Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Imaging modalities for...
3. Therapeutic modalities for...
4. Some critical notes and...
Conclusions
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2024; 14(2):571-591. doi:10.7150/thno.90187 This issue Cite
Review
Tumor-tropic Trojan horses: Using mesenchymal stem cells as cellular nanotheranostics
Ana Rosu, Behnaz Ghaemi, Jeff W.M. Bulte, Ali Shakeri-Zadeh ![]()
The Russell H. Morgan Department of Radiology and Radiological Science, Division of MR Research and Cellular Imaging Section and Vascular Biology Program, Institute for Cell Engineering, The Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Received 2023-9-14; Accepted 2023-11-21; Published 2024-1-1
Abstract

Various classes of nanotheranostics have been developed for enhanced tumor imaging and therapy. However, key limitations for a successful use of nanotheranostics include their targeting specificity with limited off-site tissue accumulation as well as their distribution and prolonged retention throughout the entire tumor. Due to their inherent tumor-tropic properties, the use of mesenchymal stem cells (MSCs) as a “Trojan horse” has recently been proposed to deliver nanotheranostics more effectively. This review discusses the current status of “cellular nanotheranostics” for combined (multimodal) imaging and therapy in preclinical cancer models. Emphasis is placed on the limited knowledge of the signaling pathways and molecular mechanisms of MSC tumor-tropism, and how such information may be exploited to engineer MSCs in order to further improve tumor homing and nanotheranostic delivery using image-guided procedures.
Keywords: Cancer, Nanoparticles, Mesenchymal stem cells, Theranostics, Image-guided therapy
1. Introduction
Cancer theranostics can be defined as “see what you treat, and treat what you see” [1]. To enable personalized cancer treatment and ease patient care, it is imperative that cancer is first properly imaged and diagnosed before being precisely treated. Combining therapeutic and diagnostic properties within a single theranostic agent is a promising approach: once the theranostic agent is properly localized to allow cancer visualization, therapy can be subsequently performed with minimal side effects.
Theranostic agents can be based on small molecules [2] or nanoparticles (NPs) [3]. NP-based theranostics or nanotheranostics are particularly attractive due to the diverse material compositions and physical structures that are available, which allow their use as agents for most cancer imaging or therapeutic modalities [4, 5]. In cases where altering the NP composition or structure is not sufficient for imaging or therapy, the necessary diagnostic or therapeutic molecule can be easily adsorbed on, covalently attached to, or carried as cargo within the NPs.
A current key limitation of nanotheranostics is that it is difficult to target, distribute, and retain NPs within the tumor. When administered intravenously (i.v.), NPs marginally accumulate within tumors in a passive fashion due to the enhanced permeability and retention (EPR) effect, with localization limited to vascularized areas [6-8]. The mechanism of the EPR effect remains under active debate. While it was initially proposed that inter-endothelial gaps in tumor vasculature were responsible for the transport of NPs into solid tumors, recent studies suggest that up to 97% of NPs enter tumors using active processes through endothelial cells [9]. Regardless, targeting NPs through the EPR effect is highly dependent on a multitude of factors, including the type and location of tumors, the degree of tumor vascularization, and the structure of the NPs themselves [10, 11]. Even if NPs reach the tumor vasculature, the tumor structure, composed of surrounding blood vessels and a necrotic core, makes it difficult for NPs to penetrate to the tumor center [12]. The theranostic use of NPs thus becomes difficult, as visualization and ablation can occur only at the tumor periphery. Further, NP localization within the tumor vasculature is temporary. Due to the high intratumoral (i.t.) pressure, NPs are easily pushed back from the tumor into the circulating blood, clearing out from the tumor within 24 to 72 hours [13]. This rapid clearance is an impediment to long-term imaging and therapy of tumors, requiring repeated administration of nanotheranostic agents.
Potential methods for improving nanotheranostic targeting to tumors include conjugation of targeting moieties to NPs, also referred to as active targeting [14], and cell-mediated delivery of NPs [15, 16]. Active targeting generally improves NP localization within tumors, as targeting moieties are designed to bind to receptors overexpressed by cancer cells and can be internalized by appropriate surface receptors. However, tumors are highly heterogeneous in receptor overexpression, making it difficult for NPs with targeting moieties to bind to all cancer cells and/or penetrate to the tumor core [17]. Cell-mediated delivery of NPs, using red blood cells, leukocytes, or stem cells, has been explored as an alternative method [15, 16]. While red blood cells and leukocytes require surface or genetic modification to target tumors, stem cells are intrinsically tumor-tropic, making them ideal delivery vehicles [18]. This review provides an overview of mesenchymal stem cell (MSC)-mediated delivery of nanotheranostics (Figure 1), also referred to as cellular nanotheranostics, with the aim of summarizing current progress and defining future directions within the field. Imaging and therapeutic modalities that have been successfully used with cellular nanotheranostics are discussed, with a focus on the potential of MSC-mediated delivery to improve the targeting, distribution, and retention of nanotheranostics within tumors.
Concept of cellular nanotheranostics following either systemic or intratumoral injection. 1) MSCs are first pre-labeled in vitro with NPs. 2) Labeled MSCs are then injected either systematically or intratumorally. 3) The tumor is imaged using a modality that can detect labeled MSCs in the tumor and off-target sites for making go or no-go decisions on initiating the treatment procedure. 4) When a successful distribution of labeled MSCs is achieved throughout the entire tumor, treatment can be more effective than using “naked” NPs without MSCs.
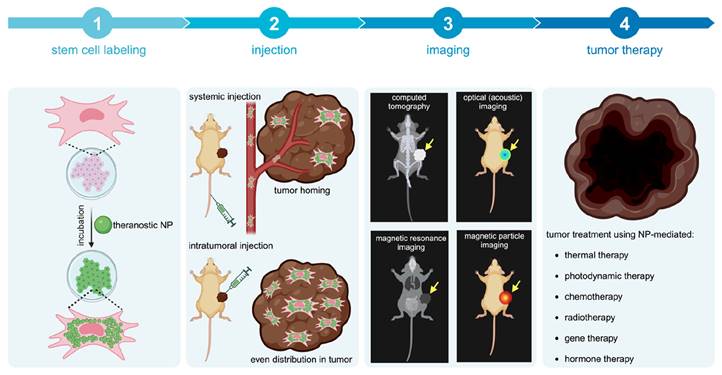
1.1. Theranostics: An emerging paradigm for imaging and treatment of cancer
Combined cancer diagnosis and therapy, as currently practiced in clinic, has several limitations. Standard imaging modalities such as magnetic resonance imaging (MRI) [3], (single photon emission computed tomography (SPECT), and positron emission tomography (PET) [19] can identify the location and some of the biological characteristics of tumors but remain poorly integrated with the administration of cancer therapies. Chemotherapy and radiotherapy still have limited specificity towards tumors, causing severe toxic side effects and a risk of cancer recurrence [4]. Theranostics offers an alternative form of cancer diagnosis and therapy. Imaging can confirm that theranostic agents have localized within tumors prior to initiating therapy, allowing for precise and personalized treatment regimens.
Most advances in theranostics have been in the field of nuclear medicine. However, theranostic radioactive molecules may have toxic side effects due to off-target localization [20] and may fail due to tumor radioresistance [4]. Thus, novel precise imaging and therapeutic modalities have been explored for theranostic applications, including fluorescence [21] and photoacoustic imaging (PAI), as well as photothermal therapy (PTT) [22] and photodynamic therapy (PDT) [23]. Together with MRI [24], gene therapy [25], and chemotherapy [26], these modalities are discussed more in detail since they can use NPs as theranostic or carrier agents.
1.2. Theranostic NPs used with MSC-mediated delivery
The composition of cancer nanotheranostics is either an NP containing a single material that has both imaging and therapeutic properties, or NPs composed of an imaging agent and a therapeutic agent [4]. For the former category, gold and magnetic metal oxide NPs have been widely used for imaging and therapy. Gold NPs (AuNPs), convert absorbed light to ultrasound waves for PAI and to heat energy for PTT [27-29]. Magnetic metal oxide NPs can serve as negative contrast agents for T2-weighted MRI [30] and magnetic particle imaging (MPI) [31] or, when exposed to intratumoral oxidation and an acidic environment, can release metal ions that serve as T1-weighted contrast agents [32]. If exposed to alternating magnetic fields, magnetic metal oxide NPs can also be used for hyperthermia, where they heat up and ablate surrounding cancer tissue [33, 34]. One particularly common species of theranostic NPs are quantum dots (QDs), used for PAI or fluorescence imaging, coated with photosensitizing molecules, which can be employed for PTT or PDT [35]. Alternatively, a theranostic NP can be enhanced by complexation with genes or chemotherapeutic agents [24].
1.3. Signaling pathways and molecular mechanisms of MSC tumor-tropism
MSCs are multipotent stem cells that can differentiate into osteocytes, chondrocytes, adipocytes, and other lineages [36]. The primary sources of MSCs for clinical applications are bone marrow, umbilical cord blood, and adipose tissue. Generally, MSCs express the markers CD90, CD73, and CD105, but the surface marker profiles of MSCs derived from different sources tend to vary [37]. Depending on their source, MSCs may have different proliferation and downstream cell differentiation rates.
MSCs are recognized for their immunomodulatory properties, making them attractive therapeutic candidates for various clinical applications. They offer several benefits over other stem cell types. Compared to induced pluripotent stem cells (iPSCs), MSCs are more cost-effective and readily available as an off-the-shelf product [38]. Furthermore, MSCs have demonstrated tumor-tropism towards a diverse range of cancer types, unlike neural stem cells (NSCs) that have been primarily used for glioma treatment [39]. Tumor-tropism, a process by which MSCs selectively migrate towards tumor sites, involves a complex and not fully understood interplay of signaling pathways mediated by various cytokines, chemokines, and growth factors (Table 1). Different cancer types express unique cytokine profiles, potentially rendering one cytokine more influential for MSC tumor-tropism in a specific cancer type than in another. Additionally, MSCs derived from varying sources may exhibit different responses to certain cytokines, a factor that has not been fully considered in previous studies.
Postulated signaling pathways and molecular mechanisms of MSC tumor-tropism.
| Signaling molecule | Signaling pathways in MSCs | Mechanism for MSC tumor-tropism |
|---|---|---|
| TNF-α | TNF-α induces VCAM-1 and VLA-1 expression on MSCs through the PI3K- and NF-kB signaling pathways. MSC VCAM-1 expression is much higher than VLA-1 [40]. VCAM-1 and VLA-1 expressing MSCs adhere to endothelial cells, although the exact biological mechanism of this remains unknown [41]. TNF-α also causes MSCs to secrete MMP-1, which cleaves extracellular IGF-2/IFGBP-2 complexes [42]. Free IGF-2 is released from the complex and binds to the MSC receptors IGF-1R or IR-A, inducing activation of PI3K-Akt/PKB and MAPK. It is unclear how these pathways interact to promote MSC migration [43]. | Enables adhesion to tumor vessel endothelial cells and cleavage of extracellular IGF-2/IGFBP-2 complexes, activating IGF-2 signaling pathways that promote MSC migration. |
| IL-6 | IL-6 binds to IL-6 receptors and GP130 on MSCs, inducing CXCL7 expression and STAT3 activation [44]. MSC-derived CXCL7 interacts with cancer cells through the CXCR2 receptor, inducing tumors to further synthesize IL-6 and IL-8. STAT3 activation, resulting from tumor-secreted IL-6 that binds to IL-6R on MSCs, induces the NF-kB-IL-6-STAT3 cascade, which leads to modest activation of ERK and cytoskeletal longitudinal organization of MSCs, enhancing the cell migratory phenotype [45]. | Induces an inflammatory cytokine feedback loop and MSC migration phenotype (ERK activation and cytoskeletal organization). |
| IL-8 | IL-8 binds to CXCR1 [46] and CXCR2 [47] on MSCs. The intracellular signaling pathway induced by CXCR1 binding is unknown, although it has been found to play a significant role in MSC tumor-tropism. CXCR2 binding leads to Akt and ERK phosphorylation, potentially activating the respective intracellular pathways and subsequent MSC migration [48]. | Activates intracellular pathways (Akt and ERK) associated with MSC migration. |
| SDF-1 | SDF-1 binds to CXCR4 on MSCs [49]. In response, MSCs secrete more SDF-1, which acts in an autocrine manner. SDF-1 binding to CXCR4 activates downstream JAK2/STAT3 [50] and ERK/MAPK [51] signaling in MSCs. FAK and paxillin signaling is then activated, which induces cytoskeletal reorganization. | Induces SDF-1 production and MSC migration phenotype (JAK2/STAT3 and ERK/MAPK pathways activated, cytoskeletal organization). |
| CXCL1 | The CXCL1 ligand binds to CXCR2 receptors on MSCs [52, 53]. The exact signaling pathways induced in tumor-tropic MSCs after CXCR2 binding are unknown, but previous studies have indicated that CXCR2 binding can activate the PI3K-Akt/PKB, ERK, MAPK, JAK2, and STAT3 pathways in other cells [54]. | Unknown role for inducing MSC migration. |
| MCP-1 | MCP-1 binds to the CCR2 receptor on MSCs, inducing unknown intracellular signaling pathways to promote migration [55]. The stromal population of cells adjacent to tumors, consisting of fibroblasts, are the main source of MCP-1 secretion. | Unknown role for inducing MSC migration. |
| TGF-β | TGF-β binds to TGF-β receptors on MSCs, upregulating CXCR4 and inducing lamellipodia protrusions [56]. Other studies indicate that SDF-1 binds to CXCR4, so it is plausible that through CXCR4 upregulation, TGF-β indirectly activates JAK2/STAT3 and ERK/MAPK signaling in MSCs, which then induces cytoskeletal reorganization [57]. | Induces CXCR4 upregulation and MSC migration phenotype (lamellipodia protrusions). |
| PDGF | PDGF binds to PDGF receptors on MSCs [58], activating the PI3K pathway and subsequent MSC migration [59]. | Activates an intracellular pathway (PI3-kinase) associated with MSC migration. |
| HIF-1α PGF CXCL16 CSF-1 | Cancer cells expressing HIF-1α secrete PGF, CXCL16 [60], and CSF-1 [61], as well as the receptors CXCR3 and CCR5. PGF and CXCL16 bind to respective VEGFR1 and CXCR6 receptors on MSCs, which cause MSCs to secrete CXCL10. CSF-1 binds to the CSF-1 receptor on MSCs, inducing CCL5 secretion. CXCL10 and CCL5 bind to CXCR3 and CCR5 receptors on cancer cells, respectively. MSCs stimulate further HIF-1α expression in cancer cells through a CXCR3-independent mechanism [62]. The intracellular signaling pathways induced by CXCR3, CXCR6, VEGFR1, and CSF-1R binding of MSCs, and how they promote tumor-tropism, remain unclear. | Induces chemokine and cytokine interactions between cancer cells and MSCs, as well as increases HIF-1α production in cancer cells. Unknown role for inducing MSC migration. |
Abbreviations: Akt=Ak strain transforming; CCR=CC motif chemokine receptor; CSF=colony stimulating factor; CXCL=chemokine (C-X-C motif) ligand; CXCR= chemokine (C-X-C motif) receptor; FAK=focal adhesion kinase; ERK=extracellular signal-regulated kinase; HIF=hypoxia inducible factor; ICAM=intercellular adhesion molecule; IGF=insulin-like growth factor; IFGBP=insulin-like growth factor binding protein; IL=interleukin; JAK=janus kinase; MAPK=mitogen-activated protein kinase; MCP=monocyte chemoattractant protein; MMP=matrix metallopeptidase; NF=nuclear factor; PDGF=platelet-derived growth factor; PGF=placenta growth factor; PI=phosphoinositide 3-kinase; PKB SDF=stromal cell-derived factor; STAT=signal transducer and transcription activator; TGF=transforming growth factor; TNF=tumor necrosis factor; VCAM=vascular cell adhesion molecule; VEGF=vascular endothelial growth factor; VLA=very late antigen.
The most studied signaling molecules involved in MSC tumor-tropism are tumour necrosis factor-α (TNF-α), interleukin (IL)-6, and stromal cell-derived factor-1 (SDF-1). After systematic administration, MSCs have been found to home towards tumors via TNF-α signaling [40]. TNF-α induces vascular cell adhesion molecule-1 (VCAM-1) and the α1β1 integrin or very late antigen-1 (VLA-1) expression on MSCs through the phosphoinositide 3-kinases (PI3Ks)- and nuclear factor kappa B (NF-κB) pathways, which then enables MSCs to adhere to endothelial cells in the tumor vasculature [41]. IL-6 secreted by tumors has been found to bind to IL-6 receptors and glycoprotein 130 (GP130) on MSCs, inducing chemokine (C-X-C motif) ligand 7 (CXCL7) expression and the signal transducer and activator of transcription 3 (STAT3) pathway activation which have respective roles in cytokine feedback loops and MSC migration [44].
CXCL7 from MSCs interacts with cancer cells via the IL-8 receptor, also known as C-X-C chemokine receptor type 2 (CXCR2), causing tumors to further synthesize IL-6 and IL-8 (another cytokine involved in tumor-tropism). Activation of the STAT3 pathway induces the NF-κB-IL-6-STAT3 cascade, leading to modest extracellular signal-regulated kinase (ERK) activation and longitudinal cytoskeletal organization in MSCs, controlling movement. Tumor-secreted SDF-1 also induces cytokine positive feedback loops, binding to C-X-C chemokine receptor type 4 (CXCR4) on MSCs and causing MSCs to secrete SDF-1 which acts in an autocrine manner [50]. SDF-1 binding to CXCR4 on MSCs activates Janus kinase 2 (JAK2)/STAT3 and ERK/ mitogen-activated protein kinase (MAPK) signaling, which further induces focal adhesion kinase (FAK) and paxillin signaling that causes cytoskeletal reorganization characteristic of migratory phenotypes [51]. Several other cytokines, chemokines, and growth factors have been identified to play roles in MSC tumor-tropism, including IL-8, CXCL1, monocyte chemoattractant protein-1 (MCP-1), transforming growth factor beta (TGF-β), platelet-derived growth factor (PDGF), hypoxia-inducible factor (HIF)-1α, placental growth factor (PGF), CXCL16, and colony stimulating factor (CSF)-1. However, more studies are needed to reveal the exact signaling pathways that promote cell migration, and, perhaps more importantly, how they interact with TNF-α, IL-6, and SDF-1. It is interesting to note that IL-6 and HIF-1, both associated with tumor hypoxia, have been shown to enhance MSC tumor-tropism [45]. Traditionally, highly hypoxic tumors are resistant to immunotherapy [63], chemotherapy, and radiotherapy [64]. The capacity of MSCs to migrate towards hypoxic tumors suggests that MSC-mediated delivery of therapeutics could address this gap in current therapies, treating tumors that would otherwise be unresponsive.
1.4. Significance of MSC-mediated delivery of nanotheranostics
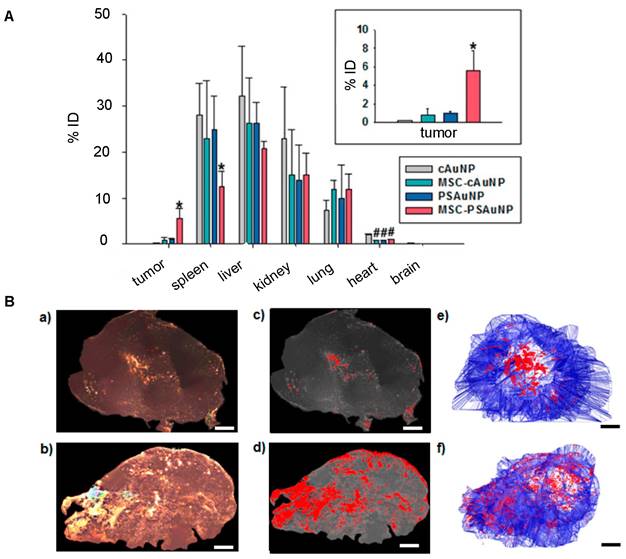
For nanotheranostics to reach full potential, it is essential that theranostic NPs accumulate, distribute, and remain within tumors to allow comprehensive imaging and precise therapy. The inherent tumor-tropism of stem cells has enabled improved NP delivery. For accumulation, there is extensive evidence from MRI cell tracking that iron and gadolinium-based NP-labeled MSCs ultimately home towards tumors [65]. Further, MSCs loaded with therapeutic NPs composed of paclitaxel (PTX) and poly (lactic-co-glycolic acid) (PLGA) have been found to localize in prostate, lung, and glioma tumors post-i.v. injection and increase survival rates [66]. The success of MSC-mediated NP delivery extends to theranostic NPs. As an example, Kang et al. achieved a 5.7-fold higher AuNP delivery to tumors for i.v. injected AuNP-labeled MSCs [67] compared to “naked” AuNPs (Figure 2A).
(A) Biodistribution of i.v. injected “naked” AuNPs and AuNP-labeled MSCs, in which AuNP-labeled MSCs achieved 5.7-fold higher AuNP delivery in the tumor compared to naked AuNPs. cAuNP and PSAuNP stand for control AuNPs and pH-sensitive AuNPs, respectively. Adapted with permission from [67], copyright 2015 ACS Publications. (B) Distribution of AuNPs post-i.t. injection, where AuNP-labeled NSCs (b, d, f) homogenously delivered AuNPs (dense bright signals) throughout the tumor, in contrast to naked AuNPs that are mostly cleared with some remaining in the center of injection (a, c, e). Adapted from [68], courtesy of ACS Publications.
Mooney et al. were able to homogeneously distribute AuNPs throughout a glioblastoma after i.t. injection of AuNP-labeled NSCs (Figure 2B) [68]. With PAI, Xu et al. [69] and Huang et al. [70] were also able to demonstrate tumor areas with increased signal after MSC-mediated NP delivery. Figure 3 illustrates the expected distribution profile of theranostic NPs within the tumor following i.t. or systemic delivery for naked NPs or NPs loaded in MSCs.
Intratumoral distribution profile of theranostic NPs using different delivery routes.
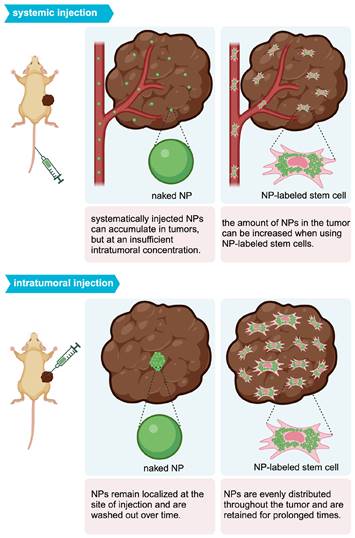
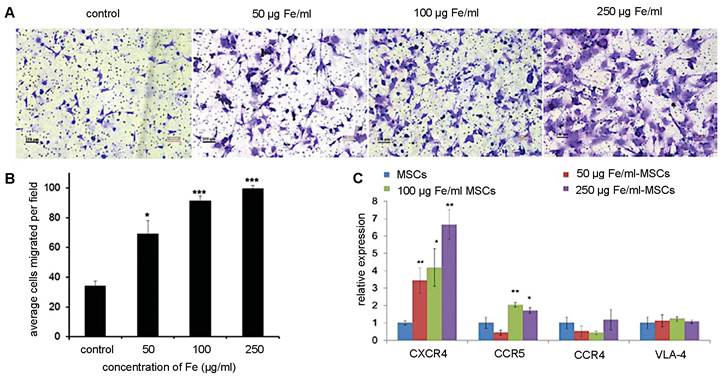
Although retention time varies with tumor type, MSC-mediated NP delivery in general prolongs NP retention compared to naked NPs. Through the EPR effect, naked NPs accumulate within and clear from tumors between 24 and 72 hours post-administration [13]. On the other hand, Layek et al. found that Cy5.5-labeled MSCs injected i.v. remained within ovarian and lung tumors for 28 and 10 days, respectively [71]. Kim et al. indicated that MSCs labeled with gadolinium-chelate NPs can be detected in vivo for at least 21 days, using a colorectal cancer model [72]. In both these studies, MSCs homed towards tumors within 24 hours of administration. As for SPIO labeling, multiple studies have demonstrated that iron uptake upregulates CXCR4 expression on the surface of hMSCs (Figure 4), providing a potential explanation for the prolonged retention of (SPIO-based) NP-labeled hMSCs within tumor sites [69, 73, 74].
(A) Trans-well pictures and (B) quantitative analysis of migrating MSCs labeled with various concentrations of magnetic nanoparticles (MNPs). MSCs labeled with increasing concentrations of MNPs have enhanced cell migration towards cancer cells. (C) Real-time PCR shows increased CXCR4 expression correlating to higher MNP concentrations used for labeling. Adapted from [73], courtesy of WILEY.
2. Imaging modalities for cellular nanotheranostics
Imaging modalities used for cellular nanotheranostics include MRI [75], PAI [76], and fluorescence imaging [77], all of which can use NPs as imaging agents. MRI is the only modality for which NPs have been clinically approved as imaging agents: superparamagnetic iron oxide (SPIO) NPs have been used for diagnosis of liver cancer [78] and lymph node metastases [79, 80]. Only one clinical study has been performed using SPIO-labeled MSCs in patients with neurodegenerative disease [81]. Small molecule Gd(III) complexes are commonly used as MRI contrast agents in the clinic, but Gd(III)-doped NPs are unlikely to be ever approved due to their slow body clearance with potential toxicity concerns.
While NPs are neither used in the clinic for PAI and fluorescence imaging, they have shown clear potential as imaging agents in pre-clinical animal studies [82-85]. AuNPs are preferred for PAI due to their high and stable signal and size- and shape-tuneable absorption rates [86]. Fluorescence imaging has been performed with a wide range of NPs that are either inherently fluorescent or loaded with fluorophores, including semiconducting polymer NPs and inorganic NP-based fluorescent probes such as QDs [77, 87].
2.1. MRI
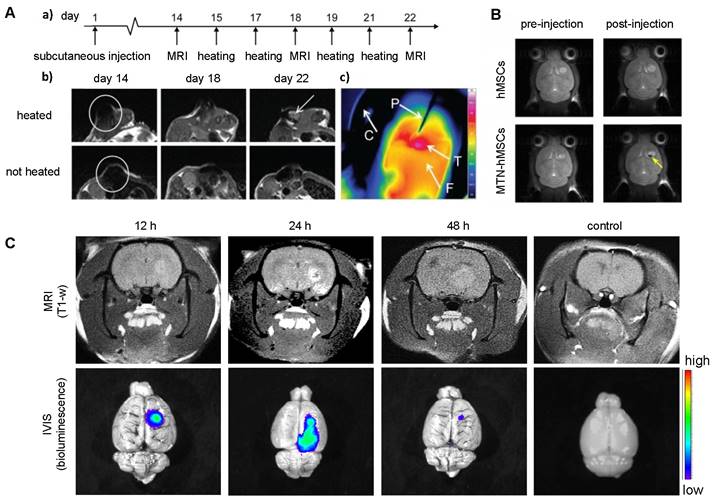
To date, all cellular theranostics studies with T1- or T2-weighted MRI have mainly used SPIO- or Gd(III)-doped NPs as imaging agents. Table 2 summarizes cellular nanotheranostics studies that used MRI as imaging modality. For most studies of MSC-mediated delivery of SPIO NPs, MRI was only applied to confirm successful tumor localization, without an attempt of therapeutic intervention. Kalber et al. visualized tumor localization of SPIO-labeled MSCs with MRI and then performed magnetic hyperthermia (Figure 5A). Surprisingly, they found no significant differences in tumor sizes after treatment, which may be due to either co-injection of SPIO-labeled MSCs with cancer cells, leading to inhomogeneous distribution of MSCs, or the low rise in surface temperature (4.3 C), which was insufficient to ablate surrounding cancer cells [88]. Huang et al. showed that MSCs can be labeled with magnetic ternary nanohybrids (MTNs) containing SPIO and TNF-related apoptosis-inducing ligand (TRAIL) DNA for MRI and subsequent suicide gene therapy (Figure 5B) [89]. Gd(III)-NP-labeled MSCs were used to target and image glioma with MRI, followed by Gd(III)-derived neutron capture therapy (Figure 5C) [90]. All other studies with MSC-mediated delivery of Gd(III)-doped NPs have been limited to imaging. MRI was performed using Mn(II) and Gd(III) NP-labeled MSCs, with the cells locating in tumors after i.v. injection [72, 91, 92].
Nanotheranostic MSC studies that have used MRI as imaging modality.
| MSC origin | Cancer type (cell line) | Animal model | Imaging agent | Injection route and n of labeled cells | Imaging paradigm | Retention time of MSC-NPs | Retention time of naked NPs | Reference | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Field strength | Contrast | Time post-injection | Control | ||||||||
| Bone marrow | Glioblastoma (C6) | Wistar rats | SPIO | I.v., 1.5E5 | 7.1 T | T2-w | 24 h | PBS | >24 h | N/A | [94] |
| N/A | Ovarian cancer (OVCAR-3) | Immunosuppressed BALB/c nu/nu mice | SPIO | I.v., 5E5 | 9.4 T | T2-w | 14,18,21 d | PBS | 22 d | N/A | [88] |
| Bone marrow | Breast cancer (4T1) | ICR mice | SPIO | I.v., N/A | N/A | T1-w | 1 h | Naked SPIO | >1 h | >1 hour | [96] |
| Bone marrow | Glioblastoma (U87MG) | Athymic nude mice | Zinc-doped SPIO | I.v., 1E6 | 7 T | T1-w, T2-w | 48 h | Unlabeled MSCs | >48 h | N/A | [74] |
| Bone marrow | Glioblastoma (U87MG) | BALB/c nude mice | PEG-SPIO | I.v., N/A | 7 T | T2-w | 7 d | Tumor-free | >7 d | N/A | [95] |
| N/A | Glioblastoma (U87MG) | Athymic nude mice | SPIO@Au NPs | I.v., 1E6 | 7 T | T2-w | 72 h | Unlabeled MSCs | >3 d | N/A | [93] |
| Bone marrow | Glioblastoma (U87MG) | BALB/c mice | Silica-Gd | I.v., 1E6 | 7.05 T | T1-w | 24 h | Naked NP | >24 h | N/A | [92] |
| Bone marrow | Colon cancer (CT26) | BALB/c nude mice | Gd(III)-chelate NPs | I.v., 1E5 | 4.7 T | T1-w | 2 h | Unlabeled MSCs | >2 h | N/A | [72] |
| Umbilical cord | Glioma (GBM8401) | F344/NNarl rat; C57BL/6JNarl rat | Gd-SPIO | I.v., 2E6 | 3 T | T2-w | 48 h | Naked NP | >48 h | N/A | [90] |
| Umbilical cord | Melanoma (B16F10) | C57BL/6 mice | Mn(II) and Gd(III) co-doped CuInS2-ZnS nanocrystals | I.v., 1E6 | 1.5 T | T1-w, T2-w | 6 h | PBS | >6 h | N/A | [91] |
Abbreviations: I.t.=Intratumoral; I.v.=Intravenous; MSC=Mesenchymal stem cell; N/A=Not available; NP=Nanoparticle; PEG=Polyethylene glycol; SPIO=Superparamagnetic iron oxide
(A) T2-weighted MRI of co-injected ovarian cancer cells and SPIO-labeled MSCs 14 days post-injection (a, b) and heatmap of subsequent hyperthermia (c). Adapted from [88], courtesy of Dove Medical Press. (B) T2-weighted MRI of magnetic ternary hybrids complexed with TRAIL (MTN) 24 hours post intracerebral injection of MTN-labeled hMSCs. Yellow arrow indicates signal from labeled MSCs. Adapted from [89], courtesy of Ivyspring International Publisher. (C) T1-weighted MRI of Gd-containing NP-labeled MSCs 12, 24, and 48 hours post i.v. injection, with accompanying bioluminescence images. Adapted from [90], courtesy of Nature Springer.
Multiple studies indicate that NP-labeled MSCs accumulate in tumors within 24 hours after i.v. injection [74, 92-94] and that they can be retained for up to 22 days with a visible MRI signal [88]. However, the 22 day-study used a different approach: injection of an ovarian cancer cell line that was mixed with SPIO-labeled hMSCs to form subcutaneous tumors in mice [88]. Other studies using i.v. injection observed that MSCs were retained for up to a week [95]. Only two studies directly compared NP-labeled MSC accumulation and retention with naked NPs. Hao et al. injected SPIO NP-labeled MSCs and naked SPIOs i.v. in a breast cancer model, then performed MRI 1 hour post-injection - however, 1 hour is likely too short to achieve the full benefit from MSC-mediated delivery [96]. Lai et al. performed MRI 48 hours post-i.v. injection of Gd(III)-containing NP-labeled MSCs and naked NPs in a glioma model, demonstrating that naked NPs could not reach the glioma while NP-labeled MSCs enabled MRI (Figure 5C) [90]. No studies using i.v. or i.t. injection reported on the retention of NP-labeled MSCs for longer than one week, and hence, more studies on the long-term dynamics of NP-labeled MSCs are warranted. It would also be desirable to use MRI for comparison of the accumulation and retention of NP-labeled MSCs vs. naked NPs, as most studies thus far used only PBS or unlabeled MSCs as controls.
2.2. PAI
Cellular nanotheranostics imaged with PAI as a diagnostic modality have all used gold-based nanotheranostic agents (Table 3), including AuNPs in the form of nanostars [70], nanocages [97], nanorods [69, 98], and coated nanospheres [93]. AuNPs are widely used as PAI contrast agents due to their strong and tunable optical absorption. When AuNPs interact with light, conduction electrons on the NP surface are driven by the incident electric field into collective oscillations, known as localized surface plasmon resonance (LSPR). After illumination, plasmons can decay nonradioactively or radioactively, resulting in absorption or light scattering, respectively [99]. With PAI, the absorption cross-section of NPs can be measured after NIR light exposure, enabling deep tissue imaging and therapy guidance [27]. In some studies, AuNPs were complexed with SPIO to allow dual-mode PAI and MRI of the same nanotheranostic agent [69, 93].
Nanotheranostic MSC studies that have used PAI as imaging modality.
| MSC origin | Cancer type (cell line) | Animal model | Imaging agent | Injection route and n of labeled cells | Imaging paradigm | Retention time of MSC-NPs | Retention time of naked NPs | Distribution area of MSC-NPs | Distribution area of naked NPs | Reference | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Wavelength | Time (post- injection) | Control | ||||||||||
| Umbilical cord | Prostate (PC-3) | Nude mice | AuNS (anisotropic) | I.t., 1E5 | 780 nm | 3 d | Naked AuNS | >3 d | >3 d | 0.073 cm2 | 0.022 cm2 | [70] |
| Bone marrow | Glioblastoma (U87-MG) | Athymic nude mice | AuNC (anisotropic) | I.v., 1E5 | 638 nm | 7 d | N/A | >7 d | N/A | 200 um2 | N/A | [97] |
| Bone marrow | Glioblastoma (U87-MG) | Athymic nude mice | SPIO@Au | I.v., 1E6 | 810 nm | 3 d | Unlabeled MSCs | >3 d | N/A | N/A | N/A | [93] |
| Umbilical cord | Negative breast cancer (MDA-MB-231) | BALB/c athymic nude mice | LDGI | I.t., 1E5 | 820 nm | 3 d | Naked LDGI | >3 d | >3 d | 0.0945 cm2 | 0.0225 cm2 | [69] |
| N/A | Breast cancer (MCF-7) | BALB/c nude mice | AuNR@HPMOs-PTX | I.t., 1E6 | 700 nm | 24 h | AuNR@HPMOs-PTX | >24 h | >24 h | N/A | N/A | [98] |
Abbreviations: AuNC=Gold nanocages; AuNR@HPMOs-PTX=Gold nanorod hollow periodic mesoporous organosilica nanospheres with paclitaxel; AuNS=Gold nanostar; I.t.=Intratumoral; I.v.=Intravenous; LDGI=Lipid-Doxorubicin-Gold-Iron oxide nanocluster; MSC=Mesenchymal stem cell; N/A=Not available; NP=Nanoparticle; SPIO=Superparamagnetic iron oxide
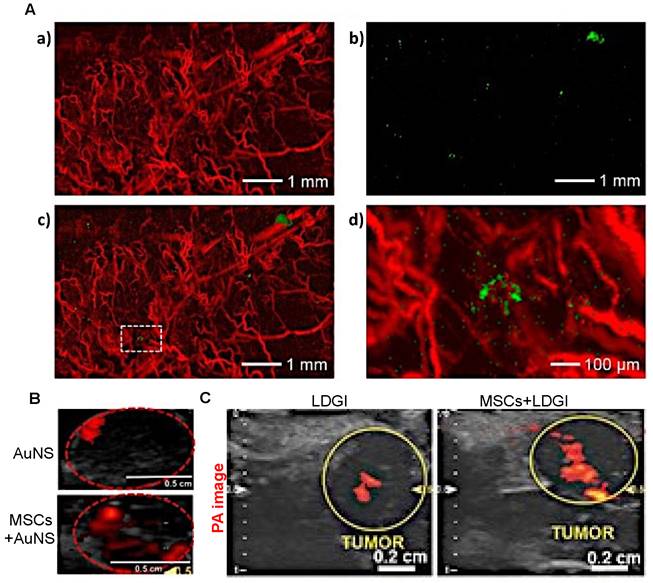
When imaged with PAI, i.v.-injected AuNP-labeled MSCs were observed to remain in tumors up to one week (Figure 6A) [97]. Unfortunately, there are no comparison data available for retention of “naked” AuNPs for more than three days post-i.v. or i.t. injection and hence, it is not clear from these studies whether MSC-mediated delivery can improve long-term retention of PAI agents. However, with PAI, it was observed that MSC-mediated delivery markedly improved the distribution of AuNPs throughout tumors three days post i.t. injection, with a 3.3-fold (Figure 6B) [70] or 4.2-fold increase in area containing signal (Figure 6C) [69]. Thus, MSC-mediated delivery can enable imaging of tumor areas that would otherwise not be visible with naked nanotheranostics.
(A) AuNP-labeled MSCs (green) injected i.v. in a U87 brain tumor model are retained up to one-week post-injection. PA images show the presence of blood vessels (red) in the tumor acquired at a wavelength of 532 nm (a) and gold nanocage (AuNC)-labeled hMSCs injected i.v. that homed to the tumor region acquired at a wavelength of 638 nm (b). The images are superimposed in (c) with the inset shown at higher magnification in (d). Adapted from [97], courtesy of Ivyspring International Publisher. (B) PAI of gold nanostar (AuNS) and AuNS-labeled MSC distribution three days post i.t. injection, showing a 3.3-fold increase in signal area for MSC-mediated delivery. Adapted from [70], courtesy of Ivyspring International Publisher. (C) PAI of i.t.-injected nanoclusters composed of lipids, doxorubicin, gold nanorods, and iron oxide (LDGI) or LDGI-labeled MSCs, showing a 4.2-fold increase in signal area for MSC-mediated delivery. Adapted [69], courtesy of WILEY.
2.3. Fluorescence imaging
Fluorescence imaging of cellular nanotheranostics has been primarily performed with NPs containing a photosensitizer including Ce6 [100-102], dibenzocyclooctyne (DBCO)-fluorophores [71], and Cr(III) doped luminescent NPs [103]. Alternatively, a few studies have used non-conventional fluorescent agents, such as Bi2Se3 NPs [104], Gd(III)-chelate NPs [72], and Mn(II) and Gd(III) co-doped CuInS2-ZnS nanocrystals [91] (Table 4).
Nanotheranostic studies that have used fluorescence imaging as imaging modality.
| MSC origin | Cancer type (cell line) | Animal model | Imaging agent | Injection route and n of labeled cells | Imaging paradigm | Retention of MSC-NPs | Reference | ||
|---|---|---|---|---|---|---|---|---|---|
| Fluorescence imaging system | Time (post- injection) | Control | |||||||
| Skin tissue | Lung carcinoma (LLC) | C57BL/6 mice | Ce6-CdSe/ZnS QD | S.c., 1E6 | UVP iBox Scientia | 24 h | Naked Ce6-QD | >24 h | [101] |
| N/A | Lung carcinoma (LLC) | BALB/c nude mice | Ce6-MnO2 NP | I.v., 1E6 | Bruker | 24 h | Post-injection mice | >24 h | [100] |
| Bone marrow | Breast cancer (4T1) | BALB/c mice | Ce6@MSV NP | I.v., 1E6 | IVIS | 72 h | Unlabeled MSCs | >72 h | [102] |
| Adipose tissue | Lung carcinoma (A549) | BALB/c mice | Bi2Se3 NP | I.t., N/A | IVIS | 24 h | Naked Bi2Se3 NPs | N/A | [104] |
| Bone marrow | Colon cancer (CT26) | BALB/c mice | Gd(III)-chelate NP | I.v., 1E6 | 12-bit CCD camera | 2 h | Unlabeled MSCs | >2 h | [72] |
| Umbilical cord | Melanoma (B16F10) | C57BL/6 mice | Mn(II) and Gd(III) co-doped CuInS2-ZnS NP | I.v., 1E6 | IVIS | 6 h | PBS | >6 h | [91] |
| Bone marrow | Lung cancer (A549) | SCID Beige mice | PTX-PLGA NP | I.v., 2.5E5 | IVIS | 8 d | Unlabeled MSCs | >8 d | [108] |
Abbreviations: I.t.=Intratumoral; I.v.=Intravenous; MSC=Mesenchymal stem cell; MSV=Multistage silicon vector; N/A=Not available; NP=Nanoparticle; S.c.=Subcutaneous; PLGA=Poly lactide-co-glycolic acid; PTX=Paclitaxel; QD=Quantum dot
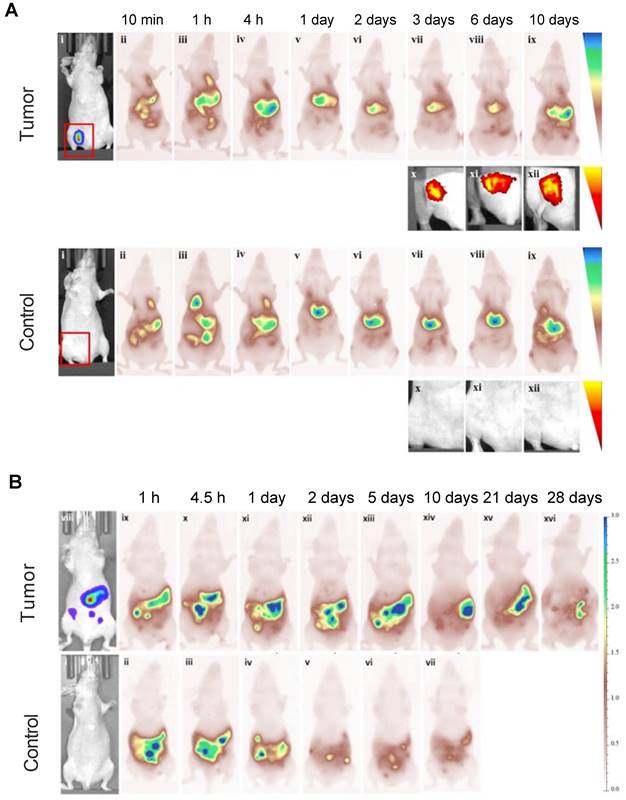
Studies performed with MSC-mediated delivery found that MSCs were retained within tumors for at least 10 or 28 days after i.v. and i.p. injection, respectively. At 10 days, the fluorescence intensity of Cy5.5-labeled MSCs was barely detectable in a lung carcinoma model, indicating that the majority of the Cy5.5 fluorophores were cleared by the liver and spleen (Figure 7A). A similar phenomenon was observed in an ovarian cancer model. On days 21 and 28 post i.p. injection, there was a small but detectable fluorescence signal, with most of the Cy5.5-labeled MSCs having cleared out the tumor (Figure 7B) [71].
(A) Cy5.5-labeled MSCs injected i.v. in a lung carcinoma model, indicating tropism and retention of MSCs within tumors up to 10 days, along with non-specific cell distribution in the liver and spleen. Tumor-free mice were used as control. (B) Ovarian carcinoma model with i.p. injected Cy5.5-labeled MSCs, showing retention of Cy5.5-labeled MSCs within tumors up to 28 days. Tumor-free mice were used as control. Adapted with permission from [71], copyright 2016 Elsevier.
Only one study has directly compared the distribution of Ce6-modified CdSe/ZnS QD (QD-Ce6)-labeled MSCs with QD-Ce6 alone for fluorescence imaging, reporting that there was a 7-fold higher fluorescence intensity when MSCs served as delivery vehicles for QD-Ce6 [101]. Overall, MSC-mediated delivery may significantly prolong the retention of nanotheranostics used for fluorescence imaging, enabling long-term monitoring of tumors during treatment. However, more studies are needed to compare the intratumoral distribution of nanotheranostic-labeled MSCs vs. naked nanotheranostics.
2.4. Other potential imaging modalities
Other imaging modalities may be used in the near future to monitor the biodistribution of cellular nanotheranostics, including MPI, magneto-motive ultrasound imaging (MMUS), and PET. MPI uses similar SPIO imaging agents as used in MRI, where they act as tracers instead of contrast agents [31]. Unlike MRI, MPI can specifically quantify the amount of SPIO-labeled MSCs post administration [105, 106]. MMUS can also use SPIO as an imaging agent [107], and hence has potential to detect SPIO-labeled MSCs as well. Finally, therapeutic NPs can be labeled with SPECT or PET tracers, but to the best of our knowledge no theranostic studies have been performed yet with NP-labeled MSCs.
3. Therapeutic modalities for cellular nanotheranostics
The primary therapeutic modalities used with cellular nanotheranostics include PTT [69, 70, 98] and PDT [100, 102], chemotherapy [71, 98, 108], and gene therapy [89, 103]. PTT is performed with a near-infrared (NIR) laser beam directed at the tumor causing intratumoral NPs to heat up and ablate the tumor tissue [109]. PDT is performed with an NIR laser as well, but it interacts with a PDT agent to produce single reactive oxygen species (ROS) and free radicals that lead to apoptosis, necrosis, and autophagy [110]. Using appropriate photosensitizers, PDT has been used in the clinic for over forty years to treat a variety of tumors. Laser ablation without PTT agents has also been used clinically, although it has not reached large clinical trials. Neither PTT nor PDT with NPs has been clinically approved yet [109].
NP-based delivery of chemotherapeutics has been widely studied. NP vehicles currently undergoing clinical trials include liposomes, polymeric micelles, protein-drug NPs, and dendrimers [111]. The efficacy of NPs as chemotherapeutic carriers to tumors depends on the EPR effect and rate of controlled drug release [112]. Therapeutic genes can be delivered with viral vectors or NPs. NPs have advantages as delivery agents due to their comparatively lower toxicity and higher carrying capacity [25]. While gene therapy has not been clinically approved as a form of cancer therapy, many studies have indicated that NPs are promising delivery agents in experimental animal models [113, 114]. In the following sections, we provide an overview of current approaches towards tumor therapy employing MSC-mediated delivery of therapeutic agents.
3.1. PTT and PDT
Cellular nanotheranostics with PTT as a therapeutic modality have primarily used Au-based NPs as photothermal agents. To enhance PTT heating, the shape and surrounding materials of AuNPs can be optimized, as demonstrated for gold nanostars (AuNS) [70], gold nanorod-embedded hollow periodic mesoporous organsilica nanospheres (AuNR-HPMOs) [98], and gold nanorod (AuNR) and iron oxide (LDGI) nanostructures [69]. With AuNPs being an excellent imaging agent for PAI, it is not surprising that all cellular nanotheranostic PTT studies have used PAI as an imaging modality (Table 5).
Nanotheranostic studies that have used PTT and PDT as therapeutic modality.
| MSC origin | Cancer type (cell line) | Animal model | Imaging modality | Injection route and n of labeled cells | PTT agent | PTT paradigm | Temperature difference (C) for MSC-NPs | Temperature difference (C) for naked NPs | Tumor volume change for MSC-NPs | Tumor volume change for naked NPs | Reference | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Laser condition | Time (post- injection) | Control | |||||||||||
| Umbilical cord | Prostate (PC-3) | Nude mice | PAI | I.t., 1E5 | AuNS (anisotropic) | 808 nm laser, 1.5 W/cm2, 10 m | 3 d | Naked AuNS | 10.9 | 8.1 | 200% | 250% | [70] |
| Umbilical cord | Negative breast cancer (MDA-MB-231) | BALB/c athymic nude mice | PAI | I.t., 1E5 | LDGI | 808 nm, 1.5 W/cm2, 10 m | 3 d | Naked LDGI | 23 | 10 | 0% | 600% | [69] |
| N/A | Breast cancer (MCF-7) | BALB/c nude mice | PAI | I.t., 1E6 | AuNR@HPMOs-PTX | 808 nm, 1.3 W/cm2, 5 m | 24 h | Naked AuNR@HPMOs-PTX | 16 | 16 | 150% | 160% | [98] |
| N/A | Lung carcinoma (LLC) | BALB/c nude mice | Fluorescence | I.v., 1E6 | Ce6-MnO2 NP | 633 nm, 0.6 W/cm2, 5 m | 24 h | Naked Ce6-MnO2 NP | N/A | N/A | 0% | 500% | [100] |
| Bone marrow | Breast cancer (4T1) | BALB/c mice | Fluorescence | I.v., 1E6 | Ce6@MSV NP | 405 nm laser, 100 mW, 15 m | 0 h | Unlabeled MSCs | N/A | N/A | 120% | 150% | [102] |
Abbreviations: AuNR@HPMOs-PTX=Gold nanorod hollow periodic mesoporous organosilica nanospheres with paclitaxel; AuNS=Gold nanostar; I.t.=Intratumoral; I.v.=Intravenous; LDGI=Lipid-Doxorubicin-Gold-Iron oxide nanocluster; MSC=Mesenchymal stem cell; MSV=Multistage silicon vector; N/A=Not available; NP=Nanoparticle; PAI=Photoacoustic imaging; PDT=Photodynamic therapy; PTT=Photothermal therapy.
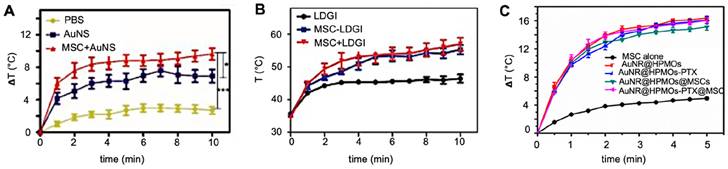
Huang et al. found a comparative temperature increase of approximately 2°C three days post i.t. injection for MSC-mediated delivery of AuNS (Figure 8A) [70], while Xu et al. found an increase of no less than 13°C three days post i.v. injection of AuNR-labeled MSCs (Figure 8B) [69]. Xu et al. also found that tumors treated with AuNR-labeled MSCs and PTT had ultimately no increase in volume, while tumors injected with naked AuNRs had a six-fold volume increase [69]. Despite the disparity in temperature difference, both Huang's and Xu's team used similar PTT conditions, including time post-injection, number of hMSCs, PTT agent injected, and laser power. The primary difference here was the injection route, i.e., i.v. vs. i.t. When naked AuNRs are injected i.v., fewer particles are expected to reach the tumor compared to AuNRs internalized and delivered by tumor-tropic hMSCs. Wu et al. exposed tumors to a PTT laser 24 hours post-i.t. injection and observed similar temperature increases for AuNRs delivered by MSCs vs. alone as naked AuNRs (Figure 8C). It is possible that at the 24-hour time point, neither nanotheranostic was cleared out yet from the tumor, making them both efficient PTT agents [98].
(A) Temperature changes during irradiation of prostate tumors post-i.t. injection of naked gold nanostars (AuNS) and AuNS-labeled MSCs, showing a differential temperature increase of approximately 2°C for MSC-mediated delivery. Adapted from [70], courtesy of Ivyspring International Publisher. (B) Temperature changes during irradiation of breast tumors post-i.v. injection of nanoclusters composed of lipids, doxorubicin, gold nanorods, and iron oxide (LDGI), LDGI-labeled MSCs, and LDGI-labeled MSCs, with a differential temperature increase of approximately 13°C for MSC-mediated delivery. Adapted from [69], courtesy of WILEY. (C) Temperature changes during irradiation of breast tumors post-i.t. injection of gold nanorod-embedded hollow periodic mesoporous organosilica NPs loaded with paclitaxel (AuNR@HPMOs-PTX) and AuNR@HPMO-PTX-labeled MSCs. Adapted with permission from [98], copyright 2016 ACS Publications.
Cellular nanotheranostics using PDT as therapeutic modality have all used Ce6 as the photosensitizing molecule with fluorescence imaging as modality. Similar to PTT, the observed tumor volume decrease was most pronounced for MSC-mediated delivery when PDT was performed after i.v. injection or delayed after i.t. injection. PDT performed 24- and 48 hours post-i.t. injection resulted in a 4-fold reduced tumor size for MSC-mediated delivery compared to naked NPs [101]. In contrast, only a 1.25-fold reduced tumor size for MSC-mediated delivery vs. naked NPs was observed when PDT was performed immediately after i.t. injection [102]. Overall, the longer the delay between i.t. injection and therapy, the more efficient MSC-delivered therapy becomes compared to naked NPs. This suggests that MSC-mediated delivery of nanotheranostics may require only a single injection, which can be used for multiple rounds of therapy spread out over time.
3.2. Chemotherapy
An overview of cellular nanotheranostic agents used in tandem with chemotherapy as therapeutic modality is listed in Table 6. All nanotheranostics incorporated the chemotherapeutic drug paclitaxel (PTX). Imaging agents include SPIO [115], DBCO-labeled fluorophores [71, 108], and AuNR@HPMOs [98], using MRI, fluorescence imaging, and PAI, as imaging modalities, respectively. Overall, MSC-mediated delivery of PTX-loaded nanotheranostics increased mice survival rates compared to PTX alone, although it should be noted that in some studies MSC-PTX was combined with hyperthermia [115] and PTT, respectively [98], making this an improper comparison. MSC-mediated delivery of nanochemotheranostic agents remains promising (Figure 9) but further studies are needed on its overall efficacy.
Nanotheranostic MSC studies that have used chemotherapy as therapeutic modality.
| MSC origin | Cancer type (cell line) | Animal model | Imaging modality | Injection route and n of labeled cells | Chemotherapy agent | Chemotherapeutic regimen | Cumulative survival days for MSC-NPs | Cumulative survival days for naked NPs | Tumor volume change for MSC-NPs | Tumor volume change for naked NPs | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose | Control | |||||||||||
| Adipose tissue | Brain astrocytoma (ALTS1C1) | C57BL/6JNarl mice | N/A | I.v., N/A | PLGA-PTX-SPIO NP | PTX dose of 2.5 mg/kg | Naked NP | 35 d | 25 d | N/A | N/A | [115] |
| N/A | Lung carcinoma (A549); ovarian cancer (MA148) | Athymic nude mice | Fluorescence | I.v., 5E5 | PTX-PLGA NP | PTX dose of 0.2 mg | Naked NP | >70 d | 55 d | N/A | N/A | [71] |
| N/A | Breast cancer (MCF-7) | BALB/c nude mice | PAI | I.t., 1E6 | AuNR@HPMOs-PTX | N/A | Naked AuNR@HPMOs-PTX | N/A | N/A | 150% | 160% | [98] |
| Bone marrow | Lung cancer (A549) | SCID Beige mice | Fluorescence | I.v., 2.5E5 | PTX-PLGA NP | PTX dose of 5 ug | PTX | 160 d | 110 d | 0% | 0% | [108] |
Abbreviations: AuNR@HPMOs-PTX=Gold nanorod hollow periodic mesoporous organosilica nanospheres with paclitaxel; I.t.=Intratumoral; I.v.=Intravenous; MSC=Mesenchymal stem cell; N/A=Not available; NP=Nanoparticle; PLGA=Poly lactide-co-glycolic acid; PTX=Paclitaxel.
(A) Survival rates of brain tumor-bearing mice injected i.v. with naked SPIO/paclitaxel (PTX)-loaded polymeric NPs (SPNPs) and SPNP-loaded MSCs, with and without hyperthermia. Adapted with permission from [115], copyright 2017 Elsevier. (B) Survival rates of ovarian carcinoma-bearing mice injected i.p. with paclitaxel-loaded, DBCO surface functionalized NPs (DBCO-NPs) and DBCO-NP-labeled glycoengineered MSCs. Adapted with permission from [71], copyright 2016 Elsevier. (C) Relative breast tumor volume of mice injected i.t. with gold nanorod-embedded hollow periodic mesoporous organosilica NPs loaded with paclitaxel (AuNR@HPMOs-PTX) and AuNR@HPMO-PTX-labeled MSCs. Adapted with permission from [98], copyright 2016 ACS Publications.
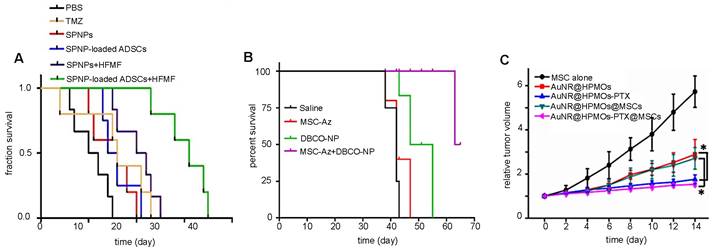
3.3. Gene therapy
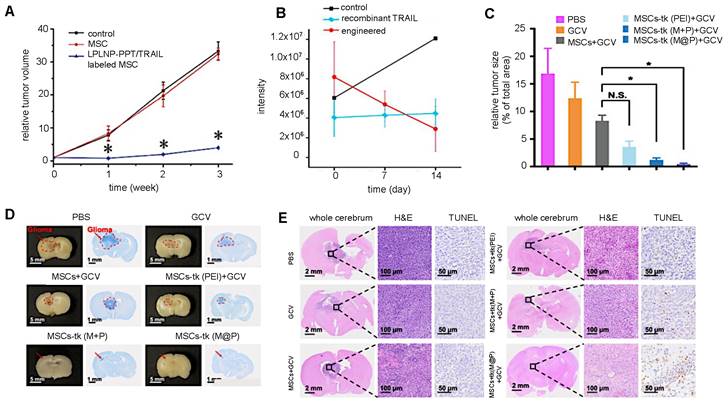
Most cellular nanotheranostic agents using gene therapy are composed of magnetic NPs using MRI as the imaging modality (Table 7). The structural composition of these NPs included magnetosome-like ferrimagnetic iron oxide nanochains (MFIONs) [116], ZnFe2O4 magnetic core and mesoporous silica shell (MCNPs) [117], and a magnetic ternary nanohybrid (MTN) system comprised of cationic materials, nucleic acids, and hyaluronic acid-decorated SPIO [89]. Only one study used fluorescence imaging as the imaging modality with dual-functional persistent luminescent nanocomposites (LPLNP-PTT/TRAIL) [103]. Here, hMSCs were transfected with TRAIL DNA (Figure 10A). Once localized in tumors, TRAIL-hMSCs secrete TRAIL, which binds to the death receptor 4 (DR4) or DR5 of tumor cells, causing apoptosis [118]. Only Yin et al. compared MSC-mediated delivery of MCNP-TRAIL plasmid complexes with intraperitoneal (i.p.) injection of TRAIL alone (Figure 10B) [117]. Ovarian tumors treated with MSCs engineered by MCNP-TRAIL plasmid complexes had a volume decrease of over 50% after two weeks, while tumors that received a single dose of TRAIL had no volume decrease. This may be explained by the short half-life of TRAIL, which typically requires high daily doses (1-10 mg/kg) to be effective [119]. No comparison was done for treatment efficacy of naked MCNP-TRAIL plasmid complexes. All studies used TRAIL for gene therapy, except for Li et al. who elected to use the herpes simplex virus thymidine kinase (HSV-tk)/ganciclovir (GCV) suicide gene for glioma treatment (Figure 10C-E) [116].
Nanotheranostic studies that have used gene therapy as therapeutic modality.
| MSC origin | Cancer type (cell line) | Animal model | Imaging modality | Injection route and n of labeled cells | Gene therapy agent | Transfection method | Gene therapy paradigm | Cumulative survival days for MSC-NPs | Tumor volume change for MSC-NPs | Tumor volume size for gene therapy control | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose | Control | |||||||||||
| Placental | Glioma (C6) | Sprague-Dawley rats | N/A | I.v., 3E5 | HSV-tk/GCV | MFION | 100 mg/kg GCV solution | GCV solution | 14 d | 2% | 17% | [116] |
| Adipose tissue | Ovarian (A2780) | Athymic nu/nu mice | N/A | I.p., 5E5 | TRAIL | ZnFe2O4 MNP-silica-PEI/TRAIL | 5 mg/kg recombinant TRAIL | Recombinant TRAIL | N/A | 50% | 100% | [117] |
| Bone marrow | Glioblastoma (U87 MG) | BALB/c nude mice | PAI | I.t., 1E5 | TRAIL | Luminescence nanocomposite/TRAIL | N/A | Unlabeled MSCs | N/A | 500% | N/A | [103] |
| N/A | Glioblastoma (U87MG) | BALB/cAnN.Cg-Foxn1nu/CrlNarl nude mice | MRI | I.c., 2E5 | TRAIL | MTN-SPIO/TRAIL | N/A | Unlabeled MSCs | 52 d | 60-fold, 60,000% | N/A | [89] |
Abbreviations: HSV-tk/GCV=Herpes simplex virus thymidine kinase/ganciclovir; I.c.= Intracranial; I.p.=Intraperitoneal; I.t.=Intratumoral; I.v.=Intravenous; MFION= Magnetosome-like ferrimagnetic iron oxide nanochain; MNP=Magnetic nanoparticle; MRI=Magnetic resonance imaging; MSC=Mesenchymal stem cell; MTN=Magnetic ternary nanohybrid; N/A=Not available; NP=Nanoparticle; PAI=Photoacoustic imaging; PEI=Polyethyleneimine; SPIO=Superparamagnetic iron oxide; TRAIL=TNF-related apoptosis-inducing ligand.
(A) Relative brain tumor volumes of mice injected i.t. with MSCs and MSCs labeled with long persistent luminescence NPs complexed with TRAIL (LPLNP-PPT/TRAIL). Adapted with permission from [103], copyright 2017 WILEY. (B) Luminescence intensity quantification of luciferase-expressing ovarian tumor injected i.p. with recombinant TRAIL protein and magnetic NP-PEI/TRAIL plasmid complexes. Adapted with permission from [117], copyright 2016 Elsevier. (C, D, and E) Relative tumor size and brain sections stained with Nissl, H&E, and TUNEL for gliomas injected i.v. with herpes simplex virus thymidine kinase (HSV-tk)-transduced MSCs. Ganciclovir (GCV) was administered to kill tumor cells after phosphorylation by HSV-tk. Adapted from [116], courtesy of Ivyspring International Publisher.
Although combining cellular nanotheranostics with gene therapy has so far only been applied for TRAIL and HSV-tk/GCV genes, previous studies have indicated that MSCs can be engineered to express a wide range of genes for therapeutic purposes. MSCs expressing immunomodulatory cytokines such as IFN-β [120] and IL-12 [121, 122] inhibited tumor growth in melanoma, renal cell carcinoma, and cervical cancer murine models. Genes for the anti-angiogenic protein thrombospondin (TSP-1) [123] and the suicide gene cytosine deaminase-uracil phosphoribosyl transferase (CD-URBT) [124-126] prevented cancer progression in glioma, melanoma, prostate, and colon cancer murine models. In future studies, the use of genetically engineered cellular nanotheranostics may be used for further improvement of NP delivery.
3.4. Radiation therapy
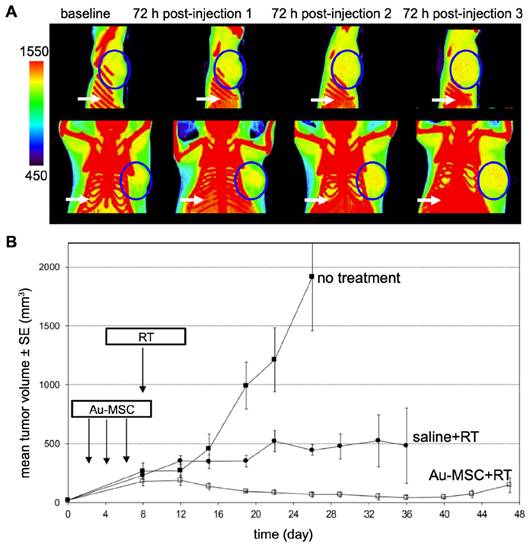
Nanoparticle-mediated radiotherapy (RT) has considerable potential for revolutionizing cancer treatment [127], particularly when combined with MSCs for targeted NP tumor delivery [128]. The strategic integration of MSCs enhances precision in cancer radiotherapy by providing a specialized vehicle for the transport of radiosensitizing NPs to the tumor. When guided by either MRI or CT, the entire procedure can be personalized. Lai et al. [90] used MSCs to deliver GD(III)-based NPs across the blood-brain-barrier to gliomas after i.v. injection. MRI and gadolinium neutron capture therapy was then performed, reducing tumor volume 4-fold and increasing median survival 2.5-fold compared to gadolinium NPs alone. Xiao et al. [129] investigated the potential of tumor-tropic adipose-derived MSCs for targeted RT of NSCLC using bismuth selenide (Bi2Se3) NPs. Key findings included the ability of MSCs to selectively deliver Bi2Se3 NPs to NSCLC tumors following i.v. injection, the enhancement of radiation-induced cell death, the significant reduction in tumor growth and prolongation of animal survival after RT, the increased radiation-induced apoptosis and reduced tumor angiogenesis associated with the therapeutic efficacy, and the favorable safety profile of MSCs-mediated Bi2Se3 NP delivery. In another study, Pullambhatla et al. [128] demonstrated that RT is more effective when paired with AuNP-labeled MSCs. Since AuNPs can enhance CT image contrast, mice bearing MDA-MB-231 breast tumors were imaged with CT before injection of labeled MSCs and at 3 different time points after (Figure 11). Following imaging at baseline, mice were injected i.v. with labeled MSCs on days 0, 3, and 6 for a total of 3 injections administered 72 hours apart. Labeled MSCs demonstrated accumulation at the tumor site at 72 hours post injection, which increased progressively after successive injections of labeled MSCs (Figure 11A). Two days after injection of labeled MSCs had been completed (day 8), mice were irradiated under CT guidance. Without treatment the mean tumor volume was 2,000 mm3 at day 24 following tumor inoculation, while mice treated with labeled MSCs in combination with radiotherapy exhibited the greatest delay in tumor growth, with a mean tumor volume of only 15 mm3 at day 45 post tumor inoculation (Figure 11B).
(A) CT images of mice bearing MDA-MB-231 tumors taken 72 hours after each i.v. injection of AuNP-labeled MSCs. Tumors are indicated by blue circles, with red pixels within the circles representing the accumulation of high-density particles at the tumor site. CT images show the accumulation of labeled MSCs in the liver and spleen (white arrows). (B) Treatment outcome in NOD/SCID mice bearing subcutaneous MDA-MB-231 tumors. Adapted from [128], courtesy of MDPI.
Although more studies are required, these initial reports suggest that the utilization of imaging-guided nanoparticle-mediated RT combined with MSCs as delivery vehicle presents a promising new treatment strategy.
3.5. Other potential therapeutic modalities
Cellular nanotheranostics may be further engineered based on other modes of cancer therapy. Stem cells engineered for cancer immunotherapy are a promising avenue for cellular nanotheranostics [130]. For instance, Zhang et al. incorporated methylene blue as a photosensitizer in MSCs transfected with a plasmid encoding IL-12 and were able to demonstrate an enhanced immune response after PDT [131]. Furthermore, it has been shown that after RT-induced tumor ablation, there is a release of tumor-associated antigens which modulates the efficacy of immunotherapy, including cytotoxic T-lymphocyte associated protein 4 (CTLA-4) checkpoint blockade [132] and anti-programmed cell death ligand 1 (PD-L1)-based immunotherapy [133]. Hence, NPs could be potentially targeted to multiple tumor metastases if delivered using MSCs, allowing extensive RT and enhancing subsequent immunotherapy.
Hematopoietic stem cell transplantation (HSCT) has been widely used for haematological malignancies and solid tumors, allowing patients to effectively regenerate hematopoietic cells after high-dose chemotherapy [134, 135] or RT [136]. Cluster of differentiation 19 (CD19) CAR-T cell therapy has been combined with HSCT, where CD19 CAR-T cell therapy eradicates leukemia cells and B cells, and HSCs promote the amplification and survival of CD19 CAR-T cells [137, 138]. HSCs could be labeled with nanotheranostic agents to simultaneously visualize their localization at the transplantation site and other tissues.
Finally, hormone therapy, which acts by blocking the receptors of endocrine cancers and prevent further cell proliferation, has been combined with NPs for enhanced imaging and therapy of cancer [139, 140]. MSCs could be possible genetically engineered to increase hormone production while also labeled with NPs.
4. Some critical notes and challenges to move forward
The future clinical use of cellular nanotheranostics faces many challenges. As for optimal timing for performing PTT, hyperthermia, and/or RT for example, the (long-term) duration of the retention of MSCs within tumors has been evaluated only a few studies. Near all studies followed up for only a few hours post-injection, with a maximum of one week. However, some studies reported that MSCs, once homed to tumors, can remain there for 3-4 weeks. Most studies only compared MSCs alone with MSC-mediated delivery of nanotheranostics. Since MSCs (without labeling) may be effective cellular therapeutics by themselves, using them as the only control is not a proper way to validate their promise as delivery vehicles of nanotheranostics [141-143].
Each cancer, depending on where it originated, has a different molecular signature, vascularization, and aggressiveness, which may influence MSC homing [144]. Only one study has made a direct comparison of the efficacy of cellular nanotheranostics towards different tumor types, reporting that Cy5.5-labeled MSCs were retained in lung cancer models for up to 10 days and in ovarian cancer models for up to 28 days [71]. Also, it remains unclear what the effect of tumor size is on stem cell tropism, although past studies demonstrated that stem cells can successfully 'trail' small glioma metastases [145].
After i.v. injection, MSCs can home into the tumor but also in other tissues that are wounded or renewing quickly, a hallmark of natural MSC repair [146, 147]. To maximize their therapeutic potential, methods for enhancing MSC tumor-tropism must be further developed so that MSCs can more specifically target tumors. Such approaches include genetic engineering to overexpress the chemokine receptors CXCR1-4 [148, 149] IL-8 [150], and interferon-β [120]. In the last study, a therapeutic effect was not observed for systemic delivery of IFN-β or by IFN-β produced by MSCs injected subcutaneously at a site distant from the tumors. Another example how cells can be engineered to improve their initial docking to target tissue is to transfect them with the very late antigen-4 (VLA-4), which specifically binds to the vascular cellular adhesion molecule 1 (VCAM-1) expressed on inflamed endothelium [151]. This facilitates their extravasation and passage into the brain parenchyma [152]. Of note, it has been demonstrated that pre-treating tumors with RT can improve MSC recruitment [153-155].
It remains controversial whether MSCs promote tumor progression and metastasis [156, 157] or enhance pathways that suppress both proliferation and apoptosis [158, 159]. To safely administer cellular nanotheranostics for cancer, it is essential to avoid MSCs becoming pro-carcinogenic. For example, for the same breast cancer cell line MDA-MB-231, injected MSCs have been reported to either promote tumor metastasis [62, 160] or inhibit primary tumor progression to metastasis [161]. Several studies which claimed tumorigenic effects of MSCs in vitro and in vivo have been retracted due to cross-contamination with cancer cell lines, making a conclusive evaluation difficult [162]. MSCs have been reported to inhibit cancer progression in glioblastoma [158], leukemia [163], and hepatoma [159] murine models. Thus, further investigations are needed to determine the clinical efficacy and safety of MSC-mediated nanotheranostic delivery for cancer therapy.
Finally, MSC-derived extracellular vesicles (EVs) may be a potential alternative to using parental MSCs as nanotheranostic delivery agents by incorporating imaging agents [164]. It is not yet clear if such an approach would eliminate the risks associated with intact MSC delivery, as related to MSCs promoting tumor progression [66]. However, a key drawback of using EVs instead include the difficulty of loading them and the lack of standardized production and purification methods [165].
Conclusions
MSC-based cellular nanotheranostics with follow-up imaging of delivery and retention may become a promising new method for treating cancer. Recent progress has demonstrated this to work well in pre-clinical cancer models for a diverse set of imaging and therapeutic modalities, including those that are clinically practiced such as MRI and chemotherapy. MRI has confirmed successful targeting of tumors when cellular nanotheranostics are employed, an objective that is difficult to achieve with traditional “naked” nanotheranostics. PAI and fluorescence imaging have also demonstrated that cellular nanotheranostics are able to deliver and retain nanotheranostic agents within tumors, enabling long-term comprehensive imaging. The enhanced delivery, distribution, and retention accomplished with cellular nanotheranostics will lead to an improved therapeutic outcome. PTT and PDT performed after MSC-mediated delivery of nanotheranostics indicate reduced tumor volumes and increased survival rates compared to administration of naked nanotheranostics. Chemotherapy and gene therapy can be used to increase survival rates when combined with cellular nanotheranostics.
Furthermore, MSC-based cellular nanotheranostics may target hypoxic tumors that are otherwise resistant to chemotherapy, RT, and immunotherapy. However, phase I clinical trials have indicated that a key challenge for MSC-mediated delivery may be an insufficient accumulation of MSCs within tumors post-i.v. administration. When delivered via an intraperitoneal catheter, MSCs successfully targeted ovarian tumors [166], but when administered systematically, which is clinically preferred, MSCs did not reach primary prostate tumors [167]. To achieve the full potential of MSC-based cellular nanotheranostics, it is imperative for future studies to elucidate the relative significance of cytokines, chemokines, and growth factors implicated in MSC tumor-tropism in inducing signaling pathways associated with cell migration. Additionally, a comprehensive understanding is needed of the temporal coordination of the signaling pathways involved in MSC tumor-tropism. Once the mechanism of MSC tumor-tropism is much better understood, MSCs may be further genetically modified to enhance tumor targeting, and thus their efficacy as delivery agents of nanotheranostics.
Acknowledgements
The graphical abstract and Figures 1 and 3 were created with BioRender software (https://biorender.com).
Funding
The authors are supported by grants from the Maryland Stem Cell Research Fund (MSCRFD-5416 and MSCRFD-6135) and the National Institutes of Health (P41 EB024495, R01 EB030376, R01 CA257557, and UH3 EB028904).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Duan H, Iagaru A, Aparici CM. Radiotheranostics - Precision Medicine in Nuclear Medicine and Molecular Imaging. Nanotheranostics. 2022;6:103-17
2. Ballinger JR. Theranostic radiopharmaceuticals: established agents in current use. Br J Radiol. 2018;91:20170969
3. Chen X, Wong S. Cancer Theranostics: Elsevier Science; 2014
4. Wong XY, Sena-Torralba A, Álvarez-Diduk R, Muthoosamy K, Merkoçi A. Nanomaterials for Nanotheranostics: Tuning Their Properties According to Disease Needs. ACS Nano. 2020;14:2585-627
5. Xie J, Lee S, Chen X. Nanoparticle-based theranostic agents. Adv Drug Deliv Rev. 2010;62:1064-79
6. Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2013;4:81-9
7. Nakamura Y, Mochida A, Choyke PL, Kobayashi H. Nanodrug Delivery: Is the Enhanced Permeability and Retention Effect Sufficient for Curing Cancer? Bioconjug Chem. 2016;27:2225-38
8. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nature Nanotechnology. 2007;2:751-60
9. Sindhwani S, Syed AM, Ngai J, Kingston BR, Maiorino L, Rothschild J. et al. The entry of nanoparticles into solid tumours. Nature Materials. 2020;19:566-75
10. Meng H, Leong W, Leong KW, Chen C, Zhao Y. Walking the line: The fate of nanomaterials at biological barriers. Biomaterials. 2018;174:41-53
11. Wu J. The Enhanced Permeability and Retention (EPR) Effect: The Significance of the Concept and Methods to Enhance Its Application. J Pers Med. 2021 11
12. Danhier F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? Journal of Controlled Release. 2016;244:108-21
13. Yu M, Zheng J. Clearance Pathways and Tumor Targeting of Imaging Nanoparticles. ACS Nano. 2015;9:6655-74
14. Tian H, Zhang T, Qin S, Huang Z, Zhou L, Shi J. et al. Enhancing the therapeutic efficacy of nanoparticles for cancer treatment using versatile targeted strategies. Journal of Hematology & Oncology. 2022;15:132
15. Anselmo AC, Mitragotri S. Cell-mediated delivery of nanoparticles: taking advantage of circulatory cells to target nanoparticles. J Control Release. 2014;190:531-41
16. Ayer M, Klok HA. Cell-mediated delivery of synthetic nano- and microparticles. J Control Release. 2017;259:92-104
17. Rosenblum D, Joshi N, Tao W, Karp JM, Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nature Communications. 2018;9:1410
18. Stuckey DW, Shah K. Stem cell-based therapies for cancer treatment: separating hope from hype. Nature Reviews Cancer. 2014;14:683-91
19. Jelercic S, Rajer M. The role of PET-CT in radiotherapy planning of solid tumours. Radiol Oncol. 2015;49:1-9
20. Gomes Marin JF, Nunes RF, Coutinho AM, Zaniboni EC, Costa LB, Barbosa FG. et al. Theranostics in Nuclear Medicine: Emerging and Re-emerging Integrated Imaging and Therapies in the Era of Precision Oncology. Radiographics. 2020;40:1715-40
21. Yang RQ, Lou KL, Wang PY, Gao YY, Zhang YQ, Chen M. et al. Surgical Navigation for Malignancies Guided by Near-Infrared-II Fluorescence Imaging. Small Methods. 2021;5:e2001066
22. Liu Y, Bhattarai P, Dai Z, Chen X. Photothermal therapy and photoacoustic imaging via nanotheranostics in fighting cancer. Chem Soc Rev. 2019;48:2053-108
23. Ji B, Wei M, Yang B. Recent advances in nanomedicines for photodynamic therapy (PDT)-driven cancer immunotherapy. Theranostics. 2022;12:434-58
24. Anani T, Rahmati S, Sultana N, David AE. MRI-traceable theranostic nanoparticles for targeted cancer treatment. Theranostics. 2021;11:579-601
25. Zaimy MA, Saffarzadeh N, Mohammadi A, Pourghadamyari H, Izadi P, Sarli A. et al. New methods in the diagnosis of cancer and gene therapy of cancer based on nanoparticles. Cancer Gene Ther. 2017;24:233-43
26. Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov. 2021;20:101-24
27. Li W, Chen X. Gold nanoparticles for photoacoustic imaging. Nanomedicine (Lond). 2015;10:299-320
28. Vines JB, Yoon JH, Ryu NE, Lim DJ, Park H. Gold Nanoparticles for Photothermal Cancer Therapy. Front Chem. 2019;7:167
29. Li Z, Lopez-Ortega A, Aranda-Ramos A, Tajada JL, Sort J, Nogues C. et al. Simultaneous Local Heating/Thermometry Based on Plasmonic Magnetochromic Nanoheaters. Small. 2018;14:e1800868
30. Bulte JW, Kraitchman DL. Iron oxide MR contrast agents for molecular and cellular imaging. NMR Biomed. 2004;17:484-99
31. Bulte JWM. Superparamagnetic iron oxides as MPI tracers: A primer and review of early applications. Adv Drug Deliv Rev. 2019;138:293-301
32. Liu Y, Jia Q, Guo Q, Wei W, Zhou J. Simultaneously activating highly selective ratiometric MRI and synergistic therapy in response to intratumoral oxidability and acidity. Biomaterials. 2018;180:104-16
33. Blanco-Andujar C, Walter A, Cotin G, Bordeianu C, Mertz D, Felder-Flesch D. et al. Design of iron oxide-based nanoparticles for MRI and magnetic hyperthermia. Nanomedicine (Lond). 2016;11:1889-910
34. Healy S, Bakuzis AF, Goodwill PW, Attaluri A, Bulte JWM, Ivkov R. Clinical magnetic hyperthermia requires integrated magnetic particle imaging. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2022;14:e1779
35. Matea CT, Mocan T, Tabaran F, Pop T, Mosteanu O, Puia C. et al. Quantum dots in imaging, drug delivery and sensor applications. Int J Nanomedicine. 2017;12:5421-31
36. Lan T, Luo M, Wei X. Mesenchymal stem/stromal cells in cancer therapy. J Hematol Oncol. 2021;14:195
37. Lv FJ, Tuan RS, Cheung KM, Leung VY. Concise review: the surface markers and identity of human mesenchymal stem cells. Stem Cells. 2014;32:1408-19
38. Neofytou E, O'Brien CG, Couture LA, Wu JC. Hurdles to clinical translation of human induced pluripotent stem cells. The Journal of Clinical Investigation. 2015;125:2551-7
39. Bagó JR, Sheets KT, Hingtgen SD. Neural stem cell therapy for cancer. Methods. 2016;99:37-43
40. Uchibori R, Tsukahara T, Mizuguchi H, Saga Y, Urabe M, Mizukami H. et al. NF-κB Activity Regulates Mesenchymal Stem Cell Accumulation at Tumor Sites. Cancer Research. 2013;73:364-72
41. Teo GS, Ankrum JA, Martinelli R, Boetto SE, Simms K, Sciuto TE. et al. Mesenchymal stem cells transmigrate between and directly through tumor necrosis factor-α-activated endothelial cells via both leukocyte-like and novel mechanisms. Stem Cells. 2012;30:2472-86
42. Guan SP, Lam ATL, Newman JP, Chua KLM, Kok CYL, Chong ST. et al. Matrix metalloproteinase-1 facilitates MSC migration via cleavage of IGF-2/IGFBP2 complex. FEBS Open Bio. 2018;8:15-26
43. Ho IA, Chan KY, Ng WH, Guo CM, Hui KM, Cheang P. et al. Matrix metalloproteinase 1 is necessary for the migration of human bone marrow-derived mesenchymal stem cells toward human glioma. Stem Cells. 2009;27:1366-75
44. Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F. et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. 2011;71:614-24
45. Rattigan Y, Hsu JM, Mishra PJ, Glod J, Banerjee D. Interleukin 6 mediated recruitment of mesenchymal stem cells to the hypoxic tumor milieu. Exp Cell Res. 2010;316:3417-24
46. Kim SM, Kim D-S, Jeong CH, Kim DH, Kim JH, Jeon HB. et al. CXC chemokine receptor 1 enhances the ability of human umbilical cord blood-derived mesenchymal stem cells to migrate toward gliomas. Biochemical and Biophysical Research Communications. 2011;407:741-6
47. Ma X, Chen J, Liu J, Xu B, Liang X, Yang X. et al. IL-8/CXCR2 mediates tropism of human bone marrow-derived mesenchymal stem cells toward CD133+/CD44+ Colon cancer stem cells. Journal of Cellular Physiology. 2021;236:3114-28
48. Liang-kuan B, Nan Z, Cheng L, Fu-Ding L, Tian-Xin L, Xu-Jun X. et al. Kidney cancer cells secrete IL-8 to activate Akt and promote migration of mesenchymal stem cells. Urologic Oncology: Seminars and Original Investigations. 2014;32:607-12
49. Lin G, Yang R, Banie L, Wang G, Ning H, Li LC. et al. Effects of transplantation of adipose tissue-derived stem cells on prostate tumor. Prostate. 2010;70:1066-73
50. Gao H, Priebe W, Glod J, Banerjee D. Activation of signal transducers and activators of transcription 3 and focal adhesion kinase by stromal cell-derived factor 1 is required for migration of human mesenchymal stem cells in response to tumor cell-conditioned medium. Stem Cells. 2009;27:857-65
51. Lourenco S, Teixeira VH, Kalber T, Jose RJ, Floto RA, Janes SM. Macrophage migration inhibitory factor-CXCR4 is the dominant chemotactic axis in human mesenchymal stem cell recruitment to tumors. J Immunol. 2015;194:3463-74
52. Senst C, Nazari-Shafti T, Kruger S, Höner Zu Bentrup K, Dupin CL, Chaffin AE. et al. Prospective dual role of mesenchymal stem cells in breast tumor microenvironment. Breast Cancer Res Treat. 2013;137:69-79
53. Kasashima H, Yashiro M, Nakamae H, Kitayama K, Masuda G, Kinoshita H. et al. CXCL1-Chemokine (C-X-C Motif) Receptor 2 Signaling Stimulates the Recruitment of Bone Marrow-Derived Mesenchymal Cells into Diffuse-Type Gastric Cancer Stroma. The American Journal of Pathology. 2016;186:3028-39
54. Korbecki J, Kupnicka P, Chlubek M, Gorący J, Gutowska I, Baranowska-Bosiacka I. CXCR2 Receptor: Regulation of Expression, Signal Transduction, and Involvement in Cancer. Int J Mol Sci. 2022 23
55. Dwyer RM, Potter-Beirne SM, Harrington KA, Lowery AJ, Hennessy E, Murphy JM. et al. Monocyte Chemotactic Protein-1 Secreted by Primary Breast Tumors Stimulates Migration of Mesenchymal Stem Cells. Clinical Cancer Research. 2007;13:5020-7
56. Shinojima N, Hossain A, Takezaki T, Fueyo J, Gumin J, Gao F. et al. TGF-β mediates homing of bone marrow-derived human mesenchymal stem cells to glioma stem cells. Cancer Res. 2013;73:2333-44
57. Li M, Zeng L, Liu S, Dangelmajer S, Kahlert UD, Huang H. et al. Transforming Growth Factor-β Promotes Homing and Therapeutic Efficacy of Human Mesenchymal Stem Cells to Glioblastoma. Journal of Neuropathology & Experimental Neurology. 2019;78:315-25
58. Watts TL, Cui R, Szaniszlo P, Resto VA, Powell DW, Pinchuk IV. PDGF-AA mediates mesenchymal stromal cell chemotaxis to the head and neck squamous cell carcinoma tumor microenvironment. J Transl Med. 2016;14:337
59. Salha S, Gehmert S, Brébant V, Anker A, Loibl M, Prantl L. et al. PDGF regulated migration of mesenchymal stem cells towards malignancy acts via the PI3K signaling pathway. Clin Hemorheol Microcirc. 2018;70:543-51
60. Jung Y, Kim JK, Shiozawa Y, Wang J, Mishra A, Joseph J. et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun. 2013;4:1795
61. Chaturvedi P, Gilkes DM, Takano N, Semenza GL. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proceedings of the National Academy of Sciences. 2014;111:E2120-E9
62. Chaturvedi P, Gilkes DM, Wong CC, Luo W, Zhang H, Wei H. et al. Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signaling promotes metastasis. J Clin Invest. 2013;123:189-205
63. Kopecka J, Salaroglio IC, Perez-Ruiz E, Sarmento-Ribeiro AB, Saponara S, De Las Rivas J. et al. Hypoxia as a driver of resistance to immunotherapy. Drug Resist Updat. 2021;59:100787
64. Chen Z, Han F, Du Y, Shi H, Zhou W. Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Signal Transduction and Targeted Therapy. 2023;8:70
65. Singh A, Jain S, Senapati S, Verma RS, Sahoo SK. Magnetic nanoparticles labeled mesenchymal stem cells: a pragmatic solution toward targeted cancer theranostics. Advanced Healthcare Materials. 2015;4:2078-89
66. Wu H-H, Zhou Y, Tabata Y, Gao J-Q. Mesenchymal stem cell-based drug delivery strategy: from cells to biomimetic. Journal of Controlled Release. 2019;294:102-13
67. Kang S, Bhang SH, Hwang S, Yoon JK, Song J, Jang HK. et al. Mesenchymal Stem Cells Aggregate and Deliver Gold Nanoparticles to Tumors for Photothermal Therapy. ACS Nano. 2015;9:9678-90
68. Mooney R, Roma L, Zhao D, Van Haute D, Garcia E, Kim SU. et al. Neural stem cell-mediated intratumoral delivery of gold nanorods improves photothermal therapy. ACS Nano. 2014;8:12450-60
69. Xu C, Feng Q, Yang H, Wang G, Huang L, Bai Q. et al. A Light-Triggered Mesenchymal Stem Cell Delivery System for Photoacoustic Imaging and Chemo-Photothermal Therapy of Triple Negative Breast Cancer. Adv Sci (Weinh). 2018;5:1800382
70. Huang L, Xu C, Xu P, Qin Y, Chen M, Feng Q. et al. Intelligent photosensitive mesenchymal stem cells and cell-derived microvesicles for photothermal therapy of prostate cancer. Nanotheranostics. 2019;3:41
71. Layek B, Sadhukha T, Prabha S. Glycoengineered mesenchymal stem cells as an enabling platform for two-step targeting of solid tumors. Biomaterials. 2016;88:97-109
72. Kim KS, Park W, Na K. Gadolinium-chelate nanoparticle entrapped human mesenchymal stem cell via photochemical internalization for cancer diagnosis. Biomaterials. 2015;36:90-7
73. Singh A, Jain S, Senapati S, Verma RS, Sahoo SK. Magnetic Nanoparticles Labeled Mesenchymal Stem Cells: A Pragmatic Solution toward Targeted Cancer Theranostics. Adv Healthc Mater. 2015;4:2078-89
74. Huang X, Zhang F, Wang Y, Sun X, Choi KY, Liu D. et al. Design considerations of iron-based nanoclusters for noninvasive tracking of mesenchymal stem cell homing. ACS Nano. 2014;8:4403-14
75. Zhao Z, Li M, Zeng J, Huo L, Liu K, Wei R. et al. Recent advances in engineering iron oxide nanoparticles for effective magnetic resonance imaging. Bioact Mater. 2022;12:214-45
76. Lemaster JE, Jokerst JV. What is new in nanoparticle-based photoacoustic imaging? Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017 9
77. Hesemans E, Buttiens K, Manshian BB, Soenen SJ. The Role of Optical Imaging in Translational Nanomedicine. J Funct Biomater. 2022 13
78. Weissleder R. Liver MR imaging with iron oxides: toward consensus and clinical practice. Radiology. 1994;193:593-5
79. Kim D, Kim J, Park YI, Lee N, Hyeon T. Recent Development of Inorganic Nanoparticles for Biomedical Imaging. ACS Cent Sci. 2018;4:324-36
80. Lee N, Hyeon T. Designed synthesis of uniformly sized iron oxide nanoparticles for efficient magnetic resonance imaging contrast agents. Chem Soc Rev. 2012;41:2575-89
81. Karussis D, Karageorgiou C, Vaknin-Dembinsky A, Gowda-Kurkalli B, Gomori JM, Kassis I. et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Archives of neurology. 2010;67:1187-94
82. Gerosa C, Crisponi G, Nurchi VM, Saba L, Cappai R, Cau F. et al. Gold Nanoparticles: A New Golden Era in Oncology? Pharmaceuticals (Basel). 2020 13
83. Longmire M, Choyke PL, Kobayashi H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine (Lond). 2008;3:703-17
84. Luke GP, Myers JN, Emelianov SY, Sokolov KV. Sentinel lymph node biopsy revisited: ultrasound-guided photoacoustic detection of micrometastases using molecularly targeted plasmonic nanosensors. Cancer Res. 2014;74:5397-408
85. Han S, Bouchard R, Sokolov KV. Molecular photoacoustic imaging with ultra-small gold nanoparticles. Biomed Opt Express. 2019;10:3472-83
86. Li K, Liu B. Polymer-encapsulated organic nanoparticles for fluorescence and photoacoustic imaging. Chem Soc Rev. 2014;43:6570-97
87. Wolfbeis OS. An overview of nanoparticles commonly used in fluorescent bioimaging. Chemical Society Reviews. 2015;44:4743-68
88. Kalber TL, Ordidge KL, Southern P, Loebinger MR, Kyrtatos PG, Pankhurst QA. et al. Hyperthermia treatment of tumors by mesenchymal stem cell-delivered superparamagnetic iron oxide nanoparticles. Int J Nanomedicine. 2016;11:1973-83
89. Huang RY, Lin YH, Lin SY, Li YN, Chiang CS, Chang CW. Magnetic ternary nanohybrids for nonviral gene delivery of stem cells and applications on cancer therapy. Theranostics. 2019;9:2411-23
90. Lai Y-H, Su C-Y, Cheng H-W, Chu C-Y, Jeng L-B, Chiang C-S. et al. Stem cell-nanomedicine system as a theranostic bio-gadolinium agent for targeted neutron capture cancer therapy. Nature Communications. 2023;14:285
91. Chetty SS, Praneetha S, Vadivel Murugan A, Govarthanan K, Verma RS. Human Umbilical Cord Wharton's Jelly-Derived Mesenchymal Stem Cells Labeled with Mn(2+) and Gd(3+) Co-Doped CuInS(2)-ZnS Nanocrystals for Multimodality Imaging in a Tumor Mice Model. ACS Appl Mater Interfaces. 2020;12:3415-29
92. Huang X, Zhang F, Wang H, Niu G, Choi KY, Swierczewska M. et al. Mesenchymal stem cell-based cell engineering with multifunctional mesoporous silica nanoparticles for tumor delivery. Biomaterials. 2013;34:1772-80
93. Qiao Y, Gumin J, MacLellan CJ, Gao F, Bouchard R, Lang FF. et al. Magnetic resonance and photoacoustic imaging of brain tumor mediated by mesenchymal stem cell labeled with multifunctional nanoparticle introduced via carotid artery injection. Nanotechnology. 2018;29:165101
94. Yudintceva N, Lomert E, Mikhailova N, Tolkunova E, Agadzhanian N, Samochernych K. et al. Targeting Brain Tumors with Mesenchymal Stem Cells in the Experimental Model of the Orthotopic Glioblastoma in Rats. Biomedicines. 2021 9
95. Hsu FT, Wei ZH, Hsuan YC, Lin W, Su YC, Liao CH. et al. MRI tracking of polyethylene glycol-coated superparamagnetic iron oxide-labelled placenta-derived mesenchymal stem cells toward glioblastoma stem-like cells in a mouse model. Artif Cells Nanomed Biotechnol. 2018;46:S448-s59
96. Hao X, Xu B, Chen H, Wang X, Zhang J, Guo R. et al. Stem cell-mediated delivery of nanogels loaded with ultrasmall iron oxide nanoparticles for enhanced tumor MR imaging. Nanoscale. 2019;11:4904-10
97. Zhang YS, Wang Y, Wang L, Wang Y, Cai X, Zhang C. et al. Labeling human mesenchymal stem cells with gold nanocages for in vitro and in vivo tracking by two-photon microscopy and photoacoustic microscopy. Theranostics. 2013;3:532
98. Wu J, Liu Y, Tang Y, Wang S, Wang C, Li Y. et al. Synergistic Chemo-Photothermal Therapy of Breast Cancer by Mesenchymal Stem Cell-Encapsulated Yolk-Shell GNR@HPMO-PTX Nanospheres. ACS Appl Mater Interfaces. 2016;8:17927-35
99. Fan M, Han Y, Gao S, Yan H, Cao L, Li Z. et al. Ultrasmall gold nanoparticles in cancer diagnosis and therapy. Theranostics. 2020;10:4944-57
100. Cao W, Liu B, Xia F, Duan M, Hong Y, Niu J. et al. MnO(2)@Ce6-loaded mesenchymal stem cells as an "oxygen-laden guided-missile" for the enhanced photodynamic therapy on lung cancer. Nanoscale. 2020;12:3090-102
101. Dapkute D, Pleckaitis M, Bulotiene D, Daunoravicius D, Rotomskis R, Karabanovas V. Hitchhiking Nanoparticles: Mesenchymal Stem Cell-Mediated Delivery of Theranostic Nanoparticles. ACS Appl Mater Interfaces. 2021;13:43937-51
102. Näkki S, Martinez JO, Evangelopoulos M, Xu W, Lehto VP, Tasciotti E. Chlorin e6 Functionalized Theranostic Multistage Nanovectors Transported by Stem Cells for Effective Photodynamic Therapy. ACS Appl Mater Interfaces. 2017;9:23441-9
103. Wu S-Q, Yang C-X, Yan X-P. A Dual-Functional Persistently Luminescent Nanocomposite Enables Engineering of Mesenchymal Stem Cells for Homing and Gene Therapy of Glioblastoma. Advanced Functional Materials. 2017;27:1604992
104. Xiao J, Zeng L, Ding S, Chen Y, Zhang X, Bian XW. et al. Tumor-Tropic Adipose-Derived Mesenchymal Stromal Cell Mediated Bi(2) Se(3) Nano-Radiosensitizers Delivery for Targeted Radiotherapy of Non-Small Cell Lung Cancer. Adv Healthc Mater. 2022;11:e2200143
105. Bulte JW, Walczak P, Gleich B, Weizenecker J, Markov DE, Aerts HC. et al. MPI Cell Tracking: What Can We Learn from MRI? Proc SPIE Int Soc Opt Eng. 2011;7965:79650z
106. Bulte JW, Walczak P, Janowski M, Krishnan KM, Arami H, Halkola A. et al. Quantitative "Hot Spot" Imaging of Transplanted Stem Cells using Superparamagnetic Tracers and Magnetic Particle Imaging (MPI). Tomography. 2015;1:91-7
107. Kubelick KP, Mehrmohammadi M. Magnetic particles in motion: magneto-motive imaging and sensing. Theranostics. 2022;12:1783-99
108. Layek B, Sadhukha T, Panyam J, Prabha S. Nano-Engineered Mesenchymal Stem Cells Increase Therapeutic Efficacy of Anticancer Drug Through True Active Tumor Targeting. Mol Cancer Ther. 2018;17:1196-206
109. Li X, Lovell JF, Yoon J, Chen X. Clinical development and potential of photothermal and photodynamic therapies for cancer. Nat Rev Clin Oncol. 2020;17:657-74
110. Hou YJ, Yang XX, Liu RQ, Zhao D, Guo CX, Zhu AC. et al. Pathological Mechanism of Photodynamic Therapy and Photothermal Therapy Based on Nanoparticles. Int J Nanomedicine. 2020;15:6827-38
111. Lee MS, Dees EC, Wang AZ. Nanoparticle-Delivered Chemotherapy: Old Drugs in New Packages. Oncology (Williston Park). 2017;31:198-208
112. Amreddy N, Babu A, Muralidharan R, Panneerselvam J, Srivastava A, Ahmed R. et al. Recent Advances in Nanoparticle-Based Cancer Drug and Gene Delivery. Adv Cancer Res. 2018;137:115-70
113. Mainini F, Eccles MR. Lipid and Polymer-Based Nanoparticle siRNA Delivery Systems for Cancer Therapy. Molecules. 2020 25
114. Cullis PR, Hope MJ. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol Ther. 2017;25:1467-75
115. Huang WC, Lu IL, Chiang WH, Lin YW, Tsai YC, Chen HH. et al. Tumortropic adipose-derived stem cells carrying smart nanotherapeutics for targeted delivery and dual-modality therapy of orthotopic glioblastoma. J Control Release. 2017;254:119-30
116. Li A, Zhang T, Huang T, Lin R, Mu J, Su Y. et al. Iron oxide nanoparticles promote Cx43-overexpression of mesenchymal stem cells for efficient suicide gene therapy during glioma treatment. Theranostics. 2021;11:8254
117. Yin PT, Shah S, Pasquale NJ, Garbuzenko OB, Minko T, Lee KB. Stem cell-based gene therapy activated using magnetic hyperthermia to enhance the treatment of cancer. Biomaterials. 2016;81:46-57
118. Zhong H-h, Wang H-y, Li J, Huang Y-z. TRAIL-based gene delivery and therapeuticstrategies. Acta Pharmacologica Sinica. 2019;40:1373-85
119. Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA. et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155-62
120. Studeny M, Marini FC, Champlin RE, Zompetta C, Fidler IJ, Andreeff M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62:3603-8
121. Gao P, Ding Q, Wu Z, Jiang H, Fang Z. Therapeutic potential of human mesenchymal stem cells producing IL-12 in a mouse xenograft model of renal cell carcinoma. Cancer Lett. 2010;290:157-66
122. Seo SH, Kim KS, Park SH, Suh YS, Kim SJ, Jeun SS. et al. The effects of mesenchymal stem cells injected via different routes on modified IL-12-mediated antitumor activity. Gene Ther. 2011;18:488-95
123. van Eekelen M, Sasportas LS, Kasmieh R, Yip S, Figueiredo JL, Louis DN. et al. Human stem cells expressing novel TSP-1 variant have anti-angiogenic effect on brain tumors. Oncogene. 2010;29:3185-95
124. Cavarretta IT, Altanerova V, Matuskova M, Kucerova L, Culig Z, Altaner C. Adipose tissue-derived mesenchymal stem cells expressing prodrug-converting enzyme inhibit human prostate tumor growth. Mol Ther. 2010;18:223-31
125. Kucerova L, Altanerova V, Matuskova M, Tyciakova S, Altaner C. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res. 2007;67:6304-13
126. Kucerova L, Matuskova M, Pastorakova A, Tyciakova S, Jakubikova J, Bohovic R. et al. Cytosine deaminase expressing human mesenchymal stem cells mediated tumour regression in melanoma bearing mice. J Gene Med. 2008;10:1071-82
127. Brown N, Rocchi P, Carmès L, Guthier R, Iyer M, Seban L. et al. Tuning ultrasmall theranostic nanoparticles for MRI contrast and radiation dose amplification. Theranostics. 2023;13:4711
128. Pullambhatla M, Rowe SP, Lisok A, Wang Y, Todd GP, Danilkovitch A. et al. Enhancement of radiotherapy with human mesenchymal stem cells containing gold nanoparticles. Tomography. 2020;6:373-8
129. Xiao J, Zeng L, Ding S, Chen Y, Zhang X, Bian Xw. et al. Tumor-tropic adipose-derived mesenchymal stromal cell mediated Bi2Se3 nano-radiosensitizers delivery for targeted radiotherapy of non-small cell lung cancer. Advanced Healthcare Materials. 2022;11:2200143
130. Li Y-R, Zhou Y, Kramer A, Yang L. Engineering stem cells for cancer immunotherapy. Trends in Cancer. 2021;7:1059-73
131. Zhang H, Feng Y, Xie X, Song T, Yang G, Su Q. et al. Engineered Mesenchymal Stem Cells as a Biotherapy Platform for Targeted Photodynamic Immunotherapy of Breast Cancer. Adv Healthc Mater. 2022;11:e2101375
132. Chen Q, Chen J, Yang Z, Xu J, Xu L, Liang C. et al. Nanoparticle-Enhanced Radiotherapy to Trigger Robust Cancer Immunotherapy. Adv Mater. 2019;31:e1802228
133. Guan X, Sun L, Shen Y, Jin F, Bo X, Zhu C. et al. Nanoparticle-enhanced radiotherapy synergizes with PD-L1 blockade to limit post-surgical cancer recurrence and metastasis. Nat Commun. 2022;13:2834
134. Frickhofen N, Berdel W, Opri F, Haas R, Schneeweiss A, Sandherr M. et al. Phase I/II trial of multicycle high-dose chemotherapy with peripheral blood stem cell support for treatment of advanced ovarian cancer. Bone marrow transplantation. 2006;38:493-9
135. Nieto Y, Shpall EJ, McNiece IK, Nawaz S, Beaudet J, Rosinski S. et al. Prognostic analysis of early lymphocyte recovery in patients with advanced breast cancer receiving high-dose chemotherapy with an autologous hematopoietic progenitor cell transplant. Clinical Cancer Research. 2004;10:5076-86
136. Nabhan C, Mehta J, Tallman MS. The role of bone marrow transplantation in acute promyelocytic leukemia. Bone Marrow Transplantation. 2001;28:219-26
137. Yu C, Cai B, Wang Y, Wu Z, Hu K, Sun Q. et al. Co-infusion of high-dose haploidentical donor cells and CD19-targeted CART cells achieves complete remission, successful donor engraftment and significant CART amplification in advanced ALL. Therapeutic Advances in Medical Oncology. 2020;12:1758835920927605
138. Zhang C, Kong P-Y, Li S, Chen T, Ni X, Li Y. et al. Donor-derived CAR-T cells serve as a reduced-intensity conditioning regimen for haploidentical stem cell transplantation in treatment of relapsed/refractory acute lymphoblastic leukemia: case report and review of the literature. Journal of Immunotherapy. 2018;41:306-11
139. Cheng TM, Chang WJ, Chu HY, De Luca R, Pedersen JZ, Incerpi S. et al. Nano-Strategies Targeting the Integrin αvβ3 Network for Cancer Therapy. Cells. 2021 10
140. Mehrotra N, Kharbanda S, Singh H. Peptide-based combination nanoformulations for cancer therapy. Nanomedicine (Lond). 2020;15:2201-17
141. Liu T, Zhu K, Ke C, Yang S, Yang F, Li Z. et al. Mesenchymal stem cells inhibited development of lung cancer induced by chemical carcinogens in a rat model. Am J Transl Res. 2017;9:2891-900
142. Yulyana Y, Ho IA, Sia KC, Newman JP, Toh XY, Endaya BB. et al. Paracrine factors of human fetal MSCs inhibit liver cancer growth through reduced activation of IGF-1R/PI3K/Akt signaling. Mol Ther. 2015;23:746-56
143. Kidd S, Caldwell L, Dietrich M, Samudio I, Spaeth EL, Watson K. et al. Mesenchymal stromal cells alone or expressing interferon-beta suppress pancreatic tumors in vivo, an effect countered by anti-inflammatory treatment. Cytotherapy. 2010;12:615-25
144. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nature Reviews Clinical Oncology. 2018;15:81-94
145. Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang W. et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci U S A. 2000;97:12846-51
146. Rustad KC, Gurtner GC. Mesenchymal Stem Cells Home to Sites of Injury and Inflammation. Adv Wound Care (New Rochelle). 2012;1:147-52
147. Devine SM, Cobbs C, Jennings M, Bartholomew A, Hoffman R. Mesenchymal stem cells distribute to a wide range of tissues following systemic infusion into nonhuman primates. Blood. 2003;101:2999-3001
148. Kim DS, Kim JH, Lee JK, Choi SJ, Kim JS, Jeun SS. et al. Overexpression of CXC chemokine receptors is required for the superior glioma-tracking property of umbilical cord blood-derived mesenchymal stem cells. Stem Cells Dev. 2009;18:511-9
149. Park SA, Ryu CH, Kim SM, Lim JY, Park SI, Jeong CH. et al. CXCR4-transfected human umbilical cord blood-derived mesenchymal stem cells exhibit enhanced migratory capacity toward gliomas. Int J Oncol. 2011;38:97-103
150. Honeth G, Staflin K, Kalliomäki S, Lindvall M, Kjellman C. Chemokine-directed migration of tumor-inhibitory neural progenitor cells towards an intracranially growing glioma. Exp Cell Res. 2006;312:1265-76
151. Gorelik M, Orukari I, Wang J, Galpoththawela S, Kim H, Levy M. et al. Use of MR cell tracking to evaluate targeting of glial precursor cells to inflammatory tissue by exploiting the very late antigen-4 docking receptor. Radiology. 2012;265:175-85
152. Jablonska A, Shea DJ, Cao S, Bulte JW, Janowski M, Konstantopoulos K. et al. Overexpression of VLA-4 in glial-restricted precursors enhances their endothelial docking and induces diapedesis in a mouse stroke model. J Cereb Blood Flow Metab. 2018;38:835-46
153. Zielske SP, Livant DL, Lawrence TS. Radiation increases invasion of gene-modified mesenchymal stem cells into tumors. Int J Radiat Oncol Biol Phys. 2009;75:843-53
154. Klopp AH, Spaeth EL, Dembinski JL, Woodward WA, Munshi A, Meyn RE. et al. Tumor irradiation increases the recruitment of circulating mesenchymal stem cells into the tumor microenvironment. Cancer Res. 2007;67:11687-95
155. Schug C, Kitzberger C, Sievert W, Spellerberg R, Tutter M, Schmohl KA. et al. Radiation-Induced Amplification of TGFB1-Induced Mesenchymal Stem Cell-Mediated Sodium Iodide Symporter (NIS) Gene (131)I Therapy. Clin Cancer Res. 2019;25:5997-6008
156. Walter M, Liang S, Ghosh S, Hornsby PJ, Li R. Interleukin 6 secreted from adipose stromal cells promotes migration and invasion of breast cancer cells. Oncogene. 2009;28:2745-55
157. Zhang T, Lee YW, Rui YF, Cheng TY, Jiang XH, Li G. Bone marrow-derived mesenchymal stem cells promote growth and angiogenesis of breast and prostate tumors. Stem Cell Res Ther. 2013;4:70
158. Ho IA, Toh HC, Ng WH, Teo YL, Guo CM, Hui KM. et al. Human bone marrow-derived mesenchymal stem cells suppress human glioma growth through inhibition of angiogenesis. Stem Cells. 2013;31:146-55
159. Qiao L, Xu Z, Zhao T, Zhao Z, Shi M, Zhao RC. et al. Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res. 2008;18:500-7
160. El-Haibi CP, Bell GW, Zhang J, Collmann AY, Wood D, Scherber CM. et al. Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy. Proc Natl Acad Sci U S A. 2012;109:17460-5
161. Meleshina AV, Cherkasova EI, Shirmanova MV, Klementieva NV, Kiseleva EV, Snopova L. et al. Influence of mesenchymal stem cells on metastasis development in mice in vivo. Stem Cell Res Ther. 2015;6:15
162. Rühle A, Huber PE, Saffrich R, Lopez Perez R, Nicolay NH. The current understanding of mesenchymal stem cells as potential attenuators of chemotherapy-induced toxicity. International Journal of Cancer. 2018;143:2628-39
163. Zhu Y, Sun Z, Han Q, Liao L, Wang J, Bian C. et al. Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK-1. Leukemia. 2009;23:925-33
164. Arifin DR, Witwer KW, Bulte JW. Non-Invasive imaging of extracellular vesicles: Quo vaditis in vivo? Journal of Extracellular Vesicles. 2022;11:12241
165. Meng W, He C, Hao Y, Wang L, Li L, Zhu G. Prospects and challenges of extracellular vesicle-based drug delivery system: considering cell source. Drug Deliv. 2020;27:585-98
166. Olson A, Marini F, Westin S, Coleman R, Thall P, Al Jhadhami V. et al. A Phase I Trial of Mesenchymal Stem Cells Transfected with a Plasmid Secreting Interferon Beta in Advanced Ovarian Cancer. Biology of Blood and Marrow Transplantation. 2018;24:S473
167. Schweizer MT, Wang H, Bivalacqua TJ, Partin AW, Lim SJ, Chapman C. et al. A Phase I Study to Assess the Safety and Cancer-Homing Ability of Allogeneic Bone Marrow-Derived Mesenchymal Stem Cells in Men with Localized Prostate Cancer. Stem Cells Transl Med. 2019;8:441-9
Author contact
![]() Corresponding author: ashaker3edu.
Corresponding author: ashaker3edu.