Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Author contributions
Ethics approval
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2022; 12(12):5389-5403. doi:10.7150/thno.72614 This issue Cite
Research Paper
Subpial delivery of adeno-associated virus 9-synapsin-caveolin-1 (AAV9-SynCav1) preserves motor neuron and neuromuscular junction morphology, motor function, delays disease onset, and extends survival in hSOD1G93A mice
Shanshan Wang1,2 ![]() *, Taiga Ichinomiya1,2,7*, Paul Savchenko1,2, Dongsheng Wang1,2, Atsushi Sawada5, Xiaojing Li1,2, Tiffany Duong1,2, Wenxi Li1,2, Jacqueline A. Bonds1,2, Eun Jung Kim1,2,6, Atsushi Miyanohara2, David M. Roth1,2, Hemal H. Patel1,2, Piyush M. Patel1,2, Takahiro Tadokoro2,4, Martin Marsala2,3, Brian P. Head1,2
*, Taiga Ichinomiya1,2,7*, Paul Savchenko1,2, Dongsheng Wang1,2, Atsushi Sawada5, Xiaojing Li1,2, Tiffany Duong1,2, Wenxi Li1,2, Jacqueline A. Bonds1,2, Eun Jung Kim1,2,6, Atsushi Miyanohara2, David M. Roth1,2, Hemal H. Patel1,2, Piyush M. Patel1,2, Takahiro Tadokoro2,4, Martin Marsala2,3, Brian P. Head1,2 ![]()
1. Department of Anesthesiology, VA San Diego Healthcare System, San Diego, CA, USA
2. Department of Anesthesiology, University of California San Diego, La Jolla, CA, USA
3. Neuroregeneration Laboratory, Department of Anesthesiology, University of California San Diego, La Jolla, CA, USA
4. Department of Anesthesiology, University of the Ryukyus, Okinawa, Japan
5. Department of Anesthesiology, Sapporo Medical University School of Medicine, Sapporo, Japan
6. Department of Anesthesiology and Pain Medicine, Yonsei University College of Medicine, Seoul, South Korea
7. Department of Anesthesiology, Nagasaki University Hospital, Nagasaki, Japan
* Shanshan Wang and Taiga Ichinomaya share co-1st authorship.
Received 2022-3-4; Accepted 2022-6-28; Published 2022-7-11
Abstract

Elevating neuroprotective proteins using adeno-associated virus (AAV)-mediated gene delivery shows great promise in combating devastating neurodegenerative diseases. Amyotrophic lateral sclerosis (ALS) is one such disease resulting from loss of upper and lower motor neurons (MNs) with 90-95% of cases sporadic (SALS) in nature. Due to the unknown etiology of SALS, interventions that afford neuronal protection and preservation are urgently needed. Caveolin-1 (Cav-1), a membrane/lipid rafts (MLRs) scaffolding and neuroprotective protein, and MLR-associated signaling components are decreased in degenerating neurons in postmortem human brains. We previously showed that, when crossing our SynCav1 transgenic mouse (TG) with the mutant human superoxide dismutase 1 (hSOD1G93A) mouse model of ALS, the double transgenic mouse (SynCav1 TG/hSOD1G93A) exhibited better motor function and longer survival. The objective of the current study was to test whether neuron-targeted Cav-1 upregulation in the spinal cord using AAV9-SynCav1 could improve motor function and extend longevity in mutant humanized mouse and rat (hSOD1G93A) models of familial (F)ALS.
Methods: Motor function was assessed by voluntary running wheel (RW) in mice and forelimb grip strength (GS) and motor evoked potentials (MEP) in rats. Immunofluorescence (IF) microscopy for choline acetyltransferase (ChAT) was used to assess MN morphology. Neuromuscular junctions (NMJs) were measured by bungarotoxin-a (Btx-a) and synaptophysin IF. Body weight (BW) was measured weekly, and the survival curve was determined by Kaplan-Meier analysis.
Results: Following subpial gene delivery to the lumbar spinal cord, male and female hSOD1G93A mice treated with SynCav1 exhibited delayed disease onset, greater running-wheel performance, preserved spinal alpha-motor neuron morphology and NMJ integrity, and 10% increased longevity, independent of affecting expression of the mutant hSOD1G93A protein. Cervical subpial SynCav1 delivery to hSOD1G93A rats preserved forelimb GS and MEPs in the brachial and gastrocnemius muscles.
Conclusion: In summary, subpial delivery of SynCav1 protects and preserves spinal motor neurons, and extends longevity in a familial mouse model of ALS without reducing the toxic monogenic component. Furthermore, subpial SynCav1 delivery preserved neuromuscular function in a rat model of FALS. The latter findings strongly indicate the therapeutic applicability of SynCav1 to treat ALS attributed to monogenic (FALS) and potentially in sporadic cases (i.e., SALS).
Keywords: caveolin-1, membrane/lipid raft (MLRs), gene therapy, hSOD1G93A, amyotrophic lateral sclerosis, motor neuron, neuromuscular junction
Introduction
Amyotrophic lateral sclerosis (ALS) derives its name from denervational atrophy of the muscles in the tongue, oropharynx, and limbs (amyotrophy), degenerating corticospinal axons resulting in thinning and scarring (sclerosis) of the lateral spinal cord, and thinning of the ventral roots. ALS is a progressive, paralytic disease due to loss of motor neurons (MN) in the brain and spinal cord. ALS signs include diffuse muscle weakness, fasciculations (spontaneous muscle twitching), hyperreflexia (upper), and paralysis (lower), and ultimately reducing life expectancy to 2-5 years [1, 2]. In addition to these motor-associated pathologies, 15-20% of ALS cases also exhibit cognitive deficits as a result of degeneration of the frontal and temporal lobes (frontotemporal dementia or FTD) [3]. Incidence of ALS is 3-5 per 100,000 persons globally and is present in both dominantly inherited familial (FALS, 5-10%) and sporadic (SALS, 90-95%) forms [2]. Typical age-of-onset is 58-63 years and 47-52 years for SALS and FALS, respectively, with both being phenotypically similar. Unfortunately, the current FDA-approved treatments for ALS including Rilutek (riluzole, a glutamate antagonist) [1] and Radicava (edaravone, a free radical scavenger) [4] only provide limited extension of life; the former acts by attenuating excessive motor neuron firing and the latter works by decreasing oxidative stress.
For FALS, genetic interventions focused on knocking down key genes such as mutant form of super oxide dismutase 1 (SOD1) [5], however, due to the lesser-known etiology of SALS, a successful therapy for ALS will most likely require combinatorial neuroprotective interventions. What is known about ALS, including FALS and SALS, is that they are highly complex, heterogeneous neuropathological conditions resulting from motor neuronal loss due to a highly neurotoxic environment [6-10]. Therefore, interventions that provide neuroprotection may preserve motor neuron survival and neuromuscular function even in the presence of known monogenic mutations or unknown sporadic etiology.
Previously we showed that neuron-targeting of the membrane/lipid raft (MLR) protein caveolin-1 (using a synapsin-driven Cav-1 engineered construct termed SynCav1) promotes hippocampal synaptic and neuroplasticity [11], significantly improves learning and memory in aged mice, and preserves learning and memory in an Alzheimer's disease (AD) mouse model independent of reducing amyloid plaques [12]. Moreover, when crossing our SynCav1 transgenic mouse (TG) with the hSOD1G93A mouse model of ALS, we observed preserved body weight (BW), longer survival, better running wheel (RW) performance, preserved spinal cord α (alpha) motor neurons, and more MLR-associated neurotrophin receptors (NTRs) in the spinal cord. In terms of human clinical relevance, a seminal finding from the United Kingdom (University of Sheffield) revealed decreased Cav-1 protein expression and disrupted MLRs in samples from sporadic human ALS cases due to mutations in the Cav-1 DNA enhancer region, suggestive of a novel ALS-associated risk variant [13].
One of the major challenges of treating neurodegenerative diseases, including ALS and AD, is to find the most effective manner by which to deliver vectors while minimizing the off-target effect. Although some have demonstrated penetration into the central nervous system and spinal cord using intrathecal or intraventricular delivery using high titers of genetic material, which unfortunately limits the effectiveness and safety of using AAV9-based vectors to treat afflicted spinal cord and brain tissue in ALS [14-17]. To achieve broader tissue distribution with greater efficacy, the current study utilized the novel spinal subpial (SP) vector delivery route developed by the Marsala laboratory [14].
Over the past few years, the predominant approach to treat ALS is to target known monogenic causes of FALS [15, 18-23]. Instead of targeting the genes or proteins attributed to the gain of toxicity, the present study tested the hypothesis that subpial delivery of neuroprotective AAV9-SynCav1 to the spinal cord of hSOD1G93A mice may mitigate disease progression, protect motor neurons, maintain neuromuscular junctions (NMJs), and extend survival, independent of targeting mutant hSOD1G93A protein. We now show for the first time that a one-time subpial delivery of AAV9-SynCav1 to hSOD1G93A mice at lumbar section significantly extends longevity and preserves BW in both male and female mice, preserves lumbar motor neuron morphology and NMJ morphology in the lower limb muscle, and maintains better RW performance independent of affecting expression of mutant hSODG93Aprotein. This study further extends the neuroprotective and therapeutic potential of SynCav1 gene therapy to another distinct neurodegenerative condition (i.e., ALS), irrespective of disease etiology, and thus SynCav1 has the potential to treat a variety of neurodegenerative conditions attributed to either familial or sporadic forms. Furthermore, SynCav1 gene therapy may work synergistically in combination with other interventions (e.g., antisense oligonucleotides, antibodies, small molecules) that specifically target monogenic familial forms of ALS and other neurodegenerative conditions.
Materials and Methods
Animal
Human mutant superoxide dismutase-1 (hSOD1G93A)-transgenic (Tg) and C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and bred in-house. Wild type (WT) littermates were used as control. All animal protocols were approved by the Veterans Administration San Diego Healthcare System Institutional Animal Care and Use Committee (#A20-030). Mice were reared (3-5/cage) with free access to food and water. WT, and hSOD1G93A mice were allocated to 4 groups randomly: WT, hSOD1G93A naïve (no surgery), hSOD1G93A + control (subpial surgery with same volume of AAV9-SynNull or saline), and hSOD1G93A + SynCav1 to receive subpial lumbar vector injections. Spinal cord tissue was processed for biochemistry, histopathology, and immunofluorescence at the indicated time points.
Sprague-Dawley rats were treated in compliance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health) in the Marsala lab and approved by UCSD. Transgenic rats expressing ALS-linked human SOD1 mutant protein (hSOD1G93A) exhibit an ALS-like neurodegenerative phenotype characterized by diminished body weight from muscle atrophy, weakness/paralysis, fasciculations, and early death, along with reductions in functional cortical alpha motor neurons and ventral horn alpha motor neurons that can be observed histologically [24]. The hSOD1G93A transgenic male rats (Marsala lab, UCSD) were bred with WT female rats to produce WT and hSOD1G93A positive male rats.
hSOD1G93A Genotyping
hSOD1G93A mice and rats were confirmed by genomic DNA extraction and PCR using the Qiagen DNeasy Blood and Tissue Kit (69504; Qiagen, Valencia, CA, USA). PCR was performed for the hSOD1 genes by using the following protocol: denaturation at 94 °C for 2 min, followed by 10 cycles at 94 °C for 20 s, 65 °C for 15 s (- 0.5 per cycle decrease), and 68 °C for 10 s, then followed by 28 cycles at 94 °C for 15 s, 60 °C for 15 s, 72 °C for 10 s, then 72 °C for 2 min and hold at 10 °C. All primers were purchased from Integrated DNA Technologies (Coralville, IA, USA). hSOD1G93A-positive mice were confirmed using the following primers: primer 1, hSOD1 Forward [CAT CAG CCC TAA TCC ATC TGA (oIMR0113)]; primer 2, hSOD1 Reverse [CGC GAC TAA CAA TCA AAG TGA (oIMR0114)]; primer 3, Internal control Forward [CTA GGC CAC AGA ATT GAA AGA TCT (oIMR7338)]; and primer 4, Internal control Forward [GTA GGT GGA AAT TCT AGC ATC ATC C (oIMR7339)]. These primers were used to detect a product size of 800 for mouse endogenous SOD1 and 600 base pair (bp) for human SOD1 as previously described [25]. Transgenic rats were genotyped by PCR amplification for the hSOD1G93A. PCR products were separated on a 1% agarose gel (35 min at 135 volts).
Viral vectors and SynCav1 Construct
A self-complementary AAV (scAAV) construct expressing neuron specific synapsin (Syn) promoter with the Cav-1 cDNA was generated at the UCSD Viral Vector core as previously described [12]. To link the neuron specific synapsin (Syn) promoter with the Cav-1 cDNA, an XbaI-SalI DNA fragment containing the Syn promoter was inserted into the NheI-SalI sites of pEGFP-N1 (Clontech). The resulting plasmid was designated pSyn-EGFP. A 537 bp Cav-1 cDNA was isolated from the pCRII-TOPO vector (Invitrogen) by digestion with PmeI-NotI and inserted into the SmaI-NotI site of the pSyn-EGFP to generate pSyn-Cav-1, in which the EGFP gene was replaced with Cav-1 cDNA. The Syn-promoter-Cav-1 cassette was isolated from pSyn-Cav-1. The scAAV vector construct, expressing Cav-1 or RFP driven by the Syn promoter, was made by DNA synthesis of the Syn promoter (480 bp) through Cav-1 (537 bp) or enhanced RFP (700 bp) and cloning it into an scAAV backbone plasmid. The scAAV9-SynCav1, scAAV9-SynRFP, or scAAV9-SynNull vectors were produced by transient transfection of HEK293T cells with each vector plasmid, pRep2/Cap9 and pAd-Helper plasmid. Helper virus-free AAV vectors were produced by Polyethylenimine (PEI)-mediated transient co-transfection of HEK293T cells with three plasmids [pVector, pRep2/CapX (X stands for each serotype) and pAd-Helper]. Cells were collected at 72 hrs post-transfection and cell lysates were made by 3x freeze/thaw cycle. AAV vectors in the cell lysates were purified by combination of sucrose-cushion ultracentrifugation and anion-exchange column chromatography. Virus titers were measured by real-time Q-PCR to determine genome copy (g.c.) number in the vector preparations (g.c./ml) as a measure of AAV particles with full genome content. Final virus titers for all three vectors are ~2.0 x1013 g.c./ml.
Subpial surgery and AAV administration
All surgical procedures were done under sterile conditions and approved by the local IACUCs. Mouse subpial procedure: Lumbar (L1-L3) subpial vector injection in mice was performed as previously described [14]. Briefly, mice were anesthetized with 2-3% isoflurane, the surgical area was shaved and cleaned, and a skin incision was made at the thoracic 12 (T12)-L1 vertebral level. Under the dissecting microscope, a dorsal laminectomy of the thoracic (T13) vertebrae was performed, and the dura overlying the lumbar (L1) spinal segments was cut open using a 31- gauge (G) needle. The pia mater was then punctured using a 36-G pia-penetrating needle, followed by the insertion of a blunt 36-G injection needle containing the scAAV9 vector. Both the pia-penetrating and the subpial injection needles were mounted on fine XYZ manipulator with needle holder (SM-15HE, Narishige, Japan). Rat subpial procedure: Wild type (WT) or hSOD1G93A Sprague Dawley rats were anesthetized by isoflurane with 2% in room air and placed into a Lab Standard Stereotaxic frame (Stoelting, Wood Dale, IL). Rats were elevated 3 cm by placing on a plastic box. A homeothermic heating blanket which was set at 37 °C with feedback from a rectal thermometer (Harvard Apparatus, Holliston, MA) was used to maintain body temperature of rats during the procedure. Rats were then placed in Spine Adaptors (Stoelting, Wood Dale, IL) and a C4-5 laminectomy was performed to expose the dorsal surface of spinal segment C4-5 using an air-powered dental drill and binocular microscope. The dura and pia mater were carefully incised using a 27- G needle and a polyethylene tubing catheter, of which its size was 0.008 ID x 0.02 OD, (SAI infusion technologies, Lake Villa, IL) was inserted into the subpia-space. Injections were made using 100-μL Hamilton Gas Tight syringe (Hamilton, Reno, NV). Mice received a total of 10 μl to the lumbar spinal cord and rats received a total of 20 μl of AAV9-SynCav1 (~2.0 x 1013 genome copies (g.c.)/ml) vector to the cervical spinal cord by subpial delivery (5 μl bilateral to the lumbar spinal cord, 10 μl total over 600 s for mice; 10 μl bilateral to the cervical spinal cord, 20 μl total over 600 s for rats) using a 50 μl Hamilton syringe and digital infusion pump (Microinjector MINJ-PD, Tritech Research). After vector delivery, the muscle and skin were closed using 7/0 Prolene and suture clips. Before recovery from anesthesia, the animals received subcutaneous fluids, antibiotics (cefazolin sodium 50 mg/kg) and the initial dose of pain medication (slow release Buprenex 0.05 mg/kg). Control group received laminectomy and subpial injection of or equal volume of either AAV9-SynNull or sterilized saline.
Assessment of disease onset and survival
Disease onset and late disease were retrospectively defined as the first sign of weight loss from peak body weight and 10% weight decline from peak body weight, respectively [18, 26, 27]. Survival analyses were performed using Kaplan-Meier analyses as previously described [25]. Survival endpoint is defined by the inability of the mouse to adjust itself within 30 seconds (s) when placed on its back, a humane endpoint that guarantees that animals are euthanized before they are unable to eat or drink.
Voluntary running wheel (RW)
At 8 and 16-week-age, mice were housed individually in plastic cages measuring 40 x 40 x 35 cm (length x width x height) with a 11.5 cm diameter running wheel mounted on the cage side (SuperFlex Open Field, Omnitech Electronics, Inc., Ohio, USA). The mice had free access to food and water ad libitum and were checked daily for health and wellness. The mice had open access to running wheels under conditions of a 12 h light/dark cycle (lights off from 6 AM to 6 PM). The duration of each experiment was 36 h including 2 dark cycles. The mice will be placed in the cages at least 24h prior to data collection. A sensory beam, connected to a desktop computer via a software (Fusion v6.5 r9999 SuperFlex Edition, Omnitech Electronics, Inc., Ohio, USA), was used to record wheel rotations. Running velocity (m/min), and total distance (m) were used for analysis.
Motor Evoked Potential (MEP)
Rats were anesthetized by intramuscular injection of ketamine (100 mg/kg) for motor-evoked potentials (MEP) recordings in the branchial or gastrocnemius muscles. Two 30-G stimulating electrodes were placed subcutaneously overlying the left and right motor cortex. In all rats, MEP of upper forelimb (brachial) and lower limb (gastrocnemius) were recorded from the muscles elicited by transcranial electrical stimulation with a pulse duration of 200 μs at 15 mA. Recording electrodes were connected to an active head stage (Warner Instruments, Hamden, CT), and the recorded signal is amplified using DP-311 differential amplifier (Warner Instruments, Hamden, CT). The amplified signal was acquired by the PowerLab 8/30 data acquisition system (ADInstruments, Australia) at sampling frequency of 20 kHz, digitized and stored using LabChart 7 (ADInstruments, Australia) in PC for analysis.
Grip Strength
Cohorts of age-matched transgenic rats and littermate controls were tested for loss of forelimb grip strength by using a grip strength meter (AMETEK, Inc., Berwyn, PA) at 8 wk (before subpial-injection) and weekly from 10 to 20 wk. Rats were held in front of a horizontal bar, such that only the forelimb paws were able to grasp the bar. The rats were gently pulled back with steady force until both paws released the bar. Each rat was given 6 attempts and the mean grip strength was recorded for analysis.
Immunofluorescence (IF) Microscopy
Motor neuron morphology and Cav-1 target validation was assessed by IF. Specifically, mice were perfused with saline and the spinal cord was dissected followed by 48 hrs post-fix in z-fix (ANATECH, #170) and dehydration in 30% sucrose. 30 µm thickness coronal spinal cord section was then incubated with Alexa Fluor 488 conjugated anti-choline acetyltransferase (ChAT) (abcam, ab225263, 1:400) and Alexa Fluor 594 conjugated caveolin-1 antibody (7C8) (Novus biologicals, NB100-615AF594, 1:1000) for overnight at 4 °C. Nuclei were counterstained with antifade DAPI mounting medium (ProLong Gold antifade mounting medium with DAPI, P36931; Invitrogen, Waltham, MA, USA). A minimum of three coronal spinal cord sections per mouse were used for quantitation of motor neuron.
For NMJ analysis, 30 µm thickness horizontal gastrocnemius muscle section was stained with 488-conjugated bungarotoxin-a (Btx-a) (B13422; 1:250; Invitrogen) to label post-synaptic structure and synaptophysin (ab32594; 1:200, abcam) to label pre-synaptic vesicles followed by 1 h incubation of secondary (Alexa Fluor 568, 1:200; B13422, Invitrogen, Waltham, MA, USA) at room temperature. Occupancy of synaptophysin labeled presynaptic structure in Btx-a labeled postsynaptic was analyzed to assess NMJ morphology [28]. Slides were imaged using Keyence All in One microscopy (BZ-X700, Keyence corporation of America, IL, USA) and confocal laser microscopy (A1; Nikon, Tokyo, Japan), Z stack with 10um was taken and projected to a single image for analysis.
Biochemical Characterization of Membrane/Lipid Rafts (MLRs) and Immunoblot (IB) assay
Thoracic (T12) to lumbar (L2) spinal cord tissue were homogenized at 4 ºC in 500 mM sodium carbonate (pH 11.0) (containing protease and phosphatase inhibitors) and then sonicated 3x for 15 s. Samples (0.5 mg/ml) were subjected to sucrose density gradient fractionation as previously described [29]. Homogenates and fractions were immunoblotted using primary antibodies Cav-1 (Cell Signaling #3268; 1:1000), B8H10 (Medimabs MM-0070-P, 1:500), ChAT (Millipore, AB144P, 1:1000), and GAPDH (Cell Signaling #2118s; 1:1000) overnight at 4 ºC followed by incubation with IR-dye labeled secondary antibody for 1 hour. Membranes were scanned by LiCor Odyssey followed by densitometric analysis.
Statistical analysis
Sample size is indicated under each section of the Methods and in the figure legends. Before determining statistical significance, data were checked for normal distribution. Data were analyzed by Student's t test, one-way analysis of variance (ANOVA) or 2-way ANOVA followed by Fisher's LSD, Tukey's, Šídák's, or Bonferroni's multiple comparisons post hoc analyses as appropriate using Prism 7 software (GraphPad, La Jolla, CA, USA). The Kaplan-Meier survival curve was analyzed using a log-rank test. Mean and median survival were analyzed by an unpaired Student's t-test. Significance was assumed when P < 0.05. All experimental groups were blinded, and code was broken only for analysis. For correlation analysis, a simple linear regression with 95% confidence intervals was performed.
Results
AAV9-SynCav1 delivery to the lumbar spinal cord increases Cav-1 protein expression in the lumbar and thoracic cord
Previous work from other groups have reported that the occurrence of symptomatic onset varies from 8 to 12 weeks (wk) [30] in the hSOD1 G93A mice using different methods and parameters [31, 32]. For example, motor deficits and motor unit loss in lower limb muscle due to selective preferential vulnerability of large, fast motor units, innervated by large motor neurons was observed in 8 wk hSOD1G93A mice, several weeks prior to the standard measurable onset of overt clinical symptoms in this mouse model [33-35]. To achieve effective AAV9 transduction of spinal cord motor neurons and peak expression of the Cav-1 transgene (which takes approximately 3-4 wk post AAV delivery), we chose to deliver AAV9-SynCav1 at week 8 via subpial delivery, a highly effective route of administration used to accomplish effective strong transgene expression in the mammalian spinal cord [11, 12]. As shown in Figure 1, lumbar subpial delivery of AAV9-SynRFP resulted in a strong expression of RFP in the lower thoracic up to lumbar (T11 to L5), with minimal expression detected in upper thoracic and cervical regions. Our previous study demonstrated that SynCav1+/ hSOD1G93A+ transgenic mice exhibit slowing of disease progression, we therefore initially performed a dose-dependent study for subpial AAV9-SynCav1 delivery to determine the optimal dose necessary to achieve Cav-1 protein expression in the spinal cord similar to that measured in SynCav1+ transgenic mice [25]. As shown in Figure S1, the optimal dose of AAV9-SynCav1 to achieve 4-fold Cav-1 overexpression in the thoracic and lumbar spinal cord was 2 x 1011 total g.c. (10 μl of 2 x 1013 g.c./ml). This dose was used in all the following studies.
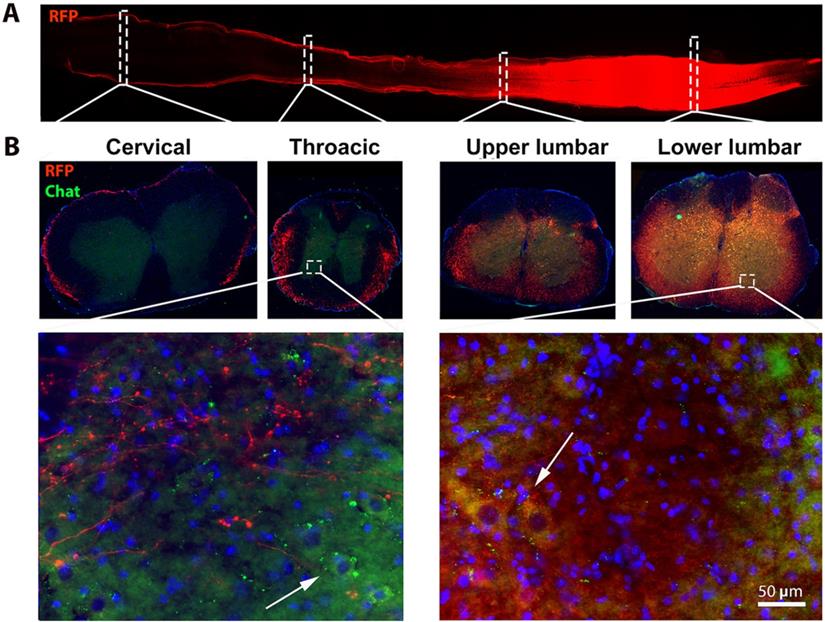
Subpial AAV9-SynRFP delivery to the lumbar spinal cord (L1) resulted in RFP transgene expression across entire lumbar cords. Spinal cords were collected 30 days post-AAV9-SynRFP. (A) Horizontal section of spinal cords showed strong RFP expression around lumbar cords. (B) Coronal sections of spinal cord were stained with motor neuron marker ChAT. Representative images showed strong RFP expression in the lumbar segment including ChAT positive lower motor neuron at the ventral horn area, while cervical and thoracic exhibit weak RFP signal predominantly in white matter.
AAV9-SynCav1 significantly delays BW loss and disease onset, and extends survival in hSOD1G93A mice
We next assessed whether subpial delivery of AAV9-SynCav1 could provide therapeutic benefit to the hSOD1G93A mouse model of ALS. As shown in Figure 2A and 2B, compared to hSOD1G93A + control, hSOD1G93A + AAV9-SynCav1 mice showed significantly delayed BW loss in both sexes. As shown in Figure 2C and 2D, average disease onset of hSOD1G93A + AAV9-SynCav1 mice was 18 wk for male (vs 15 wk hSOD1G93A + control male) and female (vs 16 wk hSOD1G93A + control female), an approximately 15% delay compared to the control group. In terms of disease progression, late disease onset (i.e., 10% decrease from peak BW) [26] was measured at 20 wk for both male and female hSOD1G93A + control mice, while AAV9-SynCav1-treated hSOD1G93A female mice exhibited delayed late disease onset until 23 wk (~11% BW decline); AAV9-SynCav1-treated hSOD1G93A male mice only exhibited ~4% BW decline at 22 wk (end of body weight record due to loss of multiple objects in the hSOD1G93A + control groups).
AAV9-SynCav1 subpial delivery significantly delays disease onset, preserves BW and extends survival in hSOD1G93A mice. (A-B) Body weight (BW), (C-D) Average disease onset timepoint (defined by peak BW) and (E-F) Kaplan-Meier survival curve for male and female mice. Groups: hSOD1G93A naïve (no surgery), hSOD1G93A + control and hSOD1G93A + SynCav1 mice. BW was expressed as mean ± SEM, Two-way ANOVA with Šídák's multiple comparisons test was used for A and B. One-way ANOVA with Dunnett's multiple comparison test was used for C and D (all compared to hSOD1G93A + control group). Kaplan-Meier Curve was expressed as median days (d) survival and analyzed using a Mantel-Cox Log-rank test (***p < 0.005, ****p < 0.0001).
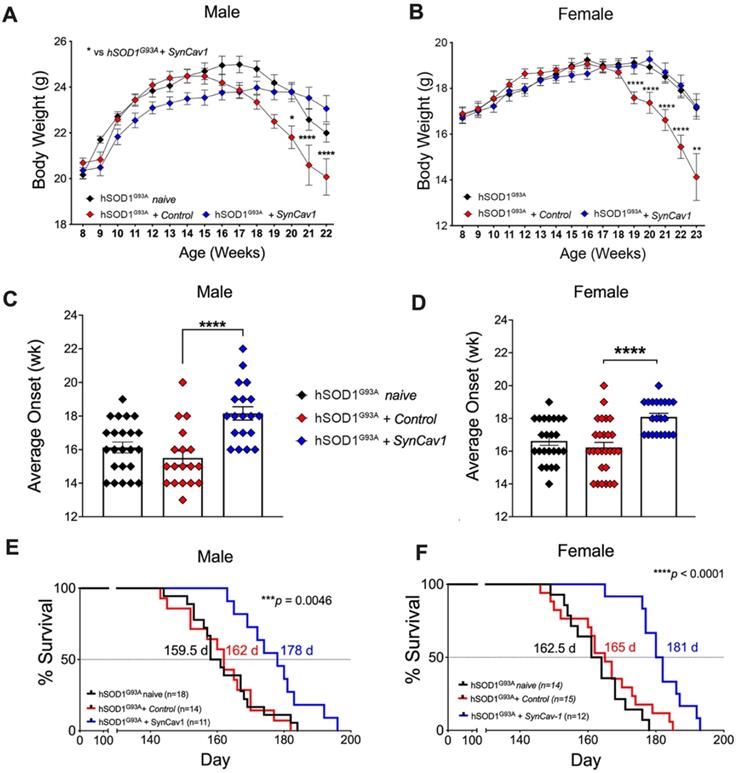
In terms of survival, although subpial surgery had no effect on survival rates for hSOD1G93A + control versus hSOD1G93A naïve, we observed a greater decline in BW in the sham surgery control group compared to naïve hSOD1G93A mice between 18-22 wk (Figure 2A and 2B), indicating that subpial surgery alone appeared to impact disease progression. Because of this effect caused by the subpial procedure alone, for all subsequent assays (RW, histological quantitation of MN and NMJ morphology), Student's t-test comparison was performed only within the non-surgery (i.e., WT vs hSOD1G93A naïve) and the surgery group (i.e., hSOD1G93A + control vs hSOD1G93A + SynCav1) to remove the confounding effects from subpial surgery on any measurable efficacy afforded by SynCav1 gene delivery. As shown in Figure 2E-2F, AAV9-SynCav1 gene delivery significantly extended survival in male and female (10%) hSOD1G93A mice when compared to hSOD1G93A + control mice (Figure 2E, median male survival days (d): hSOD1G93A + SynCav1 (178 d) vs hSOD1G93A + control (162 d), ***p = 0.0046, Kaplan-Meier; n = 11-18; Figure 2F, median female survival: hSOD1G93A + SynCav1 (181 d) vs hSOD1G93A + control (165 d); ****p < 0.0001, Kaplan-Meier; n = 12-15). These findings are direct evidence that SynCav1 significantly delays disease onset and extends longevity (i.e., slowing % BW loss) in hSOD1G93A mice.
Subpial AAV9-SynCav1 delivery preserves Cav-1 localization to lumbar spinal cord MN plasmalemmal MLRs in hSOD1G93A mice
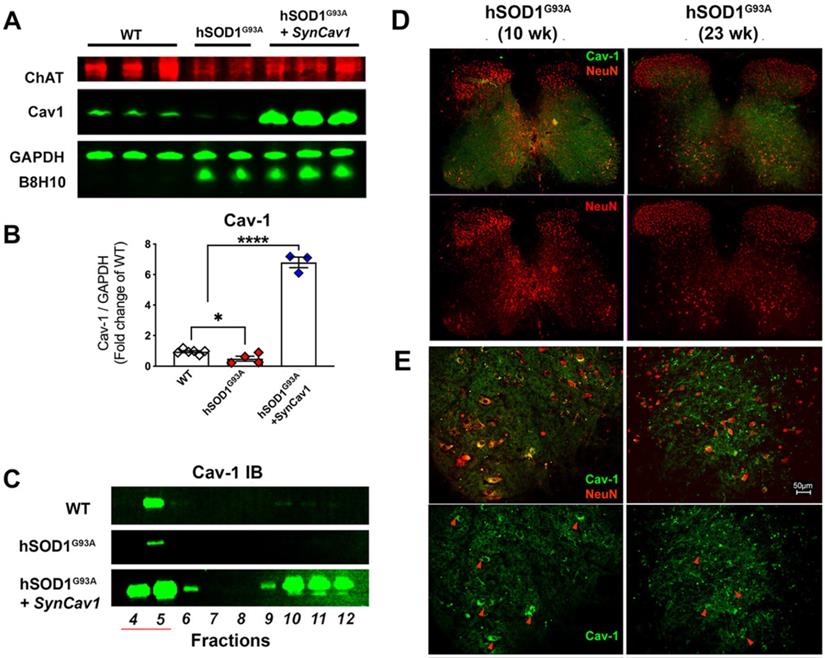
As shown in Figure 3A-3B, IB assays of lumbar spinal cord tissue homogenates from terminal stage 23 wk hSOD1G93A mice (terminal stage as defined by 162 d median survival in Figure 2C and 2D for non-treated hSOD1G93A mice) showed elevated B8H10 (mutant and WT human SOD protein) and decreased Cav-1 expression. IB of MLR fractions revealed that Cav-1 was predominantly localized to MLR fractions 4-5 (Figure 3C) in normal WT mice, which is consistent with previously published work in brain and spinal cord tissue [11, 25, 29]. However, Cav-1 expression decreased in MLR fractions from the lumbar spinal cords of 23 wk (end stage) hSOD1G93A mice. Subpial AAV9-SynCav1 delivery to 8 wk hSOD1G93A mice significantly increased Cav-1 expression and subcellular localization of Cav-1 to MLR fractions, with no effect on expression of mutant SOD protein (B8H10). One limitation to IB assays is that when performed on whole spinal cord (or brain) tissue, one is incapable of distinguishing cell-specific protein changes (i.e., neuronal versus non-neuronal cells). To better elucidate the decreased Cav-1 expression as measured by IB, we additionally performed IF on lumbar spinal cord tissue from 10 wk (early symptomatic) and 23 wk (end stage) using neuronal marker NeuN and Cav-1 (Figure 3D-3E). While Cav-1 expression exhibited a polarized plasma membrane localization in NeuN-positive large MN cell bodies at the ventral horn of 10 wk hSOD1G93A mice, a distribution pattern similar to WT mice (Figure 3E, red arrow heads); lumbar sections from 23 wk hSOD1G93A mice confirmed that the decreased Cav-1 expression specifically occurred in ventral horn degenerated MN.
Decreased neuronal Cav-1 expression in the lumbar spinal cord MN from end-stage hSOD1G93A mice. (A-B) Lumbar spinal cords from end-stage hSOD1G93A mice (21-23 wk) were subjected to IB assays to measure Cav-1, ChAT (motor neuron marker), and mutant hSOD protein (B8H10). While Cav-1 expression was significantly decreased in the end-stage naïve hSOD1G93A mice, subpial delivery of AAV9-SynCav1 resulted a 6-8 folds increase of Cav-1 protein expression, with no significant change in expression of hSOD1G93A. (C) Representative IB of lumbar spinal cord homogenates showed decreased MLR-localized Cav-1 (i.e., buoyant fractions 4-5) in naïve hSOD G93A mice compared to WT and hSOD G93A + SynCav1. (D) IF microscopy was performed to measure Cav-1 and NeuN-positive MN in lumbar sections of 10 wk (early symptomatic) and 23 wk (end-stage) hSOD G93A mice. (E) Higher magnification of ventral horn area revealed polarized Cav-1 expression on large NeuN-positive MN cell bodies at 10 wk (red arrowheads in left panels). At 23 wk, both Cav-1 expression on NeuN-positive MN and the size of NeuN-positive MN cell bodies were decreased in the ventral horn region (arrowheads in right panels) from hSOD G93A mice. Data are presented as mean ± SEM. Data were analyzed using One-way ANOVA with Fisher's LSD multiple comparisons post hoc test. (n = 3-6; *p < 0.05). Scale bar, 50 μm.
AAV9-SynCav1 treated hSOD1G93A mice exhibit better RW endurance
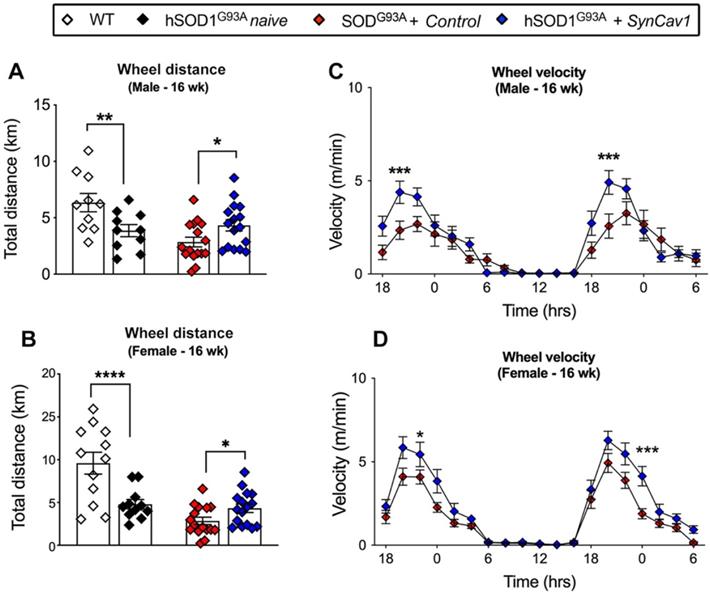
At 8 wk (before subpial surgery), mice from all groups were trained for voluntary RW test. hSOD1G93A mice were then randomly allocated to naïve, control and SynCav1 groups. Motor function was assessed by voluntary RW test again at week 16. Consistent with our previous study, both sexes of hSOD1G93A naïve mice ran significantly less total distance compared with WT (Figure 4A, male, *p = 0.02, Student's t-test, n = 10; Figure 4B, female, **p = 0.003 Student's t-test, n = 11-12). hSOD1G93A + control mice ran slightly less than hSOD1G93A naive with no significant difference between these two mice at 16 wk. hSOD1G93A + SynCav1 mice exhibited significantly greater total distance run compared with control mice (Figure 4A, male, **p = 0.03, Student's t-test, n = 10; Figure 4B, female, **p = 0.004 Student's t-test, n = 11-12). Furthermore, hSOD1G93A + SynCav1 mice exhibited significantly higher RW velocity compared with hSOD1G93A + control mice (Figure 4C, male; Figure 4D, female; 2-way ANOVA, Fisher's LSD, n = 16-19). These data demonstrate that subpial AAV9-SynCav1 delivery preserved motor function as measured by RW performance.
AAV9-SynCav1 treated hSOD1G93A mice perform better on voluntary running wheel (RW) motor test. The 16 wk male and female total distance (km) (A, B) and mean RW velocity (m/s) (C, D) measured for 36 h. Data are expressed as means ± SEM. (n = 10-19/group). Student's t-test was used for A and B. 2-way ANOVA with Fisher's LSD multiple comparison test was used for C and D (*p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.0001).
AAV9-SynCav1 preserves alpha-motor neuron morphology in the spinal cord lumbar ventral horn and NMJ morphology in the gastrocnemius muscle of hSOD1G93A mice
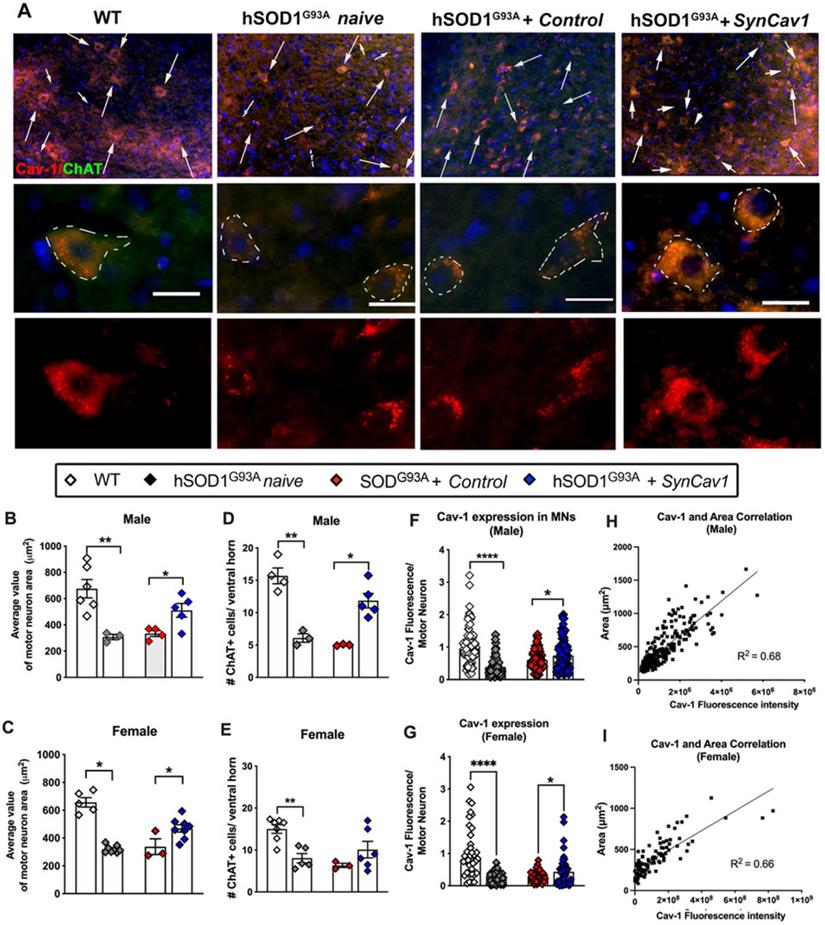
We next assayed for lower motor neuron (MN) cell counts and morphology in the lumbar ventral horn region (L3-L5) after subpial SynCav1 delivery (Figure 5A). As shown in Figure 5B-5E, the total number and cell body (i.e., soma) area of ChAT+ MNs were significantly decreased in both 19 wk male and female hSOD1G93A naïve mice compared to WT mice, consistent with previous work that showed ALS pathology preferentially affects larger alpha motor neurons innervating faster muscle fibers [36]. In contrast, SynCav1 maintained MN numbers and normal morphology and prevented loss of spinal lumbar ventral horn MNs compared to control hSOD1G93A mice. Further analysis confirmed that the decreased Cav-1 in the hSOD1G93A naïve spinal cord is mostly due to the downregulated Cav-1 in the ChAT+ MNs, while SynCav1 preserved Cav-1 expression of the ChAT+ MNs compared to hSOD1G93A + control mice (Figure 5F-5G, ****p < 0.0001, Student's t-test, n = 3-8 mice/group). Noticeably, simple linear regression correlation analysis revealed a significant positive correlation between Cav-1 expression and ChAT positive MN area (Figure 5H-5I, p < 0.0001, R2 = 0.68). Together, these results indicate that SynCav1 gene delivery affords significant neuroprotection in hSOD1G93A spinal cord MNs in both Sexes.
SynCav1 prevented lumbar ventral horn motor neuron loss in 19 wk hSOD1G93A mice. (A) Motor neuron (MN) was identified by the MN marker choline acetyl transferase (ChAT) in the lumbar ventral horn. Scale bar, 20 μm. (B-E) Number and average cell body area (μm2) of MNs per animal. A minimal of 3 images was used to calculate the average value. (F-G) Cav-1 expression was measured in individual ChAT+ somas and normalized to its area. Data are presented as mean ± SEM. Student's t-test (n = 3-8 mice/group; *p < 0.05, **p < 0.01, ****p < 0.001). (H-I) Simple linear regression with Pearson's r correlation analysis was performed comparing MN soma area with Cav-1 expression.
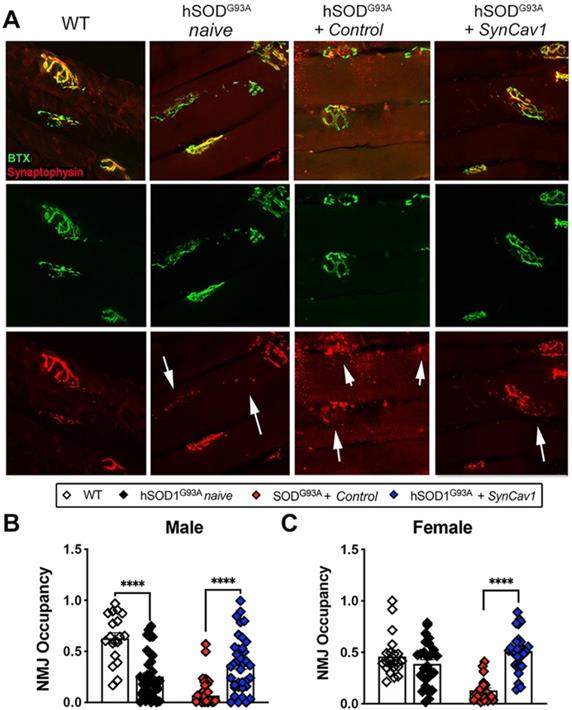
Finally, because we observed better motor function and preservation of MNs at lower lumbar level with SynCav1 delivery, we analyzed NMJ morphology in the gastrocnemius muscle, one of the larger lower limb muscles receiving synaptic innervation from L5-S1 spinal cord MN axons (Figure 6A). At 19 wk, mice from both hSOD1G93A naïve and hSOD1G93A + control exhibited a significant decrease in NMJ occupancy (Figure 6B-6C, male, ****p < 0.0001, n = 3-5 mice/group; female, ****p < 0.0001, n = 3-5 mice/group; Student's t-test), with only 30% and 20% of the NMJs occupancy measured in naïve and control hSOD1G93A mice respectively, indicating severe denervation of the GA muscle [37]. Noticeably, NMJ occupancy in hSOD1G93A + control mice were significantly lower compared to naïve hSOD1G93A mice, suggesting an effect from subpial surgery alone on NMJ integrity. In contrast, the NMJ occupancy of synaptophysin in the hSOD1G93A + SynCav1 mice was significantly greater in both sexes (****p < 0.0001, n = 3-5 mice/group; Student's t-test) compared to hSOD1G93A + controls, results that in line with the greater RW performance observed in the hSOD1G93A + SynCav1 group.
SynCav1 delivery preserved NMJ morphology in the gastrocnemius muscle. (A) Representative images of neuromuscular junction (NMJ) identified by bungarotoxin-alpha (Btx-α, post-synaptic) and synaptophysin (pre-synaptic vesicles). (B, C) NMJ occupancy is defined by pre-synaptic area overlaid with post-synaptic area (%). Student's t-test was performed for B and C (n = 3-5 mice/group; *p < 0.05, ****p < 0.0001).
AAV9-SynCav1 treated hSOD1G93A rats exhibit better grip strength and MEPs (larger amplitude and lower latency)
We next tested whether subpial AAV9-SynCav1 delivery in a larger animal model (i.e., hSOD1G93A rat model) could mitigate ALS-like disease onset. Transgenic negative or positive rats (Figure S2A) received control vector (WT + control or hSOD1G93A + control) or AAV9-SynCav1 hSOD1G93A + SynCav1) by cervical (C4/C5) subpial delivery at 2 m. At 4 m (2 m post vector delivery), IB and IF of spinal cord tissue confirmed strong Cav-1 expression in MN from SynCav1 administered rats (Figure S2B-S2C). We next measured neuromuscular function by using forelimb grip strength (GS) and MEPs in the brachial (upper forelimb) and gastrocnemius muscles (lower calf). GS and MEPs were initially measured at 8 wk (prior to subpial AAV delivery) and then measured weekly from 10-20 wk. hSOD1G93A + SynCav1 rats exhibited significantly greater peak force (g) grip strength at 13, 14, and 15 wk compared to hSOD1G93A + control rats (Figure S3A; n = 17-18 rats/group; **p < 0.01, ***p < 0.001, 2-way ANOVA, Bonferroni's multiple comparisons test). MEP recordings revealed significant greater amplitude (mV) at 18 wk (Figure S3B) and shorter latency (millisecond, ms) at 15 wk (Figure S3C) in the upper brachial forelimb muscle; lower limb MEP recordings revealed significant greater amplitude (mV) at 13, 14, and 15 wk (Figure S3D) and lower latency (ms) at 20 wk (Figure S3E) in the gastrocnemius muscle of hSOD1G93A + SynCav1 rats compared to hSOD1G93A + control (n = 8 rats/group; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, 2-way ANOVA, Bonferroni's multiple comparisons test). These data from hSOD1G93A rats treated with SynCav1 builds upon the findings already demonstrated in hSOD1G93A mice and provides further direct evidence of the therapeutic potential properties afforded by SynCav1 in a larger ALS animal model.
Discussion
The present study tested a neuroprotective, non-monogenic targeting gene delivery in the hSOD1G93A mouse model of ALS. These results show that subpial delivery of AAV9-SynCav1 delays disease onset and early disease progression, preserves ventral horn spinal motor neurons in a familial mouse model of ALS, independent of reducing the toxic monogenic protein engineered to cause the ALS-like neuropathology. Delivering genes that augment neuroprotective proteins may mitigate spinal motor neurodegeneration, prolong survival in individuals afflicted with ALS. The present study showed AAV9-SynCav1 subpial delivery significantly delays disease onset and extends survival in both male and female hSOD1G93A mice, results that are comparable to previous studies that used ASOs to target mutant monogenic hSOD1 in hSOD1G93A rodents [26]. Importantly, SynCav1 gene therapy proved efficacious in both sexes, independent of affecting expression of mutant hSOD1 protein. This latter finding suggests that SynCav1 possesses the therapeutic potential to combat other forms of FALS (e.g., SOD, FUS, C9orf72, TDP-43) [6-8, 38, 39] as well as SALS attributed to unknown etiology. Additional work is needed to test whether SynCav1 can work synergistically with other biologics or small molecules designed to knockdown known monogenic links to FALS.
A limitation of the current study is that the extended survival achieved by exogenous vector delivery is slightly lower (10% extended survival) than that observed when crossing SynCav1 transgenic mice with hSOD1G93A mice (19% extended survival) [25]. This discrepancy between the two studies may be attributed to the following reason: while SynCav1 TG mice overexpress neuronal Cav-1 throughout the whole CNS, the physical and anatomical restrictions of lumbar subpial AAV9-SynCav1 delivery in the small rodent limits broad CNS tissue AAV9 distribution and transduction and subsequent Cav-1 transgene expression. As shown in Figure 1 and Figure S1, Cav-1 transgene expression with SynCav1 predominantly distributed in the lumbar cord (~ 4-fold) and to a less degree in the thoracic cord (~ 2-fold), with no over-expression in the cervical cord after subpial delivery. Failure to target the cervical spinal cord [14], specifically the cervical plexus (C3-C5) which projects motor innervation necessary to regulate diaphragm muscle function, most likely restricted our ability to mitigate respiratory failure in these hSOD1G93A mice, the most common cause of death from ALS. However, even with this limitation of achieving cervical AAV9 spinal cord distribution/transduction and subsequent Cav-1 transgene expression, the therapeutic efficacy afforded by subpial SynCav1 gene delivery is comparable to approaches targeting the known monogenic cause of the mutant SOD1 neuropathology [40]. For example, a recent study by Powell et al. 2022 [18] delivered their AAV at wk 8 (similar time point in the current study) using CRISPR technology (AAV-RfxCas13d) to reduce mutant hSOD1 by 65% throughout the entire spinal cord (cervical to lumbar) of hSOD1G93A mice, which demonstrated a similar extension of survival. A combined therapy consisting of SynCav1 with approaches targeting knockdown of monogenic causes of ALS may work in concert to treat ALS more effectively.
On a cellular level, Cav-1 regulates cellular endocytosis and vesicular trafficking [41-43], subcellular localization of NTRs and other synaptic signaling components [11, 12, 29], preserves mitochondrial function in the brain of AD (PSAPP) mice [44] and mitochondrial morphology in the spinal cord of ALS (hSOD1G93A) mice [25]. It is well known that mutations in several genes linked to ALS (e.g., Tar DNA binding protein gene (TARDBP) which encodes for TDP-43 [6], fused in sarcoma (FUS) [39], and the C9 open-reading frame 72 genes (C9orf72) [7, 8]) impair axonal transport and alter mitochondrial function and dynamics (fission/fusion and trafficking) [9, 10, 45, 46]. Although not investigated in the present study, the neuronal protection and preservation afforded by SynCav1 may in part be through the maintenance/restoration of functional axonal transport of cellular cargoes such as NTR signaling endosomes [47, 48] and mitochondria [49]. Using novel in vitro model systems consisting of human motor neurons co-cultured with human myotubes in specialized microfluidic chambers [50-53] exposed to neurotoxic dipeptide repeats (DPRs) commonly found in FALS [8], will allow the ability to measure axonal transport of organelles and key cellular cargoes [9, 49, 54, 55] to better elucidate the molecular mechanisms underlying the neuroprotective properties of SynCav1.
Preservation of NMJ morphology is essential for preserving motor function and life quality. NMJs are unique synapses that facilitate communication between MNs and the effector muscles. Due to their long axons, MNs are extremely susceptible to retraction during pathological conditions such as ALS, thus resulting in disrupted communication between the MN and effector muscles. Initially, the MNs try to compensate for the retraction by inducing axonal sprouting and collateral innervation. However, with disease progression, this compensation fails and results in final MN death, a neuropathological event known as the “dying back” mechanism [56]. The subsequent NMJ disruption causes significant skeleton muscle atrophy, respiratory insufficiency, and death [57, 58]. The present study revealed that AAV9-SynCav1 preserved NMJ integrity and improved running wheel performance in both hSOD1G93A male and female mice. Could SynCav1 be applied at this MN 'compensatory stage' to prevent or overcome the “dying back” mechanism and maintain functional NMJ synapses? As noted above, the use of specialized in vitro models consisting of human motor neurons forming functional NMJs with human myotubes could optimize the temporal window at which to deliver SynCav1 in the presence of otherwise toxic dipeptide species to overcome this “dying back” cellular event. Another limitation to the current study is the lack of more sensitive neuromuscular measurements of functional NMJ physiology in the upper or lower limbs (e.g., MEPs, grip strength) performed specifically in the hSOD1G93A mouse ALS model. However, using the larger hSOD1G93A rat ALS model, subpial cervical spinal cord SynCav1 delivery resulted in significantly better forelimb grip strength and MEPs (greater amplitude and decreased latency) in both upper limb brachial and lower limb gastrocnemius muscles, further evidence of SynCav1's efficacy using two different ALS model species.
The current study delivered SynCav1 at week 8 (pre-symptomatic stage) to the lumbar cord which exhibited similar folds increase as observed in the healthy wild type mice. However, vector dosing needed to achieve efficacious transgene expression levels may depend upon disease stages [59]. If vector is delivered at a later stage, Cav-1 expression in the lumbar cord neuron may fail to achieve the same level of fold increase since the neurodegenerative spinal cord may not hold the capacity to express an exogenous transgene to the same levels as healthy spinal cord, an important pre-clinical finding to consider when formulating human equivalent dosing to achieve clinical efficacy [60-63]. Interestingly, we measured adverse effects from the subpial surgery on BW at the late stage of disease compared to the naïve hSODG93A mice, with no effect on disease onset or survival. A possible explanation is that the surgery may have exacerbated back muscle weakness, which in turn could have affected the ability to retrieve food located on top of the cage for late stage hSOD1G93A mice. This inability to retrieve food would result in a more rapid BW decline compared to naïve hSOD1G93A mice. The fact that we did not measure any difference in survival, MN morphology or MN count between hSOD1G93 control and hSOD1G93 naïve mice, indicate that the adverse effect from subpial surgery on BW is not through affecting MN survival directly. Nevertheless, since the lumbar subpial surgery involves an invasive laminectomy, which is difficult to perform successfully on older, severely symptomatic hSOD1G93A mice. Exploration of alternative routes of SynCav1 administration (i.e., intrathecal and intracerebroventricular) are worthy of investigation.
In summary, the present study demonstrates that subpial SynCav1 gene delivery to the lumbar spinal cord of hSOD1G93A mice prolongs survival and maintains body weight in both males and females, preserves alpha motor neurons in the ventral horn with a direct correlation to soma size and Cav-1 expression, and preserves NMJs in the lower limbs, all of which occurred independent of affecting expression of mutant hSOD1. These data suggest that SynCav1 might serve as a novel gene therapy for neurodegenerative conditions in ALS and other forms of CNS disease of unknown etiology. Further studies are needed to determine the effect of SynCav1 on disease progression when delivered at later stages of the disease.
Abbreviations
ALS: Amyotrophic lateral sclerosis; AAV9: adeno-associated virus serotype 9; Btx-α: bungarotoxin-α; ChAT: choline acetyltransferase; FALS: familial ALS; GS: grip strength; SALS: sporadic ALS; SOD: super oxide dismutase; SynCav1: synapsin-promoted caveolin-1 genetic construct; MEPs: motor evoked potentials; MLRs: membrane/lipid rafts; MN: motor neuron; NMJ: neuromuscular junction.
Supplementary Material
Supplementary figures.
Author contributions
S.W. guided the overall study design, writing the manuscript, IB and IF microscopy; T.I. performed subpial gene delivery, behavioral tests and IF microscopy; P.S. and D.W. assisted with animal breeding and behavior tests; A.S. performed all rat subpial vector delivery and motor tests; X.L, T.D., and W.L assisted with data analysis; J.A.B. and E.K. assisted with editing of the manuscript; A.M. generated AAV vectors (UCSD Viral Vector Core); D.M.R., H.H.P. and P.M.P. assisted with manuscript editing; T.T and M.M. provided subpial surgical guidance; B.P.H. guided the overall study design, scientific concept, edited the manuscript, and funded the project (VA SPiRE award 5I21RX003324).
Ethics approval
All animal protocols were approved by the Veterans Administration San Diego Healthcare System Institutional Animal Care and Use Committee (#A20-030).
Competing Interests
D.M.R., H.H.P., P.M.P. and B.P.H. hold equity and are non-paid consultants with Eikonoklastes Therapeutics LLC.
References
1. O'Connor DM, Boulis NM. Gene therapy for neurodegenerative diseases. Trends Mol Med. 2015;21:504-12
2. Brown RH, Al-Chalabi A. Amyotrophic Lateral Sclerosis. N Engl J Med. 2017;377:162-72
3. Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386:1672-82
4. Hardiman O, van den Berg LH. Edaravone: a new treatment for ALS on the horizon? Lancet Neurol. 2017;16:490-1
5. Ajroud-Driss S, Siddique T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim Biophys Acta. 2015;1852:679-84
6. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130-3
7. Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V. et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23:3579-95
8. Haeusler AR, Donnelly CJ, Rothstein JD. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci. 2016;17:383-95
9. Guo W, Stoklund Dittlau K, Van Den Bosch L. Axonal transport defects and neurodegeneration: Molecular mechanisms and therapeutic implications. Semin Cell Dev Biol. 2020;99:133-50
10. Wood A, Gurfinkel Y, Polain N, Lamont W, Lyn Rea S. Molecular Mechanisms Underlying TDP-43 Pathology in Cellular and Animal Models of ALS and FTLD. Int J Mol Sci. 2021;22(9):4705
11. Egawa J, Zemljic-Harpf AE, Mandyam CD, Niesman IR, Lysenko LV, Kleschevnikov AM. et al. Neuron-targeted caveolin-1 promotes ultrastructural and functional hippocampal synaptic plasticity. Cerebral Cortex. 2018;28(9):1-12
12. Wang S, Leem JS, Podvin S, Hook V, Kleschevnikov N, Savchenko P. et al. Synapsin-caveolin-1 gene therapy preserves neuronal and synaptic morphology and prevents neurodegeneration in a mouse model of AD. Molecular Therapy - Methods and Clinical Development. 2021;21:434-450
13. Cooper-Knock J, Zhang S, Kenna KP, Moll T, Franklin JP, Allen S. et al. Rare variant burden analysis within enhancers identifies CAV1 as an ALS risk gene. Cell Rep. 2021;34:108730
14. Bravo-Hernandez M, Tadokoro T, Navarro MR, Platoshyn O, Kobayashi Y, Marsala S. et al. Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS. Nat Med. 2020;26:118-30
15. Stoica L, Todeasa SH, Cabrera GT, Salameh JS, ElMallah MK, Mueller C. et al. Adeno-associated virus-delivered artificial microRNA extends survival and delays paralysis in an amyotrophic lateral sclerosis mouse model. Ann Neurol. 2016;79:687-700
16. Foust KD, Salazar DL, Likhite S, Ferraiuolo L, Ditsworth D, Ilieva H. et al. Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol Ther. 2013;21:2148-59
17. Biferi MG, Cohen-Tannoudji M, Cappelletto A, Giroux B, Roda M, Astord S. et al. A New AAV10-U7-Mediated Gene Therapy Prolongs Survival and Restores Function in an ALS Mouse Model. Mol Ther. 2017;25:2038-52
18. Powell JE, Lim CKW, Krishnan R, McCallister TX, Saporito-Magrina C, Zeballos MA. et al. Targeted gene silencing in the nervous system with CRISPR-Cas13. Sci Adv. 2022;8:eabk2485
19. Mueller C, Berry JD, McKenna-Yasek DM, Gernoux G, Owegi MA, Pothier LM. et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N Engl J Med. 2020;383:151-8
20. Rochat C, Bernard-Marissal N, Kallstig E, Pradervand S, Perrin FE, Aebischer P. et al. Astrocyte-targeting RNA interference against mutated superoxide dismutase 1 induces motoneuron plasticity and protects fast-fatigable motor units in a mouse model of amyotrophic lateral sclerosis. Glia. 2022;70:842-57
21. Gaj T, Ojala DS, Ekman FK, Byrne LC, Limsirichai P, Schaffer DV. In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci Adv. 2017;3:eaar3952
22. Duan W, Guo M, Yi L, Liu Y, Li Z, Ma Y. et al. The deletion of mutant SOD1 via CRISPR/Cas9/sgRNA prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Ther. 2020;27:157-69
23. Iannitti T, Scarrott JM, Likhite S, Coldicott IRP, Lewis KE, Heath PR. et al. Translating SOD1 Gene Silencing toward the Clinic: A Highly Efficacious, Off-Target-free, and Biomarker-Supported Strategy for fALS. Mol Ther Nucleic Acids. 2018;12:75-88
24. Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B. et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A. 2002;99:1604-9
25. Sawada A, Wang S, Jian M, Leem J, Wackerbarth J, Egawa J. et al. Neuron-targeted caveolin-1 improves neuromuscular function and extends survival in SOD1(G93A) mice. FASEB J. 2019;33:7545-54
26. Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, Hung G. et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116:2290-6
27. Lim CKW, Gapinske M, Brooks AK, Woods WS, Powell JE, Zeballos CM. et al. Treatment of a Mouse Model of ALS by In vivo Base Editing. Mol Ther. 2020;28:1177-89
28. Pratt SJP, Valencia AP, Le GK, Shah SB, Lovering RM. Pre- and postsynaptic changes in the neuromuscular junction in dystrophic mice. Front Physiol. 2015;6:252
29. Mandyam CD, Schilling JM, Cui W, Egawa J, Niesman IR, Kellerhals SE. et al. Neuron-Targeted Caveolin-1 Improves Molecular Signaling, Plasticity, and Behavior Dependent on the Hippocampus in Adult and Aged Mice. Biol Psychiatry. 2017;81:101-10
30. Barneoud P, Lolivier J, Sanger DJ, Scatton B, Moser P. Quantitative motor assessment in FALS mice: a longitudinal study. Neuroreport. 1997;8:2861-5
31. Vinsant S, Mansfield C, Jimenez-Moreno R, Del Gaizo Moore V, Yoshikawa M, Hampton TG. et al. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part II, results and discussion. Brain Behav. 2013;3:431-57
32. Ligon LA, LaMonte BH, Wallace KE, Weber N, Kalb RG, Holzbaur EL. Mutant superoxide dismutase disrupts cytoplasmic dynein in motor neurons. Neuroreport. 2005;16:533-6
33. Wooley CM, Sher RB, Kale A, Frankel WN, Cox GA, Seburn KL. Gait analysis detects early changes in transgenic SOD1(G93A) mice. Muscle Nerve. 2005;32:43-50
34. Hayworth CR, Gonzalez-Lima F. Pre-symptomatic detection of chronic motor deficits and genotype prediction in congenic B6.SOD1(G93A) ALS mouse model. Neuroscience. 2009;164:975-85
35. Hegedus J, Putman CT, Gordon T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2007;28:154-64
36. Nijssen J, Comley LH, Hedlund E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017;133:863-85
37. Pratt SJP, Iyer SR, Shah SB, Lovering RM. Imaging Analysis of the Neuromuscular Junction in Dystrophic Muscle. Methods Mol Biol. 2018;1687:57-72
38. Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806-19
39. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C. et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205-8
40. Leyton-Jaimes MF, Kahn J, Israelson A. AAV2/9-mediated overexpression of MIF inhibits SOD1 misfolding, delays disease onset, and extends survival in mouse models of ALS. Proc Natl Acad Sci U S A. 2019;116:14755-60
41. Liu P, Rudick M, Anderson RG. Multiple functions of caveolin-1. J Biol Chem. 2002;277:41295-8
42. del Pozo MA, Balasubramanian N, Alderson NB, Kiosses WB, Grande-Garcia A, Anderson RG. et al. Phospho-caveolin-1 mediates integrin-regulated membrane domain internalization. Nat Cell Biol. 2005;7:901-8
43. Head BP, Patel HH, Tsutsumi YM, Hu Y, Mejia T, Mora RC. et al. Caveolin-1 expression is essential for N-methyl-D-aspartate receptor-mediated Src and extracellular signal-regulated kinase 1/2 activation and protection of primary neurons from ischemic cell death. FASEB J. 2008;22:828-40
44. Wang S, Ichinomiya T, Terada Y, Wang D, Patel HH, Head BP. Synapsin-Promoted Caveolin-1 Overexpression Maintains Mitochondrial Morphology and Function in PSAPP Alzheimer's Disease Mice. Cells. 2021;10:2487
45. Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995-1007
46. Dafinca R, Barbagallo P, Talbot K. The Role of Mitochondrial Dysfunction and ER Stress in TDP-43 and C9ORF72 ALS. Front Cell Neurosci. 2021;15:653688
47. Chen XQ, Sawa M, Mobley WC. Dysregulation of neurotrophin signaling in the pathogenesis of Alzheimer disease and of Alzheimer disease in Down syndrome. Free Radic Biol Med. 2018;114:52-61
48. Pearn ML, Schilling JM, Jian M, Egawa J, Wu C, Mandyam CD. et al. Inhibition of RhoA reduces propofol-mediated growth cone collapse, axonal transport impairment, loss of synaptic connectivity, and behavioural deficits. Br J Anaesth. 2018;120:745-60
49. Fumagalli L, Young FL, Boeynaems S, De Decker M, Mehta AR, Swijsen A. et al. C9orf72-derived arginine-containing dipeptide repeats associate with axonal transport machinery and impede microtubule-based motility. Sci Adv. 2021;7:3013
50. Campenot RB. Local control of neurite development by nerve growth factor. Proc Natl Acad Sci U S A. 1977;74:4516-9
51. Bellmann J, Goswami RY, Girardo S, Rein N, Hosseinzadeh Z, Hicks MR. et al. A customizable microfluidic platform for medium-throughput modeling of neuromuscular circuits. Biomaterials. 2019;225:119537
52. Santhanam N, Kumanchik L, Guo X, Sommerhage F, Cai Y, Jackson M. et al. Stem cell derived phenotypic human neuromuscular junction model for dose response evaluation of therapeutics. Biomaterials. 2018;166:64-78
53. Stoklund Dittlau K, Krasnow EN, Fumagalli L, Vandoorne T, Baatsen P, Kerstens A. et al. Human motor units in microfluidic devices are impaired by FUS mutations and improved by HDAC6 inhibition. Stem Cell Reports. 2021;16:2213-2227
54. Guo W, Naujock M, Fumagalli L, Vandoorne T, Baatsen P, Boon R. et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat Commun. 2017;8:861
55. Fazal R, Boeynaems S, Swijsen A, De Decker M, Fumagalli L, Moisse M. et al. HDAC6 inhibition restores TDP-43 pathology and axonal transport defects in human motor neurons with TARDBP mutations. EMBO J. 2021;40:e106177
56. Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248-64
57. Dadon-Nachum M, Melamed E, Offen D. The "dying-back" phenomenon of motor neurons in ALS. J Mol Neurosci. 2011;43:470-7
58. Murray LM, Talbot K, Gillingwater TH. Review: neuromuscular synaptic vulnerability in motor neurone disease: amyotrophic lateral sclerosis and spinal muscular atrophy. Neuropathol Appl Neurobiol. 2010;36:133-56
59. Tang F, Wong H, Ng CM. Rational Clinical Dose Selection of Adeno-Associated Virus-Mediated Gene Therapy Based on Allometric Principles. Clin Pharmacol Ther. 2021;110:803-7
60. Federici T, Taub JS, Baum GR, Gray SJ, Grieger JC, Matthews KA. et al. Robust spinal motor neuron transduction following intrathecal delivery of AAV9 in pigs. Gene Ther. 2012;19:852-9
61. Rosenberg JB, Kaplitt MG, De BP, Chen A, Flagiello T, Salami C. et al. AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer's Disease. Hum Gene Ther Clin Dev. 2018;29:24-47
62. Bey K, Deniaud J, Dubreil L, Joussemet B, Cristini J, Ciron C. et al. Intra-CSF AAV9 and AAVrh10 Administration in Nonhuman Primates: Promising Routes and Vectors for Which Neurological Diseases? Mol Ther Methods Clin Dev. 2020;17:771-84
63. Hinderer C, Katz N, Dyer C, Goode T, Johansson J, Bell P. et al. Translational Feasibility of Lumbar Puncture for Intrathecal AAV Administration. Mol Ther Methods Clin Dev. 2020;17:969-74
Author contact
![]() Corresponding author: shw049edu and bheaducsd.edu
Corresponding author: shw049edu and bheaducsd.edu