Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(16):7995-8007. doi:10.7150/thno.50990 This issue Cite
Research Paper
Ablation of lncRNA Miat attenuates pathological hypertrophy and heart failure
Liu Yang1,2,3, Jianxin Deng1, Wenxia Ma1, Aijun Qiao1, Shiyue Xu1, Yang Yu2, Chan Boriboun1, Xiang Kang1, Dunzheng Han1, Patrick Ernst1, Lufang Zhou1,4, Jiawei Shi1, Eric Zhang1, Tao-Sheng Li5, Hongyu Qiu6, Shinichi Nakagawa7, Seth Blackshaw8, Jianyi Zhang1, Gangjian Qin1,2 ![]()
1. Molecular Cardiology Program, Department of Biomedical Engineering, University of Alabama at Birmingham, School of Medicine and School of Engineering, Birmingham, AL 35294, USA.
2. Feinberg Cardiovascular Research Institute, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
3. Department of Cardiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
4. Department of Medicine, University of Alabama at Birmingham, Birmingham, AL 35294, USA
5. Department of Stem Cell Biology, Atomic Bomb Disease Institute, Nagasaki University, Nagasaki 852-8523, Japan.
6. Center for Molecular and Translational Medicine, Institute of Biomedical Science, Georgia State University, Atlanta, GA 30303, USA.
7. Faculty of Pharmaceutical Sciences, Hokkaido University, Sapporo 060-0812, Japan.
8. Solomon H. Snyder Department of Neuroscience, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA.
Received 2020-7-22; Accepted 2021-4-29; Published 2021-7-6
Abstract

Rationale: The conserved long non-coding RNA (lncRNA) myocardial infarction associate transcript (Miat) was identified for its multiple single-nucleotide polymorphisms that are strongly associated with susceptibility to MI, but its role in cardiovascular biology remains elusive. Here we investigated whether Miat regulates cardiac response to pathological hypertrophic stimuli.
Methods: Both an angiotensin II (Ang II) infusion model and a transverse aortic constriction (TAC) model were used in adult WT and Miat-null knockout (Miat-KO) mice to induce pathological cardiac hypertrophy. Heart structure and function were evaluated by echocardiography and histological assessments. Gene expression in the heart was evaluated by RNA sequencing (RNA-seq), quantitative real-time RT-PCR (qRT-PCR), and Western blotting. Primary WT and Miat-KO mouse cardiomyocytes were isolated and used in Ca2+ transient and contractility measurements.
Results: Continuous Ang II infusion for 4 weeks induced concentric hypertrophy in WT mice, but to a lesser extent in Miat-KO mice. Surgical TAC for 6 weeks resulted in decreased systolic function and heart failure in WT mice but not in Miat-KO mice. In both models, Miat-KO mice displayed reduced heart-weight to tibia-length ratio, cardiomyocyte cross-sectional area, cardiomyocyte apoptosis, and cardiac interstitial fibrosis and a better-preserved capillary density, as compared to WT mice. In addition, Ang II treatment led to significantly reduced mRNA and protein expression of the Ca2+ cycling genes Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a) and ryanodine receptor 2 (RyR2) and a dramatic increase in global RNA splicing events in the left ventricle (LV) of WT mice, and these changes were largely blunted in Miat-KO mice. Consistently, cardiomyocytes isolated from Miat-KO mice demonstrated more efficient Ca2+ cycling and greater contractility.
Conclusions: Ablation of Miat attenuates pathological hypertrophy and heart failure, in part, by enhancing cardiomyocyte contractility.
Keywords: lncRNA, Miat, cardiac hypertrophy, heart failure, RNA splicing, cardiomyocytes
Introduction
Cardiovascular disease is the leading cause of death worldwide [1]. Under biomechanical stress such as hemodynamic overload or high levels of neurohormonal mediators, cardiomyocytes undergo hypertrophic growth to meet the environmental demands. However, sustained pathological cardiac hypertrophy can lead to heart failure [2], and the process is accompanied by a complex spectrum of pathophysiological changes, including cardiomyocyte Ca2+ mishandling, hypertrophic growth and death, vascular insufficiency, and interstitial fibrosis [3]. Currently there is no specific treatment to effectively reverse cardiac pathological hypertrophy and reduce the morbidity and mortality of heart failure. Thus, uncovering new molecular mechanisms of cardiac remodeling has the potential to identify novel therapeutic targets [4].
Heart pump function is governed by cellular Ca2+ and excitation-contraction coupling [5]. Upon pathological hypertrophic stimuli (e.g., pressure overload), cardiomyocytes increase contractility by augmenting the release of Ca2+ from the sarcoplasmic reticulum (SR), the major intracellular Ca2+ store, into the cytoplasm through RyRs (at systole) as well as the uptake of Ca2+ back into SR through SERCA2a (at diastole) [6]. When the demand is not met, the cardiomyocytes compensate by synthesizing excess contractile proteins (hypertrophy), which jeopardizes the structural balance between subcellular organelles for efficient Ca2+ signaling, contraction/relaxation, and energy metabolism, and causes insufficiency of vascular supply. These changes collectively contribute to cardiomyocyte cell death including apoptosis, which in turn triggers a pro-inflammatory response, fibrosis, and abnormal accumulation of extracellular matrix proteins (e.g., collagens); excessive accumulation of collagen stiffens the ventricles, which further impairs contraction and relaxation and reduces capillary density. This adverse structural remodeling of ventricular walls increases oxygen diffusion distances, leading to myocardial ischemia that contributes to the transition from hypertrophy to failure [7]. It is well recognized that impaired RyR2 function (e.g., Ca2+ leak) or decreased SERCA2a expression/activity can lead to reduced pump function and serial compensatory responses, including chronic activation of neurohormonal pathways and hypertrophic gene programming, which ultimately results in decompensation and heart failure [8]. Thus, restorations of the activity and integrity of SERCA2a and RyR2 are promising therapeutic strategies [9, 10].
LncRNAs are an emerging class of regulatory molecules with no or limited protein-coding capacity [11]. At present, only a small proportion of lncRNAs have been functionally validated in cardiovascular biology, including heart development [12, 13] and hypertrophy [14-16]. Miat, also known as Retinal Non-Coding RNA 2 (RNCR2) or Gomafu, is an intergenic lncRNA highly conserved across species including mouse and human [17, 18]. It is ~9 kb long, accumulated within the nucleus, and expressed in the central nervous system, heart, lung, and spleen [19, 20]. In a large-scale genome-wide association study, Miat was identified to contain multiple single-nucleotide-polymorphisms (SNPs) in 6 loci that are strongly associated with increased morbidity of MI [19]. Subsequently Miat has been suggested as a potential biomarker for left ventricular dysfunction in patients with MI and type 2 diabetes [21]. Experimental evidence in rodents suggest that Miat promotes post-MI fibrosis [22] and cardiac hypertrophy [23-25] and contributes to diabetic microvascular dysfunction [26]. However, due to the lack of a genetic Miat model, the function of Miat in cardiovascular disease has not been clearly defined.
In this study we examined the role of Miat in cardiac hypertrophy in newly-generated Miat KO mice. We found that KO of Miat protects hearts from Ang II-induced pathological hypertrophy and TAC-induced heart failure, and that this beneficial effect is at least partially attributable to the enhanced calcium handling and contractility of cardiomyocytes.
Materials and Methods
Mice
All the animal studies and operation procedures in this report were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Alabama at Birmingham and performed in compliance with the ''Guide for the Care and Use of Laboratory Animals'' (NIH publication) and all relevant ethical regulations for animal testing and research. The Miat KO mice were generated by deleting the entire Miat gene, including the two splicing variants, plus 70 bp upstream and 1 bp downstream sequences and genotyped as we previously described [20]. Male, 8-10 week-old homozygote Miat-KO mice and their WT littermates on C57BL/6 background were used for all the experiments unless specified. Sample sizes were predetermined by power calculation with 80% power.
Ang II infusion model
WT and Miat-KO mice received Ang II (2 mg/kg BW/day, Sigma Aldrich) or saline (control) continuously via Alzet mini-osmotic pumps (model 2004; Durect Corporation) for up to 28 days. Pumps were prepared and subcutaneously implanted in the mice by following the manufacturer's instructions.
TAC model
TAC model was performed using a clip method [27]. Briefly, mice were anesthetized by continuous inhalation of 2% isoflurane. A skin incision was made at the level of the suprasternal notch, followed by a longitudinal cut of ~3 mm in length in the proximal portion of the sternum. A metal clip calibrated to a 29-gauge needle was placed in the aortic arch between the innominate artery and the left common carotid artery. For the Sham group, the procedure was identical except the placement of the clip to induce constriction. Post-operative care was performed by following our approved animal study protocol. For pain management, Buprenex (0.05-0.2 mg/kg) was subcutaneously injected every 12 h for 48 h. In addition, Metacam (1 mg/kg) was subcutaneously injected at the end of surgery and continued daily for 3 d. Mice were euthanized 6 weeks later, and the tissues were isolated for histological and biochemical analysis.
Echocardiography
Cardiac function was recorded and analyzed before and after Ang II infusion or TAC surgery, using a Vevo 2100 high-resolution ultrasound system (VisualSonics Inc) as we previously described [28]. For systolic function, mice were conscious and secured with surgical tapes to the platform in supine position. Data were collected after three days of training. Parasternal long-axis view, short-axis view at the papillary muscle level and 2-D guided M-mode images were recorded. Left ventricular fractional shortening (FS), ejection fraction (EF), stroke volume (SV) and cardiac output (CO) were calculated. For diastolic function, mice were lightly anesthetized via 2% isoflurane inhalation, and heart rates were maintained at 350-450 beats per minutes. The transmitral left ventricular outflow tract Doppler spectra (E, A) was recorded from an apical four chamber view. Tissue Doppler was used to measure the lateral E' spectra, and to calculate the E/E' ratio. Measurements were performed by an individual who was blinded to the treatment assignments.
Blood pressure (BP) measurement
Systolic and diastolic arterial BP in WT and Miat-KO mice were measured by a noninvasive tail-cuff BP analyzer (CODA System, Kent Scientific) as we described previously [29]. Mice received 5 days of training before data collection. For each mouse, twenty measurements were collected and averaged.
Histology
Both formalin-fixed paraffin-embedded (FFPE) and fixed frozen (FF) sections were used in this study. For FFPE sections, cardiac tissues were fixed in 10% formalin overnight and embedded in paraffin, as described previously [30]. For FF sections, tissues were fixed in 4% paraformaldehyde for 4 h, dehydrated in 30% sucrose and embedded in OCT compound followed by cryosectioning. Three sections per heart and 6 fields per section were examined. The HE staining (ThermoFisher Scientific) was performed by following standard procedures. The Sirius Red/Fast Green (Chondrex), WGA (ThermoFisher Scientific) and TUNEL (Sigma Aldrich) staining were performed according to manufacturer's instructions. Anti-CD31 (Abcam, 1:50), Anti-cardiac troponin I (Abcam, 1:50) antibodies were used in the fluorescent immunohistology. All histological assessment and analyses were performed by an individual blinded to treatment assignments.
Isolation of neonatal mouse cardiomyocytes
Cardiomyocytes were isolated from neonatal WT or Miat-KO mice after euthanasia. Briefly, the beating hearts were excised and transferred into ice-cold HEPES-buffered saline solution (Mg2+ and Ca2+ free). After removing the atria and large vessels, hearts were washed and minced into small pieces less than or equal to 1 mm3. Tissues were then digested 5 times by incubating at 37 °C in HEPES-buffered saline solution containing collagenase type II. After each digestion step, the supernatant was filtered (100 micron) and collected in DMEM/F12 supplemented with 20% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were then centrifuged, resuspended and plated into T25 tissue-culture flasks at 37 °C. After 4 h, the non-adhesive cardiomyocytes were re-plated and cultured in DMEM/F12 supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.1 mM bromodeoxyuridine.
Lentiviral Miat-shRNA vector and Miat overexpression plasmid
Viral vectors encoding Miat short hairpin RNA (shRNA) were produced by co-transfection of 293FT cells with plasmid coding for Miat shRNA (pLKO.1-shRNAMiat-puro) or control scrambled shRNA (pLKO.1-shRNAScrambled-puro), pMD2.G and psPAX2, as previously described [31]. Medium was collected 48-72 h after transfection and concentrated by ultracentrifugation (2 h, 4 °C at 2,5000 rpm). Then the concentrated virus was carefully resuspended in PBS. For infection, the virus was suspended in fresh medium containing 8 µg/mL polybrene and applied onto HL-1 cells with a multiplicity of infection at 2 to 1. Three days later, the transduced cells were selected in 5 ug/mL puromycin and continued for 1 week. The shRNA sequences are: Miat shRNA: 5'-CCG GCC TAG AAA CCT GAT GTA GAC TCG AGT CTA CAT CAG GTT TCT AGG TTT TTG-3'; Control Scrambled shRNA: 5'-CCG GCC TAA GGT TAA GTC GCC CTC GCT CGA GCG AGG GCG ACT TAA CCT TAG GTT TTT G-3'. For Miat overexpression, plasmid pCAG-Miat-GFP coding for mouse Miat [17] or control plasmid pCAG-GFP was transfected into HL-1 cells using Amaxa Nucleofector (Lonza), following the manufacturer's instructions. The level of Miat expression was confirmed by qRT-PCR.
Immunocytochemistry
Cells were cultured on chamber slides and fixed with cold 4% paraformaldehyde, then stained with primary antibodies specific for cardiac Troponin T (Abcam,1:100). Pertinent secondary antibodies were conjugated with Alexa Fluor 594. Nuclei were counterstained with DAPI (Vector Laboratories, Burlingame, CA). Sections were examined under a Nikon confocal microscope.
Quantitative real-time RT-PCR (qRT-PCR)
qRT-PCR was performed as we described previously [32]. Briefly, total RNA was extracted from heart tissues using the E.N.Z.A Total RNA Kit (Omega) and reverse transcribed into cDNA using PrimeScript RT Reagent Kit (TAKARA). The values were normalized to the levels of 18S. The sequences of all primers are reported in Table S1.
Western Blotting
Western Blotting was performed via standard techniques as we described previously [32], by using antibodies against RyR (Abcam, Cambridge, UK), Serca 2a (Abcam, Cambridge, UK), and GAPDH (Cell Signaling Technology, Danvers, MA, USA).
RNA sequencing (RNA-seq) and bioinformatics analyses
Left ventricular tissues were collected from WT or Miat-KO mice that received Ang II or saline treatment for 7 days. Total RNA was extracted using TRIzol (ThermoFisher Scientific). RNA samples were then sequenced as described elsewhere [33]. Briefly, quality control testing was performed on all the RNA samples using the Agilent 2100 Bioanalyzer, followed by polyadenylate positive (poly A+) selection and conversion to cDNA. After library construction, paired-end sequencing runs were performed on the Illumina HiSeq 2500. All RNA-seq fastq Reads were aligned to the reference genome mouse mm10. Genes with a fold change greater than 2 (regardless of isoforms) and p<0.05 were used for Gene Ontology (GO) analyses (http://david.abcc.ncifcrf.gov/). Gene isoforms were analyzed by MISO (https://miso.readthedocs.io/en/fastmiso/) and MATS (http://rnaseq-mats.sourceforge.net/).
Isolation of adult mouse cardiomyocytes
Adult mouse left ventricular cardiomyocytes were isolated as described [34]. Briefly, hearts were excised from the heparinized and anesthetized mice, cannulated via the aorta and connected to a Langendorff perfusion system (Radnoti). Hearts were perfused for about 5 min with calcium-free perfusion solution, followed by digestion buffer containing 1 mg/mL type II collagenase (Worthington) for about 15 min. Both atria and right ventricle were trimmed off from the heart. Cells were then dissociated from the left ventricle to form a single cell suspension, followed by calcium reintroduction.
Fractional shortening (FS) and intracellular calcium measurements
Immediately after isolation, cardiomyocytes were used to measure FS and intracellular calcium transients as we previously reported [35]. Briefly, cells were loaded with 5 µM Fluo-4 AM (ThermoFisher Scientific, F14201, dissolved in DMSO containing 20% pluronic F-127) for 10 min at 37 °C and then perfused with Tyrode's solution (137 mM NaCl, 1.2 mM MgCl2, 1.2 mM NaH2PO4, 20 mM HEPES, 5.4 mM KCl, 1.8 mM CaCl2, 10 mM glucose, pH 7.4) in a heated chamber (37 °C) on the stage of an inverted fluorescent microscope (Olympus, Fv1000) to remove the dye and complete de-esterification. After the cardiomyocytes reached a steady state with field stimulations (1 Hz), line-scan images were acquired along the longitudinal axis of the cells. The F0 was measured right before stimulation and the maximal Fluo-4 fluorescence (F) was measured at peak amplitude. Calcium amplitude (∆F/F0, ∆F=F-F0) was calculated. FS of cardiomyocytes was measured with edge detection.
Statistics
All values are reported as mean ± standard error of the mean (SEM). Unpaired two-tailed Student's t test was used to compare means of two groups. One-way or two-way analysis of variance (ANOVA) were used in multiple (> 2) group comparisons with one or two independent variables, respectively. p < 0.05 was considered statistically significant.
Results
Deletion of Miat attenuates concentric cardiac hypertrophy
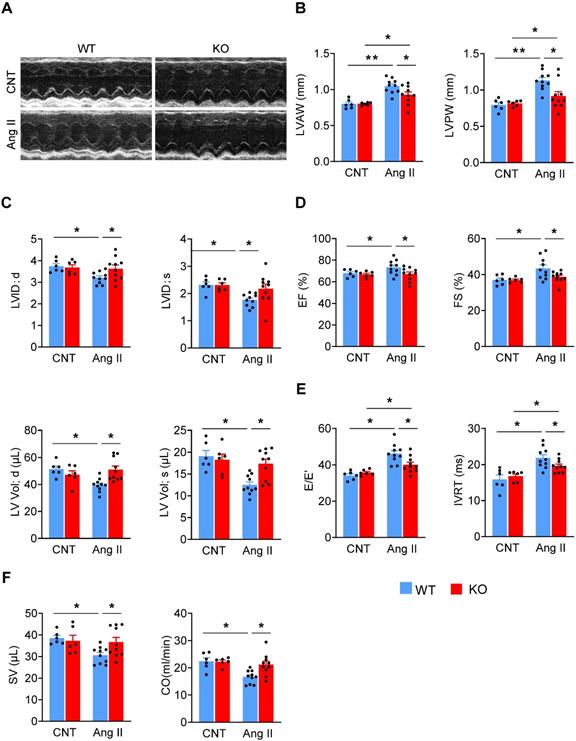
We firstly treated mice with Ang II (2 mg/kg/d) via a subcutaneously-implanted mini-osmotic pump, and at various time points analyzed Miat expression in the left ventricle (LV). Miat was significantly increased (~3 fold) at Day 7, then declined to baseline at Day 14 and 28 (Figure S1). To determine the role of Miat in cardiac hypertrophy, we used Miat-KO mice in which the entire Miat gene plus 71 bp of surrounding sequences have been removed (Figure S2A-B) [22]. These KO mice appear developmentally normal and show no difference in gross appearance, body weight, or histological characteristics of major organs including the heart, as compared to WT littermates (Figure S2C-F). Ang II treatments dramatically increased the systolic and diastolic blood pressure in both WT and KO mice to similar levels (Figure S3). Notably, while Ang II induced a significant increase in the LV wall thickness (Figure 1A-B, Table S2) and decrease in the LV internal diameter and volume (Figure 1C) in WT mice, these structural changes were significantly attenuated in KO mice, indicating that Miat-deletion reduces Ang II-induced concentric hypertrophy. Interestingly, the attenuated hypertrophy was not observed until Day 21 of Ang II infusion in Miat-KO mice (Figure S4). Although WT mice displayed a slight increase in their ejection fraction (EF) and fractional shortening (FS) (Figure 1D), their diastolic function was markedly impaired after Ang II treatment, as indicated by increased E/E' and isovolumic relaxation time (IVRT) (Figure 1E). The overall cardiac function of WT mice was significantly reduced as indicated by the reduced stroke volume and cardiac output (Figure 1F); in contrast, these functional impairments were markedly attenuated in KO mice.
Deletion of Miat attenuates Ang II-induced cardiac dysfunction. Miat-KO mice and WT littermates were treated with Ang II (2 mg/kg BW/day) or saline control (CNT) via a subcutaneously-implanted mini-osmotic pump for 28 days, then cardiac functions were evaluated by echocardiography. (A) Representative image of echocardiography. (B) Thicknesses of LV anterior wall (LVAW) and posterior wall (LVPW). (C) LV internal diameter (LVID; upper panels) and volume (LV Vol; lower panels) at diastole (d) and systole (s). (D) LV ejection fraction (EF) and fractional shortening (FS). (E) Ratio between early mitral inflow velocity and mitral annular early diastolic velocity (E/E'; left panel) and isovolumic relaxation time (IVRT; right panel). (F) stroke volume (SV; left panel) and cardiac output (CO; right panel). *p < 0.05, **p < 0.01.
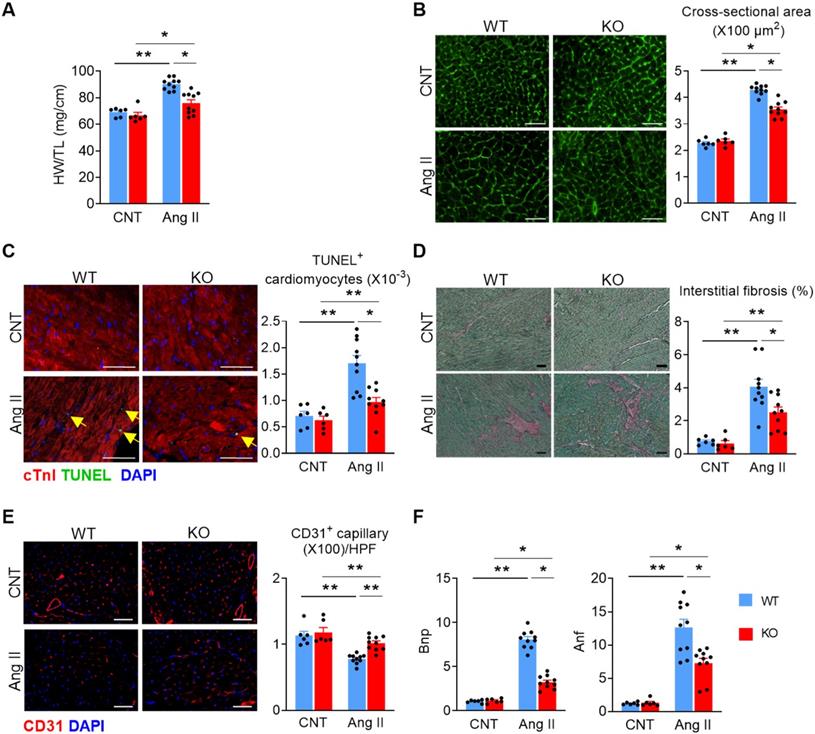
We then dissected the hearts from WT and KO mice to evaluate cardiac remodeling histologically. Ang II treatment significantly increased heart-weight to tibia-length ratio (Figure 2A) and cardiomyocyte cross-sectional area (Figure 2B), which was attenuated in KO mice. KO mice displayed significantly reduced cardiomyocyte apoptosis (Figure 2C), interstitial fibrosis (Figure 2D), and a better-preserved capillary density after Ang II treatment (Figure 2E). Furthermore, the mRNA levels of brain natriuretic peptide (Bnp) and atrial natriuretic factor (Anf) were significantly increased in the heart tissue of both WT and KO mice, but to a lesser extent in KO mice (Figure 2F). These data indicate that Miat deletion or inhibition alleviates Ang II-induced adverse cardiac remodeling. Furthermore, deletion of Miat attenuated Ang II-induced hypertrophy of primary cardiomyocytes in vitro (Figure S5), and knockdown (KD) of Miat in the HL-1 cardiomyocyte cell line diminished Ang II-induced expression of Bnp and Anf. Notably, overexpression (OE) of Miat in HL-1 cells increased, while restoration of Miat expression in Miat-KD HL-1 cells partially rescued, Ang II-induced expression of the cardiac hypertrophy genes (Figure S6 & Figure S7). Thus, Miat promotes cardiomyocyte hypertrophic growth autonomously.
Deletion of Miat attenuates Ang II-induced cardiac pathological hypertrophy. At 28 days after continuous Ang II (2 mg/Kg BW/day) or saline treatment, mice were euthanized and examined. (A) Heart weight (HW) versus tibia length (TL) ratio. (B) Representative images of wheat germ agglutinin (WGA) staining (left panel) and quantification of cardiomyocyte cross-section areas (right panel). (C) Representative images of TUNEL and cTnI staining (left panel) and quantification of apoptotic cardiomyocytes (right panel, % of nuclei). Yellow arrows indicate TUNEL+ cardiomyocytes. Scale bar = 50µm. (D) Representative images of Sirius Red/Fast Green staining (left panel) and quantification of interstitial fibrosis areas (right panel). (E) Representative images of CD31 staining (left panel) and quantification of capillary density (right panel). HPF, high-powered field. (F) qRT-PCR analyses of the mRNA levels of Bnp and Anf in the heart tissue. *p < 0.05, **p < 0.01.
Deletion of Miat attenuates eccentric cardiac hypertrophy and heart failure
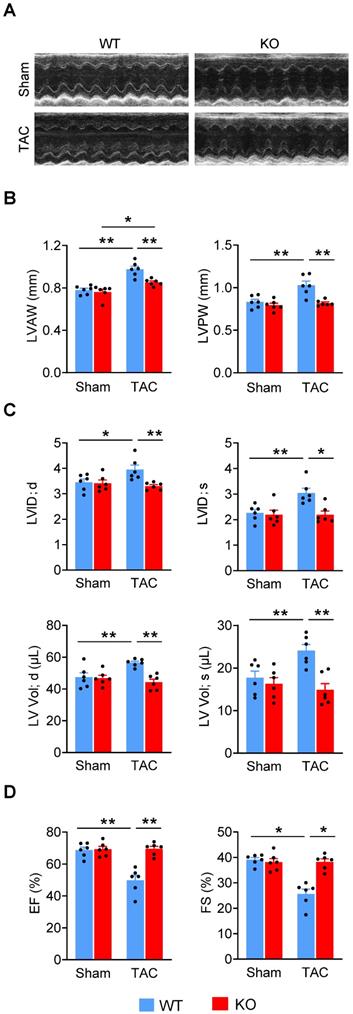
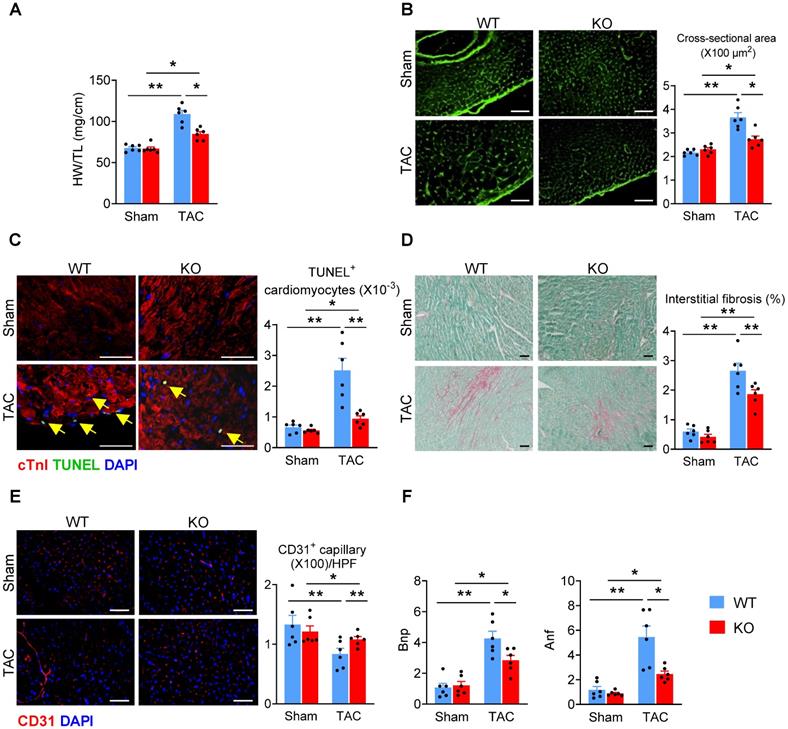
Since adverse cardiac remodeling and hypertrophy leads to heart failure, we examined the effects of Miat in heart failure by using the TAC model in Miat-KO and WT mice. Six weeks after surgery, although the LV wall was still thicker than baseline (Figure 3A-B, Table S3), the heart chamber (Figure 3C) was markedly dilated in WT mice, but not in Miat-KO mice. These structural changes in WT mice are consistent with a significant impairment in systolic cardiac function, as evidenced by a reduction in EF and FS (Figure 3D). KO mice displayed much better cardiac performance than the WT group (Figure 3A-D). In the gross and histological examinations, both heart-weight to tibia-length ratio and cardiomyocyte cross-sectional area were dramatically increased in WT mice, but to a lesser extent in KO mice (Figure 4A-B). Consistently, Miat deficiency results in reduced cardiomyocyte apoptosis (Figure 4C), interstitial fibrosis (Figure 4D), and a better-preserved capillary density (Figure 4E). The levels of Bnp and Anf were also significantly lower in KO mice (Figure 4F). Collectively, these data suggest that Miat deletion protects the heart from pressure overload-induced pathological hypertrophy and heart failure.
Deletion of Miat attenuates TAC-induced heart failure. Cardiac functions were evaluated with echocardiography in Miat-KO mice and WT littermates 6 weeks after TAC or Sham surgery. (A) Representative image of echocardiography. (B) Thicknesses of LV anterior wall (LVAW) and posterior wall (LVPW). (C) LV internal diameter (LVID; upper panels) and volume (LV Vol; lower panels) at diastole (d) and systole (s). (D) LV ejection fraction (EF) and fractional shortening (FS). *p < 0.05, **p < 0.01.
Deletion of Miat attenuates TAC-induced cardiac adverse remodeling. At 6 weeks after TAC or Sham surgery, mice were euthanized and examined. (A) Heart-weight (HW) to tibia-length (TL) ratio. (B) Representative images of WGA staining (left panel) and quantification of cardiomyocyte cross-section areas (right panel). (C) Representative images of TUNEL and cTnI staining (left panel) and quantification of apoptotic cardiomyocytes (right panel, % of nuclei). Yellow arrows indicate TUNEL+ cardiomyocytes. Scale bar = 50 µm. (D) Representative images of Sirius Red/Fast Green staining (left panel) and quantification of interstitial fibrosis areas (right panel). (E) Representative images of CD31 staining (left panel) and quantification of capillary density (right panel). HPF, high-powered field. (F) qRT-PCR analyses of the mRNA levels of Bnp and Anf in the heart tissue. *p < 0.05, **p < 0.01.
Deletion of Miat blunts Ang II-induced cardiac hypertrophic gene expression
To gain molecular insights into the regulation of cardiac remodeling by Miat, we isolated RNAs from LV of WT and Miat-KO mice that had been continuously treated with Ang II or a saline control for 7 days and performed paired-end RNA sequencing. In the basal state, the KO mice displayed altered expressions of genes associated with a number of biological functions, including gene transcription, cell cycle, protein phosphorylation, DNA damage response, intracellular transduction, chromatin modification and RNA splicing (Table S4, left). With Ang II treatment, the number of genes with altered expression levels was much higher in each of these functional categories (Table S4, right). In addition, KEGG pathway analyses revealed that the Ang II-induced differentially expressed genes between WT and KO mice are highly enriched in the PI3K-Akt and MAPK signaling pathways (Table S5).
Then we generated a heatmap of genes related to cardiac hypertrophy and contractility (Figure 5A). There were slight differences in the expression levels of these genes between WT and Miat-KO in the basal state. Ang II treatment led to drastic changes in their expression in the WT mice, including an elevation in the pro-hypertrophic genes (Mef2d, Gata4, Nfatc2, Hand2, Mapk7, Mapk3, Akt1, and Cib1) and a reduction in the anti-hypertrophic genes (Foxo3, Mlip, and Corin). However, these changes were largely diminished in the KO mice (Figure 5A, left). While Ang II treatment dramatically altered the expression of cardiac contractile genes in WT mice, including those involved in ion channels and calcium handling (Atp1a2, Cacnb1, S100a1, Fxyd1, Atp2a2, Casq2, Asph, RyR2, Slc8a1), myofibrils (Mybpc3, Pdlim5, Tnnt2), sarcolemma (Caveolin 1, Snta1, Mylk3) and cell junctions (Desmin, Jup), their changes are minimal in KO mice (Figure 5A, right). Furthermore, we confirmed the mRNA and protein expression of a number of key genes, including Serca2a (also known as Atp2a2), RyR2, Asph, Cib 1, Des, Jup and Cav 1 (Figure 5B-C). While the expression levels of Serca2a and RyR2 were comparable between WT and KO mice at baseline, Ang II treatment induced a marked downregulation of Serca2a and RyR2 in WT mice but not in KO mice (Figure 5B-C, Figure S8). Thus, our results clearly indicate that the Ang II-induced changes were blunted in Miat-KO mice, consistent with their attenuated hypertrophic response and enhanced contractility. Furthermore, in Miat-KD HL-1 cells, the level of Serca2a and RyR2 expression was better preserved than in HL-1 cells with Ang II treatment (Figure S7), and this protective effect was partially impaired by restoration of Miat expression (Figure S7).
Deletion of Miat blunts Ang II-induced hypertrophic gene program in the myocardium. WT and Miat-KO mice were treated with Ang II (WTA, KOA) or saline control (WTC, KOC) for 7 days, then RNAs were isolated from LV and sequenced. (A) Heatmaps of genes associated with cardiac hypertrophy (left panel) and contractility (right panel). ATP2A2 = SERCA2a. (B & C) validation of gene expression levels by qRT-PCR (B) and Western blotting (C, n = 3-6). *p < 0.05, **p < 0.01.
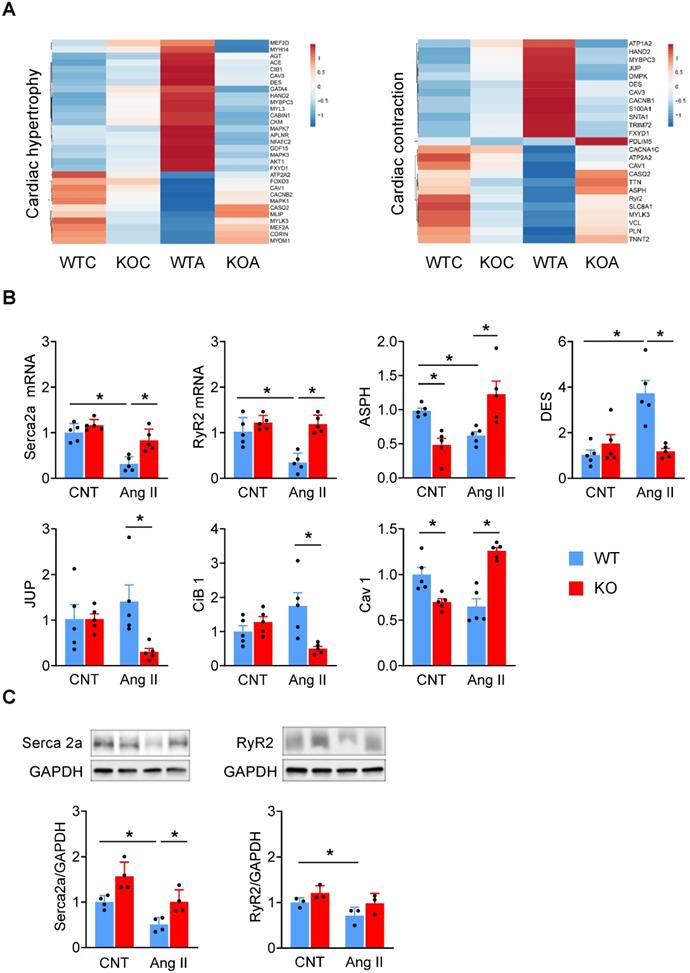
In addition, bioinformatics analyses revealed that Ang II induced ~1000 significant splicing events in WT mice and strikingly only about 300 in Miat-KO mice, suggesting that the gene-splicing activities are significantly reduced in the absence of Miat (Figure S9A). We further analyzed the number of different types of splicing events. Miat KO led to a decreased number of splicing events in each type, most dramatically in skipped exon (SE) and alternative 3' splicing site (A3SS) (Figure S9A). Gene ontogeny (GO) analyses revealed that genes related to ventricular cardiac muscle tissue morphogenesis and cardiac muscle contraction were alternatively spliced due to the depletion of Miat (Figure S9B). In addition, the genes with changed constitution of isoforms regulate a number of biological processes, including heart contraction, mitochondria, cell junction and the oxidation-reduction process (Table S6). For example, phospholamban, a muscle-specific SR Ca2+-ATPase inhibitor highly expressed in cardiac muscle, was identified as two different isoforms in our dataset (Figure S9C). Although the function for each isoform is currently not known, our data demonstrate a significant decrease in the percentage of isoform 1 after Ang II treatment in WT mice (from 56% to 38%), but not in the KO mice (from 68% to 67%), suggesting that Miat is important for Ang II-induced alternative splicing of phospholamban. In addition, Cav 1.2 and Camk2b are key regulators of cardiac contraction and hypertrophy; while Ang II treatment resulted in decreased inclusion of Exon 10 in Cav 1.2 and increased inclusion of Exon 13 in Camk2b in the heart of WT mice, these Ang II-induced splicing alterations were blunted in KO mice (Figure S9D-E). Collectively, these data support that Miat regulates the expression and alternative splicing of genes related to hypertrophy and contractility.
Deletion of Miat enhances the contractility of cardiomyocytes
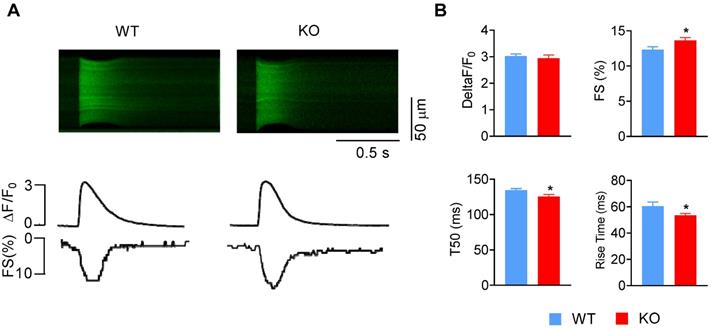
Accordingly, we investigated whether Miat regulates the contractility of cardiomyocytes. Cardiomyocytes were isolated from adult WT and Miat-KO mice, and their fractional shortening (FS) and calcium transients were recorded (Figure 6A-B). KO cardiomyocytes display a slightly increased cell contractility, reduced rise time and half-time of decay (T50) of the Ca2+ transient, but comparable fold change of calcium amplitude, which suggests enhanced SR efficiency during Ca2+ cycling. The resting calcium concentration is unaltered (Figure S10). Collectively, these functional changes in KO cardiomyocytes are consistent with the altered gene expression and attenuated hypertrophic response in the KO mice.
Deletion of Miat increases cardiomyocyte contractility. Cardiomyocytes were isolated from adult WT and Miat-KO mice. (A) Representative confocal line-scan images of Ca2+ transients (upper panels) along with their spatial averages and cell contractility by Fractional shortening (FS) (lower panels) in response to field stimulation (1 Hz). (B) Average calcium amplitude (ΔF/F0), cell fraction shortening, half-time of decay (T50) and the rise time of Ca2+ transient. *p < 0.05, **p < 0.01. n = 41-88.
Discussion
In this study, we have identified a previously unrecognized function of Miat in adverse cardiac remodeling during mechanic stress. Genetic deletion of Miat attenuates pressure overload-induced pathological hypertrophy and heart failure, which is associated with a favorable global change in hypertrophic gene expression in the stressed myocardium and enhanced calcium handling and contractility of cardiomyocytes. Although several recent reports have suggested potential involvements of Miat in cardiac pathologies [23-25, 36], our study for the first time demonstrates that endogenous Miat suppresses cardiomyocyte contractile function to contribute to the development of pathological hypertrophy and heart failure.
We used two cardiac hypertrophy mouse models, revealing distinct pathophysiological conditions. The Ang II model features a decreased ventricular chamber size and impaired diastolic function, which mimics heart failure with preserved EF (HFpEF) in clinical settings. The TAC model indicates typical heart failure with reduced EF (HFrEF), characterized by dramatic decrease in EF and FS and a dilated ventricular chamber. Although ACE inhibitors [37] /ARBs [38] /ARNI [39], β-blockers [40] and aldosterone antagonists [41] have been proven to improve the clinical outcome of HFrEF, none of them have shown remarkably beneficial effects in the treatment of HFpEF. It is exciting that our data indicates ablation of Miat protects against both systolic and diastolic cardiac dysfunction. This has brought the clinical significance for Miat as a potential therapeutic target in HF, especially in HFpEF. Further studies are warranted to test the role of Miat in other HFpEF models.
Our results reveal that, while the expression levels of Serca2a and RyR2 are comparable between WT and Miat-KO mice in the basal state, Ang II treatment induces a marked downregulation of Serca2a and RyR2 in WT mice but not in KO mice, suggesting that endogenous Miat is responsible for Serca2a and RyR2 downregulation during the hypertrophic stress. The beneficial roles of Serca2a and RyR2 expression in cardiomyocyte calcium handling during cardiac hypertrophy and heart failure have been well established [42-44]: while RyR2 activity results in rapid Ca2+-release from the sarcoplasmic reticulum (SR) and transient increase of intracellular Ca2+ concentration to trigger cell contraction, Serca2a removes Ca2+ from the sarcoplasm through Ca2+ reuptake into the SR to ensure cell relaxation. Knockout of Serca2a in mice leads to heart dysfunction [45], heterozygous Serca2a mice display accelerated heart failure under pressure overload [46], while cardiac-specific overexpression of a high Ca2+-affinity mutant of Serca2a attenuates pressure overload-induced cardiac hypertrophy [47]. Similarly, knockout of RyR2 is embryonic lethal with abnormal cardiomyocytes [48], and mutation of Ryr2 results in early cardiac hypertrophy and lethality [49]. Thus, our data support that Miat aggravates stress-induced Ca2+ mishandling through Serca2a and RyR2 downregulation. Notably, the better preserved Serca2a and RyR2 expression in Miat-KO mice precedes the alleviation of cardiac hypertrophy, suggesting a causal role of Miat-mediated Serca2a and RyR2 downregulation in the pathogenesis of cardiac hypertrophy.
We found attenuated cardiomyocyte apoptosis and interstitial fibrosis in the heart of KO mice, which is consistent with the enhanced systolic and diastolic function in these mice and suggests that depletion of Miat suppresses the cardiac remodeling during cardiac hypertrophy and failure. In addition, we observed a better-preserved capillary density in Miat-KO mice. While it is well known that cardiac hypertrophic pathology is associated with reduced vascular density, dysfunctional coronary vasculature has also been shown to contribute to the pathogenesis of cardiac hypertrophy [50]. Interestingly, Yan et al. showed that knockdown of Miat attenuates diabetes-associated deregulation of retinal microvasculature by protecting high glucose-induced apoptosis of endothelial cells [26]. Although the biological context in our study is different, and our RNA-seq data from the whole heart did not specifically indicate a prominent molecular signature of neovascularization, potential effects of Miat in the coronary circulation and function cannot be ruled out and warrants further investigation.
In our RNA-seq dataset from Ang II-stressed hearts, MMP2 and galectin-3 levels were lowered, while myeloperoxidase, TNF-α, and IL-6 levels were unaltered in Miat knockout mice compared to WT mice. Although galectin-3 is predominantly expressed by activated macrophages and a promising biomarker for patients with heart failure, it does not appear to affect the survival, systolic and diastolic dysfunction, cardiac fibrosis, and cardiomyocyte hypertrophy in the pressure-overloaded heart [51]. On the other hand, targeted deletion of MMP2 ameliorates myocardial remodeling in mice with chronic pressure overload [52], thus the reduced MMP2 expression in Miat-KO mice may have also potentially contributed to the attenuated cardiac fibrosis and remodeling.
Notably, we found that Ang II induces a marked increase in the splicing events in the myocardium of WT mice, which is largely blunted in Miat-KO mice. Although Miat has been shown to be involved in RNA splicing in neuronal and renal cells [18, 53-55], such as by interaction with the Qki and Srsf1 splicing factors to alter the splicing of Disc1 and ErbB4 in schizophrenia [56, 57], our study for the first time shows that Miat profoundly contributes to the RNA splicing in hypertrophic hearts. Recently, the roles of Qki and Srsf1 in cardiac [58, 59] and vascular [60-62] biology are increasingly appreciated. Further studies are underway to investigate whether Miat regulates cardiac RNA splicing through Qki and Srsf1 and what the functions of our identified splicing isoforms are in the pathogenesis of cardiac hypertrophy.
In conclusion, our results suggest that in the stressed heart, endogenous Miat jeopardizes cardiomyocyte calcium handling and contractile function, contributing to adverse myocardial remodeling and hypertrophy. Thus, in-depth investigation of Miat may lead to identification of new therapeutic targets.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Dr. Mary Flowers Braswell for her generous donation of the Vevo 2100 VisualSonics Fujifilm Imaging System. We thank Drs. Michael Crowley and David Crossman (Heflin Center for Genomic Science Core Laboratories at UAB) for the technical assistance with RNA sequencing and data analyses.
Sources of Funding
This work was supported by the National Institute of Health (HL113541, HL130052, HL131110, HL138990 to G.Q.; HL142291 to H.Q & G.Q.); American Heart Association (13POST17350000 to Y.Y.; 16POST29820001 to L.Y.; 18POST34070088 to S.X., 18PRE34080358 to E.Z.; 19CDA34630052 to A.Q.; and 19TPA34910227 to G.Q.); American Diabetes Association (1-15-BS-148 to G.Q.); and the startup funds from Northwestern University and University of Alabama at Birmingham (to G.Q.).
Data availability statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
Author Contributions
LY and GQ conceptualized the study; LY, JD, WM, AQ, SX, YY, CB, XK, DH, PE, JS, and EZ collected data; GQ, HQ, SN, SB, LZ, and JZ contributed to key resources; TL and JZ made intellectual contributions and assisted in data interpretations; LY and GQ wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP. et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation. 2020;141:e139-e596
2. Hill JA, Olson EN. Cardiac plasticity. The New England journal of medicine. 2008;358:1370-80
3. Tham YK, Bernardo BC, Ooi JY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Archives of toxicology. 2015;89:1401-38
4. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nature reviews Cardiology. 2018;15:387-407
5. Luo M, Anderson ME. Mechanisms of altered Ca(2)(+) handling in heart failure. Circulation research. 2013;113:690-708
6. Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E. et al. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601-5
7. Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128:191-227
8. Lehnart SE, Maier LS, Hasenfuss G. Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart Fail Rev. 2009;14:213-24
9. Dulhunty AF, Beard NA, Hanna AD. Regulation and dysregulation of cardiac ryanodine receptor (RyR2) open probability during diastole in health and disease. J Gen Physiol. 2012;140:87-92
10. Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circulation research. 2012;110:1646-60
11. Uszczynska-Ratajczak B, Lagarde J, Frankish A, Guigo R, Johnson R. Towards a complete map of the human long non-coding RNA transcriptome. Nat Rev Genet. 2018;19:535-48
12. Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML. et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570-83
13. Grote P, Wittler L, Hendrix D, Koch F, Wahrisch S, Beisaw A. et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Developmental cell. 2013;24:206-14
14. Viereck J, Kumarswamy R, Foinquinos A, Xiao K, Avramopoulos P, Kunz M. et al. Long noncoding RNA Chast promotes cardiac remodeling. Sci Transl Med. 2016;8:326ra22
15. Wang Z, Zhang XJ, Ji YX, Zhang P, Deng KQ, Gong J. et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nature medicine. 2016;22:1131-9
16. Han P, Li W, Lin CH, Yang J, Shang C, Nuernberg ST. et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102-6
17. Rapicavoli NA, Poth EM, Blackshaw S. The long noncoding RNA RNCR2 directs mouse retinal cell specification. BMC Dev Biol. 2010;10:49
18. Tsuiji H, Yoshimoto R, Hasegawa Y, Furuno M, Yoshida M, Nakagawa S. Competition between a noncoding exon and introns: Gomafu contains tandem UACUAAC repeats and associates with splicing factor-1. Genes Cells. 2011;16:479-90
19. Ishii N, Ozaki K, Sato H, Mizuno H, Saito S, Takahashi A. et al. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. Journal of human genetics. 2006;51:1087-99
20. Ip JY, Sone M, Nashiki C, Pan Q, Kitaichi K, Yanaka K. et al. Gomafu lncRNA knockout mice exhibit mild hyperactivity with enhanced responsiveness to the psychostimulant methamphetamine. Sci Rep. 2016;6:27204
21. Vausort M, Wagner DR, Devaux Y. Long noncoding RNAs in patients with acute myocardial infarction. Circulation research. 2014;115:668-77
22. Qu X, Du Y, Shu Y, Gao M, Sun F, Luo S. et al. MIAT Is a Pro-fibrotic Long Non-coding RNA Governing Cardiac Fibrosis in Post-infarct Myocardium. Sci Rep. 2017;7:42657
23. Li Y, Wang J, Sun L, Zhu S. LncRNA myocardial infarction-associated transcript (MIAT) contributed to cardiac hypertrophy by regulating TLR4 via miR-93. Eur J Pharmacol. 2018;818:508-17
24. Li Z, Liu Y, Guo X, Sun G, Ma Q, Dai Y. et al. Long noncoding RNA myocardial infarctionassociated transcript is associated with the microRNA1505p/P300 pathway in cardiac hypertrophy. International journal of molecular medicine. 2018;42:1265-72
25. Zhu XH, Yuan YX, Rao SL, Wang P. LncRNA MIAT enhances cardiac hypertrophy partly through sponging miR-150. Eur Rev Med Pharmacol Sci. 2016;20:3653-60
26. Yan B, Yao J, Liu JY, Li XM, Wang XQ, Li YJ. et al. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circulation research. 2015;116:1143-56
27. Wende AR, O'Neill BT, Bugger H, Riehle C, Tuinei J, Buchanan J. et al. Enhanced cardiac Akt/protein kinase B signaling contributes to pathological cardiac hypertrophy in part by impairing mitochondrial function via transcriptional repression of mitochondrion-targeted nuclear genes. Molecular and cellular biology. 2015;35:831-46
28. Tang YL, Zhu W, Cheng M, Chen L, Zhang J, Sun T. et al. Hypoxic preconditioning enhances the benefit of cardiac progenitor cell therapy for treatment of myocardial infarction by inducing CXCR4 expression. Circulation research. 2009;104:1209-16
29. Zhou J, Zhu Y, Cheng M, Dinesh D, Thorne T, Poh KK. et al. Regulation of vascular contractility and blood pressure by the E2F2 transcription factor. Circulation. 2009;120:1213-21
30. Wu M, Zhou J, Cheng M, Boriboun C, Biyashev D, Wang H. et al. E2F1 suppresses cardiac neovascularization by down-regulating VEGF and PlGF expression. Cardiovascular research. 2014;104:412-22
31. Cheng M, Zhou JL, Wu M, Boriboun C, Thorne T, Liu T. et al. CXCR4-Mediated Bone Marrow Progenitor Cell Maintenance and Mobilization Are Modulated by c-kit Activity. Circulation research. 2010;107:1083-U78
32. Cheng M, Zhou J, Wu M, Boriboun C, Thorne T, Liu T. et al. CXCR4-mediated bone marrow progenitor cell maintenance and mobilization are modulated by c-kit activity. Circulation research. 2010;107:1083-93
33. Arnone B, Chen JY, Qin G. Characterization and analysis of long non-coding rna (lncRNA) in In Vitro- and Ex Vivo-derived cardiac progenitor cells. PloS one. 2017;12:e0180096
34. Li D, Wu J, Bai Y, Zhao X, Liu L. Isolation and culture of adult mouse cardiomyocytes for cell signaling and in vitro cardiac hypertrophy. Journal of visualized experiments: JoVE. 2014
35. Deng J, Wang G, Huang Q, Yan Y, Li K, Tan W. et al. Oxidative stress-induced leaky sarcoplasmic reticulum underlying acute heart failure in severe burn trauma. Free radical biology & medicine. 2008;44:375-85
36. Zhou X, Zhang W, Jin M, Chen J, Xu W, Kong X. lncRNA MIAT functions as a competing endogenous RNA to upregulate DAPK2 by sponging miR-22-3p in diabetic cardiomyopathy. Cell death & disease. 2017;8:e2929
37. Group CTS. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The New England journal of medicine. 1987;316:1429-35
38. Cohn JN, Tognoni G, Valsartan Heart Failure Trial I. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. The New England journal of medicine. 2001;345:1667-75
39. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR. et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. The New England journal of medicine. 2014;371:993-1004
40. Effect of metoprolol CR/XL in chronic heart failure. Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet. 1999;353:2001-7
41. Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A. et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. The New England journal of medicine. 1999;341:709-17
42. Park WJ, Oh JG. SERCA2a: a prime target for modulation of cardiac contractility during heart failure. BMB Rep. 2013;46:237-43
43. Belevych AE, Radwanski PB, Carnes CA, Gyorke S. 'Ryanopathy': causes and manifestations of RyR2 dysfunction in heart failure. Cardiovascular research. 2013;98:240-7
44. Zhihao L, Jingyu N, Lan L, Michael S, Rui G, Xiyun B. et al. SERCA2a: a key protein in the Ca(2+) cycle of the heart failure. Heart Fail Rev. 2020;25:523-35
45. Andersson KB, Birkeland JA, Finsen AV, Louch WE, Sjaastad I, Wang Y. et al. Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. Journal of molecular and cellular cardiology. 2009;47:180-7
46. Ji Y, Lalli MJ, Babu GJ, Xu Y, Kirkpatrick DL, Liu LH. et al. Disruption of a single copy of the SERCA2 gene results in altered Ca2+ homeostasis and cardiomyocyte function. The Journal of biological chemistry. 2000;275:38073-80
47. Nakayama H, Otsu K, Yamaguchi O, Nishida K, Date MO, Hongo K. et al. Cardiac-specific overexpression of a high Ca2+ affinity mutant of SERCA2a attenuates in vivo pressure overload cardiac hypertrophy. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2003;17:61-3
48. Takeshima H, Komazaki S, Hirose K, Nishi M, Noda T, Iino M. Embryonic lethality and abnormal cardiac myocytes in mice lacking ryanodine receptor type 2. The EMBO journal. 1998;17:3309-16
49. Yamaguchi N, Takahashi N, Xu L, Smithies O, Meissner G. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca release channel. The Journal of clinical investigation. 2007;117:1344-53
50. Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y. et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444-8
51. Frunza O, Russo I, Saxena A, Shinde AV, Humeres C, Hanif W. et al. Myocardial Galectin-3 Expression Is Associated with Remodeling of the Pressure-Overloaded Heart and May Delay the Hypertrophic Response without Affecting Survival, Dysfunction, and Cardiac Fibrosis. The American journal of pathology. 2016;186:1114-27
52. Matsusaka H, Ide T, Matsushima S, Ikeuchi M, Kubota T, Sunagawa K. et al. Targeted deletion of matrix metalloproteinase 2 ameliorates myocardial remodeling in mice with chronic pressure overload. Hypertension. 2006;47:711-7
53. Zhou L, Xu DY, Sha WG, Shen L, Lu GY, Yin X. Long non-coding MIAT mediates high glucose-induced renal tubular epithelial injury. Biochemical and biophysical research communications. 2015;468:726-32
54. Spadaro PA, Flavell CR, Widagdo J, Ratnu VS, Troup M, Ragan C. et al. Long Noncoding RNA-Directed Epigenetic Regulation of Gene Expression Is Associated With Anxiety-like Behavior in Mice. Biol Psychiatry. 2015;78:848-59
55. Ishizuka A, Hasegawa Y, Ishida K, Yanaka K, Nakagawa S. Formation of nuclear bodies by the lncRNA Gomafu-associating proteins Celf3 and SF1. Genes Cells. 2014;19:704-21
56. Chung DW, Volk DW, Arion D, Zhang Y, Sampson AR, Lewis DA. Dysregulated ErbB4 Splicing in Schizophrenia: Selective Effects on Parvalbumin Expression. Am J Psychiatry. 2016;173:60-8
57. Barry G, Briggs JA, Vanichkina DP, Poth EM, Beveridge NJ, Ratnu VS. et al. The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Molecular psychiatry. 2014;19:486-94
58. Gupta SK, Garg A, Bar C, Chatterjee S, Foinquinos A, Milting H. et al. Quaking Inhibits Doxorubicin-Mediated Cardiotoxicity Through Regulation of Cardiac Circular RNA Expression. Circulation research. 2018;122:246-54
59. Ji X, Ding W, Xu T, Zheng X, Zhang J, Liu M. et al. MicroRNA-31-5p attenuates doxorubicin-induced cardiotoxicity via quaking and circular RNA Pan3. Journal of molecular and cellular cardiology. 2020;140:56-67
60. Xie N, Chen M, Dai R, Zhang Y, Zhao H, Song Z. et al. SRSF1 promotes vascular smooth muscle cell proliferation through a Delta133p53/EGR1/KLF5 pathway. Nature communications. 2017;8:16016
61. Cochrane A, Kelaini S, Tsifaki M, Bojdo J, Vila-Gonzalez M, Drehmer D. et al. Quaking Is a Key Regulator of Endothelial Cell Differentiation, Neovascularization, and Angiogenesis. Stem Cells. 2017;35:952-66
62. Caines R, Cochrane A, Kelaini S, Vila-Gonzalez M, Yang C, Eleftheriadou M. et al. The RNA-binding protein QKI controls alternative splicing in vascular cells, producing an effective model for therapy. Journal of cell science. 2019 132
Author contact
![]() Corresponding author: Gangjian Qin, MD, FAHA, Department of Biomedical Engineering, School of Medicine & School of Engineering, University of Alabama at Birmingham, 1720 2nd Ave S, Volker Hall G094N, Birmingham, AL 35294, USA. Tel: (205) 934-6690; Fax: (205) 934-9101; Email: gqinedu
Corresponding author: Gangjian Qin, MD, FAHA, Department of Biomedical Engineering, School of Medicine & School of Engineering, University of Alabama at Birmingham, 1720 2nd Ave S, Volker Hall G094N, Birmingham, AL 35294, USA. Tel: (205) 934-6690; Fax: (205) 934-9101; Email: gqinedu