Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Pathology of coronary...
Coagulation
The circulating endothelial...
Role of MQC in coronary...
Cardioprotective strategies on...
Pharmacological interventions...
Concluding remarks and...
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(14):6766-6785. doi:10.7150/thno.60143 This issue Cite
Review
Coronary microvascular injury in myocardial infarction: perception and knowledge for mitochondrial quality control
Xing Chang1,2, Amanda Lochner3, Hsueh-Hsiao Wang4, Shuyi Wang5,6, Hang Zhu1, Jun Ren5, Hao Zhou1,5 ![]()
1. Chinese PLA General Hospital, Medical School of Chinese PLA, Beijing 100853, China.
2. Guang'anmen Hospital of Chinese Academy of Traditional Chinese Medicine, Beijing, China.
3. Department of Biomedical Sciences, Faculty of Health Sciences, University of Stellenbosch, Stellenbosch, South Africa.
4. Department of Medicine, Mackay Medical College, New Taipei City, 252, Taiwan.
5. University of Wyoming College of Health Sciences, Laramie, WY 82071, USA.
6. Shanghai University School of Medicine, Shanghai 200044, China.
Received 2021-3-6; Accepted 2021-4-14; Published 2021-5-3
Abstract

Endothelial cells (ECs) constitute the innermost layer in all blood vessels to maintain the structural integrity and microcirculation function for coronary microvasculature. Impaired endothelial function is demonstrated in various cardiovascular diseases including myocardial infarction (MI), which is featured by reduced myocardial blood flow as a result of epicardial coronary obstruction, thrombogenesis, and inflammation. In this context, understanding the cellular and molecular mechanisms governing the function of coronary ECs is essential for the early diagnosis and optimal treatment of MI. Although ECs contain relatively fewer mitochondria compared with cardiomyocytes, they function as key sensors of environmental and cellular stress, in the regulation of EC viability, structural integrity and function. Mitochondrial quality control (MQC) machineries respond to a broad array of stress stimuli to regulate fission, fusion, mitophagy and biogenesis in mitochondria. Impaired MQC is a cardinal feature of EC injury and dysfunction. Hence, medications modulating MQC mechanisms are considered as promising novel therapeutic options in MI. Here in this review, we provide updated insights into the key role of MQC mechanisms in coronary ECs and microvascular dysfunction in MI. We also discussed the option of MQC as a novel therapeutic target to delay, reverse or repair coronary microvascular damage in MI. Contemporary available MQC-targeted therapies with potential clinical benefits to alleviate coronary microvascular injury during MI are also summarized.
Keywords: coronary microvasculature, ECs, myocardial infarction, mitochondrial quality control
Introduction
Coronary microcirculation is usually present in vessels with diameters ranging 200-400 μm and is invisible under coronary angiography. Unlike macrocirculation which is mainly carried by epicardial arteries (diameter > 400 µm), coronary microvasculature consists of small arteries (diameter < 400 µm), arterioles (diameter < 100 µm) and capillaries (diameter < 10 µm). The function of macrocirculation encompasses the delivery of fresh blood, oxygen and nutrients as well as the removal of carbon dioxide. Coronary microcirculation is widely employed by human body to control of distribution of blood flow, with an important role in the regulation of vascular resistance. Endothelial cells (ECs), vascular smooth muscle cell (VSMC) and pericytes are the main components of coronary microvasculature. The coronary microvasculature regulatory control mechanisms were recently systematically reviewed by Heusch and colleagues [1]. The interior surface of coronary microcirculation is lined by a monolayer of ECs and acts as a conduit for transporting blood and nutrients. ECs also serve as a defensive barrier against infiltration of micro-organisms, immune cells, and coagulation components, thereby reducing risk of thrombosis. Vascular endothelium is a dynamic endocrine organ consisting of multiple populations of ECs with different architectural and functional properties that synergistically promotes an anti-thrombotic and anti-inflammatory environment and maintains the tissue perfusion. The coronary microvascular tone is regulated by the balance between vasodilators such as bradykinin and nitric oxide (NO) and vasoconstrictors such as endothelin, both of which are released by ECs. Endothelial dysfunction and/or coronary microvascular impairment due to inflammation or oxidative stress is the main cause of several cardiovascular disorders [2]. For example, myocardial infarction (MI) is caused by coronary thrombosis (coronary microvascular spasm or occlusion) (Figure 1). In the heart, ECs and cardiomyocytes form crosstalk with each other through the release of several cell signaling transmitters [3]. However, excessive EC-cardiomyocyte signaling exchange may prompt a vicious cycle that causes cardiomyocyte injury and death.
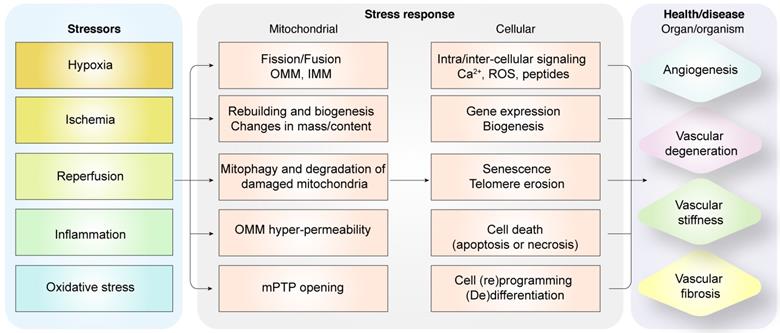
Diagrammatic representation shows the relationship between pathological alterations in vasculature during myocardial infarction and mitochondrial quality control mechanisms. Intrinsic and extrinsic stress signals such as hypoxia, ischemia, reperfusion, inflammation or oxidative stress activate mitochondrial fission or fusion. Mitophagy promotes degradation of damaged mitochondria. Mitochondrial biogenesis regulates synthesis of mitochondrial DNA, proteins, and lipids to restore optimal mitochondrial mass. Outer mitochondrial membrane permeabilization (OMM) is induced and mitochondrial permeability transition pore (mPTP) opening is enhanced when mitochondrial damage becomes excessive. OMM promotes cellular apoptosis, whereas, mPTP opening induces necrosis or necroptosis. Dysregulated mitochondrial quality control (MQC) mechanisms induce aberrant calcium signaling, excessive ROS, changes in gene expression of proteins related to bioenergetics, senescence, telomere erosion, metabolic reprogramming, (de)differentiation, or cell death (apoptosis or necrosis) in the coronary microvascular endothelial cells. These changes in endothelial cells, coronary arteries or cardiac microcirculation induce vascular degeneration, stiffness, or fibrosis and reduce angiogenesis.
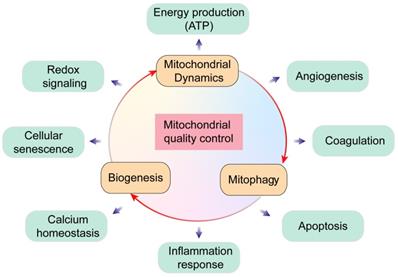
Mitochondria are cellular powerhouses that produce ATP through oxidative phosphorylation. The relative mitochondrial mass compared to total cytoplasmic volume is 2-6% in ECs and ~32% in cardiomyocytes. Moreover, ECs are more dependent on glycolysis as energy source and are less dependent on oxidative phosphorylation. ECs with acquired mitochondrial abnormalities demonstrate a cancer-like phenotype including increased pyruvate dehydrogenase kinase (PDK) and pyruvate kinase muscle isoform 2 (PKM2) levels and activity associated with uncoupled glycolysis [4]. Therefore, ECs would require much less oxygen from the coronary microcirculation, thereby ensuring sufficient oxygen supply to cardiomyocytes. In addition to the pseudo-hypoxic state, ECs activate the pentose phosphate pathway, a major source of cellular reducing power in the form of NADPH [5]. The ECs utilize pentose phosphate pathway as the predominant anti-oxidative mechanism to protect ECs from oxidative damage and maintain cellular redox balance. In addition to metabolic switch capacity, mitochondria modulate their structure, function, and mass to meet the metabolic needs of the heart through mitochondrial quality control (MQC) mechanisms including fission, fusion, mitophagy, and biogenesis. MQC machinery plays a critical role in cardiac health and disease, while pathological changes in MQC may trigger onset and development of cardiovascular disorders including MI (Figure 2).
Overview of mitochondrial quality control. Mitochondria regulate several key biological processes in the endothelial cells including energy production, redox signaling, cellular senescence, calcium homeostasis, angiogenesis, coagulation, apoptosis and inflammation response.
In this review, we will first describe unique features of healthy coronary microvasculature and currently perceived pathological mechanisms that damage coronary microcirculation in MI. Then, we will highlight the distinct roles of various MQC mechanisms including fission, fusion, mitophagy, and biogenesis in coronary microvascular homeostasis under normal physiological and pathophysiological conditions such as MI. In the end, we will summarize mitochondria- and endothelium-targeted pharmacological agents with therapeutic potentials for MI.
Pathology of coronary microvascular ECs damage in MI
Clinical features of coronary microvascular damage in MI
MI is caused by sudden occlusion of the coronary arteries that results in reduced blood supply to the myocardium and ischemic damage to the heart. Rapid restoration of coronary flow to the ischemic myocardium (reperfusion) is the primary therapeutic strategy for MI. However, successful recanalization to restore blood flow and oxygen supply to the myocardium is associated with increased risk of reperfusion injury. Therefore, coronary microvascular injury in MI results in myocardial damage because of both ischemic and reperfusion injuries. After 180 minutes of coronary occlusion, cardiomyocytes in the ischemic myocardium show intracellular edema, swollen mitochondria with amorphous dense bodies, absence of glycogen stores, and numerous breaks in the sarcolemmal membrane [6]. These alterations suggest that ischemia-mediated myocardial damage is mainly due to a transient shortage of oxygen and nutrients as well as then accumulation of breakdown products.
Ultrastructural changes in the ECs within ischemic myocardium are less prominent compared with those noted in cardiomyocytes during ischemia. The most prominent change in ECs during early ischemia is the loss of pinocytotic vesicles [7]. As ischemia persists, ECs exhibit localized areas of swelling and bleb formation on the intraluminal surface, gaps between adjacent ECs, and occasional foci of hemorrhage [8]. However, these changes in ECs can only be observed in 40% of the microvessels mainly localized in the sub-endocardial zones that contain irreversibly damaged cardiomyocytes after 180 minutes of coronary occlusion. This suggests that ECs in the coronary microvasculature are less prone to ischemic insult compared with cardiomyocytes and coronary microvascular damage in ECs lags behind cardiomyocyte injury [9]. Although the duration of ischemia determines the extent of myocardial damage, infarcted size is also defined by the number and localization of occluded coronary arteries.
Percutaneous coronary intervention (PCI) and coronary artery bypass surgery (CABG) are the main streams treatments currently available to revascularize the occluded coronary arteries. However, more than 50% of patients with coronary microvascular damage receiving PCI or CABG displayed a no-reflow phenomenon due to inadequate myocardial reperfusion [10]. In contrast to ischemic injury, both coronary microvascular dysfunction and endothelial cell death contribute to cardiac reperfusion injury [11, 12]. Nonetheless, the precise molecular mechanisms underlying coronary microcirculation reperfusion injury are not well studied compared to the perceived knowledge of ischemic injury [13].
The signs of the no-reflow phenomenon despite open epicardial coronary artery in MI patients include decreased ventricular function, lower ejection fraction, and poorer clinical outcomes. Therefore, reperfusion therapeutic strategy must take into consideration to restore the functional status of myocardial microvasculature in addition to epicardial coronary artery patency [14].
During reperfusion, capillary lumen in the coronary ischemic zone is completely filled with endothelial protrusions and membrane-bound bodies including degranulated platelets [6]. Moreover, Heusch and coworkers [15, 16] reported accumulation of atherothrombotic debris and soluble substances (such as serotonin, thromboxane B2, and TNFα), released from the ruptured atherosclerotic plaque in epicardial coronary artery, all of which with a pivotal role in perturbing microvascular perfusion during myocardial infarction [17].
Unlike coronary microvascular ECs, cardiomyocytes exhibit intracellular edema, relaxed myofibrils, swollen mitochondria with fractured cristae, and nuclear changes during both reperfusion and ischemic injuries. Prolonged reperfusion causes significant changes in both cardiomyocytes and coronary microcirculation. Following 3.5 h of reperfusion, myocardium displays a diffused necrosis band blended with hyper-contracted sarcomeres [18]. Post-ischemic myocardium exhibits massive intravascular accumulation of neutrophils. In areas of impaired flow, a 20-fold increase in neutrophils and red blood cell stasis is observed due to plugging of capillaries by neutrophils [19]. Moreover, extravascular red blood cells are frequently observed in gaps within the endothelial lining [19]. In canine MI model, no-reflow zone was nearly three times greater 3.5 h later in comparison with the no-reflow zone after 2 mins of reperfusion [19]. Similarly, in a rabbit MI model, the size of no-reflow zone was nearly three-fold larger after 2 h reperfusion compared to the no-reflow zone after 30 min reperfusion [20]. Moreover, the rise in the size of no-reflow zone limited myocardial blood flow in the area of risk to ~44% of the baseline flow observed prior to the initiation of myocardial ischemia [21]. These data favor a time-dependent worsening of coronary microvascular damage during reperfusion injury coinciding with cardiac dysfunction.
In addition to irreversible damage evoked by cardiac reperfusion injury, recognition of the role for myocardial stunning and hibernation has greatly broadened our understanding of myocardial ischemia/reperfusion injury. Myocardial stunning refers to reversible, yet slowly recovering, contractile dysfunction which follows the brief periods of myocardial ischemia. At the molecular levels, stunned myocardium following MI is not considered a cardioprotective tune-down of contractile force, but rather a core feature of post-ischemic injury. The mechanisms of myocardial stunning have been elegantly and comprehensively addressed by Bolli and Marbán [22, 23] decades ago and were carefully recapitulated by Heusch [24]. Abnormal calcium signal and excessive oxidative stress have been identified as the potential mediators of myocardial stunning [22-24]. In brief, ROS formation is enhanced as a result of injured mitochondrial function and activated NADPH oxidase [25]. Besides, myocardial ischemia/reperfusion also induces calcium overload through increased sodium-proton exchange, reverse mode sodium-calcium exchange, and defective sarcoplasmic reticulum [26]. More importantly, elevated cytoplasmic calcium load turns on NADPH oxidase to foster mitochondrial ROS generation and, in turn, ROS impairs sarcoplasmic reticulum function [27]. Finally, the interplay between calcium and oxidative stress further promotes oxidative modification and proteolysis of myofibrillar proteins [28], resulting in blunted calcium responsiveness in cardiomyocytes. Short-term myocardial hibernation is characterized by reduced regional contractile function and blood flow, which both recover after reperfusion or revascularization [29]. In fact, myocardial hibernation is involved in the activation of adaptive responses to sustain myocardial viability with reduced blood flow. Unlike myocardial stunning, the precise molecular basis underneath myocardial hibernation remains unclear. Abnormal calcium homeostasis [30] and defective endogenous nitric oxide [31] are perceived possible culprit factors for the occurrence of myocardial hibernation.
Altogether, MI involves irreversible and/or reversible ischemic damage and reperfusion injury. Although cardiomyocytes are deemed the primary targets for cardioprotection, damage in coronary microvascular ECs also exacerbates MI. However, molecular machineries underneath coronary microvascular damage are not well understood and require in-depth scrutiny given its prevalent role in the pathophysiology of MI.
Oxidative stress in ECs
The basic function of cardiac coronary circulation is to optimize the delivery of oxygen and nutrients to cardiomyocytes and regulate contractile function in particular relaxation. Oxygen consumption by ECs is significantly lower when compared to that of cardiomyocytes. Therefore, greater percentage of oxygen in the blood is available to perivascular cardiomyocytes. Reactive oxygen species (ROS) are by-products of metabolism which function as secondary messengers to transmit physiological signals [32, 33]. However, excessive ROS production compromises intracellular components such as phospholipids, proteins, and DNA. MI is accompanied by an imbalance between ROS production and detoxification by antioxidant enzymes. ECs are sensitive to hypoxia, especially evident during hypoxic coronary vasoconstriction. Mitochondrial complexes I and III are primary sources of ROS generation under pathological conditions. Furthermore, nicotinamide adenine dinucleotide phosphate oxidases (NOXs) are major enzymes to generate mitochondrial ROS in ECs. ECs express four different NOX isoforms-superoxide-generating NOX1, NOX2, and NOX5 as well as hydrogen peroxide-generating NOX4 [34]. The main downstream targets of NOX-derived ROS in the ECs include NF-κB, activated protein-1 (AP1), hypoxia-inducible factor-1 (HIF-1), p53, p38, c-Jun N-terminal kinase, and Src kinases [35]. NOX-derived ROS promotes coronary microvascular damage, which then causes aberrant vasoconstriction, apoptosis, inflammation, fibrosis, hypertrophy, and aging [36]. The upregulation of NOX enzymes during hypoxia increases ROS levels in the ECs and promotes angiotensin II type -1 (AT-1) receptor-mediated vasoconstriction [37]. Although hypoxia-related coronary contraction is an adaptive response to accelerate blood flow under normal conditions, narrowed coronary artery such as in MI fails to deliver sufficient oxygen and glucose to cardiomyocytes. Moreover, angiotensin II stimulates endothelin-1 (ET-1), which induces cardiac hypertrophy and fibrosis [38]. This finding validates a crosstalk between oxidative stress-related changes in the ECs and cardiac hypertrophy.
Coronary artery spasms are also provoked by blunted endothelium-dependent vasorelaxation, predominantly mediated by NO. The endothelium-derived NO diffuses across cellular membranes, entering into vascular smooth muscle cells (VSMCs), and increases cyclic guanosine monophosphate (cGMP) levels through soluble guanylate cyclase. Increased cGMP levels help to decrease intracellular calcium levels, en route to vasodilation. NOX-derived ROS oxidize NO to peroxynitrite, a highly reactive and vasoconstrictive ROS species to reduce the bioavailability of NO. Besides, upregulation of NOX-mediated redox signaling also disrupts endothelial barrier function [39]. This suggests a link between oxidative stress and endothelial barrier integrity.
Dyslipidemia, a condition with high circulating levels of low-density lipoprotein (LDL) and low levels of high-density lipoprotein (HDL), has been considered an underlying cause of coronary artery disease or MI. Interestingly, high LDL levels upregulate NOX activity, which promotes ROS accumulation and senescence of endothelial progenitor cells [40].
Inflammation in ECs
Coronary microvascular ECs are both active participants and regulators of inflammation in MI. ECs possess an anti-inflammatory role in the cardiovascular system under normal physiological conditions to repel neutrophils in circulation. However, in response to cardiac tissue injury, especially myocardial ischemia, ECs upregulate leukocyte-adhesion molecules and attract neutrophils to remove necrotic tissue in infarcted zones [41]. In a canine model of acute myocardial ischemia injury, neutrophil accumulation occurs within 3-6 h upon activation of ECs, and peaks at around 48 h after coronary artery occlusion [42]. Following reflow of fresh blood into the ischemic myocardium, rapid accumulation and initiation of neutrophil infiltration occurs within 3 minutes and peaks within 2 to 3 h [43]. Neutrophil adhesion triggers the release of ROS and a positive feedback loop to further activate host inflammatory response during their transmigration into the endothelium [44]. In non-inflamed cardiac tissues, ECs do not express adhesion molecules or thrombogenic factors on their surface. However, once damaged, ECs release vascular endothelial growth factor (VEGF) and stromal-derived factor 1 (SDF-1), which recruit endothelial progenitor cells (EPCs) to repair and regenerate blood vessels by promoting proliferation, differentiation, and angiogenesis of ECs [45]. Damaged ECs also upregulate several adhesion molecules such as intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion molecule 1 (VCAM1), which attract pro-inflammatory cells such as neutrophils and macrophages [46]. These immune cells secrete pro-inflammatory cytokines such as interleukin 6 (IL-6), tumor necrosis factor α (TNFα), and monocyte chemo-attractant protein 1 (MCP1), which induce expression of cyclo-oxygenases (COXs) in various tissues and cells, including ECs [47]. Elevated levels of pro-inflammatory factors, especially COX, are tightly associated with vasoconstriction. Shreeniwas and coworkers reported that hypoxia enhances adherence of leukocytes to the cultured monolayers of human ECs [48]. In addition, ample pro-inflammatory factors such as IL-6, TNFα, and MCP1 directly and indirectly trigger endothelial dysfunction and microvascular damage. These pro-inflammatory factors prompt synthesis of prostaglandins (PGs) including prostaglandin E2 (PGE2) [49]. Elevated PGE2 levels and activity promotes platelet activation and adhesion to the damaged ECs, thereby further limiting blood flow in the coronary microcirculation [50].
Higher P-selectin expression in activated platelets facilitates adhesion of circulating monocytes to the walls of blood vessels [51]. The damaged ECs express CD40, which interacts with CD40 ligand (CD40L) and promotes secretion of von Willebrand factor (vWF) from the endothelial Weibeo-Palade bodies [52]. Moreover, interaction between endothelium and platelets through CD40 and CD40L triggers activation of macrophages and T-cells on the vessel wall and accelerates the inflammatory response [53].
Elevated ROS levels in the ECs from MI patients significantly increased levels of oxidized low-density lipoprotein (oxLDL). Circulating ox-LDL is primarily taken up by macrophages, which then migrate through damaged endothelium into sub-endothelial connective tissues. Next, macrophages differentiate into foam cells, which are wrapped by smooth muscle cells to trigger atherosclerotic plaque formation [54]. In this context, EC dysfunction and EC-mediated ox-LDL generation are both involved in macrophage activation and atherosclerotic plaque formation. Tian and associates reported that endothelium-specific anti-inflammatory therapies such as nicorandil or shexiang tongxin dropping pills enhanced myocardial blood flow and cardiac function in patients with coronary microvascular diseases [55]. Furthermore, drugs that reduce endothelial inflammation attenuate MI-related cardiac damage [56]. These results highlight that inflammation promotes EC damage during MI.
Coagulation
Under physiological conditions, the glycocalyx layer made up of proteoglycans and glycoproteins acts as an anti-coagulation barrier between ECs and blood. The glycocalyx regulates permeability of the ECs and inhibits platelets from adhering to ECs given their negative charge (Figure 3). Moreover, prostacyclins (a product of arachidonic acid metabolism in endothelial cells) with vasodilating property, inhibit platelet aggregation by elevating cAMP levels in the platelets. Furthermore, NO produced by ECs promotes increased synthesis of GMP and reduces intracellular calcium levels, GPIIb/IIIa platelet receptor transformation, and binding of integrin to fibrinogen in the platelets, thereby inhibiting their activation. Tissue factor pathway inhibitors (TFPI) located on the surface of ECs suppress the initiation of coagulation through interrupting the generation of factor VIIa-tissue-factor complexes. ECs also express thrombomodulin and specific proteoglycans to activate protein C and thus inhibit the coagulation cascade by limiting proteolysis of factors VIIIa (FVIIIa) and Va (FVa). Once thrombin is formed, ECs release tissue plasminogen activator (tPA) to break down blood clots and restore blood flow.
The coagulation process in endothelial cells.
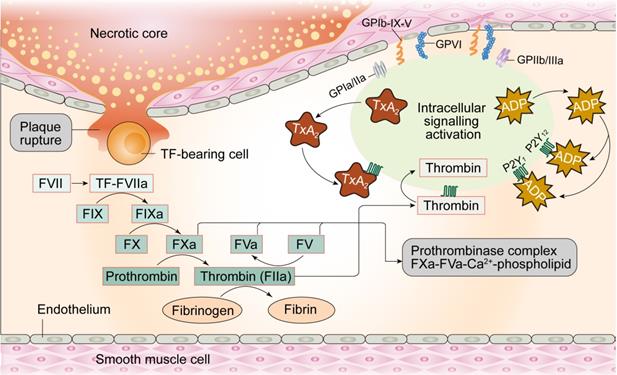
Considering the role of intact endothelium acts as a natural barrier against thrombogenesis, damaged ECs stimulate thrombus formation. Physical loss of endothelium exposes the sub-endothelial extracellular matrix and promotes adhesion of platelets, release of tissue factors, and local depletion of PGI2 and plasminogen activators. Thrombosis initiation can occur courtesy of perturbations in the dynamic balance between pro- and anti-thrombotic activities in the endothelium, independent of denuded or physically disrupted endothelium [57]. Under pathological conditions, dysfunctional ECs upregulate pro-coagulant factors such as platelet adhesion molecules, tissue factors, plasminogen activator inhibitor-1 and downregulate anti-coagulant factors such as thrombomodulin, PGI2, and t-PA [58], creating a pro-thrombotic microenvironment. For example, damaged ECs produce TXA2, which turns on platelet activation, adhesion and aggregation onto the vascular walls [59]. Activated ECs express ADAM-15, a transmembrane cell-surface protein and member of the ADAM (a disintegrin and metalloproteinase) family, prior to its binding and activation of platelets through the GIIb/IIIa receptor in the face of MI [60]. Taken together, endothelium damage-mediated platelet activation followed by coronary artery thrombosis are the major perceived pathogenic mechanisms underscoring MI.
Several therapies have surfaced in MI targeting the complex interactions between platelets and ECs. The primary therapies to prevent thrombosis and reduce the risk of acute MI include classical anti-thrombotic agents such as aspirin and clopidogrel as well as novel more potent drugs such as prasugrel and ticagrelor [61]. Aspirin suppresses platelet activation by inactivating cyclooxygenase-1 (COX-1), thus retarding formation of thromboxane A2 (TXA2), a potent platelet agonist and vasoconstrictor. In addition, oxidative stress and inflammation in ECs could be attenuated by aspirin [62]. Supplementation of aspirin is also capable of stimulating the endothelial NO-GMP cascade to favor microvascular relaxation [63]. The integrity of endothelial gap junctions could be sustained by aspirin, an effect that is followed by an inhibition of inflammatory cells infiltration [64].
Similar to aspirin, ticagrelor is a novel therapeutic reagent with proven clinical benefits compared with the classical anti-thrombotic medications [65]. Ticagrelor interrupts adenosine reuptake in erythrocytes and improves cardiac blood flow in a canine model of coronary blood flow [66]. Ticagrelor also promotes ATP release from red blood cells and enhances discharge of EC-related vasodilatory factors such as prostacyclin and NO into microcirculation [67]. Furthermore, supplementation of ticagrelor improves brachial artery vascular tone in acute coronary syndrome (ACS) patients by enhancing nitroglycerin-mediated dilation [68]. Bonello et al. reported that ticagrelor treatment increased the numbers of circulating EPCs in patients with MI [69]. These results convincingly support that drug-induced endothelial protection may prevent thrombosis and is a plausible therapeutic approach for MI patients.
The circulating endothelial compartment
During MI, damaged ECs shed from the micro-vessel wall and enter the circulation. MI patients show ~4.5 fold higher levels of circulating ECs (CECs) compared with healthy individuals [70]. Furthermore, detachment of ECs release endothelial-derived microvesicles or microparticles (EMPs) [71]. Thus, CECs and EMPs are deemed potential biomarkers of acute/chronic endothelial dysfunction in cardiovascular diseases, especially MI. EMPs derived from remodeling of the plasma membrane and externalization of phosphatidylserine are shed as submicrometric vesicles from stressed or damaged ECs [72]. Moreover, apoptotic and/or necrotic CECs promote microvascular inflammation response by interacting with the healthy endothelium. EMPs are a potential source of thrombus formation due to pro-coagulation factors within EMPs such as phosphatidylserine and tissue factor (TF) [73].
Compared with the control group, total numbers of CECs are significantly elevated in patients with unstable angina in the absence of necrotic cardiomyocytes (as indicated by troponin or creatine kinase MB-fraction assays) [74]. Furthermore, when compared to healthy volunteers, MI patients with occlusion of left anterior descending artery (LAD) express significantly higher levels of CD31+/CD42- EMPs comparable to troponin T levels, although such effects did not correlate with the estimated CK-MB values [75]. Numbers of CD31+/CD42- EMPs correlate well with ischemic area at risk (MaR), but not infarct size (IS) in patients with LAD infarctions, as evaluated using magnetic resonance imaging [75]. Increased numbers of CECs are associated with a higher prevalence of adverse cardiac events and cardiac remodeling in MI patients [76]. In MI patients receiving anti-thrombotic therapies such as aspirin and clopidogrel, reduced levels of CECs correlate with improved left ventricular function [77]. However, increased numbers of CECs are not associated with classical markers for myocardial necrosis such as creatine kinase or troponin in MI patients [77]. Therefore, increased numbers of CECs should not be used as a marker for cardiomyocyte damage, but rather represent an early sign of active peri-plaque rupture that eventually leads to acute athero-thrombotic occlusion of the entire afflicted vessel and ultimate MI.
Pericytes
Pericytes are vascular mural cells embedded within the microvascular basement membrane, enwrapping capillaries, venules and terminal arterioles [78]. Coronary ECs maturation and stability are under the control of pericytes, suggesting a role of pericytes in regulating coronary microvascular function [79]. Deficiency in pericytes results in perturbed vascular development and the formation of microaneurysms in heart [80]. During MI, a variety of evidence indicates that dysregulated cardiac pericytes may lead to adverse effects via interference with myocardial blood flow. Abnormal pericytes contraction causes stenosis of cardiac valves and no-reflow in capillaries [81, 82], resulting in decreased coronary blood flow, which may be reversed by administration of the pericyte relaxant adenosine [81]. In addition to vasoconstriction, pericytes is also involved in the post-infarction inflammatory response through distinct mechanisms including 1) suppression of inflammation response through releasing the inflammation inhibitory factors such as leukemia inhibitory factor (LIF), cyclooxygenase-2 (COX-2), and heme oxygenase-1 (HMOX-1) [83]; 2) attenuation of leukocytes recruitment and transendothelial trafficking through release of chemokines [84]; 3) inhibition of the secretion of ECM and proliferation of fibrotic cells through production of matrix metalloproteinases [85]; and 4) interference with inflammatory activities of ECs [86].
Given their roles in angiogenesis, coronary microcirculation regulation, and anti-inflammation property, pericytes are also perceived as a promising avenue to foster the repair of microvasculature in infarcted heart. Intramyocardial administration of pericytes is capable of delaying or reversing unfavorable remodeling of infarcted heart through promotion of angiogenesis and inhibition of ECs apoptosis [87]. In line with this finding, Katare and colleagues further noted that engraftment of saphenous vein-derived pericytes into infarcted heart drastically improved systolic dysfunction through augmentation of the secretion of angiogenic factors such as VEGF and angiopoietin-1 [88]. In addition to the soluble cytokines, microRNAs such as miR-132, derived from pericytes after transplantation into the infarcted heart, also restored cardiac function and increased the density of capillary [88]. Recently, it was reported that the crosstalk between pericytes and ECs may serve as a necessary bridge for early vasculogenesis. Pericytes are involved in the ECs recruitment, differentiation, maturation and vessel stability through various mechanism including PDGF-B/PDGFRB2, S1P/EDG-1, ANG1/2/TIE2, Cadherin, and Notch signals [89, 90]. Nonetheless, further studies are warranted to further elucidate the complex impact of pericyte-endothelial cross-talk in the regulation of coronary microvascular damage and repair following MI.
Role of MQC in coronary microvascular ECs injury during MI
Mitochondrial dynamics
Mitochondria are highly dynamic cellular organelles that continuously undergo fusion (two individual mitochondria join together to become one) and fission (one mitochondrion divides into two) (Figure 4). In ECs, mitochondria exist as filamentous networks under normal physiological conditions and are fragmented during hypoxia [91].
A mitochondrial-centric view of coronary endothelial homeostasis. Ischemia/hypoxia promotes enhanced phosphorylation of Drp1. Subsequently, phosphorylated Drp1 (p-Drp1) oligomerizes around the mitochondrial outer membrane after being recruited and stabilized by mitochondrial receptors such as Fis1 and Mff and induces mitochondrial fission. In general, enhanced p-Drp1-mediated mitochondrial fission is accompanied by increased mitophagy and mitochondrial biogenesis. Fragmented mitochondria with low membrane potential induce PINK1 localization into the mitochondria. PINK1 recruits Parkin to the mitochondria. Concurrently, Fundc1, the mitochondria-localized mitophagy receptor, is activated through post-transcriptional phosphorylation. Mitochondria interact with LC3 on the lysosomes through Parkin and Fundc1, and form autophagosomes. Mitophagy promotes degradation of fragmented mitochondria to sustain mitochondrial homeostasis. Mitochondrial biogenesis is activated in response to mitochondrial fission or mitophagy. PGC1α is a major regulator of mitochondrial biogenesis that acts as a compensatory mechanism to mitochondrial fission or mitophagy. The other transcription factors involved in mitochondrial biogenesis are NRF1, NRF2, ERR, and PPAR. Mitochondrial biogenesis upregulates OXPHOS, TCA cycle, mtDNA and TFAM levels. Note: Drp1, dynamin-related protein 1; Fis1, mitochondrial fission factor 1; Mff, mitochondrial fission factor; NRF, nuclear respiratory factor; OXPHOS, oxidative phosphorylation; PGC1α, peroxisome proliferator-activated receptor g coactivator 1α; PINK1, phosphatase and tensin homolog-induced putative kinase 1; ROS, reactive oxygen species; TCA, tricarboxylic acid; TFAM, mitochondrial transcription factor A.
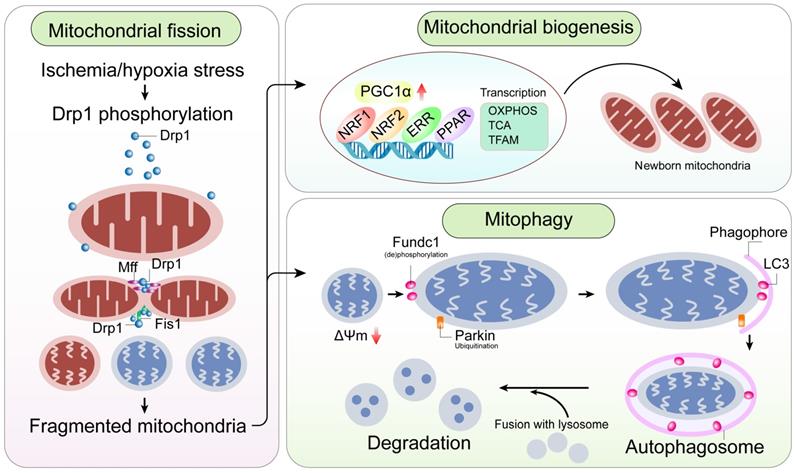
Mitochondrial fission is a multi-step process that involves constriction and scission of the mitochondrial inner membrane through a process mediated by dynamin-related protein 1 (Drp1) and its binding partners, including fission protein 1 (Fis1), mitochondrial fission factor (Mff), mitochondrial dynamics protein of 49 kDa (MiD49), and mitochondrial dynamics protein of 51 kDa (MiD51) (Table 1). Drp1 is a cytoplasmic protein lacking a membrane-anchoring domain. It assembles and oligomerizes into rings which wrap around mitochondria, in concert with its binding partners promotes mitochondrial fission. During normal physiological conditions, mitochondrial fission coordinates proliferation of ECs so that adequate number of mitochondria are received by daughter cells. However, pathological mitochondrial fission may turn on unfavorable mitochondrial apoptotic pathway in the ECs.
Post-translational modifications of mammalian mitochondrial dynamics-related proteins
| Protein substrate | Regulators | Full name | Post-transcriptional modification | Effects of the modifications | References |
|---|---|---|---|---|---|
| Drp1 | CDK1 | Cyclin-dependent kinase 1 | Phosphorylation of Ser 616 | Activation | [184] |
| PKA | Protein kinase A | Phosphorylation of Ser637 | Inactivation | [185] | |
| DNA-PKcs | DNA-dependent protein kinase, catalytic subunit | Phosphorylation of Ser616 | Activation | [186] | |
| O-GlcNAc transferase or OGT | uridine diphospho-N-acetylglucosamine:polypeptide β-N-acetylglucosaminyltransferase | O-GlcNAcylation of Drp1 at Thr585/586 | Activation | [187] | |
| MAPL | Mitochondria-associated protein ligase | SUMOylation of Drp1 | Activation | [188] | |
| SENP5 | SUMO-specific Peptidase 5 | De-SUMOylation of Drp1 | Inactivation/stabilization | [189] | |
| March5 | Membrane associated RING finger protein 5 | Ubiquitination of Drp1 | Degradation | [190] | |
| Mfn1 | ERK | Extracellular-signal-regulated kinase 1/2 | Phosphorylation of T562 | Inactivation | [191] |
| March5 | Membrane associated RING finger protein 5 | Ubiquitin of Mfn1 | Degradation | [192] | |
| Mfn2 | PINK1 | PTEN-induced kinase 1 | Phosphorylation of Thr111 and Ser442 | Signaling | [193] |
| March5 | Membrane associated RING finger protein 5 | Ubiquitin of Mfn2 | Activation | [194] | |
| Mfn1/2 | USP30 | Ubiquitin specific peptidase 30 | Deubiquitin of Mfn1/2 | Inactivation | [195] |
| Mff | NR4A1 | Nuclear receptor subfamily 4 group A member 1 | Phosphorylation of Ser146 | Activation | [196] |
Mitochondrial fusion is controlled by mitofusin proteins (Mfn1/2) and optic atrophy 1 (OPA1), which are located on the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM), respectively (Table 1). Mfn1 and Mfn2 form homodimers or heterodimers to induce OMM fusion, whereas, integration of IMM is controlled by OPA1. Fusion of two mitochondria would promote the mixture of mitochondrial contents including mitochondrial DNA, metabolites, and mitochondria-resident proteins, and is deemed a necessary step to remodel structural organization and restore functional capacity of mitochondria for damaged DNA, membranes, or proteins.
Mitochondrial fission
Endothelial cell senescence and dysfunction in high glucose conditions is associated with increased mitochondrial fission [92]. Mouse coronary ECs from diabetic mice exhibit elevated ROS levels, fragmented and shorter mitochondria, and increased cell death when grown in high glucose (hyperglycemic) conditions [92]. In a mouse model of type 1 diabetes, mitochondria show fragmented morphology in the coronary ECs because of increased expression of Drp1 and decreased levels of OPA1[93]. Shenouda and colleagues noted increased mitochondrial fragmentation, mitochondrial ROS levels, and Fis1 expression in the venous ECs isolated from diabetic patients compared to those from healthy subjects [94]. Furthermore, siRNA-mediated knockdown of Fis1 or Drp1 inhibited mitochondrial fragmentation and impaired production of NO in the ECs [94]. This suggested that mitochondrial fission played a central role in EC dysfunction. Diabetes is an independent risk factor for the onset and development of atherosclerosis and MI. Anti-diabetic drugs such as metformin alleviate diabetes-mediated atherosclerosis through inhibition of Drp1-dependent mitochondrial fission, subsequently improving EC function and reducing vascular inflammation [95]. Moreover, human endothelial cells (HUVEC) treated with a single dose of hydrogen peroxide to induce oxidative stress demonstrate transient changes in mitochondrial morphology and mtDNA damage [96].
In post-ischemic or reperfused hearts, increased Mff protein expression and phosphorylation induces mitochondrial fission [97-99]. Following ischemia/reperfusion, levels of dual-specificity protein phosphatase 1 (DUSP1) are significantly downregulated in coronary ECs, thus activating JNK, prior to translocation to the nucleus to promote Mff transcription [99]. During post-ischemic reperfusion, nuclear receptor subfamily 4 group A member 1 (NR4A1) is upregulated and the serine/threonine kinase, casein kinase2 α (CK2α), is activated in coronary ECs [98]. Subsequently, CK2α phosphorylates Mff at Ser-146 to promote its affinity to Drp1. Transcriptional modification and post-transcriptional phosphorylation of Mff enhances Drp1 recruitment onto the surface of mitochondria, leading to excessive mitochondrial fission in coronary ECs during myocardial ischemia/reperfusion injury. In a model of renal ischemia/reperfusion injury, RNA-binding protein Pumilio2 (Pum2) was significantly downregulated in renal tubular cells, thus enhancing the stability and translation of Mff mRNAs [100]. However, this novel regulatory mechanism of Mff remains to be examined in coronary ECs.
Mechanical forces also participate in the regulation of mitochondrial dynamics. Breton-Romero and associates demonstrated that alteration in shear stress, a known inducer of atherosclerotic plaque formation or rupture, may promote Drp1 translocation from the cytosol to the mitochondrial membranes to favor mitochondrial fission in coronary ECs [101]. This finding suggests that mitochondria respond to signals transduced by changes in fluidic mechanical forces of shear stress in the vascular bed through induction of mitochondrial fission. Along the same line, Chen et al noted that mitochondrial fission inhibitor, mdivi-1, suppressed endothelin-1-induced vasoconstriction [102].
Microvascular outgrowth by enhancing myocardial blood perfusion is a protective mechanism against MI. However, angiogenic property of coronary ECs is blunted by myocardial ischemia/reperfusion through complex mechanisms. Treatment with mdivi-1 (an inhibitor of mitochondrial fission) prior to myocardial ischemia/reperfusion challenge ameliorates cardiac anomaly and promotes angiogenesis by upregulating the gap junction protein connexin 43, chemotactic factor CXCL10, and endothelial cell-specific receptor tyrosine kinase [103]. Reminiscent to Mdivi-1, calpain inhibition is able to prevent hyperglycaemia-mediated mitochondrial fission while restoring mitophagy to prevent simulated ischemia-reperfusion injury [104]. Increased mitochondrial fission correlates with mtDNA damage and reduced proliferative capacity of the ECs, which may be related to impaired MI-related angiogenesis [96]. These findings suggest that inhibition of mitochondrial fission may serve as a potential therapeutic avenue for patients with chronic MI through augmentation of angiogenesis.
Besides, it was reported that other mitochondrial functions, such as respiration, ATP formation, and control of calcium kinetics may also be affected by myocardial ischemia/reperfusion injury prior to mitochondrial fission induction [105], suggesting possible mitochondrial function adaptions independent of MQC in myocardial ischemia/reperfusion injury. Therefore, it is necessary to point out that assessment of mitochondrial dysfunction and MQC in MI is rather complex due to the existence of various cross talk machineries.
Mitochondrial fusion
Compared with mitochondrial fission, mitochondrial fusion is not well studied in dysfunctional coronary ECs during MI. Makino et al reported that downregulation of OPA1 expression promoted mitochondrial fragmentation in coronary ECs isolated from diabetic mice [93]. Furthermore, high-fat diet (HFD) significantly induced atherosclerotic phenotype in apoE-deficient mice and downregulated Mfn2 and OPA1 [106]. Conversely, a fish oil enriched diet retarded the progression of atherosclerotic lesions through upregulation of Mfn2 and OPA1 and endothelium-dependent vasorelaxation response to acetylcholine [106]. These observations suggested that mitochondrial fusion prevented formation of coronary atherosclerotic lesions.
Mfn1/2 regulate mitochondrial fusion and VEGF-mediated angiogenic function in coronary ECs [107]. Jesse and team reported that VEGF overtly upregulated Mfn2 and Mfn1 levels in cultured ECs [107]. Mfn1/2 silencing increased mitochondrial fragmentation and disrupted VEGF-mediated migration and differentiation of ECs in vitro [107]. It was also reported that reduced mitochondrial fusion contributes to oxidative stress and apoptosis in ECs [108].
Downregulation of mitochondrial fusion proteins under stress has been reported in a number of clinical studies. For example, Diaz-Morales et al reported that mitochondrial fusion-related proteins such as Mfn1/2 and OPA1 were significantly downregulated in circulating ECs or EPCs isolated from diabetic patients compared to healthy volunteers [109]. Furthermore, reduced mitochondrial fusion and increased mitochondrial fission in ECs of diabetic patients correlated with elevated leukocyte-EC interactions, indicating higher degree of vascular inflammation [109]. These findings would suggest that extracellular vascular inflammation is regulated by mitochondrial fusion status in leukocytes and other cell types.
Mitophagy
Mechanism of mitophagy
Mitochondrial fission and fusion are self-regulatory mechanisms in response to stress. However, when damaged mitochondria cannot be repaired by fission or fusion, mitochondrial autophagy (mitophagy) is activated to remove damaged mitochondria in conjunction with autophagolysosomes (Figure 4). At the molecular level, damaged mitochondria are marked by several “adaptors” that are recognized and ingested by lysosomes and hydrolyzed into amino acids, glucose, fatty acids, and nucleotides, which are recycled to synthesize new mitochondrial and other cellular components. Thus, mitophagy requires specific and well-orchestrated communication between mitochondria and lysosomes, and serves as a compensatory mechanism to maintain the quality and quantity of mitochondrial network in cells.
Parkin and FUN14 domain containing 1 (Fundc1) are two specific mitophagic adaptors (Table 2). Parkin induces mitochondrial receptor-independent mitophagy, whereas, Fundc1 mediates mitochondrial receptor-dependent mitophagy [110]. Collapse in mitochondrial membrane potential and activity of presenilin-associated rhomboid-like (PARL) proteases in damaged mitochondria promotes accumulation of PTEN-induced kinase 1 (PINK1) on the outer mitochondrial membrane (OMM) and recruitment of cytoplasmic Parkin onto mitochondria via PINK1. Then, Parkin polyubiquitinates several OMM proteins such as voltage dependent-anion-selective channel 1 (VDAC1) and Mfn1/2; the polyubiquitinated proteins are recognized by p62, which interacts with light chain 3 (LC3) to form autophagosomes. Fundc1 is an OMM protein that interacts with LC3 when dephosphorylated and induces mitophagy [111].
Mitophagy adaptors and their regulatory mechanisms in endothelial cells
| Mitophagy Adaptors | Full name | Regulator | Signal | Effects | References |
|---|---|---|---|---|---|
| PINK1/Parkin | Phosphatase and tensin homologue (PTEN)-induced putative kinase 1/Parkin | Sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) | Transcriptional upregulation | Overexpression of SERCA improves endothelial viability, barrier integrity, and cytoskeleton assembly in cardiac microvascular endothelial cells under cardiac ischemia-reperfusion injury by activating Parkin-related mitophagy. | [197] |
| Ca2+/calmodulin-dependent protein kinase II (CaMKII) | Post-transcriptional phosphorylated activation | CaMKII induces loss of mitochondrial potential and energy disorder in oxidized low-density lipoprotein (ox-LDL)-treated aortic endothelial cells through activation of Parkin-mediated mitophagy. | [198] | ||
| Uncoupling protein2 (Ucp2) | Transcriptional downregulation | Loss of endothelial Ucp2 leads to excessive PINK1-induced mitophagy, inadequate mitochondrial biosynthesis, and increased apoptosis in endothelium. | [199] | ||
| Regulator of calcineurin 1-1L (Rcan1-1L) | Transcriptional upregulation | Rcan1-1L overexpression upregulates Parkin-related mitophagy and reverses growth inhibition in hypoxia-treated endothelial cells. | [117] | ||
| Bnip3 | BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 | 5' AMP-activated protein kinase (AMPK) | Transcriptional upregulation | Inhibition of AMPK attenuates the activity of mitochondrial respiration complexes I and III in endothelial cells under oxidized low-density lipoprotein treatment by blunting Bnip3-related mitophagy. | [113] |
| Fundc1 | FUN14 domain-containing 1 | Nuclear receptor subfamily 4 group A member 1 (NR4A1) | Post-transcriptional phosphorylated inactivation | NR4A1 induces inflammation and apoptosis of cardiac microvascular endothelial cells by inhibiting Fundc1-dependent mitophagy in a mouse model of cardiac ischemia-reperfusion injury. | [196] |
Mitophagy and EC protection
A vital role of mitophagy is widely reported in endothelial protection. Mitochondrial energy metabolism and endothelial function are maintained by mitophagy through a mechanism involving activation of the Sirt1/FOXO3 signaling cascade [112]. Oxidized LDL promotes EC injury, characterized by decreased mobilization, impaired vessel sprouting and blunted tube formation, although these effects are partially reversed by mitophagy induction [113]. Mitophagy was shown to retard EC senescence in the settings of hyperglycemia or hyperlipidemia by way of reduced mROS production [114]. Inhibition of mitophagy by Parkin silencing overtly lowered mitochondrial membrane potential, induced apoptosis and promoted mitochondrial fragmentation in ECs [115]. This finding favors a possible compensatory role for mitophagy in the face of deranged mitochondrial dynamics and mitochondrial fragmentation.
Angiogenesis is vital for cardiomyocyte perfusion and metabolism in the setting of MI. Mitophagy induction correlates with elevated levels of CD31 and VEGF [98]. This notion suggests that angiogenesis is positively regulated by mitophagy. Meanwhile, mitophagy protects against mitochondria damage-induced apoptosis of ECs [98]. Moreover, mitophagy induction promotes improved vascular relaxation, although the underlying molecular basis remains elusive. Another cardinal feature in MI-associated cardiac remodeling is interstitial fibrosis, which involves vascular matrix remodeling and inflammation. Mitophagy prevents activation of matrix metalloproteinases (MMPs) within the mitochondria and cytoplasm, thereby reducing MMP levels in the extracellular matrix [116]. ECs from infarcted cardiac tissues exhibited significantly higher surface levels of adhesive factors such as ICAM1 or VCAM1, the effect of which was reversed by induction of mitophagy [98]. In MI, coronary ECs present increased expression of pro-inflammatory factors such as IL-8, MMP9, and MCP1, the effects of which were also reversed by mitophagy [98]. To this end, it may be concluded an anti-inflammatory role for mitophagy in coronary ECs during MI.
In MI, mitophagy is induced upon exposure to hypoxia and acts as a hypoxic pre-conditioning mechanism. After exposure to hypoxia, mitochondrial membrane potential is reduced and Parkin translocates to the mitochondria in ECs to trigger induction of mitophagy [117]. However, reoxygenation following hypoxia suppresses mitophagy, which causes accumulation of damaged mitochondria that mediate mitochondrial apoptosis in coronary ECs [98, 118] and cardiomyocytes [119]. In sum, it is generally accepted that ischemia triggers mitophagy to attenuate mitochondrial damage, whereas, post-ischemic reperfusion inhibits mitophagy. However, regardless of ischemia or reperfusion, suppression of mitophagy induces mitochondrial dysfunction and cell death. This suggests that baseline mitophagy is indispensable for mitochondrial function and cell viability.
Mitochondrial biogenesis
Mild mitochondrial stress is rectified by mitochondrial fission or fusion, whereas severely damaged mitochondria are removed by mitophagy, resulting in significant reduction in mitochondrial mass (Figure 4). Therefore, de novo mitochondrial biogenesis is required to maintain adequate mitochondrial mass and function. Mitochondrial biogenesis involves transcription and assembly of over 1100 proteins encoded by both nuclear and mitochondrial DNA in addition to synthesis of phospholipids to generate the inner and outer mitochondrial membranes. Peroxisome proliferator activated receptor γ co-activator (PGC1α) and nuclear respiratory factor 2 (NRF2) act as transcriptional factors of most mitochondrial proteins and are considered as master regulators of mitochondrial biogenesis. Mechanistically, PGC1α activates NRF2 and promotes synthesis of nuclear-encoded mitochondrial-resident proteins including mitochondrial transcription factor A (TFAM). Level of PGC1α is tightly regulated by physiological cues including exercise, cold temperature, and fasting [120]. PGC1α-mediated mitochondrial biogenesis has been widely described in endothelial protection. Global Pgc1α knockout in mice displayed smaller arteries with deformed vascular plexus, low perfusion feature and defective adherent junctions between ECs due to lower levels of membrane-bound VE-cadherin [121]. Moreover, Pgc1α knockout ECs exhibited a pre-matured aging phenotype with elevated ROS levels [121]. In Pgc1α-knockout mice, endothelium-dependent vasodilation was suppressed due to reduced NO and ATP levels as well as delayed mitochondrial turnover rate [122]. Compared with wild-type mice, Pgc1α-knockout mice are more likely to develop atherosclerosis due to impaired lipid metabolism and defective mitochondrial biogenesis [123].
Under physiological conditions, PGC1α enhances resistance to oxidative stress by inducing hemeoxygenase-1 in ECs [124]. Exercise-induced angiogenesis is attributed to higher PGC1α expression, which enhances mitochondrial function and promotes paracrine function of VEGF including increased proliferative capacity of ECs [125]. PGC1α also promotes viability of ECs and vascular permeability [126]. Decreased PGC1α is associated with increased levels of pro-inflammatory factors including TNFα, NF-κB, MCP1 and VCAM1 in ECs [127]. Conversely, adenovirus-mediated overexpression of PGC1α reduces ROS production and suppression of NF-κB, MCP1 and VCAM1 triggered by TNFα [127]. These findings strongly favor an anti-inflammatory role for PGC1α in vascular homeostasis.
PGC1α is a repressor for oxidative stress, hyperglycemia, hyperlipidemia, and inflammation, all of which are risk factors for MI. In diabetic rats, level of PGC1α is significantly reduced in association with low capillary volume [128]. Low-intensity running was shown to enhance capillary volume and pro-angiogenic factors by augmenting PGC1α levels, thereby combating microcirculatory complications in type 2 diabetes [128]. PGC1α-related mitochondrial biogenesis may be credited for several anti-diabetic drugs such as thiazolidinediones [129]. Supplementation of thiazolidinediones attenuated hyperglycemia-induced EC dysfunction, although such these effects were abrogated in Pgc1α-deficient ECs [129]. Vascular inflammation induced by angiotensin II was also shown to be associated with reduced PGC1α levels [130]. In comparison with healthy individuals, patients with pathological left ventricular outflow tract, circulating PGC1α levels were overtly reduced correlating with increased metabolic biomarkers for mitochondrial damage (lactate, ratio lactate/pyruvate) and EC injury (asymmetric dimethylarginine and total homocysteine) [131].
A recent study (in vivo and in vitro) showed that thrombospondin mediates Drp1 signaling after ischemia/reperfusion in aged hearts. Using RAEC, an association was shown between thrombospondin and Drp1 through PGC1a [132]. In post-ischemic heart, loss of PGC1α levels facilitated mitochondrial fragmentation and promotes cardiac dysfunction [132]. This suggests that PGC1α-related mitochondrial biogenesis can potentially alleviate pathogenesis underlying mitochondrial dynamics-related disorders. The role for ROS generation in ischemia/reperfusion was demonstrated by the finding with treatment of mitochondria-targeted antioxidants with improved PGC1α-related mitochondrial biogenesis, mitochondrial respiration, endothelium-dependent vasodilation, and blood flow in ischemic hind limbs [133]. PGC1α expression also correlates with reduced angiogenesis in infarcted hearts and pressure overload-induced cardiac hypertrophy [134]. PGC1α preserves angiogenesis but not mitochondrial oxidative phosphorylation and contractile function in mice following transverse aortic constriction [134].
Taken together, PGC1α-mediated mitochondrial biogenesis plays a key role in vascular integrity through regeneration of new mitochondria to replenish aged or damaged mitochondria in the governance of mitochondrial quality control. Nonetheless, clinical trials targeting PGC1α-related mitochondrial biogenesis have not been successful thus far in patients with MI.
Cardioprotective strategies on coronary microvascular injury during MI
Ischemic preconditioning (IPC)
IPC refers to the ability of brief episodes of ischemia to improve the resistance of myocardium to subsequent ischemic attack. Ample evidence has gained to ensure the protective of IPC in ischemic myocardium. More importantly, IPC also benefits coronary vasculature, as evidenced by preserved endothelial function and decreased ECs damage [135]. With the help of IPC, flow-mediated coronary dilator response to vasodilators such as adenosine and NO was also restored to near-normal levels [136]. Post-infarction inflammation response, including increased neutrophil adherence and accumulation [136], was also attenuated by IPC, possibly due to improved ECs integrity. The protective mechanisms of IPC have been widely documented in experimental models of myocardial ischemia/reperfusion injury. IPC contributes to activation of KATP channels, leading to hyperpolarization of ECs [137]. This alteration widens the electrochemical gradient to facilitate calcium entry, leading to augmented release of NO from ECs [138]. Moreover, IPC treatment is also tied with adenosine release and activation of adenosine receptors [139], resulting in activation of KATP channels via a G protein-dependent mechanism and thus microcirculation dilation. Besides, IPC suppresses abnormal ROS production [140] and augments the release of prostaglandin E2 [141]. Levels of endothelin-1 [142] are inhibited whereas the content of eNOS is increased [143] by IPC in coronary microvasculatures.
Ischemic postconditioning (IPost)
IPost refers to a series of brief ischemia/reperfusion cycles applied immediately to the site of ischemic organ following reperfusion, resulting in decreased MI size and improved cardiac function. Intensive effort has been initiated to evaluate the therapeutic potential of IPost for its protection of coronary vasculature against MI. Microvascular obstruction is alleviated and coronary blood flow is preserved in reperfused hearts following IPost [144]. It is necessary to point out that several studies failed to display any beneficial actions of IPost on microvascular obstruction [145-147]. In addition, one study [145] did not note any reduction in MI size with IPost whereas the others [146, 147] observed a smaller MI zone with the IPost procedure. Although these conflicting findings are difficult to be explained at this time, the mechanism for IPost in coronary microvascular protection has been widely discussed. Decreased myocardial oedema and attenuated neutrophil adherence have been observed in reperfused hearts tissue after treatment with IPost [148]. Besides, levels of endothelial P-selectin are downregulated in response to IPost [148]. The mobilization of endothelial progenitor cells is also improved by IPost [149], an endogenous repair mechanism for re-endothelization and neoangiogenesis.
Pharmacological strategies
Vast efforts have been continuously engaged over the past decades in the search for pharmacological agents in the preservation of coronary vasculature against myocardial infarction. For example, angiopoietin-like peptide 4, a hypoxia-dependent factor, plays an important role in regulating vascular permeability. Administration of angiopoietin-like peptide 4 significantly reduces MI size and attenuates microvascular obstruction through regulation of endothelial gap-junction VE-cadherin complex [150]. Excessive opening of mitochondrial permeability transition pore (mPTP) has been identified as one of the key pathological mechanisms for myocardial ischemia/reperfusion injury. Cyclosporine-A (CSA), as inhibitor of mPTP, was found to interrupt reperfusion-mediated cardiomyocyte death and therefore reduce MI size [151]. However, this beneficial effect has not been verified by large animal experiments [146] and/or large clinical studies [152] given the complexity of the molecular pore structure. Melatonin, which is originally identified as a regulator of circadian rhythm, has been found to sustain coronary microvascular permeability and augment eNOS levels in reperfused heart tissues [118, 153], resulting in decreased MI size and improved cardiac function. Administration of the sodium-glucose cotransporter 2 inhibitors (SGLT2i), a new class of anti-diabetes drugs, significantly reduce MI size and sustain left ventricular mechanoenergetics [154]. Furthermore, levels of eNOS are elevated with improved endothelium-dependent vasodilation in response to SGLT2i therapy, suggesting that cardiomyocytes and coronary ECs may serve as targets for SGLT2i in myocardial infarction [154]. Although many pre-clinical data support the cardioprotective actions of these pharmacological agents, the neutral results of clinical trials may dampen the enthusiasm of researchers. The translational issues for cardioprotective agents as adjunct to reperfusion therapy with a main focus on coronary microvasculature has been comprehensively discussed by Heusch and colleagues [11, 155-158]. A better understanding of signals and molecular mechanisms that regulate coronary ECs function following MI may contribute to identify novel preventive and therapeutic strategies aimed to reduce myocardial injury.
Pharmacological interventions targeting MQC to protect against coronary microvascular ECs damage
Mitochondrial dynamics regulation
In addition to mitochondria-targeted antioxidants, several small molecules have been developed and tested to improve MQC and alleviate EC dysfunction. Angiotensin II-treated ECs showed elongated mitochondrial morphology, stabilized mitochondrial membrane potential, increased eNOS expression, decreased apoptosis, and improved migration when pre-treated with mdivi-1, a selective inhibitor of mitochondrial fission protein Drp-1. Rat thoracic aorta and mesenteric arteries exhibited overtly higher Drp1-mediated mitochondrial fission in the realm of endothelin-1-induced vasoconstriction, the effect of which was reversed by mdivi-1 treatment. Mdivi-1 also suppressed metabolic disorder-associated EC defect by increasing NO bioavailability and improving endothelium-dependent vascular relaxation [159]. Atherosclerotic mice treated with mdivi-1 showed increased glucose and decreased fatty acid uptake, reduced mitochondrial fragmentation, and decreased expression of p-Drp1Ser616 [160]. Moreover, mdivi-1 treatment reduced mitochondrial ROS production and VCAM-1 expression in thoracic aorta from atherosclerotic mice [95]. In a mouse model of myocardial ischemia/reperfusion injury, mdivi-1 treatment reduced infarct size, improved cardiac function, and induced angiogenesis [161]. In addition, mdivi-1 treatment inhibited reperfusion-mediated reduction in connexin 43 levels, upregulation of matrix metalloproteinase (MMP3) and apoptosis of ECs in the reperfused myocardium [162]. Mdivi-1 treatment also favored NO production and viability of cultured ECs subjected to shear stress or hypoxia/reoxygenation injury [163]. EC cells treated with mdivi-1 displayed improved mitochondrial dynamics and reduced levels of von Willebrand Factor and gap junction communication [164]. This demonstrates that normalization of mitochondrial dynamics is necessary to maintain the vascular barrier and anti-coagulant properties.
In contrast to mitochondrial fission, support evidence for pharmacological approaches targeting mitochondrial fusion is relatively fewer. This is mainly due to the multi-faceted roles of Mfn1/2 in the regulation of mitochondrial fusion, mitophagy, mitochondrial calcium levels, and mitochondria-ER communication. Further investigations are necessary to hunt and identify clinically effective mitochondria fusion-targeted drugs for preservation of EC function in MI patients.
Mitochondrial biogenesis modification
Regular aerobic exercise promotes mitochondrial biogenesis and is recommended as a healthy lifestyle strategy for reducing MI-related risk factors such as obesity, dyslipidemia and diabetes mellitus. Ample evidence has denoted a critical role for MQC in MI-related ECs changes. For example, exercising rats following MI was shown to overtly promote EC proliferation, VEGF level, angiogenesis, eNOS activity, and endothelium-dependent vasodilation [165]. In addition to exercise, mitochondrial biogenesis in ECs may be stimulated by various medications including statins [166], resveratrol [167], liraglutide [168], cannabidiol [169], salidroside [170], and red wine polyphenols [171]. Nonetheless, only metformin [172] and resveratrol [173] augment mitochondrial biogenesis in ECs, improved cardiac function and decreased infarct size in MI or myocardial I/R injury models.
Mitophagy induction
Trehalose is a small molecule isolated from mushrooms and honey that activates autophagy and mitophagy in ECs [174]. For example, four weeks of oral trehalose supplementation reversed age-associated decline in NO-mediated endothelium-dependent vasodilation [174, 175]. Mechanistically, Parkin and BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (Bnip3) as well as PGC1α expression levels were significantly increased in the aortic tissues after trehalose treatment [175]. Administration of trehalose for four weeks reversed cardiac remodeling and fibrosis in the wild-type MI model mice, but did not show beneficial effects in the Beclin1-knockout MI model mice [176]. Moreover, trehalose enhanced VEGF-mediated angiogenesis in the Mycobacterium marinum-infected zebrafish [177], but, these effects have not been validated in MI-related ECs.
Another natural activator of autophagy is spermidine, isolated from wheat germ, soy beans, and mushrooms [178]. Dietary intake of spermidine is inversely associated with all-cause mortality and MI risk in humans [179]. Spermidine improved age-associated decline in aortic mitochondrial respiration and reduced inflammation and atherosclerosis through mitophagy induction in hyperlipidemic mice [180]. In addition, ECs isolated from veins of diabetic patients exhibited improved NO signaling and NO-mediated endothelium-dependent vasodilation following spermidine treatment [181]. In a rat model of MI, spermidine supplement reversed cardiac dysfunction and inhibited cardiomyocyte apoptosis through mitophagy induction [182]. However, further evidence is required to demonstrate the beneficial effects of spermidine on ECs during MI through mitophagy activation.
Concluding remarks and Perspectives
Coronary microvascular injury is an often neglected topic in the field of MI. Prognosis of cardiovascular disease is worse in women as opposed to age-matched men courtesy of microvascular dysfunction. However, current therapies mainly focused on improving the viability of cardiomyocytes and effective therapies targeting coronary microvascular injury are not yet available. In ECs, mitochondria play a vital role in integrating the intracellular signal transduction pathways that regulate EC function. MQC mechanisms including mitochondrial dynamics, mitophagy and mitochondrial biogenesis play a pivotal role in regulating mitochondrial structure and function. They are also implicated in EC dysfunction and coronary microvascular injury in the infarcted heart tissues (Figure 5). Mitochondrial fusion and fission regulate the number of healthy mitochondria through changes in mitochondrial dynamics. Mitophagy is a self-clearance mechanism that removes damaged mitochondria to maintain mitochondrial function and homeostasis. Mitochondrial biogenesis is the de novo synthesis mechanism to maintain optimal mitochondrial mass, especially following reduced mitochondrial mass due to mitophagy. These three MQC mechanisms regulate mitochondrial function in the ECs. Dysregulated MQC induces oxidative stress in ECs due to excessive accumulation of damaged mitochondria followed by activation of an inflammatory response in cardiac microvasculature. Defective ECs and inflamed vascular bed lower the threshold for vasospasms and facilitate thrombogenesis, thus limiting blood flow in the myocardium during MI. Moreover, dysfunctional ECs undergo apoptosis and fibrosis because of impaired MQC. This is associated with microvascular degeneration, chronic myocardial ischemia, and negative cardiac remodeling during MI. A number of unanswered questions remain with regards to MQC in coronary microvascular injury during MI. First, it is of interest to explore whether the location of ECs (i.e., cells located at the infarcted area versus border zone) possesses any distinct role in regulating microvascular damage during or after MI. Second, it remains unclear whether MQC could be regulated by a common upstream regulator or signaling pathway in ECs. Third, it is of interest to describe the interactive actions between MQC processes. Fourth, although several drugs have been developed or validated for the treatment of coronary microvascular injury, no single drug or therapeutic approach targeting global MQC has yet been proven effective for the protection of coronary microvascular injury.
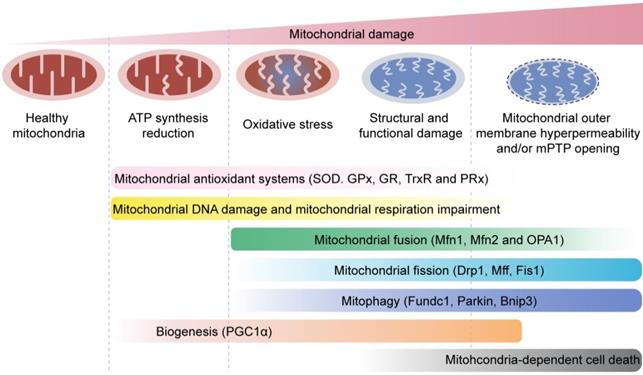
Mitochondrial quality control mechanisms. Mild mitochondrial stress is accompanied by decreased ATP production and mitochondrial oxidative stress. This stimulates mitochondrial anti-oxidative enzymes such as superoxidase dismutase (SOD), glutathione reductase (GR), glutathione peroxidases (GPXs), thioredoxin reductase (TrxR), and peroxiredoxin (PRx) to reduce the levels of mitochondrial reactive oxygen species (mROS). Mitochondrial injury is followed by mitochondrial DNA damage, which impairs transcription and translation of mitochondria-encoded electron transport chain proteins. Severe mitochondrial stress is characterized by aberrant alterations in the mitochondrial structure and function. This induces mitochondrial fusion mediated by mitofusin 1 (Mfn1), mitofusin 2 (Mfn2), and optic atrophy 1 (OPA1). Mitochondrial fusion allows mixing of damaged and healthy mitochondria to restore functional homeostasis. Meanwhile, mitochondrial fission is activated by cytosolic dynamin-1-like protein (Drp1) and its receptors, and results in segregation of damaged mitochondria that are then targeted for degradation by mitophagy through specific adaptors such as FUN14 domain-containing 1 (Fundc1), Parkin, and BCL-2/adenovirus E1B 19 kDa protein-interacting protein 3 (Bnip3). Excessive mitochondrial damage and stress promotes increased mitochondrial fission that converts reticular mitochondrial network into punctate and fragmented mitochondria. This reduces ATP production and affects cellular growth and survival. Besides, excessive mitophagy significantly reduces mitochondrial mass leading to bioenergetic crisis and cell death or apoptosis. Newer mitochondria are synthesized by mitochondrial biogenesis through specific transcription factors such as peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α). However, excessive mitochondrial damage induces cell death.
Last but not the least, this review mainly discusses and summarizes the role of coronary ECs in coronary microvascular damage in myocardial infarction. It is noteworthy to point out that coronary microcirculation, including small arteries and arterioles, is composed of vascular smooth muscle and pericytes, in addition to ECs. Although the direct contact of ECs with blood flow means that they are particularly vulnerable to the damaging molecules in the blood, possible influence from dysregulated vascular smooth muscle cells in coronary microvascular homeostasis cannot be neglected. For example, eNOS-mediated vasodilation response is initiated by ECs and executed by vascular smooth muscle cells. In addition, coronary artery spasm has been reported to interplay between endothelial dysfunction and vascular smooth muscle cell hyperreactivity [183]. It requires further investigation and summarization to obtain more complete elucidation of the role vascular smooth muscle cells in MI.
Acknowledgements
Author Statement
Hao Zhou, Hang Zhu and Xing Chang collected and analyzed all literatures. Xing Chang and Hao Zhou wrote and revised the original draft. Amanda Lochne, Hsueh-Hsiao Wang, Shuyi Wang and Jun Ren contributed to discussion and revisions of the manuscript. All authors approved the submission of this manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bassenge E, Heusch G. Endothelial and neuro-humoral control of coronary blood flow in health and disease. Reviews of physiology, biochemistry and pharmacology. 1990;116:77-165
2. Heusch G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol. 2020;17:773-89
3. Talman V, Kivelä R. Cardiomyocyte-Endothelial Cell Interactions in Cardiac Remodeling and Regeneration. Front Cardiovasc Med. 2018;5:101
4. Bausch D, Fritz S, Bolm L, Wellner UF, Fernandez-Del-Castillo C, Warshaw AL. et al. Hedgehog signaling promotes angiogenesis directly and indirectly in pancreatic cancer. Angiogenesis. 2020;23:479-92
5. Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E. et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. 2015;90:927-63
6. Kloner RA, Ganote CE, Jennings RB. The "no-reflow" phenomenon after temporary coronary occlusion in the dog. J Clin Invest. 1974;54:1496-508
7. Lindal S, Sørlie D, Jørgensen L. Endothelial cells of the cardiac microvasculature during and after cold cardioplegic ischaemia. Comparison of endothelial and myocyte damage. Scand J Thorac Cardiovasc Surg. 1988;22:257-65
8. Kloner RA, Ganote CE, Jennings RB, Reimer KA. Demonstration of the “no-reflow” phenomenon in the dog heart after temporary ischemia. Recent Adv Stud Cardiac Struct Metab. 1975;10:463-74
9. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:Iii27-32
10. Niccoli G, Montone RA, Ibanez B, Thiele H, Crea F, Heusch G. et al. Optimized Treatment of ST-Elevation Myocardial Infarction. Circ Res. 2019;125:245-58
11. Heusch G. Coronary microvascular obstruction: the new frontier in cardioprotection. Basic Res Cardiol. 2019;114:45
12. Heusch G, Skyschally A, Kleinbongard P. Coronary microembolization and microvascular dysfunction. International journal of cardiology. 2018;258:17-23
13. Heusch G. Coronary microcirculation-a neglected target of cardioprotection. Circulation journal: official journal of the Japanese Circulation Society. 2014;78:1830-1
14. Heusch G. The Coronary Circulation as a Target of Cardioprotection. Circulation research. 2016;118:1643-58
15. Bayliss AL, Sundararaman A, Granet C, Mellor H. Raftlin is recruited by neuropilin-1 to the activated VEGFR2 complex to control proangiogenic signaling. Angiogenesis. 2020;23:371-83
16. Heusch G, Kleinbongard P, Bose D, Levkau B, Haude M, Schulz R. et al. Coronary microembolization: from bedside to bench and back to bedside. Circulation. 2009;120:1822-36
17. Heusch G, Gersh B. The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge. European heart journal. 2017;38:774-84
18. Veith C, Neghabian D, Luitel H, Wilhelm J, Egemnazarov B, Muntanjohl C. et al. FHL-1 is not involved in pressure overload-induced maladaptive right ventricular remodeling and dysfunction. Basic Res Cardiol. 2020;115:17
19. Ambrosio G, Weisman HF, Mannisi JA, Becker LC. Progressive impairment of regional myocardial perfusion after initial restoration of postischemic blood flow. Circulation. 1989;80:1846-61
20. Reffelmann T, Kloner RA. Microvascular reperfusion injury: rapid expansion of anatomic no reflow during reperfusion in the rabbit. Am J Physiol Heart Circ Physiol. 2002;283:H1099-107
21. Alidoosti M, Lotfi R, Lotfi-Tokaldany M, Nematipour E, Salarifar M, Poorhosseini H. et al. Correlates of the "No-Reflow" or "Slow-Flow" Phenomenon in Patients Undergoing Primary Percutaneous Coronary Intervention. J Tehran Heart Cent. 2018;13:108-14
22. Bolli R. Mechanism of myocardial "stunning". Circulation. 1990;82:723-38
23. Bolli R, Marbán E. Molecular and cellular mechanisms of myocardial stunning. Physiological reviews. 1999;79:609-34
24. Heusch G. Myocardial stunning and hibernation revisited. Nature reviews Cardiology. 2021 doi: 10.1038/s41569-021-00506-7
25. McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312:159-63
26. Atar D, Gao WD, Marban E. Alterations of excitation-contraction coupling in stunned myocardium and in failing myocardium. J Mol Cell Cardiol. 1995;27:783-91
27. Rowe GT, Manson NH, Caplan M, Hess ML. Hydrogen peroxide and hydroxyl radical mediation of activated leukocyte depression of cardiac sarcoplasmic reticulum. Participation of the cyclooxygenase pathway. Circ Res. 1983;53:584-91
28. Przyklenk K, Kloner RA. Superoxide dismutase plus catalase improve contractile function in the canine model of the "stunned myocardium". Circ Res. 1986;58:148-56
29. Diamond GA, Forrester JS, deLuz PL, Wyatt HL, Swan HJ. Post-extrasystolic potentiation of ischemic myocardium by atrial stimulation. Am Heart J. 1978;95:204-9
30. Heusch G, Rose J, Skyschally A, Post H, Schulz R. Calcium responsiveness in regional myocardial short-term hibernation and stunning in the in situ porcine heart. Inotropic responses to postextrasystolic potentiation and intracoronary calcium. Circulation. 1996;93:1556-66
31. Heusch G, Post H, Michel MC, Kelm M, Schulz R. Endogenous nitric oxide and myocardial adaptation to ischemia. Circ Res. 2000;87:146-52
32. Hamilton S, Terentyeva R, Martin B, Perger F, Li J, Stepanov A. et al. Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS. Basic Res Cardiol. 2020;115:38
33. Wagner M, Bertero E, Nickel A, Kohlhaas M, Gibson GE, Heggermont W. et al. Selective NADH communication from α-ketoglutarate dehydrogenase to mitochondrial transhydrogenase prevents reactive oxygen species formation under reducing conditions in the heart. Basic Res Cardiol. 2020;115:53
34. Jones SA, O'Donnell VB, Wood JD, Broughton JP, Hughes EJ, Jones OT. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am J Physiol. 1996;271:H1626-34
35. Damico R, Zulueta JJ, Hassoun PM. Pulmonary endothelial cell NOX. Am J Respir Cell Mol Biol. 2012;47:129-39
36. Depoix CL, Colson A, Hubinont C, Debieve F. Impaired vascular endothelial growth factor expression and secretion during in vitro differentiation of human primary term cytotrophoblasts. Angiogenesis. 2020;23:221-30
37. Schröder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J. et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res. 2012;110:1217-25
38. Weng X, Yu L, Liang P, Li L, Dai X, Zhou B. et al. A crosstalk between chromatin remodeling and histone H3K4 methyltransferase complexes in endothelial cells regulates angiotensin II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2015;82:48-58
39. Goitre L, DiStefano PV, Moglia A, Nobiletti N, Baldini E, Trabalzini L. et al. Up-regulation of NADPH oxidase-mediated redox signaling contributes to the loss of barrier function in KRIT1 deficient endothelium. Sci Rep. 2017;7:8296
40. Li TB, Zhang JJ, Liu B, Liu WQ, Wu Y, Xiong XM. et al. Involvement of NADPH oxidases and non-muscle myosin light chain in senescence of endothelial progenitor cells in hyperlipidemia. Naunyn Schmiedebergs Arch Pharmacol. 2016;389:289-302
41. Harlan JM, Vedder NB, Winn RK, Rice CL. Mechanisms and consequences of leukocyte-endothelial interaction. West J Med. 1991;155:365-9
42. Engler RL, Dahlgren MD, Morris DD, Peterson MA, Schmid-Schönbein GW. Role of leukocytes in response to acute myocardial ischemia and reflow in dogs. Am J Physiol. 1986;251:H314-23
43. Mehta JL, Nichols WW, Mehta P. Neutrophils as potential participants in acute myocardial ischemia: relevance to reperfusion. J Am Coll Cardiol. 1988;11:1309-16
44. Schnoor M, Alcaide P, Voisin MB, van Buul JD. Crossing the Vascular Wall: Common and Unique Mechanisms Exploited by Different Leukocyte Subsets during Extravasation. Mediators Inflamm. 2015;2015:946509
45. Del Papa N, Pignataro F. The Role of Endothelial Progenitors in the Repair of Vascular Damage in Systemic Sclerosis. Front Immunol. 2018;9:1383
46. Wihastuti TA, Aini FN, Lutfiana NC, Heriansyah T, Zamrudah N. Exploration of Adhesion Molecule Expression in Cardiac Muscle of Early Atherosclerosis Dyslipidemic Sprague Dawley Rats. Open Med Chem J. 2018;12:124-9
47. Félétou M, Huang Y, Vanhoutte PM. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br J Pharmacol. 2011;164:894-912
48. Shreeniwas R, Koga S, Karakurum M, Pinsky D, Kaiser E, Brett J. et al. Hypoxia-mediated induction of endothelial cell interleukin-1 alpha. An autocrine mechanism promoting expression of leukocyte adhesion molecules on the vessel surface. J Clin Invest. 1992;90:2333-9
49. Yin J, Xia W, Zhang Y, Ding G, Chen L, Yang G. et al. Role of dihydroartemisinin in regulating prostaglandin E(2) synthesis cascade and inflammation in endothelial cells. Heart Vessels. 2018;33:1411-22
50. Gomez I, Foudi N, Longrois D, Norel X. The role of prostaglandin E2 in human vascular inflammation. Prostaglandins Leukot Essent Fatty Acids. 2013;89:55-63
51. Koyama H, Maeno T, Fukumoto S, Shoji T, Yamane T, Yokoyama H. et al. Platelet P-selectin expression is associated with atherosclerotic wall thickness in carotid artery in humans. Circulation. 2003;108:524-9
52. Han L, Dai L, Zhao YF, Li HY, Liu O, Lan F. et al. CD40L promotes development of acute aortic dissection via induction of inflammation and impairment of endothelial cell function. Aging (Albany NY). 2018;10:371-85
53. Urbich C, Dernbach E, Aicher A, Zeiher AM, Dimmeler S. CD40 ligand inhibits endothelial cell migration by increasing production of endothelial reactive oxygen species. Circulation. 2002;106:981-6
54. Zmysłowski A, Szterk A. Current knowledge on the mechanism of atherosclerosis and pro-atherosclerotic properties of oxysterols. Lipids Health Dis. 2017;16:188
55. Tian J, Zhang L, Yang X, Zuo H, Zhao X, Yong J. et al. The effect of Shexiang Tongxin Dropping Pills on coronary microvascular dysfunction (CMVD) among those with a mental disorder and non-obstructive coronary artery disease based on stress cardiac magnetic resonance images: A study protocol. Medicine (Baltimore). 2020;99:e20099
56. Lee SJ, Lee CK, Kang S, Park I, Kim YH, Kim SK. et al. Angiopoietin-2 exacerbates cardiac hypoxia and inflammation after myocardial infarction. J Clin Invest. 2018;128:5018-33
57. Koupenova M, Kehrel BE, Corkrey HA, Freedman JE. Thrombosis and platelets: an update. Eur Heart J. 2017;38:785-91
58. Bridges E, Sheldon H, Kleibeuker E, Ramberger E, Zois C, Barnard A. et al. RHOQ is induced by DLL4 and regulates angiogenesis by determining the intracellular route of the Notch intracellular domain. Angiogenesis. 2020;23:493-513
59. FitzGerald GA. Mechanisms of platelet activation: thromboxane A2 as an amplifying signal for other agonists. Am J Cardiol. 1991;68:11b-5b
60. Detter MR, Shenkar R, Benavides CR, Neilson CA, Moore T, Lightle R. et al. Novel Murine Models of Cerebral Cavernous Malformations. Angiogenesis. 2020;23:e651-666
61. Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345:494-502
62. Podhaisky HP, Abate A, Polte T, Oberle S, Schröder H. Aspirin protects endothelial cells from oxidative stress-possible synergism with vitamin E. FEBS Lett. 1997;417:349-51
63. Grosser N, Schröder H. Aspirin protects endothelial cells from oxidant damage via the nitric oxide-cGMP pathway. Arterioscler Thromb Vasc Biol. 2003;23:1345-51
64. Zhou X, Wu Y, Ye L, Wang Y, Zhang K, Wang L. et al. Aspirin alleviates endothelial gap junction dysfunction through inhibition of NLRP3 inflammasome activation in LPS-induced vascular injury. Acta Pharm Sin B. 2019;9:711-23
65. Armstrong D, Summers C, Ewart L, Nylander S, Sidaway JE, van Giezen JJ. Characterization of the adenosine pharmacology of ticagrelor reveals therapeutically relevant inhibition of equilibrative nucleoside transporter 1. J Cardiovasc Pharmacol Ther. 2014;19:209-19
66. van Giezen JJ, Sidaway J, Glaves P, Kirk I, Björkman JA. Ticagrelor inhibits adenosine uptake in vitro and enhances adenosine-mediated hyperemia responses in a canine model. J Cardiovasc Pharmacol Ther. 2012;17:164-72
67. Ohman J, Kudira R, Albinsson S, Olde B, Erlinge D. Ticagrelor induces adenosine triphosphate release from human red blood cells. Biochem Biophys Res Commun. 2012;418:754-8
68. Torngren K, Ohman J, Salmi H, Larsson J, Erlinge D. Ticagrelor improves peripheral arterial function in patients with a previous acute coronary syndrome. Cardiology. 2013;124:252-8
69. Bonello L, Frere C, Cointe S, Laine M, Mancini J, Thuny F. et al. Ticagrelor increases endothelial progenitor cell level compared to clopidogrel in acute coronary syndromes: A prospective randomized study. Int J Cardiol. 2015;187:502-7
70. Damani S, Bacconi A, Libiger O, Chourasia AH, Serry R, Gollapudi R. et al. Characterization of circulating endothelial cells in acute myocardial infarction. Sci Transl Med. 2012;4:126ra33
71. Chen CW, Wang LL, Zaman S, Gordon J, Arisi MF, Venkataraman CM. et al. Sustained release of endothelial progenitor cell-derived extracellular vesicles from shear-thinning hydrogels improves angiogenesis and promotes function after myocardial infarction. Cardiovasc Res. 2018;114:1029-40
72. Ridger VC, Boulanger CM, Angelillo-Scherrer A, Badimon L, Blanc-Brude O, Bochaton-Piallat ML. et al. Microvesicles in vascular homeostasis and diseases. Position Paper of the European Society of Cardiology (ESC) Working Group on Atherosclerosis and Vascular Biology. Thromb Haemost. 2017;117:1296-316
73. Horn P, Baars T, Kahlert P, Heiss C, Westenfeld R, Kelm M. et al. Release of Intracoronary Microparticles during Stent Implantation into Stable Atherosclerotic Lesions under Protection with an Aspiration Device. PLoS One. 2015;10:e0124904
74. Muse ED, Kramer ER, Wang H, Barrett P, Parviz F, Novotny MA. et al. A Whole Blood Molecular Signature for Acute Myocardial Infarction. Sci Rep. 2017;7:12268
75. Jung C, Sörensson P, Saleh N, Arheden H, Rydén L, Pernow J. Circulating endothelial and platelet derived microparticles reflect the size of myocardium at risk in patients with ST-elevation myocardial infarction. Atherosclerosis. 2012;221:226-31
76. Abdel Hamid M, Bakhoum SW, Sharaf Y, Sabry D, El-Gengehe AT, Abdel-Latif A. Circulating Endothelial Cells and Endothelial Function Predict Major Adverse Cardiac Events and Early Adverse Left Ventricular Remodeling in Patients With ST-Segment Elevation Myocardial Infarction. J Interv Cardiol. 2016;29:89-98
77. Li C, Wu Q, Liu B, Yao Y, Chen Y, Zhang H. et al. Detection and validation of circulating endothelial cells, a blood-based diagnostic marker of acute myocardial infarction. PLoS One. 2013;8:e58478
78. Alex L, Frangogiannis NG. Pericytes in the infarcted heart. Vasc Biol. 2019;1:H23-h31
79. Simonavicius N, Ashenden M, van Weverwijk A, Lax S, Huso D, Buckley C. et al. Pericytes promote selective vessel regression to regulate vascular patterning. Blood. 2012;120:1516-27
80. Hellström M, Kalén M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047-55
81. O'Farrell FM, Mastitskaya S, Hammond-Haley M, Freitas F, Wah WR, Attwell D. Capillary pericytes mediate coronary no-reflow after myocardial ischaemia. Elife. 2017;6:e29280
82. Weiss RM, Miller JD, Heistad DD. Fibrocalcific aortic valve disease: opportunity to understand disease mechanisms using mouse models. Circ Res. 2013;113:209-22
83. Shi Y, Hu G, Su J, Li W, Chen Q, Shou P. et al. Mesenchymal stem cells: a new strategy for immunosuppression and tissue repair. Cell Res. 2010;20:510-8
84. Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von Brühl ML. et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and 'instruct' them with pattern-recognition and motility programs. Nat Immunol. 2013;14:41-51
85. Saxena A, Chen W, Su Y, Rai V, Uche OU, Li N. et al. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol. 2013;191:4838-48
86. Su H, Cantrell AC, Zeng H, Zhu S-H, Chen J-X. Emerging Role of Pericytes and Their Secretome in the Heart. Cells. 2021;10:548
87. Avolio E, Madeddu P. Discovering cardiac pericyte biology: From physiopathological mechanisms to potential therapeutic applications in ischemic heart disease. Vascul Pharmacol. 2016;86:53-63
88. Deckelbaum RA, Lobov IB, Cheung E, Halasz G, Rajamani S, Lerner J. et al. The potassium channel Kcne3 is a VEGFA-inducible gene selectively expressed by vascular endothelial tip cells. Angiogenesis. 2020;23:179-92
89. Sweeney M, Foldes G. It Takes Two: Endothelial-Perivascular Cell Cross-Talk in Vascular Development and Disease. Front Cardiovasc Med. 2018;5:154
90. Geevarghese A, Herman IM. Pericyte-endothelial crosstalk: implications and opportunities for advanced cellular therapies. Transl Res. 2014;163:296-306
91. Ma C, Beyer AM, Durand M, Clough AV, Zhu D, Norwood Toro L. et al. Hyperoxia Causes Mitochondrial Fragmentation in Pulmonary Endothelial Cells by Increasing Expression of Pro-Fission Proteins. Arterioscler Thromb Vasc Biol. 2018;38:622-35
92. Yu T, Sheu SS, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res. 2008;79:341-51
93. Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia. 2010;53:1783-94
94. Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE. et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011;124:444-53
95. Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z. et al. Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mitochondrial Fission. Diabetes. 2017;66:193-205
96. Jendrach M, Mai S, Pohl S, Vöth M, Bereiter-Hahn J. Short- and long-term alterations of mitochondrial morphology, dynamics and mtDNA after transient oxidative stress. Mitochondrion. 2008;8:293-304
97. Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu P. et al. Mff-Dependent Mitochondrial Fission Contributes to the Pathogenesis of Cardiac Microvasculature Ischemia/Reperfusion Injury via Induction of mROS-Mediated Cardiolipin Oxidation and HK2/VDAC1 Disassociation-Involved mPTP Opening. J Am Heart Assoc. 2017;6:e00538
98. Zhou H, Wang J, Zhu P, Zhu H, Toan S, Hu S. et al. NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2alpha. Basic Res Cardiol. 2018;113:23
99. Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S. et al. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 2018;14:576-87
100. Wang J, Zhu P, Toan S, Li R, Ren J, Zhou H. Pum2-Mff axis fine-tunes mitochondrial quality control in acute ischemic kidney injury. Cell Biol Toxicol. 2020;36:365-78
101. Bretón-Romero R, Acín-Perez R, Rodríguez-Pascual F, Martínez-Molledo M, Brandes RP, Rial E. et al. Laminar shear stress regulates mitochondrial dynamics, bioenergetics responses and PRX3 activation in endothelial cells. Biochim Biophys Acta. 2014;1843:2403-13
102. Chen C, Gao JL, Liu MY, Li SL, Xuan XC, Zhang XZ. et al. Mitochondrial Fission Inhibitors Suppress Endothelin-1-Induced Artery Constriction. Cell Physiol Biochem. 2017;42:1802-11
103. Shi Y, Fan S, Wang D, Huyan T, Chen J, Chen J. et al. FOXO1 inhibition potentiates endothelial angiogenic functions in diabetes via suppression of ROCK1/Drp1-mediated mitochondrial fission. Biochim Biophys Acta Mol Basis Dis. 2018;1864:2481-94
104. Ong S, Lee W, Shao N, Ismail N, Katwadi K, Lim M. et al. Calpain Inhibition Restores Autophagy and Prevents Mitochondrial Fragmentation in a Human iPSC Model of Diabetic Endotheliopathy. Stem cell reports. 2019;12:597-610
105. Kleinbongard P, Gedik N, Kirca M, Stoian L, Frey U, Zandi A. et al. Mitochondrial and Contractile Function of Human Right Atrial Tissue in Response to Remote Ischemic Conditioning. J Am Heart Assoc. 2018;7:e009540
106. Sun R, Wang X, Liu Y, Xia M. Dietary supplementation with fish oil alters the expression levels of proteins governing mitochondrial dynamics and prevents high-fat diet-induced endothelial dysfunction. Br J Nutr. 2014;112:145-53
107. Lugus JJ, Ngoh GA, Bachschmid MM, Walsh K. Mitofusins are required for angiogenic function and modulate different signaling pathways in cultured endothelial cells. J Mol Cell Cardiol. 2011;51:885-93
108. de Jel DVC, Disch FJM, Kroon S, Mager JJ, Verdam FJ. Intranasal Efudix reduces epistaxis in hereditary hemorrhagic telangiectasia. Angiogenesis. 2020;23:271-4
109. Diaz-Morales N, Rovira-Llopis S, Bañuls C, Escribano-Lopez I, de Marañon AM, Lopez-Domenech S. et al. Are Mitochondrial Fusion and Fission Impaired in Leukocytes of Type 2 Diabetic Patients? Antioxid Redox Signal. 2016;25:108-15
110. Hughes WE, Beyer AM, Gutterman DD. Vascular autophagy in health and disease. Basic Res Cardiol. 2020;115:41
111. Zhou H, Zhu P, Wang J, Zhu H, Ren J, Chen Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2alpha-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018;25:1080-93
112. Tseng AH, Shieh SS, Wang DL. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free radical biology & medicine. 2013;63:222-34
113. Li C, Tan Y, Wu J, Ma Q, Bai S, Xia Z. et al. Resveratrol Improves Bnip3-Related Mitophagy and Attenuates High-Fat-Induced Endothelial Dysfunction. Front Cell Dev Biol. 2020;8:796
114. Wang X, Zhang JQ, Xiu CK, Yang J, Fang JY, Lei Y. Ginseng-Sanqi-Chuanxiong (GSC) Extracts Ameliorate Diabetes-Induced Endothelial Cell Senescence through Regulating Mitophagy via the AMPK Pathway. Oxid Med Cell Longev. 2020;2020:7151946
115. Liu N, Wu J, Zhang L, Gao Z, Sun Y, Yu M. et al. Hydrogen Sulphide modulating mitochondrial morphology to promote mitophagy in endothelial cells under high-glucose and high-palmitate. J Cell Mol Med. 2017;21:3190-203
116. Vacek TP, Vacek JC, Tyagi SC. Mitochondrial mitophagic mechanisms of myocardial matrix metabolism and remodelling. Arch Physiol Biochem. 2012;118:31-42
117. Sun L, Hao Y, An R, Li H, Xi C, Shen G. Overexpression of Rcan1-1L inhibits hypoxia-induced cell apoptosis through induction of mitophagy. Mol Cells. 2014;37:785-94
118. Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q. et al. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res. 2017;63:e12413
119. Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autophagy and Mitophagy in Cardiovascular Disease. Circ Res. 2017;120:1812-24
120. Viña J, Gomez-Cabrera MC, Borras C, Froio T, Sanchis-Gomar F, Martinez-Bello VE. et al. Mitochondrial biogenesis in exercise and in ageing. Adv Drug Deliv Rev. 2009;61:1369-74
121. García-Quintans N, Sánchez-Ramos C, Prieto I, Tierrez A, Arza E, Alfranca A. et al. Oxidative stress induces loss of pericyte coverage and vascular instability in PGC-1α-deficient mice. Angiogenesis. 2016;19:217-28
122. Borniquel S, Valle I, Cadenas S, Lamas S, Monsalve M. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1alpha. Faseb j. 2006;20:1889-91
123. Stein S, Lohmann C, Handschin C, Stenfeldt E, Borén J, Lüscher TF. et al. ApoE-/- PGC-1α-/- mice display reduced IL-18 levels and do not develop enhanced atherosclerosis. PLoS One. 2010;5:e13539
124. Ali F, Ali NS, Bauer A, Boyle JJ, Hamdulay SS, Haskard DO. et al. PPARdelta and PGC1alpha act cooperatively to induce haem oxygenase-1 and enhance vascular endothelial cell resistance to stress. Cardiovasc Res. 2010;85:701-10
125. Chinsomboon J, Ruas J, Gupta RK, Thom R, Shoag J, Rowe GC. et al. The transcriptional coactivator PGC-1alpha mediates exercise-induced angiogenesis in skeletal muscle. Proc Natl Acad Sci U S A. 2009;106:21401-6
126. Zheng Z, Chen H, Wang H, Ke B, Zheng B, Li Q. et al. Improvement of retinal vascular injury in diabetic rats by statins is associated with the inhibition of mitochondrial reactive oxygen species pathway mediated by peroxisome proliferator-activated receptor gamma coactivator 1alpha. Diabetes. 2010;59:2315-25
127. Kim HJ, Park KG, Yoo EK, Kim YH, Kim YN, Kim HS. et al. Effects of PGC-1alpha on TNF-alpha-induced MCP-1 and VCAM-1 expression and NF-kappaB activation in human aortic smooth muscle and endothelial cells. Antioxid Redox Signal. 2007;9:301-7
128. Kondo H, Fujino H, Murakami S, Tanaka M, Kanazashi M, Nagatomo F. et al. Low-intensity running exercise enhances the capillary volume and pro-angiogenic factors in the soleus muscle of type 2 diabetic rats. Muscle Nerve. 2015;51:391-9
129. Fujisawa K, Nishikawa T, Kukidome D, Imoto K, Yamashiro T, Motoshima H. et al. TZDs reduce mitochondrial ROS production and enhance mitochondrial biogenesis. Biochem Biophys Res Commun. 2009;379:43-8
130. Kröller-Schön S, Jansen T, Schüler A, Oelze M, Wenzel P, Hausding M. et al. Peroxisome proliferator-activated receptor γ, coactivator 1α deletion induces angiotensin II-associated vascular dysfunction by increasing mitochondrial oxidative stress and vascular inflammation. Arterioscler Thromb Vasc Biol. 2013;33:1928-35
131. Zhloba AA, Subbotina TF, Alekseevskaia ES, Moiseeva OM, Gavriluk ND, Irtuga OB. [The metabolic and protein markers of dysfunction of mitochondria in patients with cardio-vascular diseases.]. Klin Lab Diagn. 2015;60:35-41
132. Kelm NQ, Beare JE, Weber GJ, LeBlanc AJ. Thrombospondin-1 mediates Drp-1 signaling following ischemia reperfusion in the aging heart. FASEB Bioadv. 2020;2:304-14
133. Miura S, Saitoh SI, Kokubun T, Owada T, Yamauchi H, Machii H. et al. Mitochondrial-Targeted Antioxidant Maintains Blood Flow, Mitochondrial Function, and Redox Balance in Old Mice Following Prolonged Limb Ischemia. Int J Mol Sci. 2017 18
134. Pereira RO, Wende AR, Crum A, Hunter D, Olsen CD, Rawlings T. et al. Maintaining PGC-1α expression following pressure overload-induced cardiac hypertrophy preserves angiogenesis but not contractile or mitochondrial function. Faseb j. 2014;28:3691-702
135. Kaeffer N, Richard V, François A, Lallemand F, Henry JP, Thuillez C. Preconditioning prevents chronic reperfusion-induced coronary endothelial dysfunction in rats. Am J Physiol. 1996;271:H842-9
136. Kurzelewski M, Czarnowska E, Maczewski M, Beresewicz A. Effect of ischemic preconditioning on endothelial dysfunction and granulocyte adhesion in isolated guinea-pig hearts subjected to ischemia/reperfusion. J Physiol Pharmacol. 1999;50:617-28
137. Beresewicz A, Maczewski M, Duda M. Effect of classic preconditioning and diazoxide on endothelial function and O2- and NO generation in the post-ischemic guinea-pig heart. Cardiovasc Res. 2004;63:118-29
138. Lückhoff A, Busse R. Calcium influx into endothelial cells and formation of endothelium-derived relaxing factor is controlled by the membrane potential. Pflugers Arch. 1990;416:305-11
139. Bouchard JF, Lamontagne D. Mechanisms of protection afforded by preconditioning to endothelial function against ischemic injury. Am J Physiol. 1996;271:H1801-6
140. Maczewski M, Duda M, Pawlak W, Beresewicz A. Endothelial protection from reperfusion injury by ischemic preconditioning and diazoxide involves a SOD-like anti-O2- mechanism. J Physiol Pharmacol. 2004;55:537-50
141. Bouchard JF, Chouinard J, Lamontagne D. Participation of prostaglandin E2 in the endothelial protective effect of ischaemic preconditioning in isolated rat heart. Cardiovasc Res. 2000;45:418-27
142. Li Z, Jin ZQ. Ischemic preconditioning enhances integrity of coronary endothelial tight junctions. Biochem Biophys Res Commun. 2012;425:630-5
143. Kim SJ, Zhang X, Xu X, Chen A, Gonzalez JB, Koul S. et al. Evidence for enhanced eNOS function in coronary microvessels during the second window of protection. Am J Physiol Heart Circ Physiol. 2007;292:H2152-8
144. Zhao JL, Yang YJ, You SJ, Cui CJ, Gao RL. Different effects of postconditioning on myocardial no-reflow in the normal and hypercholesterolemic mini-swines. Microvasc Res. 2007;73:137-42
145. Bodi V, Ruiz-Nodar JM, Feliu E, Minana G, Nunez J, Husser O. et al. Effect of ischemic postconditioning on microvascular obstruction in reperfused myocardial infarction. Results of a randomized study in patients and of an experimental model in swine. Int J Cardiol. 2014;175:138-46
146. Zalewski J, Claus P, Bogaert J, Driessche NV, Driesen RB, Galan DT. et al. Cyclosporine A reduces microvascular obstruction and preserves left ventricular function deterioration following myocardial ischemia and reperfusion. Basic Res Cardiol. 2015;110:18
147. Skyschally A, Amanakis G, Neuhäuser M, Kleinbongard P, Heusch G. Impact of electrical defibrillation on infarct size and no-reflow in pigs subjected to myocardial ischemia-reperfusion without and with ischemic conditioning. Am J Physiol Heart Circ Physiol. 2017;313:H871-h8
148. Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA. et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579-88
149. Jung C, Sörensson P, Saleh N, Arheden H, Rydén L, Pernow J. Effects of myocardial postconditioning on the recruitment of endothelial progenitor cells. J Interv Cardiol. 2012;25:103-10
150. Liu Y, Lian K, Zhang L, Wang R, Yi F, Gao C. et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res Cardiol. 2014;109:415
151. Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM. Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovasc Res. 2002;55:534-43
152. Ottani F, Latini R, Staszewsky L, La Vecchia L, Locuratolo N, Sicuro M. et al. Cyclosporine A in Reperfused Myocardial Infarction: The Multicenter, Controlled, Open-Label CYCLE Trial. J Am Coll Cardiol. 2016;67:365-74
153. Zhou H, Ma Q, Zhu P, Ren J, Reiter RJ, Chen Y. Protective role of melatonin in cardiac ischemia-reperfusion injury: From pathogenesis to targeted therapy. J Pineal Res. 2018;64:e12471
154. Sayour AA, Korkmaz-Icöz S, Loganathan S, Ruppert M, Sayour VN, Oláh A. et al. Acute canagliflozin treatment protects against in vivo myocardial ischemia-reperfusion injury in non-diabetic male rats and enhances endothelium-dependent vasorelaxation. J Transl Med. 2019;17:127
155. Bøtker HE, Cabrera-Fuentes HA, Ruiz-Meana M, Heusch G, Ovize M. Translational issues for mitoprotective agents as adjunct to reperfusion therapy in patients with ST-segment elevation myocardial infarction. J Cell Mol Med. 2020;24:2717-29
156. Hausenloy DJ, Chilian W, Crea F, Davidson SM, Ferdinandy P, Garcia-Dorado D. et al. The coronary circulation in acute myocardial ischaemia/reperfusion injury: a target for cardioprotection. Cardiovasc Res. 2019;115:1143-55
157. Heusch G. Critical Issues for the Translation of Cardioprotection. Circ Res. 2017;120:1477-86
158. Heusch G. 25 years of remote ischemic conditioning: from laboratory curiosity to clinical outcome. Basic Res Cardiol. 2018;113:15
159. Tanner M, Wang J, Ying R, Suboc T, Malik M, Couillard A. et al. Dynamin-related protein 1 mediates low glucose-induced endothelial dysfunction in human arterioles. American journal of physiology Heart and circulatory physiology. 2017;312:H515-H27
160. Hong S-G, Shin J, Rath M, Sayoc J, Choi S-Y, Park J-Y. Drp1 inhibitor mdivi-1 attenuates disturbed flow-induced metabolic shift and prevents cell activation in endothelial cells. The FASEB Journal. 2019;33:lb470-lb
161. Ding M, Dong Q, Liu Z, Liu Z, Qu Y, Li X. et al. Inhibition of dynamin-related protein 1 protects against myocardial ischemia-reperfusion injury in diabetic mice. Cardiovascular diabetology. 2017;16:19
162. Veeranki S, Tyagi S. Mdivi-1 induced acute changes in the angiogenic profile after ischemia-reperfusion injury in female mice. Physiological reports. 2017;5:e13298
163. Giedt R, Yang C, Zweier J, Matzavinos A, Alevriadou B. Mitochondrial fission in endothelial cells after simulated ischemia/reperfusion: role of nitric oxide and reactive oxygen species. Free radical biology & medicine. 2012;52:348-56
164. Kaloya S, McCarron J, Chalmers S, McCormick C. P26 Inhibition of mitochondrial fission decreased endothelial cell von willebrand factor content and gap junction communication. Heart. 2018;104:A10-A1
165. Wang Y, Wang S, Wier WG, Zhang Q, Jiang H, Li Q. et al. Exercise improves the dilatation function of mesenteric arteries in postmyocardial infarction rats via a PI3K/Akt/eNOS pathway-mediated mechanism. Am J Physiol Heart Circ Physiol. 2010;299:H2097-106
166. Dymkowska D, Wrzosek A, Zabłocki K. Atorvastatin and pravastatin stimulate nitric oxide and reactive oxygen species generation, affect mitochondrial network architecture and elevate nicotinamide N-methyltransferase level in endothelial cells. J Appl Toxicol. 2020;19:e4094
167. Csiszar A, Labinskyy N, Pinto JT, Ballabh P, Zhang H, Losonczy G. et al. Resveratrol induces mitochondrial biogenesis in endothelial cells. Am J Physiol Heart Circ Physiol. 2009;297:H13-20
168. Chai W, Fu Z, Aylor K, Barrett E, Liu Z. Liraglutide prevents microvascular insulin resistance and preserves muscle capillary density in high-fat diet-fed rats. American journal of physiology Endocrinology and metabolism. 2016;311:E640-8
169. Hao E, Mukhopadhyay P, Cao Z, Erdélyi K, Holovac E, Liaudet L. et al. Cannabidiol Protects against Doxorubicin-Induced Cardiomyopathy by Modulating Mitochondrial Function and Biogenesis. Mol Med. 2015;21:38-45
170. Xing S, Yang X, Li W, Bian F, Wu D, Chi J. et al. Salidroside stimulates mitochondrial biogenesis and protects against H₂O₂-induced endothelial dysfunction. Oxid Med Cell Longev. 2014;2014:904834
171. Duluc L, Jacques C, Soleti R, Iacobazzi F, Simard G, Andriantsitohaina R. Modulation of mitochondrial capacity and angiogenesis by red wine polyphenols via estrogen receptor, NADPH oxidase and nitric oxide synthase pathways. Int J Biochem Cell Biol. 2013;45:783-91
172. Driver C, Bamitale KDS, Kazi A, Olla M, Nyane NA, Owira PMO. Cardioprotective Effects of Metformin. J Cardiovasc Pharmacol. 2018;72:121-7
173. Fourny N, Lan C, Sérée E, Bernard M, Desrois M. Protective Effect of Resveratrol against Ischemia-Reperfusion Injury via Enhanced High Energy Compounds and eNOS-SIRT1 Expression in Type 2 Diabetic Female Rat Heart. Nutrients. 2019 11
174. LaRocca T, Henson G, Thorburn A, Sindler A, Pierce G, Seals D. Translational evidence that impaired autophagy contributes to arterial ageing. The Journal of physiology. 2012;590:3305-16
175. LaRocca T, Hearon C, Henson G, Seals D. Mitochondrial quality control and age-associated arterial stiffening. Experimental gerontology. 2014;58:78-82
176. Sciarretta S, Yee D, Nagarajan N, Bianchi F, Saito T, Valenti V. et al. Trehalose-Induced Activation of Autophagy Improves Cardiac Remodeling After Myocardial Infarction. J Am Coll Cardiol. 2018;71:1999-2010
177. Walton EM, Cronan MR, Cambier CJ, Rossi A, Marass M, Foglia MD. et al. Cyclopropane Modification of Trehalose Dimycolate Drives Granuloma Angiogenesis and Mycobacterial Growth through Vegf Signaling. Cell Host Microbe. 2018;24:514-25.e6
178. Madeo F, Eisenberg T, Pietrocola F, Kroemer G. Spermidine in health and disease. Science (New York, NY). 2018 359
179. Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T. et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016;22:1428-38
180. Tyrrell DJ, Blin MG, Song J, Wood SC, Zhang M, Beard DA. et al. Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circ Res. 2020;126:298-314
181. Fetterman JL, Holbrook M, Flint N, Feng B, Bretón-Romero R, Linder EA. et al. Restoration of autophagy in endothelial cells from patients with diabetes mellitus improves nitric oxide signaling. Atherosclerosis. 2016;247:207-17
182. Yan J, Yan JY, Wang YX, Ling YN, Song XD, Wang SY. et al. Spermidine-enhanced autophagic flux improves cardiac dysfunction following myocardial infarction by targeting the AMPK/mTOR signalling pathway. Br J Pharmacol. 2019;176:3126-42
183. Hubert A, Seitz A, Pereyra VM, Bekeredjian R, Sechtem U, Ong P. Coronary Artery Spasm: The Interplay Between Endothelial Dysfunction and Vascular Smooth Muscle Cell Hyperreactivity. Eur Cardiol. 2020;15:e12
184. Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD, Counter CM. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol. 2011;13:1108-15
185. Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589-98
186. Zhou H, Zhu P, Wang J, Toan S, Ren J. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal transduction and targeted therapy. 2019;4:56
187. Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R. et al. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-beta-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem. 2012;287:30024-34
188. Prudent J, Zunino R, Sugiura A, Mattie S, Shore GC, McBride HM. MAPL SUMOylation of Drp1 Stabilizes an ER/Mitochondrial Platform Required for Cell Death. Mol Cell. 2015;59:941-55
189. Zunino R, Braschi E, Xu L, McBride HM. Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J Biol Chem. 2009;284:17783-95
190. Park YY, Lee S, Karbowski M, Neutzner A, Youle RJ, Cho H. Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J Cell Sci. 2010;123:619-26
191. Pyakurel A, Savoia C, Hess D, Scorrano L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol Cell. 2015;58:244-54
192. Park YY, Nguyen OT, Kang H, Cho H. MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis. 2014;5:e1172
193. Chen Y, Dorn GW 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471-5
194. Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S. et al. MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol Cell. 2013;51:20-34
195. Wang Y, Serricchio M, Jauregui M, Shanbhag R, Stoltz T, Di Paolo CT. et al. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy. 2015;11:595-606
196. Zhou H, Wang J, Zhu P, Zhu H, Toan S, Hu S. et al. NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2α. Basic Res Cardiol. 2018;113:23
197. Tan Y, Mui D, Toan S, Zhu P, Li R, Zhou H. SERCA Overexpression Improves Mitochondrial Quality Control and Attenuates Cardiac Microvascular Ischemia-Reperfusion Injury. Mol Ther Nucleic Acids. 2020;22:696-707
198. Li P, Bai Y, Zhao X, Tian T, Tang L, Ru J. et al. NR4A1 contributes to high-fat associated endothelial dysfunction by promoting CaMKII-Parkin-mitophagy pathways. Cell Stress Chaperones. 2018;23:749-61
199. Haslip M, Dostanic I, Huang Y, Zhang Y, Russell KS, Jurczak MJ. et al. Endothelial uncoupling protein 2 regulates mitophagy and pulmonary hypertension during intermittent hypoxia. Arterioscler Thromb Vasc Biol. 2015;35:1166-78
Author contact
![]() Corresponding author: Dr. Hao Zhou, E-mail: zhouhaoorg; Department of Cardiology, Chinese PLA General Hospital, Medical School of Chinese PLA, Beijing 100853, China.
Corresponding author: Dr. Hao Zhou, E-mail: zhouhaoorg; Department of Cardiology, Chinese PLA General Hospital, Medical School of Chinese PLA, Beijing 100853, China.