Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Materials
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2014; 4(8):808-822. doi:10.7150/thno.8255 This issue Cite
Research Paper
Exploration of Panviral Proteome: High-Throughput Cloning and Functional Implications in Virus-host Interactions
Xiaobo Yu1*, Xiaofang Bian1*, Andrea Throop1, Lusheng Song1, Lerys Del Moral1, Jin Park1, Catherine Seiler1, Michael Fiacco1, Jason Steel1, Preston Hunter1, Justin Saul1, Jie Wang1, Ji Qiu1, James M. Pipas2, Joshua LaBaer1 ![]()
1. The Virginia G. Piper Center for Personalized Diagnostics, Biodesign Institute, Arizona State University, Tempe, AZ 85287, USA
2. Department of Biological Sciences, University of Pittsburgh, Pennsylvania 15260, USA.
* These authors contributed equally to this work.
Received 2013-12-1; Accepted 2014-4-27; Published 2014-6-6
Abstract
Throughout the long history of virus-host co-evolution, viruses have developed delicate strategies to facilitate their invasion and replication of their genome, while silencing the host immune responses through various mechanisms. The systematic characterization of viral protein-host interactions would yield invaluable information in the understanding of viral invasion/evasion, diagnosis and therapeutic treatment of a viral infection, and mechanisms of host biology. With more than 2,000 viral genomes sequenced, only a small percent of them are well investigated. The access of these viral open reading frames (ORFs) in a flexible cloning format would greatly facilitate both in vitro and in vivo virus-host interaction studies. However, the overall progress of viral ORF cloning has been slow. To facilitate viral studies, we are releasing the initiation of our panviral proteome collection of 2,035 ORF clones from 830 viral genes in the Gateway® recombinational cloning system. Here, we demonstrate several uses of our viral collection including highly efficient production of viral proteins using human cell-free expression system in vitro, global identification of host targets for rubella virus using Nucleic Acid Programmable Protein Arrays (NAPPA) containing 10,000 unique human proteins, and detection of host serological responses using micro-fluidic multiplexed immunoassays. The studies presented here begin to elucidate host-viral protein interactions with our systemic utilization of viral ORFs, high-throughput cloning, and proteomic technologies. These valuable plasmid resources will be available to the research community to enable continued viral functional studies.
Keywords: Panviral proteome, ORFeome, Gateway® cloning, Gene synthesis, Virus-host interactions, Nucleic Acid Programmable Protein Arrays (NAPPA).
Introduction
With the rapid progression of high-throughput (HT) -omic technologies (e.g., genomics, transcriptomics and proteomics), biological information has exploded over the past decade. These HT approaches allow scientific investigations to shift from traditional reductionism to systems biology approaches to study complex interactions in biological systems with a holistic perspective [1]. As simple intracellular organisms, viruses must hijack host factors to survive and propagate at multi-stages of their life cycles in the host. Viruses have developed a variety of delicate strategies to facilitate their invasion and replication/transcription of their genomes, while at the same time silencing host immune responses by manipulating host proteins and cellular processes. The systematic characterization of the viral proteome-host interactions, and the identification of new host targets, would be invaluable in the understanding of viral invasion and replication mechanisms in infectious diseases, autoimmune diseases, and cancer [2]. The findings from these studies will greatly facilitate the detection of viral infection and progression, antiviral drug discovery, and therapeutic treatment [3-5].
Through a profound effort from the ORFeome Collaboration, a large set of sequence-verified open reading frame (ORF) clone collections (bacteria, yeast, mouse, human) have been constructed and are accessible to the research community [6-14]. Interestingly, of the 2,000 viral genomes that have been sequenced, only a small fraction of viral ORFeome plasmids have been constructed, and the molecular mechanism of most of virus-host interactions is largely unknown [2]. Pellet et al. pioneered the ViralORFeome 1.0 (http://pbildb1.univ-lyon1.fr/viralorfeome/index.php), an open-access database and management system that provides an integrated set of bioinformatics tools enabling the potential capture of viral ORFs in the Gateway® recombinational cloning system. With the ViralORFeome database, Pellet et al. collected 545 ORF clones from ~238 viral genes in Gateway® entry vectors (pDONR207 or pDONR223). In addition, this study demonstrated the phenotypic expression of 67 viral genes transfected in HEK-293T cells [15]. Unfortunately, the overall progress of viral cloning has not progressed much beyond this due to the high cost of HT cloning and the difficulty in managing a large repository in a general lab [2, 15, 16].
Since the establishment of our laboratory and the DNASU Repository at Arizona State University, we have devoted significant effort in the HT cloning, construction, and maintenance of large DNA repositories (http://dnasu.org) [17, 18]. We have successfully created tens of thousands of sequence-verified ORFs in Gateway® recombination system, ranging from human to numerous pathogens and model organisms, such as S. cerevisiae, P. aeruginosa, F. tularensis, V. cholerae and Y. pestis [17-25]. The high flexibility of Gateway® system allows researchers to rapidly transfer these collections into any compatible expression vectors that fit their biological studies [26-36]. For example, using the S. cerevisiae plasmid collection, Zhu et al. purified 89 known and predicted S. cerevisiae transcription factor proteins, and profiled their DNA binding sites using ChIP-chip. More than 2.3 million gapped and ungapped 8-bp oligo sequences were identified, which allowed the re-annotation of in vivo TF binding targets, and examination of TFs' potential effects on gene expression in about 1700 environmental and cellular conditions [23, 34]. Cooper et al. executed a genome-wide screen of 3,000 over-expressed yeast genes for the enhancers and suppressors of alpha-synuclein (αSyn). The misfolding of αSyn is associated with devastating neurodegenerative disorders such as Parkinson's disease (PD). 34 and 20 genes were identified to suppress and enhance αSyn toxicity, respectively. Rab1, the mammalian YPT1 homolog, was found to protect against αSyn-induced dopaminergic neuron loss in PD animal models [35]. We constructed human and pathogen Nucleic Acid Programmable Protein Arrays (NAPPA), with which we executed wide proteomics studies to find the antibody biomarkers that have potential value in the early detection of cancers, diabetes, autoimmune, and infectious diseases [26-33].
In this project, we plan to generate a plasmid collection that encodes all important human disease related viral proteins (~6,000). The aim of our panviral proteome is to discover and characterize virus-host interactions and determine the association of these viral proteins with various human diseases. Similar to our human and pathogen collections, we will distribute these viral ORF constructs to the research community though DNASU (http://dnasu.org) [11], including the initial collection described in this paper containing 2,035 ORF clones in Gateway® entry vector (pDNOR221) and mammalian cell-free expression vectors (Table 1). To illustrate the applications of this collection, we characterized the expression of ORF clones from three viral genomes utilizing human HeLa cell lysate-based cell-free expression system, western blot, and in-gel fluorescence. In addition, we studied rubella virus-host interactions using NAPPA arrays displaying 10,000 unique human proteins. We also analyzed the host serological response to coxsackievirus antigens using micro-fluidic multiplexed immunoassays. The explorations of our viral clone collection showed the highly efficient cell-free production of viral ORF proteins in vitro, identification of novel host-viral protein interactions, and a demonstration of how immune-dominant viral antigens could be used in the future for target drug development and detection of viral pathogen infection.
List of viral ORF clones that have been collected.
| Name | Abbre. | Family | Entry vector | Mammalian cell-free expression vectors | |
|---|---|---|---|---|---|
| Closed | Fusion | ||||
| Epstein-Barr virus | EBV | Herpesvirinae | 73 | 89 | 89 |
| Hepatitis B Virus | HBV | Hepadnaviridae | 7 | 7 | |
| Human papillomaviruses, type5 | HPV5 | Picobirnaviridae | - | 4 | 4 |
| Human papillomaviruses, type6 | HPV6 | Picobirnaviridae | - | 5 | 5 |
| Human papillomaviruses, type8 | HPV8 | Picobirnaviridae | - | 3 | 3 |
| Human papillomaviruses, type11 | HPV11 | Picobirnaviridae | - | 5 | 5 |
| Human papillomaviruses, type16 | HPV16 | Picobirnaviridae | - | 3 | 8 |
| Human papillomaviruses, type18 | HPV18 | Picobirnaviridae | - | 4 | 8 |
| Human papillomaviruses , type33 | HPV33 | Picobirnaviridae | - | 4 | 4 |
| Kaposi's sarcoma-associated herpesvirus | KSHV | Herpesviridae | 67 | 67 | 67 |
| Human cytomegalovirus | HCMV | Herpesvirinae | - | 165 | 165 |
| Herpes simplex virus type 1 | HSV1 | Herpesviridae | 94 | 94 | 94 |
| Adenovirus | Adenoviridae | - | 16 | 16 | |
| Simian virus 40 | SV40 | Polyomaviridae | 6 | 6 | 6 |
| Varicella-zoster virus | VZV | Herpesviridae | 66 | 84 | 94 |
| Vaccinia | VACV | Poxviridae | 167 | 167 | |
| Human endogenous retrovirus K | HERK | Retroviridea | 4 | 4 | 4 |
| Influenza A virus, H1N1 | Orthomyxoviridae | - | 10 | 10 | |
| Influenza A virus, H3N2 | Orthomyxoviridae | - | 10 | 10 | |
| Chikungunya virus | CHIKV | Togaviridae | 9 | 9 | 9 |
| Sindbis virus | SINV | Togaviridae | 9 | 9 | 9 |
| Semliki Forest virus | SFV | Togaviridae | 9 | 9 | 9 |
| Yellow fever virus | YFV | Flaviviridae | 11 | 11 | 11 |
| Tioman virus | Paramyxoviridae | 3 | 3 | 3 | |
| Measles virus | MeV | Paramyxoviridae | 5 | 5 | 5 |
| Coxsackievirus | Picornaviridae | - | 12 | 12 | |
| Mumps virus | MuV | Paramyxoviridae | - | 8 | 8 |
| Rubella virus | RUBV | Togaviridae | - | 5 | 5 |
| Rotavirus A | RV-A | Reoviridae | - | 12 | 12 |
| Number | 356 | 830 | 849 | ||
| Total number | 2035 | ||||
Materials and Methods
HT 2-Step PCR based cloning
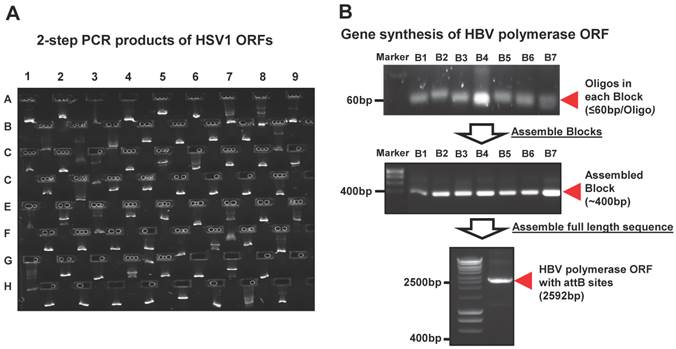
We performed the HT 2-step PCR cloning as previously described [20]. Briefly, the reference sequences of viral ORFs were uploaded into our FLEXGene Laboratory information management system (LIMS) (http://flex.asu.edu/FLEX). The gene specific primers were designed by using a nearest-neighbor algorithm, with each primer containing a gene specific region and partial attB site. Universal primers were employed to provide the remaining full length attB site and further amplification of the target gene ORF. The start codon was normalized to ATG. The fusion and closed versions of the entry clones were created with and without removal of stop codon, respectively. Two-step PCR amplifications were performed in 96-well PCR plate using Tm of 60oC with Veriti® Thermal Cycler (Life Technologies, Grand Island, NY). The 2nd PCR products were examined using E Gel® Electrophoresis System (Life Technologies, Grand Island, NY) (Figure 2A). Lastly, the PCR product was purified using PCR clean-up kit (MACHEREY-NAGEL, Bethlehem, PA).
The representative images of viral gene cloning by 2-step PCR and de novo gene synthesis. (A) Examination of the 2nd PCR products from PCR-based cloning in E-gel. Here, 72 HSV1 ORFs amplified from their templates are shown as 9 columns × 8 rows. (B) Synthesis of HBV polymerase ORF using gene synthesis. The total 84 oligos (~60bp/oligo) were designed and automatically allocated into seven blocks by Gene Design 3.0. The oligos before (~60 bp/oligo) and after assembly (~400 bp/block) are shown at their expected sizes in an agarose gel. These assembled blocks were further annealed into a full-length HBV polymerase with the size of 2592bp, including attB sites.
De novo gene synthesis
Gene design
First, attB1 (GGGGACAAGTTTGTACAAAAAAGCAGGCTCCACC) and the converse complementary oligo of attB2 (GACCCAGCTTTCTTGTACAAAGTGGTCCCC) were added to both ends of ORF reference sequences. The oligos with less than 60 base pairs (bp) were designed using oligoDesign3.0 (http://54.235.254.95/cgi-bin/gd/gdOlapDes.cgi), which can be separated into several blocks according to the length of target sequence [37, 38]. The oligos were ordered from Integrated DNA Technologies (Coralville, IA).
Preparation of oligos and primers
All oligos were diluted into the stock concentration of 30 µM. The Template Oligo Mix (TOM1) was prepared by mixing 5 µL of inner oligos of each block with appropriate nuclease-free H2O to the final concentration of 2.5 µM. Primer Oligo Mix (POM1) was prepared by mixing 10 µL of the first and last oligos of each block with appropriate H2O to the final concentration of 10 µM. Primer Oligo Mix (POM2) was prepared by mixing 10 µL of the first and last oligo at the end of target ORF reference sequence with appropriate H2O to the final concentration of 10 µM. Gateway® universal forward and reverse primers (GUPs) were prepared at 10 µM[39].
Gene synthesis
For the viral ORFs with longer sequences (>700bp), the oligos need to be separated into multi-blocks by Gene Design 3.0, and undergo several assembly/amplification cycles to synthesize full-length reference sequence, e.g. HBV polymerase (Figure 2B). For the assembly of short oligos (≤ 60bp), each block was performed in a 50 µL reaction containing 1U iProof polymerase (BioRad, Hercules, CA) and 2.5 µL of a 250 µM TOM1 oligo mix. The PCR reaction was executed by starting at 98oC for 4 minute (min), followed by 25 cycles of 30 seconds (s) at 98oC for denaturation, 45s at 59oC for annealing, and 45s at 72oC for elongation. The amplification reaction was performed in a 50 µL PCR mix containing 1U iProof polymerase (Hercules, CA), 2 µL of assembled product, and 5 µL of POM1. The PCR reaction was executed with the same conditions as stated above, except 30 cycles was used. The amplified PCR products were ran and examined on an agarose gel with Ethidium Bromide and Bioline Hyperladder I (Bioline, Taunton, MA). Once the final assembled oligos were confirmed, they were purified using QIAquick Gel Extraction Kit (QIAGEN, Valencia, CA), and quantitated using a Nanodrop spectrophotometer (Wilmington, DE).
The assembly of seven blocks of HBV polymerase ORF sequence was performed in a 50 µL reaction containing 1U iProof polymerase and 50 ng of previously annealed oligos from each block. The second assembly PCR was performed the same as the first assembly reaction, but with a 1min elongation step. The second amplification reaction was performed in a 50 µL reaction containing 1 U iProof polymerase, 2 µL of previous assembled PCR product, and 5 µL of POM2 or UOP. The PCR was run under the same conditions as the first amplification reaction, but with a 2min elongation step. Once the full-length ORF reference sequence was synthesized, it was purified and the concentration was quantitated using a Nanodrop spectrophotometer (Figure 2B).
Construction of viral entry & expression clones
The procedure for the construction of entry and expression clones using the Gateway® system has been well described previously [14, 20, 23, 40]. Briefly, all purified PCR products and synthesized ORF sequences with attB1 and attB2 sites were cloned into a Gateway® entry vector (pDONR221) using BP Clonase (Life Technologies, Grand Island, NY). The clones were colony selected from agar plates and the ORF inserts were sequenced verified using our Automatic Clone Evaluation (ACE) software. For clones to be accepted in our viral gene plasmid collection, they must not contain truncations, frameshifts, or more than one amino acid change when compared to the reference sequence. In addition, any nucleotide change in the sequences of att-sites was also not accepted because it may cause failure of BP or LR reactions [20, 21].
To construct the collection of expression clones, sequence verified viral ORFs in entry vectors were transferred into mammalian cell-free expression vectors (pANT7_cGST, pJFT7_nHalo or pJFT7_cHalo) using LR Clonase (Life Technologies, Grand Island, NY). All solution transfers were executed by the Biomek FX automation workstation, and each cloning step was assigned a specific barcode which is tracked by the FLEXGene database throughout the cloning process.
Identification of host targets for rubella virus using NAPPA arrays
Five NAPPA arrays containing 10,000 purified human cDNA plasmids were printed as previously described (~2,000 ORFs/slide) [41]. To prepare NAPPA protein arrays, the slide was blocked with Superblock solution (Pierce, Rockford, IL) for 1hour (hr) at room temperature. The slide was then covered with a hybridization chamber and 160 μL of human HeLa cell lysate-based cell-free expression system (Thermo scientific, Rockford, IL) was injected into the chamber. The cell-free expression was performed at 30oC for 1.5 hrs, and then incubated at 15oC for 0.5 h. After briefly washing with PBST (PBS, 0.2%Tween 20), the resulting NAPPA protein array was blocked with cold PPI blocking buffer (1×PBS, 1%Tween 20 and 1% BSA, pH7.4) for 2 hrs at 4oC. In parallel, the rubella viral ORFeome proteins with C-terminal Halo tag (E1, E2, P90, capsid, and the mix of 150N and 150C) were produced in 170 μL human HeLa cell lysate-based cell-free expression system for 2 hrs at 30oC with 100 ng/μL of DNA.
To identify the host targets of rubella virus using our NAPPA arrays, the NAPPA human 10K arrays were incubated with the expressed rubella viral proteins in HeLa cell lysates for 16 hrs at 4oC. After washing three times with PPI wash buffer (PBS, 5 mM MgCl2, 0.5% Tween20, 1% BSA and 0.5% DTT, pH7.4), the protein-protein complexes formed on the NAPPA arrays were detected using 12.5 µM Alexa 660 conjugated Halo-ligand (Promega, Madison, WI) and incubated for 2hrs at 4oC. The fluorescent images were obtained with a Tecan's PowerScanner (Männedorf, Switzerland). The fluorescent signal intensity was quantitated using Array-Pro Analyzer (Media Cybernetics, Bethesda, MD).
Statistical analysis
Prior to the statistical analysis, we examined all fluorescent microarray images for the spot shape, dust, and non-specific binding, in order to remove false positive signals. We then normalized the raw signal intensity of all spots to decrease the background variations from slide to slide. Normalization was performed by subtracting the background signals from non-specific binding of query proteins or Alexa660 labeled Halo ligand, which was estimated by the first quartile of the printing buffer-only control. Next, we calculated the normalized value using the raw signal intensity of each spot divided by the median background-adjusted value of all features on the array. Finally, we calculated the Z-score of each protein that was used for the target selection.
The selection of host target candidates of rubella virus was based on the following criteria: (1) Z-score ≥ 3, (2) Z-score ratio of query protein to Halo negative control ≥ 2, and (3) the targets have to meet previous criteria in two independent experiments.
Bioinformatics analysis
The protein annotation was performed with The UniProt (Universal Protein Resource) databases and PANTHER (Protein ANalysis THrough Evolutionary Relationships) Classification System. The rubella virus-host interaction network was built using Cytoscape (v2.6.3, available in http://www.cytoscape.org).
Co-localization verification of rubella virus capsid with host targets identified from NAPPA screens
The rubella virus capsid and its identified host targets were cloned into pCS Cherry DEST (Addgene) and pcDNATM6.2/N-EmGFP/YFP-DEST (Life Technologies) vectors, respectively, using LR recombinant reactions. HeLa cells were transiently co-transfected with GFP-host targets and mCherry-capsid for 20 hrs. The cells were then fixed in 4% paraformaldehyde PBS and imaged using Zeiss LSM 510 inverted fluorescence confocal microscope (Zeiss) (100x objective) and images analyzed on ImageJ software.
Serological analysis of coxsackievirus proteins using micro-fluidic multiplexed immunoassays
The micro-fluidic antigen channels were fabricated by covering an aminosilane coated glass slide with a customized PDMS mold embedded with 13 micro-channels. The PDMS mold was prepared by soft-lithography as previous descried (Figure 6A) [42]. The antigen coating of each channel was performed by injection of 5 µL expressed coxsackievirus proteins in human HeLa cell lysate-based cell-free expression system and incubated for 1h at room temperature. After washing three times with 0.2% PBST, the PDMS mold was removed and the antigen coated slide was blocked with 5% milk for 1 hr. The reaction channels were prepared by applying a new set of PDMS micro-fluidic channels that lay vertical to the antigen strips formed on the slide surface. In each reaction channel, the immobilized antigens were detected with an anti-Halo antibody and Alexa555 labeled goat anti-chicken IgG antibody (Life Technologies, Grand Island, NY) (Figure 6A and 6B).
Immunological profiling of human serological response to coxsackievirus antigens. (A) The work flow of micro-fluidic multiplexed immunoassays, (B) The fluorescent image of coxsackievirus protein expression and detection using anti-Halo antibody, (C) The fluorescent signal intensity of viral proteins coated in the antigen channels (left, y-axis) and the coefficient variations from channel to channel (right, y-axis), (D) and (E) are the fluorescent images and the heatmap of serological antibody profiling using micro-fluidic multiplexed immunoassays, respectively. 00 is buffer alone and 01-11 are the human sera samples. n.c. is HeLa cell lysate as negative control, and (F) is the validation of immune-dominant antigens (VP0 and VP1) using RAPID-ELISA.
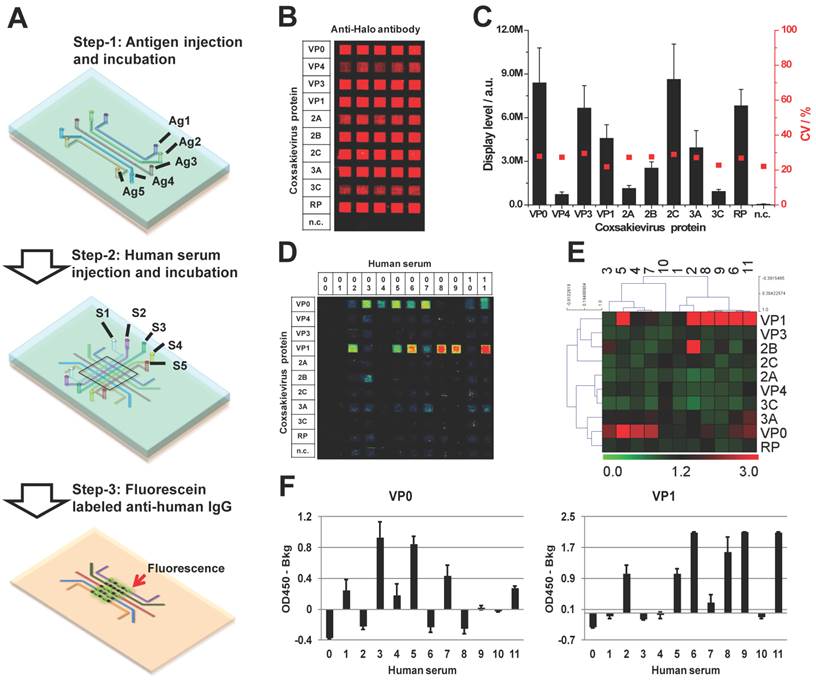
The sera samples from human subjects were employed under the approval of IRB, with the age of subjects ranging from two to thirty-one years old. To test the serological response of the subjects, the sera were diluted 10 fold in the mix of E.coli and HeLa lysates, and incubated for 3 hrs. Then 5 µL of each diluted serum was injected into each reaction channel in duplicate. After a 1 hr incubation, the reaction channels were removed and the serological antibodies were captured by the antigens, and detected with Alexa647 labeled anti-human IgG secondary antibody (Jackson Immunotech, West Grove, PA). The resulting slide was scanned using Tecan microarray scanner, and the signal was quantitated in the same manner as the NAPPA arrays. The two-way non-hierarchical cluster analysis and the generation of heat map were performed using TM4: Microarray Software Suite (http://www.tm4.org/) based on complete linkage clustering method [43, 44].
Validation of coxsackievirus immune-dominant antigens using RAPID-ELISA
The immune-dominant antigens (VP0 and VP1) were further tested in duplicate with the same sera samples using RAPID-ELISA as previously described [33]. The VP0 and VP1 proteins were produced as described above. The signals of each antigen were calculated by subtracting out the background noise produced by serum antibodies in the well without viral proteins from the original read.
Results
Construction of viral clones
We have developed a pipeline consisting of enterprise-class software and databases (FLEXGene LIMS and ACE) that are integrated with state-of-the-art robotics, including a DNA Factory (High-Res BioSolutions), BioMek FX liquid handler, and freezer storage system (Brooks Universal Biostore) [10, 17, 18, 45-48]. The streamlined integration of these systems has allowed us to manage our HT cloning projects, the DNASU plasmid repository, as well as millions of transactions in a seamless fashion (Figure 1A).
High-throughput viral gene cloning and repository construction. (A) The pipeline of high-throughput viral gene cloning, repository management and clone distribution. (B) The strategies of viral gene cloning and repository construction.
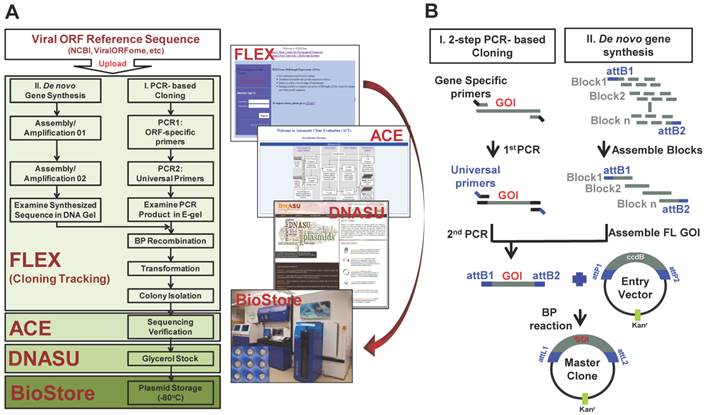
To construct our viral gene collection, we created a reference sequence library of viral genes downloaded from NCBI (http://www.ncbi.nlm.nih.gov/pubmed/) or ViralORFeome (http://pbildb1.univ-lyon1.fr/viralorfeome/index.php), and uploaded these reference sequences into our FLEXGene database (Figure 1A). FLEXGene enables us to track every construct during all stages of the cloning pipeline and storage. To capture these viral ORFs using the Gateway® cloning system, we employed two approaches: 1) HT 2-step PCR-based cloning and 2) de novo gene synthesis (Figure 1). The first approach has been frequently utilized in our lab over the past decade, and is used to clone the majority of viral genes with templates available [21, 23, 25, 40, 49-51] (Figure 1B). A representative image of 2-step PCR results is shown in Figure 2A.
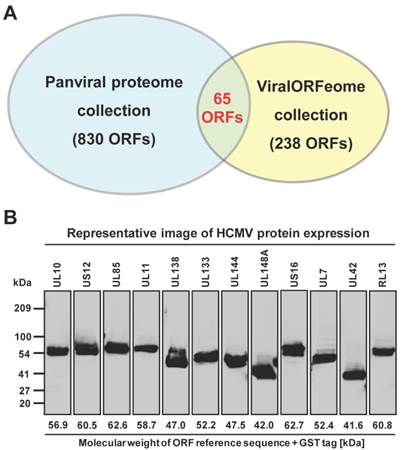
However, the transcription of a small percentage of viral genes naturally contains frameshifts and stutters, which will not produce natural gene products when used in common experimental systems. These viral ORFs have multi-fragments with various lengths at different locations in their genomes, and cannot be simply amplified by the standard 2-step PCR based cloning. In addition, there are also some viruses whose complete templates are not easily acquired because of the regulatory restrictions, high contagious risk, etc. To address these issues, we utilized de novo gene synthesis as an alternative approach to produce the viral ORF according to their documented reference sequences (Figure 1). First, we designed a series of oligos (≤ 60bp) with chunk overlaps of 20bp for the target viral ORF using oligoDesign3.0 [38]. The attB1 and reverse complementary of attB2 were added to the 5' end of the first oligo and at the end of the last oligo, respectively. Depending on the length of target sequence and user definition, the oligos can be allocated into multi-blocks. The oligos in each block were assembled and then amplified using assembly/amplification PCR (Materials and Methods). Several assembly/amplification cycles are necessary to produce the full-length viral ORFs (e.g., HBV polymerase). Each PCR product was ran on an agarose gel and examined to confirm successful synthesis (Figure 2B). After obtaining the complete ORF sequence, including the attB sites, the insert (i.e., ORF) can then be transferred into a Gateway® entry vector using the BP reaction. Next, the clones were colony selected from agar plates and the inserts were sequenced verified. The viral gene' s expected (reference) sequence was compared to its actual sequence using ACE [46], and each discrepancy (e.g., silent or missense mutations, inframe or frameshift insertions or deletions) was recorded. Only insert sequences that passed our defined criteria were accepted into our panviral plasmid collection (Materials and Methods) [20, 21]. With these approaches, we have successfully cloned 830 viral genes into the Gateway® entry vector and three mammalian expression vectors (pANT7_cGST, pJFT7_nHalo or pJFT7_cHalo) to create a total of 2,035 ORF clones (Table 1). In addition, we compared the ViralORFeome collection to ours, and found 65 genes in common (Figure 3A).
Panviral proteome has the largest viral ORF clone collection to date. (A) Comparison of the number of viral ORF clones shared by panviral proteome and viralORFeome collections. (B) Examination of the expression of HCMV ORFeome clones from panviral proteome collection using western blot. The detection of GST-protein was performed using mouse anti-GST antibody and HRP labeled sheep anti-mouse IgG antibody. The band of expressed proteins at their expected sizes was visually inspected and the results is listed in Additional file 1: Table S1.
To demonstrate the applications of our panviral proteome collection, we utilized three viral clone sets to validate protein expression, examine virus-host interactions, and monitor detection of the host antibody response to coxsackievirus antigens.
Human cytomegalovirus (HCMV) ORFeome expression
In addition to sequence verifying all clones, we sought to demonstrate the production of protein of the correct size from a subset of these clones. We selected HCMV, which has a relatively large viral genome containing 165 ORFs, and transferred the ORFs to a mammalian cell-free expression vector (pANT7_cGST). The HCMV ORFeome was expressed in vitro using a human HeLa cell lysate-based cell-free expression system, the proteins were ran on a SDS-PAGE, and the proteins were transferred onto a nitrocellulose membrane. Each expressed protein has a C-terminal GST tag, and thus can be detected using a monoclonal mouse anti-GST antibody and HRP-labeled sheep anti-mouse IgG secondary antibody (Additional file 1: Supplementary methods). A representative image of western blot detection is shown in Figure 3B. By using visual inspection, most proteins showed a band on the nitrocellulose membrane at the expected locations of their molecular weight (Additional file 1: Table S1). These results indicate that all proteins from core and tegument classes, and most proteins from other classes (capsid, secreted, envelop and membrane) as well, were successfully expressed and detected using anti-GST antibody. We failed to detect protein for six of the 165 proteins for reasons that are not clear at this time.
As an alternative method for testing protein expression, we tested the expression of rubella viral ORFeome and coxsackievirus ORFs using in-gel fluorescence, in which the newly expressed protein of interest is covalently linked to a fluorescent tag (Additional file 1: Supplementary methods). The results indicate that all ORFs from these two viruses were successfully expressed and detected with Alexa660-labeled Halo-ligand (Additional file 1: Figure S1 and Figure S3). In total we tested 182 proteins using one of these two methods, and 96.7% were detected at the correct size and at a level exceeding the background.
Rubella virus - host interactions
Rubella virus has a small genome composed of single stranded (ss+) RNA that produces only five proteins (E1, E2, capsid, P90 and P150). To demonstrate the use of our panviral proteome collection in the study of virus-host interactions, we performed an unbiased screening of potential host targets for rubella viral proteins using NAPPA arrays displaying 10,000 unique human proteins. NAPPA is an advanced cell-free protein array with high-density and wide screening ability [52, 53]. More than 2,300 cDNA constructs are printed onto an aminosiliane coated slide and are transcribed/translated in situ at the time of experimentation using a mammalian cell-free expression system. The expressed proteins are captured/displayed with high affinity and specificity by an anchored anti-tag antibody printed together with the plasmid cDNA. NAPPA has been extensively used for applications such as antibody characterization, biomarker discovery, and detection of protein-protein interactions [26, 52-54].
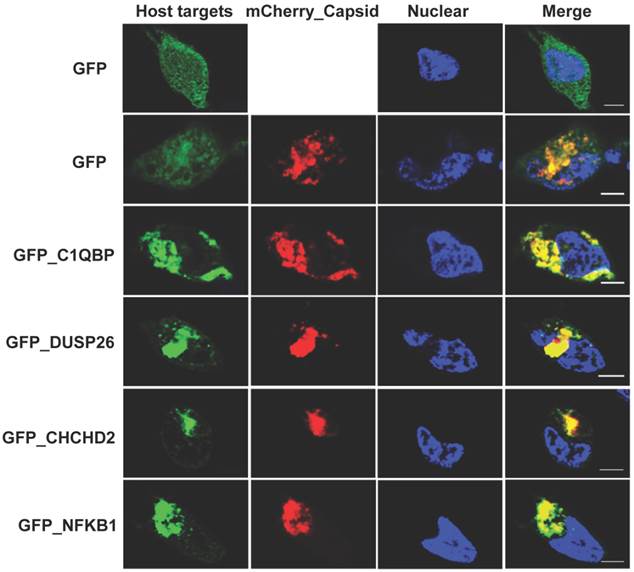
The NAPPA protein arrays were probed with each viral protein bearing an N-terminal Halo tag that can be readily detected using fluorescein labeled Halo ligand if it binds to the host protein on the arrays (Additional file 1: Figure S1). A representative image of NAPPA screening is shown in Figure 4A. The image indicates that C1QBP/p32, a known host target of capsid [55], produced a more specific and bright fluorescence spot compared to neighboring proteins and Halo protein alone as a negative control. To select additional host target candidates, we employed the following criteria: (1) Z-score ≥ 3, (2) Z-score ratio of query to negative control ≥ 2, and (3) the targets should meet the previous two criteria in two independent experiments (Materials and Methods). Using these parameters as a cut-off, a total of 56 host proteins were identified as candidate targets of rubella viral proteins (Table 3 and Figure 4). Fourteen host proteins were further selected based on their Z-score and biological relevance and validated using an independent in vitro bead based pull-down assay (Additional file 1: Supplementary methods). Among them, six viral proteins (C1QBP, CHCHD2, ZnT8, GPR1, Rab25 and Arl8A) were confirmed using in vitro based pull-down assays (Figure 4C). In addition, we transiently co-transfected HeLa cells with the mCherry tagged capsid and four GFP tagged host targets for 20 hrs, and imaged the intracellular localization of both constructs using confocal microscopy. Besides the known binding protein of C1QBP[55], we found three novel host proteins,CHCHD2, DUSP26 and NKFB1, were also co-localized with the capsid in HeLa cells (Figure 5).
Rubella virus - host interactions. (A) The representative images show the host target identified for rubella virus using NAPPA arrays displaying 10,000 human proteins. The color of the microarray spot from blue to red corresponds to the fluorescent signal from weak to strong. (B) Rubella virus - host interaction network. The host proteins with red letters are the previous known targets and solid lines indicate independent validation. The annotation of biological process was performed by the PANTHER (Protein ANalysis THrough Evolutionary Relationships) Classification System. The interaction network was constructed with Cytoscape. (C) Selective validation of host targets using bead-based pull-down assays. PD: Pull-down, WB: Western blot. Halo protein is used as a query control in both microarray and pull-down assays.
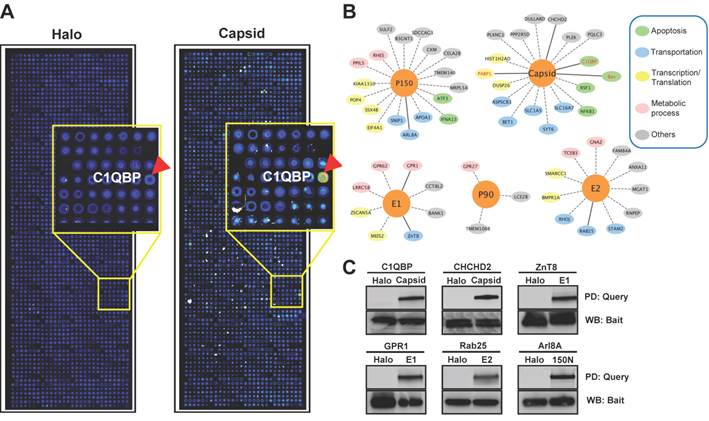
List of host target candidates for rubella virus identified from NAPPA arrays.
| No. | Host protein | Gene ID | Description | Protein class | Cellular location | Z-score | |
|---|---|---|---|---|---|---|---|
| Halo | Query | ||||||
| E1 targets | |||||||
| 1 | ZnT8 | 169026 | Zinc transporter 8 | Transporter | Cell membrane | 0.3 | 6.6 |
| 2 | CCT8L2 | 150160 | Putative T-complex protein 1 subunit theta-like 2 | Chaperonin | Cytoplasm | -0.2 | 6.3 |
| 3 | LRRC58 | 116064 | Leucine-rich repeat-containing protein 58 | Growth factor receptor | n.a. | -0.4 | 4.9 |
| 4 | GPR62 | 118442 | Probable G-protein coupled receptor 62 | G-protein coupled receptor | Cell membrane | 0.4 | 4.7 |
| 5 | MEIS2 | 4212 | Homeobox protein Meis | Homeobox transcription factor | Nucleus | 0.5 | 4.6 |
| 6 | ZSCAN5A | 79149 | Zinc finger and SCAN domain-containing protein 5A | KRAB box transcription factor | Nucleus | -0.1 | 3.8 |
| 7 | BANK1 | 55024 | B-cell scaffold protein with ankyrin repeats | n.a. | n.a. | 0.3 | 3.5 |
| 8 | GPR1 | 2825 | G-protein coupled receptor 1 | G-protein coupled receptor | Membrane | -0.5 | 3.5 |
| E2 targets | |||||||
| 9 | ANXA11 | 311 | Annexin A11 | Transfer/carrier protein | Cytoplasm; Melanosome; Nucleus | 0.9 | 9.4 |
| 10 | FAM84A | 151354 | Protein FAM84A | Acyltransferase | n.a. | -0.4 | 7.8 |
| 11 | RAB25 | 57111 | Ras-related protein Rab-25 | Small GTPase | Cell membrane; Lipid-anchor; Cytoplasmic side | 0.2 | 5.4 |
| 12 | RHOJ | 57381 | Rho-related GTP-binding protein RhoJ | Small GTPase | Cell membrane; Lipid-anchor; Cytoplasmic side | -0.2 | 5.3 |
| 13 | SMARCC1 | 6599 | SWI/SNF complex subunit SMARCC1 | Transcription cofactor | Nucleus | 0.2 | 5.0 |
| 14 | BMPR1A | 657 | Bone morphogenetic protein receptor type-1A | Serine/threonine protein kinase receptor | Membrane | 0.6 | 4.5 |
| 15 | STAM2 | 10254 | Signal transducing adapter molecule 2 | Serine/threonine protein kinase receptor | Cytoplasm | -0.1 | 4.3 |
| 16 | RNPEP | 6051 | Aminopeptidase B | Transporter, Membrane traffic protein | Secreted | -0.3 | 3.7 |
| 17 | MGAT1 | 4245 | Protein O-linked-mannose beta-1,2-N-acetylglucosaminyltransferase 1 | Glycosyltransferase | Golgi apparatus membrane | 0.1 | 3.7 |
| 18 | TCEB3 | 6924 | Transcription elongation factor B polypeptide 3 | Transcription factor | Nucleus | -0.7 | 3.5 |
| 19 | GNAZ | 2781 | Guanine nucleotide-binding protein G(z) subunit alpha | Heterotrimeric G-protein | Membrane; Lipid-anchor | 0.5 | 3.4 |
| P90 targets | |||||||
| 20 | TMEM106B | 54664 | Transmembrane protein 106B | n.a. | Late endosome membrane | -0.1 | 7.7 |
| 21 | LCE2B | 26239 | Late cornified envelope protein 2B | Structural protein | n.a. | -0.2 | 4.4 |
| 22 | GPR27 | 2850 | Probable G-protein coupled receptor 27 | G-protein coupled receptor | Membrane | 0.4 | 3.2 |
| Capsid targets | |||||||
| 23 | ASPSCR1 | 79058 | Tether containing UBX domain for GLUT4 | n.a. | Endomembrane system | 0.3 | 7.9 |
| 24 | PQLC3 | 130814 | PQ-loop repeat-containing protein 3 | n.a. | n.a. | 0.3 | 7.1 |
| 25 | PPP2R5D | 5528 | Serine/threonine-protein phosphatase 2A 56 kDa regulatory subunit delta isoform | Protein phosphatase | Cytoplasm; Nucleus | -0.5 | 5.4 |
| 26 | HIST1H2AD | 3013 | Histone H2A type 1-D | n.a. | Nucleus; Chromosome | 0.2 | 4.5 |
| 27 | RSF1 | 51773 | Remodeling and spacing factor 1 | Acetyltransferase, Transcription factor | Nucleus | 0.0 | 4.4 |
| 28 | C1QBP | 708 | Complement component 1 Q subcomponent-binding protein | Antibacterial response protein complement component | Mitochondrion matrix; Nucleus | -0.3 | 4.3 |
| 29 | NFKB1 | 4790 | Nuclear factor NF-kappa-B p105 subunit | Transcription factor | Nucleus; Cytoplasm | -0.7 | 4.3 |
| 30 | SYT6 | 148281 | Synaptotagmin-6 | Membrane trafficking regulatory protein | Cytoplasmic vesicle | 0.5 | 4.2 |
| 31 | BET1 | 10282 | BET1 homolog | SNARE protein | Golgi apparatus membrane | 0.7 | 4.1 |
| 32 | DULLARD | 23399 | CTD nuclear envelope phosphatase 1 | Serine/threonine protein phosphatase | Endoplasmic reticulum membrane | -0.7 | 3.9 |
| 33 | DUSP26 | 78986 | Dual specificity protein phosphatase 26 | Protein phosphatase | Cytoplasm; Nucleus; Golgi apparatus | -0.5 | 3.8 |
| 34 | PLEK | 5341 | Pleckstrin | Cytoskeletal protein | n.a. | -0.6 | 3.5 |
| 35 | PLXNC1 | 10154 | Plexin-C1 | Tyrosine protein kinase receptor, Protein kinase | Membrane | -0.3 | 3.4 |
| 36 | CHCHD2 | 51142 | Coiled-coil-helix-coiled-coil-helix domain-containing protein 2, mitochondrial | n.a. | Mitochondrion | 0.0 | 3.3 |
| 37 | SLC1A5 | 6510 | Neutral amino acid transporter B(0) | Cation transporter | Membrane | -0.4 | 3.1 |
| 38 | SLC16A7 | 9194 | Monocarboxylate transporter 2 | Transporter | Membrane | -0.5 | 3.1 |
| P150 targets | |||||||
| 39 | ATF3 | 467 | Cyclic AMP-dependent transcription factor ATF-3 | CREB transcription factor | Nucleus | 3.9 | 11.7 |
| 40 | RHES | 23551 | GTP-binding protein Rhes | Small GTPase | Cell membrane; Lipid-anchor | 3.3 | 9.3 |
| 41 | CKM | 1158 | Creatine kinase M-type | Amino acid kinase | Cytoplasm. | 0.0 | 7.0 |
| 42 | APOA1 | 335 | Apolipoprotein A-I | Transporter | Secreted | 2.0 | 6.4 |
| 43 | TMEM140 | 55281 | Transmembrane protein 140 | n.a. | Membrane | -0.1 | 6.0 |
| 44 | PPIL5 | 122769 | Leucine-rich repeat protein 1 | Growth factor receptor, adenylate cyclase | n.a. | 0.0 | 5.6 |
| 45 | B3GNT1 | 11041 | N-acetyllactosaminide beta-1,3-N-acetylglucosaminyltransferase | Glycosyltransferase | Golgi apparatus membrane | 1.8 | 5.6 |
| 46 | POP4 | 10775 | Ribonuclease P protein subunit p29 | Endoribonuclease | Nucleus | 1.8 | 5.5 |
| 47 | SDCCAG3 | 10807 | Serologically defined colon cancer antigen 3 | n.a. | Cytoplasm | 1.6 | 5.1 |
| 48 | SSX4B | 548313 | Protein SSX4 | Transcription factor | n.a. | 1.2 | 4.1 |
| 49 | KIAA1310 | 55683 | KAT8 regulatory NSL complex subunit 3 | n.a. | Nucleus | 0.9 | 4.0 |
| 50 | IFNA13 | 3447 | Interferon alpha-1/13 | Interferon superfamily | Secreted | 0.4 | 3.9 |
| 51 | CELA2B | 51032 | Chymotrypsin-like elastase family member 2B | Serine protease | Secreted | 0.4 | 3.5 |
| 52 | ARL8A | 127829 | ADP-ribosylation factor-like protein 8A | Small GTPase | Late endosome membrane; Lysosome membrane | 0.2 | 3.4 |
| 53 | SNIP1 | 79753 | Smad nuclear-interacting protein 1 | Transporter | Nucleus | 0.2 | 3.4 |
| 54 | EIF4A1 | 1973 | Eukaryotic initiation factor 4A-I | RNA helicase, translation initiation factor helicase | n.a. | 0.7 | 3.3 |
| 55 | SULF2 | 55959 | Extracellular sulfatase Sulf-2 | Hydrolase | Endoplasmic reticulum | 0.4 | 3.3 |
| 56 | MRPL54 | 116541 | 39S ribosomal protein L54, mitochondrial | n.a. | Mitochondrion | 0.1 | 3.2 |
*n.a.: not available.
Co-localization analysis of rubella virus capsid and its host targets in HeLa cells. The co-transfection of HeLa cells were executed for 20 hrs using the constructs of mChery tagged capsid (Red) and GFP tagged host targets (Green). The cells were fixed in 4% paraformaldehyde and imaged using confocal fluorescence microscopy. The nucleus was stained with Hoechst dye and shown as blue. Scale bar, 5µm.
Host serological response to coxsackievirus proteins
As a member of Enterovirus family, Coxsackievirus B4 is one of the six serotypes of coxsackievirus group B. The coxsackievirus B infection is common in the summer and autumn in the areas of temperate climates and all year in tropics. Adolescents less than 16 years old are the susceptible population, but infection has been also observed in adults [56]. To investigate whether coxsackievirus proteins can serve as antigens inducing a humoral response in the blood of people with potential coxsackievirus infection, we performed a serological test using micro-fluidic multiplexed immunoassays. The experimental setup is shown in Figure 6A. First, we coated 11 micro-fluidic channels with unpurified HeLa cell lysate expressing each of 10 coxsackievirus antigens, and one with HeLa cell lysate alone as negative control (n.c). The immobilization of these antigens (rows) was confirmed using anti-Halo tag antibody (columns). These results indicate that all 10 antigens were successfully coated on their corresponding channels and are observed as specific red fluorescent signals, which do not exist in the channel coated with only HeLa lysates (n.c.) (Figure 6B). Further signal quantification shows that the signal intensity of coated coxsackievirus antigens is higher than that of the negative control plus three folds of the standard deviations with an average variation of 26.3% (Figure 6C).
To detect the serological antibodies, we randomly selected 11 sera samples regardless of their disease background. The prepared antigen array was incubated using 5 µL of 1:10 diluted human serum in each channel and the detection was performed using an Alexa647 labeled anti-human IgG secondary antibody. VP1 and VP0 showed responses in the largest number of subjects, each showing responses in 50% of the subjects; although, VP1 had the strongest signal intensity (Figure 6D). Notably, there was not complete overlap in the responses to these two antigens, with some subjects responding to one but not the other. We did not observe any signal in the n.c. from either antigens or sera samples (#00), which demonstrate the specificity of these antibodies. The separation between VP0 and VP1 responders was illustrated by performing a two-way non-hierarchical cluster analysis and populating a heat-map (Figure 6E). Most of antigens (VP3, 2C, 2A, VP4, 3C and RP) did not show any response in these sera samples. The immune-dominant antigens of VP0 and VP1 were further validated using ELISA with the same sera samples (Figure 6F). Without clinical histories and with such a small sample size, we cannot draw specific conclusions regarding immunoproteomic responses to coxsackievirus B infections. Nevertheless, these data illustrate the rapid feasibility of using a microfluidic approach to address this question.
Discussion
During the past decade we have developed a HT cloning and management pipeline, which integrates customized software, databases, and state-of-art robotics (Figure 1). More specifically, the FLEXGene database is used to track the overall cloning pipeline starting from the uploading of reference sequences, through transferring ORFs into the Gateway® entry vector and destination vectors, sequence verification, preparation of glycerol stock, to the end with the submission of final information and plasmid samples to DNASU [17, 18]. ACE automates the sequence evaluation process, enabling the management of hundreds of clones simultaneously by interacting with FLEXGene and comparing each clone's actual and reference sequences using defined criteria to find discrepancies [20, 21, 46]. DNASU is a central repository of plasmids which stores over 200,000 plasmids from more than ~800 organisms [10, 14]. These plasmids are easily searchable through the website http://dnasu.org, and DNASU operations distribute these plasmids to researchers worldwide upon request. Each plasmid is stored in a 2-D barcoded tube as a glycerol stock in a fully automated -80°C Brooks Biostore freezer allowing fast retrieval upon ordering. These collections have demonstrated their great value in numerous biological studies in basic research, cancer, autoimmune and infectious disease [26, 27, 31, 34, 35, 52, 53, 57-60].
In this work, we have integrated viral cloning into the pipeline that has already been extensively used for human and bacterial gene cloning. The difference from previous non-viral cloning is that we employ gene synthesis for those viral ORFs without templates, or whose reference polypeptide sequence is not contiguous in the genome (Table 2). The cost of gene synthesis is much higher than the 2-step PCR-based approach because of the high cost of the oligos required for synthesis. Even with local synthesis of the oligos using an oligo synthesizer, which would reduce the cost, this method is likely to be more expensive than the PCR approach. By using the GeneDesign 3.0 and the protocol we developed, we successfully obtained 100% of the genes we attempted to synthesize, with DNA lengths ranging from 200 bp to 3,000 bp (Figure 2B).
Compare the PCR-based cloning and de novo gene synthesis.
| PCR-based cloning | De novo gene synthesis | |
|---|---|---|
| Target viral gene | With template | Without template or the viral ORF reference sequence is not contiguous in the genome |
| Safety requirement | High | Low |
| Throughput | High | Low |
| Procedure | 2-step PCRs | Depending on the length of target ORF, and may need multi-assembly/amplification cycles |
| Time | ~ 6 hours | ~ 6 hours /cycle |
| Cost | Low | High, but can be significantly alleviated by synthesizing oligos in the lab |
The successful translation of these viral clones to proteins is the key to study virus-host interactions, as well as their association with human diseases [2]. To illustrate this approach, we chose three viruses, two (HCMV and rubella virus) with complete ORFeomes and one (coxsackievirus) with 10/12 of ORFeome clones. We tested this virus-host interaction using a human HeLa cell lysate-based cell-free expression system. We selected this human cell-free expression system because of its inherent advantages of high efficiency and effectiveness, and its ability to produce membrane and large size proteins [61, 62]. It has been recently employed in our lab in the fabrication of high-density NAPPA arrays [62, 63]. With this system, we found 96.4% HCMV and 100% of rubella virus and coxsackievirus ORFeomes were successful expressed (Figure 3B, Additional file 1: Figure S1, Figure S3 and Table S1). This corresponds favorably to the previous results using wheat-germ and rabbit reticulocyte lysate cell-free expression systems [52, 64]. These results demonstrate the ease and efficiency of our viral collection in the production of viral proteins by combination with human cell-free expression system in vitro.
The comprehensive collection of viral ORFeome clones enables us to study virus-host interaction from a systematic standpoint, and reveals new host cellular proteins that might be hijacked by viruses to alter host cellular processes. Rubella virus is the causative agent of German measles in children and congenital rubella syndrome when the virus infection occurs during the first trimester of pregnancy. In addition, it can cause polyarthralgia, a complication of rubella infection in adult women. Humans are the only known host of rubella virus, and the development of effective vaccines have significantly reduced the incidence in developed countries [65]. However, the molecular mechanisms of rubella infection/replication in the host, and the causation of congenital rubella syndrome are still unclear. There have been a few host proteins, C1QBP/p32, Bax and RABP, identified to be involved in host-rubella interactions [55, 66-68]. In this work, we screened all rubella viral proteins on NAPPA arrays with 10,000 human proteins, and identified 55 novel candidate host targets in addition to the three previously identified interactors [66-68]. Six viral proteins were selected and validated using in vitro based pull-down, and four viral proteins were used in in vivo based cellular co-localization analysis (Figure 4C and Figure 5). In our intracellular localization studies, we found that the rubella proteins locate to different sub-cellular locations (i.e. membrane (36%), nucleus (19%), cytoplasm (16%), secreted (5%), mitochondrion (3%), endoplasmic reticulum (2%) and others (14%)) (Figure 4B, Additional file 1: Figure S2 and Table 3).
With all of these identified targets, we constructed the first systematic rubella virus-host interaction network, and analyzed the biological processes of these host targets associated in host cell (Figure 4B). The annotation results indicate that four capsid targets, C1QBP, Bax, RSF1 and NFKB1, and two P150 targets, ATF3 and IFNA13, are involved in apoptosis, which is consistent with the cytotoxicity caused by rubella virus [68, 69]. Other host targets of E1, E2, capsid and P150 are associated with transport, transcription/translation and metabolic processes. P90 has the fewest targets associated with metabolic processes [55, 70].
E1 is the main surface viral protein that might co-opt the host G-protein-coupled receptors and activate their downstream pathways through GPR1 and GRP62 [71, 72]. The functions of E2 and P90 are largely unknown; although, it was found that the glycosylation of E2 could alter the specific membrane fusion using rubella virus JR23 strain [73]. Here, we provide new evidence that E2 and P90 may participate in the viral attachment and fusion between viral envelope and the endosomal membrane through membrane proteins of GNAZ, TMEM106B and GPR27, and regulate the cytoskeleton by RhoJ [72, 74]. In addition, E2 and P150 may be involved in the viral vesicle transportation through Rab and Arl GTPases of Rab25 and Arl8A [75, 76]. Viral capsid is the only protein that has been found with multi-functions, which include the regulation of viral replication and acting as a potent inhibitor of host cell apoptosis and protein synthesis [55, 66, 68]. Here we found, besides C1QBP, that the capsid may also interact with other host cell proteins in membranes, cytoplasm, mitochondria, endoplasmic reticulum, Golgi, and nucleus, and can include such host proteins as PLXNC1, CHCHD2, BET1, NFKB1, etc. [77, 78]. These results need additional confirmation with cell-based in vivo assays; however, these results give us a first glimpse of a comprehensive view of rubella-virus interactions by physical interactions. In future functional studies, these results may reveal important host proteins involved in the regulation of rubella viral invasion, transportation, and replication.
Most of coxsackievirus B4 infection exists as asymptomatic, undifferentiated febrile illness, or mild upper respiratory symptoms. But this virus can induce a wide range of diseases such as aseptic meningitis, encephalitis, pleurodynia, myocarditis, and pericarditis [79-82]. The development of a rapid serological test and vaccine would be a valuable tool in the early diagnosis and prevention of coxsackievirus [83]. In this work, we tested the antigenicity of coxsackievirus proteins using a micro-fluidic multiplexed immunoassay which can detect up to 24 antigens simultaneously with only 0.5 µL undiluted human serum [42]. We found the unpurified coxsackievirus proteins in HeLa cell lysates can be coated on the amino modified slide surface and detected with anti-Halo tag antibody (Figure 6B and 6C). The uses of unpurified cell-free produced proteins have been frequently employed in the fabrication of pathogen microarrays to screen and validate antibody biomarkers of infectious diseases [84-87]. The key step of this approach is to block the non-specific bindings of serum antibodies to background proteins originating from HeLa cell lysates or E.coli during DNA preparation [88]. Here, we found by blocking the sera using a mix of HeLa cell and E.coli lysates in 5% milk, the background was largely decreased and the signal to noise ratio was improved. The screening of 11 sera samples using microfluidic multiplexed immunoassay and ELISA, revealed that VP0 and VP1 might be two dominant antigens which are recognized by the host immune system (Figure 6D-F). The differential antigenicity of VP0 and VP1 implies that these two antigens might be potential markers that have complementary value in the diagnostics of coxsackievirus infection as well as development of a new vaccine. However, further experiments with much larger well-characterized clinical sample sets are needed to establish this relationship.
Conclusion
In this work, we describe the initial effort of our panviral proteome with the collection of 2,035 viral OFR clones from 30 viral species. We demonstrated the uses of our viral collection in the highly efficient production of viral proteins using cell-free expression system in vitro, global identification of host targets for rubella virus using NAPPA protein arrays, and detection of host serological response using micro-fluidic multiplexed immunoassays. In the future, we will be continuing to collaborate with other labs to extend our collection, study viral invasion/evasion mechanisms, develop novel diagnostic tools, and drugs for the therapeutic treatment.
Supplementary Materials
Additional File 1Supplementary methods, Figure S1-S3 and Table S1.
Acknowledgements
This work was supported in part by JDRF funded research 5-2012-537. We thank Marc Vidal and David Hill (Dana-Farber Cancer Institute), Jürgen Haas (University of Edinburgh), Vincent Lotteau (Université de Lyon), Frédéric Tangy and Pierre Legrain (Institut Pasteur), Merja Roivainen (National Institute for Health and Welfare, Helsinki, Finland), Tom C. Hobman (University of Alberta), Biao He (University of Georgia), Grant McFadden (University of Florida) and other people for their kindness to provide RNA, DNA and ORF templates. We thank Decker Kimberly and Matthias P Machner (NICHD) for providing the pcDNATM6.2/N-EmGFP/YFP-DEST and pCS Cherry DEST vectors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ahn AC, Tewari M, Poon CS, Phillips RS. The limits of reductionism in medicine: could systems biology offer an alternative? PLoS Med. 2006;3:e208
2. Uetz P, Rajagopala SV, Dong YA, Haas J. From ORFeomes to protein interaction maps in viruses. Genome Res. 2004;14:2029-33
3. Simonis N, Rual JF, Lemmens I, Boxus M, Hirozane-Kishikawa T, Gatot JS. et al. Host-pathogen interactome mapping for HTLV-1 and -2 retroviruses. Retrovirology. 2012;9:26
4. Rozenblatt-Rosen O, Deo RC, Padi M, Adelmant G, Calderwood MA, Rolland T. et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature. 2012;487:491-5
5. Ran X, Bian X, Ji Y, Yan X, Yang F, Li F. White spot syndrome virus IE1 and WSV056 modulate the G1/S transition by binding to the host retinoblastoma protein. J Virol. 2013;87:12576-82
6. Temple G, Gerhard DS, Rasooly R, Feingold EA, Good PJ, Robinson C. et al. The completion of the Mammalian Gene Collection (MGC). Genome Res. 2009;19:2324-33
7. Yang X, Boehm JS, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R. et al. A public genome-scale lentiviral expression library of human ORFs. Nat Methods. 2011;8:659-61
8. Matsuyama A, Arai R, Yashiroda Y, Shirai A, Kamata A, Sekido S. et al. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol. 2006;24:841-7
9. Strausberg RL, Feingold EA, Klausner RD, Collins FS. The mammalian gene collection. Science. 1999;286:455-7
10. Cormier CY, Mohr SE, Zuo D, Hu Y, Rolfs A, Kramer J. et al. Protein Structure Initiative Material Repository: an open shared public resource of structural genomics plasmids for the biological community. Nucleic Acids Res. 2010;38:D743-9
11. Seiler CY, Park JG, Sharma A, Hunter P, Surapaneni P, Sedillo C. et al. DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Res. 2013
12. Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N. et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173-8
13. Lamesch P, Li N, Milstein S, Fan C, Hao T, Szabo G. et al. hORFeome v3.1: a resource of human open reading frames representing over 10,000 human genes. Genomics. 2007;89:307-15
14. Festa F, Steel J, Bian X, Labaer J. High-throughput cloning and expression library creation for functional proteomics. Proteomics. 2013;13:1381-99
15. Pellet J, Tafforeau L, Lucas-Hourani M, Navratil V, Meyniel L, Achaz G. et al. ViralORFeome: an integrated database to generate a versatile collection of viral ORFs. Nucleic Acids Res. 2010;38:D371-8
16. Navratil V, de Chassey B, Meyniel L, Delmotte S, Gautier C, Andre P. et al. VirHostNet: a knowledge base for the management and the analysis of proteome-wide virus-host interaction networks. Nucleic Acids Res. 2009;37:D661-8
17. Brizuela L, Braun P, LaBaer J. FLEXGene repository: from sequenced genomes to gene repositories for high-throughput functional biology and proteomics. Mol Biochem Parasitol. 2001;118:155-65
18. Brizuela L, Richardson A, Marsischky G, Labaer J. The FLEXGene repository: exploiting the fruits of the genome projects by creating a needed resource to face the challenges of the post-genomic era. Arch Med Res. 2002;33:318-24
19. Witt AE, Hines LM, Collins NL, Hu Y, Gunawardane RN, Moreira D. et al. Functional proteomics approach to investigate the biological activities of cDNAs implicated in breast cancer. J Proteome Res. 2006;5:599-610
20. Rolfs A, Montor WR, Yoon SS, Hu Y, Bhullar B, Kelley F. et al. Production and sequence validation of a complete full length ORF collection for the pathogenic bacterium Vibrio cholerae. Proc Natl Acad Sci U S A. 2008;105:4364-9
21. Rolfs A, Hu Y, Ebert L, Hoffmann D, Zuo D, Ramachandran N. et al. A biomedically enriched collection of 7000 human ORF clones. PLoS One. 2008;3:e1528
22. Montor WR, Huang J, Hu Y, Hainsworth E, Lynch S, Kronish JW. et al. Genome-wide study of Pseudomonas aeruginosa outer membrane protein immunogenicity using self-assembling protein microarrays. Infect Immun. 2009;77:4877-86
23. Hu Y, Rolfs A, Bhullar B, Murthy TV, Zhu C, Berger MF. et al. Approaching a complete repository of sequence-verified protein-encoding clones for Saccharomyces cerevisiae. Genome Res. 2007;17:536-43
24. Pearlberg J, Degot S, Endege W, Park J, Davies J, Gelfand E. et al. Screens using RNAi and cDNA expression as surrogates for genetics in mammalian tissue culture cells. Cold Spring Harb Symp Quant Biol. 2005;70:449-59
25. Aguiar JC, LaBaer J, Blair PL, Shamailova VY, Koundinya M, Russell JA. et al. High-throughput generation of P. falciparum functional molecules by recombinational cloning. Genome Res. 2004;14:2076-82
26. Miersch S, Bian X, Wallstrom G, Sibani S, Logvinenko T, Wasserfall CH. et al. Serological autoantibody profiling of type 1 diabetes by protein arrays. J Proteomics. 2013
27. Wright C, Sibani S, Trudgian D, Fischer R, Kessler B, LaBaer J. et al. Detection of multiple autoantibodies in patients with ankylosing spondylitis using nucleic acid programmable protein arrays. Mol Cell Proteomics. 2012;11:M9 00384
28. Manzano-Roman R, Diaz-Martin V, Gonzalez-Gonzalez M, Matarraz S, Alvarez-Prado AF, LaBaer J. et al. Self-assembled protein arrays from an Ornithodoros moubata salivary gland expression library. J Proteome Res. 2012;11:5972-82
29. Gibson DS, Rooney ME, Finnegan S, Qiu J, Thompson DC, Labaer J. et al. Biomarkers in rheumatology, now and in the future. Rheumatology (Oxford). 2012;51:423-33
30. Gibson DS, Qiu J, Mendoza EA, Barker K, Rooney ME, LaBaer J. Circulating and synovial antibody profiling of juvenile arthritis patients by nucleic acid programmable protein arrays. Arthritis Res Ther. 2012;14:R77
31. Anderson KS, Sibani S, Wallstrom G, Qiu J, Mendoza EA, Raphael J. et al. Protein microarray signature of autoantibody biomarkers for the early detection of breast cancer. J Proteome Res. 2011;10:85-96
32. Ceroni A, Sibani S, Baiker A, Pothineni VR, Bailer SM, LaBaer J. et al. Systematic analysis of the IgG antibody immune response against varicella zoster virus (VZV) using a self-assembled protein microarray. Mol Biosyst. 2010;6:1604-10
33. Ramachandran N, Anderson KS, Raphael JV, Hainsworth E, Sibani S, Montor WR. et al. Tracking humoral responses using self assembling protein microarrays. Proteomics Clin Appl. 2008;2:1518-27
34. Zhu C, Byers KJ, McCord RP, Shi Z, Berger MF, Newburger DE. et al. High-resolution DNA-binding specificity analysis of yeast transcription factors. Genome Res. 2009;19:556-66
35. Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B. et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324-8
36. Ramirez AB, Loch CM, Zhang Y, Liu Y, Wang X, Wayner EA. et al. Use of a single-chain antibody library for ovarian cancer biomarker discovery. Mol Cell Proteomics. 2010;9:1449-60
37. Richardson SM, Liu S, Boeke JD, Bader JS. Design-A-Gene with GeneDesign. Methods Mol Biol. 2012;852:235-47
38. Richardson SM, Nunley PW, Yarrington RM, Boeke JD, Bader JS. GeneDesign 3.0 is an updated synthetic biology toolkit. Nucleic Acids Res. 2010;38:2603-6
39. Park J, Labaer J. Recombinational cloning. Curr Protoc Mol Biol. 2006 Chapter 3: Unit 3 20
40. Murthy T, Rolfs A, Hu Y, Shi Z, Raphael J, Moreira D. et al. A full-genomic sequence-verified protein-coding gene collection for Francisella tularensis. PLoS One. 2007;2:e577
41. Anderson KS, Sibani S, Wallstrom G, Qiu J, Mendoza EA, Raphael J. et al. Protein Microarray Signature of Autoantibody Biomarkers for the Early Detection of Breast Cancer. J Proteome Res. 2010
42. Song L, Zhang Y, Wang W, Ma L, Liu Y, Hao Y. et al. Microfluidic assay without blocking for rapid HIV screening and confirmation. Biomed Microdevices. 2012;14:631-40
43. Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N. et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374-8
44. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863-8
45. Hu Y, Labaer J. Tracking gene-disease relationships for high-throughput functional studies. Surgery. 2004;136:504-10
46. Taycher E, Rolfs A, Hu Y, Zuo D, Mohr SE, Williamson J. et al. A novel approach to sequence validating protein expression clones with automated decision making. BMC Bioinformatics. 2007;8:198
47. Zuo D, Mohr SE, Hu Y, Taycher E, Rolfs A, Kramer J. et al. PlasmID: a centralized repository for plasmid clone information and distribution. Nucleic Acids Res. 2007;35:D680-4
48. Cormier CY, Park JG, Fiacco M, Steel J, Hunter P, Kramer J. et al. PSI:Biology-materials repository: a biologist's resource for protein expression plasmids. J Struct Funct Genomics. 2011;12:55-62
49. Park J, Hu Y, Murthy TV, Vannberg F, Shen B, Rolfs A. et al. Building a human kinase gene repository: bioinformatics, molecular cloning, and functional validation. Proc Natl Acad Sci U S A. 2005;102:8114-9
50. Marsischky G, LaBaer J. Many paths to many clones: a comparative look at high-throughput cloning methods. Genome Res. 2004;14:2020-8
51. Labaer J, Qiu Q, Anumanthan A, Mar W, Zuo D, Murthy TV. et al. The Pseudomonas aeruginosa PA01 gene collection. Genome Res. 2004;14:2190-200
52. Ramachandran N, Raphael JV, Hainsworth E, Demirkan G, Fuentes MG, Rolfs A. et al. Next-generation high-density self-assembling functional protein arrays. Nat Methods. 2008;5:535-8
53. Ramachandran N, Hainsworth E, Bhullar B, Eisenstein S, Rosen B, Lau AY. et al. Self-assembling protein microarrays. Science. 2004;305:86-90
54. Wright C, Sibani S, Trudgian D, Fischer R, Kessler B, Labaer J. et al. Detection of multiple autoantibodies in patients with ankylosing spondylitis using nucleic acid programmable protein arrays. Mol Cell Proteomics. 2010
55. Beatch MD, Everitt JC, Law LJ, Hobman TC. Interactions between rubella virus capsid and host protein p32 are important for virus replication. J Virol. 2005;79:10807-20
56. Knowlton KU. CVB infection and mechanisms of viral cardiomyopathy. Curr Top Microbiol Immunol. 2008;323:315-35
57. Seton-Rogers SE, Lu Y, Hines LM, Koundinya M, LaBaer J, Muthuswamy SK. et al. Cooperation of the ErbB2 receptor and transforming growth factor beta in induction of migration and invasion in mammary epithelial cells. Proc Natl Acad Sci U S A. 2004;101:1257-62
58. Thanawastien A, Montor WR, Labaer J, Mekalanos JJ, Yoon SS. Vibrio cholerae proteome-wide screen for immunostimulatory proteins identifies phosphatidylserine decarboxylase as a novel Toll-like receptor 4 agonist. PLoS Pathog. 2009;5:e1000556
59. Zhang L, Villa NY, Rahman MM, Smallwood S, Shattuck D, Neff C. et al. Analysis of vaccinia virus-host protein-protein interactions: validations of yeast two-hybrid screenings. J Proteome Res. 2009;8:4311-8
60. Gonzalez-Malerva L, Park J, Zou L, Hu Y, Moradpour Z, Pearlberg J. et al. High-throughput ectopic expression screen for tamoxifen resistance identifies an atypical kinase that blocks autophagy. Proc Natl Acad Sci U S A. 2011
61. Reckel S, Sobhanifar S, Durst F, Lohr F, Shirokov VA, Dotsch V. et al. Strategies for the cell-free expression of membrane proteins. Methods Mol Biol. 2010;607:187-212
62. Festa F, Rollins SM, Vattem K, Hathaway M, Lorenz P, Mendoza EA. et al. Robust microarray production of freshly expressed proteins in a human milieu. Proteomics Clin Appl. 2013;7:372-7
63. Wang J, Barker K, Steel J, Park J, Saul J, Festa F. et al. A versatile protein microarray platform enabling antibody profiling against denatured proteins. Proteomics Clin Appl. 2013;7:378-83
64. Goshima N, Kawamura Y, Fukumoto A, Miura A, Honma R, Satoh R. et al. Human protein factory for converting the transcriptome into an in vitro-expressed proteome. Nat Methods. 2008;5:1011-7
65. Reef SE, Strebel P, Dabbagh A, Gacic-Dobo M, Cochi S. Progress toward control of rubella and prevention of congenital rubella syndrome--worldwide, 2009. J Infect Dis. 2011;204(Suppl 1):S24-7
66. Ilkow CS, Mancinelli V, Beatch MD, Hobman TC. Rubella virus capsid protein interacts with poly(a)-binding protein and inhibits translation. J Virol. 2008;82:4284-94
67. Beatch MD, Hobman TC. Rubella virus capsid associates with host cell protein p32 and localizes to mitochondria. J Virol. 2000;74:5569-76
68. Ilkow CS, Goping IS, Hobman TC. The Rubella virus capsid is an anti-apoptotic protein that attenuates the pore-forming ability of Bax. PLoS Pathog. 2011;7:e1001291
69. Duncan R, Esmaili A, Law LM, Bertholet S, Hough C, Hobman TC. et al. Rubella virus capsid protein induces apoptosis in transfected RK13 cells. Virology. 2000;275:20-9
70. Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science. 2009;324:1327-30
71. Sodhi A, Montaner S, Gutkind JS. Viral hijacking of G-protein-coupled-receptor signalling networks. Nat Rev Mol Cell Biol. 2004;5:998-1012
72. Zhen Z, Bradel-Tretheway B, Sumagin S, Bidlack JM, Dewhurst S. The human herpesvirus 6 G protein-coupled receptor homolog U51 positively regulates virus replication and enhances cell-cell fusion in vitro. J Virol. 2005;79:11914-24
73. Wu B, Liu X, Wang Z. Effects of E2 and E1 glycosylation on specific membrane fusion in rubella virus strain JR23. Intervirology. 2009;52:68-77
74. Ho H, Soto Hopkin A, Kapadia R, Vasudeva P, Schilling J, Ganesan AK. RhoJ modulates melanoma invasion by altering actin cytoskeletal dynamics. Pigment Cell Melanoma Res. 2013;26:218-25
75. Kessler D, Gruen GC, Heider D, Morgner J, Reis H, Schmid KW. et al. The action of small GTPases Rab11 and Rab25 in vesicle trafficking during cell migration. Cell Physiol Biochem. 2012;29:647-56
76. Hofmann I, Munro S. An N-terminally acetylated Arf-like GTPase is localised to lysosomes and affects their motility. J Cell Sci. 2006;119:1494-503
77. Kung CP, Raab-Traub N. Epstein-Barr virus latent membrane protein 1 modulates distinctive NF- kappaB pathways through C-terminus-activating region 1 to regulate epidermal growth factor receptor expression. J Virol. 2010;84:6605-14
78. Zhang T, Hong W. Ykt6 forms a SNARE complex with syntaxin 5, GS28, and Bet1 and participates in a late stage in endoplasmic reticulum-Golgi transport. J Biol Chem. 2001;276:27480-7
79. Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med. 1998;4:781-5
80. Huber S, Ramsingh AI. Coxsackievirus-induced pancreatitis. Viral Immunol. 2004;17:358-69
81. Chia JK. The role of enterovirus in chronic fatigue syndrome. J Clin Pathol. 2005;58:1126-32
82. Tan EL, Wong AP, Poh CL. Development of potential antiviral strategy against coxsackievirus B4. Virus Res. 2010;150:85-92
83. Xu F, He D, He S, Wu B, Guan L, Niu J. et al. Development of an IgM-capture ELISA for Coxsackievirus A16 infection. J Virol Methods. 2011;171:107-10
84. Lee SJ, Liang L, Juarez S, Nanton MR, Gondwe EN, Msefula CL. et al. Identification of a common immune signature in murine and human systemic Salmonellosis. Proc Natl Acad Sci U S A. 2012;109:4998-5003
85. Vigil A, Chen C, Jain A, Nakajima-Sasaki R, Jasinskas A, Pablo J. et al. Profiling the humoral immune response of acute and chronic Q fever by protein microarray. Mol Cell Proteomics. 2011;10:M110 006304
86. Trieu A, Kayala MA, Burk C, Molina DM, Freilich DA, Richie TL. et al. Sterile protective immunity to malaria is associated with a panel of novel P. falciparum antigens. Mol Cell Proteomics. 2011;10:M111 007948
87. Kunnath-Velayudhan S, Salamon H, Wang HY, Davidow AL, Molina DM, Huynh VT. et al. Dynamic antibody responses to the Mycobacterium tuberculosis proteome. Proc Natl Acad Sci U S A. 2010;107:14703-8
88. Davies DH, Liang X, Hernandez JE, Randall A, Hirst S, Mu Y. et al. Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proc Natl Acad Sci U S A. 2005;102:547-52
Author contact
![]() Corresponding author: J. L. (jlabaeredu).
Corresponding author: J. L. (jlabaeredu).