Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
State-of-the-art: assays showing...
Benefits brought by functional...
Overview of CSRA clinical...
Expanding the field of...
Regulatory considerations and...
Concluding remarks
Abbreviations
Author Contributions
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2021; 11(19):9538-9556. doi:10.7150/thno.55954 This issue Cite
Review
Benefits of functional assays in personalized cancer medicine: more than just a proof-of-concept
Christophe Bounaix Morand du Puch1, Mathieu Vanderstraete1, Stéphanie Giraud1, Christophe Lautrette1, Niki Christou2,3, Muriel Mathonnet2,3 ![]()
1. Oncomedics SAS, 1 avenue d'ESTER, F-87069 Limoges, France.
2. Centre hospitalier universitaire Dupuytren, Service de chirurgie digestive, générale et endocrinienne, 2 avenue Martin Luther King, F-87042 Limoges, France.
3. EA3842/CAPTuR (Contrôle de l'Activation cellulaire, Progression Tumorale et Résistance thérapeutique), Faculté de Médecine, 2 rue du Docteur Marcland, F-87025 Limoges cedex, France.
Received 2020-11-15; Accepted 2021-5-16; Published 2021-9-21
Abstract

As complex and heterogeneous diseases, cancers require a more tailored therapeutic management than most pathologies. Recent advances in anticancer drug development, including the immuno-oncology revolution, have been too often plagued by unsatisfying patient response rates and survivals. In reaction to this, cancer care has fully transitioned to the “personalized medicine” concept. Numerous tools are now available tools to better adapt treatments to the profile of each patient. They encompass a large array of diagnostic assays, based on biomarkers relevant to targetable molecular pathways. As a subfamily of such so-called companion diagnostics, chemosensitivity and resistance assays represent an attractive, yet insufficiently understood, approach to individualize treatments. They rely on the assessment of a composite biomarker, the ex vivo functional response of cancer cells to drugs, to predict a patient's outcome. Systemic treatments, such as chemotherapies, as well as targeted treatments, whose efficacy cannot be fully predicted yet by other diagnostic tests, may be assessed through these means. The results can provide helpful information to assist clinicians in their decision-making process. We explore here the most advanced functional assays across oncology indications, with an emphasis on tests already displaying a convincing clinical demonstration. We then recapitulate the main technical obstacles faced by researchers and clinicians to produce more accurate, and thus more predictive, models and the recent advances that have been developed to circumvent them. Finally, we summarize the regulatory and quality frameworks surrounding functional assays to ensure their safe and performant clinical implementation. Functional assays are valuable in vitro diagnostic tools that already stand beyond the “proof-of-concept” stage. Clinical studies show they have a major role to play by themselves but also in conjunction with molecular diagnostics. They now need a final lift to fully integrate the common armament used against cancers, and thus make their way into the clinical routine.
Keywords: Cancer, chemosensitivity, CSRA, functional assay, personalized medicine
Introduction
It is widely accepted that, for diseases with complex and heterogeneous molecular backgrounds such as cancers, “one-size-fits-all” treatment strategies are no longer desirable. This is best illustrated by the unsatisfying response and survival rates observed on unselected patient cohorts for most classes of drugs. To propose and sort patients according to their predicted response to a cure, personalized medicine - or, more accurately, subpopulation medicine - requires reliable tests to stratify and ultimately retain relevant groups of responsive patients. Such tests measure specific response biomarkers in adequate samples. They are formally gathered under the companion diagnostics (CDx) designation [1].
True CDx are currently defined by the US Food and Drug Administration (FDA) as “a medical device, often an in vitro device, which provides information that is essential for the safe and effective use of a corresponding drug or biological product [and which] can identify patients who are most likely to benefit from a particular therapeutic product” [2]. The European Medicines Agency (EMA) provides a very similar definition [3]. Result of the test is usually a sufficient condition for giving or not a linked medication or group of medications [4]. Within the CDx family falls a subtype of assays called complementary diagnostics (CoDx). Instead of rigidly directing a patient to a treatment, CoDx rather provide information about the potentially enhanced benefits of receiving a drug. They do not make a specific drug mandatory, though, and a negative result does not disqualify the linked drug. Hence, the main difference between the two types of tests is the freedom of decision for the physicians regarding the choice of treatment for their patient [5].
The history, current role and perspectives of common CDx and CoDx have been extensively reviewed [5-8]. So far, such tests have been mostly developed in cancer indications and already possess a crucial role in personalized medicine. A growing number of treatments are now dependant not only on broad diagnosis, but also on the results of tests that identify actionable characteristics (molecular diagnostics). This justifies the requirement for concomitant safety and effectiveness assessments of both the drug candidate and its CDx during development steps [9]. Their role will expand in the future: indeed, predictive biomarkers are now integrated early in most drug development programs in oncology [10], with the encouragement of the FDA and a significant impact on drug approval, as exemplified in BIO's Clinical Development Success Rates 2006-2015 [11].
The definitions for CDx and CoDx are broad and understood as associating the presence of a specific biomarker (whether a single or a group of mutations, or a protein product) in a patient's body with a specific drug. This refers to the diseased tissue's history [12]. Nevertheless, another type of CoDx is frequently overlooked, namely functional assays. These are the mere transposition of preclinical in vitro assays, led on selected models to study the response to a drug candidate, to clinically-applicable ex vivo assays: indeed, they test on a patient's own cells, upstream of treatment initiation, the arsenal of therapies available for a specific indication. Instead of identifying the roots of a diseased phenotype, they capture the final, and as such clinically relevant, response to a drug produced by the interplay between all biological variables. This outcome acts as a composite, surrogate biomarker for drugs with no known indicator of susceptibility or resistance. Functional assays may hence provide a personalized medicine approach for systemic treatments, including chemotherapies, which have not been clinically associated with single or groups of biomarkers yet.
Predictive functional assays, especially chemosensitivity and resistance assays (CSRA), have been pursued for several decades [13]. They rely on the ex vivo modelling of a patient's tumour from pathologically-qualified samples obtained during a medical procedure (Figure 1): diagnosis biopsy, exeresis fragment of primary lesion or metastases, effusion, ascites, blood containing circulating tumour cells… Modelling protocols vary across a wide range, but described workflows share several common features. First, tumour material is processed to two-dimension (2D)/three-dimension (3D) primary cultures retaining the tumour cells' original characteristics. After in vitro exposure to treatments of interest, the biological response is analysed through a relevant endpoint to provide a functional profile (chemo-sensitivity or -resistance, DNA repair capacities…), which may mirror that of the original cancer lesions. This profile is ultimately integrated into the clinical decision-making process to individualize the treatment(s) a cancer patient will receive.
The role of ex vivo chemosensitivity and chemoresistance assays (CSRA) in cancer care, a virtuous cycle. Such assays use qualified patient samples and primary culture technologies to directly test the activity of relevant anticancer drugs on a patient's own tumour cells. The resulting chemosensitivity profile is usable by physicians to fine-tune treatments.
Despite long-standing efforts, an efficient protocol still has to obtain recognition by the biomedical community [14], let alone approval by regulatory authorities. The most recent assessment guidelines of CSRA by the American Society of Clinical Oncology (ASCO) came out less than a decade ago [15]. They followed a 2004 initial article on the matter [16], and we might infer a similar, updated evaluation might be published within the next 12-24 months. In these two articles, recommendations remained unchanged: “The use of CRSA to select chemotherapeutic agents for individual patients is not recommended outside of the clinical trial setting. Oncologists should make chemotherapy treatment recommendations on the basis of published reports of clinical trials and a patient's health status and treatment preferences. Because the in vitro analytic strategy has potential importance, participation in clinical trials evaluating these technologies remains a priority”. This statement enough shows the great potential the ASCO sees in CSRA, albeit they have not fulfilled clinicians' expectations yet.
In this review, we present the most advanced functional assays for treatment individualization in oncology. We then show through a meta-analysis of technical and clinical performances gathered so far that the notion of CSRA is already way beyond “proof-of-concept”. Challenges faced by functional assays, especially in their development along with the expansion of the anticancer drug modes of action (MoA), are presented. Finally, we discuss quality management of the environment of functional assays, as well as regulatory considerations framing their approval, for they provide both hurdles and handrails for the successful implementation of these assays within the clinical routine.
State-of-the-art: assays showing significant predictive capacities
Pioneer chemosensitivity assays were designed in the late 1970s. They were primarily based on clonogenic properties of tumours and yet showed promising results [17,18]. Since then, numerous chemosensitivity assays have been developed to predict ex vivo the action of a drug or a combination of drugs on a patient's tumour. Cell culture methods and readouts varied throughout the years, and techniques improved to reach high accuracy levels. In their vast majority, these functional assays follow common steps: (i) dissociation of a tumour specimen and isolation of tumour cells, (ii) primary cell culture in presence of chemotherapies, (iii) assay cell viability/mortality, and (iv) data analysis to produce a chemosensitivity profile (Table 1).
Overview of main chemosensitivity assays developed over the past five decades
| Functional assay | Culture method | Endpoint(s) | First study | Ref |
|---|---|---|---|---|
| Clonogenic assay | 3-D matrix culture of tumour cells | Colony formation and counting | 1970s | 17 |
| DiSC | Cell monolayer | Counting of cell mortality using light microscopy | 1983 | 19 |
| HDRA | 3D (collagen sponge) | MTT/[3H] thymidine incorporation | 1986 | 23 |
| FCA | Tumour fragments | Esterase-driven formation of fluorescein | 1988 | 27 |
| EDRA | Tumour fragments | [3H] thymidine incorporation | 1990 | 21 |
| MiCK | Cell monolayer | Measure of cell apoptosis by spectrophotometry | 1994 | 30-34 |
| CD-DST | 3D (collagen droplets) | MTT/ATP bioluminescence | 1996 | 26 |
| ATP-CRA | Cell monolayer | ATP bioluminescence | 1997 | 22 |
| ChemoFx® | Cell monolayer | Counting of cell number by fluorescence microscopy | 2002 | 36 |
| The Oncogramme® | Cell monolayer | Counting of cell mortality using light microscopy | 2010 | 38-40 |
| ChemoID® | Cell monolayer | Measure of cell proliferation using WST-8 | 2014 | 35 |
| CANScriptTM | Tumour fragments | Multiple | 2015 | 37 |
Innovative chemosensitivity assays were developed in the late 1980s by three American physicians and scientists, Robert Nagourney, David Kern and Larry Weisenthal. From their work were developed two non-clonogenic assays: the differential staining cytotoxicity assay (DiSC), and the Extreme Drug Resistance Assay (EDR), also known as the Kern assay. The DiSC assay is based on the counting of global cell death among the tumour cell population following drug treatment. Cell death observation relies on the loss of cell membrane integrity, which is observed by dye exclusion using Fast Green. Viable cells are then counterstained with hematoxylin/eosin, then cell mortality is evaluated using a light microscope [19]. This technique demands a high level of expertise, since it requires the ability to accurately recognize tumour cells from normal somatic cells. Interestingly, differential staining also applies to dead endothelial cells, allowing the observation of the effect of anti-angiogenics molecules such as the anti-VEGF bevacizumab [20]. The EDR assay relies on the measure of cell proliferation by counting the incorporation of 3H-thymidine. Following 72 h of drug treatment, cells are incubated with 3H-thymidine for another 48 h, allowing its incorporation during S-phase. Results of the assay are categorized as high, intermediate or low drug-resistance, by comparison with untreated controls [21]. EDR assay has been commercialized in the USA by Oncotech; however, this CSRA seems no longer available.
Numerous studies investigated the relevance of applying cell metabolism measurement techniques to primary tumour chemosensitivity. The first example is the ATP-Chemotherapy Response Assay (ATP-CRA). This technique relies on total ATP dosage by bioluminescence [22]. Tumour tissues harvested following surgical resection or biopsy are cut into small pieces, then cells are separated by enzymatic digestion. A given number of cells are cultured in presence of drugs, then the amounts of ATP are measured using a luminometer. Any drug impairing cell growth or proliferation will ultimately decrease the total amount of ATP within cells. From these results, a cell death rate is calculated. The high sensitivity of ATP bioluminescence allows to miniaturize the assay, which necessitates little tumour material.
Another cell metabolism assay designed for chemosensitivity assessment purpose is MTT. This technique has been extensively used for the development of the Histoculture Drug Response Assay (HDRA). Unlike ATP-CRA, HDRA is based on the culture of small pieces of the tumour (1-2 mm) on collagen-coated matrices [23]. Fragments are treated with chemotherapies for 48 to 96 h, then cell viability is evaluated. Another system named ITRA (Integrative Tumor Response Assay) was developed to determine the efficacy of second line treatments [24,25]. It consists in two successive HDRAs, the latter being performed on cells that survived the first round of chemotherapy. Another technique, called Collagen gel Droplet embedded culture Drug Sensitivity Test (CD-DST) has been widely developed. The CD-DST general concept relies on the embedding of tumour cells in collagen droplets to mimic the in vivo situation [26]. One advantage of this method is that collagen droplets gather tumour cells as well as non-tumoral cells and ECM components. Following droplet formation, cells are cultured in presence of anticancer drugs, and cell viability is assessed by estimating neutral red uptake and by measured growth inhibition rate (IR). IR is expressed as T/C ratio, with T being the optical density of treated collagen droplets, and C being that of the control group. In vitro drug sensitivity is then determined by implementing a growth inhibition rate threshold, which is mostly above 50%.
Although less developed, several other academic initiatives are worth mentioning, notably Fluorescent Cytoprint (FCA) and Sulforhodamine B (SRB) assays. FCA measures the activity of cytosolic esterases, which convert non-fluorescent fluorescein monoacetate into fluorescein. Small tumour fragments, named micro-organs, are cultured on collagen-coated metal grids. Drug-induced cytotoxicity is measured by comparing cytoprints, i.e. fluorescent microscopy pictures, before and after drug treatment [27]. Sulforhodamine B assay is a colorimetric assay that consists in quantifying the total amount of proteins, which reflects total cell number. To our knowledge, only two publications mentioned this assay as a putative CSRA [28,29].
Apart from these academic initiatives, several proprietary assays were also developed by biotechnology companies. Below are detailed five of the most advanced techniques:
- The MicroCulture-Kinetic assay (MiCK) is a drug-induced apoptosis assay. The history of this assay relies on the observation that most chemotherapies induce cell death via apoptosis [30]. Membrane blebbing and nucleus condensation are hallmarks of apoptosis: as such, they participate in a rise of optical density. Then the principle of MiCK assay is to measure OD600 following drug treatment [31]. Using a proprietary algorithm, the MiCK assay converts OD changes into Kinetic Units (KU), which indicate tumour chemosensitivity. First described in 1994, the MiCK assay is a precursor CSRA documented with numerous technical and clinical studies. Initially designed for leukaemia cells, the MiCK assay was later applied on solid tumour samples, including ovary, breast, lung and endometrium [32-34]. It is now commercialized by Pieran Biosciences as ChemoINTEL™;
- The ChemoID® assay has been recently commercialized by US-based company Cordgenics. It is atypical since it aims at providing separate bulk tumour cell and cancer stem-like cell (CSC) responses to chemotherapies. Procedures include the following steps: tumour dissociation, CSC enrichment in a bioreactor, cell sorting by flow cytometry, chemotherapy treatment, and finally a WST-8 cell proliferation assay [35]. This innovative technology looks to overcome CSC chemotherapy resistance occurring in some cancers, and to prevent recurrence. Randomized, assay-directed clinical trials are currently ongoing against glioblastoma and epithelial ovarian cancer, with overall response rate as primary outcome measure (ClinicalTrials.gov numbers: NCT03632798, NCT03949283 and NCT03632798);
- The ChemoFX® assay has been developed by the US company Helomics and is dedicated to gynaecological cancers [36]. Its endpoint is total DNA quantification. Global procedure includes primary culture of 1 mm3 tumour fragments until confluency, then trypsinization and subculture into 384-well plates with drugs. Serial dilutions are tested and, following a 72-h treatment, cells are stained with DAPI then counted by fluorescent microscopy. Tumour response is evaluated by measuring the area under the curve.
- The CANscript® technology has been developed by Indian company Mitra Biotech. This technique recreates the native tumour environment by culturing thin tumour explants into a 3D matrix, in presence of autologous serum [37]. Prediction of clinical outcomes is achieved by combining several readouts such as viability, proliferation and apoptosis using a machine learning proprietary algorithm;
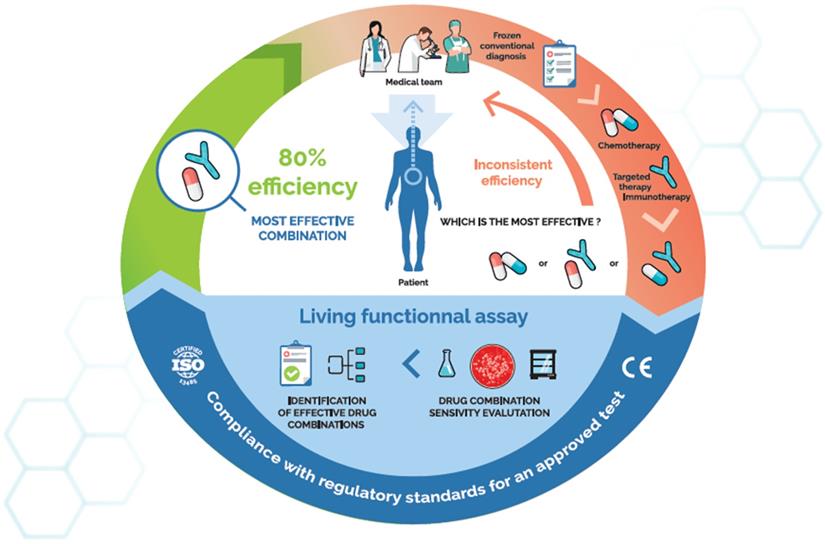
- The Oncogramme® has been developed by the French company Oncomedics to which belong/belonged some of the authors of this review. It consists in the measurement of drug-induced cellular mortality using fluorescence microscopy. Its main advantage lies in the use of a fully standardized, serum-free cell culture medium, allowing both optimal reliability and high culture success rates [38-40]. Moreover, the Oncogramme® was designed to measure the effect of drug combinations routinely used in cancer patient care, such as FOLFOX or FOLFIRI. The Oncogramme® was originally developed against metastatic colon cancer (ClinicalTrials.gov number: NCT02305368), and its use in breast and ovary cancers is currently under investigation (ClinicalTrials.gov number: NCT02910622). The Oncogramme® is, to our knowledge, the only CE-marked functional assay, allowing its distribution within the European Union. A randomized, assay-directed phase 3 clinical trial is currently ongoing to evaluate its capacity to improve 1-year progression free survival (PFS) of metastatic colon cancer patients (ClinicalTrials.gov number: NCT03133273).
Benefits brought by functional assays across indications: a meta-analysis
Criteria used to evaluate CSRA technical performances and results
Numerous proof-of-concept studies have been performed on a wide panel of malignant pathologies using methods described above. Table 2 and Table 3 summarize studies in which clinical and technical performances have been evaluated.
Overview of the analytical performances of the major chemosensitivity assays described in the literature against a large array of solid cancers
| Assay | Cancer | Success rate | Accuracy | PPV | NPV | Cohort size (number of patients) | Sensitivity | Specificity | Authors | References |
|---|---|---|---|---|---|---|---|---|---|---|
| ATP | Gastrointestinal | 85.0% | 84.0% | - | - | 25 | 64.0% | 100.0% | Kawamura et al., 1997 | [41] |
| ATP | Ovary | 89.0% | 71.0% | 66.0% | 89.0% | 93 | 95.0% | 44.0% | Konecny et al., 2000 | [42] |
| ATP | Ovary | 85.0% | 85.0% | 50.0% | 100.0% | 33 | 100.0% | 82.0% | Ng TY et al., 2000 | [43] |
| ATP | Ovary | - | 70.7% | 83.0% | 56.5% | 161 | 68.8% | 74.3% | O'Meara et al., 2001 | [44] |
| ATP | Lung | 90.6% | 90.0% | 100.0% | 80.0% | 31 | 83.3% | 100.0% | Kim BS et al., 2004 | [45] |
| ATP | Lung | 90.6% | - | - | - | 53 | - | - | Kang et al., 2005 | [22] |
| ATP | Lung | 43.8% | 68.8% | 61.1% | 78.6% | 34 | - | - | Moon YW et al., 2007 | [46] |
| ATP | Breast | 93.0% | 85.0% | 100.0% | 66.7% | 43 | 78.6% | 100.0% | Kim et al., 2008 | [47] |
| ATP | Ovary | 69.0% | 90.0% | 94.1% | - | 29 | 94.1% | - | Han et al., 2008 | [48] |
| ATP | Gastrointestinal | 95.8% | 77.8% | 85.7% | 75.9% | 36 | 46.2% | 95.7% | Kim JH et al., 2010 | [49] |
| ATP | Colon | 79.0% | - | 94.0% | 38.0% | 62 | - | - | Lee et al., 2011 | [50] |
| ATP | Bladder | 96.3% | 74.3% | 83.7% | 66.7% | 54 | 97.6% | 20.0% | Ge et al., 2012 | [51] |
| ATP | Ovary | - | - | 83.0% | 84.8% | 80 | 88.6% | 77.8% | Zhang et al., 2015 | [52] |
| HDRA | Head and Neck | 88.0% | 74.0% | 83.0% | 64.0% | 26 | 71.0% | 78.0% | Robbins et al., 1994 | [53] |
| HDRA | Gastric & Colon | 96.3% | - | 66.7% | 100.0% | 38 | 100.0% | 90.6% | Furukawa et al., 1995 | [54] |
| HDRA | Ovary | 97.0% | 87.0% | 88.0% | 86.0% | 15 | 88.0% | 86.0% | Ohie et al., 2000 | [55] |
| HDRA | Breast | 98.8% | 80.0% | 100.0% | 70.0% | 15 | 62.5% | 100.0% | Tanino H et al., 2001 | [56] |
| HDRA | Head and Neck | 97.6% | - | - | - | 42 | - | - | Singh et al., 2002 | [57] |
| HDRA | Head and Neck | - | 91.7% | 90.0% | 100.0% | 19 | 79.0% | 66.7% | Ariyoshi et al., 2003 | [58] |
| HDRA | Head and neck | - | 77.8% | 76.9% | 80.0% | 49 | 90.9% | 57.1% | Hasegawa et al., 2007 | [59] |
| HDRA | Head and Neck | 91.0% | 74.0% | 69.0% | 80.0% | 57 | 79.0% | 71.0% | Pathak et al., 2007 | [60] |
| HDRA | Lung | 97.4% | 83.0% | 73.2% | 100.0% | 343 | 100.0% | 68.1% | Yoshimasu et al., 2007 | [61] |
| HDRA | Ovary | - | - | 62.0% | 81.0% | 61 | 90.0% | 43.0% | Neubauer et al., 2008 | [62] |
| HDRA | Oesophagus | 89.3% | - | - | - | 53 | 66.7% | 55.6%-66.7% | Fujita et al., 2009 | [63] |
| HDRA | Glioma | 94.0% | - | - | - | 33 | 100.0% | 60.0% | Gwak H et al., 2011 | [64] |
| HDRA | Colon | - | 66.3% | - | - | 86 | 72.7% | 54.7% | Yoon et al., 2012 | [65] |
| ITRA | Colon | - | 61.9% | 57.1% | 64.3% | 42 | 44.4% | 75.0% | Yoon et al., 2017 | [24] |
| ITRA | Ovary | - | 44.4% | 40.0% | 66.7% | 18 | 85.7% | 18.2% | Kim et al., 2019 | [25] |
| CD-DST | Multiple | 80.0% | 91.0% | 80.0% | 100.0% | 11 | 100.0% | 86.0% | Kobayashi et al., 1997 | [66] |
| CD-DST | Breast | 84.3% | 87-94.4% | 83.3% | 95.5-100% | 70 | 92.9% | 62.5-95.5% | Takamura et al., 2002 | [67] |
| CD-DST | Mesothelioma | - | 50.0% | - | - | 26 | 100.0% | 36.0% | Higashiyama et al., 2008 | [68] |
| CD-DST | NSCLC | - | 70.0% | 50.0% | 92.0% | 81 | 88.0% | 63.0% | Higashiyama et al., 2010 | [69] |
| CD-DST | Gastric | 80.0% | - | - | - | 64 | - | - | Naitoh et al., 2014 | [70] |
| CD-DST | OSCC | 81.8% | 92.3% | 90.9% | 100.0% | 14 | - | - | Sakuma et al., 2017 | [71] |
| FCA | Multiple | - | - | 85.0% | 97.0% | 73 | 98.0% | 81.0% | Leone et al., 1991 | [27] |
| The Oncogramme® | Colorectal | 97.4% | 63.6% | 64.7% | 60.0% | 19 | 84.6% | 33.3% | Bounaix Morand du Puch et al., 2016 | [39] |
| CANScript™ | Multiple | - | - | 93.9% | 100.0% | 55 | - | - | Majumder et al., 2015 | [37] |
| ChemoFX® | Ovary | - | - | 63.6% | 100.0% | 18 | - | - | Ness et al., 2002 | [72] |
| ChemoFX® | Breast | 83.9% | - | - | - | 62 | - | - | Mi et al., 2008 | [73] |
| ChemoFX® | Head and Neck | 72.7% | - | 81.8% | - | 22 | - | - | Jamal et al., 2017 | [74] |
| ChemoID® | Glioblastoma | - | - | 54.6% | 100.0% | 11 | 100.0% | 50.0% | Claudio et al., 2017 | [75] |
| MICK | Endometrium | 78.9% | - | - | - | 19 | - | - | Ballard et al., 2010 | [32] |
| Mean | 86.6% | 76.1% | 76.7% | 82.0% | 51.8 | 84.2% | 68.3% | |||
| Median | 89.2% | 77.8% | 82.4% | 81.0% | 40.0 | 88.0% | 72.7% |
Meta-analysis of studies that explored through clinical investigation the capacity of chemosensitivity assays in improving patient outcomes
| Reference | Authors | Pathology | Assay | Cohort size | Randomisation | Treatment | Outcomes |
|---|---|---|---|---|---|---|---|
| [76] | Strickland et al., 2013 | AML | MiCK | 109 | No | According to physicians | MiCK assay results correlate well with clinical outcome of patients in terms of OS and response rate. |
| [67] | Takamura et al., 2002 | Breast | CD-DST | 70 | No | According to physicians | No differences in OS between drug-sensitive and resistant patients.Longer TTP in drug-sensitive patients (15.6 vs. 2.5 months, p < 0.005). |
| [77] | Bosserman et al., 2015 | Breast | MiCK | 30 | No | CSRAs results to be used at physician's discretion | The use of the MiCK assay led to a higher response rate (38.1% vs. 0%, p = 0.04), and longer TTP (7.4 vs. 2.2 months, p < 0.01). |
| [78] | Kim et al., 2014 | Breast | HDRA | 50 | No | According to physicians | No correlation between breast cancer subtype and chemoresponse found using HDRA. |
| [79] | Shinden et al., 2016 | Breast | HDRA | No | Paclitaxel | Paclitaxel inhibition rate is significantly associated with DFS (p = 0.036). | |
| [80] | Mekata et al., 2013 | Colon | CD-DST | 151 | No | According to physicians | No differences in OS for patients found with high- and low-sensitivity for 5-FU.Significant differences in 5-year RFS (p = 0.04). |
| [81] | Ji et al., 2017 | Colon | HDRA | 89 | No | 5-FU | Better 5-year PFS in chemosensitive group.No significant improvement of OS. |
| [82] | Hur et al., 2012 | Colorectal liver metastasis | CD-DST | 63 | Yes | According to CSRA results or physician's choice | Better treatment response. |
| [83] | Kubota et al., 1995 | Gastric cancer | HDRA | 128 | No | Mitomycin C and tegafur | OS and DFS are longer in the HDRA-sensitive group for both drugs. |
| [70] | Naitoh et al., 2014 | Gastric cancer | CD-DST | 64 | No | According to CSRA results | Higher survival rate in patients found as drug sensitive (p = 0.019). Longer time to progression (p = 0.023). |
| [84] | Tanigawa et al., 2016 | Gastric cancer | CD-DST | 206 | No | S-1 | Better relapse-free survival in drug-responder subgroup (p = 0.0014). |
| [85] | Howard et al., 2017 | Glioblastoma | ChemoID® | 41 | No | According to physicians | Longer OS and recurrence time in patients with positive stem cell chemoprofile. |
| [57] | Singh et al., 2002 | Head and Neck | HDRA | 41 | No | 5-FU, cisplatin | Correlation between HDRA chemoresponse and clinical outcome. |
| [86] | Wilbur et al., 1992 | Lung | DiSC | 45 | No | According to physicians | Improved OS in drug-sensitive patient subgroup (p = 0.04). |
| [46] | Moon et al., 2009 | Lung | ATP | 120 | No | CSRA-guided treatment | No significant differences in PFS and OS between both groups.Higher response rate in ATP subgroup (71% vs. 38%, p = 0.023). |
| [87] | Akazawa et al., 2017 | Lung | CD-DST | 39 | No | platinum-based adjuvant chemotherapy | Better 5-year DFS in chemotherapy-sensitive patients (p = 0.037).No differences in OS. |
| [88] | Inoue et al., 2017 | Lung | CD-DST | 87 | No | According to phyisicians | No differences in OS and 5-year DFS. |
| [89] | Chen et al., 2017 | Lung | ATP | 120 | No | According to physicians | Improved PFS and OS in chemosensitive groups (p = 0.046 and p = 0.041, respectively). |
| [90] | Ugurel et al., 2006 | Melanoma | ATP | 53 | ¨No | Assay-directed chemotherapy | Chemosensitive patients showed improved OS (14.6 months vs. 7.4, p = 0.041). Progression arrest in more patients (59.1% vs. 22.6%, p = 0.01). |
| [33] | Bosserman et al., 2012 | Multiple | MiCK | 40 | No | CSRAs results to be used at physician's discretion | Increased response rates when physicians used MICK assay (44% vs. 6.7%, p > 0.02). |
| [91] | Kurbacher et al., 1998 | Ovary | ATP | 55 | No | According to CSRA results | Higher overall response rate (64% vs. 37%, p = 0.04) in Assay group.Better PFS in platinum-refractory patients (p = 0.004). |
| [92] | Gallion et al., 2006 | Ovary | ChemoFX® | 256 | No | According to physicians | Correlation of ChemoFX assay results with Progression-Free Interval. |
| [93] | Cree et al., 2007 | Ovary | ATP | 147 | Yes | According to CSRA results or physician's choice | No significant differences for OS, RR or PFS. |
| [94] | Herzog et al., 2010 | Ovary | ChemoFX® | 192 | No | According to physicians | Correlation of ChemoFX assay results with median OS. |
| [34] | Salom et al., 2012 | Ovary | MiCK | 150 | No | According to physicians | Longer OS and RFP in stage III and IV patients that received the best chemotherapy (p > 0.01 and p = 0.03, respectively). |
| [95] | Jung et al., 2013 | Ovary | HDRA | 104 | No | According to physicians | Longer PFS in chemosensitive patients (34.0 vs. 16.0 months, p = 0.03). |
| [96] | Park et al., 2016 | Pancreas | ATP | 57 | No | Gemcitabine | Better disease-free survival in gemcitabine-sensitive patients (p = 0.017) |
A total of 42 studies have been included, from 1991 to 2019. Clinical studies older than 1990 and/or related to clonogenic assays were deliberately omitted.
First interesting observation is the high primary culture success rate obtained in most studies. It ranges from 43.8% to 98.8%, with a mean of 86.6%. Of note, the lower limit of this range, obtained on lung cancer [47], was the only value below 72.7% among the 28 we report here. When excluded, the mean rises to 88.2%, better accounting for the success rate of primary cultures. This clearly demonstrates the ability of all these methods to culture primary tumour cells from tumour explants. Primary cell culture is often optimized using serum-containing media, which favour cell proliferation, but do not allow assay standardization. Notably, ATP-CRA and HDRA protocols usually add 5 to 20% serum to culture media [47,70]. Noteworthy, this hindrance has been overcome in several techniques such as the Oncogramme®, without affecting success rate [39].
In the context of CSRA, sensitivity represents a key parameter to study, since it measures the proportion of responders properly identified as such by the assay [97]. In other words, sensitivity is an index of prediction efficacy. Sensitivities calculated in studies and listed in Table 2 vary from 44.4% to 100%, with a median of 98.0%. No method shows any greater efficiency, as mean sensitivities for all techniques are all above 80% [9]. Noticeably, the ITRA method has lower sensitivity percentages. However, this exploratory technique aims at evaluating tumour response to second-line agents, and therefore could hardly be compared with other CSRA. As a result of such good sensitivity indexes, positive predictive value (PPV), which measures the proportion of accurate predictions among all positive calls, is also high and reaches a median of 83.0% (Table 2). This high percentage emphasizes the global precision of chemosensitivity assays.
Opposite to sensitivity, specificity represents the percentage of true negative, i.e. the proportion of non-responders among the population found as such by the assay. In studies listed in Table 2, specificity is significantly lower than sensitivity, but still reaches 72.7%, with percentages varying from 18.2 to 100%. In that context, specificity actually corresponds to chemoresistance measurement, which is not the primary objective of chemosensitivity assays. Besides, dedicated assays have been developed to measure chemoresistance, the most advanced one being the Extreme Drug Resistance Assay (EDRA) [21,98,99]. Negative predictive value (NPV), the index of false negatives among all negative calls, has a median value of 82.9%, suggesting that in most cases CSRA are also able to accurately identify non-responding patients. Finally, accuracy (measured as the percentage of true positive and true negative patients among total population) represents the best index of CSRA effectiveness, as it measures the probability of a correct prediction. In this meta-analysis, accuracy ranges from 44.4% to 94.4% and reaches a median value of 77.8%.
Taken together, these results demonstrate the accuracy of chemosensitivity assays, with an ability to predict positive outcomes of around 90%. Despite being high, prediction of non-responders is still perfectible in most cases, as specificity is almost systematically lower than sensitivity. However, results must be taken with care, since many of the abovementioned studies were conducted using heterogenous samples, i.e. which already underwent chemotherapeutic treatments, or from diverse histologic grades. Because of this lack of consistency, one can hardly determine whether one chemosensitivity assay gives better results than another. To our knowledge, only few comparative studies were performed on the same tumour material. In ovarian cancer, it has been shown that DiSC, ATP tumour chemosensitivity and MTT assays correlate well [100]. Further comparative studies will be needed to answer about this question.
Overview of CSRA clinical performances
To ensure the reliability of CSRA, technical performances must be linked to clinical benefits. Among 27 clinical studies listed in our meta-analysis (Table 3), only two of them failed to find significant benefits for the patients [88,93]. Most of these studies were retrospective evaluations and allowed to correlate chemo-responses to clinical outcomes. In various studies, patients treated with a regimen for which they have been found sensitive showed better clinical outcomes in terms of PFS, time to progression (TTP) or overall survival (OS). As an example, patients with advanced ovarian cancer found as paclitaxel- and carboplatin-sensitive using HDRA showed a significantly longer PFS than patients categorized as resistant (34 vs. 16 months, p = 0.025) [95]. These encouraging results were obtained in studies covering a wide range of solid malignancies including breast, colon, lung or gastric cancers.
So far, seven studies were conducted in a prospective manner, with treatments being guided by CSRA results. Again, patients who benefited from CSRA showed better clinical outcomes. OS was statistically improved in one study (14.6 vs. 7.4 months, p = 0.041) [90]. In addition, several other studies put into evidence longer PFS [91], time to progression [77] or response rates [33,46,77,81,82,91]. To our knowledge, only two randomized study investigated the contribution of CSRA-guided chemotherapy [82,93]. Some other blinded, randomized studies are currently ongoing in stage IV colon cancer (the Oncogramme®, NCT03133273), glioblastoma (ChemoID®, NCT03632135) and ovarian cancer (ChemoID®, NCT03632798).
Besides clinical cohort studies, case reports also highlighted the usefulness of chemosensitivity testing before choosing a chemotherapy [101-108]. Among others, ATP-based and CD-DST assays were used to help treatment decision for rare pathologies such as colorectal choriocarcinoma [101], Stewart-Treves syndrome [106], or parathyroid carcinoma [108].
Taken together, this meta-analysis clearly demonstrates the efficacy and usefulness of CSRA in various cancer types. Beyond satisfying technical performances, clinical studies also showed that the use of CSRA could lead to clinical benefits for patients. However, as urged by the ASCO, this tendency must be confirmed with more solid interventional, randomized studies, with patients being treated according to the results of the assays.
Expanding the field of functional assays
In the previous sections, we discussed the most advanced functional assays, having reached a high degree of prediction through clinical evidence. Each assay has its own advantages and limitations, leaving room for substantial improvement in ex vivo modelling and measurement of functional response to an ever-expending array of drug classes. Several parameters should be taken into consideration for a continuous adaptation to biology and medical practice. In this section, we pinpoint those we identified as the most relevant. In addition, we discuss other technologies dedicated to treatment response prediction, with varying degrees of development and clinical implementation.
Accurate cancer models are more crucial than ever, both at the drug development and clinical practice levels, as illustrated by the despised role of historical cell lines [109]. Heterogeneity and microenvironment influence are main targets in the development of relevant ex vivo models. Intrinsic heterogeneity is one of the characteristics of cancer, deriving from both genomic instability, one of its so-called hallmarks, and immunoedition [110-112]. A first degree of tumour heterogeneity is reflected in metastasis. Indeed, advanced solid tumours spread through metastasis from a subset of cells displaying a more aggressive and mobile phenotype. Throughout the course of carcinogenesis, it is hypothesized these resistant cells progressively overcome other tumour cells, notably through Darwinian selection induced by selection pressure [113], immunoedition [114] and tumour repopulation following chemoradiation treatment [115]. They then produce distant lesions whose characteristics may greatly differ from the primary lesion. The purpose of chemotherapy in adjuvant setting is to control the primary lesion and avoid the occurrence of metastases. In the neo-adjuvant setting, its aim is to reduce the size of metastases and bring them to resecability. For advanced/metastatic solid cancers, one may thus wonder whether it is appropriate to target only one lesion for chemosensitivity assessment, especially if it is the primary tumour. Chemosensitivity comparison between primary and distant lesions has been investigated. Results actually suggest different profiles, with metastases being frequently more resistant to chemotherapy than primary lesions [116-118]. However, the true clinical impact of this difference remains to be established: a randomized study on CRC using ATP-CRA showed that treatment selection upon chemosensitivity profiles deduced from primary lesions still increased liver metastasis resecability rate [82]. It appears the best scenario would be for clinicians to receive comprehensive information about the highest possible number of a patient's lesions to finely tune their therapeutic approach. This could lead to the destruction of all tumour cells, independently of their degree of aggressiveness. However, in most cases, every lesion will not be accessible for sampling. Sampling being selective in nature, there is a non-negligible probability to miss during this procedure a tumour zone containing more aggressive subclones [111]. This sampling bias can occur both at the tumour resection and histopathological analysis steps. In addition, if several samples must be tested for each patient, this may significantly raise the complexity and costs of the whole procedure, making it economically less relevant. Fine cost calculations should be performed once the final test format is set, and appropriate resources mobilized accordingly to ensure full accessibility to patients. As metastases are the main target of chemotherapy, their functional analysis should be prioritized over primary lesions. Also, despite arguably being the most crucial switch in anticancer therapy, tumour invasion and metastasis blockades has received less attention than cytotoxic therapies [119]. Future investigations will hopefully demonstrate whether treatment individualization may also be envisioned in that area.
Metastasis spreading through rare, mobile cells illustrates the importance of capturing functional response at the single cell level, instead of basing recommendations on a global response of mixed cell populations. Current CSRA do not differentiate between cancer cell subpopulations. Consequently, they are not able to identify treatment-resistant cells, usually present in very low number, before they generate disease recurrence. Most assays select tumour cells from the large number of cells extracted from sampled primary or distant lesions, whether by differential centrifugation, functionalized microbeads, FACS or selective culture conditions. These assays hence rely on a more homogenous subpopulation to derive clinically useful sensitivity profiles. However, rarer, more aggressive cells still end up lost within the dynamic range. Either label-free cell sorting technologies [120] or co-staining protocols coupled to image analysis and detection algorithms may improve the specific detection of rare cells at any relevant step of the CSRA workflow. Circulating tumour cells remain easily accessible and represent a target of choice in that regard. Their chemosensitivity prediction role has proven possible on various cancer samples from molecular signatures [121]. Their transferability to ex vivo assays, however, may require sufficient expansion of this rare material, with spheroids as the final testable product [122,123]. Additionally, effusion-derived cancer cells, accessible and more abundant, have also proven another useful source of material for chemosensitivity prediction in a large array of cancers [124].
Another bias that hampers representativeness of ex vivo models results from the molecular drift that may occur during the primary culture phase. This has indeed been a concern for patient-derived xenografts (PDX) models [125, 126] discussed later in this section, although overall stability throughout passages has actually been demonstrated in colon PDX collections [127]. CSRA used in a clinical environment aim for a reduced turnaround time for practicality reasons. Consequently, short-time primary culture, with very limited number of passages, should limit this effect and preserve representativeness [128]. Mid- or high-throughput molecular biology technologies such as qPCR-based assays (already implemented into the clinical setting for diagnosis purpose) or next-generation sequencing methods (making their way through the clinical laboratory), combined with pathology-specific gene panels, may help ensuring the most relevant molecular alterations are maintained in the cultivated cells of a specific model [129]. Once again for practical reasons and reduced costs, such approaches can be utilized at the research and development stage of a CSRA, to establish the model's quality. The continuous clinical implementation of advanced molecular technologies may later allow their direct use during clinical implementation of the CSRA, as a quality control tool.
Tumour heterogeneity is also reflected in tumour microenvironment (TME). Two-dimension (2D) models obtained from dissociation of tumour tissue have been favoured so far. Obviously, such models are not comprehensively representative of tumour contexture, lacking key elements from the TME that cannot be fully compensated for with specifically formulated culture media. 3D models receive more and more attention to study the impact of drugs on tumour growth, neo-angiogenesis, or interaction between immune cells and tumours [130,131]. 2D models do not preserve tumour architecture, both at the tissue and cellular levels, nor do they systematically maintain the expression of relevant signalling intermediates [132]; they may also alter tumour cell proliferation compared to their patient counterpart [133]. 3D models, on the other hand, are thought to better recapitulate actual pharmacokinetics, with gradients of drug accessibility within the 3D structure, and an inner core that might stay safe from cytotoxic activity. Such models encompass a large array of technical approaches: tumour cells/fibroblast cocultures with direct contact (examples reviewed in [130]), spheroids derived from either single cell or multicellular structures [134], organoids [135], tissue fragments [136]. This list can be extended with more recent bio-printing techniques: they use layer-by-layer deposition of bio-inks to combine tissue spheroids or cell pellets with de-cellularized extra cellular matrix; effective vascularization through a computer-aided pre-designed structure allows generating viable 3D constructs [137, 138]. 3D models also increase the diversity of measurable endpoints in space and time, from metabolic activity to biomarker immunodetection [131]. Some assays have been brought to ex vivo/in vivo comparison to study the predictive capacities of the model, a few examples of which follow. First, short-term culture of tumour fragments on poly-2-hydroxyethylmetacrylate (PolyHEMA) was used to investigate the chemosensitivity of various patient samples, including liver metastases, to irinotecan active metabolite SN-38 [139]. This coating produced 3D nodules, while preventing fibroblast invasion during culture. A proliferation index was measured on nodule sections. Comparison with response evaluation criteria in solid tumours (RECIST) [140] measured in a small patient subset suggested a trend, albeit statistically insignificant, towards effective response prediction to SN-38 for this model. It does not seem to have been pursued further yet to evaluate clinical significance. A more ambitious study demonstrated the representativity of patient-derived organoid models of CRC and gastroesophageal cancers [141]. It was shown that spatiotemporal molecular and expression heterogeneity was preserved for different lesion sites and over treatment course; in addition, excellent chemosensitivity performances were observed. Exploiting similar models, the TUMOROID clinical trial studied the capacity of a CRC patient-derived tumour organoid-based assay to identify non responders to first- and second-line chemotherapy [142]. While being hampered by only a 63% culture success rate, it demonstrated excellent predictivity, although for irinotecan-based therapies only. In CRC-induced peritoneal carcinomatosis, chemotherapy is a major instrument at various level of disease management [143], notably intraperitoneal chemotherapy associated to hyperthermia (HIPEC) and pressurized intraperitoneal aerosol chemotherapy (PIPAC). Organoids generated from CRC patient-derived xenograft (PDX) models have proven effective to study the efficacy against peritoneal metastases of chemotherapeutic protocols involving HIPEC [144]. Building on these seemingly relevant models from Fanny Jaulin's laboratory, a clinical study termed ORGANOTREAT-01 should start soon, investigating the predictive capacity of organoids from digestive cancer samples against a large drug panel. All these models involve 3D structures obtained from non-adherent culture conditions, which, contrary to gel-like matrices, allow better addition and washout of drugs. However, they do not accurately mimic the in vivo drug bioavailability influenced by, among other factors, administration route, plasma clearance, extracellular matrix and cancer-associated fibroblasts (CAF). A thorough reconstitution of TME is illustrated by the CANscriptTM platform, which uses explants cultivated within a tumour grade-matched protein matrix supplemented with autologous patient serum [37,145,146]. Still, such assays complexify the whole procedure, especially when autologous supporting material (serum, fibroblasts) is required. This may negatively influence time-to-results and costs, thus restricting test execution to specialized laboratories.
Desirable characteristics to improve models include miniaturization, tight control of culture conditions over time, throughput enhancement, and study of orthogonal functional response parameters [37]. Borrowing to other “hard sciences”, more complex culture systems couple these characteristics with targeted imaging or electromagnetic endpoints: technologies such as microfluidics [147] and organ-on-a-chip, also termed as “microscale cell culture analogues” [148], are used to measure chemosensitivity. Numerous tumours can be effectively cultured, such as lung, bone marrow, brain, breast, urinary system (kidney, bladder and prostate), intestine and liver. Co-culturing of multi-tissue types in tumour-on-a-chip systems, and specifically heart-liver-intestine co-culture [132], which allow studying interdependdent effects of multiple miniaturized organs, offer the most realistic models to recapitulate the tumour in vivo-like microenvironment. Label-free, non-destructive biophysical treatment sensitivity biomarkers are also investigated, hence mass accumulation rates determined at the single-cell level [149,150], or shifts in impedance spectra of melanoma fragments measured by impedance spectroscopy [151]. Applicability to every cancer, being “solid” or “liquid”, of all these modelling technologies, whether 3D or biophysical, remains to be demonstrated [142].
Besides accurate tumour modelling, to be predictive an assay should measure: (i) how efficiently a drug induces cancer cell killing, as it is the foundation of tumour shrinkage; and (ii) which cancer cells are killed among a heterogeneous subpopulation; as previously stated, the latter is particularly important to predict recurrence. Chemoradiation therapy induces cell death through a large variety of mechanisms [152,153]. A true measure of cell death, including all its forms and without losing information due to assay conditions [154], should best predict the effect of a cytotoxic drug [153,155,156]. Future assays should be more adaptable to specific drug MoA. A solid biomarker hypothesis is required for trustable CDx. It appears less necessary in the case of functional assays, provided the chosen endpoint is clinically meaningful and appropriately measured. Standardization is currently an aim, especially when developing cost- and time-effective assays. This, however, leads to the oversimplification of ex vivo models we previously emphasized, making it challenging to reproduce pharmacokinetics of drug exposure in vitro. Depending on the drug's MoA, it may rather be required to finely tune its conditions to fully study its effects, then predict a clinical response. This could be done by controlling total duration of assay and specific duration of drug exposure, as well as measuring cell response at different time points and concentrations, in presence of the drug or after a recovery time. Microfluidics-based technologies, previously cited, may bring the required plasticity.
The essential and ambiguous role of immune contexture in modulating both clonal evolution patterns of cancer through immunoedition [114] and patient's prognosis [157,158] has now been clearly demonstrated. The therapeutic revolution brought by immuno-oncology drugs, especially immune checkpoints inhibitors (ICI), has nevertheless produced inconsistent clinical benefits. Outcome has been linked to the extent of cancer-induced immune priming, which in turn may be predicted by biomarkers, e.g. tumour mutation burden (TMB), PD-1/PDL-1, and tumour-infiltrating lymphocytes (TIL). Percentages of response are variable and, to some extent, fail to be accurately predicted by biomarkers [159]. Primary culture of cancer cells to produce a relevant ex vivo model is challenging; adding another level of complexity, that is to say coculturing cancer cells with autologous immune cells and measuring a response, is even more challenging. Yet, several functional assays are being developed [160], using various readouts. A so-called gold standard technique is the chromium 51 release assay, which has been used for decades [161]. Easier to implement, biophysical parameters such as real-time cellular impedance can be converted into cytolysis measurement to study the in vitro activity of most immunotherapy drug classes and immune effector cells [162]. Closer to bedside, changes in immune activity against tumour cells have been measured in melanoma explant models through the histological identification of relevant actors and subsequent measurement of both immune infiltration and inter-cell distance, pre- and post-treatment with anti-PD-1 nivolumab [153]. Patient-specific responses to ICI have also been observed using the CANscriptTM platform [163]. Regarding the specific targets PD-1/PD-L1, the ability of CoDx to predict tumour sensibility to immunotherapies has been inconsistent so far, especially due to the variable expression of these markers in space and time [7]. It may be expected such difficulty will also be encountered in ex vivo sensitivity assays aiming at predicting tumour response to ICI targeting this pair, as well as, more generally, any other non-static target with a fluctuating and microenvironment-dependant presence.
Another category of anticancer drugs, anti-angiogenics, are challenging to test using conventional 2D/3D ex vivo models, especially because they would require a longer time to allow tumour model vascularization. Preclinical models exist but are not easily amenable to the clinic [152], and their value would probably be inferior to that of functional assays dedicated to drugs that have a direct effect on tumour cells. A surrogate approach investigated the neutralizing effect of bevacizumab on circulating VEGF using a VEGF-dependent cell line subjected to sera of cancer patients treated with bevacizumab suggested potential to predict clinical benefit [164]. Chick chorioallantoic membrane (CAM) assay has also established as an interesting model to study tumorigenesis and antiangiogenic treatment effects [165,166]. This approach, easier to implement than PDX, may thus be considered as well for tumour response prediction studies. The embryonated egg is a highly vascularized, nutrients- and growth factors-enriched environment, in which cells from primary tumours or cell lines can be easily grafted. This in vivo-model is easier, time- and cost-competitive to generate as compared to mouse models. The whole process allows obtaining usable tumours within 10-13 days of grafting; it thus appears suitable for clinical applications. CAM has hence been used for testing chemosensitivity or chemotoxicity [167]. Its accessibility allows performing topical and intravenous administration of anticancer drugs, as well as determining optimal irradiation conditions for photodynamics therapies in multiple tumour samples [168]. More recently, tumour grafting models and functional testing were developed on zebrafish models: Fior et al. showed that xenografts in zebrafish larvae have enough resolution to measure interpatient and intrapatient heterogeneity in chemotherapy response in 4 days [169], and to predict tumour response to radiotherapy [170].
DNA repair capacities of cancer cells can be studied in vitro from cell extracts and represent a promising tool to predict response to drugs targeting DNA damage response pathways [171], especially PARP inhibitors [172]. In addition, microsatellite instability (MSI), TMB and neoantigen load are the consequences of DNA repair impairment. As such, they play an important role in clinical response to ICI [159], whose activity might also be predicted by DNA repair functional assays. In vitro detection of DNA damage may be approached through analytical chemistry, molecular or immunological methodologies [173]. So far, this has been especially beneficial to predict radiotherapy-induced adverse events from peripheral blood lymphocytes (PBL) [174]. Nevertheless, the Comet assay, commonly employed in genotoxicity studies to measure lethal DNA strand breaks, was used to assess radiosensitivity in bladder cancer [175] and sensitivity to topoisomerase I inhibitor irinotecan in CRC [176]. Interestingly, in the latter work, predictivity was based on ex vivo results obtained on more easily accessible PBL rather than directly on tumour cells, suggesting a surrogate target for measuring specific anticancer activity. The Comet assay has also proven useful to measure alkylation and addition products generated by chemotherapies, suggesting a transferability to CSRA [177]. A recent clinical study showed no advantage of either irinotecan- or oxaliplatin-based regimen against mismatch repair-deficient/MSI metastatic CRC [178]. It would be interesting to investigate with a CSRA if this absence of difference, visible within an all-comer cohort, is also reflected at the individual level.
For drug discovery purposes, PDX-based in vivo models appear scientifically sound to preserve the tumour's characteristics, recreate its environment and study the response of a whole sample instead of individualized cells or clonal groups [141,179,180]. However, several hurdles may impair their implementation into the clinical setting for treatment individualization: (i) availability of sufficiently humanized animals; (ii) low throughput; (iii) unpredictable graft take rate; (iv) heterogeneous growth rate, resulting in variable and potentially unacceptable time-to-results; (v) model drift through sample fragmentation and passages [181]; (vi) costs, sanitary and ethical issues associated with the use of laboratory animals. In addition, one can expect the host would introduce biases altering the sensitivity profile of the grafted sample [182]. Chemosensitivity prediction through the use of PDX models has been tried on heterogeneous cohorts of patients with solid cancers [183,184]. Yet, because of the previously listed drawbacks, especially take rate and propagation time, we question their current applicability within the clinical setting.
Because of their demonstrated predictive capacities on patient-derives models, functional assays are interesting tools for drug preclinical validation. Some contract research organisations (CRO) rely on such business model. Following in vitro assessment of a drug's MoA, functional assays allow preparing in vivo and first-in human studies by identifying and/or confirming disease subtypes most susceptible to respond to the drug candidate. This additional data investigating the biological hypothesis of a drug might reinforce the Investigational New Drug (IND) dossier. Additionally, it is worth noting that functional assays have another role to play in drug clinical development. Since they rely on a specific type of biomarker, functional assays may also help shaping clinical trials, much like CDx are doing with umbrella and basket trials [185]. However, similarly to biomarkers approved across several indication, functional assays are employed on several diseases. Hence, for a drug utilized against different cancers, they may require disease-specific sensitivity thresholds.
Two-dimension models currently offer the easiest path to clinical implementation of CSRA. This is due to both their adaptability to various clinical laboratory settings, and shorter turnaround time. However, the diversity of approaches overviewed here offers much promise to generate more complex and more predictive models. Continued integration of live cell culture technologies within clinical laboratories should facilitate their routine use. Moreover, technological advances, especially in image analysis and high content screening [133,186], allow to combine an array of endpoints. This allows better monitoring complex biological processes, either at the individual cell phenotype or tissue architecture level. This will undoubtedly be key to definitely bridge bench and bedside, by offering clinically useful functional assays.
Regulatory considerations and quality management of functional assays
CDx and CoDx may directly influence a patient's course of treatment. Consequently, they fall under strict regulation, and their manufacturing and execution require a stringent quality management framework. In our introduction, we pointed out CDx and CoDx have received, respectively, official and unofficial definitions. Functional assays, because of their nature, definitely are CoDx: they recommend specific treatment among the available list against a given indication; the medical team ultimately decide the best strategy to apply, based on this information but also on pathology's characteristics and overall patient's status. In addition, functional assays predict treatment response concomitantly for several drug classes, contrary to conventional CDx or CoDx; they may thus require a specific definition. In any case, the regulatory framework applicable to CDx also applies to functional assays.
Regulations in Europe and USA differ in the way they consider CDx as a whole, and functional assays dedicated to treatment individualization in particular. In the US, CDx fall within “class III” products, presenting a potential, unreasonable risk of illness or injury. As such, they require clinical investigation under an Investigational Device Exemption (IDE) before premarket approval (PMA). PMA is issued by the FDA's Center for Devices and Radiobiological Health (CDRH). Approval from the FDA allows commercialisation of CDx, which must be performed in an environment certified under the Clinical Laboratory Improvement Amendments (CLIA). Historically, some functional assays have started as laboratory developed tests (LDT) in clinical institutions [187,188]. However, the FDA noted this may lead to medical use of products with unproven performance and insufficient manufacturing controls. Guidance from the FDA issued in 2014 now gives LDT a more traditional validation track to ultimately allow their use in “making medical treatment decision” [189]. Manufacturers are required to notify the FDA and provide “basic information” about their product.
Marketing of MD in the European Union is conditioned to CE marking. In that respect, IVD-MD used to fall under the 98/79 EC directive. It allowed manufacturers to self-certify their diagnostics products for CE marking. Also, CDx were considered as “low-risk devices”, which, given their destination, was not appropriate [6]. IVD-MD are now delimited by the 2017/746 regulation, coming into full enforcement in May 2022. It places CDx into “class C” devices, presenting “high personal risk” and “moderate to low risk” to public health. This regulation entails manufacturers to produce assays complying with a set of harmonized requirements regarding their performances. Assays are also expected to be supported by an appropriate QMS. Most recent ISO standards specifically applicable to functional assays as IVD-MD are ISO 13485:2016 (general requirements of QMS for regulatory purposes) and ISO 14971:2012 (risk management). In 2018, the FDA announced its intent to modernize the quality system regulations for medical devices. This decision includes a transition away from 21 CFR 820 towards the ISO 13485 standards. The proposed rule has yet to be issued. It will create opportunities to harmonize global practices in the medical device industry. Although differences are considered minor, the transition for companies is expected to stretch over a few years, following a thorough gap analysis. Assessment of CE compliance is performed by notified bodies (NB). European NBs are currently restructuring to comply with the new regulation.
Inherent to CDx and CoDx is the dependency of risks and benefits of the selected treatment(s) upon the intrinsic performances of the assays. Rigorous demonstration of the assay robustness, reproducibility and clinical benefit must be conducted to achieve sufficient reliability. The three pillars of assay validation are: analytical validation, clinical validation and clinical utility [12]. Analytical validation rests on several parameters: repeatability (agreement between successive measures of the same sample), reproducibility (agreement between measures of several samples of the same measurand), precision (proximity of measurement results to the true value), accuracy (degree of repeatability of measurements) and limit of detection (e.g. minimal quantity of material to assess for producing a reliable result). In most advanced functional assays, patient's clinical response acts as the gold standard for benchmarking the patient's cell in vitro response. Hence, most of the aforementioned parameters must be derived from studies directly involving patients. There is no common rule regarding reproducibility of biomarker assays as a whole; however, coefficients of variation inferior to 15 % were cited as an aim to achieve [190]. Accuracy can be studied in early development phases on readily available in vitro models such as cells lines [191]. Thorough standard operating procedures (SOP), adequate training of operators, quality of assay material (batch control of consumables and reagents, restriction to standardized reagents with an entirely controlled formulation [192]), proper instrument controls and maintenance, are other factors that positively influence reproducibility. If not mandatory per se, automation (liquid handling, endpoint measurement) is highly desirable to ensure a throughput compatible with sample turnover at a clinical diagnostics scale, as well as robustness and reproducibility of the whole procedure, thus facilitating compliance with regulatory requirements. For 3D models grown in matrices, liquid handling remains challenging, hence limiting the adaptability of such approaches [133]. Laboratories currently offering functional testing services rarely communicate on the technical performances of their technology: Helomics published the positive impact of automation on their workflow for ChemoFX®, with improved accuracy and precision [191].
Sample collection, processing and logistics are key steps in the functional assay flowchart, since such tests necessarily deal with live cells or cell extracts [171]. Specific media and procedures may be developed to ensure standardization, and thus control, of the whole process [38-40,192]. Sample quality depends on available material; it should contain enough tumour tissue, while undesirable zones (necrotic, fibrous, adipose, mucinous, or containing blood clots) are reduced to minimum. The major hurdle in logistics is clearly the time frame between sampling at bedside and processing at the IVD laboratory. Our repeated observations show that: (i) maximum delay for preserved sample integrity is 72 hours; (ii) best sample integrity is obtained with samples processed no later than 48 hours following sampling; (iii) sample cryopreservation is undesirable, as it both lowers the number of available cells to perform the assay and attenuates their response to cytotoxic drugs (unpublished data from our group). This constrained time frame prevents sample shipping across long distances and borders because of obvious technical and legal hazards. It either limits technologies to a domestic market or requires their direct implantation in target countries. It also extends the number of operating days of the laboratory to six per week, to ensure Friday despatch is possible [38-40].
Following proper design controls and performance measurements, clinical validity of the IVD-MD is assessed. It aims at demonstrating its ability to “identify, measure, or predict the presence or absence of a medical condition or predisposition” for which the device is intended [12]. Key parameters such as diagnostic sensitivity (proportion of positive patients correctly identified as such), diagnostic specificity (proportion of negative patients correctly identified as such), positive predictive value (PPV; proportion of assay-positive patients that are actually positive) and negative predictive value (NPV; proportion of assay-negative patients that are actually negative) are measured at this stage [193,194]. For functional assays, endpoint cut-off is crucial, as it is the limit between predicted responders and non-responders to a specific drug. Integration of assay's constraints, especially regarding sampling and logistics, should also be fully evaluated at that stage. Finally, clinical utility is the demonstration that assay's results improve the therapeutic benefits patients obtain from treatment personalization versus a more systematic use. In accordance with the latest recommendations of the ASCO [15], this parameter requires large, randomized, prospective clinical trials investigating patient's outcome (response rates, progression-free survival, or, even better, overall survival at appropriate timepoint) after assay-directed treatment or oncologist-chosen treatment. The results of such trials are yet to be published. Their design is not trivial, as multi-drug regimens make it difficult to gain an insight into the effect of individual drug components.
As previously discussed, the proliferation of experimental or clinically implemented chemosensitivity assays directed against a large array of indications has been accompanied with the development of greatly varying protocols. Although understandable from a technological differentiation standpoint, the major pitfall of such situation is already illustrated by anti-PD-1/PD-L1 antibodies: each marketed antibody has its own CDx, with clinical trials constructed in such a way that they lead to drug-specific cut-offs to declare efficacy [6]. Assays cannot be substituted to one another. They require laboratories to be able to perform the whole array of tests to adapt to a physician's preference for one drug or the other. Still, it may be envisioned that ICI CDx will be amenable to interchangeability, because they rely on a common endpoint [195]. Because of their extreme technical heterogeneity, however, this will not be possible for functional assays. It may ultimately happen that, for a given pathology, the best predictive model wins it all.
Concluding remarks
As stated in a founding review [12], CSRA are a significant player in next-generation functional diagnostics, a concept that should not be solely identified with genomics. Indeed, molecular data come in huge volumes and are intricate. Their interpretation is frequently impaired by the difficulty of relating a patient's genotype with the actual behaviour of their tumour. And even with their genome or transcriptome deciphered, few patients will benefit from an already approved molecularly targeted drug [196]. In addition, insight into off-label use has been disappointing [197]. Identifying the right treatment thus necessitates the exploration of a different level of biological information. The armamentarium already available to predict tumour responses indeed encompasses complementary technologies (high-throughput genomics, immunoprofiling, functional testing) whose concomitant use may provide clinicians with a complete, multilevel profile for each patient, including particular susceptibilities to drug toxicity [198]. Such comprehensive data could allow a completely personalized approach in deciding which therapies would best treat each cancer. In that context, CSRA should keep their role as CoDx, prioritizing drug combinations rather than selecting or disqualifying them. Several groups have come to this conclusion [14,179] and some companies include this perspective into their business model.
The main criticism raised against CSRA is the lack of translatability of ex vivo response into clinical response [14,199]. Despite the recent level of evidence not being considered sufficient to recommend a routine clinical use yet, there is a growing body of cues to show it is actually possible to predict a patient's response to several classes of drugs. Most importantly, this information is useful to improve their chances for prolonged survival and a better quality of life. Besides improved response rates and survival, there are other benefits to be gained from CSRA: (i) reduction in deleterious side effects, due to shortened therapeutic cycles or the avoidance of inefficient drugs; (ii) decrease in costs of global care, due to optimized patient management and a reduced use of expensive drugs.
The intense development of targeted therapies might have caused some to forget that chemotherapies still stand as standards-of-care against cancer. Obviously, their response rates remain insufficient. Use of these “historical” anticancer treatments would benefit from personalized approaches, too, which has been the foundation of CSRA. Failure of chemoresistance prediction now allows efforts to concentrate of chemosensitivity prediction, which clearly holds a much higher clinical utility [199].
While this may remain a pious wish because of industrial reasons, the recent experience gained from the development of “me-too” drugs and CDx in immuno-oncology, with thresholds that are not comparable from one assay to the other while they are supposed to measure the same endpoint, leads us to suggest the creation of working groups and workshops dedicated to functional assays. They would gather the main actors in the field to discuss technical (especially endpoints) and regulatory considerations to help structuring assays.
Personalized medicine offers the perspective of tailoring therapeutic approaches for every patient. Yet, not every patient harbours an already known actionable drug target. Blending biological knowledge with technological development to better grasp the behaviour of every patient's cancer should ensure that none is left in the ditch along the road.
Abbreviations
2D: two-dimension; 3D: three-dimension; ASCO: American Society of Clinical Oncology; CAF: cancer-associated fibroblast; CAM: chick chorioallantoic membrane; CDRH: Center for Devices and Radiobiological Health; CDx: companion diagnostics; cGMP: current good manufacturing practices; CLIA: Clinical Laboratory Improvement Amendments; CSRA: chemosensitivity and resistance assay; CoDx: complementary diagnostics; CRC: colorectal cancer; CRO: contract research organisation;; EMA: European Medicines Agency; FDA: Food and Drug Administration; ICI: immune checkpoint inhibitor; HIPEC: intraperitoneal chemotherapy associated to hyperthermia; IVD: in vitro diagnostics; IVD-MD: in vitro diagnostics medical device; LDT: laboratory developed tests; MD: medical device; MoA: mode of action; MSI: microsatellite instability; NB: notified body; NPV: negative predictive value; OS: overall survival; PBL: peripheral blood lymphocyte; PDX: patient-derived xenograft; PFS: progression-free survival; PIPAC: pressurized intraperitoneal aerosol chemotherapy; PMA: Premarket Approval; PPV: positive predictive value; QMS: quality management system; RECIST: response evaluation criteria in solid tumours; SOP: standard operating procedures; TIL: tumour-infiltrating lymphocyte; TMB: tumour mutation burden; TME: tumour microenvironment.
Author Contributions
Pr. M. Mathonnet, Dr. C. Bounaix Morand du Puch, Dr. N. Christou, Dr. S. Giraud and Dr. C. Lautrette conceived the original idea. Dr. C. Bounaix Morand du Puch and Dr. M. Vanderstraete drafted the manuscript with equal contributions. Pr. M. Mathonnet revised the manuscript.
Competing Interests
CBMP and MVA are former employees of Oncomedics SAS. SGI and CLA are founders of Oncomedics SAS and have an equity position in the company. MM and NC declare no competing interests.
References
1. Papadopoulos N, Kinzler KW, Vogelstein B. The role of companion diagnostics in the development and use of mutation-targeted cancer therapies. Nat Biotechnol. 2006;24(8):985-95
2. FDA. Companion diagnostics 2018. https://www.fda.gov/medical-devices/vitro-diagnostics/companion-diagnostics
3. EMA. Companion diagnostics ('in-vitro diagnostics') 2019. https://www.ema.europa.eu/en/human-regulatory/overview/medical-devices#companion-diagnostics-('in-vitro-diagnostics')-section
4. FDA. Developing and labeling in vitro companion diagnostic devices for a specific group of oncology therapeutic products - Guidance for industry 2020. https://www.fda.gov/media/120340/download
5. Scheerens H, Malong A, Bassett K, Boyd Z, Gupta V, Harris J. et al. Current status of companion and complementary diagnostics: strategic considerations for development and launch. Clin Transl Sci. 2017;10:84-92
6. Jørgensen JT. Companion and complementary diagnostics: clinical and regulatory perspectives. Trends Cancer. 2016;2:706-12
7. Dracopoli NC, Boguski MS. The evolution of oncology companion diagnostics from signal transduction to immuno-oncology. Trends Pharmacol Sci. 2017;38:41-54
8. Jørgensen JT. Companion and Complementary Diagnostics. 1st ed. Academic Press. 2019
9. Fridlyand J, Simon RM, Walrath JC, Roach N, Buller R, Schenkein DP. et al. Considerations for the successful co-development of targeted cancer therapies and companion diagnostics. Nat Rev Drug Disc. 2013;12:743-55
10. Milne CP, Cohen JP, Chakravarthy R. Market watch: where is personalized medicine in industry heading? Nat Rev Drug Discov. 2015;14:812-3
11. International Council for Harmonisation (1997). ICH harmonised tripartite guideline: E8 general considerations for clinical trials. Thomas DW, Burns J, Audette J, Carroll A, Dow-Hygelund C, Hay M (2016) Clinical development success rates. 2006-2015. BIO Industry Analysis.
12. Friedman AA, Letai A, Fisher DE, Flaherty KT. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer. 2015;15:747-56
13. Byfield JE, Stein JJ, Bennett LR. In vitro assay of chemosensitivity in gynecologic malignancies: Technique and preliminary results using a radiochemical microassay. Oncology. 1971;25:55-65
14. Jenks S. Chemosensitivity assays: still eyeing the clinic. J Natl Cancer Inst. 2012;104:1775-7
15. Burstein HJ, Mangu PB, Somerfield MR, Schrag D, Samson D, Holt L. et al. American Society of Clinical Oncology clinical practice guideline update on the use of chemotherapy sensitivity and resistance assays. J Clin Oncol. 2011;29:3328-30
16. Schrag D, Garewal HS, Burstein HJ, Samson DJ, Von Hoff DD, Somerfield MR. American Society of Clinical Oncology technology assessment: chemotherapy sensitivity and resistance assays. J Clin Oncol. 2004;22:3631-8
17. Salmon SE, Hamburger AW, Soehnlen B, Durie BGM, Alberts DS, Moon TE. Quantitation of Differential Sensitivity of Human-Tumor Stem Cells to Anticancer Drugs. N Engl J Med. 1978;298:1321-7
18. Bertelsen CA, Sondak VK, Mann BD, Korn EL, Kern DH. Chemosensitivity testing of human solid tumors. A review of 1582 assays with 258 clinical correlations. Cancer. 1984;53:1240-5
19. Weisenthal LM. Differential staining cytotoxicity assay: a review. Methods Mol Biol. 2011;731:259-83
20. Weisenthal LM, Patel N, Rueff-Weisenthal C. Cell culture detection of microvascular cell death in clinical specimens of human neoplasms and peripheral blood. J Intern Med. 2008;264:275-87
21. Won DJ, Ji YL, Jong HK, Hang JY, Hyun JR, Park JY. et al. Efficacy of taxane and platinum-based chemotherapy guided by extreme drug resistance assay in patients with epithelial ovarian cancer. J Gynecol Oncol. 2009;20:96-100
22. Kang SM, Park MS, Chang J, Kim SK, Kim H, Shin D-H. et al. A feasibility study of adenosine triphosphate-based chemotherapy response assay (ATP-CRA) as a chemosensitivity test for lung cancer. Cancer Res Treat. 2005;37:223-7
23. Suksawat M, Klanrit P, Phetcharaburanin J, Namwat N, Khuntikeo N, Titapun A. et al. In vitro and molecular chemosensitivity in human cholangiocarcinoma tissues. PLoS One. 2019;14:e0222140
24. Yoon YS, Kim CW, Roh SA, Cho DH, Kim TW, Kim MB. et al. Development and applicability of integrative tumor response assays for metastatic colorectal cancer. Anticancer Res. 2017;37:1297-1303
25. Kim JH, Yoon YS, Kim JC, Kim YM. Assessment of the applicability of integrative tumor response assays in advanced epithelial ovarian cancer. Anticancer Res. 2019;39:313-8
26. Kobayashi H. Development of a new in vitro chemosensitivity test using collagen gel droplet embedded culture and image analysis for clinical usefulness. Recent Results Cancer Res. 2003;161:48-61
27. Leone LA, Meitner PA, Myers TJ, Grace WR, Gajewski WH, Fingert HJ. et al. Predictive value of the fluorescent cytoprint assay (FCA): a retrospective correlation study of in vitro chemosensitivity and individual responses to chemotherapy. Cancer Invest. 1991;9:491-503
28. Arienti C, Tesei A, Verdecchia GM, Framarini M, Virzì S, Grassi A. et al. Peritoneal carcinomatosis from ovarian cancer: chemosensitivity test and tissue markers as predictors of response to chemotherapy. J Transl Med. 2011;9:94. doi: 10.1186/1479-5876-9-94
29. Arienti C, Tesei A, Verdecchia GM, Framarini M, Virzì S, Grassi A. et al. Role of conventional chemosensitivity test and tissue biomarker expression in predicting response to treatment of peritoneal carcinomatosis from colon cancer. Clin Colorectal Cancer. 2013;12:122-7
30. Kravtsov VD, Greer JP, Whitlock JA, Koury MJ. Use of the microculture kinetic assay of apoptosis to determine chemosensitivities of leukemias. Blood. 1998;92:968-80
31. Bosserman L, Prendergast F, Herbst R, Fleisher M, Salom E, Strickland S. et al. The microculture-kinetic (MiCK) assay: The role of a drug-induced apoptosis assay in drug development and clinical care. Cancer Res. 2012;72:3901-5
32. Ballard KS, Hornesley HD, Hodson C, Presant CA, Rutledge J, Hallquist A. et al. Endometrial carcinoma in vitro chemosensitivity testing of single and combination chemotherapy regimens using the novel microculture kinetic apoptosis assay: Implications for endometrial cancer treatment. J Gynecol Oncol. 2010;21:45-9
33. Bosserman LD, Rajurkar SP, Rogers K, Davidson DC, Chernick M, Hallquist A. et al. Correlation of drug-induced apoptosis assay results with oncologist treatment decisions and patient response and survival. Cancer. 2012;118:4877-83
34. Salom E, Penalver M, Homesley H, Burrell M, Garrett A, Presant CA. et al. Correlation of pretreatment drug induced apoptosis in ovarian cancer cells with patient survival and clinical response. J Transl Med. 2012;10:162. doi: 10.1186/1479-5876-10-162
35. Kelly SE, Di Benedetto A, Greco A, Howard CM, Sollars VE, Primerano DA. et al. Rapid selection and proliferation of CD133(+) cells from cancer cell lines: chemotherapeutic implications. PLoS One. 2010;5:e10035
36. Richard S, Wells A, Connor J, Price F. Use of chemofx® for identification of effective treatments in epithelial ovarian cancer. PLoS Curr. 2015Jul13;7:ecurrents.eogt.8b0b6fffc7b999b34bc4c8152edbf237
37. Majumder B, Baraneedharan U, Thiyagarajan S, Radhakrishnan P, Narasimhan H, Dhandapani M. et al. Predicting clinical response to anticancer drugs using an ex vivo platform that captures tumour heterogeneity. Nat Commun. 2015;6:6169. doi: 10.1038/ncomms7169
38. Giraud S, Loum E, Bessette B, Fermeaux V, Lautrette C. Oncogramme, a new promising method for individualized breast tumour response testing for cancer treatment. Anticancer Res. 2011;31:139-45
39. Bounaix Morand du Puch C, Giraud S, Lautrette C, Nouaille M, Labrunie A, Luce S. et al. Chemotherapy outcome predictive effectiveness by the oncogramme: pilot trial on stage-IV colorectal cancer. J Transl Med. 2016;14:10. doi: 10.1186/s12967-016-0765-4
40. Giraud S. Oncogramme, an adapted method for individualized tumour response testing of ovary cancer treatments. J Cancer Res Therap Oncol. 2014;2:1-9
41. Kawamura H, Ikeda K, Takiyama I, Terashima M. The usefulness of the ATP assay with serum-free culture for chemosensivity testing of gastrointestinal cancer. Eur J Cancer. 1997;33:960-6
42. Konecny G, Crohns C, Pegram M, Felber M, Lude S, Kurbacher C. et al. Correlation of drug response with the ATP tumorchemosensitivity assay in primary FIGO stage III ovarian cancer. Gynecol Oncol. 2000;77:258-63
43. Ng TY, Ngan HYS, Cheng DKL, Wong LC. Clinical applicability of the ATP cell viability assay as a predictor of chemoresponse in platinum-resistant epithelial ovarian cancer using nonsurgical tumor cell samples. Gynecol Oncol. 2000;76:405-8
44. O'Meara AT, Sevin BU. Predictive value of the ATP chemosensitivity assay in epithelial ovarian cancer. Gynecol Oncol. 2001;83:334-42
45. Kim BS, Ahn YM, Kim J, Yoon EJ, Choi SH. Clinical utility of adenosine triphosphate-based chemosensitivity response assay (ATP-CRA) in non-small cell lung cancer: preliminary study. J Clin Oncol. 2004;22(14_suppl):7259-7259
46. Yong WM, Sung HC, Yong TK, Joo HS, Chang J, Se KK. et al. Adenosine triphosphate-based chemotherapy response assay (ATP-CRA)-guided platinum-based 2-drug chemotherapy for unresectable nonsmall-cell lung cancer. Cancer. 2007;109:1829-35
47. Kim HA, Yom CK, Moon BI, Choe KJ, Sung SH, Han WS. et al. The use of an in vitro adenosine triphosphate-based chemotherapy response assay to predict chemotherapeutic response in breast cancer. Breast. 2008;17:19-26
48. Han SS, Choi SH, Lee YK, Kim JW, Park NH, Song YS. et al. Predictive value of individualized tumor response testing by ATP-based chemotherapy response assay in ovarian cancer. Cancer Invest. 2008;26:426-30
49. Kim JH, Lee KW, Kim YH, Lee KH, Oh DY, Kim J. et al. Individualized tumor response testing for prediction of response to paclitaxel and cisplatin chemotherapy in patients with advanced gastric cancer. J Korean Med Sci. 2010;25:684-90
50. Lee JH, Kim MC, Oh SY, Kwon HC, Kim SH, Kwon KA. et al. Predictive value of in vitro adenosine triphosphate-based chemotherapy response assay in advanced gastric cancer patients who received oral 5-fluorouracil after curative resection. Cancer Res Treat. 2011;43:117-23
51. Ge WQ, Pu JX, Zheng SY. Clinical application of the adenosine triphosphate-based response assay in intravesical chemotherapy for superficial bladder cancer. Asian Pac J Cancer Prev. 2012;13:689-92
52. Zhang J, Li H. Heterogeneity of tumor chemosensitivity in ovarian epithelial cancer revealed using the adenosine triphosphate-tumor chemosensitivity assay. Oncol Lett. 2015;9:2374-80
53. Robbins KT, Connors KM, Storniolo AM, Hanchett C, Hoffman RM. Sponge-gel-supported histoculture drug-response assay for head and neck cancer: correlations with clinical response to cisplatin. Arch Otolaryngol Head Neck Surg. 1994;120:288-92
54. Furukaw T, Kubota T, Hoffman RM. Clinical applications of the histoculture drug response assay. Clin Cancer Res. 1995;1:305-11
55. Ohie S, Udagawa Y, Kozu A, Komuro Y, Aoki D, Nozawa S. et al. Cisplatin sensitivity of ovarian cancer in the histoculture drug response assay correlates to clinical response to combination chemotherapy with cisplatin, doxorubicin and cyclophosphamide. Anticancer Res. 2000;20:2049-54
56. Tanino H, Oura S, Hoffman RM, Kubota T, Furukawa T, Arimoto J. et al. Acquisition of multidrug resistance in recurrent breast cancer demonstrated by the histoculture drug response assay. Anticancer Res. 2001;21:4083-6
57. Singh B, Li R, Xu L, Poluri A, Patel S, Shaha AR. et al. Prediction of survival in patients with head and neck cancer using the histoculture drug response assay. Head Neck. 2002;24:437-42
58. Ariyoshi Y, Shimahara M, Tanigawa N. Study on chemosensitivity of oral squamous cell carcinomas by histoculture drug response assay. Oral Oncol. 2003;39:701-7
59. Hasegawa Y, Goto M, Hanai N, Ijichi K, Adachi M, Terada A. et al. Evaluation of optimal drug concentration in histoculture drug response assay in association with clinical efficacy for head and neck cancer. Oral Oncol. 2007;43:749-56
60. Pathak KA, Juvekar AS, Radhakrishnan DK, Deshpande MS, Pai VR, Chaturvedi P. et al. In vitro chemosensitivity profile of oral squamous cell cancer and its correlation with clinical response to chemotherapy. Indian J Cancer. 2007;44:142-6
61. Yoshimasu T, Oura S, Hirai I, Tamaki T, Kokawa Y, Hata K. et al. Data acquisition for the histoculture drug response assay in lung cancer. J Thorac Cardiovasc Surg. 2007;133:303-8
62. Neubauer H, Stefanova M, Solomayer E, Meisner C, Zwirner M, Wallwiener D. et al. Predicting resistance to platinum-containing chemotherapy with the ATP tumor chemosensitivity assay in primary ovarian cancer. Anticancer Res. 2008;28:949-55
63. Fujita Y, Hiramatsu M, Kawai M, Nishimura H, Miyamoto A, Tanigawa N. Histoculture drug response assay predicts the postoperative prognosis of patients with esophageal cancer. Oncol Rep. 2009;21:499-505
64. Gwak HS, Park HJ, Yoo H, Youn SM, Rhee CH, Lee SH. Chemotherapy for malignant gliomas based on histoculture drug response assay: a pilot study. J Korean Neurosurg Soc. 2011;50:426-33
65. Yoon YS, Kim CW, Roh SA, Cho DH, Kim GP, Hong YS. et al. Applicability of histoculture drug response assays in colorectal cancer chemotherapy. Anticancer Res. 2012;32:3581-6
66. Kobayashi H, Tanisaka K, Doi O, Kodama K, Higashiyama M, Nakagawa H. et al. An in vitro chemosensitivity test for solid human tumors using collagen gel droplet embedded cultures. Int J Oncol. 1997;11:449-55
67. Takamura Y, Kobayashi H, Taguchi T, Motomura K, Inaji H, Noguchi S. Prediction of chemotherapeutic response by collagen gel droplet embedded culture-drug sensitivity test in human breast cancers. Int J Cancer. 2002;98:450-5
68. Higashiyama M, Oda K, Okami J, Maeda J, Kodama K, Takami K. et al. In vitro-chemosensitivity test using the collagen gel droplet embedded culture drug test (cd-dst) for malignant pleural mesothelioma: Possibility of clinical application. Ann Thorac Cardiovasc Surg. 2008;14:355-62
69. Higashiyama M, Oda K, Okami J, Maeda J, Kodama K, Imamura F. et al. Prediction of chemotherapeutic effect on postoperative recurrence by in vitro anticancer drug sensitivity testing in non-small cell lung cancer patients. Lung Cancer. 2010;68:472-7
70. Naitoh H, Yamamoto H, Murata S, Kobayashi H, Inoue K, Tani T. Stratified phase II trial to establish the usefulness of the collagen gel droplet embedded culture-drug sensitivity test (CD-DST) for advanced gastric cancer. Gastric Cancer. 2014;17:630-7
71. Sakuma K, Tamura R, Hanyu S, Takahashi H, Sato H, Oneyama T. et al. Clinical study on collagen gel droplet-embedded culture drug sensitivity test for multidrug combination chemotherapy and super selective intra-arterial infusion chemoradiotherapy in oral squamous cell carcinoma. Mol Clin Oncol. 2017;7:1021-26
72. Ness RB, Wisniewski SR, Eng H, Christopherson W. Cell viability assay for drug testing in ovarian cancer: in vitro kill versus clinical response. Anticancer Res. 2002;22:1145-9
73. Mi Z, Holmes FA, Hellerstedt B, Pippen J, Collea R, Backner A. et al. Feasibility assessment of a chemoresponse assay to predict pathologic response in neoadjuvant chemotherapy for breast cancer patients. Anticancer Res. 2008;28:1733-40
74. Jamal BT, Grillone GA, Jalisi S. Chemoresponse assay in head and neck cancer patients: A three-year follow up. J Clin Diagn Res. 2017;11:XC01-XC03
75. Claudio PP, Mathis SE, Nande R, Lawrence L, Alberico A, Julien TD. et al. ChemoID assay for glioblastoma. J Clin Oncol. 2015 33(15_suppl, e1):3028-3028
76. Strickland SA, Raptis A, Hallquist A, Rutledge J, Chernick M, Perree M. et al. Correlation of the microculture-kinetic drug-induced apoptosis assay with patient outcomes in initial treatment of adult acute myelocytic leukemia. Leuk Lymphoma. 2013;54:528-34
77. Bosserman L, Rogers K, Willis C, Davidson D, Whitworth P, Karimi M. et al. Application of a drug-induced apoptosis assay to identify treatment strategies in recurrent or metastatic breast cancer. PLoS One. 2015;10:e0122609
78. Kim KY, Chung BW, Yang I, Kim MB, Hoffman RM. Independence of cytotoxic drug sensitivity profiles and receptor subtype of invasive ductal breast carcinoma demonstrated by the histoculture drug response assay (HDRA). Anticancer Res. 2014;34:7197-201
79. Shinden Y, Kijima Y, Hirata M, Arima H, Nakajyo A, Tanoue K. et al. Clinical significance of the histoculture drug response assay in breast cancer. Anticancer Res. 2016;36:6173-78
80. Mekata E, Sonoda H, Shimizu T, Tatsuta T, Yamaguchi T, Endo Y. et al. Clinical predictive value of in vitro anticancer drug sensitivity test for the therapeutic effect of adjuvant chemotherapy in patients with stage II-III colorectal cancer. Mol Clin Oncol. 2013;1:763-7
81. Ji WB, Um JW, Ryu JS, Hong KD, Kim JS, Min BW. et al. Clinical significance of 5-fluorouracil chemosensitivity testing in patients with colorectal cancer. Anticancer Res. 2017;37:2679-82
82. Hur H, Kim NK, Kim HG, Min BS, Lee KY, Shin SJ. et al. Adenosine triphosphate-based chemotherapy response assay-guided chemotherapy in unresectable colorectal liver metastasis. Br J Cancer. 2012;106:53-60
83. Kubota T, Sasano N, Abe O, Nakao I, Kawamura E, Saito T. et al. Potential of the histoculture drug-response assay to contribute to cancer patient survival. Clin Cancer Res. 1995;1:1537-43
84. Tanigawa N, Yamaue H, Ohyama S, Sakuramoto S, Inada T, Kodera Y. et al. Exploratory phase II trial in a multicenter setting to evaluate the clinical value of a chemosensitivity test in patients with gastric cancer (JACCRO-GC 04, Kubota memorial trial). Gastric Cancer. 2016;19:350-60
85. Howard CM, Valluri J, Alberico A, Julien T, Mazagri R, Marsh R. et al. Analysis of chemopredictive assay for targeting cancer stem cells in glioblastoma patients. Transl Oncol. 2017;10:241-54
86. Wilbur DW, Camacho ES, Hilliard DA, Dill PL, Weisenthal LM. Chemotherapy of non-small cell lung carcinoma guided by an in vitro drug resistance assay measuring total tumour cell kill. Br J Cancer. 1992;65:27-32
87. Akazawa Y, Higashiyama M, Nishino K, Uchida J, Kumagai T, Inoue T. et al. Impact of in vitro chemosensitivity test-guided platinum-based adjuvant chemotherapy on the surgical outcomes of patients with p-stage IIIA non-small cell lung cancer that underwent complete resection. Mol Clin Oncol. 2017;7:327-35
88. Inoue M, Maeda H, Takeuchi Y, Fukuhara K, Shintani Y, Funakoshi Y. et al. Collagen gel droplet-embedded culture drug sensitivity test for adjuvant chemotherapy after complete resection of non-small-cell lung cancer. Surg Today. 2018;48:380-87
89. Chen Z, Zhang S, Ma S, Li C, Xu C, Shen Y. et al. Evaluation of the in vitro chemosensitivity and correlation with clinical outcomes in lung cancer using the ATP-TCA. Anticancer Agents Med Chem. 2018;18:139-45
90. Ugurel S, Schadendorf D, Pföhler C, Neuber K, Thoelke A, Ulrich J. et al. In vitro drug sensitivity predicts response and survival after individualized sensitivity-directed chemotherapy in metastatic melanoma: a multicenter phase II trial of the dermatologic cooperative oncology group. Clin Cancer Res. 2006;12:5454-63
91. Kurbacher CM, Cree IA, Bruckner HW, Brenne U, Kurbacher JA, Müller K. et al. Use of an ex vivo ATP luminescence assay to direct chemotherapy for recurrent ovarian cancer. Anticancer Drugs. 1998;9:51-7
92. Gallion H, Christopherson WA, Coleman RL, Demars L, Herzog T, Hosford S, et al. Progression-free interval in ovarian cancer and predictive value of an ex vivo chemoresponse assay.Int J Gynecol Cancer. 2006;16:194-201
93. Cree IA, Kurbacher CM, Lamont A, Hindley AC, Love S, Cree IA. et al. A prospective randomized controlled trial of tumour chemosensitivity assay directed chemotherapy versus physician's choice in patients with recurrent platinum-resistant ovarian cancer. Anticancer Drugs. 2007;18:1093-101
94. Herzog TJ, Krivak TC, Fader AN, Coleman RL. Chemosensitivity testing with ChemoFx and overall survival in primary ovarian cancer. Am J Obstet Gynecol. 2010;203:68.e1-6
95. Jung PS, Kim DY, Kim MB, Lee SW, Kim JH, Kim YM. et al. Progression-free survival is accurately predicted in patients treated with chemotherapy for epithelial ovarian cancer by the histoculture drug response assay in a prospective correlative clinical trial at a single institution. Anticancer Res. 2013;33:1029-34
96. Park JS, Kim JK, Yoon DS. Correlation of early recurrence with in vitro adenosine triphosphate based chemotherapy response assay in pancreas cancer with postoperative gemcitabine chemotherapy. J Clin Lab Anal. 2016;30:804-10
97. Wong HB, Lim GH. Measures of diagnostic accuracy: sensitivity, specificity, PPV and NPV. Proc Sing Healthcare. 2011;20:316-8
98. Eltabbakh GH, Piver MS, Hempling RE, Recio FO, Lele SB, Marchetti DL. et al. Correlation between extreme drug resistance assay and response to primary paclitaxel and cisplatin in patients with epithelial ovarian cancer. Gynecol Oncol. 1998;70:392-7
99. Mechetner E, Brünner N, Parker RJ. In vitro drug responses in primary and metastatic colorectal cancers. Scand J Gastroenterol. 2011;46:70-8
100. Tatar B, Boyraz G, Selçuk İ, Doğan AK, Usubütün A, Tuncer ZS. In vitro chemosensitivity in ovarian carcinoma: comparison of three leading assays. J Turk Ger Gynecol Assoc. 2016;17:35-40
101. Maehira H, Shimizu T, Sonoda H, Mekata E, Yamaguchi T, Miyake T. et al. A rare case of primary choriocarcinoma in the sigmoid colon. World J Gastroenterol. 2013;19:6683-8
102. Sakuma K, Tamura R, Noda N, Mizutani M, Yamaguchi A, Tanaka A. Collagen gel droplet-embedded culture drug sensitivity testing in hard palate cancer-predicted antitumor efficacy of cetuximab: a case report. Mol Clin Oncol. 2017;7:637-41
103. Gu F, Ma Y, Fan Y, Lang R, Ding T, Hao X. et al. Synovial sarcoma individual chemotherapy directed by collagen gel droplet embedded culture drug sensitivity test: a case report. Oncol Lett. 2010;1:885-8
104. Jung WH, Yoon AP, Eun JJ, Kang YL, Ji EK, Sohn SK. Complete remission of unresectable colon cancer after preoperative chemotherapy selected by adenosine triphosphate-based chemotherapy response assay. J Korean Med Sci. 2008;23:916-9
105. Rios Perez MV, Dai B, Koay EJ, Wolff RA, Fleming JB. Regression of stage IV pancreatic cancer to curative surgery and introduction of a novel ex-vivo chemosensitivity assay. Cureus. 2015;7:e423
106. Breidenbach M, Rein D, Schmidt T, Heindel W, Kolhagen H, Mallmann P. et al. Intra-arterial mitoxantrone and paclitaxel in a patient with Stewart-Treves syndrome: selection of chemotherapy by an ex vivo ATP-based chemosensitivity assay. Anticancer Drugs. 2000;11:269-73
107. Srouji IA, Resouly A, Cree IA. Case of thymic parathyroid carcinoma in a haemodialysis patient: Application of tumour chemosensitivity testing. J Laryngol Otol. 2004;118:162-4
108. Qian KQ, Ye H, Xiao YW, Bao YY, Qi CJ. Tumor lysis syndrome associated with chemotherapy in primary retroperitoneal soft tissue sarcoma by ex vivo ATP-based tumor chemo-sensitivity assay (ATP-TCA). Int J Gen Med. 2009;2:1-4
109. Ledford H. US cancer institute to overhaul tumour cell lines. Nature. 2016;530:391
110. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646-74
111. Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J. et al. A big bang model of human colorectal tumor growth. Nat Genet. 2015;47:209-16
112. Galon J, Bruni D. Tumor immunology and tumor evolution: intertwined histories. Immunity. 2020;52:55-81
113. Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822-9
114. Angelova M, Mlecnik B, Vasaturo A, Bindea G, Fredriksen T, Lafontaine L. et al. Evolution of metastases in space and time under immune selection. Cell. 2018;175:751-65
115. Jiang MJ, Gu DN, Dai JJ, Huang Q, Tian L. Dark side of cytotoxic therapy: chemoradiation-induced cell death and tumor repopulation. Trends Cancer. 2020;6:419-31
116. Takebayashi K, Mekata E, Sonoda H, Shimizu T, Shiomi H, Naka S. et al. Differences in chemosensitivity between primary and metastatic tumors in colorectal cancer. PLoS One. 2013;8:e73215
117. Higashiyama M, Okami J, Maeda J, Tokunaga T, Fujiwara A, Kodama K. et al. Differences in chemosensitivity between primary and paired metastatic lung cancer tissues: in vitro analysis based on the collagen gel droplet embedded culture drug test (CD-DST). J Thorac Dis. 2012;4:40-7
118. Maniwa Y, Yoshimura M, Hashimoto S, Takata M, Nishio W. Chemosensitivity of lung cancer: differences between the primary lesion and lymph node metastasis. Oncol Lett. 2010;1:345-9
119. Meirson T, Gil-Henn H, Samson AO. Invasion and metastasis: the elusive hallmark of cancer. Oncogene. 2020;39:2024-6
120. Lacroix A, Deluche E, Zhang LY, Dalmay C, Mélin C, Leroy J. et al. A new label-free approach to glioblastoma cancer stem cell sorting and detection. Anal Chem. 2019;91:8948-57
121. Gazzaniga P, Naso G, Gradilone A, Cortesi E, Gandini O, Gianni W. et al. Chemosensitivity profile assay of circulating cancer cells: prognostic and predictive value in epithelial tumors. Int J Cancer. 2010;126:2437-47
122. Yang C, Xia BR, Jin WL, Lou G. Circulating tumor cells in precision oncology: clinical applications in liquid biopsy and 3D organoid model. Cancer Cell Int. 2019;19:341. doi: 10.1186/s12935-019-1067-8
123. Hamilton G, Rath B. Applicability of tumor spheroids for in vitro chemosensitivity assays. Expert Opin Drug Metab Toxicol. 2019;15:15-23
124. Szulkin A, Szatmári T, Hjerpe A, Dobra K. Chemosensitivity and resistance testing in malignant effusions with focus on primary malignant mesothelioma and metastatic adenocarcinoma. Pleura Peritoneum. 2016;1:119-33
125. Shi J, Li Y, Jia R, Fan X. The fidelity of cancer cells in PDX models: characteristics, mechanism and clinical significance. Int J Cancer. 2020;146:2078-88
126. Mondal AM, Ma AH, Li G, Krawczyk E, Yuan R, Lu J. et al. Fidelity of a PDX-CR model for bladder cancer. Biochem Biophys Res Commun. 2019;517:49-56
127. Julien S, Merino-Trigo A, Lacroix L, Pocard M, Goéré D, Mariani P. et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res. 2012;18:5314-28
128. Meijer TG, Naipal KA, Jager A, van Gent DC. Ex vivo tumor culture systems for functional drug testing and therapy response prediction. Future Sci OA. 2017;3:FSO190
129. Ben-David U, Beroukhim R, Golub TR. Genomic evolution of cancer models: perils and opportunities. Nat Rev Cancer. 2019;19:97-109
130. Fong ELS, Harrington DA, Farach-Carson MC, Yu H. Heralding a new paradigm in 3D tumor modeling. Biomaterials. 2016Nov;108:197-213
131. Powley IR, Patel M, Miles G, Pringle H, Howells L, Thomas A. et al. Patient-derived explants (PDEs) as a powerful preclinical platform for anti-cancer drug and biomarker discovery. Br J Cancer. 2020Mar;122:735-44
132. Morgan MM, Johnson BP, Livingston MK, Schuler LA, Alarid ET, Sung KE. et al. Personalized in vitro cancer models to predict therapeutic response: challenges and a framework for improvement. Pharmacol Ther. 2016;165:79-92
133. Booij TH, Price LS, Danen EHJ. 3D cell-based assays for drug screens: challenges in imaging, image analysis, and high-content analysis. SLAS Discov. 2019;24:615-27
134. Weiswald LB, Bellet D, Dangles-Marie V. Spherical cancer models in tumor biology. neoplasia. 2015;17:1-15
135. Van De Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A. et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933-45
136. Koerfer J, Kallendrusch S, Merz F, Wittekind C, Kubick C, Kassahun WT. et al. Organotypic slice cultures of human gastric and esophagogastric junction cancer. Cancer Med. 2016;5:1444-53
137. Datta P, Dey M, Ataie Z, Unutmaz D, Ozbolat IT. 3D bioprinting for reconstituting the cancer microenvironment. NPJ Precis Oncol. 2020;4:18. doi: 10.1038/s41698-020-0121-2
138. Ma X, Liu J, Zhu W, Tang M, Lawrence N, Yu C. et al. 3D bioprinting of functional tissue models for personalized drug screening and in vitro disease modeling. Adv Drug Deliv Rev. 2018;132:235-51
139. Brouquet A, Taleb P, Lot AS, Beauchet A, Julie C, Prevost G. et al. A model of primary culture of colorectal cancer and liver metastasis to predict chemosensitivity. J Surg Res. 2011;166:247-54
140. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228-47
141. Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernández-Mateos J, Khan K. et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;359:920-6
142. Ooft SN, Weeber F, Dijkstra KK, McLean CM, Kaing S, van Werkhoven E. et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci Transl Med. 2019;11:eaay2574
143. Abboud K, André T, Brunel M, Ducreux M, Eveno C, Glehen O. et al. Management of colorectal peritoneal metastases: expert opinion. J Visc Surg. 2019;156:377-9
144. Roy P, Canet-Jourdan C, Annereau M, Zajac O, Gelli M, Broutin S. et al. Organoids as preclinical models to improve intraperitoneal chemotherapy effectiveness for colorectal cancer patients with peritoneal metastases: preclinical models to improve HIPEC. Int J Pharm. 2017;531:143-52
145. Rajan A, Radhakrishnan P, Ghosh Mehrotra D, Padiernos E, Achhal El Kadmiri MA, Maity TK. et al. Evaluation of drug sensitivity and tumor heterogeneity in thoracic cancers using CANScript, a novel ex-vivo tumor platform. J Clin Oncol. 2018;36:e24246
146. Kumar N, Prasad P, Jash E, Jayasundar S, Singh I, Alam N. et al. CAMP regulated EPAC1 supports microvascular density, angiogenic and metastatic properties in a model of triple negative breast cancer. Carcinogenesis. 2018;39:1245-53
147. Silva A, Jacobson T, Meads M, Distler A, Shain K. An organotypic high throughput system for characterization of drug sensitivity of primary multiple myeloma cells. J Vis Exp. 2015(101):e53070
148. Kashaninejad N, Nikmaneshi MR, Moghadas H, Oskouei AK, Rismanian M, Barisam M. et al. Organ-tumor-on-a-chip for chemosensitivity assay: a critical review. Micromachines (Basel). 2016;7:130. doi: 10.3390/mi7080130
149. Stevens MM, Maire CL, Chou N, Murakami MA, Knoff DS, Kikuchi Y. et al. Drug sensitivity of single cancer cells is predicted by changes in mass accumulation rate. Nat Biotechnol. 2016;34:1161-7
150. Cetin AE, Stevens MM, Calistri NL, Fulciniti M, Olcum S, Kimmerling RJ. et al. Determining therapeutic susceptibility in multiple myeloma by single-cell mass accumulation. Nat Commun. 2017;8:1613. doi: 10.1038/s41467-017-01593-2
151. Jahnke HG, Poenick S, Maschke J, Kendler M, Simon JC, Robitzki AA. Direct chemosensitivity monitoring ex vivo on undissociated melanoma tumor tissue by impedance spectroscopy. Cancer Res. 2014;74:6408-18
152. Martins I, Raza SQ, Voisin L, Dakhli H, Allouch A, Law F. et al. Anticancer chemotherapy and radiotherapy trigger both non-cell-autonomous and cell-autonomous death article. Cell Death Dis. 2018;9:716. doi: 10.1038/s41419-018-0747-y
153. Rello-Varona S, Herrero-Martín D, Lopez-Alemany R, Muñoz-Pinedo C, Tirado OM. “(Not) all (dead) things share the same breath”: identification of cell death mechanisms in anticancer therapy. Cancer Res. 2015;75:913-7
154. Weisenthal LM, Dill PL, Kurnick NB, Lippman ME. Comparison of dye exclusion assays with a clonogenic assay in the determination of drug-induced cytotoxicity. Cancer Research. 1983;43:258-64
155. Eastman A. Improving anticancer drug development begins with cell culture: misinformation perpetrated by the misuse of cytotoxicity assays. Oncotarget. 2017;8:8854-66
156. Méry B, Guy JB, Vallard A, Espenel S, Ardail D, Rodriguez-Lafrasse C. et al. In vitro cell death determination for drug discovery: a landscape review of real issues. J Cell Death. 2017;10:1179670717691251
157. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C. et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960-4
158. Pagès F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C. et al. International validation of the consensus immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391:2128-39
159. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19:133-50
160. Cattaneo CM, Dijkstra KK, Fanchi LF, Kelderman S, Kaing S, van Rooij N. et al. Tumor organoid-T-cell coculture systems. Nat Protoc. 2020;15:15-39
161. Lawrence BP. Measuring the activity of cytolytic lymphocytes. Curr Protoc Toxicol. 2005;18(Chapter):Unit 18.6
162. Cerignoli F, Abassi YA, Lamarche BJ, Guenther G, Ana DS, Guimet D. et al. In vitro immunotherapy potency assays using real-time cell analysis. PLoS One. 2018;13:e0193498
163. Radhakrishnan P, Goldman A, Ulaganathan B, Thaya Kumar A, Maciejko L, Gertje H. et al. Predicting tumor-immune response to checkpoint inhibitors using a novel patient-derived live tumor explant model. J Clin Oncol. 2017;35:e20035
164. Wentink MQ, Broxterman HJ, Lam SW, Boven E, Walraven M, Griffioen AW. et al. A functional bioassay to determine the activity of anti-VEGF antibody therapy in blood of patients with cancer. Br J Cancer. 2016;115:940-8
165. Mélin C, Perraud A, Christou N, Bibes R, Cardot P, Jauberteau MO. et al. New ex-ovo colorectal-cancer models from different SdFFF-sorted tumor-initiating cells. Anal Bioanal Chem. 2015;407:8433-43
166. Ciolofan A, Creţu OM, Mogoantă SŞ, Ciucă EM. The tyrosine kinase inhibitors effects on metastatic tumor graft in the chick chorioallantoic membrane assay. Rom J Morphol Embryol. 2017;58:1257-62
167. Ribatti D. The chick embryo chorioallantoic membrane (CAM) assay. Reprod Toxicol. 2017;70:97-101
168. DeBord LC, Pathak RR, Villaneuva M, Liu HC, Harrington DA, Lewis MT. et al. The chick chorioallantoic membrane (CAM) as a versatile patient-derived xenograft (PDX) platform for precision medicine and preclinical research. Am J Cancer Res. 2018;8:1642-60
169. Fior R, Póvoa V, Mendes RV, Carvalho T, Gomes A, Figueiredo N. et al. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proc Natl Acad Sci USA. 2017;114:E8234-43
170. Costa B, Ferreira S, Póvoa V, Cardoso MJ, Vieira S, Stroom J. et al. Developments in zebrafish avatars as radiotherapy sensitivity reporters — towards personalized medicine. EBioMedicine. 2020;51:102578
171. Forestier A, Sarrazy F, Caillat S, Vandenbrouck Y, Sauvaigo S. Functional DNA repair signature of cancer cell lines exposed to a set of cytotoxic anticancer drugs using a multiplexed enzymatic repair assay on biochip. PLoS ONE. 2012;7:e51754
172. Guillot C, Favaudon V, Herceg Z, Sagne C, Sauvaigo S, Merle P. et al. PARP inhibition and the radiosensitizing effects of the PARP inhibitor ABT-888 in in vitro hepatocellular carcinoma models. BMC Cancer. 2014;14:603-12
173. Vidya G, Gladwin V, Chand P. A comprehensive review on clinical applications of comet assay. J Clin Diagn Res. 2015;9:GE01-5
174. Riou O, Bourgier C, Brengues M, Bonnefoi N, Michaud HA, Castan F. et al. Predictive assays for responses of tumors and normal tissues in radiation oncology. Cancer Radiother. 2019;23:666-73
175. Bowman KJ, Al-Moneef MM, Sherwood BT, Colquhoun AJ, Goddard JC, Griffiths TRL. et al. Comet assay measures of DNA damage are predictive of bladder cancer cell treatment sensitivity in vitro and outcome in vivo. Int J Cancer. 2014;134:1102-11
176. Wood JP, Smith AJO, Bowman KJ, Thomas AL, Jones GDD. Comet assay measures of DNA damage as biomarkers of irinotecan response in colorectal cancer in vitro and in vivo. Cancer Med. 2015;4:1309-21
177. McKenna DJ, McKeown SR, McKelvey-Martin VJ. Potential use of the comet assay in the clinical management of cancer. Mutagenesis. 2008;23:183-90
178. Tougeron D, Sueur B, Zaanan A, de la Fouchardiére C, Sefrioui D, Lecomte T. et al. Prognosis and chemosensitivity of deficient MMR phenotype in patients with metastatic colorectal cancer: An AGEO retrospective multicenter study. Int J Cancer. 2020;147:285-96
179. Kreahling JM, Altiok S. Special technologies for ex vivo analysis of cancer. Cancer Control. 2015;22:226-31
180. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M. et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21:1318-25
181. Lee JY, Kim SY, Park C, Kim NKD, Jang J, Park K. et al. Patient-derived cell models as preclinical tools for genome-directed targeted therapy. Oncotarget. 2015;6:25619-30
182. Fuse E, Tanii H, Kurata N, Kobayashi H, Shimada Y, Tamura T. et al. Unpredicted clinical pharmacology of UCN-01 caused by specific binding to human α1-acid glycoprotein. Cancer Res. 1998;58:3248-53
183. Hidalgo M, Bruckheimer E, Rajeshkumar NV, é Garrido-Laguna I, De Oliveira E, Rubio-Viqueira B. et al. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther. 2011;10:1311-6
184. Izumchenko E, Paz K, Ciznadija D, Sloma I, Katz A, Vasquez-Dunddel D. et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann Oncol. 2017;28:2595-605
185. Wulfkuhle JD, Spira A, Edmiston KH, Petricoin EF. Innovations in clinical trial design in the era of molecular profiling. Methods Mol Biol. 2017;1606:19-36
186. Ahonen I, Åkerfelt M, Toriseva M, Oswald E, Schüler J, Nees M. A high-content image analysis approach for quantitative measurements of chemosensitivity in patient-derived tumor microtissues. Sci Rep. 2017;7:6600. doi: 10.1038/s41598-017-06544-x
187. Weisenthal LM, Su YZ, Duarte TE, Nagourney RA. Non-clonogenic, in vitro assays for predicting sensitivity to cancer chemotherapy. Prog Clin Biol Res. 1988;276:75-92
188. Nagourney RA. Ex vivo programmed cell death and the prediction of response to chemotherapy. Curr Treat Options Oncol. 2006;7:103-10
189. FDA. Framework for regulatory oversight of laboratory developed tests (LDTs) - draft guidance 2014. https://www.fda.gov/media/89841/download
190. Kraus VB. Biomarkers as drug development tools: discovery, validation, qualification and use. Nat Rev Rheumatol. 2018;14:354-62
191. Heinzman JM, Rice SD, Corkan LA. Robotic liquid handlers and semiautomated cell quantification systems increase consistency and reproducibility in high-throughput, cell-based assay. JALA. 2010;5:7-14
192. Loum E, Giraud S, Bessette B, Battu S, Mathonnet M, Lautrette C. Oncogramme, a new individualized tumor response testing method: application to colon cancer. Cytotechnology. 2010;62:381-8
193. Saah AJ, Hoover DR. “Sensitivity” and “specificity” reconsidered: the meaning of these terms in analytical and diagnostic settings. Ann Intern Med. 1997;126:91-4
194. Simon R. Sensitivity, specificity, PPV, and NPV for Predictive Biomarkers. J Natl Cancer Inst. 2015;107(8):djv153
195. Torlakovic E, Lim HJ, Adam J, Barnes P, Bigras G, Chan AWH. et al. “Interchangeability” of PD-L1 immunohistochemistry assays: a meta-analysis of diagnostic accuracy. Mod Pathol. 2020;33:4-17
196. Massard C, Michiels S, Ferté C, Le Deley MC, Lacroix L, Hollebecque A. et al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov. 2017;7:586-95
197. Le Tourneau C, Delord JP, Gonçalves A, Gavoille C, Dubot C, Isambert N. et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16:1324-34
198. Mini E, Nobili S. Pharmacogenetics: implementing personalized medicine. Clin Cases Miner Bone Metab. 2009;6:17-24
199. Schink JC, Copeland LJ. Point: chemosensitivity assays have a role in the management of recurrent ovarian cancer. J Natl Compr Canc Netw. 2011;9:115-20
Author contact
![]() Corresponding author: Muriel Mathonnet , E-mail: muriel.mathonnetfr; Phone : 33 555 056 713; Fax : 33 555 056 725.
Corresponding author: Muriel Mathonnet , E-mail: muriel.mathonnetfr; Phone : 33 555 056 713; Fax : 33 555 056 725.