Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results and Discussion
Conclusions
Experimental Section
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(17):5035-5048. doi:10.7150/thno.35418 This issue Cite
Research Paper
Zwitterionic Polymer-Gated Au@TiO2 Core-Shell Nanoparticles for Imaging-Guided Combined Cancer Therapy
Tao Zheng1, Wentao Wang2, Fan Wu2, Ming Zhang1,2 ![]() , Jian Shen2
, Jian Shen2 ![]() , Yi Sun1
, Yi Sun1 ![]()
1. Department of Health Technology, Technical University of Denmark, Kongens Lyngby, 2800, Denmark;
2. Jiangsu Collaborative Innovation Center for Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing 210023, P. R. China.
Received 2019-4-2; Accepted 2019-6-14; Published 2019-7-9
Abstract

With advances in nanoparticle (NP) synthesis and engineering, nanoscale agents with both therapeutic and diagnostic functions have been increasingly exploited for cancer management. Herein, we synthesized a new type of zwitterionic polymer-gated Au@TiO2 core-shell nanoparticles, which showed that they could selectively target and efficiently eliminate cancer cells via photothermal therapy (PTT), photodynamic therapy (PDT), pH/NIR-induced drug release, and cationic therapy.
Methods: In the present study, the multifunctional therapeutic agent [Mn@P(CitAPDMAEMA)@Au@TiO2@DOX] was prepared to treat cancer with imaging-guided combination method. Firstly, Au@TiO2 core-shell nanoparticles (NPs) were synthesized. Taking advantage of broad and strong photoabsorption and reactive oxygen species (ROS) generation, Au@TiO2 core-shell NPs facilitated the single light-induced PTT and PDT. Next, a chemotherapy drug doxorubicin (DOX) was loaded into Au@TiO2 core-shell NPs. Then, a biocompatible zwitterionic polymer P(CitAPDMAEMA) was grafted to improve the hemocompatibility of NPs and prolong the circulation time. The polymer also served as a capping or switching material for pH-triggered drug release. In addition, the cationic nature of P(CitAPDMAEMA) eased the binding to human cervical cancer (HeLa) cells and effectively inhibited their growth in acidic environments (termed cationic therapy). Moreover, with Mn2+ ions immanently chelated, Mn@P(CitAPDMAEMA)@Au@TiO2@DOX NPs were able to provide enhanced contrast under T1- or T2-weighted magnetic resonance imaging (MRI).
Results: The in vitro and in vivo anticancer experiments demonstrated the tumor was effectively inhibited with minimal side effects by the multifunctional NPs.
Conclusions: As far as we know, this is the first presentation of four therapeutic methods into one nanomaterial, which will open up a new dimension for the design of combined treatment.
Keywords: Au@TiO2 core-shell NPs, photothermal, photodynamic, cationic therapy, chemotherapy, magnetic resonance imaging
Introduction
Traditional cancer treatment such as chemotherapy can lead to serious side effects, as the anticancer drugs kill both cancerous and normal cells due to the lack of cell specificity [1-5]. In the last few years, nanomaterial-based therapeutic methods such as photothermal therapy (PTT), photodynamic therapy (PDT) and controlled drug delivery system have attracted a lot of attention. PTT ablates malignant tissues by utilizing external light-induced hyperthermia. It causes negligible damage to normal tissues, and is convenient, non-invasive, remote-controllable and safe [6-8]. Various photothermal agents such as Au nanoparticles (AuNPs) [9], copper NPs [10], carbon derivatives [11], transition metal sulfides [12], black titania [13], and black phosphorus (BPs) [14] have been intensively explored. Among them, AuNPs are preferred due to their high absorption ability in the near-infrared (NIR) region and excellent biocompatibility [9]. However, for AuNPs to be good PTT agents, their localized surface plasmon resonance (LSPR) absorption must be tuned to the 550-900 nm region to allow for NIR laser treatment. This could be done by carefully controlling the size and shape of AuNPs, but the synthesis process was complicated. Alternatively, the LSPR coupling between AuNPs and semiconductor has been proposed as a novel approach [15]. Interestingly, we found that the titania (TiO2) shell modified with AuNPs is one of the most effective methods to shift their LSPR absorption to a longer wavelength. The condensed free-space optical field within the sub-wavelength regions adjacent to the surface of the AuNPs enables significant electric field enhancement under resonant excitation, which increases the efficiency of photothermal conversion.
On the other hand, PDT relies on photosensitizers (PSs) to release the reactive oxygen species (ROS) and cytotoxic free radicals when they absorb light at appropriate wavelengths [16-18]. Organic PSs are the most widely used PS types. They mainly release singlet oxygen (1O2) which is produced by spin inversion of triplet oxygen to singlet oxygen (type II PDT). Some special organic PSs can also form hydrogen peroxide (H2O2) and hydroxyl radicals (·OH) by ionization events (type I PDT) [19,20]. Despite these advantages, the organic PSs depend on oxygen for cytotoxic ROS production, which limits PDT effectiveness in hypoxic conditions, such as many solid tumors [21]. Moreover, in-situ oxygen in PDT is rapidly depleted, creating transient hypoxia microenvironment, which further reduces the therapeutic effects [22]. Consequently, it is necessary to design simple and biocompatible PSs that can evolve oxygen continuously without exogenous activation [23]. The semiconductor material TiO2 has been considered as a new type of PS, as they are able to catalyze H2O2 into O2. It is reported that H2O2 is abundant in cancer microenvironment and is an appropriate source for the production of O2 within tumors. However, the application of TiO2 as PSs has been hampered by the requirement of UV light excitation and the low quantum efficiency [24]. In this study, we innovatively combined TiO2 with AuNPs to create a novel Au@TiO2 core-shell nanohybrid. Coupling TiO2 with AuNPs results in enhanced electron-hole separation under red light irradiation, which enables the utilization of PTT and PDT for cancer treatment at NIR region.
The in vivo applications of NPs are often hindered by the challenges such as short circulation time, and biological protein adsorption. One effective way to address these problems is to coat the NPs with appropriate polymers. Recently, zwitterionic polymers have attracted a lot of attention as stealth materials in drug delivery systems. Conjugates of zwitterionic polymers with therapeutic agents (i.e. PSs, drugs or proteins) have been demonstrated [25, 26]. Due to their limited interactions with biological proteins [27, 28], they showed excellent biocompatibility and could escape from the reticuloendothelial system, achieving long circulation time [30, 31]. We synthesized a pH-sensitive polymer with quaternary ammonium groups according to a previous method [32], which was zwitterionic at physiological pH and was converted to a cationic species in acidic environments. Herein, we loaded doxorubicin (DOX) into the Au@TiO2 core-shell, then grafted the zwitterionic polymer onto the NP for combined cancer therapy. As the cancer cells' metabolism and the immune response contribute to an acidity tumor microenvironment [33, 34], the zwitterionic polymers convert to a cationic state when in proximity of the tumor tissues. The cationic nature of the polymer, not only enhances cellular uptake due to the strengthened NP-cellular membrane interactions, but also inhibits the cell growth. In addition, the pH-sensitive charge-conversion results in the falling of the polymer chains, making it possible for “on-demand” drug release at the acidic tumor sites.
In the present study, a novel, multifunctional DOX-loaded, manganese ion (Mn2+)-chelated complex, and zwitterionic polymer [P(CitAPDMAEMA)]-grafted Au@TiO2 ore-shell NPs [Mn@P(CitAPDMAEMA)@Au@TiO2@DOX] was reported, which could selectively target and eliminate cancer cells via PTT, PDT, pH/NIR-induced drug release and cationic therapy (Scheme 1). We have synthesized a new Au@TiO2 core-shell through in-situ growth and the Ostwald ripening process of TiO2 shells on the preformed AuNPs (Scheme 1A). The DOX as a chemotherapeutic agent was loaded to the Au@TiO2 core-shell NPs, which was then coated by the zwitterionic polymer P(CitAPDMAEMA) (Scheme 1B). We also chelated Mn2+ to offer an obvious contrast in T1-weighted MRI. The synergetic cancer therapy could be achieved under 635 nm laser irradiation (Scheme 1C). Au@TiO2 core-shell NPs had both photothermal and photodynamic effects with high ROS generating efficiency and photothermal conversion efficiency. The Au@TiO2 core-shell NPs exhibited improved photodynamic performance compared to those of commercial TiO2, Au/TiO2 hybrid, or Au@TiO2 core-shell NPs. The biocompatible P(CitAPDMAEMA) improved the hemocompatibility of the NPs. Meanwhile, the cationic nature of P(CitAPDMAEMA) facilitated the binding to human cervical cancer (HeLa) cells and inhibited their growth effectively in an acidic environment (Scheme S1). P(CitAPDMAEMA) also served as capping or switching materials for Au@TiO2@DOX NPs. The drug was only released when the NPs reached the low pH environment and red light irradiation. In vitro and in vivo studies demonstrated that the Mn@P(CitAPDMAEMA)@Au@TiO2@DOX NPs could be used as superior theranostic agents for multimodality image-guided synergistic cancer therapy.
Schematic illustration of the synthesis process of the (A) Au@TiO2 core-shell NPs and (B) P(CitAPDMAEMA)@Au@TiO2@DOX. (C) Schematic representation of P(CitAPDMAEMA)@Au@TiO2@DOX-based delivery system enhancing the cationic therapy, chemotherapy, PDT, and PTT under red-light irradiation.
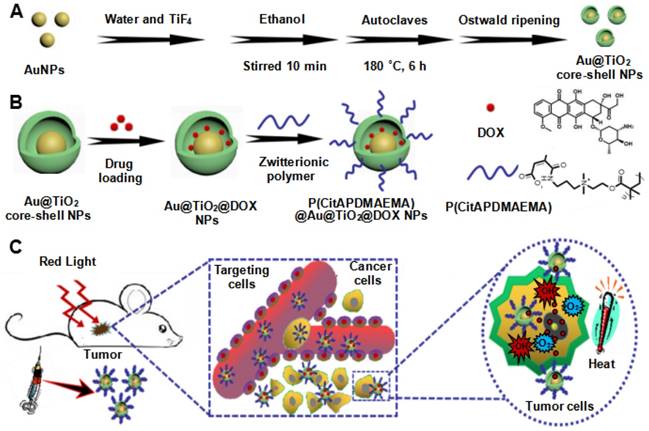
Results and Discussion
Preparation and Characterization of Au@TiO2 Core-shell NPs
The Au@TiO2 core-shell NPs were prepared according to previous methods with some modification [35]. The overall procedure is shown in Scheme S2. The micro-morphology of the AuNPs (Figure S1A) and Au@TiO2 core-shell NPs (Figure S1B) were studied by transmission electron microscopy (TEM). Figure 1A shows the high-resolution TEM (HR-TEM) images of Au@TiO2 core-shell NPs. Due to the differential electron densities of Au and TiO2, the darker spots were the Au core while lighter areas were TiO2 shells. It is worth noting that AuNPs were surrounded by a TiO2 shell. Furthermore, the hollow space between the core and the shell in each nanohybrid could be observed clearly. The Au core could move within the TiO2 shell, producing an eccentric shape of the Au@TiO2 core-shell NPs. The Au@TiO2 core-shell NPs exhibited a uniform diameter of 108 nm (Figure S2A), with an average Au core size of 30 ± 1.7 nm and an average TiO2 shell thickness of 32 ± 3.5 nm. The dynamic light scattering (DLS) results indicated that the average hydrodynamic particle size of Au@TiO2 core-shell NPs was 135 ± 3.8 nm (Figure S2B), revealing that they were stable in deionized water without aggregation. HR-TEM images revealed that the TiO2 shell was polycrystalline with many small and randomly oriented crystallites. The lattice fringes with a d-spacing of 0.23 nm could be indexed to the (111) plane of Au (Figure 1B), whereas the d-spacing of 0.35 nm agreed with the (101) plane of TiO2 [35]. Moreover, Energy-dispersive X-ray (EDS) elemental mapping was also applied to characterize the functionalization of Au@TiO2 core-shell NPs. Figure 1C shows the uniform elemental distribution (Ti, Au, and O). The Ti element mapping was derived from the TiO2 layer and the Au element mapping, suggesting the existence of AuNPs.
(A) TEM and (B) HRTEM images of Au@TiO2 core-shell NPs. (C) STEM image and corresponding two-dimensional elemental maps of Au@TiO2 core-shell NPs. (D) XPS survey scan for AuNPs and Au@TiO2 core-shell NPs. (E) XRD spectra of AuNPs and Au@TiO2 core-shell NPs. (F) N2 adsorption-desorption isotherms and pore size distribution (inset) of Au@TiO2 core-shell NPs.
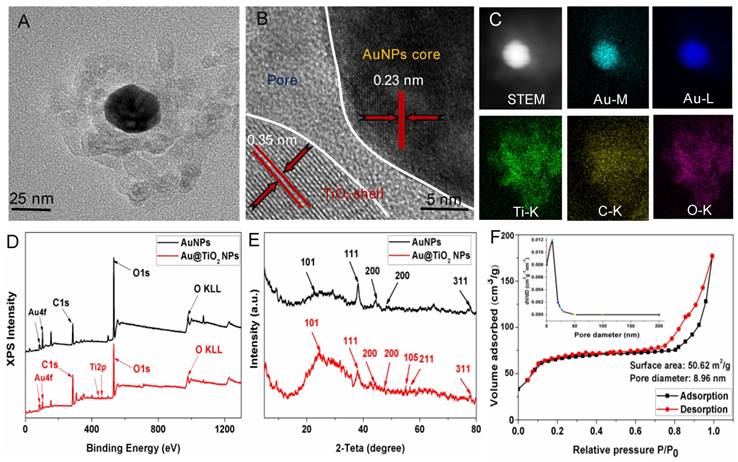
The components of the Au@TiO2 core-shell structure were studied by X-ray photoelectron spectroscopy (XPS). Figure 1D shows that the Au 4f doublet peaks appeared at 83.3 and 86.9 eV [35], where the splitting of 4.5 eV further indicated the metallic nature of Au (Figure S3A). The Ti 2p peaks for TiO2 were located at about 458.4 and 464.2 eV with the splitting of the 2p doublet being at 5.8 eV (Figure 1D and Figure S3B), confirming a Ti4+ state in octahedral coordination with oxygen [36]. The XPS spectra peak of O 1 s at 532.1 eV was attributed to the Ti-O bonds in the TiO2 lattice (Figure S3C). In addition, X-ray diffraction (XRD) was used to study two different crystal nanostructures. Figure 1E shows that all the samples displayed two series of peaks, which belongs to the anatase-TiO2 structure and the face-centered-cubic Au. The peaks at 2θ of 25.3, 47.9, 53.8, and 54.7 could be ascribed to the (101), (200), (105), and (211) planes of anatase-TiO2 [37], respectively, while the left peaks were corresponding to the (111), (200), (220), and (311) faces of face-centered-cubic Au. Thus, the presence of Au and TiO2 in the Au@TiO2 core-shell NPs were confirmed.
Nitrogen physisorption measurements were also used to study the properties of the Au@TiO2 core-shell NPs structure. Figure 1F displays the N2 adsorption-desorption isotherms and the respective pore size distribution curves of samples. The Au@TiO2 core-shell NPs displayed a type IV isotherm with a well-defined hysteresis loop, which indicated well-developed mesoporous characteristics. The BET surface areas of the Au@TiO2 core-shell NPs were estimated to be 50.62 m2/g, and its average pore diameters were about 8.96 nm, indicating that the Au@TiO2 core-shell NPs could provide more active sites and easier passage for the drug into the shell.
Drug Loading
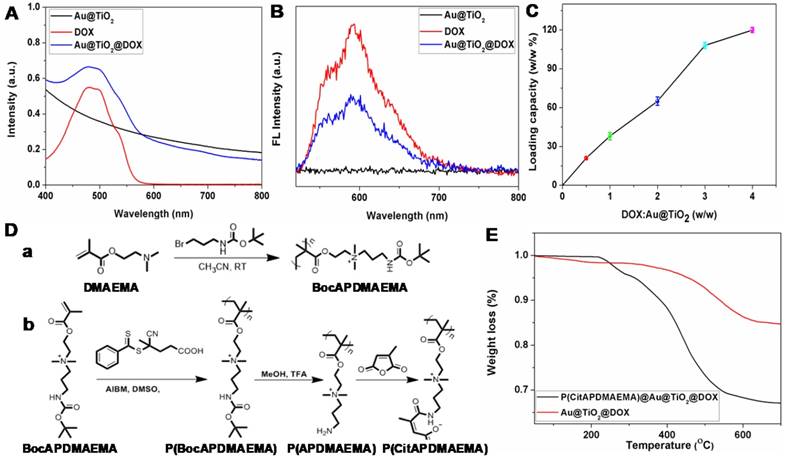
In this study, we selected DOX as the model drug to test the feasibility of Au@TiO2 core-shell NPs as a drug delivery carrier. The conjugation was confirmed by zeta potential, UV-vis spectra and fluorescence spectra [38]. The zeta potential of Au@TiO2 core-shell NPs changed from -18.9 to -1.2 mV (Figure S4A-B) after DOX loading, indicating that the negatively charged Au@TiO2 core-shell NPs bonded with positively charged DOX (Figure S4C). A notable difference of Au@TiO2@DOX core-shell NPs was found in the spectra where the UV-vis absorption peak located at 490 nm (Figure 2A). It was caused by the absorption of DOX. Moreover, the fluorescence of DOX was quenched after DOX loading on the Au@TiO2 core-shell NPs (Figure 2B), verifying an interaction between the two components. By increasing the weight ratio (feeding DOX/Au@TiO2 core-shell NPs), the drug loaded on the NPs also increased (Figure 2C). The maximum loading capacity reached ~120%. There are two factors contributing to the high drug-loading capacity of the Au@TiO2 core-shell NPs. One is that, the Au@TiO2 core-shell NPs could bind to DOX molecules via different types of interactions, such as electrostatic attraction and hydrogen bonding. The other reason is that the unique structure of the Au@TiO2 core-shell NPs with a large central hollow cavity provided enough space for loading the drug. Thus, the distributed DOX molecules could be retained and kept in the Au@TiO2 carriers to form DOX-loaded Au@TiO2 NPs.
(A) UV-vis and (B) fluorescence spectra of Au@TiO2 core-shell NPs, DOX, and Au@TiO2@DOX NPs. (C) Quantification of DOX loading at different DOX: Au@TiO2 core-shell NPs ratios. (n = 3, mean ± s.d.) (D) Synthetic route for the preparation of (a) BocAPDMAEMA and (b) P(CitAPDMAEMA). (E) TGA curves of Au@TiO2@DOX NPs and P(CitAPDMAEMA)@Au@TiO2@DOX NPs.
Synthesis of P(CitAPDMAEMA@Au@TiO2@DOX NPs
As mentioned previously, it is desirable to develop high-performance biocompatible anticancer agents for cancer treatment, it is necessary to cap or switch nanocarriers with porous structures [39]. In light of that, the smart pH-sensitive polymer P(CitAPDMAEMA) was synthesized [29], as shown in Figure 2D. Characterization of these materials was shown in Figure S5. P(CitAPDMAEMA) was coated to the surface of Au@TiO2@DOX NPs, through a physicochemical interactions, such as π-π stacking, hydrophobic and electrostatic interactions. The Fourier transform infrared (FT-IR) spectra (Figure S6) of P(CitAPDMAEMA)@Au@TiO2@DOX NPs confirmed the efficient surface modification of P(CitAPDMAEMA) on the Au@TiO2@DOX NPs [39]. The resultant P(CitAPDMAEMA)@Au@TiO2@DOX NPs showed no remarkable changes in morphology (Figure S7A) as compared to Au@TiO2 NPs. Compared to Au@TiO2 NPs, the larger diameter of P(CitAPDMAEMA)@Au@TiO2@DOX NPs was likely due to the presence of P(CitAPDMAEMA) loaded on the surface of the Au@TiO2@DOX NPs (Figure S7B). As illustrated in Figure S8A, the P(CitAPDMAEMA)@Au@TiO2@DOX NPs exhibited high stability in phosphate buffered solution (PBS), fetal bovine serum (FBS), and Dulbecco's modified Eagle medium (DMEM).
The P(CitAPDMAEMA) content in the P(CitAPDMAEMA)@Au@TiO2@DOX NPs was evaluated by thermal gravimetric analysis (TGA) (Figure 2E). The different weight loss between the P(CitAPDMAEMA)@Au@TiO2@DOX NPs and the Au@TiO2@DOX NPs at 700 °C indicated that the P(CitAPDMAEMA) grafting amount was about 18.4%. Full nitrogen adsorption isotherms (Figure S8B) were observed in order to obtain information about the specific surface areas of the P(CitAPDMAEMA)@Au@TiO2@DOX NPs. The BET surface area and the total pore volume of the P(CitAPDMAEMA)@Au@TiO2@DOX NPs were found to be 9.34 m2/g and 0.112 cm3/g, respectively, which were smaller than those of the Au@TiO2@DOX NPs (41.17 m2/g and 0.674 cm3/g, respectively). These results indicated that P(CitAPDMAEMA)@Au@TiO2@DOX NPs not only showed a considerable absence of porosity but also had important pore blocking because of the valve-like function of P(CitAPDMAEMA).
Photothermal Properties of P(CitAPDMAEMA)@Au@TiO2@DOX NPs
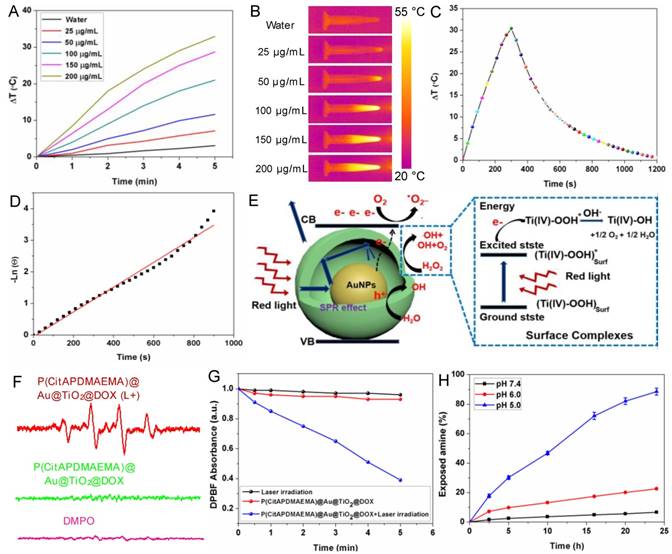
Another important property of P(CitAPDMAEMA)@Au@TiO2@DOX NPs was their adjustable photothermal effects. Τhe temperature changes in P(CitAPDMAEMA)@Au@TiO2@DOX NPs aqueous solutions with different concentrations under 635 nm laser irradiation at 2.0 W/cm2 were initially investigated (Figure 3A-B and Figure S9). The initial temperature of water and P(CitAPDMAEMA)@Au@TiO2@DOX solution is 25 °C. The solution temperature was able to be increased precisely by 32.5 °C (200 μg/mL). On the contrary, the temperature of pure water only increased by 2.8 °C. Therefore, cancer cells incubated with P(CitAPDMAEMA)@Au@TiO2@DOX (200 μg/mL) could be easily heated to temperatures higher than 50 °C within 5 min under laser irradiation at 2.0 W/cm2, thereby efficiently eliminating the cancer cells [40, 41]. In order to further study the photothermal conversion efficiency of P(CitAPDMAEMA)@Au@TiO2@DOX, the temperature changes of the solution (200 μg/mL, 1.0 mL) under continuous 635 nm laser irradiation (2.0 W/cm2) as a function of time were recorded until the solution reached a stable temperature (Figure 3C). According to the results, the photothermal conversion efficiency could reach 20.4% (Figure 3D).
(A) Temperature curves of P(CitAPDMAEMA)@Au@TiO2@DOX NPs at different concentrations. (B) IR images of water and P(CitAPDMAEMA)@Au@TiO2@DOX NPs solutions under 635 nm laser irradiation (2.0 W/cm2, 5 min). (C) Photothermal effect of P(CitAPDMAEMA)@Au@TiO2@DOX NPs solution (200 µg/mL) under 635 nm laser irradiation (2.0 W/cm2, 5 min). (D) Linear relationship between time and Ln (θ) obtained from the cooling time of (C). (E) Mechanism illustration for the enhanced photodynamic activity of Au@TiO2 core-shell NPs. The hollow structure could increase the optical path through multiple reflections and scattering, which enhanced light capturing inside the core-shell NPs and boosts light utilization. (F) EPR spectra (DMPO/·OH) of P(CitAPDMAEMA)@Au@TiO2@DOX NPs with or without laser irradiation. (G) Time-based absorption intensity reduction of DPBF. (H) Degradation of citraconic amide of P(CitAPDMAEMA)@Au@TiO2@DOX NPs under different pH. (n = 6, mean ± s.d.).
Photodynamic Properties of P(CitAPDMAEMA)@Au@TiO2@DOX NPs
A proposed principle for the improved photodynamic performance of Au@TiO2 core-shell NPs was described in Figure 3E. The LSPR effect of AuNPs could extend the optical absorption into the red region [42]. Therefore, upon red-light excitation, photo-electrons could be produced and transferred from the AuNPs to the conduction band of TiO2, leaving holes in the AuNPs. The reactive electrons combined with oxygen to form superoxide radicals (·O2-) and the holes could react with H2O to release hydroxyl radicals (·OH), which played important roles in PDT. Meanwhile, H2O2 could be easily adsorbed on the TiO2 surface and gave rise to surface complexes, which extended the photoresponse of TiO2 to the red region. The production of ·OH radicals showed the appearance of an electron-transfer process between the surface complexes and the TiO2 conduction band under red-light irradiation [35, 43]. Consequently, the proposed reaction mechanism of H2O2 and TiO2 was as follows (Figure 3E): the H2O2 molecule was adsorbed onto the TiO2 surface and formed surface titanium-(IV) hydrogen peroxide complexes (TiIV-OOH), which extended the photoresponse to the visible light region and could be activated by visible light. The active surface complex (TiIV-OOH)* injected an electron to the conduction band of the Au@TiO2 core-shell NPs and generated e- and TiIV-·OOH, which might further give rise to TiIV-OH and 1/2O2 [44]. Therefore, the elevated concentration of O2 was helpful to yield ·O2-, which further enhanced the efficacy of PDT. Moreover, the conduction band electron further reacted with the adsorbed H2O2 and released the ·OH radical on the TiO2 surface. These highly reactive groups could be helpful to generate ROS on the surface of TiO2 and Au. It should be noted that in the core-shell NPs, most of the AuNPs surface was exposed to the medium. Therefore, both TiO2 and AuNPs could provide plenty of active sites to form ROS.
In addition, to explore the performance of P(CitAPDMAEMA)@Au@TiO2@DOX NPs in PDT, the ROS production capability of P(CitAPDMAEMA)@Au@TiO2@DOX NPs were investigated. Electron spin resonance (ESR) spectroscopy was applied to detect ·OH and ·O2_ generation by using 5,5'-dimethylpyrroline-1-oxide (DMPO) as a spin trap agent. According to the ESR spectrum, P(CitAPDMAEMA)@Au@TiO2@DOX NPs proved the characteristics of DMPO/·OH (Figure 3F) and DMPO/·OOH adduct (Figure S10A), which suggested the release of ·OH and ·O2- and agreed with the literature [44]. As a comparison, no ·OH and ·O2- generation was detected in only DMPO or P(CitAPDMAEMA)@Au@TiO2@DOX NPs without laser irradiation. Interestingly, the intensity of the signal increased sharply upon 635 nm laser irradiation, confirming that P(CitAPDMAEMA)@Au@TiO2@DOX NPs could efficiently support the conversion of laser energy to chemical energy and release ROS for PDT. In order to quantify ROS generation, a photochemical method using 1,3-diphenylisobenzofuran (DPBF) as ROS probe in acetonitrile was employed [45]. In the presence of P(CitAPDMAEMA)@Au@TiO2@DOX NPs, an obvious reduction in the absorption concentration of DPBF as a function of the irradiation time under 635 nm laser was observed. While in the control group, a slight decrease was shown (Figure 3G) when treated only with 635 nm laser, indicating the ability of P(CitAPDMAEMA)@Au@TiO2@DOX NPs to generate ROS under red-light irradiation. Furthermore, the ROS probe 2,7-dichlorodihydrofluorescein diacetate (H2DCFHDA) could generate green fluorescence after being oxidized by ROS, which was used to further prove intracellular ROS production under 635 nm laser irradiation. As shown in Figure S10B, a very weak fluorescence signal was presented for the resistant HeLa incubated with P(CitAPDMAEMA)@Au@TiO2@DOX NPs without irradiation. In contrast, the cells after irradiated with 635 nm laser exhibited a strong green fluorescence, demonstrating the intracellular production of ROS. As a result, P(CitAPDMAEMA)@Au@TiO2@DOX NPs could work as a new multifunctional theranostic nanoplatform for PTT and PDT when irradiated by a single 635 nm laser.
Hydrolysis of Citraconic Amide of P(CitAPDMAEMA)@Au@TiO2@DOX NPs
Amides are acid labile when they have β-carboxylic acid groups, and primary amines can be exposed during their degradation. P(CitAPDMAEMA)@Au@TiO2 NPs was thought to experience conversion from zwitterionic to cationic P(CitAPDMAEMA)@Au@TiO2@DOX NPs due to citraconic amide degradation in acidic conditions. The process gave P(CitAPDMAEMA)@Au@TiO2@DOX NPs effective cancer cell targeting ability in acidic conditions. An amine-reactive fluorescence dye, fluorescamine, which reacts with the primary amine to generate fluorescent products [29]. As such, with the fluorescamine method, the pH-dependent decomposition rate of the citraconic amide of P(CitAPDMAEMA)@Au@TiO2@DOX NPs could be detected by comparing the fluorescence before and after incubation. According to the results demonstrated in Figure 3H, about 97.2% of polymers retained the zwitterionic property in the physiological environment (pH 7.4), but they exposed the primary amine and quaternary ammonium under the acidic conditions, due to the removal of the -COO- group in citraconic acid. The surface zeta potential of P(CitAPDMAEMA)@Au@TiO2@DOX NPs also changed after P(CitAPDMAEMA) hydrolysis (Figure S11).
Hemolytic Properties of P(CitAPDMAEMA)@Au@TiO2@DOX NPs
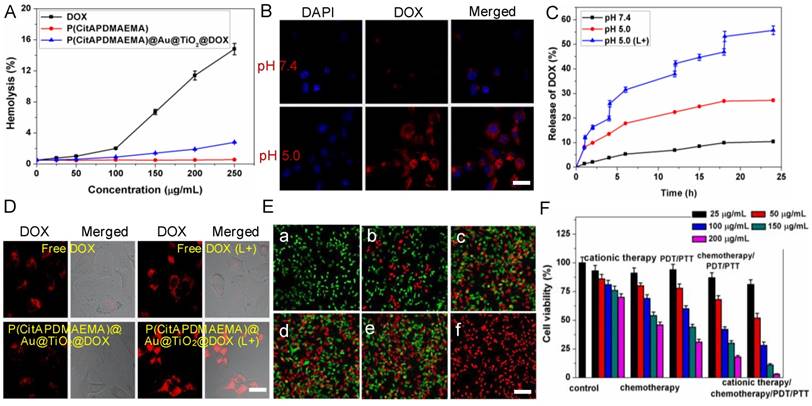
For potential clinical applications, anticancer agents should have no detrimental effects on the human body. To determine the cytotoxicity of anticancer agents that are particularly relevant to the intravenous application, hemolysis is a standard method [46]. Fresh whole rabbit blood was applied to measure the hemotoxicity of P(CitAPDMAEMA), DOX, and P(CitAPDMAEMA)@Au@TiO2@DOX NPs (Figure 4A). Among the tested concentrations, few red blood cells (RBCs) were lysed, indicating a very low hemolytic activity of P(CitAPDMAEMA). Meanwhile, no detectable hemolytic activity of P(CitAPDMAEMA)@Au@TiO2 NPs on RBCs during the entire experimental DOX concentration range was found. It should be noted that DOX concentration could decide the hemolytic activity of soluble DOX. Hemolysis was not measured when the tested concentration of DOX below 100 μg/mL, while at 250 μg/mL concentration, 14.3% hemolysis was detected. These results indicated that the P(CitAPDMAEMA)@Au@TiO2@DOX NPs were safe for blood-contact usage and held potential for intravenous administration.
(A) Hemolytic rates of the DOX, P(CitAPDMAEMA) and P(CitAPDMAEMA)@Au@TiO2@DOX NPs. (n = 3, mean ± s.d.) (B) pH-dependent interaction between P(CitAPDMAEMA)@Au@TiO2@DOX NPs and HeLa cells. Scale bar: 30 μm (C) Drug release profile of P(CitAPDMAEMA)@Au@TiO2@DOX NPs at different conditions (pH 7.5, pH 5.5, and pH 5.5 +laser irradiation) within 24 h. (n = 3, mean ± s.d.) (D) CLSM images of HeLa cells incubated with free DOX and P(CitAPDMAEMA)@Au@TiO2@DOX NPs with or without laser irradiation (L+). Scale bar: 25 μm. (E) Fluorescence images of calcein AM/PI-stained HeLa cells incubated with various media: a) laser only; b) P(CitAPDMAEMA) (pH 5.0, cationic therapy); c) P(CitAPDMAEMA)@Au@TiO2@DOX NPs (pH 5.0, cationic therapy/chemotherapy); d) Au@TiO2 core-shell NPs (laser irradiation, PTT/PDT; e) Au@TiO2@DOX NPs (laser irradiation, chemotherapy/PTT/PDT); f) P(CitAPDMAEMA)@Au@TiO2@DOX NPs (ationic therapy /chemotherapy/ PTT/PDT). Scale bar: 100 μm (F) Cytotoxicity of HeLa cells after different treatments. (n = 6, mean ± s.d.).
pH-dependent Binding of P(CitAPDMAEMA)@Au@TiO2@DOX NPs to HeLa cells
The zwitterionic biocompatible P(CitAPDMAEMA)@Au@TiO2@DOX NPs were thought to change into cationic and bind to the surface of cancer cells in the acidic condition. The binding of P(CitAPDMAEMA)@Au@TiO2@DOX NPs to cancer cells under acidic condition was evaluated by confocal laser scanning microscopy (CLSM), and HeLa cells were applied as the model cancer strain. P(CitAPDMAEMA)@Au@TiO2@DOX NPs were firstly labeled with DOX and then incubated with HeLa cells in PBS with different pH for 1 h. From Figure 4B, we could see the strong fluorescence in a group of HeLa cells at pH 5.0, while a low fluorescent signal was detected in the group at pH 7.4.
DOX Releasing
In order to evaluate the properties of P(CitAPDMAEMA) as drug release tools, the in vitro release of DOX from the P(CitAPDMAEMA)@Au@TiO2@DOX NPs was monitored by applying the dialysis membrane against PBS. In order to investigate the pH-dependent drug release, the released DOX was collected by dialyzing P(CitAPDMAEMA)@Au@TiO2@DOX NPs in PBS with different pHs. After 24 h, about 27.2% of DOX was released from the nanoparticles in pH 5.0, in contrast to 10.4% of DOX that was released in pH 7.4 (Figure 4C). Next, it was investigated whether red light-induced photothermal heating could also induce DOX release. P(CitAPDMAEMA)@Au@TiO2@DOX NPs in PBS (pH 5.0 or 7.4) were irradiated by 635 nm laser at 2.0 W/cm2 for 5 min at different time points. As compared to the DOX amount released in dark without laser irradiation, red light-stimulus dramatically increased the DOX release. Besides, with the same laser irradiation, the DOX release seemed to be more apparent in the lower pH environment compared to that in the physiological pH condition. These results indicated that the P(CitAPDMAEMA) molecules could be effectively applied as tools to instantly turn on/off the drug release, controlled by an environmental pH and red light.
In addition, it was examined whether the red-light induced drug release action of P(CitAPDMAEMA)@Au@TiO2@DOX NPs also exists inside cells [47]. Due to the fluorescence quenching of DOX after loading on the nano-carriers, reoccurrence of the DOX fluorescence could become an indicator for drug release. As shown in the CLSM images (Figure 4D), no dramatic discrepancy was detected in free DOX-treated cells after laser irradiation, and the drug molecules were evenly dispersed in the cytoplasm and nuclei. By comparison, obvious improved DOX fluorescence was detected in cells incubated with P(CitAPDMAEMA)@Au@TiO2@DOX NPs after laser irradiation, suggesting DOX was release from the nano-carriers. Analysis by ImageJ software on LSCM images disclosed the quantitative evidence that NIR laser irradiation promoted drug release from P(CitAPDMAEMA)@Au@TiO2@DOX NPs in cells (Figure S12A). These results collectively proved that red-light irradiation could effectively trigger intracellular drug release from P(CitAPDMAEMA)@Au@TiO2@DOX NPs.
Cell Toxicity
The in vitro cytotoxicity of as-prepared P(CitAPDMAEMA) and Au@TiO2 core-shell NPs were then evaluated with HeLa cells as a model by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay (Figure S12B). No obvious cytotoxicity to the P(CitAPDMAEMA) or Au@TiO2 core-shell NPs-transfected HeLa cells was observed as the P(CitAPDMAEMA) and Au@TiO2 core-shell NPs concentration ranged from 50 to 150 μg/mL, while a slight reduction of cell viability appeared at extremely high concentrations (200 μg/mL), indicating their good biocompatibility.
Subsequently, the efficacy of the cationic therapy/chemotherapy/PDT/PTT and the cytotoxicity of the produced P(CitAPDMAEMA)@Au@TiO2@DOX NPs were investigated in vitro. Normal and dead cells were determined by applying calcein AM (green) and PI (red) staining, respectively. In the laser-only (2.0 W/cm2) groups, no dead cells was observed, due to green fluorescence displayed by all cells (Figure 4E,a). This phenomenon showed the inability of the heat generated from pure water under irradiation to cause cancer cell death. Since P(CitAPDMAEMA) is able to bind to HeLa cells and can be converted to cationic P(CitAPDMAEMA) in the acidic environment, it was thought to obtain anticancer efficacy. In Figure S13, it was observed that P(CitAPDMAEMA) pretreated at pH 7.4, but did not show any effect on the growth of the HeLa cells, which was due to its zwitterionic nature at neutral pH. However, in the case of P(CitAPDMAEMA) pretreated at pH 5.0 (Figure 4E,b), the growth of the HeLa cells was obviously restrained. This result could be explained that P(CitAPDMAEMA) effectively absorbed the negative charge on the membrane of HeLa cells in the acidic environment, which inhibited the growth and metastasis of HeLa cells. Meanwhile, some cells got damage after incubating with 200 μg/mL Au@TiO2@DOX NPs (Figure 4E,c) because of the release of DOX. After laser irradiation at 2.0 W/cm2 and incubation with 200 μg/mL Au@TiO2 core-shell NPs (Figure 4E,d) and Au@TiO2@DOX NPs (Figure 4E,e), most of the cells were destroyed. On the contrary, after laser irradiation at 2.0 W/cm2 and incubation with 200 μg/mL P(CitAPDMAEMA)@Au@TiO2@DOX NPs (Figure 4E,f), almost all cells were killed, as demonstrated by the obtained intense homogeneous red fluorescence. The result was attributed to the fact that P(CitAPDMAEMA)@Au@TiO2@DOX NPs were able to simultaneously release DOX at high power doses, converting back to cationic P(CitAPDMAEMA), and generate ·OH/·O2-, and produce heat to efficiently kill cells by combined cationic therapy/ chemotherapy/PDT/PTT using a single red laser. Moreover, the cationic therapy/chemotherapy/PDT/ PTT efficacy of the P(CitAPDMAEMA)@Au@TiO2@DOX NPs was quantitatively assessed by the MTT assay. Among cationic therapy, chemotherapy, PDT/PTT, and chemotherapy/PDT/PTT groups, cell viability decreased with the increasing dose of the sample, while some cells remained alive even at a high sample concentration (200 μg/mL). However, in the cationic therapy/chemotherapy/PDT/PTT group, a mortality rate of ~98% was obtained, indicating that the simultaneous effects of cationic therapy/chemotherapy/PDT/PTT were able to effectively kill cancer cells (Figure 4F). These results agreed with the calcein AM and PI staining findings.
MR Imaging
It is known that the carboxyl and hydroxyl groups can easily chelate different kinds of metal ions, such as Cu2+, Mn2+, and Fe3+ [47, 48]. Among these metal ions, Mn2+ has been widely utilizing as a contrast agent for MRI. Based on the present system, Mn2+ was successfully loaded on P(CitAPDMAEMA)@Au@TiO2@DOX NPs by intrinsically chelating with carboxyl and hydroxyl groups on the surface of NPs. Interestingly, the obtained Mn@P(CitAPDMAEMA)@Au@TiO2@DOX NPs demonstrated a significant intensity-dependent brightening function under T1-weighted MRI (inset of Figure 5A), as well as a darkening function under T2-weighted MRI (inset of Figure 5B). The r1 and r2 relaxivities of the Mn@P(CitAPDMAEMA)@Au@TiO2@DOX NPs were detected at 11.1 mM-1s-1 and 40.8 mM-1s-1, respectively.
The (A) T1/(B) T2 relaxation rates and T1/T2-MR images (insets) of n@P(CitAPDMAEMA)@Au@TiO2@DOX NPs solutions with different Mn2+ concentrations. (C) In vivo T1/T2-MR images of HeLa-tumor-bearing mice taken 0 h and 24 h post i.v. injection with Mn@P(CitAPDMAEMA)@Au@TiO2@DOX NPs. (D) The fluorescence images of HeLa-tumor-bearing mice after i.v. injected with P(CitAPDMAEMA)@Au@TiO2@DOX NPs. The tissues were exploited for ex vivo fluorescence images after 24 h i.v. injection. (E) Blood circulation of Mn@P(CitAPDMAEMA)@Au@TiO2@DOX NPs after i.v. injection (n = 5, mean ± s.d.). (F) IR thermal images of HeLa-tumor-bearing mice injected with (a) Au@TiO2 core-shell NPs, (b) Au@TiO2@DOX NPs, (c) P(CitAPDMAEMA)@Au@TiO2@DOX NPs, and (d) PBS under 635 nm laser irradiation (2.0 W/cm2, 5 min).
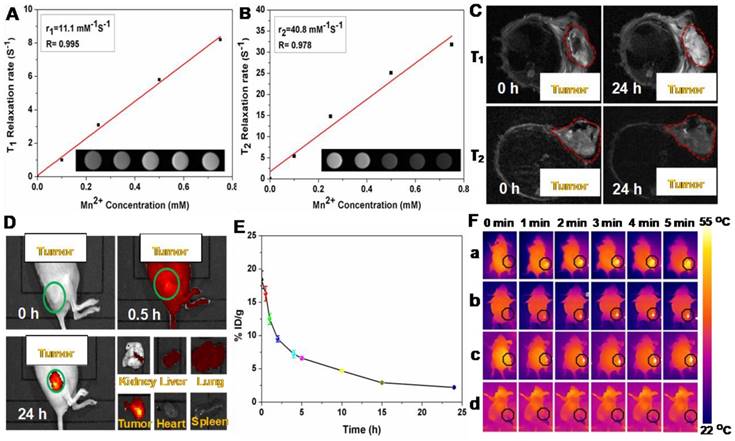
Therefore, the developed Mn@P(CitAPDMAEMA)@Au@TiO2@DOX NPs could be applied as a contrast agent for both T1-weighted and T2-weighted MRI. Furthermore, in vivo MRI was applied on mice. Mice bearing HeLa tumors were initially intravenously (i.v.) injected with Mn@P(CitAPDMAEMA)@Au@TiO2@DOX NPs and then imaged on a 3.0 T MRI system. Significant brightening or darkening effects were observed in the tumor region of mice compared with the pre-injected image (Figure 5C), suggesting high tumor accumulation of those NPs after systemic administration. Furthermore, the stronger T1- and T2-weighted MRI signal could be observed in the tumor region at 24 h post-injection. ImageJ was used to measure the region-of-interest (ROI) quantification (Figure S14) which further confirmed the enhancement of signal intensity in the tumor region after i.v. injection of Mn@P(CitAPDMAEMA)@Au@TiO2@DOX NPs. These results indicated that the effective tumor accumulation of Mn@P(CitAPDMAEMA)@Au@TiO2@DOX NPs via EPR effect and blood circulation.
Fluorescent Imaging
The whole-body fluorescence imaging was used to detect the biodistribution of DOX in HeLa tumor-bearing mice at specific time points following intravenous injection of P(CitAPDMAEMA)@Au@TiO2@DOX NPs (5 mg/kg). As shown in Figure 5D, P(CitAPDMAEMA)@Au@TiO2@DOX NPs could spread into the body of mice. Interestingly, the drug delivery nanosystem based on the P(CitAPDMAEMA)@Au@TiO2@DOX NPs was accumulated in the tumor at 24 h. This result was validated by the quantitative detection of DOX intensity in the normal tissue and the tumor regions (Figure S15). It was speculated that P(CitAPDMAEMA)@Au@TiO2 NPs were captured by the liver, spleen, and lung tissues, which prolonged the blood-circulation lifetime. As a result, this progress increased the efficiency of the P(CitAPDMAEMA)@Au@TiO2@DOX NPs to recognize and gather at the tumor site [49]. In particular, the gathering of P(CitAPDMAEMA)@Au@TiO2@DOX NPs in the heart decreased, indicating that the nanosystem could decrease cardiac muscle toxicity caused by DOX. These findings showed that P(CitAPDMAEMA)@Au@TiO2@DOX NPs were able to recognize the tumor, accumulate in the tumor, and decrease the side effects of DOX.
Blood Circulation and Biodistribution
Prior to combination therapy, the in vivo behavior of the P(CitAPDMAEMA)@Au@TiO2@DOX NPs were examined. Female Balb/c mice were i.v. injected with P(CitAPDMAEMA)@Au@TiO2@DOX NPs (5 mg/kg, 200 μL). Blood was collected from the mice at every time point. After being dispersed in lysis buffer and extracted with HCl/isopropanol, DOX was measured by fluorescence in order to assess its intensity in the blood [47]. As seen in Figure 5E, the signals of DOX gradually decreased over time following a two-compartment model, where the first (t1/2(α)) and second (t1/2(β)) phases of circulation half-lives were calculated to be 1.92 ± 0.24 h and 9.86 ± 0.52 h, respectively. Furthermore, half of P(CitAPDMAEMA)@Au@TiO2@DOX NPs were rapidly excreted through the urine and feces route 24 h after injection due to their nanoscale (Figure S16). After 24 h intravenous injection, a large part of P(CitAPDMAEMA)@Au@TiO2@DOX NPs appeared in the liver and kidney because of the reticuloendothelial system clearance. Around 23.7% of the P(CitAPDMAEMA)@Au@TiO2@DOX NPs were accumulated into the tumor through the EPR effect and the cationic targeting effect (Figure S17). It could also modify the targeted ligands to improve its accumulation efficiency. P(CitAPDMAEMA)@Au@TiO2@DOX NPs could be not only passively target the tumor tissue, but also evaluate the tumor site/dimension/morphology by fluorescent/infrared thermal/MR treble-modal imaging which was able to guide the simultaneous PTT and PDT.
In Vivo Photothermal Imaging
In the light of good photothermal conversion of P(CitAPDMAEMA)@Au@TiO2@DOX NPs, we evaluated in vivo photothermal efficiency in the tumor model. Female Balb/c mice were i.v. injected with Au@TiO2 core-shell NPs, Au@TiO2@DOX NPs, P(CitAPDMAEMA)@Au@TiO2@DOX NPs, and PBS. After 6 h, tumor regions of mice were irradiated by 635 nm laser with a power density of 2.0 W/cm2 for 5 min. The change of temperature during irradiation was detected by an IR thermal camera (Figure 5F and Figure S18). The temperatures of tumor in the mice injected with Au@TiO2@DOX NPs, Au@TiO2 core-shell NPs, and P(CitAPDMAEMA)@Au@TiO2@DOX NPs demonstrated a rapid increase and then maintained at 54 °C during 635 nm laser irradiation. In contrast, little change was measured in the tumor temperature of the mice injected with P(CitAPDMAEMA)@Au@TiO2 NPs and PBS under 635 nm laser irradiation using the same parameters.
In Vivo Combined Therapy
Based on the mentioned in vivo imaging results, in vivo cancer treatment with P(CitAPDMAEMA)@Au@TiO2@DOX NPs was carried out (Scheme 1). Female Balb/c mice were subcutaneously injected into their backs with 1×106 HeLa cells. After one week, the tumors grew to 60 mm3 and the mice were divided into 6 groups: P(CitAPDMAEMA)@Au@TiO2 NPs (1), P(CitAPDMAEMA)@Au@TiO2@DOX NPs (2), PBS (plus Laser) (3), Au@TiO2@DOX NPs (plus Laser) (4), Au@TiO2 core-shell NPs (plus Laser) (5), and P(CitAPDMAEMA)@Au@TiO2@DOX NPs (plus Laser) (6). The drug and drug-loaded NPs were i.v. injected into each mouse through the tail vein. After the treatment by laser, the tumor volume was measured by a digital caliper every two days for a total of two weeks (Figure 6A-B). The combination therapy group of the P(CitAPDMAEMA)@Au@TiO2@DOX NPs with laser irradiation, demonstrated an obvious inhibition of tumor growth. The Au@TiO2 core-shell NPs and Au@TiO2@DOX NPs with laser irradiation presented a small inhibitory function during the early days, but grew normally later. The P(CitAPDMAEMA)@Au@TiO2 NPs and P(CitAPDMAEMA)@Au@TiO2@DOX NPs treatment groups seemed to have a minimal influence on tumor growth. These results were also confirmed by representative tumor images (Figure 6C) at day 14. Slices of tumors stained with hematoxylin and eosin (HE) and TUNEL staining (Figure 6D) were obtained, in order to further assess the therapeutic function of the various treatments. It was shown that the combination treatment group P(CitAPDMAEMA)@Au@TiO2@DOX NPs with laser irradiation showed the most effective damage on most of the tumor cells, while the other five groups depicted little or no damage on tumor cells, which had normal membrane morphology and nuclear structures.
(A) Tumor growth curves for mice with various treatment. (n = 5, mean ± s.d., **P<0.01, and ***P<0.001). (B) The digital photographs of mice 14th days with various treatments. (C) Representative photographs of the excised tumors 14th days with various treatments. (up to bottom: PBS (L+), P(CitAPDMAEMA)@Au@TiO2 NPs, P(CitAPDMAEMA)@Au@TiO2@DOX NPs, Au@TiO2 core-shell NPs (L+), Au@TiO2@DOX NPs (L+), and P(CitAPDMAEMA)@Au@TiO2@DOX NPs (L+)). (D) TUNEL and HE staining of tumor slices collected from different mice groups after 14 days treatment. (E-J) Blood biochemistry analysis (LDH, CK, AST, ALT, UA, and Creatinine) of the different mice groups after 14 days treatment. (n = 5, mean ± s.d.).
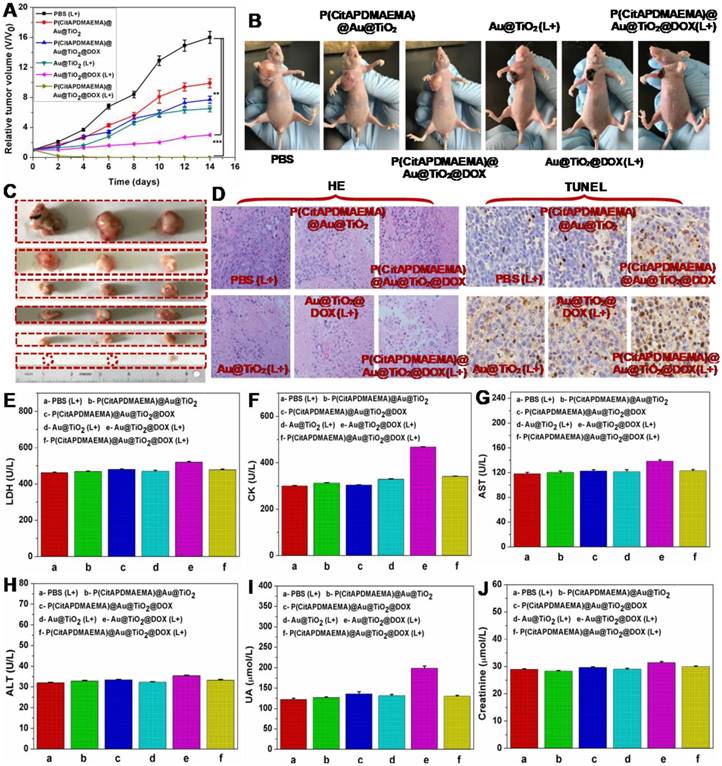
In order to investigate whether these treatments would induce toxicity, the body weight of the mice was monitored (Figure S19) and histology of organ samples including lung, liver, spleen, kidney, and heart was performed (Figure S20). The body weight of the mice in the treated groups was about the same as those in the control group. No loss of body weight or other serious toxic effects were observed. Furthermore, the histological analysis did not reveal any serious irreversible pathological alterations or injuries in the organs of the mice of all groups. Moreover, the toxicity of the P(CitAPDMAEMA)@Au@TiO2@DOX NPs with laser irradiation was detected by blood biochemical analysis. As can be seen in Figure 6E-F, the P(CitAPDMAEMA)@Au@TiO2@DOX NPs with laser irradiation group demonstrated no apparent increase in lactate dehydrogenase (LDH) and creatine kinase (CK) levels, which were necessary parameters for heart function. In addition, the analysis of liver function (Figure 6G-H) [aspartate aminotransferase (AST) and alanine aminotransferase (ALT)] and kidney function (Figure 6J-I) [uric acid (UA) and creatinine] parameters did not show any damage to liver and kidney in the mice treated with P(CitAPDMAEMA)@Au@TiO2@DOX NPs with laser irradiation. This phenomenon suggested that the P(CitAPDMAEMA)@Au@TiO2@DOX NPs combination treatment did not induce any toxicity. All results above clearly showed that the P(CitAPDMAEMA)@Au@TiO2@DOX NPs could serve as powerful in vivo anticancer agents.
Conclusions
In conclusion, we have successfully designed and constructed a multifunctional nanohybrid for imaging-guided combination treatment of cancer. The resultant P(CitAPDMAEMA)@Au@TiO2@DOX NPs possessed the following features: (1) rapid binding to cancer cells under acidic conditions; (2) high photothermal conversion efficiency under 635 nm laser irradiation; (3) efficient ROS release upon 635 nm laser irradiation; (4) induced DOX release by external red-light and at intracellular pH values. In addition, those NPs possessed obvious contrasts in MRI after the chelation with Mn2+. An imaging-guided cancer therapy, which combined with those functional parts, was shown in mouse tumor model experiments. The cationic therapy/ chemotherapy/PTT/PDT delivered by the P(CitAPDMAEMA)@Au@TiO2@DOX NPs showed a significant combined effect in destructing tumors using. Moreover, the tiny size of the nanoparticles allowed for partial renal clearance and high tumor uptake in vivo, making the theranostic agent a promising candidate for future clinical translation.
Experimental Section
Chemicals
2-(Dimethylamino)ethyl methacrylate (DMAEMA, 98%), citraconic anhydride (Cit, 98%), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 98%) and 4-cyanopentanoic acid dithiobenzoate (CPADB, 98%) were purchased from Sinopharm Chemical Reagent CO, Ltd., China. Doxorubicin (DOX) were purchased from Tianjin Biolite Biotech Co., Ltd. The dialysis tube membrane with molecular weight cutoff (MWCO) of 1000 was purchased from Peg Bio, Suzhou, China. All aqueous solution used in experiment was deionized water (DI, 18.2 MΩ·cm) obtained from Milli-Q water purification system.
Instruments and characterization
The infrared spectrum was performed on a Nexus 670 FTIR type (Nicolet). The X-ray diffraction (XRD) analysis was performed using a D/Max 2500V/PC diffractometer (Rigaku Corporation, Japan). UV-Vis spectroscopy measurements were performed on a Cary 5000 UV-Vis-NIR spectrometer (Varian). The surface composition and element analysis of the samples were recorded using X-ray photoelectron spectroscopy (XPS, EscaLab-250, Thermo, USA). The hydrodynamic size and zeta-potential were measured on a Malvern ZEN 3600 Zetasizer (Malvern Instruments, UK). The transmission electron microscopy (TEM) images were acquired on a JEM-2100F TEM. The fluorescent images of cells were acquired by Confocal Scanning Laser Microscope (CSLM, TI-E-A1R, Nikon, Japan). The infrared thermal images were recorded with a PTT monitoring system MG33 (Shanghai Magnity Electronics Co. Ltd.). The methods used for material characterization are displayed in the experimental section of the supporting information.
Supplementary Material
Supplementary experimental section, schemes and figures.
Acknowledgements
This project was supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and the Villum Fonden, Denmark, Project No. 13153. M.Z. and T.Z. would like to thank the China Scholarship Council (CSC) for its generous support. We would like to thank Dr. Jon Ashley for helping with the manuscript proof-reading.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lv R, Yang P, He F, Gai S, Yang G, Lin J. Hollow Structured Y2O3:Yb/Er-CuxS Nanospheres with Controllable Size for Simultaneous Chemo/Photothermal Therapy and Bioimaging. Chem Mater. 2015;27:483-96
2. Zhang M, Wang W, Zhou N, Yuan P, Su Y, Shao M. et al. Near-infrared light triggered phototherapy, in combination with chemotherap using magnetofluorescent carbon quantum dots for effective cancer treating. Carbon. 2017;118:752-64
3. Dong Z, Gong H, Gao M, Zhu W, Sun X, Feng L. et al. Polydopamine Nanoparticles as a Versatile Molecular Loading Platform to Enable Imaging-guided Cancer Combination Therapy. Theranostics. 2016;6:1031-42
4. Guo W, Wang F, Ding D, Song C, Guo C, Liu S. TiO2-x Based Nanoplatform for Bimodal Cancer Imaging and NIR-Triggered Chem/Photodynamic/Photothermal Combination Therapy. Chem Mater. 2017;29:9262-74
5. Wang H, Zhu W, Liu J, Dong Z, Liu Z. pH-Responsive Nanoscale Covalent Organic Polymers as a Biodegradable Drug Carrier for Combined Photodynamic Chemotherapy of Cancer. ACS Appl Mater Interfaces. 2018;10:14475-82
6. Zhang M, Wang W, Cui Y, Zhou N, Shen J. Near-infrared light-mediated photodynamic/photothermal therapy nanoplatform by the assembly of Fe3O4 carbon dots with graphitic black phosphorus quantum dots. Int J Nanomedicine. 2018;13:2803-19
7. Akhavan O, Meidanchi A, Ghaderi E, Khoei S. Zinc ferrite spinel-graphene in magneto-photothermal therapy of cancer. J Mater Chem B. 2014;2:3306-14
8. Liu B, Li C, Chen G, Liu B, Deng X, Wei Y. et al. Synthesis and Optimization of MoS2@Fe3O4-ICG/Pt(IV) Nanoflowers for MR/IR/PA Bioimaging and Combined PTT/PDT/Chemotherapy Triggered by 808 nm Laser. Adv Sci. 2017;4:1600540
9. Sun X, Wang C, Gao M, Hu A, Liu Z. Remotely Controlled Red Blood Cell Carriers for Cancer Targeting and Near-Infrared Light-Triggered Drug Release in Combined Photothermal-Chemotherapy. Adv Funct Mater. 2015;25:2386-94
10. Zhu D, Liu M, Liu X, Liu Y, Prasad PN, Swihart MT. Au-Cu2-xSe heterogeneous nanocrystals for efficient photothermal heating for cancer therapy. J Mater Chem B. 2017;5:4934-42
11. Huang Y, Lai Y, Shi S, Hao S, Wei J, Chen X. Copper sulfide nanoparticles with phospholipid-PEG coating for in vivo near-infrared photothermal cancer therapy. Chem Asian J. 2015;10:370-76
12. Ge J, Jia Q, Liu W, Guo L, Liu Q, Lan M. et al. Red-Emissive Carbon Dots for Fluorescent, Photoacoustic, and Thermal Theranostics in Living Mice. Adv Mater. 2015;27:4169-77
13. Yang T, Wang Y, Ke H, Wang Q, Lv X, Wu H. et al. Protein-Nanoreactor-Assisted Synthesis of Semiconductor Nanocrystals for Efficient Cancer Theranostics. Adv Mater. 2016;28:5923-30
14. Mou J, Lin T, Huang F, Chen H, Shi J. Black titania-based theranostic nanoplatform for single NIR laser induced dual-modal imaging-guided PTT/PDT. Biomaterials. 2016;84:13-24
15. Lv R, Yang D, Yang P, Xu J, He F, Gai S. et al. Integration of Upconversion Nanoparticles and Ultrathin Black Phosphorus for Efficient Photodynamic Theranostics under 808 nm Near-Infrared Light Irradiation. Chem Mater. 2016;28:4724-34
16. Huang J, Zhu Y, Liu C, Zhao Y, Liu Z, Hedhili MN. et al. Fabricating a Homogeneously Alloyed AuAg Shell on Au Nanorods to Achieve Strong, Stable, and Tunable Surface Plasmon Resonances. Small. 2015;11:5214-21
17. Zhang M, Wang W, Cui Y, Chu X, Sun B, Zhou N. et al. Magnetofluorescent Fe3O4/carbon quantum dots coated single-walled carbon nanotubes as dual-modal targeted imaging and chemo/photodynamic/photothermal triple-modal therapeutic agents. Chem Eng J. 2018;338:526-38
18. Dong Z, Feng L, Hao Y, Chen M, Gao M, Chao Y. et al. Synthesis of Hollow Biomineralized CaCO3-Polydopamine Nanoparticles for Multimodal Imaging-Guided Cancer Photodynamic Therapy with Reduced Skin Photosensitivity. J Am Chem Soc. 2018;140:2165-78
19. Lu F, Yang L, Ding Y, Zhu J-J. Highly Emissive Nd3+-Sensitized Multilayered Upconversion Nanoparticles for Efficient 795 nm Operated Photodynamic Therapy. Adv Funct Mater. 2016;26:4778-85
20. Dai Y, Cheng S, Wang Z, Zhang R, Yang Z, Wang J. et al. Hypochlorous Acid Promoted Platinum Drug Chemotherapy by Myeloperoxidase-Encapsulated Therapeutic Metal Phenolic Nanoparticles. ACS Nano. 2018;12:455-63
21. Li W, Cao L, Han L, Xu Q, Ma Q. Superoxide dismutase promotes the epithelial-mesenchymal transition of pancreatic cancer cells via activation of the H2O2/ERK/NF-kappaB axis. Int J Oncol. 2015;46:2613-20
22. Jovanovic SP, Syrgiannis Z, Markovic ZM, Bonasera A, Kepic DP, Budimir MD. et al. Modification of Structural and Luminescence Properties of Graphene Quantum Dots by Gamma Irradiation and Their Application in a Photodynamic Therapy. ACS Appl Mater Interfaces. 2015;7:25865-74
23. Ge J, Jia Q, Liu W, Lan M, Zhou B, Guo L. et al. Carbon Dots with Intrinsic Theranostic Properties for Bioimaging, Red-Light-Triggered Photodynamic/Photothermal Simultaneous Therapy In Vitro and In Vivo. Adv Healthc Mater. 2016;5:665-75
24. Zhang P, Huang H, Huang J, Chen H, Wang J, Qiu K. et al. Noncovalent Ruthenium(II) Complexes-Single-Walled Carbon Nanotube Composites for Bimodal Photothermal and Photodynamic Therapy with Near-Infrared Irradiation. ACS Appl Mater Interfaces. 2015;7:23278-90
25. Zhou Z, Shen Y, Tang J, Fan M, Van Kirk EA, Murdoch WJ. et al. Charge-Reversal Drug Conjugate for Targeted Cancer Cell Nuclear Drug Delivery. Adv Funct Mater. 2009;19:3580-9
26. Khatoon S, Han HS, Lee M, Lee H, Jung DW, Thambi T. et al. Zwitterionic mesoporous nanoparticles with a bioresponsive gatekeeper for cancer therapy. Acta Biomater. 2016;40:282-92
27. Hao L, Lin L, Zhou J. pH-Responsive Zwitterionic Copolymer DHA-PBLG-PCB for Targeted Drug Delivery: A Computer Simulation Study. Langmuir. 2019;35:1944-53
28. Deng H, Zhao X, Liu J, Deng L, Zhang J, Liu J. et al. Reactive oxygen species (ROS) responsive PEG-PCL nanoparticles with pH-controlled negative-to-positive charge reversal for intracellular delivery of doxorubicin. J Mater Chem B. 2015;3:9397-408
29. Li F, Yang H, Bie N, Xu Q, Yong T, Wang Q. et al. Zwitterionic Temperature/Redox-Sensitive Nanogels for Near-Infrared Light-Triggered Synergistic Thermo-Chemotherapy. ACS Appl Mater Interfaces. 2017;9:23564-73
30. Jin Q, Chen Y, Wang Y, Ji J. Zwitterionic drug nanocarriers: a biomimetic strategy for drug delivery. Colloids Surf B Biointerfaces. 2014;124:80-6
31. Mizuhara T, Saha K, Moyano DF, Kim CS, Yan B, Kim YK. et al. Acylsulfonamide-Functionalized Zwitterionic Gold Nanoparticles for Enhanced Cellular Uptake at Tumor pH. Angew Chem Int Ed Engl. 2015;54:6567-70
32. Liu P, Xu G, Pranantyo D, Xu LQ, Neoh K-G, Kang E-T. pH-Sensitive Zwitterionic Polymer as an Antimicrobial Agent with Effective Bacterial Targeting. ACS Biomater Sci Eng. 2017;4:40-6
33. Sun H, Chang MYZ, Cheng WI, Wang Q, Commisso A, Capeling M. et al. Biodegradable zwitterionic sulfobetaine polymer and its conjugate with paclitaxel for sustained drug delivery. Acta Biomater. 2017;64:290-300
34. Zhang P, Sun F, Tsao C, Liu S, Jain P, Sinclair A. et al. Zwitterionic gel encapsulation promotes protein stability, enhances pharmacokinetics, and reduces immunogenicity. Proc Natl Acad Sci U S A. 2015;112:12046-51
35. Sun H, He Q, She P, Zeng S, Xu K, Li J. et al. One-pot synthesis of Au@TiO2 yolk-shell nanoparticles with enhanced photocatalytic activity under visible light. J Colloid Interface Sci. 2017;505:884-91
36. Klug MT, Dorval Courchesne N-M, Lee YE, Yun DS, Qi J, Heldman NC. et al. Mediated Growth of Zinc Chalcogen Shells on Gold Nanoparticles by Free-Base Amino Acids. Chem Mater. 2017;29:6993-7001
37. He F, Yu W, Fan X, Jin B. In Vitro Cytotoxicity of Biosynthesized Titanium Dioxide Nanoparticles in Human Prostate Cancer Cell Lines. Trop J Pharm Res. 2018;16:2793-9
38. Yadav HM, Thorat ND, Yallapu MM, Tofail SAM, Kim J-S. Functional TiO2 nanocoral architecture for light-activated cancer chemotherapy. J Mater Chem B. 2017;5:1461-70
39. Wang H, Mu Q, Revia R, Wang K, Zhou X, Pauzauskie PJ. et al. Chitosan-Gated Magnetic-Responsive Nanocarrier for Dual-Modal Optical Imaging, Switchable Drug Release, and Synergistic Therapy. Adv Healthc Mater. 2017:6 1601080
40. Wang X, Li H, Liu X, Tian Y, Guo H, Jiang T. et al. Enhanced photothermal therapy of biomimetic polypyrrole nanoparticles through improving blood flow perfusion. Biomaterials. 2017;143:130-41
41. Peng H, Tang J, Zheng R, Guo G, Dong A, Wang Y. et al. Nuclear-Targeted Multifunctional Magnetic Nanoparticles for Photothermal Therapy. Adv Healthc Mater. 2017:6 1601289
42. Zhang XD, Wu D, Shen X, Chen J, Sun YM, Liu PX. et al. Size-dependent radiosensitization of PEG-coated gold nanoparticles for cancer radiation therapy. Biomaterials. 2012;33:6408-19
43. Deepagan VG, You DG, Um W, Ko H, Kwon S, Choi KY. et al. Long-Circulating Au-TiO2 Nanocomposite as a Sonosensitizer for ROS-Mediated Eradication of Cancer. Nano Lett. 2016;16:6257-64
44. Yin W, Bao T, Zhang X, Gao Q, Yu J, Dong X. et al. Biodegradable MoOx nanoparticles with efficient near-infrared photothermal and photodynamic synergetic cancer therapy at the second biological window. Nanoscale. 2018;10:1517-31
45. Liu Y, Liu Y, Bu W, Cheng C, Zuo C, Xiao Q. et al. Hypoxia Induced by Upconversion-Based Photodynamic Therapy: Towards Highly Effective Synergistic Bioreductive Therapy in Tumors. Angew Chem Int Ed Engl. 2015;54:8105-9
46. Zhang M, Wu F, Wang W, Shen J, Zhou N, Wu C. Multifunctional Nanocomposites for Targeted, Photothermal, and Chemotherapy. Chem Mater. 2018;31:1847-59
47. Liu J, Wang C, Wang X, Wang X, Cheng L, Li Y. et al. Mesoporous Silica Coated Single-Walled Carbon Nanotubes as a Multifunctional Light-Responsive Platform for Cancer Combination Therapy. Adv Funct Mater. 2015;25:384-92
48. Liu Y, Ai K, Lu L. Polydopamine and its derivative materials: synthesis and promising applications in energy, environmental, and biomedical fields. Chem Rev. 2014;114:5057-115
49. Yu K, Liang B, Zheng Y, Exner A, Kolios M, Xu T. et al. PMMA-Fe3O4 for Internal Mechanical Support and Magnetic Thermal Ablation of Bone Tumors. Theranostics. 2019;9:4192-4207
Author contact
![]() Corresponding authors: Ming Zhang, mzhandk; Jian Shen, jshenedu.cn; Yi Sun, suyidk
Corresponding authors: Ming Zhang, mzhandk; Jian Shen, jshenedu.cn; Yi Sun, suyidk