Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(13):3952-3965. doi:10.7150/thno.30814 This issue Cite
Research Paper
Non-classical estrogen signaling in ovarian cancer improves chemo-sensitivity and patients outcome
Dapeng Hao1*, Jingjing Li1*, Jianlin Wang1, Yuan Meng1, Zhiqiang Zhao1, Chao Zhang1, Kai Miao1, Chuxia Deng1, Benjamin K. Tsang3, Li Wang1,2 ![]() , Li-jun Di1
, Li-jun Di1 ![]()
1. Cancer Center, Faculty of health Sciences, University of Macau, Macau, SAR of the People's Republic of China
2. Metabolism Core, Faculty of Health Sciences, University of Macau, Macau, SAR of the People's Republic of China
3. Department of Obstetrics and Gynecology and Cellular and Molecular Medicine, University of Ottawa, Ottawa, Ontario K1N 6N5, Canada
* Equal contribution
Received 2018-10-20; Accepted 2019-3-29; Published 2019-5-31
Abstract

Deficiency in homologous recombination repair (HRR) is frequently associated with hormone-responsive cancers, especially the epithelial ovarian cancer (EOC) which shows defects of HRR in up to half of cases. However, whether there are molecular connections between estrogen signaling and HRR deficiency in EOC remains unknown.
Methods: We analyzed the estrogen receptor α (ERα) binding profile in EOC cell lines and investigated its association with genome instability, HRR deficiency and sensitivity to chemotherapy using extensive public datasets and in vitro/in vivo experiments.
Results: We found an inverse correlation between estrogen signaling and HRR activity in EOC, and the genome-wide collaboration between ERα and the co-repressor CtBP. Though the non-classical AP-1-mediated ERα signaling, their targets were highly enriched by HRR genes. We found that depleting ERα in EOC cells up-regulates HRR activity and HRR gene expression. Consequently, estrogen signaling enhances the sensitivity of ovarian cancer cells to chemotherapy agents in vitro and in vivo. Large-scale analyses further indicate that estrogen replacement and ESR1 expression are associated with chemo-sensitivity and the favorable survival of EOC patients.
Conclusion: These findings characterize a novel role of ERα in mediating the molecular connection between hormone and HRR in EOC and encourage hormone replacement therapy for EOC patients.
Keywords: Ovarian cancer, Estrogen signaling, Deficiency of homologous-recombination, Chemotherapy, Hormone replacement
Introduction
Epithelial ovarian cancer (EOC), especially the high-grade serous EOC (HGSOC), is characterized by positive ERα status in the vast majority of tumors, regardless of tumor subtypes [1, 2], which is consistent with its estrogen etiology as shown by large-scale epidemiological studies [3]. ERα has been well studied for its transcriptional regulation in response to estrogen in breast cancer [4]. The ligand bound ERα enters nucleus and can be either an activator or a repressor, depending on its interacting co-factors. Previous studies have demonstrated the activating function of ERα by collaborating with co-activators such as SRC-1 family, p300/CBP, SWI/SNF complex, TRAP complex and other histone modifiers [5], and the repressive function via interacting with CtBP, LCoR, Rip140, ZNF366 and HDACs [6-8].
ERα regulates target genes through either classical model or non-classical model. In the classical model that accounts for the majority of ERα bindings on DNA in breast cancer cells, such as MCF7, ERα recognizes the estrogen response elements (EREs) to regulate gene transcription with the assistance of two pioneering factors, GATA3 and FOXA1 [4, 9]. In the non-classical model, ERα forms transcriptional complex with other DNA binding factors such as AP-1 and SP1, and is recruited to the binding sites of these factors [10]. While the majority of ERα bindings in breast cancer cells rely on GATA3 and FOXA1, these two pioneering factors are likely to be breast cancer specific markers [11]. Consequently, how ERα behaves in EOC is elusive.
In breast cancer, patients with ERα+ tumor have much better survival than patients with ERα- tumor owing to the success of adjuvant hormone therapy using agents that block the mitogenic effect of estrogen, such as tamoxifen, fulvestrant and aromatase inhibitors [12, 13]. However, the anti-estrogen therapies have been disappointing in the treatment of EOC [14]. On the contrary, hormone replacement therapy (HRT), as an optional choice to alleviate the symptoms associated with oophorectomy, shows a beneficial effect on the survival of EOC patients, as demonstrated by clinical trial [15] and retrospective study [16], although inconsistent data may also exist [17-19].
EOC is characterized by DNA repair defects [20], especially the deficiency in homologous recombination repair (HRR) [21]. The core HRR genes including RAD51, ATM/ATR, CHEK1/CHEK2, BRCA1/BRCA2, MRN complex and Faconia anemia (FA) genes, are frequently altered in EOC and other hormone-related cancers [22, 23]. Since cells deficient in these genes are vulnerable to replicative stress and double-strand breaks, platinum-based chemotherapy is still the primary choice for the treatment of EOC [24]. Previous studies have suggest that hormone therapy has no significant effect on chemotherapy in breast cancer [25, 26]. However, the observation that HRR deficient tumors frequently originate from hormone enriched tissues points to a possibility that HRR may have a molecular connection with hormones [27]. It has been found that estrogen increases the genome instability in ERα+ EOC cells [28]. Here, we explore the ERα transcriptional programme on a global scale in EOC cells. We show that ERα represses HRR activity by direct bindings on HRR genes via interacting with CtBP in EOC cells, and suggest that estrogen replacement has the potential to benefit EOC patients from chemotherapy.
Materials and Methods
Cell lines, chemicals and antibodies
SKOV3 and HO8910 cells were used as representative ovarian cancer cell lines (see Supplementary materials for discussion of ovarian cancer cell lines). They were originally obtained from NICLR (National infrastructure of cell line resource) with certificates. SKOV3 and HO8910 were cultured in regular DMEM supplemented with 10% (v/v) FBS, penicillin-streptomycin (Thermo Fisher). Cisplatin, β-Estradiol (estrogen), fulvestrant, doxycycline (Dox), olaparib were all purchased from Sigma Aldrich. The anti-CtBP, anti-ERα, anti-Rad51, anti-GAPDH, anti-β-actin and anti-c-JUN antibodies were purchased from Santa Cruz (USA). The anti-caspase3, anti-PARP and the anti-γH2AX antibody was from Millipore (USA). Anti-CtBP recognizes both CtBP1 and CtBP2 unless otherwise indicated. Unless specified in the manuscript, cells were cultured by normal media with physiological level of estrogen.
Expression vectors and gene knockdown vectors
The CtBP1 and CtBP2 coding sequence were cloned into pCMV-script expression vector with or without HA tag and FLAG tag respectively. The pLVX-tight-puro lentivirus vector (Clontech) was also used for cloning of CtBP1 and CtBP2 as lentivirus expression vector. ERα coding sequence was amplified from the pCI-nGL1-HEGO and further cloned into pLVX-Tight-Puro. For lentivirus production, PLVX-Tight-Puro-CtBP (or ERα)-GFP and pLVXTet-On-Advanced (Clontech) plasmids were co-transfected into 293FT cells for lentivirus package. The lentivirus containing supernatant was harvested post transfection for 72 hours, and spun down, filtered with 0.45μM syringe filter, then infected the target cells and selected with Puromycin and G418. Dox was added to the cell culture medium to induce inserting gene expression. All the gene knockdown experiments were through the lentivirus vector pLKO1. The shRNAs targeting CtBP (Sense 5'-CCGGAGGGAGGACCTGGAGAAGTTCCTCGAGGAACTTCTCCAGGTCCTC-3' and anti-sense 5'-AATTCAAAAAAGGGAGGACCTGGAGAAGTTCCTCGAGGAACTTCTCCAGGTCCTCCCT-3') were cloned into pLKO1 for virus packaging in 293T cells and the supernatant were used for transduction directly or further concentration. ERα knockdown is through siRNA according to Liang et al. [29]. All the luciferase assay was performed using the pGL3 series of vectors from Promega.
ChIP and ChIPseq
After treatments, cells were cross-linked with 1% (w/v) formaldehyde for 5 min at room temperature. Ice cold glycine (125mM) was applied to quench formaldehyde. Then the cells were washed twice with ice cold PBS and collected. Cross-linked cells were resuspended in 1 ml immunoprecipitation (IP) buffer (150mMNaCl, 50mM Tris-HCl (pH 7.5), 5mM EDTA, NP-40(0.5%), Triton X-100 (1%), and added cocktail proteinase inhibitor (Sigma). Cell lysate were sonicated for 10 x 30 s with 30 s break using Qsonica Q700 sonicator. Then the sonicated cells were centrifuged and the supernatant was performed for immunoprecipitation. Each antibody was incubated with lysate overnight with rotation at 4 °C. And then the lysate was incubated with pre-blocked protein G beads with rotation for 10h at 4 °C. Then the beads were rinsed with high salt IP buffer supplemented with 500mM NaCl, IP buffer and finally resuspended in TE buffer (pH 8.0). Then Proteinase K (Qiagen #1018832) was added in DNA-protein complex for digestion overnight at 65 °C. Finally the DNA was purified by phenol-chloroform extraction and ethanol precipitation with the presence of glycogen (Ambion #AM9510). The purified DNA was used for real-time PCR.
Bioinformatics analysis
Sequences generated by the Illumina genome analyzer were aligned against genome version hg19. Binding sites of transcriptional factors were enriched by comparing the ChIP samples to input. Prognostic value was determined by Cox proportional hazards model in each datasets individually and integrated using fixed-effects meta-analysis. Expression data were downloaded from GEO, processed using RMA method and were quantile normalized. All the statistical analyses were performed in R software version 3.31. For more details, please refer to supplemental materials and methods.
Western Blot
The cells were lysed on ice using RIPA Buffer (Thermo # 89901) with the presence cocktail protease inhibitor (Sigma #SRE0055). Total protein (20 μg) from each sample was separated by SDS-PAGE in SDS running buffer (TAKARA #T9101) at 150 V for 1h at room temperature, and transferred to PVDF membranes at 300mA for 3h at 4°C. Blots were then probed with primary antibody at 1:1000 overnight at 4°C. Then the membrane was washed and incubated with HRP-conjugated secondary antibody (Santa Cruz) at 1:5000 dilution. After being washed for 5 times in PBST, the membrane was incubated with ECL detection reagent (#RPN2235) and then visualized with ChemiDoc Touch Imaging system (Bio-Rad).
γH2AX and RAD51 foci assay
Cells were grown on cover-slips. Wash cells twice with PBS and fix in 3.5% paraformaldehyde. Wash cells with PBS for 3 times. Cells are permeablized in PBS supplemented with 10% goat serum and 0.3% Triton X-100 for 15mins. Wash cells twice by PBS. Then cells were treated with 3% H2O2 for 10mins and wash for twice. Block cells with blocking buffer(10% goat serum in 1xPBS) for 1hr. Incubate cells with primary antibody(1:50) in blocking buffer for 1.5hr. Wash cells for 3 times by PBS. For γH2AX staining, the cells were incubated with Alexa Fluor® 488 Goat Anti-Mouse IgG for 1hr. After washing for 3 times, mount cover slips using VECTASHIELD with DAPI. For CtBP1 and CtBP2 staining, the Cells were incubated with EnVision+ System- HRP Labelled Polymer Anti-mouse (Dako, Carpinteria, CA, K4000) for 1 hr. After washing for 3 times, cells were incubated with Liquid DAB+ (Dako) for 3 minutes, and wash twice again. Then the cells were counterstained in Hematoxylin (Vector) for 30 sec, twice. Sequentially wash cells with water(twice, 5mins), 95% ethanol(2min) and 100% ethanol(2min). Finally, dip cells in xylene and mount with Permount onto a slide for microscopy imaging.
MTT assay
Cells were seeded in 96-well flat-bottom culture plates. After incubation with indicated treatment, the medium was aspirated and cells were treated with MTT (M5655, Sigma) containing medium for 4h. Then, the unreduced MTT solution was discarded, and DMSO (0.130 ml) was added into each well of the reduced MTT solution to dissolve the purple formazan precipitate, then OD values were detected with 550 nm filter of Victor X5 (Perkin Elmer, US).
HRR efficiency assay
ISceI-GR and pDRGFP plasmids [30] were co-transfected into cells, then treated cells with indicated conditions. HRR efficiency was measured with the ratio of GFP-positive cells out of all cells.
Engrafted tumor assay
HO8910 was used as the cell model over SKOV3 because of its high capacity of forming engrafted tumors and the fact that SKOV3 is Cisplatin resistant. Female ovariectomized NOD-SCID mice (6-8 weeks old) were subcutaneously injected 5×106 HO8910 cells [31] in each hind limb and randomly divided into three groups 5 weeks after cancer cells injection. Mice in control group were administrated with 3mg/kg cisplatin (S1552, Beyotime, China) only by intraperitoneal injection every 3 days; Mice in Estrogen combined with Cisplatin group were planted [32] with 17β-estradiol tablets (SE121, Innovative Research American), and also administrated with 3mg/kg cisplatin by intraperitoneal injection every 3 days; Mice in Fulvestrant group were administrated with Fulvestrant 30mg/kg by local injection [33] and 3mg/kg cisplatin by intraperitoneal injection every 3 days.
In another set of female NOD-SCID mice (6-8 weeks old), 5×106 HO8910 cells with empty lentiviral construct transduced or inducible ER or CtBP overexpression lentiviral construct transduced, were subcutaneously injected [31] in each hind limb and randomly divided into three groups 5 weeks after cancer cells injection. Mice in control group were administrated with 3mg/kg cisplatin (S1552, Beyotime, China) only by intraperitoneal injection every 3 days; Mice in CtBP and ERα overexpression group were given Dox (1.5 mg/ml plus 50 mg/ml sucrose) containing drinking water [34] and replaced every 3 days to make sure the water fresh, as well as 3mg/kg cisplatin by intraperitoneal injection every 3 days; Tumors were surgically removed after various administrations lasted for 15 days.
Data access
The RNA-seq data and ChIP-seq data from this study have been uploaded to the Gene Expression Omnibus (GEO) database (GSE116018).
Results
Genome-wide mapping of ERα binding reveals RAD51 as a direct target in EOC cell lines
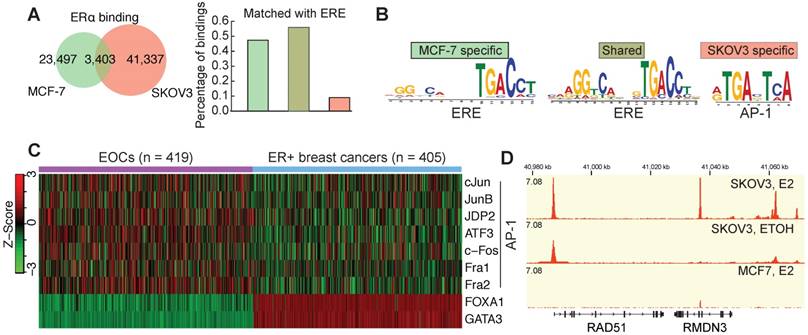
ERα in EOCs shows a strong expression compared to other cancers (Figure S1A) and a mild overexpression compared to ovary surface epithelium (Figure S1B), whereas its expression is decreased in most other cancers (Figure S1C). This is consistent with the previous finding that ERα is strongly expressed in EOC [35]. The availability of genomic data has led to the dispute over the representative EOC cell lines [36]. We used the most updated genomic data and confirmed that SKOV3 is a representative cell line by having the characterized TP53 mutation and a wildtype and strong expressing ERα (Figure S1D). ChIP-seq analysis identified 44,770 ERα binding sites in SKOV3 cells, of which only 7.6% are overlapped with the binding sites in MCF7 cells (Figure 1A), which might imply a tissue-specific ERα bindings. As expected, motif analysis of ERα bindings in MCF7 indicated a classical model characterized by the enrichment of classical ERE and GATA3 motif (Figure 1A and Figure S2A). However, in SKOV3 AP-1 motif is highly enriched in ERα bindings, implying the predominant non-classical regulatory mechanism of ERα. De novo motif discovery further confirmed this difference (Figure 1B). To confirm this in tumor samples, we analyzed the corresponding gene expressions and found that the pioneer factors of classical model [37], GATA3 and FOXA1, are silenced in EOCs, whereas AP-1 coding genes are overexpressed in EOCs in comparison to ERα+ breast cancer (Figure 1C and Figure S2B). To understand the biological meaning underlying this difference, we performed the genomic region enrichment analysis, which showed a highly distinct functional enrichment of ERα bindings in SKOV3 (Figure S2E). Interestingly, AP-1 mediated transcriptional regulation and DNA damage response were significantly enriched. Given the clinical importance of HRR in EOC, we specifically examined the core HRR genes, and surprisingly found that most of them have the ERα binding on the promoter region, including RAD51, ATR, BRCA1, PALB2, and FA genes. Figure 1D shows an example of estrogen-inducible ERα binding at the transcriptional start site (TSS) of RAD51, which has been confirmed by ChIP-qPCR in SKOV3 and another high-grade serous ovarian cancer (HGSC) cell line HO8910 (Figure S2C-D). However, no such ERα binding was observed in MCF7 cells (Figure 1D and Figure S2C).
Enrichment of the non-classical mode of ERα signaling in EOC by the binding profiles. A. The venn diagram showing the fraction of MCF7-specific bindings, shared bindings and SKOV3-specific bindings. The bars on the left indicate the fraction of each category of bindings that are matched with an ERE. B. The motifs identified by de novo motif analysis (MEME). C. Heatmap showing the expression of AP-1 members, FOXA1 and GATA3 in EOCs and ER+ breast cancers. Agilent microarray data of TCGA are used. D. ERα binding at the RAD51 locus in unstimulated and E2-treated SKOV3 cells, and in E2-treated MCF7 cells.
ERα is involved in DNA damage response and represses HRR activity
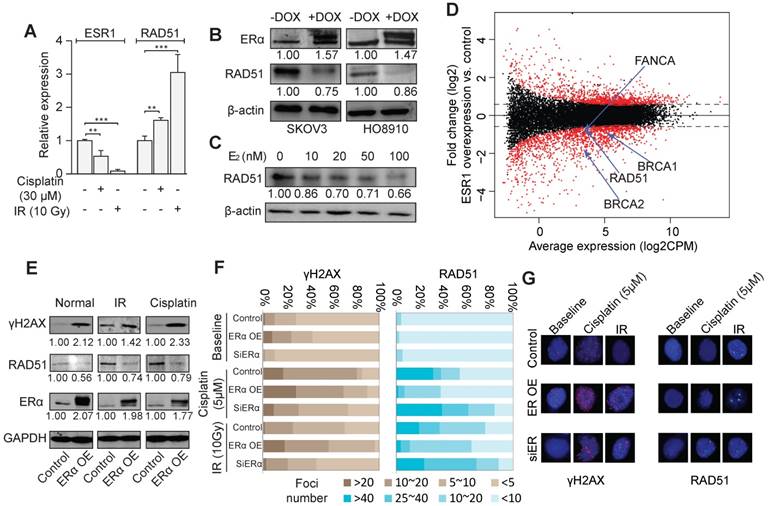
The treatment of irradiation (IR) and cisplatin led to a significant down-regulation of ERα and an up-regulation of RAD51 in SKOV3 and HO8910 cells (Figure 2A and Figure S3A-B). In stable cell lines with inducible ERα overexpression, RAD51 expression was remarkably decreased (Figure 2B). Also, Rad51 showed dose-dependent repression by estrogen (Figure 2C), including the condition of 10nM of estrogen, the closest dosage to endogenous estrogen level. Knocking down of ERα also resulted in the abolishment of estrogen induction of RAD51 expression in SKOV3 cells (Figure S3C). Furthermore, a significant inverse correlation of the protein abundance between ERα and RAD51 was observed in TCGA datasets (Spearman rho = -0.25, p < 10-6; Figure S3D). We then assessed the effect of ERα on global gene expression by RNA-seq of SKOV3 cells with or without ERα overexpression. The RNA-seq data revealed that many HRR genes including RAD51, ATR, BRCA1/2 and FA genes were down regulated by ERα overexpression (Figure 2D). Interestingly, we found that ERα was more likely to be a repressor, as indicated by the 769 downregulated genes versus 326 upregulated genes (fold-change > 2 and FDR < 0.05). We further confirmed some of the downregulated genes by qPCR in SKOV3 and HO8910 cells (Figure S3E). Overexpression of ERα increased the cellular level of γΗ2ΑΧ and decreased RAD51 (Figure 2E), no matter whether the cells were treated by IR or cisplatin, demonstrating a substantial role of ERα in governing the genome stability of EOC cells. Consistently, overexpression of ERα resulted in the increased formation of γΗ2ΑΧ foci and decreased RAD51 foci after the treatment of cisplatin or IR, whereas knockdown of ERα resulted in less γΗ2ΑΧ foci and more RAD51 foci (Figure 2F-G).
ERα represses HRR activity. A. Expression of ERα and RAD51 under the treatment of cisplatin (30 μM) or irradiation (IR, 10Gy) in SKOV3 cells detected by RT-PCR. B. Western blot showing the expression of ERα and RAD51 with or without the DOX-induced ERα expression in SKOV3 and HO8910 cells. The numbers indicate the average quantitation and the SD from three independent experiments. C. The expression of RAD51 in SKOV3 treated with different dosages of E2. D. M-A plot for the transcriptome expression changes comparing ER α overexpressing SKOV3 cells (DOX+) with control cells (DOX-). X-axis corresponds to the average expression of genes across all the samples, and Y-axis indicates the fold change of gene expression. Several significantly altered DNA damage repair genes are also shown. E. Western blot showing the expression of RAD51, ERα and γH2AX with or without the DOX-induced ERα expression in SKOV3 cells with or without treatment by cisplatin (5 μM) or IR (10 Gy). F. Quantification of γH2AX foci and RAD51 foci in SKOV3 cells transfected with exogenous ERα or siRNA of ERα, with or without treatment of cisplatin or IR. SKOV3 cells are fixed for immunofluorescence staining after treated with Cisplatin 5uM for 24h or 2 hours later after 10Gy irradiation. Cells are divided into four groups according to the number of foci and the percentage of each group is indicated. G. Examples of γH2AX foci and RAD51 foci in SKOV3 cells.
CtBP is recruited by ERα and is correlated with clinical outcome
We found that ERα bindings in SKOV3 are highly overlapped with the targets of a transcriptional corepressor, C-terminal binding protein (CtBP) that have been reported in a previous study [38]. Moreover, we found many common interacting proteins of CtBP and ERα according to STRING database [39], such as NRIP1, CREBBP, HDAC1/2, BRCA1, ZNF217 and SP1, implying a high probability of functional collaboration.
Therefore, we performed an in vivo Co-IP experiment and observed an estrogen-dependent interaction between CtBP and ERα in SKOV3 cells after 24 hours treatment of estrogen (Figure 3A and Figure S4A). Immunofluorescence staining of ERα and CtBP also supported the colocalization of these two proteins in nucleus (Figure S4B). We also found an ERα binding site at the promoter of CtBP (Figure S4C). To test that if CtBP could be regulated by ERα, we validated the recruitment of ERα at CtBP promoter by ChIP-qPCR and observed the regulatory activity of ER on CtBP promoter by luciferase assay (Figure 3B-C). Moreover, the expression of CtBP was upregulated by estrogen dose dependently (Figure 3D). Fulvestrant, however, repressed the expression of CtBP and TFF1, a known target gene of ERα ( Figure 3E). Ectopic overexpression of ERα also increased CtBP expression (Figure 3F). However, the regulation effect is relatively modest in cell lines. To further explore the association between CtBP and ERα, we quantified their expression in serous ovarian cancer tissue arrays, and found a significant coexpression between CtBP and ERα (Pearson's r = 0.54, p < 10-6; Figure 3G). The coexpression was further supported by public resources (Figure S4D). In addition, we found significantly higher expression of CtBP in estrogen-responsive tissues and tumors (Figure S4E-H).
Recruitment of CtBP by ERα in EOC. A. Results from co-IP experiments in SKOV3 cells. ERα-containing protein complex is immunoprecipitated from E2- and EtOH-treated SKOV3 cells using anti-ERα or IgG, followed by western blot using antibodies against ERα and CtBP. B. ChIP-qPCR results testing the binding of ERα at the TSS of CtBP in SKOV3 cells. The enrichment of ERα is shown as percentage to input. C. Luciferase assay of ERα regulation of CTBP1 promoter activity. Top panel shows ERα expression with different dosages of ERα vector for transfection, and bottom panel shows the promoter activity relative to vehicle. Significance is determined by comparing to the group with 0 vector using T test. D. Western blot showing the expression of CtBP in SKOV3 cells treated with different dosages of E2. E. qPCR results for the expression of CTBP1 and TFF1 in SKOV3 cells treated with different dosages of fulvestrant. F. Western blot showing the expression of CtBP and ERα in SKOV3 and HO8910 cells transduced with DOX inducible ERα expression lentiviral vector. G. Coexpression between CtBP and ERα across serous ovarian cancer tissues (n = 74). The array images were acquired using Leica Microsystems at 40X. Three representative areas of each spot were analyzed for the staining signal by MIPAR. The average intensity of the three areas was calculated for each spot for both CtBP and ERα staining. H. Frequency of amplification of CtBP genes (GISTIC) across TCGA cancer types. Uterine cancers are divided into serous-like (HRR deficient) tumors and other tumors. Significance is determined by hypergeometric distribution test using pan-cancer amplification as background. I. Kaplan-Meier overall survival curves. Patients are separated into EOCs with (red) and without CtBP amplification (green). Right panel shows the relative fraction of sensitive and resistant EOCs to chemotherapy for EOCs with CtBP amplification and EOCs without CtBP amplification (p < 0.05, Fisher exact test).
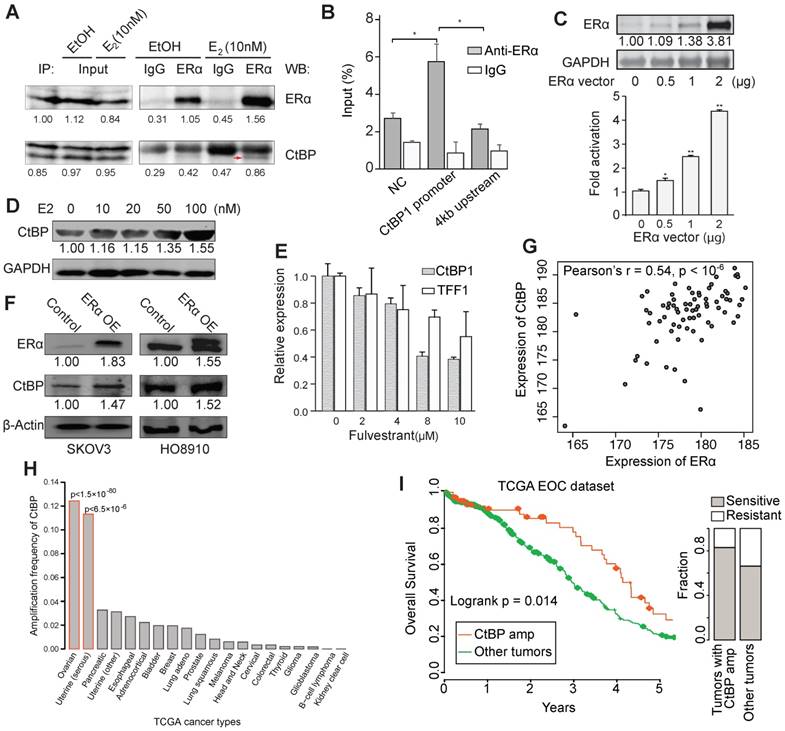
It has been found that CtBP could target multiple HRR genes including BRCA1, ATR, PALB2, FANCD2, FANCM, and RAD51C [38]. A mild but significant inverse correlation between the expression of CtBP and HRR genes was observed across EOCs (Figure S5A-B). Given the overexpression of CtBP in EOC tumors compared to normal ovary tissues (Figure S4H), we next tested whether CtBP plays a functional role in EOC. By analyzing 80 whole-genome sequenced EOCs [20], we found that high CtBP expression is correlated with more somatic mutations and structural variants (Figure S5C-D). In accordance with this, we found that CtBP genes (CTBP1 and CTBP2) are selectively amplified in HRR deficient tumors including EOC and serous-like uterine cancer (Figure 3H), and the amplification is associated with more somatic mutations (Figure S5E-F). Importantly, the amplification of CtBP is associated with improved survival (log-rank test, p < 0.02) and less chemoresistance (Fisher exact test, p < 0.05) (Figure 3I). Although there are many chemo-resistant mechanisms, the expression of CtBP, in some cases, could be reduced while its targeting HRR genes were upregulated during the acquirement of chemo-resistance, as shown in EOC cells (Figure S5G). This suggests that CtBP could be involved in the process of chemo-resistance. We also found that in a whole-genome sequenced EOC cohort, CtBP is among the top genes associated with the response to chemotherapy (Figure S5H).
ERα represses gene expression via genome-wide collaboration with CtBP
ChIP-seq data between CtBP and ERα revealed that about two-third of ERα bindings are overlapped with CtBP in SKOV3 cells (Figure 4A). These overlapping sites are significantly enriched for the promoter bindings of DNA repair genes (FDR < 0.01, Table S1). To investigate whether ERα mediates CtBP recruitment, we mapped CtBP bindings globally to identify the significant changes induced by ERα inhibition via 24 hours treatment of fulvestrant. This demonstrated a globally redistributed CtBP binding profile at the ERα-CtBP-shared binding events (Figure 4B-C). Overall, 62% of ERα-CtBP-shared bindings had decreased binding affinity (Weaker sites, FDR < 0.05 and fold change > 2), and 38% did not change (No change sites).
Genome-wide collaboration between ERα and CtBP. A. Venn diagrams showing the overlapping between CtBP and ERα binding sites (top) and binding genes (middle). Bottom panel shows the enriched functions of promoter-binding genes for each category. B. M-A plot of differential binding affinity (DBA) analysis (EdgeR) of CtBP-ERα-shared bindings in Fulvestrant treated vs. untreated cells. Two-third of CtBP-ERα-shared bindings showed significant decreases in affinity at a FDR < 0.05. The red dots below 0 represent Weaker bindings under treatment of fulvestrant, whereas the black dots around x-axis represent No change bindings under treatment of fulvestrant. C. Representative examples of the redistributed CtBP bindings in fulvestrant treated cells. D. Genome-wide bindings of CtBP and ERα in SKOV3 cells and their influence on gene expression. (Left) Heatmaps of ChIP-seq data for ERα bindings and CtBP bindings, and (right) activating/repressive regulation predictions using BETA algorithm. E. Integrated view of DEGs and the four categories of CtBP or ERα bindings. DEGs are divided into three clusters according to hierarchical clustering of gene expression (left). Also indicated is the information about whether each gene has an binding event within the 10kb window centered at TSS (right panel, the color of bars corresponds to the colors of the four categories of binding sites in D).
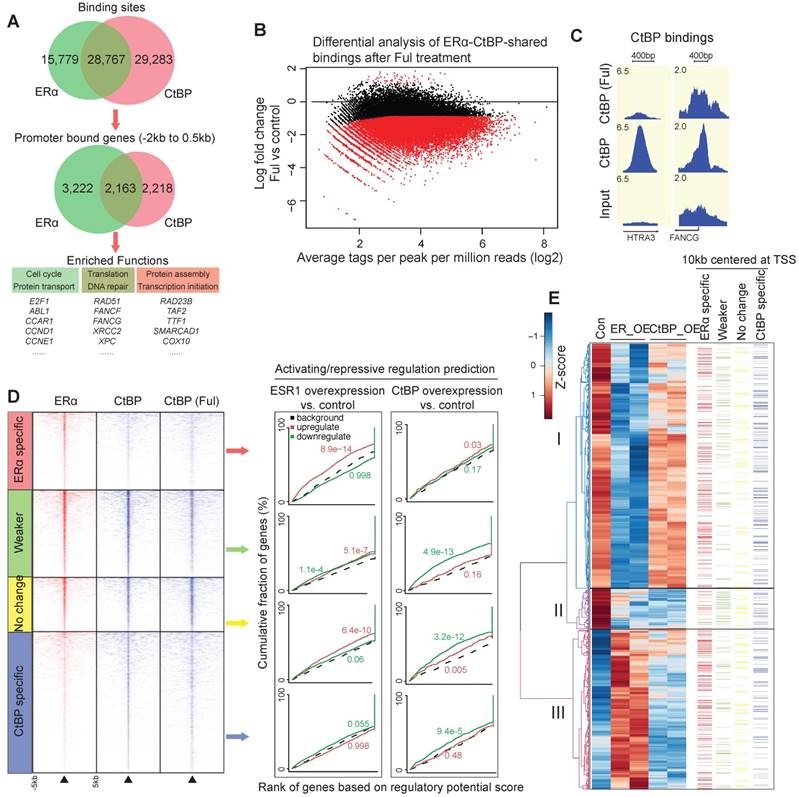
To unravel the genome-wide regulation on gene expression by CtBP and ERα, we also measured the transcriptome changes by RNA-seq. We used the software BETA to estimate the regulatory potential by considering the bindings within 100kb of the TSS, and then generated a cumulative distribution to determine the activating or repressive function of the transcription factor [40]. From the analysis (Figure 4D), we had the following observations at the genome-wide level: i) CtBP is repressive at all the categories having CtBP binding events including the ERα-CtBP-shared binding; ii) ERα is a potential activator in the ERα-specific sites and No change sites; and iii) ERα only has repressive function at the Weaker sites, where CtBP recruitment is likely to be modulated by ERα. De novo motif analysis of the 50bp flanking sequence by the center of Weaker sites, demonstrated a centrally distributed AP-1 motif. However, we did not find any centrally enriched motif for the No change sites. This further highlights the difference between the Weaker and the No change sites, and suggests that the non-classical estrogen-signaling is required to recruit CtBP for gene repression.
Of note, overexpression of CtBP and ERα resulted in a significant overlap of differentially expressed genes (DEGs) that were downregulated (hypergeometric test, p <10-100). We focused on the DEGs of ERα and correlated them with the four categories of binding events using a 10kb window of the TSS of genes in a visual map presented in Figure 4E. The DEGs were divided into three clusters, including the DEGs repressed strongly by ERα but not or only modestly by CtBP (cluster I, n = 803), the DEGs repressed strongly by both ERα and CtBP (cluster II, n = 132) and the DEGs activated by ERα (cluster III, n = 526). Cluster III is enriched with ERα-specific bindings but short of CtBP-specific bindings, whereas cluster II is enriched with the Weaker bindings but short of ERα-specific bindings (p < 0.05 at all cases, χ2-square test). These observations suggest that in cluster I and II, both ERα and CtBP are required for gene regulation, whereas in cluster III, ERα binding is dominant to activate gene expression. Taken together, our results indicate a genome-wide transcriptional collaboration between CtBP and ERα.
ERα and CtBP improve the response to chemotherapy agents
CtBP also binds at the promoter of RAD51 in EOC cells, which we further validated using ChIP-qPCR (Figure 5A). EOC cells with CtBP overexpression exhibited reduced RAD51 expression (Figure 5B). Using HRR efficiency reporter assay, we confirmed that both ERα and CtBP displayed the ability of repressing HRR, whereas the knockdown of CtBP attenuates the inhibition of ERα on HRR efficiency (Figure 5C). Given that an AP-1 motif was found within the CtBP-ERα-shared binding site at RAD51 promoter, we specifically silenced c-Jun (a subunit of AP-1) in EOC cells using RNAi and observed that silencing of AP-1 caused the loss of CtBP and ERα recruitment at the binding site (Figure 5D and Figure S6A-B), suggesting the non-classical model of ERα in the regulation of RAD51.
Sensitivity to chemotherapy agents through CtBP- and ERα-mediated repression of HRR. A. CtBP binding at the gene locus of RAD51 (left) and the validation by ChIP-qPCR in SKOV3 cells (right). B. Western blot showing the expression of RAD51 in SKOV3 and HO8910 cells upon induced CtBP overexpression by DOX. Over-expression of CtBP is DOX-induced and cells untreated with DOX are used as control. C. pDR-GFP vector based HRR efficiency reporter assay in SKOV3 cells with the conditions of ERα overexpression, CtBP overexpression or ERα overexpression plus CtBP knockdown. D. ChIP-qPCR validation for AP-1-mediated CtBP and ERα binding at the TSS of RAD51 in SKOV3 cells. Unspecified asterisks indicate the significance comparing with corresponding IgG. E. Dosage-dependent SKOV3 cell viability on cisplatin upon treatment by ERα overexpression or knockdown. Values are normalized to concentration 0 of each group. F. Cell viability assay for SKOV3 cells transfected with indicated vector or siRNA, after the treatment of cisplatin (3μM) for 72h. G. Cell viability assay for SKOV3 cells after different treatments as indicated for 24h (Cisplatin: 3μM; Olaparib: 10μM). Error bars in the figure indicate standard deviation of 3 replicates.
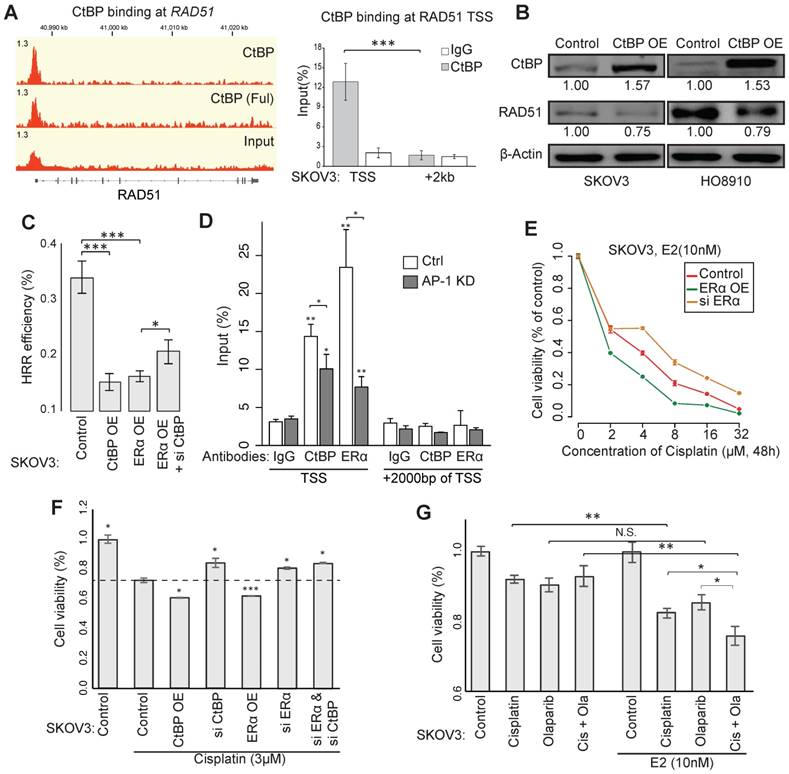
HRR deficiency predicts sensitivity to chemotherapy, as shown by the favorable outcome of patients carrying BRCA1/2 mutations [41]. We found that estrogen, ERα and CtBP had the ability to increase the sensitivity to cisplatin (Figure 5E and Figure S6C). Knockdown of CtBP or ERα significantly enhanced the cell viability under treatment of cisplatin in SKOV3 (Figure 5F and Figure S6D). Moreover, the estrogen treatment improved the response of SKOV3 cells to Cisplatin, Olaparib and their combination (Figure 5G and Figure S6G-H). Given that SKOV3 is a Cisplatin resistant cell line, more profound results have been observed for other EOC cell lines (Figure S6D-G).
Effects of hormone replacement in xenografts and in EOC patients
EOC xenografts using HO8910 cells were established and used to test the effect of ERα on tumor growth inhibition in vivo. We found that co-administration of estrogen increased the tumor inhibition effect of cisplatin whereas co-administration of fulvestrant blocked the growth inhibition effect of cisplatin (Figure 6A). As expected, overexpressing ERα or CtBP greatly improved the response to cisplatin of xenografts. IHC analysis of PARP also revealed significant increased DNA damages associated with ERa and CtBP overexpression (Figure S7A), indicating that the tumor cells may start to count on the alternative PARP dependent DNA repair pathway for survival. The cellular apoptosis of tumors was evaluated by detecting the cleaved PARP and cleaved caspase 3. Consistently, we observed the increased apoptosis in tumors treated with estrogen (Figure S7A).
Effect of HRT on xenografts and EOC patients. A. Measurement of HO8910 engrafted tumors subjected to cisplatin treatment (3mg/kg, 3 days interval) in combination with ER or CtBP overexpression or E2 (subcutaneously implanted estrogen pellet) or fulvestrant treatment (30mg/kg, 3 days interval). Tumor weights are compared between different groups using Wilcoxon rank-sum test. B, C. Forest plots for meta-analysis of OS (B) and DFS (C). The analysis was performed using software RevMan 5.3. CIs are set at 95% and shown as horizontal lines. Solid vertical lines indicate the no-difference point between HRT and control group. Pooled HRs are determined by the Fixed effect model.
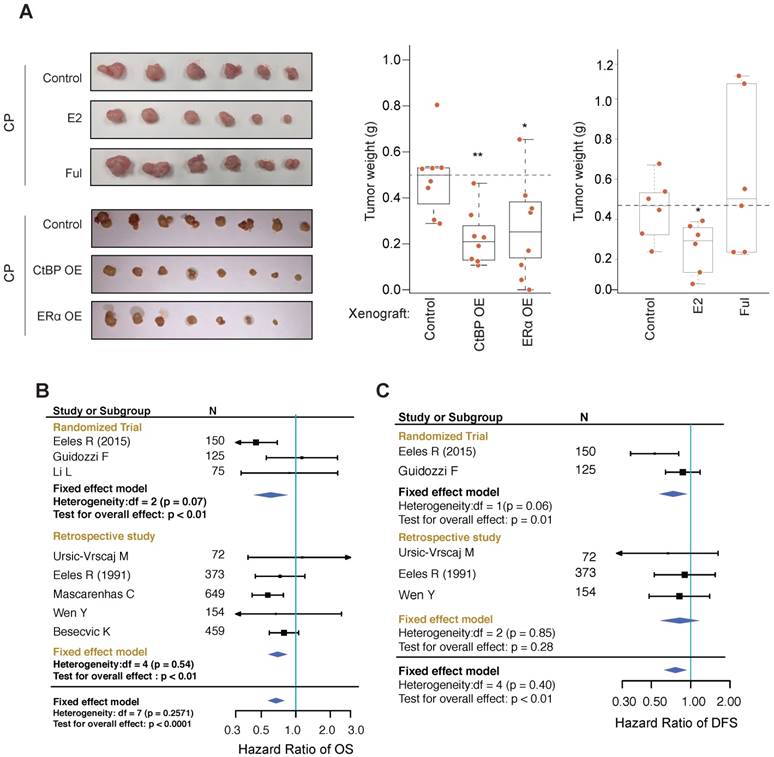
EOC patients suffered from menopausal syndromes may occasionally be administrated by hormone replacement therapy (HRT). Since most of the EOC patients receive chemotherapy, this provides a unique chance to test our model. By searching the Medline database using designed MeSH terms, 1,911 articles were retrieved. After manual screening, 9 studies were included in our analysis [15-18, 42-46], involving in total 837 patients who received HRT post-diagnosis of EOC or were users of HRT at the time of investigation, and 1,900 patients who didn't receive HRT (Table S2 and Figure S7B). The endpoint data for meta-analysis include overall survival (OS), disease-free survival (DFS), death events and recurrence events. Meta-analysis of these studies revealed a significant lower hazard ratio (HR) of OS and a significant lower odds ratio (OR) of disease-associated deaths among patients who received HRT than the patients who didn't receive HRT (Figure 6B and Figure S7C; HR of OS: 0.67 [95% CI, 0.57-0.80], p<0.0001 and OR of death: 0.58 [95% CI, 0.48-0.70], p<0.0001). We also noticed that HRT was significantly associated with a lower HR of disease free survival (DFS) and a lower OR of recurrences (Figure 6C and Figure S7D; HR = 0.76 [95% CI, 0.62-0.94], p<0.01 and OR = 0.57 [95% CI, 0.38-0.86], p<0.01). These results suggest that, whereas pre-diagnosis hormone use has been found to increase the risk of EOC, post-diagnosis HRT improves the outcome of EOC and reduces disease recurrences.
ESR1 is a favorable prognostic factor in EOC and associated with chemo-sensitivity
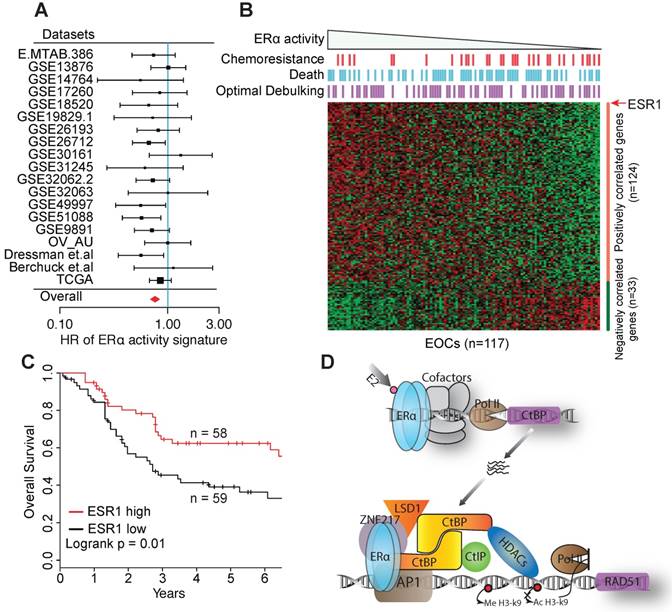
Compared to breast cancer, EOC tumors express quite similar levels of ESR1 (Figure S8A), which presents a challenge for the prognostic evaluation of ERα in individual cohorts, and led to inconsistent conclusions in previous studies [1, 47]. We therefore collected 19 public gene expression datasets, including 2,652 primary EOC tumors in total (Table S3). Our inclusion criterion requires at least 40 samples with continuous overall survival (OS) time accurate to days and censoring status. These datasets, when evaluated individually, show that ERα expression is associated with favorable outcome, though nonsignificant in most datasets. To get a consistent result, we leveraged the 19 datasets using meta-analysis and revealed a significant association between ESR1 expression and improved overall survival of EOC patients after controlling for stage, grade, age and histological subtypes (HR = 0.83 [95% CI, 0.75-0.93], p < 0.01, Figure S8B), indicating that ERα is an independent prognostic factor of EOC.
To further overcome the challenge by the similar expression levels of ESR1, we developed an ERα signature by combining the expression of ESR1 and genes consistently co-expressed with ESR1 across EOC cohorts and human tissues, to reflect ERα activity more robustly against expression noise (see Supplementary Methods and Figure S8C). The signature exhibits a lower HR than ESR1 expression in most datasets, including the TCGA dataset. By meta-analysis, the ERα signature exhibits a stronger association with favorable survival in univariable model (HR = 0.77 [95% CI, 0.69-0.85], p < 0.01), and in multivariable model controlling for age, stage, grade and histological subtypes (HR = 0.78 [95% CI, 0.70-0.87], p < 0.01, Figure 7A). We also repeated the analysis by limiting to HGSOCs, and observed a similar result (HR = 0.76 [95% CI, 0.68-0.85], p < 0.01). The dataset of Dressman et.al. shows a significant HR for both ESR1 expression and the ERα signature and is one of the few datasets providing response information to chemotherapy. To further investigate this dataset, we correlated the response to chemotherapy to the ERα signature, and found that ERα activity is independent of residual tumor size but significantly associated with the response to chemotherapy (High vs. low ERα activity group, p < 0.01 by Fisher exact test, Figure 7B). Consequently, ESR1 expression is a significant prognostic factor of ovarian cancer patients (Figure 7C). In TCGA dataset, high expression of ESR1 or ERα is also significantly associated with improved response to chemotherapy (Figure S8D).
The prognostic value of ERα in EOC. A. Forest plot visualing the hazard ratios (HRs) of ERα activity in each dataset evaluated by the multivariate Cox proportional hazards model. Squares show HR estimates of gene expression. The sizes of square are determined by the weights in meta-analysis summaries. Segments show 95% CIs, and the red diamonds show the fixed-effects meta-analysis summaries of HRs over all the datasets. B. Heatmap of the 157 signature genes of ERα activity in the Dressman et.al dataset. Bins above the heatmap indicate the clinical information of corresponding patients in the heatmap. Samples are ordered by the value of the signiture of ERα activity. C. Kaplan-Meier overall survival curves. Patients are divided into two groups by the expression of ESR1. D. A proposed model for the collaboration between ERα and CtBP on inhibiting core genes of HRR pathway.
Discussion
EOC is the most lethal gynecologic malignancy [48], largely due to the developed resistance to neoadjuvant chemotherapies [49]. Oophorectomy, which is the general operation during the first line treatment of ovarian cancer, significantly reduces the estrogen synthesis after removal of ovary. However, due to the estrogen etiology of ovarian cancer [3], estrogen replacement is not comprehensively recommended for most patients. Chemotherapy performed afterward is mostly on a lack-of-estrogen background. Due to the data limitation, it is almost impossible for a strict evaluation to what extent might the lack of estrogen affect the response to on-going chemotherapy. Nevertheless, the observation that patients receiving HRT have a better outcome deserves a deeper clinical investigation. Previous studies have suggested that HRT may improve the outcome by the relief of symptoms from oophorectomy [15, 16]. Our results provide an alternative explanation by establishing a molecular connection between estrogen signaling and HRR. Notably, we also observed a significantly lower risk of disease recurrence for patients taking HRT, which is more likely due to the improved response to chemotherapy. In addition, we observed that the expression of ERα in pre-treatment primary tumors is associated with improved response to chemotherapy, further confirming that the beneficial effect of HRT could come from improved response to chemotherapy. Of note, the prognostic significance of ERα expression is highly underestimated due to the lack of estrogen in EOC patients. Nevertheless, it indicates a great potential of using HRT to improve the response to chemotherapy and the quality of life for EOC patients.
Previous knowledge indicated that DNA damage repair (DDR) pathways are not direct targets of ERα in breast cancer models [50]. To our surprise, our results actually identified the DDR pathway as one of the top targeted functions of ERα in ovarian cancer cells. Further characterization also validated the regulation of ERα on these DDR genes, in particular the central player RAD51. ERα has quite different binding profiles in EOC cells compared to breast cancer cell line MCF7, probably due to the non-classical versus classical regulatory mechanism. Usually, ERα recruitment to ERE requires GATA3 and FOXA1 as pioneer factors to create the accessible chromatin domain [4, 9], and ERα+ breast cancer is characterized by the high expression of GATA3 and FOXA1. However, in EOC, these two critical factors are actually absent, indicating that our understanding of ERα function, mainly acquired from breast cancer studies, does not fit to ovarian cancer.
AP-1 is a heterodimer formed by Fos and Jun proteins [51], and is required for estrogen-responsive cellular functions [52]. Notably, although AP-1 promotes cell proliferation, high expression of c-Fos was found to be associated with favorable outcomes in EOC patients received platinum-based chemotherapy [53], which is in consistent with what we have observed for ERα. These results are in contrast to the mainstream concept of the oncogenic function of AP-1 and ERα, but could be explained by their interplay with the response to chemotherapy.
We have previously shown that CtBP globally repress DNA repair genes in breast cancer cells [38]. Interestingly, AP-1 motif was enriched in the CtBP binding sites, suggesting that CtBP recruitment to its target genes relies on AP-1 in breast cancer too. In fact, many DDR genes have one or more AP-1 binding motifs in their promoters, including RAD51 [54]. In ovarian cancer cells, we speculate that ERα represses DNA repair by forming complex with AP-1 and CtBP. The genome-wide co-binding between ERα and CtBP, and the enrichment of AP-1 motif in the co-binding sites further supports this hypothesis. Therefore, our results suggest a model in which ERα recruits CtBP for inhibition of HRR genes through an AP-1-mediated nonclassical model in EOC (Figure 7D). Our results together with previous studies have revealed that the expression of ERα, CtBP and AP-1 are all associated with a survival benefit in EOC [20, 53], which is consistent with the previous finding that the regulation of DNA repair activity is strongly associated with outcomes and response to chemotherapy in EOC [55]. Although our results are only preliminary to fully delineate ERα function and the proposed model in ovarian cancer, they have revealed the potential of a combinational therapy using platinum drugs and hormone replacement for the treatment of ovarian cancer patients.
Supplementary Material
Supplementary methods and figures.
Supplementary tables.
Acknowledgements
This work is supported by the Science and Technology Development Fund (FDCT) of Macao SAR to LD (FDCT/0014/2018/A1, FDCT/0117/2018/A3), the Multi-Year Research Grant from the University of Macau to LD (MYRG2018-00158-FHS), and the National Natural Science Foundation of China (NSFC 81772980) to LD. This work is also supported by the Multi-Year Research Grant from the University of Macau to LW (MYRG2016-00251-FHS). Dapeng Hao was supported by National Natural Science Foundation of China (grant no. 31701153). Benjamin K Tsang is supported by a grant from the Canadian Institutes of Health Research (MOP-126144).
Author contributions
DH and LD conceived the project; DH, JL, JW, YM, ZZ, and CZ performed the research; CD, BT provided reagents, technique and writing assistance; DH, LW and LD wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sieh W, Kobel M, Longacre TA, Bowtell DD, deFazio A, Goodman MT. et al. Hormone-receptor expression and ovarian cancer survival: an Ovarian Tumor Tissue Analysis consortium study. The Lancet Oncology. 2013;14:853-62
2. Hogdall EV, Christensen L, Hogdall CK, Blaakaer J, Gayther S, Jacobs IJ. et al. Prognostic value of estrogen receptor and progesterone receptor tumor expression in Danish ovarian cancer patients: from the 'MALOVA' ovarian cancer study. Oncol Rep. 2007;18:1051-9
3. Collaborative Group On Epidemiological Studies Of Ovarian C, Beral V, Gaitskell K, Hermon C, Moser K, Reeves G. et al. Menopausal hormone use and ovarian cancer risk: individual participant meta-analysis of 52 epidemiological studies. Lancet. 2015;385:1835-42
4. Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43:27-33
5. McKenna NJ, Xu J, Nawaz Z, Tsai SY, Tsai MJ, O'Malley BW. Nuclear receptor coactivators: multiple enzymes, multiple complexes, multiple functions. J Steroid Biochem Mol Biol. 1999;69:3-12
6. Cavailles V, Dauvois S, L'Horset F, Lopez G, Hoare S, Kushner PJ. et al. Nuclear factor RIP140 modulates transcriptional activation by the estrogen receptor. EMBO J. 1995;14:3741-51
7. White JH, Fernandes I, Mader S, Yang XJ. Corepressor recruitment by agonist-bound nuclear receptors. Vitam Horm. 2004;68:123-43
8. Lopez-Garcia J, Periyasamy M, Thomas RS, Christian M, Leao M, Jat P. et al. ZNF366 is an estrogen receptor corepressor that acts through CtBP and histone deacetylases. Nucleic Acids Res. 2006;34:6126-36
9. Theodorou V, Stark R, Menon S, Carroll JS. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013;23:12-22
10. Gaub MP, Bellard M, Scheuer I, Chambon P, Sassone-Corsi P. Activation of the ovalbumin gene by the estrogen receptor involves the fos-jun complex. Cell. 1990;63:1267-76
11. Davis DG, Siddiqui MT, Oprea-Ilies G, Stevens K, Osunkoya AO, Cohen C. et al. GATA-3 and FOXA1 expression is useful to differentiate breast carcinoma from other carcinomas. Human pathology. 2016;47:26-31
12. Early Breast Cancer Trialists' Collaborative G. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet. 2015;386:1341-52
13. Tryfonidis K, Zardavas D, Katzenellenbogen BS, Piccart M. Endocrine treatment in breast cancer: Cure, resistance and beyond. Cancer Treat Rev. 2016;50:68-81
14. Simpkins F, Garcia-Soto A, Slingerland J. New insights on the role of hormonal therapy in ovarian cancer. Steroids. 2013;78:530-7
15. Eeles RA, Morden JP, Gore M, Mansi J, Glees J, Wenczl M. et al. Adjuvant Hormone Therapy May Improve Survival in Epithelial Ovarian Cancer: Results of the AHT Randomized Trial. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2015;33:4138-44
16. Eeles RA, Tan S, Wiltshaw E, Fryatt I, A'Hern RP, Shepherd JH. et al. Hormone replacement therapy and survival after surgery for ovarian cancer. Bmj. 1991;302:259-62
17. Guidozzi F, Daponte A. Estrogen replacement therapy for ovarian carcinoma survivors: A randomized controlled trial. Cancer. 1999;86:1013-8
18. Ursic-Vrscaj M, Bebar S, Zakelj MP. Hormone replacement therapy after invasive ovarian serous cystadenocarcinoma treatment: the effect on survival. Menopause. 2001;8:70-5
19. Hein A, Thiel FC, Bayer CM, Fasching PA, Haberle L, Lux MP. et al. Hormone replacement therapy and prognosis in ovarian cancer patients. European journal of cancer prevention: the official journal of the European Cancer Prevention Organisation. 2013;22:52-8
20. Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S. et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489-94
21. Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609-15
22. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179-204
23. Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475-85
24. Lu J, Wu D, Li C, Zhou M, Hao D. Correlation between gene expression and mutator phenotype predicts homologous recombination deficiency and outcome in ovarian cancer. Journal of molecular medicine. 2014;92:1159-68
25. Early Breast Cancer Trialists' Collaborative G. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687-717
26. Sledge GW Jr, Hu P, Falkson G, Tormey D, Abeloff M. Comparison of chemotherapy with chemohormonal therapy as first-line therapy for metastatic, hormone-sensitive breast cancer: An Eastern Cooperative Oncology Group study. J Clin Oncol. 2000;18:262-6
27. Walsh CS. Two decades beyond BRCA1/2: Homologous recombination, hereditary cancer risk and a target for ovarian cancer therapy. Gynecologic oncology. 2015;137:343-50
28. Stopper H, Schmitt E, Gregor C, Mueller SO, Fischer WH. Increased cell proliferation is associated with genomic instability: elevated micronuclei frequencies in estradiol-treated human ovarian cancer cells. Mutagenesis. 2003;18:243-7
29. Xiong J, Yu D, Wei N, Fu H, Cai T, Huang Y. et al. An estrogen receptor alpha suppressor, microRNA-22, is downregulated in estrogen receptor alpha-positive human breast cancer cell lines and clinical samples. FEBS J. 2010;277:1684-94
30. Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633-8
31. Zhang H, Zhang X, Ji S, Hao C, Mu Y, Sun J. et al. Sohlh2 inhibits ovarian cancer cell proliferation by upregulation of p21 and downregulation of cyclin D1. Carcinogenesis. 2014;35:1863-71
32. Ingberg E, Theodorsson A, Theodorsson E, Strom JO. Methods for long-term 17beta-estradiol administration to mice. Gen Comp Endocrinol. 2012;175:188-93
33. Robertson JF. Fulvestrant (Faslodex) - how to make a good drug better. Oncologist. 2007;12:774-84
34. Yu HM, Liu B, Chiu SY, Costantini F, Hsu W. Development of a unique system for spatiotemporal and lineage-specific gene expression in mice. Proc Natl Acad Sci U S A. 2005;102:8615-20
35. Lindgren PR, Cajander S, Backstrom T, Gustafsson JA, Makela S, Olofsson JI. Estrogen and progesterone receptors in ovarian epithelial tumors. Mol Cell Endocrinol. 2004;221:97-104
36. Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun. 2013;4:2126
37. Manavathi B, Samanthapudi VS, Gajulapalli VN. Estrogen receptor coregulators and pioneer factors: the orchestrators of mammary gland cell fate and development. Front Cell Dev Biol. 2014;2:34
38. Di LJ, Byun JS, Wong MM, Wakano C, Taylor T, Bilke S. et al. Genome-wide profiles of CtBP link metabolism with genome stability and epithelial reprogramming in breast cancer. Nat Commun. 2013;4:1449
39. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic acids research. 2015;43:D447-52
40. Wang S, Sun H, Ma J, Zang C, Wang C, Wang J. et al. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nature protocols. 2013;8:2502-15
41. Chetrit A, Hirsh-Yechezkel G, Ben-David Y, Lubin F, Friedman E, Sadetzki S. Effect of BRCA1/2 mutations on long-term survival of patients with invasive ovarian cancer: the national Israeli study of ovarian cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:20-5
42. Wernli KJ, Newcomb PA, Hampton JM, Trentham-Dietz A, Egan KM. Hormone therapy and ovarian cancer: incidence and survival. Cancer causes & control: CCC. 2008;19:605-13
43. Besevic J, Gunter MJ, Fortner RT, Tsilidis KK, Weiderpass E, Onland-Moret NC. et al. Reproductive factors and epithelial ovarian cancer survival in the EPIC cohort study. British journal of cancer. 2015;113:1622-31
44. Wen Y, Huang H, Huang H, Wu M, Shen K, Pan L. The safety of postoperative hormone replacement therapy in epithelial ovarian cancer patients in China. Climacteric: the journal of the International Menopause Society. 2013;16:673-81
45. Li L, Pan Z, Gao K, Zhang W, Luo Y, Yao Z. et al. Impact of post-operative hormone replacement therapy on life quality and prognosis in patients with ovarian malignancy. Oncology letters. 2012;3:244-9
46. Mascarenhas C, Lambe M, Bellocco R, Bergfeldt K, Riman T, Persson I. et al. Use of hormone replacement therapy before and after ovarian cancer diagnosis and ovarian cancer survival. International journal of cancer Journal international du cancer. 2006;119:2907-15
47. van Kruchten M, van der Marel P, de Munck L, Hollema H, Arts H, Timmer-Bosscha H. et al. Hormone receptors as a marker of poor survival in epithelial ovarian cancer. Gynecologic oncology. 2015;138:634-9
48. Kroeger PT Jr, Drapkin R. Pathogenesis and heterogeneity of ovarian cancer. Curr Opin Obstet Gynecol. 2017;29:26-34
49. Bitler BG, Watson ZL, Wheeler LJ, Behbakht K. PARP inhibitors: Clinical utility and possibilities of overcoming resistance. Gynecol Oncol. 2017
50. Caldon CE. Estrogen signaling and the DNA damage response in hormone dependent breast cancers. Front Oncol. 2014;4:106
51. Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859-68
52. Dahlman-Wright K, Qiao Y, Jonsson P, Gustafsson JA, Williams C, Zhao C. Interplay between AP-1 and estrogen receptor alpha in regulating gene expression and proliferation networks in breast cancer cells. Carcinogenesis. 2012;33:1684-91
53. Mahner S, Baasch C, Schwarz J, Hein S, Wolber L, Janicke F. et al. C-Fos expression is a molecular predictor of progression and survival in epithelial ovarian carcinoma. British journal of cancer. 2008;99:1269-75
54. Christmann M, Kaina B. Transcriptional regulation of human DNA repair genes following genotoxic stress: trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res. 2013;41:8403-20
55. Kang J, D'Andrea AD, Kozono D. A DNA repair pathway-focused score for prediction of outcomes in ovarian cancer treated with platinum-based chemotherapy. Journal of the National Cancer Institute. 2012;104:670-81
Author contact
![]() Corresponding authors: Di li-jun at lijundiedu.mo and liwangedu.mo. Room 4009, Faculty of Health Sciences (E12), University of Macau, Avenida da Universidade, Taipa, Macau, China. Tel. 853-88224497; Fax. 853-88222314
Corresponding authors: Di li-jun at lijundiedu.mo and liwangedu.mo. Room 4009, Faculty of Health Sciences (E12), University of Macau, Avenida da Universidade, Taipa, Macau, China. Tel. 853-88224497; Fax. 853-88222314