Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Experimental
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2013; 3(9):677-686. doi:10.7150/thno.6535 This issue Cite
Research Paper
Surface Passivation of Carbon Nanoparticles with Branched Macromolecules Influences Near Infrared Bioimaging
Lina Wu 1, 2*, Micah Luderer 1*, Xiaoxia Yang 1, Corban Swain 1, Huiying Zhang 1, Kate Nelson 3, Allen J. Stacy 1, Baozhong Shen 2 ![]() , Gregory M. Lanza1, Dipanjan Pan1
, Gregory M. Lanza1, Dipanjan Pan1 ![]()
1. Division of Cardiology, Washington University School of Medicine, 4320 Forest Park Avenue, Saint Louis, MO 63108, USA.
2. Key Laboratory of Molecular Imaging in College of Heilongjiang Province, Department of Radiology, the 4th Affiliated Hospital of Harbin Medical University, Harbin, 150001, China.
3. Nano Research Facility (NNIN-NSF), Washington University, St. Louis, MO 63130, USA.
* Authors equal contribution
Received 2013-4-22; Accepted 2013-5-31; Published 2013-8-14
Abstract

A superior and commercially exploitable 'green synthesis' of optically active carbon nanoparticle (OCN) is revealed in this work. The naked carbon particles (<20 nm) were derived from commercial food grade honey. The fluorescence properties of these particles were significantly enhanced by utilizing hyberbranched polymer for surface passivation. A dramatic increase in near infrared emission was achieved compared to a linear polymer (PEG) coated carbon nanoparticles. Interestingly, as passivating agent becomes more extensively branched (pseudo generation 2 to 4), the average radiant efficiency amplifies considerably as a direct result of the increasing surface area available for light passivation. The particles showed negligible loss of cell viability in presence of endothelial cells in vitro. Preliminary in vivo experiment showed high contrast enhancement in auxiliary lymphnode in a mouse model. The exceptionally rapid lymphatic transport of these particles suggests that such an approach may offer greater convenience and reduced procedural expense, as well as improved surgical advantage as the patient is positioned on the table for easier resection.
Keywords: Carbon nanoparticle, hyperbranched polymer, near infrared imaging, contrast agent, surface passivation.
Introduction
Fluorescence sensing and imaging techniques use exogenous (fluorescent probes) or endogenous (reporter genes) agents excited at particular wavelengths for in vitro detection of molecules and in vivo bio-imaging [1-5]. Unfortunately, the performance and sensitivity of these laser-induced fluorescence techniques are limited by the long-standing barrier of fluorescent probes. Organic fluorochromes are easily photo-bleached during the time scale of observation, leading to reduced sensitivity and decreased tracking time of the targets. Large fluorescent tags also can perturb the labeled biomolecules, causing artificial movement within cells and changed protein interactions. Additionally, small Stokes shifts of organic fluorochrome can cause self-quenching and measurement error by excitation and scattered light [6]. Semiconductor quantum dots exhibit excellent photophysical properties, which include size-tunable narrow emissions, large Stokes shifts and minimal photo bleaching. Unfavorably, the synthesis of quantum dots involves harsh conditions and toxic precursors, which make them less prone to surface passivation and may impart severe long term toxicity. Aggregation-induced emission organic nanoparticles, such as Ln3+ doped HAp nanoparticles and polydopamine nanoparticles have been developed for cell imaging. However their relatively bigger size (~90 nm) puts a question over their future clinical translation [7-10]. Fluorescent emission from small gold nanoclusters in solution has been disclosed [11-13]. Few-atom gold nanoclusters have sizes comparable to the Fermi wavelength, resulting in molecule-like behavior including discreet electronic states and size-dependent fluorescence [14,15]. However, the optical properties of gold are highly dependent on expensive and complicated surface chemistry and their clinical use is discouraged by the unpredictable high cost.
Luminescent carbon nanoparticles have emerged as a promising approach showing high potential in biological labeling, bio-imaging and various optoelectronic device applications [16-22]. Furthermore, their inherent chemical inertness and biocompatibility make them more advantageous over conventional cadmium based quantum dots [23]. A wide range of synthetic approaches have been pursued to produce carbon nanoparticles including arc discharge, laser ablation, electrochemical oxidation, candle burning and combustion of natural gas burners [24-28]. These methods lack the suitability for commercial production and often suffer low yield, insolubility of as-synthesized particles, complex purification, and weak photo luminescence. The mechanistic insight into the origin of fluorescence from carbon nanoparticles is unclear. Our understanding on the microstructure and the organic passivation with surface ligands is inadequate to influence the design of these nanoparticles for near infrared light emission. Evidently, a more translatable approach is required, which may address production expenditure, long term stability, biosafety and offer further insight into the mechanism of surface passivation. Consequently, a superior synthetic design is an unmet need for these agents leading to improved optical properties of these particles to achieve successful in vivo imaging within the near infrared region.
Towards this aim, the present work represents a facile synthesis of optically active carbon nanoparticles (OCN or OCNs), whose surface is passivated with branched organic macromolecules to produce strong photoluminescence characteristics (Figure 1). Interestingly, the carbon nanoparticles are derived from food grade honey using a solvent-less 'green' chemistry approach. To the best of our knowledge, this is the first known examples of a fluorescent carbon probe produced from honey for in vivo near infrared (NIR) fluorescence imaging. It was further hypothesized that an extensively branched polymer could spawn a greater surface area for interaction when light passivates the particle surface. We observed that a densely packed OCN surface with hyperbranched polymer engenders superior optical properties in comparison to the linear macromolecules.
Synthesis of OCN: (a) commercial grade honey (neat), passivating agent: hyper-branched polymer or polyethyleneglycol (MW = 400 Da), microwave, isolation of particles; (b) chemical structures of the macromolecules used for surface passivation; (c) representative solubility pattern of polyethyleneglycol coated OCN; (d) table summarizing the synthetic conditions and OCN physical properties.
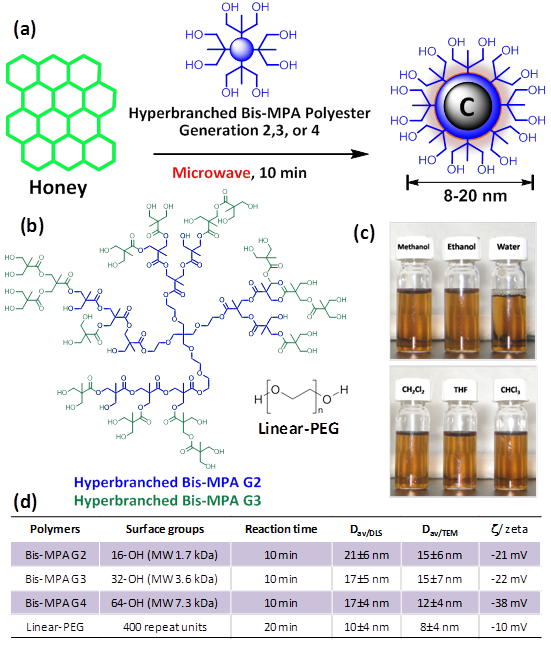
Experimental
General Experimental Procedure
Unless otherwise listed, all solvents and reagents were purchased from Aldrich Chemical Co. (St. Louis, MO) and used as received. Argon (Ultra High Purity: UHP, 99.99%) was used for storage of materials. The Spectra/Por membrane (Cellulose MWCO: 20 000 Da) used for dialysis was obtained from Spectrum Medical Industries, Inc. (Laguna Hills, CA).
Synthesis of luminescent carbon nanoparticles from honey
Commercial food grade honey (Great Value™ Clover Honey 1 wt%; batch composition- fructose: 38% glucose: 31%, maltose: 7.1%, sucrose: 1.3% higher sugars: 1.5%, water: 17.2%) is suspended with an organic macromolecular passivating agent (8wt%; G2-G4 hyperbranched bis-MPA polyester hydroxyl polymers (HBP) or PEG400), (neat or dissolved in minimum volume of solution) purged with argon and heated in a domestic microwave oven for 10 min. Microwave power was set at 1200 W with an output power: 50%. The product was visually changed from light yellow to dark brown to black. The as synthesized particles were centrifuged at 12000 g for 20 min, followed by collecting the supernatant by filtering through a 0.22 μm syringe filter.
Dynamic light scattering (DLS) measurements
Hydrodynamic diameter distribution and distribution averages for the OCN in aqueous solutions were determined by dynamic light scattering. Hydrodynamic diameters were determined using a Brookhaven Instrument Co. (Holtsville, NY) Model Zeta Plus particle size analyzer. Nanoparticles were dialyzed into water prior to analysis. Scattered light was collected at a fixed angle of 90°. A photomultiplier aperture of 400 mm was used, and the incident laser power was adjusted to obtain a photon counting rate between 200 and 300 kcps. Only measurements for which the measured and calculated baselines of the intensity autocorrelation function agreed to within +0.1% were used to calculate nanoparticle hydrodynamic diameter values. All determinations were made in multiples of five consecutive measurements.
Electrophoretic potential measurements (Zeta Potential) and measurement of isoelectric point
Zeta potential (ζ) values for the OCN were determined with a Brookhaven Instrument Co. (Holtsville, NY) model Zeta Plus zeta potential analyzer. Measurements were made following dialysis (MWCO 20 kDa dialysis tubing, Spectrum Laboratories, Rancho Dominguez, CA) of nanoparticles suspensions into water. Data were acquired in the phase analysis light scattering (PALS) mode following solution equilibration at 25 °C.
The zeta potentials of very dilute carbon NPs aqueous suspensions with different pH values were measured at 25 ºC with a zeta potential analyzer (Brookhaven Instrument Co., Holtsville, NY). HCl (0.5 M) and NaOH (0.5 M) solutions were employed as the acid-base regulator. The tests were repeated three times in order to obtain accurate results.
Raman Spectroscopy
Raman measurements were performed with a Renishaw InVia spectrometer using the 532 nm wavelength of an Ar+ laser. The instrument was calibrated using an internal silicon standard (521 cm-1). The spectrum was obtained using a dynamic scan for 5 accumulations at 1 s exposure time each at 10% laser intensity (23 mW). The Raman spectrum of pure Si was subtracted from the spectrum of collected carbon nanoparticles.
Scanning electron microscopy (SEM) imaging and Energy dispersive X-ray (EDX) analysis
The samples on Si wafer were imaged using a Nova NanoSEM 230 EDAX Genesis in high vacuum mode at 10 kV and at a working distance of 5 mm. Samples were prepared as follows: One drop of OCN suspension in nanopure water was deposited on a piece of clean Si wafer (Si wafer was rinsed and sonicated in nanopure water, ethanol and acetone respectively). After 1 min, excess liquid was removed by touching the edge of the liquid bead using a piece of filter paper. The sample was allowed to air dry for 1 hour before sputter coating, and then was sputter coated with Au-Pd for 15 seconds at 25 mA.
Fourier transform infrared (FT-IR) Spectroscopy
A Thermo Nicolet Nexus470 FTIR with SmartPerformer - ATR sampler with a Ge crystal was used for FTIR measurements. The system was purged with dry air during collection of the baseline spectrum (air) and during the analysis. The Ge crystal was covered with the carbon nanoparticle sample and 12 psi of pressure was applied to the sample during the analysis.
Transmission electron microscopy (TEM) Measurements
10 μL aliquot of the diluted OCN stock solution was placed on 400 mesh copper grid. After 1 minute, excess fluid was removed by a piece of filter paper. Samples were viewed using a JEOL 1200EX electron microscope operating at 80 kV. High-resolution samples were viewed and analyzed with a Philips Tecnai F20 Field Emission Gun electron microscope operating at 200 kV.
Atomic force microscopy (AFM) Measurements
A Digital Instruments Dimension 3000 series AFM and standard Veeco tapping mode silicon probes with platinum-iridium (PtIr) coating were used for scanning the samples. Typically, aqueous suspensions of carbon nanoparticle samples were dried in a class 10000-clean room on a clean glass slide for 3h. Once dried, samples were placed on the AFM and scanned. Pertinent scanning parameters were as follows: Resonant frequency (probe): 60-80 kHz; Example of tip velocity: (4 um/s for 2 um), (15 um/s for 5 um), (30 um/s for 10 um). Aspect ratio: 1:1; Resolution: 512 samples/line, 256 lines.
Photoluminescence and photostability study
The photoluminescence and UV-visible (UV/vis) absorption spectra of carbon NPs suspension were recorded at ambient temperature by using calibrated Horiba JY Fluoromax 4P fluorescence spectrometer and Cary 4000 spectrophotometer, respectively.
The carbon NPs suspension was exposed to UV irradiation in a UV chamber (CL-1000 Ultraviolet corsslinker, UVP) at the intensity of 100×100 μJ/cm2 for 960 min (16h) to determine the fluorescence stability of the sample.
Animal Research Studies
Guidelines on the care and the use of laboratory animals at Washington University in St. Louis were followed for all animal experiments. C57 black mice were exposed intravenously of OCN (100 μL, Cv= 300 mg/mL) or 0.9% NaCl aqueous solution (nontoxic control). At 2 and 24 hours post injection, mice were sacrificed and blood was taken from heart for serum chemistry analysis. It should be noted that the concentration used in this research for OCN was their volume concentration, which was nominally calculated based on the weight of OCN samples per unit volume of solvent.
In vivo fluorescence images were acquired with a Xenogen IVIS imaging system (Caliper LifeSciences, Hopkinton, MA, USA). Guidelines on the care and the use of laboratory animals at Washington University in St. Louis were followed for all animal experiments. Carbon NPs suspension (Cv=100 mg/mL, 1 ml/kg) was administered intradermally into the hindpaw of the mouse. Images were acquired dynamically for 30 min connectively. Biodistributions of carbon NPs were studied at 2 hours and 24 hours post I.V. and I.D. injection, respectively. Dissected organs were subjected to NIR imaging. Fluorescence emission was normalized to photons per second per centimeter squared per steradian (p/sec/cm2/sr), deducted the influence of backgrounds. During image acquisition, anesthesia was maintained with isoflurane (1 L/min oxygen and 0.75% isoflurane, Euthanex Corp.) and vital signs and pulse oximetry (NONIN Medical INC., 8600V) was monitored continuously. Hydration was maintained by infusing 8 ml of 0.9% saline subcutaneously into the animal at a distal site as needed. After image acquisition, the animals were euthanized by pentobarbital overdose.
Results and Discussion
Three generations of hyperbranched bis-MPA polyester hydroxyl polymers (HBP) were chosen as the precursor for surface passivation. Pseudo second, third and fourth generation (G2, G3, G4) hyperbranched polymers corresponded to 16, 32, and 64 peripheral hydroxyl groups, respectively. Additionally, polyethylene glycol (linear PEG, MW 400 Da) was used for comparison. The synthesis of the optically active carbon nanoparticles is economical and involved only a domestic microwave heating step. In a typical synthesis, commercial grade honey (Great Value™ Clover Honey 1 wt%; batch composition; fructose: 38%, glucose: 31%, maltose: 7.1%, sucrose: 1.3% higher sugars: 1.5%, water: 17.2%) was suspended with the passivating agent (8 wt %), purged with argon and heated in a domestic microwave oven for 10 min. A 1:8 ratio (w/w) of honey-to-polymer was found to be the ideal composition. Other ratios (e.g. 1:2 and 1:4) were also explored but resulted in poor OCN solubility; this was likely attributed to inadequate coating of the carbon core by polymer. In addition, a 1:16 ratio was also investigated, but with no noticeable improvement in solubility or optical properties. Additionally, extended reaction time (20-30 min) resulted in insoluble charred material apparently due to instability of the hyperbranched polymer under that condition.
DLS measurement, shown in Figure 2a and in a tabular form in Figure 1d, revealed that the OCNs are monodispersed and the hydrodynamic size in aqueous suspension is within 8-20 nm. Significant variation in hydrodynamic sizes was not observed with increasing generation. The typical anhydrous morphologies of G4-HBP capped OCNs are exhibited in Figure 2b. TEM image showed that the as synthesized OCN appeared as spherical particles with good monodispersity. The core sizes of the OCNs were calculated as 5.3±0.3 nm. As they are capped within the hyperbranched polymers, each OCN revealed a bright corona around the particle with a thickness ranging from ca 0.5-1.6 nm. Figure 2e shows the AFM image of OCNs as uniform spherical nanoparticles with measured height values of 10±1 nm, which is in good agreement with the TEM and DLS analysis. The obtained OCNs can be dispersed in aqueous media and some other organic solvents (methanol, ethanol etc.). The colloidal suspensions remained stable for several months without noticeable precipitation with respect to particle size, polydispersity and zeta potential. This probably can be ascribed to its zeta potential of -38.12 mV in de-ionized water (Figure 2f) providing high electrostatic repulsion as the polymeric passivating agents creates steric barriers. Influence of pH on the zeta potential of G4-HBP capped OCN was studied. The isoelectric point of OCN particles was calculated to be ca. pH 4. Results indicate that OCN particles are highly stable within a pH range 6-12, which is suitable for in vivo administration in a clinical setting. Particles may tend to coagulate or flocculate at a lower pH ranges (Figure 2f).
Physico-chemical characterizations of OCNs. (a) hydrated state number-averaged hydrodynamic diameter (dynamic light scattering) of as-synthesized G4-HBP-OCN dispersed (0.2 μM) in deionized water; (b) anhydrous state TEM images of G4-HBP-OCN drop deposited over nickel grid (scale bar 200 nm and 100 nm respectively); (c) particle size distribution from TEM; (d) SEM image of the as-synthesized OCN sample (gold/palladium coated, scale bar 2 μm); (e) particle height distribution (Hav) from AFM; (f) variation of zeta potential with pH; (g) FT-IR spectroscopy signals of as-synthesized OCN; (h) Raman spectrum of G4-HBP-OCN (baseline corrected).
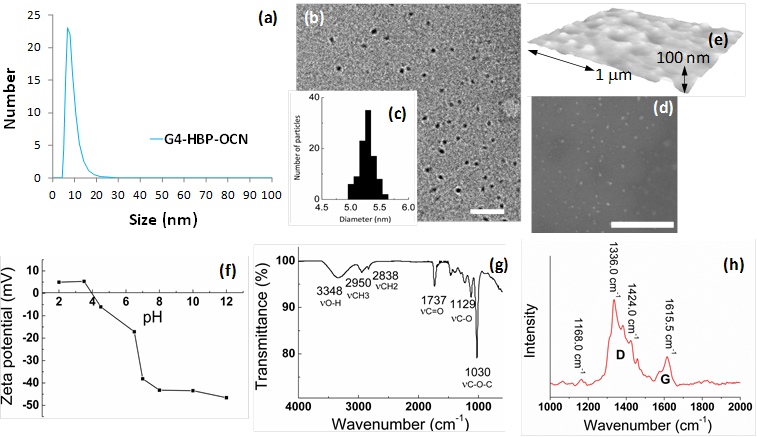
The FT-IR spectrum (Figure 2g) exhibited asymmetric stretching band corresponding to methyl functionalities (νaCH3) at 2950 cm-1, the methylene symmetric stretching band (νsCH2) at 2838 cm-1, and the C=O stretching band at 1737 cm-1. The broad peak centered at 3348 cm-1 and the bands in the range 1000-1400 cm-1 indicated the presence of a large amount of residual hydroxyl groups and C-O groups. These hydrophilic groups govern the solubility of the nanoparticles and promote the biocompatibility of the system. FT-IR analyses of the free polyethylene glycol and carbon nanopaticles indicate that the polyethylene glycol is strongly associated with the carbon nanoparticles. As shown in Supplementary Material: Figure S2, compared to the free polyethylene glycol, L-PEG-OCNs show a shift in FT-IR absorption. In the FT-IR curve of polyethylene glycol, the peaks at 1456, 1136 and 951 cm-1 are assigned to the O-H in-plane deformation, C-O-C stretching vibration and C-OH stretching, respectively. After the L-PEG-OCNs are formed, these peaks show a blue shift.
Thermo gravimetric analyses (TGA) were performed on a TGA Q 5000 (TA Instruments) in presence of nitrogen atmosphere. The TGA curve is shown in Supplementary Material: Figure S3. The first weight loss below 200 °C corresponded to the evaporation of the adsorbed water. The second step, which represents the most significant weight loss, could be attributed to the removal of polyethylene glycol groups. The weight percentages of L-PEG and OCNs calculated using the TGA results were 88% and 9.2%, respectively.
The Raman spectrum (Figure 2h) of G4-HBP capped carbon nanoparticles clearly showed the characteristic D band (ca. 1336 cm-1) and G-band (ca.1615 cm-1). The D (defect/disorder) band originates from a hybridized vibrational mode related to graphene edges indicating the presence of sp3 defects. The G band is related to the in-plane vibration of sp2 carbon atoms. Compared to other reported carbon materials [25,29], G4-HBP capped OCN exhibited a higher intensity of D band indicating towards high proportions of surface defects as well as the larger surface-to-volume ratio of small-sized carbon nanoparticles. The Raman peaks of G4-HBP were also observed. The band at 1424 cm-1 is due to symmetric stretching of COO- group and the band at 1168 cm-1 can be assigned to CH2 rocking. These results further suggest that the carbon nanoparticles have been successfully capped with hyperbranched polymers. Figure 3a shows the UV-Vis absorption spectra of the OCNs passivated with different macromolecules. All the OCNs show a characteristic absorption peak at ~320 nm, which is recognized to the n-π* transition of the C=O band and the π-π* transition of the conjugated C=C band. The photoluminescence (PL) of the OCNs exhibits a λex dependence of the emission wavelength and intensity (Figure 3b and Supplementary Material: Figure S1), which is typical of carbon nanoparticles [28]. As the λex increased from 350 nm to 700 nm, the emission peak of the OCNs is shifted from 521 nm to 751 nm accordingly. The OCNs exhibited considerable photostability, exemplifying only a 7% decrease in emission intensity after 16 h of continuous UV excitation (Figure 3c).
(a) UV-vis absorption spectra of OCNs (G2, G3 and G4 coated); (b) the corresponding normalized emission spectra at different excitation wavelengths; (c) PL spectra comparison for freshly prepared G4 HBP-OCN and after continuous irradiation for 16h (λex=350); (d) parameters for quantum yield calculated for OCNs.
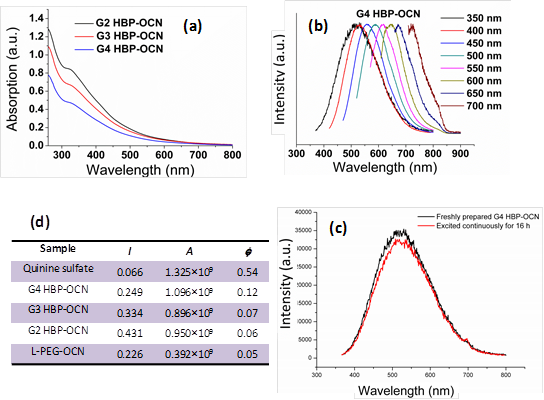
Mechanistically, OCNs have no classical band-gap absorptions, thus the photoluminescence in OCNs is thought to be related to the passivated surface defects of the carbon cores. The surface passivation stabilizes the surface energy traps and makes them emissive [30-32]. Interestingly, the hyperbranched macromolecular ligands have little influence on the absorption and emission wavelength, which may indicate that the polymers with different molecular weights and same functional groups produce the OCNs with similar core sizes and surface defects. But the ligands with a higher molecule weight (pseudo G4) show an enhancement on their quantum yield (QY). Using quinine bisulfate (QY 0.54 in 0.1M H2SO4) as the reference, the QY of the OCNs in aqueous solution was measured to be 0.06 (for G2-OCNs), 0.08 (for G3-OCNs), and 0.13 (for G4-OCNs), respectively (Figure 3d). Most of the previously reported methods for carbon nanoparticles produced a QY of less than 0.01. Although some methods can produce QYs of 0.05 to 0.2, these methods often require a sophisticated high energy radiation-based syntheses followed by complicated surface functionalization and size separation [25, 33, 34]. The high QY for G4-OCN (0.13) is presumably due to the nesting by the high-density hyper branched Bis-MPA polyester ligands from quenchers in solution and extensively branched polymer spawning a greater surface area for interaction.
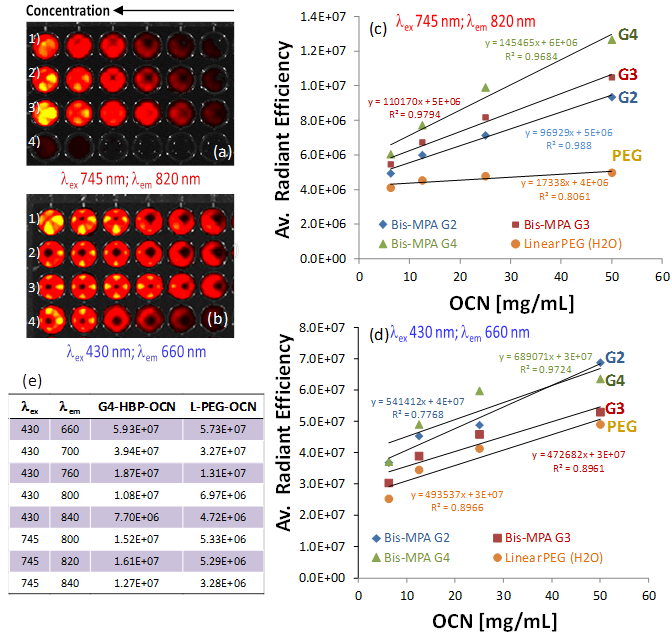
Figure 4 summarizes results from a fluorescence imaging study performed with a Xenogen IVIS system. Four serially diluted OCN samples (1: G2-HBP-OCN; 2: G3-HBP-OCN; 3: G4-HBP-OCN; 4: Linear-PEG-OCN) were filled in Corning® 384 Well Optical Imaging Flat Clear Bottom Black Polystyrene TC-Treated microplates. Interestingly, in the NIR range (λex 745 nm; λem 820 nm), significant signal enhancement was observed (Figure 4e) from G4-HBP-OCN and other hyperbranched polymer capped OCNs in comparison to linear PEG-coated OCN. The experiment was repeated in the visible range (λex 430 nm; λem 660 nm) and an average radiant efficiency against OCN concentration at the visible range revealed only a moderate to no considerable escalation (Figure 4d). Figure 4c shows the overlaid average radiant efficiencies of linear and hyperbranched polymer coated carbon nanoparticles. The data shown in Figure 4c demonstrates that in the NIR range, as passivating agent becomes more extensively branched (G2 to G4), the average radiant efficiency increases considerably. This likely is a direct result of the increasing surface area available for light passivation as the OCN coating becomes more extensively branched. Additionally, all three HBP OCNs (G2, G3 and G4) exemplified higher average radiant efficiencies compared to their linear PEG equivalent, indicating improved optical properties are likely a result of the hyperbranched coating. Near infrared imaging is a powerful diagnostic tool with the potential to serve as a minimally invasive, nonionizing method for sensitive, deep tissue imaging. Near-infrared light has a longer wavelength than visual light, allowing it to travel deeper into tissue. This potential is further realized with the use of highly branched fluorescent carbon nanoparticles.
Fluorescence imaging of OCN in suspension with Xenogen-IVIS system. Fluorescence signals generated from a cell culture well plate filled with serially diluted OCN at 1:2, 1:4, 1:8, 1:16, 1:32 and the original volume concentration for column one is 200 mg/mL (1: G2-HBP-OCN; 2: G3-HBP-OCN; 3: G4-HBP-OCN; 4: Linear-PEG-OCN), a) excited at 745 nm and emission at 820 nm; b) excited at 430 nm and emission at 660 nm; c) average radiant efficiency obtained by ROI linear regression analysis against OCN concentration showing a dramatic increase in fluorescence efficiency at NIR range for hyperbranched polymer capped OCNs; d) average radiant efficiency against OCN concentration at the visible range showing moderate to no considerable enhancement; e) comparative summary of excitation and emission wavelengths for G4-HBP-OCN and linear PEG-OCN.
A preliminary biocompatibility assessment of the hyperbranched polymer capped OCN nanoparticles was undertaken, which measured the effects of OCN exposure on cellular viability over time and clinical pathology in vivo. The initial cell viability assessment of OCN, while limited in scope, is a critical first step toward the identification of potentially detrimental contacts between biological species and OCN. The cell viabilities in the presence of the OCN were evaluated by AlamarBlue® cytotoxicity assay in endothelial 2F2B cells, which monitors enzymatic activity of a cell organelle (in this case mitochondria). Results indicated that OCN behave similarly at time points 24 and 72 h respectively with de-ionized water. No loss of cell viability was observed at a sufficiently high concentration. This suggests the use of OCN even at very high concentrations (100 mg/ml) would not be prohibited in vitro (Figure 5a). Serum chemistry analyses exhibited normal kidney performance in OCN injected mice (n=3). Liver enzymes and kidney function were evaluated after injection of high doses of OCN. The data showed (Figure 5b) that there is an initial mild influence on mice liver function 2 hours post injection of OCN, which then begins to resolve towards normal physiological levels 24 hours post injection. This suggests the burden of OCN on the liver and kidneys is an acute response that resolves as the OCN are subsequently metabolized and secreted.
(a) cell viability of OCN in 2F2B endothelial cells; (b) clinical pathology and serum chemistry results for OCN and untreated mice; non-invasive in vivo fluorescence imaging of SLN in mouse: fast accumulation of OCN in popliteal lymph node post injection (c) 1 min and (d) 30 min; (e) excised lymphnode showing the accumulation of dark deposit of carbon particles (yellow arrow), inset: a control excised lymphnode from untreated mice showing no visible accumulation of OCN; qualitative tissue distribution of OCN: 2h (f-g) and 24h (h-i) tissue accumulation of intradermally and intravenously injected OCN.
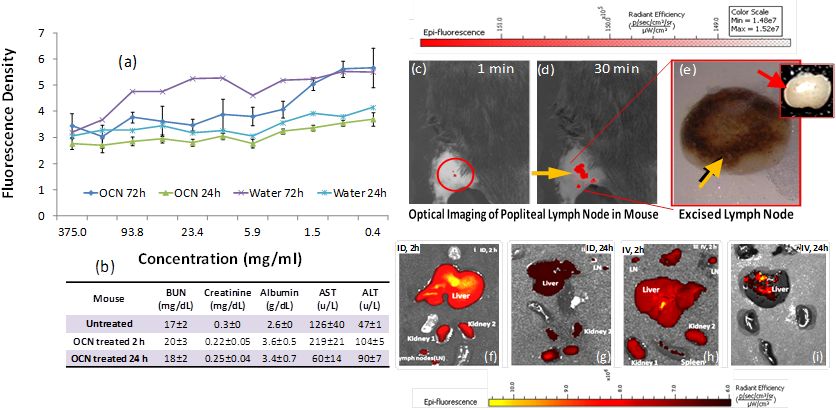
These promising preliminary results prompted us to explore the feasibility of using OCN for in vivo near infrared imaging. The non-invasive detection of axilliary lymph nodes could minimize resection time of sentinel lymph node biopsy by providing precise spatial localization of the SLN. In clinical practice, this is poorly achieved with current optical and nuclear techniques. Prior to image acquisition, the axillary surface of a mouse was shaved for optical imaging. Following intradermal L-PEG-OCN injection, the optical image of the same area was acquired dynamically for 30 minutes. Within 1 min post injection of OCN, lymph node (Figure 5c) was clearly visible with ca. 200% contrast enhancement. The lymph node was monitored further over time, which exhibited contrast enhancement of the lymph node for 30 minutes (Figure 5c). Figure 5e depicts the excised lymphnode removed following completion of the optical imaging. The accumulation of the carbon particles was clearly evident from the presence of discolorations.
A preliminary and qualitative tissue distribution study was undertaken in a mouse model. It was noticed that OCN, like other nanoparticulates, were presumably being cleared by the macrophages of the reticuloendothelial system (RES) through the liver and the spleen. The predominant accumulation of OCN was seen in liver, spleen and kidney. This is consistent with a renal excretion pathway. At 24 hours, the level of fluorescence signal intensity from both the intradermally and intravenously injected mice was obviously decreased, suggesting the rapid clearance of OCN in vivo (Figure 5f-i). Further in-depth bio-distribution studies for all the candidate agents are warranted to obtain quantitative results.
Conclusions
In summary, we report a simple and commercially amenable 'green syntheses' of carbon nanoparticles. The fluorescence properties of these particles were significantly enhanced by tuning the surface passivating agents. By utilizing a hyberbranched polymer for surface passivation, a dramatic increase in near infrared emission was achieved compared to a linear PEG coated OCN. The particles showed negligible loss of cell viability in presence of endothelial cells. Preliminary in vivo experiment showed high contrast enhancement in auxiliary lymphnode in a mouse model. The exceptionally rapid signal enhancement of the SLN suggests that such an approach may offer greater convenience and reduced procedural expense, as well as improved surgical advantage as the patient is positioned on the table for easier resection.
The surface passivation with hyperbranched polymer and exploitation of the surface defect to demonstrate significant improvement in the NIR fluorescence properties suitable for in vitro as well as in vivo application has never been explored. As contrary to some of the reported carbon nanoparticles, which showed excellent fluorescence characteristics in the blue/green region, these particles produced excellent fluorescence properties in the NIR region. These early in vitro and in vivo evaluations are crucial to direct further in depth applications.
Supplementary Material
Fig.S1 -S3.
Acknowledgements
The financial support from the AHA 0835426N, NIH under the Grants NS059302, CA119342, R01 EB000712, R01 EB008085, R01 CA134539 is greatly appreciated. L.W. and B.S. appreciate the financial support of National Natural Science Foundation of China (No. 81101087, 81130028 and 31210103913), China Scholarship Council (2011823056), Science and Technology Research Project of Heilongjiang Education Department (No. 12511325), China Postdoctoral Science Foundation (2012M510992) and Heilongjiang Postdoctoral Foundation (LBH-Z11054).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Berezin MY, Achilefu S. Fluorescence Lifetime Measurements and Biological Imaging. Chem Rev. 2010;110:2641-2684
2. Ntziachristos V. Going deeper than microscopy: the optical imaging frontier in biology. Nat Methods. 2010;7:603-614
3. Choi HS, Gibbs SL, Lee JH. et al. Targeted zwitterionic near-infrared fluorophores for improved optical imaging. Nat Biotechnol. 2012;31:148-153
4. Razansky DC. Deliolanis N, Vinegoni C, et al. Deep Tissue Optical and Optoacoustic Molecular Imaging Technologies for Pre-Clinical Research and Drug Discovery. Curr Pharm Biotechnol. 2012;13:504-522
5. Wu L, Cai X, Nelson K. et al. A green synthesis of carbon nanoparticle from honey for real-time photoacoustic imaging. Nano Res. 2013;6:312-325
6. Andrews DL, Demidov AA. An introduction to laser spectroscopy: Springer. 2002.
7. Qin W, Ding D, Liu J. et al. Biocompatible Nanoparticles with Aggregation-Induced Emission Characteristics as Far-Red/Near-Infrared Fluorescent Bioprobes for In Vitro and In Vivo Imaging Applications. Adv Funct Mater. 2012;22:771-779
8. Zhang X, Wang S, Xu L. et al. Biocompatible polydopamine fluorescent organic nanoparticles: facile preparation and cell imaging. Nanoscale. 2012;4:5581-5584
9. Zhang X, Zhang X, Wang S. et al. Surfactant modification of aggregation-induced emission material as biocompatible nanoparticles: Facile preparation and cell imaging. Nanoscale. 2013;5:147-150
10. Hui J, Zhang X, Zhang Z. et al. Fluoridated HAp:Ln3+ (Ln = Eu or Tb) nanoparticles for cell-imaging. Nanoscale. 2012;4:6967-6970
11. Wang Z, Wu L, Cai W. Size-Tunable Synthesis of Monodisperse Water-Soluble Gold Nanoparticles with High X-ray Attenuation. Chem Eur J. 2010;16:1459-1463
12. Qian H, Zhu M, Wu Z. et al. Quantum Sized Gold Nanoclusters with Atomic Precision. Acc Chem Res. 2012;45:1470-9
13. Wang Z, Wu L, Cai W. et al. Luminescent Au11 nanocluster superlattices with high thermal stability. J Mater Chem. 2012;22:3632-3636
14. Pan D, Pramanik M, Senpan A. et al. Near infrared photoacoustic detection of sentinel lymph nodes with gold nanobeacons. Biomaterials. 2010;31:4088-4093
15. Swierczewska M, Lee S, Chen X. The design and application of fluorophore-gold nanoparticle activatable probes. Phys Chem Chem Phys. 2011;13:9929-9941
16. Liu Z, Liang X-J. Nano-Carbons as Theranostics. Theranostics. 2012;2:235
17. Swierczewska M, Choi KY, Mertz EL. et al. A Facile, One-Step Nanocarbon Functionalization for Biomedical Applications. Nano Lett. 2012;12:3613-3620
18. Liu Z, Robinson JT, Tabakman SM. et al. Carbon materials for drug delivery & cancer therapy. Mater Today. 2011;14:316-323
19. Zhu Y, Li J, Li WX. et al. The Biocompatibility of Nanodiamonds and Their Application in Drug Delivery Systems. Theranostics. 2012;2:302-312
20. Cao L, Yang ST, Wang X. et al. Competitive performance of carbon “quantum” dots in optical bioimaging. Theranostics. 2012;2:295-301
21. Zhang X, Wang S, Zhu C. et al. Carbon-dots derived from nanodiamond: Photoluminescence tunable nanoparticles for cell imaging. J Colloid Interface Sci. 2013;397:39-44
22. Zhang J, Shen W, Pan D. et al. Controlled synthesis of green and blue luminescent carbon nanoparticles with high yields by the carbonization of sucrose. New J Chem. 2010;34:591-593
23. Tao H, Yang K, Ma Z. et al. In Vivo NIR Fluorescence Imaging, Biodistribution, and Toxicology of Photoluminescent Carbon Dots Produced from Carbon Nanotubes and Graphite. Small. 2012;8:281-290
24. Tian L, Ghosh D, Chen W. et al. Nanosized carbon particles from natural gas soot. Chem Mater. 2009;21:2803-2809
25. Baker SN, Baker GA. Luminescent carbon nanodots: emergent nanolights. Angew Chem Int Ed. 2010;49:6726-6744
26. Huang P, Lin J, Wang X. et al. Light-Triggered Theranostics Based on Photosensitizer-Conjugated Carbon Dots for Simultaneous Enhanced-Fluorescence Imaging and Photodynamic Therapy. Adv Mater. 2012;24:5104-5110
27. Liu H, Ye T, Mao C. Fluorescent carbon nanoparticles derived from candle soot. Angew Chem Int Ed. 2007;46:6473-6475
28. Sun YP, Zhou B, Lin Y. et al. Quantum-sized carbon dots for bright and colorful photoluminescence. J Am Chem Soc. 2006;128:7756-7757
29. Lu J, Yang JX, Wang J. et al. One-pot synthesis of fluorescent carbon nanoribbons, nanoparticles, and graphene by the exfoliation of graphite in ionic liquids. ACS Nano. 2009;3:2367-2375
30. Jia X, Li J, Wang E. One-pot green synthesis of optically pH-sensitive carbon dots with upconversion luminescence. Nanoscale. 2012;4:5572-5575
31. Liu JH, Yang ST, Chen XX. et al. Fluorescent Carbon Dots and Nanodiamonds for Biological Imaging: Preparation, Application, Pharmacokinetics and Toxicity. Curr Drug Metab. 2012;13:1046-1056
32. Wang X, Cao L, Yang ST. et al. Bandgap-Like Strong Fluorescence in Functionalized Carbon Nanoparticles. Angew Chem Int Ed. 2010;122:5438-5442
33. Peng H, Travas-Sejdic J. Simple Aqueous Solution Route to Luminescent Carbogenic Dots from Carbohydrates. Chem Mater. 2009;21:5563-5565
34. Bhunia SK, Saha A, Maity AR. et al. Carbon Nanoparticle-based Fluorescent Bioimaging Probes. Sci Rep. 2013;3:1473
Author contact
![]() Corresponding author: E-mail: dipanjanedu and shenbzhsina.com.
Corresponding author: E-mail: dipanjanedu and shenbzhsina.com.