabetes Metab Syndr Obes. 2014;7:211-23
2. Wang H, Desai KD, Dong H, Owzarski S, Romagnuolo J, Morgan KA. et al. Prior surgery determines islet yield and insulin requirement in patients with chronic pancreatitis. Transplantation. 2013;95:1051-7
3. Naziruddin B, Iwahashi S, Kanak MA, Takita M, Itoh T, Levy MF. Evidence for instant blood-mediated inflammatory reaction in clinical autologous islet transplantation. Am J Transplant. 2014;14:428-37
4. Kanak MA, Takita M, Itoh T, SoRelle JA, Murali S, Kunnathodi F. et al. Alleviation of instant blood-mediated inflammatory reaction in autologous conditions through treatment of human islets with NF-kappaB inhibitors. Transplantation. 2014;98:578-84
5. Itoh A, Ridgway WM. Targeting innate immunity to downmodulate adaptive immunity and reverse type 1 diabetes. Immunotargets Ther. 2017;6:31-8
6. Tai N, Wong FS, Wen L. The role of the innate immune system in destruction of pancreatic beta cells in NOD mice and humans with type I diabetes. J Autoimmun. 2016;71:26-34
7. Kim HS, Lee MS. Role of innate immunity in triggering and tuning of autoimmune diabetes. Curr Mol Med. 2009;9:30-44
8. Mandrup-Poulsen T, Bendtzen K, Nerup J, Dinarello CA, Svenson M, Nielsen JH. Affinity-purified human interleukin I is cytotoxic to isolated islets of Langerhans. Diabetologia. 1986;29:63-7
9. Schwarznau A, Hanson MS, Sperger JM, Schram BR, Danobeitia JS, Greenwood KK. et al. IL-1beta receptor blockade protects islets against pro-inflammatory cytokine induced necrosis and apoptosis. J Cell Physiol. 2009;220:341-7
10. Yamada K, Takane N, Otabe S, Inada C, Inoue M, Nonaka K. Pancreatic beta-cell-selective production of tumor necrosis factor-alpha induced by interleukin-1. Diabetes. 1993;42:1026-31
11. Mandrup-Poulsen T, Bendtzen K, Dinarello CA, Nerup J. Human tumor necrosis factor potentiates human interleukin 1-mediated rat pancreatic beta-cell cytotoxicity. J Immunol. 1987;139:4077-82
12. Pukel C, Baquerizo H, Rabinovitch A. Destruction of rat islet cell monolayers by cytokines. Synergistic interactions of interferon-gamma, tumor necrosis factor, lymphotoxin, and interleukin 1. Diabetes. 1988;37:133-6
13. Andersen NA, Larsen CM, Mandrup-Poulsen T. TNFalpha and IFNgamma potentiate IL-1beta induced mitogen activated protein kinase activity in rat pancreatic islets of Langerhans. Diabetologia. 2000;43:1389-96
14. Baraldo S, Balestro E, Bazzan E, Tiné ME, Biondini D, Turato G. et al. Alpha-1 Antitrypsin Deficiency Today: New Insights in the Immunological Pathways. Respiration. 2016;91:380-5
15. de Serres F, Blanco I. Role of alpha-1 antitrypsin in human health and disease. J Intern Med. 2014;276:311-35
16. Ehlers MR. Immune-modulating effects of alpha-1 antitrypsin. Biol Chem. 2014;395:1187-93
17. Sorrells S, Camprubi S, Griffin R, Chen J, Ayguasanosa J. SPARTA clinical trial design: exploring the efficacy and safety of two dose regimens of alpha1-proteinase inhibitor augmentation therapy in alpha1-antitrypsin deficiency. Respir Med. 2015;109:490-9
18. McElvaney NG, Burdon J, Holmes M, Glanville A, Wark PA, Thompson PJ. et al. Long-term efficacy and safety of α1 proteinase inhibitor treatment for emphysema caused by severe α1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE). Lancet Respir Med. 2017;5:51-60
19. Hubbard RC, Crystal RG. Alpha-1-antitrypsin augmentation therapy for alpha-1-antitrypsin deficiency. Am J Med. 1988;84:52-62
20. Lewis EC, Shapiro L, Bowers OJ, Dinarello CA. Alpha1-antitrypsin monotherapy prolongs islet allograft survival in mice. Proc Natl Acad Sci U S A. 2005;102:12153-8
21. Lewis EC, Mizrahi M, Toledano M, Defelice N, Wright JL, Churg A. et al. alpha1-Antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc Natl Acad Sci U S A. 2008;105:16236-41
22. Wang J, Sun Z, Gou W, Adams DB, Cui W, Morgan KA. et al. alpha-1 Antitrypsin Enhances Islet Engraftment by Suppression of Instant Blood-Mediated Inflammatory Reaction. Diabetes. 2017;66:970-80
23. Wang LH, Rothberg KG, Anderson RG. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol. 1993;123:1107-17
24. Zhou X, Liu Z, Shapiro L, Yang J, Burton GF. Low-density lipoprotein receptor-related protein 1 mediates alpha1-antitrypsin internalization in CD4+ T lymphocytes. J Leukoc Biol. 2015;98:1027-35
25. Marsh M, Helenius A. Virus entry: open sesame. Cell. 2006;124:729-40
26. Abderrahmani A, Niederhauser G, Favre D, Abdelli S, Ferdaoussi M, Yang JY. et al. Human high-density lipoprotein particles prevent activation of the JNK pathway induced by human oxidised low-density lipoprotein particles in pancreatic beta cells. Diabetologia. 2007;50:1304-14
27. Ferdaoussi M, Abdelli S, Yang JY, Cornu M, Niederhauser G, Favre D. et al. Exendin-4 protects beta-cells from interleukin-1 beta-induced apoptosis by interfering with the c-Jun NH2-terminal kinase pathway. Diabetes. 2008;57:1205-15
28. Cataldo LR, Mizgier ML, Bravo Sagua R, Jaña F, Cárdenas C, Llanos P. et al. Prolonged Activation of the Htr2b Serotonin Receptor Impairs Glucose Stimulated Insulin Secretion and Mitochondrial Function in MIN6 Cells. PLoS One. 2017;12:e0170213
29. Papas KK, Pisania A, Wu H, Weir GC, Colton CK. A stirred microchamber for oxygen consumption rate measurements with pancreatic islets. Biotechnol Bioeng. 2007;98:1071-82
30. Villena JA. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015;282:647-72
31. Oropeza D, Jouvet N, Bouyakdan K, Perron G, Ringuette LJ, Philipson LH. et al. PGC-1 coactivators in β-cells regulate lipid metabolism and are essential for insulin secretion coupled to fatty acids. Mol Metab. 2015;4:811-22
32. Petrache I, Fijalkowska I, Medler TR, Skirball J, Cruz P, Zhen L. et al. α-1 Antitrypsin Inhibits Caspase-3 Activity, Preventing Lung Endothelial Cell Apoptosis. Am J Pathol. 2006;169:1155-66
33. Zhang B, Lu Y, Campbell-Thompson M, Spencer T, Wasserfall C, Atkinson M. et al. Alpha1-antitrypsin protects beta-cells from apoptosis. Diabetes. 2007;56:1316-23
34. Sohrab S, Petrusca DN, Lockett AD, Schweitzer KS, Rush NI, Gu Y. et al. Mechanism of alpha-1 antitrypsin endocytosis by lung endothelium. FASEB J. 2009;23:3149-58
35. Aggarwal N, Korenbaum E, Mahadeva R, Immenschuh S, Grau V, Dinarello CA. et al. α-Linoleic acid enhances the capacity of α-1 antitrypsin to inhibit lipopolysaccharide-induced IL-1β in human blood neutrophils. Mol Med. 2016;22:680-93
36. Aldonyte R, Tunaitis V, Surovas A, Suriakaite K, Jarmalaviciute A, Magnusson KE. et al. Effects of major human antiprotease alpha-1-antitrypsin on the motility and proliferation of stromal cells from human exfoliated deciduous teeth. Regen Med. 2010;5:633-43
37. Park S, Hong SM, Lee JE, Sung SR, Kim SH. Chlorpromazine attenuates pancreatic beta-cell function and mass through IRS2 degradation, while exercise partially reverses the attenuation. J Psychopharmacol. 2008;22:522-31
38. Papas KK, Bellin MD, Sutherland DE, Suszynski TM, Kitzmann JP, Avgoustiniatos ES. et al. Islet Oxygen Consumption Rate (OCR) Dose Predicts Insulin Independence in Clinical Islet Autotransplantation. PLoS One. 2015;10:e0134428
39. Marcondes AM, Karoopongse E, Lesnikova M, Margineantu D, Welte T, Dinarello CA. et al. α-1-Antitrypsin (AAT)-modified donor cells suppress GVHD but enhance the GVL effect: a role for mitochondrial bioenergetics. Blood. 2014;124:2881-91
40. Xiong S, Mu T, Wang G, Jiang X. Mitochondria-mediated apoptosis in mammals. Protein Cell. 2014;5:737-49
41. Cook SJ, Stuart K, Gilley R, Sale MJ. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. 2017;284:4177-95
42. Holohan C, Szegezdi E, Ritter T, O'Brien T, Samali A. Cytokine-induced beta-cell apoptosis is NO-dependent, mitochondria-mediated and inhibited by BCL-XL. J Cell Mol Med. 2008;12:591-606
43. Scarim AL, Heitmeier MR, Corbett JA. Heat shock inhibits cytokine-induced nitric oxide synthase expression by rat and human islets. Endocrinology. 1998;139:5050-7
44. Gurzov EN, Eizirik DL. Bcl-2 proteins in diabetes: mitochondrial pathways of β-cell death and dysfunction. Trends Cell Biol. 2011;21:424-31
45. Jedicke N, Struever N, Aggrawal N, Welte T, Manns MP, Malek NP. et al. α-1-antitrypsin inhibits acute liver failure in mice. Hepatology. 2014;59:2299-308
46. Lockett AD, Van Demark M, Gu Y, Schweitzer KS, Sigua N, Kamocki K. et al. Effect of cigarette smoke exposure and structural modifications on the α-1 Antitrypsin interaction with caspases. Mol Med. 2012;18:445-54
47. Abecassis A, Schuster R, Shahaf G, Ozeri E, Green R, Ochayon DE. et al. alpha1-antitrypsin increases interleukin-1 receptor antagonist production during pancreatic islet graft transplantation. Cell Mol Immunol. 2014;11:377-86
48. Cui W, Angsana J, Wen J, Chaikof EL. Liposomal formulations of thrombomodulin increase engraftment after intraportal islet transplantation. Cell Transplant. 2010;19:1359-67
49. Tai MM. A mathematical model for the determination of total area under glucose tolerance and other metabolic curves. Diabetes Care. 1994;17:152-4
50. Bergin D, Reeves E, Hurley K, Wolfe R, Jameel R, Fitzgerald S. et al. The Circulating Proteinase Inhibitor α-1 Antitrypsin Regulates Neutrophil Degranulation and Autoimmunity. Sci Transl Med. 2014;6(217):217ra1
Author contact
![]() Corresponding author: Hongjun Wang, Ph.D. Department of Surgery, Medical University of South Carolina, CRI 410, 173 Ashley Avenue, Charleston, SC 29425. Tel: 843-792-1800, Fax: 843-792-3315, Email: wanghoedu.
Corresponding author: Hongjun Wang, Ph.D. Department of Surgery, Medical University of South Carolina, CRI 410, 173 Ashley Avenue, Charleston, SC 29425. Tel: 843-792-1800, Fax: 843-792-3315, Email: wanghoedu.