Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Materials and methods
3. Results
4. Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(8):805-817. doi:10.7150/thno.11520 This issue Cite
Research Paper
Bioluminescence-Activated Deep-Tissue Photodynamic Therapy of Cancer
Yi Rang Kim1,2,3, Seonghoon Kim1, Jin Woo Choi4,5, Sung Yong Choi2, Sang-Hee Lee2, Homin Kim2, Sei Kwang Hahn6, Gou Young Koh1,2, Seok Hyun Yun1,5,7 ![]()
1. Graduate School of Nanoscience and Technology (WCU), Korea Advanced Institute of Science and Technology, 291 Daehak-Ro, Yusong-Gu, Daejon 305-701, Korea
2. Graduate School of Medical Science and Engineering, Korea Advanced Institute of Science and Technology, 291 Daehak-Ro, Yusong-Gu, Daejon 305-701, Korea
3. Department of Oncology, Asan Medical Center, Univ. Ulsan College of Medicine, Seoul , Korea
4. Department of Pharmacology, Wonkwang Institute of Dental Research, School of Dentistry, Wonkwang University, Iksan, Chonbuk, 570-749, Korea
5. Wellman Center for Photomedicine, Massachusetts General Hospital, 65 Landsdowne St. UP-525, Cambridge, MA 02139, USA
6. Department of Materials Science and Engineering, Pohang University of Science and Technology (POSTECH), 77 Cheongam-ro, Nam-gu, Pohang, Gyeongbuk 790-784, Korea.
7. Department of Dermatology, Harvard Medical School, Massachusetts General Hospital, 40 Blossom St. Boston, MA 02114, USA
Received 2015-1-7; Accepted 2015-3-23; Published 2015-4-18
Abstract

Optical energy can trigger a variety of photochemical processes useful for therapies. Owing to the shallow penetration of light in tissues, however, the clinical applications of light-activated therapies have been limited. Bioluminescence resonant energy transfer (BRET) may provide a new way of inducing photochemical activation. Here, we show that efficient bioluminescence energy-induced photodynamic therapy (PDT) of macroscopic tumors and metastases in deep tissue. For monolayer cell culture in vitro incubated with Chlorin e6, BRET energy of about 1 nJ per cell generated as strong cytotoxicity as red laser light irradiation at 2.2 mW/cm2 for 180 s. Regional delivery of bioluminescence agents via draining lymphatic vessels killed tumor cells spread to the sentinel and secondary lymph nodes, reduced distant metastases in the lung and improved animal survival. Our results show the promising potential of novel bioluminescence-activated PDT.
Keywords: Photodynamic Therapy, Bioluminescence, Photosensitizers, Resonance Energy Transfer, Cancer, Photobiology, Photomedicine
1. Introduction
A variety of endogenous, administered, or genetically incorporated molecules can be excited or altered upon absorption of a single nonionizing photon (450-700 nm). The light-induced chemical processes are thermodynamically robust, because of the high activation energy, and energy-efficient because the downstream biochemical events, such as enzymatic reactions, can effectively amplify their biological effects. Numerous treatment methods based on the photochemical processes have been developed, which include photodynamic therapy (PDT) [1-3], low-level light therapy [4] and collagen crosslinking [5], as well as optogenetics [6]. Despite the growing biomedical applications of the light-based techniques, a major common challenge has been the difficulty of delivering the activation light deep into the tissue. Owing to its intrinsic absorption and scattering [7], the penetration depth of visible light is no more than a several mm's in tissue.[8] To date, the clinical use of optical techniques has been limited to superficial layers, such as the skin and retina, or the epithelial surfaces of organs that are accessible by catheters or endoscopes.
An intriguing solution to this problem arises from the concept of internal light sources. Implanting fiber-optic light sources [9] and light emitting diodes [10, 11] could be a viable approach in some settings, but it requires invasive procedures. Among potential candidates [12], bioluminescence (BL) offers an attractive alternative to solid-state optoelectronic devices because the source molecules (i.e. enzymes and substrates) can be delivered minimal- invasively. BL has been widely used in optical imaging, but its potential in therapies [13, 14] has been only recently recognized [15-18]. Theodossiou et al. has used a cancer cell line expressing firefly luciferase and observed efficient cell death in vitro when both photosensitizers and luciferin were administered into the cytosol [13], but this conclusion was later disputed [14]. Recently, Hsu et al. [15] obtained a reduction of tumor growth by using self-illuminating quantum dots (QDs) [19] and attributed the effect to bioluminescence resonance energy transfer (BRET) [20-24] to photosensitizers in the cytoplasm. Despite these encouraging results, however, the mechanism of cell killing has been speculative [16], and it remained controversial whether BL-induced PDT (“BL-PDT” hereinafter) can be effective given its much lower energy than the energy typically required for laser illumination in conventional PDT. Yuan et al. reported significant treatment effects of tumor [18] and fungus [25] by using a chemiluminescent molecule, luminol. While luminol has biocompatibility issues, such as their adverse effects on DNA and high affinity to serum albumin [26-28], bioluminescent molecules generally offer higher biocompatibility.
Here, using self-illuminating conjugates of Rluc8 (Luc) and QD (hereinafter Luc-QD) [19] and photosensitizer chlorine e6 (Ce6) [15], we describe our finding that BRET-induced activation of Ce6 at the cell membrane, rather than inside cytosol, can lead to significant cytotoxicity. Our experimental data provide insights into the mechanism, distribution, and therapeutic efficacy of BL-PDT in direct comparison to conventional PDT using laser irradiation-induced activation [29]. By intratumoral delivery of agents, we investigate the effects of BL-PDT on tumor growth in mice in vivo for three different cancer cell lines: CT26 colorectal cancer cells, B16F10 melanoma cells, and LLC lung cancer cells. Furthermore, we demonstrate the feasibility of novel peritumoral BL-PDT, where the therapeutic agents are delivered to local lymph nodes (LNs) to ablate the cancer cells spread to the LNs located at depths not accessible by external illumination of light. This work shows the benefit of BL-PDT for suppressing distant organ metastasis and extending animal survival.
2. Materials and methods
2.1. Synthesis of Luc-QD conjugates
RLuc8 proteins were produced in E.coli LMG 194 strain from pBAD-RLuc8 plasmid supplied from Dr. Gambhir in Stanford University [30]. RLuc8 proteins were purified and extracted by immobilized metal ion affinity chromatography and anion exchange chromatography. N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (EDC, Fluka) was used to conjugate RLuc8 and carboxylated 655-nm quantum dots (QD-655, Life Technologies), mixed with a ratio of 40:1 in solution [31]. The emission spectrum of QD-655 overlaps well with the absorption spectrum of Ce6. QD-655 has an amphiphilic polymer coating with a functionalized polyethylene glycol (PEG) linker. The Luc-QD conjugates were selected by centrifuging (2.7 kg, 3 min, 4 °C) several times with 100 kD cutoff centrifugal filters (Pall). For consistent and maximum energy efficiency, Luc-QD was synthesized within a few hours before use.
2.2. Transmission electron microscopy
For imaging Luc-QD conjugates, glow-discharged carbon-coated 400 mesh copper grids (Electron Microscopy Science) either unstained or negatively stained with 0.75% uranyl formate solution were used. For cell imaging, at 30 min after CTZ injection CT-26 cells were fixed with 3% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) containing 0.1% CaCl2 for 2 h at room temperature. The cells were washed five times with 0.1 M cacodylate buffer at 4 °C and then post-fixed with 1% OsO4 in 0.1 M cacodylate buffer containing 0.1% CaCl2 for 2 h at 4 °C. After rinsing with cold distilled water, the cells were dehydrated slowly with a series of ethanol and propylene oxide at 4 °C. Then, the samples were embedded in Embed 812 resin (Electron Microscopy Science). After polymerization at 70 °C for 36 h, serial sections were cut by an ultramicrotome (Ultracut UC7, Leica) and mounted on formvar-coated slot grids (Electron Microscopy Science). The sections were stained with 4% uranyl acetate for 10 min and lead citrate for 7 min. Images were recorded with a FEI Tecnai T12 Bio-TWIN TEM operated at 120 kV with a 67,000× or 110,000× nominal magnification.
2.3. BL-PDT of cells in vitro
Murine Lewis lung cancer (LLC) and B16F10 melanoma cells (ATCC) tagged with GFP were grown in DMEM with 10% FBS and 1% penicillin and streptomycin. CT26 murine colon cancer cells (ATCC) were grown in RPMI media containing 10% FBS and 1% penicillin and streptomycin. Typically, 2 x105 cells were transferred to each well containing 200-µl FBS-free medium in a 48-well plate. After 12 hours of incubation, the medium was changed with 200 µl FBS-free media, added with Ce6 (10 µg/ml, Santa Cruz Biotechnology), incubated for 3 h for accumulation of Ce6 into the cells, and then changed to fresh FBS-free DMEM after washing twice with PBS. Conventional PDT was conducted [32] by using a single-spatial-mode laser (Shanghai Sanctity laser technology) with a center wavelength of 660 nm. A custom-built telescope was used to expand the laser beam to illuminate each well uniformly. The intensity level was adjusted by using neutral density filters. For BL-PDT, Luc-QD conjugates were administrated to the media and incubated for 5 min, and then h-coelenterazine (CTZ; Prolume) dissolved in 1% propylene glycol (Junsei) with cell specific media and 50-mM (2-hydroxypropyl)-β-cyclodextrin was administered to the well.
2.4. In vitro analysis of cytotoxicity
For cell viability imaging, Hoechst stain dye (500 µg/ml, Sigma-Aldrich) was added to the medium at 4 h after PDT, and cells were incubated for 5 min at 37°C. Then, the cells were incubated with propidium iodide (25 µg/ml, Sigma-Aldrich) for 5 min at room temperature. The excitation laser power used in confocal microscopy was set to <1 mW at the sample to avoid any photo-induced effects on cell viability by the imaging light. MTT assay was carried out according to the previously reported protocols [33, 34]. MTT (Invitrogen) was dissolved in pH 7.4 PBS and stored at 4°C. After PDT, the well plate was incubated at 37°C for 8 h. Then, the media was changed to 200 µl FBS-free media, 20 µl of 5 mg/ml MTT solutions were added to each well, and the well plate was incubated for 1 h at 37°C. After removing the media, 200 µl DMSO was added and stirred vigorously by pipetting to dissolve violet color formazan crystal completely in 20 min, and 100-µl aliquots of the solution were transferred to a well. The absorbance at 540 nm was measured with an ELISA reader.
2.5. Analysis of intracellular ROS
For SOSG imaging, 100-µg SOSG reagent vial (Invitrogen) was dissolved in 660 µl methanol to make 250 µM stock solution [35], and 12 µl of stock solution was added in the cell culture well to reach 15 µM final working concentration. After incubation for 5 min, BL- or laser-PDT was conducted as described above, and confocal images were taken during the procedure. For DCFH-DA (Cell Biolabs) and lipid peroxidation measurement (Abcam) [36, 37], cells in a 48 well plate were washed three times gently with DPBS. Pre-made 20× DCFH-DA solution was mixed with FBS-free RPMI medium. The 1× DCFH-DA/RPMI solution as added to the well and incubated at 37 °C for 40 min. After removing the 1× DCFH-DA/RPMI solution, RPMI was added to the washed cell culture, and PDT was performed. After 30 min, the treated cells were washed, and 200 µl media and 200-µl 2× cell-lysis buffer were added and mixed thoroughly. After 5 min incubation, 150 µl of mixture was transferred to a microplate reader (Spectramax plus384), and the fluorescence at 530 nm (480 ex.) was measured in comparison to standard DCF concentrations. For lipid peroxidation study, cells before treatment and 10- and 30-min after treatment were homogenized on ice with mixture of 300 µl MDA lysis buffer and 3 µl 100× BHT. The solution was centrifuged to remove insoluble material for 10 min (13 kg). 200 µl of the supernatant was mixed with 600 µl TBA solution and incubated for 60 min at 95 °C and cooled down to room temperature in ice bath for 10 min. 200 µl of the sample was transferred to the microplate reader, and the fluorescence intensity at 553 nm (ex. at 532 nm) was measured in comparison to various concentrations of MDA standard solution.
2.6. Intratumoral BL-PDT in vivo
All animal experiments were approved from the Institutional Animal Care and Use Committee (IACUC) at KAIST and performed in compliance with the institutional guidelines. 2x106 CT-26 cells were injected subcutaneously into the flanks of 8-week-old BALB/cAnN.Cg-Foxn1 nu/CrliOri mice (Balb/c-nu), as previously described [38]. 1.5x106 LLC cancer cells or B16F10 cells were injected to wild-type C57BL/6 mice (8 weeks old). At 4 hours after injection of Ce6 (10 mg/kg) into the tail vein, the mouse was anesthetized. For laser PDT, 660 nm laser light was irradiated on the tumor through the skin. For BL-PDT, Luc-QD (50 pmol) was injected intratumorally, and CTZ dissolved with Fuel-Inject (prolume) were administrated intravenously. Luminescence from CTZ was imaged with IVIS spectrum (Xenogen). The first treatment session was conducted at day 9, when the tumor volume became about 50 mm3. The size of the tumor was measured by using electric calipers, as described previously [39]. For histology and immunohistochemistry, mice were sacrificed two days after BL-PDT, and the tumors were resected and fixed at 4% paraformaldehyde (PFA) for 3 h. Paraffin-embedded tumors were stained with hematoxylin and eosin (H&E) according to standard protocol. The other tumors were rinsed several times with PBS and stored with 30% sucrose for overnight for dehydration. For immunohistochemistry (IHC), the tumors were embedded in OCT solution and stored at -80 °C. Frozen sections were made with a thickness of 20 µm by using a microtome. Hamster CD31 antibody (1:200, Santa Cruz) and rabbit caspase-3 antibody (1:300, R&D Systems) were used as primary antibody for IHC. Hamster FITC (1:1000, Santa Cruz) and rabbit cy-3 (1:1000, Santa Cruz) were used as secondary antibody. The tissue slides were stained with APR-648 (BioActs), which is a Cy-5 labelled phosphatidylserine recognizing peptide, and 4',6-diamidino-2-phenylindole (DAPI, Life Technologies).
2.7. Peritumoral BL-PDT to treat LN metastases (CT26 and LLC)
To make LN metastasis models, 1.5x106 CT26 cells or LLC cells were injected into the right-side footpad of 8-week-old Balb/c-nu or C57BL/6 mice, respectively. At day 11 after tumor injection, 12 µg Ce6, 30 pmol Luc-QD conjugates and 70 nmol CTZ were injected sequentially with 10 min intervals into the dorsum of the foot. Two days after the treatment, the popliteal LN was isolated from the mouse, fixed with 4% PFA in PBS, and paraffinized. TdT-mediated dUTP Nick-End Labeling (TUNEL) assay (Promega) was conducted. For in situ apoptosis detection (ApopTag; Chemicon), hydrated tissues after de-parafinization were incubated with proteinase K. Reaction buffer containing biotinyl-UTP, deoxyribonucleotide transferase, and streptavidin-rhodamin was sequentially added. For immunostaining, antigen retrieval was achieved by boiling the tissues three times with 0.1% citrate buffer in a microwave oven. To minimize non-specific reaction, the tissues were incubated with H2O2 and then with PBS-based blocking buffer containing 4% BSA and 0.1% triton X-100. For detecting metastatic tumor cells in LN, anti-pan cytokeratin antibody (1:200, Abcam) and anti-mouse cy3 antibody (1:1000, Santa Cruz) were used. Cellular nuclei were stained with DAPI at the concentration of 0.1mg/ml. Two weeks after BL-PDT, or day 25, the mice were sacrificed, and both lungs were isolated and fixed with 4% PFA. After fixation, lungs of CT26 bearing mice were embedded with paraffin, and the paraffinized tissues were sectioned at interval of 100 µm. The number of metastases at six sectioned slides around mid-section lungs per mouse was counted and analyzed statistically. The surface lung metastases of LLC bearing mice were manually counted, and the weight of lungs was measured [40].
2.8. In vivo treatment of lymph-node metastases (B16F10)
1.5x106 B16F10 melanoma cells were injected subcutaneously into the footpad of 8-week-old C57BL/6 mice. BL-PDT was performed at day 21 after implantation by injecting 12 µg Ce6, 30 pmol Luc-QD conjugates and 70 nmol CTZ sequentially with 10 min intervals into the peritumoral dorsum of the foot. Two days after the treatment, the popliteal LNs of the mouse were fixed with 1% PFA in PBS and dehydrated with 20% sucrose. After rinse with PBS, resected LNs were embedded in tissue freezing medium (Leica). Mid-sectioned LNs were incubated with blocking solution containing goat serum (Jackson ImmunoResearch). The sectioned tissues were incubated overnight with one of more of the following primary antibodies (1:200): anti-vascular endothelial growth factor receptor 3, goat polyclonal (VEGFR3: R&D); anti-CD31, hamster monoclonal (Millipore); anti-Melan-A, mouse polyclonal (Abcam); and anti-caspase-3, rabbit polyclonal (R&D). After several rinse with PBS, the sectioned tissues were incubated for 2 h with one or more of the following secondary antibodies (1:1000): anti-goat cy3 (invitrogen), anti-hamster cy5 (Jackson ImmunoResearch), anti-mouse cy3 (Jackson ImmunoResearch), and anti-rabbit FITC (Jackson ImmunoResearch).
2.9. Statistical analysis
Error bars represent standard deviation (s.d.) from the mean. Data were analyzed by Student t-test, ANOVA with SPSS version 18. P-values of <0.05 were considered statistically significant.
3. Results
3.1. Mechanism of activation of photosensitizers
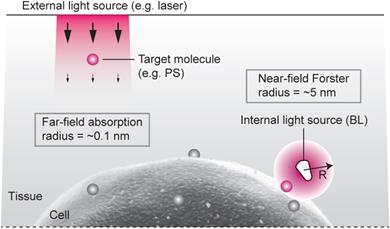
Although external illumination offers convenience, there are two fundamental drawbacks. One is light propagation loss. To induce meaningful photochemical effects, irradiance greater than 1 mW/cm2 is required at the target. The intrinsic optical attenuation in the skin is 5-10 dB per in the visible spectrum (see Supplementary Material). Considering the maximum permissible exposure level (1 W/cm2) at the skin surface, the effective therapeutic depth is less than several mm's, which is consistent with experiments. The second drawback is the relatively weak interaction between light and molecule (Fig. 1). The absorption cross-section (σa) of Ce6, an efficient photosensitizer, is 2.3x10-16 cm2 at λ=660 nm (calculated from the extinction coefficient of 59,000 M-1 cm-1). This means that, at 1 mW/cm2, each Ce6 molecule absorbs, statistically, less than one of incoming 106 photons per sec in a minimum diffraction-limited area of 0.1 µm2.
Radiation- vs. resonant energy transfer-induced activation of photosensitizers. Conventional light delivery by external illumination suffers from superficial therapeutic depths due to the small absorption cross-section of a molecule and the optical propagation loss in tissue. By contrast, the resonance energy transfer from an injectable, or biologically producible, bioluminescence source can activate a target molecule more effectively due to strong near-field interaction, in the regions inaccessible by the external light.
The electric field, Ε, at a distance, r, from an excited BL molecule can be expressed as:
(1)
where k=2π/λ and p is the dipole moment of the light source [41]. The first term in the parenthesis describes far-field radiation. The second term represents the near-field non-radiative field with intensity decaying with 1/r6, which is responsible for Förster resonance energy transfer (FRET). The efficiency of FRET, η, is expressed as [42]:
(2)
where QY is the quantum yield of the donor and R0 is a Förster radius, which is typically 5-10 nm. From Eq. (1), the intensity ratio of the non-radiative to the radiative field (the ratio of the ensemble square of the first and second terms) can be shown to be 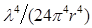 , where
, where 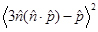 = 2/3 was used [42]. For r <10 nm, the intensity ratio is much greater than unity; for example, the ratio is 1.3x105 for r=5 nm and λ=660 nm.
= 2/3 was used [42]. For r <10 nm, the intensity ratio is much greater than unity; for example, the ratio is 1.3x105 for r=5 nm and λ=660 nm.
For η=100%, one substrate-enzyme reaction—i.e. one “BL photon”—is sufficient to activate one Ce6 molecule. (For η=50%, two BL photons can activate one Ce6 molecule.) Therefore, the near-field interaction by BRET energy sources can solve the drawbacks of the conventional approach using far-field radiation (Fig. 1). Considering that the probability of one BL photon is absorbed at a distance of R0 is 50%, the effective non-radiative absorption cross-section of a FRET acceptor may be defined to be 2πR02, whereas the radiative cross-section is σa. Therefore, their ratio 2πR02/σa is ~6830, which represents the advantage in the efficiency of FRET-induced over radiation-induced activation.
3.2. Measurement of BRET energy
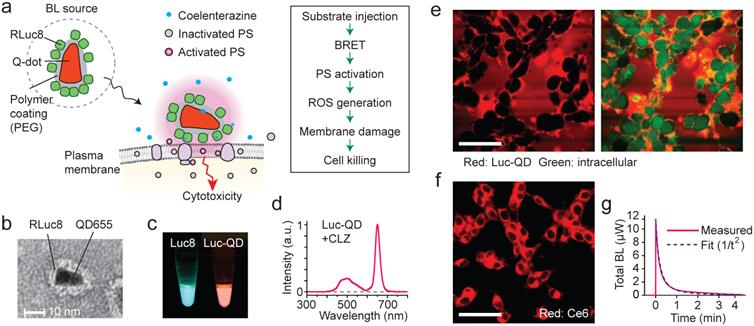
We have designed an experiment using Luc-QD as the BL source and amphiphilic Ce6. Our hypothesis is that, upon administration of CTZ, the generated BL energy is transferred to nearby Ce6 and, subsequently, the activated Ce6 triggers membrane damage that results in cell killing [43, 44] (Fig. 2a). This cascade of FRET events does not involve radiative emission of photons (Supplementary Fig. 1). The Luc-QD conjugates were synthetized by covalently conjugating 10-20 RLuc8 molecules onto the polymeric coat of QD, as confirmed by TEM (Fig. 2b and Supplementary Fig. 2a). When mixed with CTZ, Luc-QD solution emits red fluorescence, whereas the original BL from RLuc8 has a blue-green color (Fig. 2c). The BRET efficiency from RLuc8 to QD was 60-65% (Fig. 2d). Time-lapse confocal fluorescence microscopy showed that Luc-QD conjugates were highly concentrated around the external surface of cells without entering the cytoplasm (Fig. 2e and Supplementary Fig. 3), as consistent with TEM images (Supplementary Figs. 2b-d). The high affinity, dense distribution, and short distance of Luc-QD to the cell membrane are critical for activating Ce6 at the cell membrane (Fig. 2f). When a culture well containing 2x105 murine colorectal cancer CT26 cells in monolayer (1 cm2) was illuminated with 660 nm laser light with 2.2 mW, the difference of transmission through Ce6-incubated versus non-incubated cells was 13 µW (Supplementary Fig. 4a). This means that a total 2x108 photons/s are absorbed per cell. From the absorption cross-section of Ce6, the concentration of Ce6 is calculated to be about 100 µM (6x107 Ce6 per cell), and these molecules collectively are activated 4x107 times per min by the laser flux.
The design and principle of BL-PDT. a, Illustration of the proposed mechanism based on resonance energy transfer at cellular membrane. b, TEM image of Luc-QD conjugate. c, Luminescence from RLuc8 and Luc-QD. d, Emission spectrum of Luc-QD. The BRET ratio was calculated from the ratio of the area under curve of RLuc8 emission between 390 and 600 nm and that of QD emission between 600 and 710 nm. e, Confocal images of red fluorescence from QD, showing the distribution of Luc-QD outside cells (CT26 cells; green: intracellular dye). f, Confocal fluorescence image of Ce6, showing its distribution in the cytosol. g, Luminescence power generated after administration of 28 nmol CTZ. Scale bars in e and f, 50 µm.
The average intermolecular distance of randomly distributed Ce6 is ~50 nm in the cell. Modeling a cell as a sphere with a radius (R) of 10 µm with uniform concentration of Ce6, within 10 nm (R0) from the external surface of the cell membrane we estimate that 0.33% (=0.33 R0/R) or 2x105 molecules are present. 50 pmol (3x1013) Luc-QD conjugates were administered into the medium. Due to the high concentration of Luc-QD surrounding the cells, the majority of Ce6 at the cell membrane is expected to be within a FRET radius (<10 nm) from one or more donor molecules. After the incubation of Ce6 for 3 h and a subsequent 5-min incubation of Luc-QD, 28 nmol (1.7x1016) CTZ were added. Luminescence appeared immediately but decreased rapidly (Fig. 2g and Supplementary Fig. 4b). According to our simple model (Supplementary Fig. 5), the initial 1/t2 type decay (0-1 min) may indicate a degradation of Luc-QD as they react with CTZ. This accounts for our observation that a secondary injection of additional CTZ generated weaker luminescence. The total emitted luminescence for 1 min was about 180 µJ (6x1014 photons). The difference of total luminescence from Ce6-incubated vs. non-incubated cells was 14 µJ. This corresponds to 70 pJ or 3x108 “BL photons” per cell, which represents the total energy transferred to nearby Ce6. The number of activated Ce6 is thus 3x108 per min, more than 4x107 per min calculated above for laser illumination for 1 min at 2.2 mW/cm2. Therefore, this quantitative analysis suggests that BRET energy in the order of 100 µJ could generate stronger photochemical activation in the cellular membrane than laser energy in the order of 100 mJ.
3.3. BL-PDT induces cytotoxicity at the cellular membrane
We used MTT assay to measure the cytotoxicity induced by BL-PDT on CT26 cells in vitro. The cell death increased with increasing amount of CTZ (Fig. 3a). Negligible cytotoxicity was measured with Ce6 only or without Ce6 (Luc-QD and CTZ), or when Rluc8 replaced Luc-QD. For comparison, CT26 cells in monolayer were treated with a 660 nm laser for 3 min at various intensity levels (Fig. 3b). Little cytotoxicity was observed at intensities lower than 0.1 mW/cm2. At 2.2 mW/cm2, the treatment effect with 3-min exposure was similar to that of BL-PDT (28 nmol CTZ), which is consistent with our estimation above. Similar result was obtained for B16F10 melanoma cells tagged with GFP (Supplementary Fig. 6). Time-lapse microscopy showed typical swelling and rounding of cells and damaged mitochondrial granules during BL- and laser-PDT (Fig. 3c and 3d). These changes continued for 30 min, longer than the duration of BL emission (Supplementary Fig. 7). Imaging with DNA-binding membrane-permeable Hoechst and impermeable propidium iodide (PI) dyes revealed some striking features of BL-PDT (Fig. 3e). The damaged cells indicated by PI-positive nuclei were surrounded by large amount of Luc-QD. By contrast, PI-negative cells had negligible Luc-QD around their cellular membrane. Unlike BL-PDT, laser-PDT did not cause nucleus disintegration (Supplementary Fig. 8). TEM taken 30 min after BL-PDT showed membrane rupture (Fig. 3f).
Effects of BL-PDT on cancer cells in vitro. a, MTT analysis of CT26 cells treated with BL-PDT. b, The efficiency of conventional PDT at different laser powers. c, Time-lapse images of LLC-GFP cells during BL-PDT. d, Time-lapse images of LLC-GFP cells during laser-PDT. Control cells without laser irradiation showed no change in GFP fluorescence. e, Confocal image of LLC cells 4 h after BL-PDT. Damaged cells with PI-positive nuclei are surrounded heavily by Luc-QD (squares), whereas live cells with intact PI-negative nuclei have no or less Luc-QD around their cell surfaces (arrows). f, TEM image showing membrane disruption and leak of intracellular components after BL-PDT. g, DCF assay to measure intracellular ROS level. h, Lipid peroxidation assay. i, Time-lapse confocal images of SOSG and Luc-QD. Cells surrounded by more Luc-QD show earlier infiltration of SOSG. ANOVA test p values: * <0.05, ** <0.01. Error bars, s.d. Scale bars, 50 µm in c and d; 100 µm in e and i.
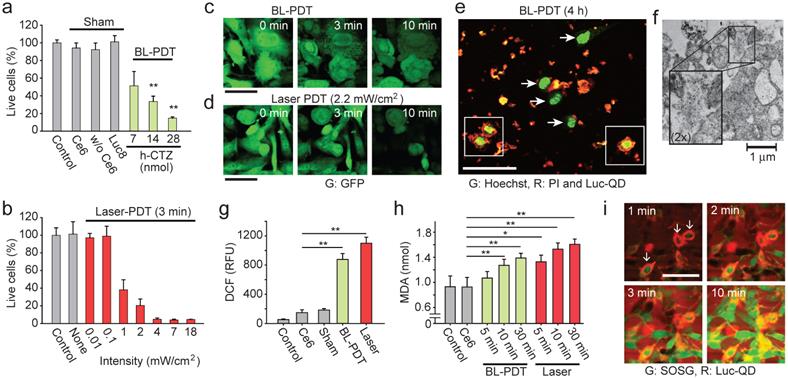
Generation of singlet oxygen and reactive oxygen species (ROS) is a hallmark of PDT. We expect that the activation Ce6 by BRET generates ROS and causes lipid peroxidation at the cell membrane, and subsequent cell death signaling following the membrane damage generates a significant amount of ROS in the cytosol that can be detected by standard cellular assays. Indeed, significant intracellular ROS was measured by dichloro-dihydro-fluorescein diacetate (DCFH-DA) in BL-PDT, as well as laser-PDT (Fig. 3g). Measurement of malondialdehyde (MDA) confirmed lipid peroxidation (Fig. 3h), evidencing ROS-induced membrane damages [45]. The amount of MDA increased with time after the treatments. Time-lapse microscopy (with 1 min interval) using membrane-impermeable probe, singlet oxygen sensor green (SOSG), suggested that significant membrane damage occurred within the first 1 min after CTZ injection, starting with the cells with more Luc-QD at their membranes (Fig. 3i). After 10 min, most treated cells, but not controls, showed strong SOSG signals in the cytoplasm and round morphology (Supplementary Fig. 9). The relatively extended duration of ROS generation and cellular changes compared to the duration of BL emission (<1 min) suggests the involvement of intracellular signaling pathways, such as ROS-induced ROS release by mitochondria [46].
3.4. BL-PDT suppresses tumor growth in mice in vivo
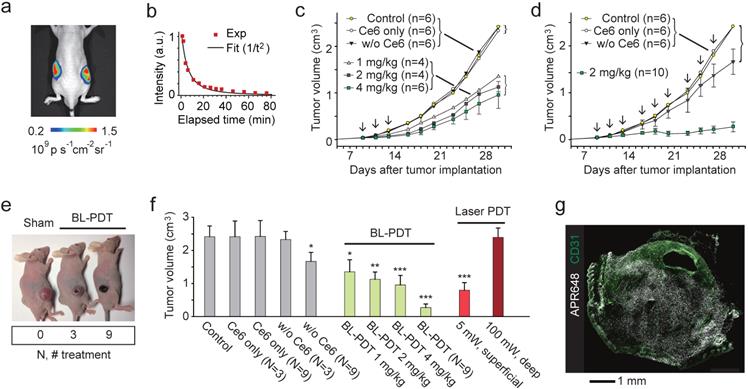
To test the effect of BL-PDT on tumor growth, we used tumor implant models. CT26 cells were injected into Balb/c nude mice at both flanks subcutaneously. When the tumor volume reached at about 50 mm3, the first treatment was conducted by systemic delivery of Ce6. After 4 hour of incubation, Luc-QD (3x1013) was injected directly into the tumor, and after 5-10 min, CTZ (60-300 nmol) was injected intravenously (Fig. 4a). The luminescence was visible for more than one hour after CTZ injection (Fig. 4b). This emission duration is much longer than in vitro settings (Fig. 2h) presumably because of the longer diffusion time of CTZ in the tumor tissue. In the treatment group that received three sessions of BL-PDT over a week, but not the untreated and sham-treated groups, showed the decrease of tumor growth. The growth inhibition increased, but not statistically significantly, with the administered amount of CTZ from 1 to 4 mg/kg (Fig. 4c). A nine-session (N=9) treatment resulted in a near complete inhibition of tumor growth (Fig. 4d). The regression and necrosis of tumor was visibly apparent (Fig. 4e). H&E staining of the major organs, including the liver, kidney and bladder, did not show any apparent adverse effect of BL-PDT. The growth inhibition was similar or better than conventional PDT (three sessions; N=3) by transdermal illumination (660 nm, 5 mW) for 3 min (Fig. 4f). When the laser light was illuminated through a 5-mm-thick pad of soft tissue, no therapeutic effect was observed even with higher laser powers (Fig. 4f). Apoptotic cell death was observed across the entire tumor (Fig. 4g and Supplementary Fig. 10)
Effects of BL-PDT on tumors in vivo. a, Luminescence (575-650nm) from tumors injected with Luc-QD (50 pmol i.t.) and CTZ (28 nmol i.v.). b, Luminescence intensity from the tumor over time. c, Tumor growth curves for various treatment conditions. Arrows indicate the three sessions of BL-PDT. d, Tumor growth curves for extended BL-PDT protocols with 2-3 day treatment intervals (arrows). e, Photos of mice (at day 23) treated 0, 3, and 9 times, respectively. f, Tumor sizes measured at day 30 for control, sham-treated, BL-, and laser-PDT-treated mice (calculated from the data in b: N=3 or N=9 indicates groups that received three or nine treatment sessions.) g, Immunohistology with APR648 indicating apoptotic cell death within a tumor. Scale bars, 1 mm in (g). Error bars, mean +/- s.d. Two-sided Student test p values: * <0.05, ** <0.01, *** <0.001.
3.5. Treatment of lymph-node metastases by local delivery
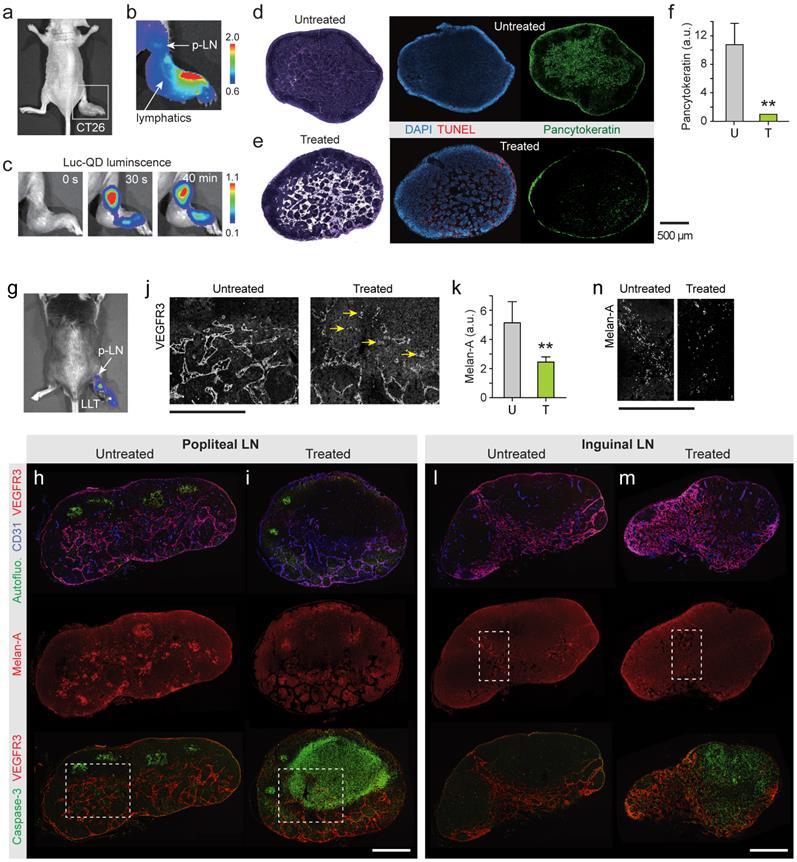
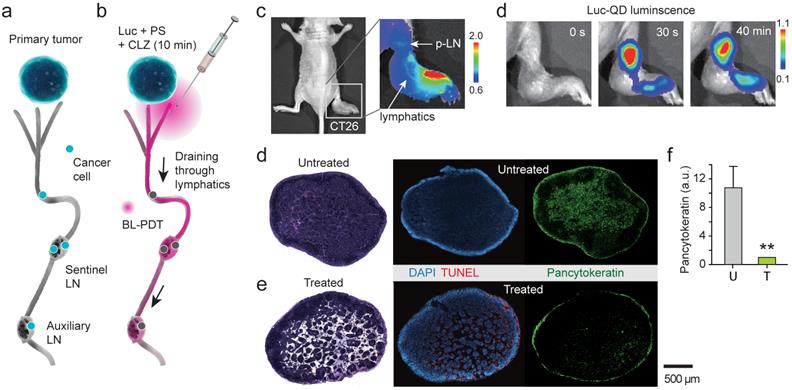
Cancer cells spread to local and regional LNs and also in-transit cells in peritumoral lymphatic vessels increase the risk of recurrence in the breast. It is challenging to eradicate these cells by conventional PDT because of the limited light penetration. BL-PDT offers a potential to treat LN metastasis by local delivery of the agents via the peritumoral lymphatic draining system and activation by subsequent administration of CTZ (Fig. 5a). To test the therapeutic effect of BL-PDT on LN metastases, we used footpad implant models, in which the popliteal LN (p-LN) is the sentinel LN. 1.5x106 CT26 cells were injected into the footpad of Balb/c nude mice. Eleven days after tumor cell inoculation, the primary tumor was grown to a size of several millimeters at the injection site (Fig. 5b), and tumor cells were found in the p-LN with almost 100% efficiency. At day 11, Ce6 (50 nmol) was injected into the dorsum of the foot in the vicinity of the tumor, which allows effective draining of the agents to the p-LN through the lymphatic vessels (Fig. 5c). After 10 min, Luc-QD (30 pmol) was injected into the peritumoral region, which immediately drained to the LN. After 10 min, CTZ (70 nmol) was injected, which generated luminescence almost instantly. The luminescence intensity decreased slowly over time (Fig. 5d and Supplementary Fig. 11). In addition to the sentinel LN, depending on the injection condition, it was possible to deliver the agents to the secondary LN, such as lumbar and inguinal LNs, through lymphatic drainage (Supplementary Fig. 12). Histologic analysis confirmed the enlarged size and lymphangiogenesis in the p-LN and increased pancytokeratin positive metastatic tumor cells at day 13 in untreated mice (Fig. 5e). BL-PDT resulted in destruction of the lymphatic structure in the LN and cell death indicated by in situ TUNEL along the lymphatic vessels (Fig. 5f). The pancytokeratin positive metastatic tumor cells were 10 fold decreased after BL-PDT (Fig. 5g).
Treatment of lymph-node metastases. a, A schematic of LN metastasis and BL-PDT procedure. b, Photo of a mouse with CT-26 tumor grown in the footpad at day 11. c, Fluorescence image after injection of Ce6 into the foot. Draining Ce6 visualizes the lymphatic vessel and the popliteal LN (p-LN). d, Time-lapse luminescence images after CTZ injection into the dorsum of the foot. Strong luminescence is generated from the p-LN. e, Histology images of untreated LN sections harvested at day 13, stained with H&E, TUNEL, and pancytokeratin. f, Images of LN sections harvested at day 13, two days after BL-PDT, stained with H&E, TUNEL, and pancytokeratin. Cell death along the expanded lymphatic vessels is evident. f, Integrated pancytokeratin signals within the untreated and treated LN's.
3.6. Reduction of melanoma cells in the sentinel and secondary LNs
To test BL-PDT for orthotopic melanoma, 1.5x106 B16F10 cells were injected into the footpad of wild-type C57BL/6 mice. BL emission from the p-LN was confirmed, although it was less bright than in the CT-26 bearing nude mice due to the presence of hair and high melanin in the tumor cells (Fig. 6a). BL-PDT was performed 21 days after implantation. At day 23, the popliteal and inguinal LNs were harvested from both treated and untreated groups, sectioned, and examined by immunohistochemistry. The untreated p-LN had a considerable number of tumor cells, indicated by melan-A surface antigen, scattered in multiple groups throughout the paracortex and medulla (Fig. 6b). The treated p-LN exhibited several distinct consequences of BL-PDT including the remarkable increase of apoptotic cell deaths throughout the cortex and paracortex (Fig. 6c). In these regions, the tumor vessels and lymphatic sinuses appear to have been severely disintegrated (Fig. 6d). The amount of melan-A signal decreased to a half after the treatment in the p-LNs (Fig. 6e). The effects of BL-PDT were not limited to sentinel LN. Tumor cells were also found at inguinal LNs in the untreated mouse, particularly along the lymphatic vessels (Fig. 6f). At inguinal LN in the treated mouse (Fig. 6g), considerable apoptosis signals were measured in the paracortex and medulla. Notably, the number of melanoma cells at inguinal LN was significantly reduced in the treated compared to untreated mice (Fig. 6h).
Treatment of lymph-node metastases. a, BL image of a C57/BL6 mouse with B16F10 tumor in the footpad (day 21), after injection of Luc-QD and CTZ into the foot. b, IHC images of p-LN sections harvested from an untreated mouse at day 23, stained for blood vessels (CD31), lymphatics (VEGFR3/CD31), melanoma cells (melan-A), and apoptosis (caspase-3). Strong green autofluorescence indicates germinal centers. c, Images of p-LN sections harvested from a treated mouse at day 23, two days after BL-PDT. d, Images of the regions marked in b and c. Arrows indicate damaged blood vessels. e, Integrated melan-A fluorescence magnitudes of the untreated (U) and treated (T) p-LNs. f, Inguinal LN sections harvested from the untreated mouse. g, Inguinal LN sections harvested from the treated mouse. h, The regions marked in f and g. Scale bars, 500 µm. Error bars, s.d. **, Kruskal-Wallis test p <0.01.
3.7. LN PDT reduces distant metastasis and improves animal survival
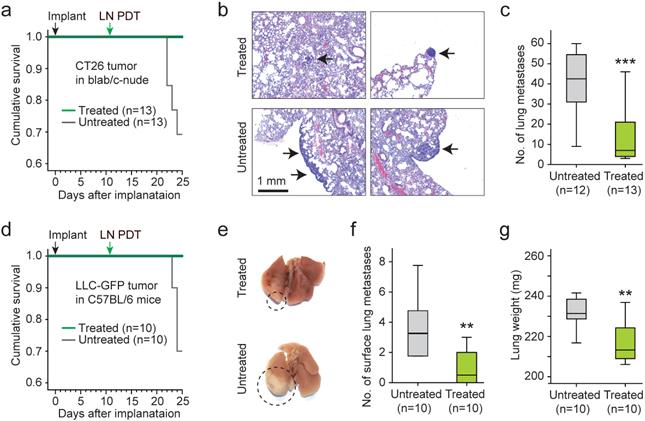
To assess the effects of the LN treatment on distant metastasis, the lungs from treated (n=13) and untreated (n=12) animals were harvested 25 days after implantation of CT26 cells and 14 days after LN BL-PDT. Four mice of the untreated group died by day 25, where as 100% of the treated mice survived (Fig. 7a). The increase of survival time for the treated animals was statistically significant. Histology of the lung tissues showed that the metastatic lung nodules in the treated mice were substantially smaller than those in the untreated survived mice (Fig. 7b). The number of lung nodules was significantly smaller in the treated group (Fig. 7c). We also performed the same experiment with LLC bearing mice in a treated (n=10) and untreated (n=10) groups. By day 25, 3 untreated mice died whereas none died from the treated group (Fig. 7d). The apparent size of the tumor nodules in the surface of the lung was significantly less in the treated animals (Fig. 7e and Supplementary Fig. 13). The number of visible metastasis in the lung surface was less in the treated mice (Fig. 7f). The weight of the lung was also smaller in the treated mice (Fig. 7g). These results indicate that LN PDT effectively reduced distant lung metastasis and extended the survival time of tumor-bearing animals.
Therapeutic effects of LN BL-PDT on animal survival and lung metastasis. a. Kaplan-Meier survival curve of CT26 bearing mice. b. H&E stained images of lung harvested 14 days after LN PDT. Arrows indicate small nodules in treated mice (top) and larger nodules with the sign of metastatic infiltration (bottom). Scale bar, 1 mm. c. The total number of metastatic lung nodules found from each animal group (6 lung sections per mouse, 12 mice). d. Kaplan-Meier survival curve of LLC-GFP bearing mice. e. Photographs of the lungs harvested at day 25. Circles indicate macroscopic metastatic nodules. f. The numbers of lung surface metastases. g. The weights of the lungs. *, Kruskal-Wallis test p <0.05; **, p <0.01.
4. Discussion
Clinically, conventional PDT has not been used to treat macroscopic tumors and metastases mainly due to the need of light activation. BL-PDT can solve this limitation by allowing the photochemical activation energy to be delivered by the transport of molecules rather than electro-magnetic radiation. Our results showed that BRET-induced activation at the cell membrane induced significant cytotoxicity. Like conventional PDT, the cytotoxicity mechanism of BL-PDT is based on the generation of reactive oxygen species and singlet oxygen from photosensitizers. This mechanism is distinctively different from those employed in chemotherapy, which attacks cancer cells by interrupting processes, or inhibiting substances, necessary for cellular replication. Although the general mechanisms of drug resistance would apply to PDT, the degree of resistance is far less than those encountered in chemotherapy, and no cross-resistance between PDT and chemotherapy has been reported [47]. For example, a protective mechanism such as repair to DNA damage, which can cause chemo-resistance, is limited in PDT [48, 49]. Because of the minimal cross-resistance, PDT is synergistic with chemotherapy [50, 51] and has shown to be effective in treating chemo-resistant [48, 52, 53] and multidrug resistant cancer [54-56]. Therefore, BL-PDT may be a modality in combinational therapy to treat chemo-resistant triple-negative breast cancer [57] or radiation-resistant cancers, such as metastatic melanoma.
Ce6 localizes in the cell membrane, intracellular vesicles and other organelles including mitochondria and lysosomes [58-60]. In the BL-PDT technique demonstrated here, the primary site of activation of Ce6 is the cell membrane. The activated Ce6 generates ROS, and the generated ROS induce lipid peroxidation of the cell membrane. Lipid peroxidation destructs the integrity of cellular membrane, which disrupts the intracellular homeostasis. Consequently, necrosis is induced as a cell death process. In conventional laser-induced PDT, it has been shown that it is sufficient to kill cancer cells with laser PDT by using photosensitizers located only within cell membranes [61, 62] or extracellular photosensitizers attached to cellular membranes [43, 63]. Conventional PDT using membrane-targeted genetically encoded photosensitizers has shown more effective and faster cytotoxicity through lipid peroxidation compared to conventional PDT using mitochondria targeted genetically encoded photosensitizers [61, 64].
Although the cell membrane is an effective target for BL-PDT, we expect that simultaneous intracellular activation of photosensitizers would enhance cytotoxicity. Cell death mechanisms depending on the subcellular localization of photosensitizers have been extensively studies, including the biological effects following mitochondrial photosensitization [65-67]. For more efficient BL-PDT, one attractive approach is to develop conjugates of bioluminescent molecules and photosensitizers as direct BRET pairs. A number of luciferases and photosensitizers with reasonably good spectral overlaps are available, such as RLuc8.6 (emission: 490-590 nm) [68] and Rose Bengal (absorption 520-560 nm) [69]. These molecules can be chemically conjugated and delivered together. Such conjugates can maximize the BRET-induced activation of photosensitizers.
The BL-PDT agents used here and previously do not have designed-in specificity to cancer cells. A variety of tumor-targeted drug delivery techniques developed for PDT and other molecular therapies can be adopted for BL-PDT. The BL-PDT agents may be combined with monoclonal antibodies (mAbs) against specific biomarkers or RGD peptides to enable targeted BRET-induced photoimmunotherapy (PIT) [43, 70, 71]. More sophisticated designs, such as protease-cleavable quenchers, may be employed to increase tumor specificity. Some BL molecules require cofactors, such as Ca2+ and adenosine triphosphate (ATP), which may offer methods to couple with specific signaling pathways and metabolic activities. The dependence of BRET efficiency on distance (~r-6) may be used to target specific molecular interactions [20-24]. Since BL-PDT uses non-toxic agents prior to activation, unlike chemotherapeutic agents, strategies based on molecular transport and pharmacokinetics, such as optimizing the time delay between the luciferase and luciferin injections, may enhance the target specificity, minimizing damages to normal tissues. Advanced drug delivery systems may be employed to enable systemic delivery of the BL molecules.
Resection of peritumoral LNs is frequently performed during surgery, but this procedure can cause complications such as lymphedema, a lifelong chronic condition that is caused by the disruption of the normal drainage pattern [72]. BL-PDT offers an alternative strategy to manage LN metastases without disrupting the LNs, and this treatment can be particularly useful in conjunction with diagnostic BL imaging [73]. In our protocol of LN PDT, the primary tumor is little affected because the therapeutic agents are administered at the peritumoral dorsum of the foot. Our data showed a considerable reduction of cancer cells spread to both sentinel and secondary regional LNs. This effect is likely to be responsible for the significant reduction of distant metastasis and the improvement of animal survival, although it is not yet clear whether this result is solely due to cell deaths occurred in situ by BL-PDT or contributed by reduced metastasis of tumor cells during the two days after the treatment possibly owing to any reduction in afferent lymphatic drainage to the LNs. Whole-body BL imaging conducted four days after BL-PDT showed a partial destruction of upstream lymphatic vessels around the primary tumor. (Supplementary Fig. 14). This also suggests the possibility of eradicating in-transit cancer cells in the lymphatic vessels by BL-PDT [74]. Cancer-targeted BL-PDT agents are expected to minimize the damage to the lymphatic vessels and ensure the recovery of the normal function of the lymphatic system. The ability to eradicate remaining cancer cells more thoroughly with minimal damage to normal tissue is highly significant because it can lower local recurrence, mortality, morbidity, and cost-burden of breast cancer patients. Further research to test the potential of BL-PDT as adjuvant therapy to treat LN metastasis is warranted.
In conclusion, BRET energy-induced activation is a new paradigm of how we generate photochemical effects in deep regions that cannot be reached by external optical illumination. The use of internal energy source can have far-reaching applications beyond PDT. Deep-tissue photochemical activation via BL molecules may offer new possibilities in the field of Photomedicine for a wide range of applications from cancer treatment, to immune and brain stimulation, and to in-body sensing and genetic control.
Supplementary Material
Supplementary Figures 1-14.
Acknowledgements
We thank S. Kim for technical help on TEM, and Dr. Tayyaba Hasan at Wellman Center and Drs. Myung-Gyu Choi and Jae-Myung Park at Seoul St. Mary's Hospital for discussions. This work was funded by the Korea National Research Foundation of Korea (R31-2008-000-10071-0), the Bio & Medical Technology Development Program (2012M3A9C6049791), and Mid-career Researcher Program (2012R1A2A2A06045773) of the National Research Foundation (NRF) funded by the Korean government (MEST), and also by MGH ECOR Fund for Medical Discovery and the US National Institutes of Health (U54CA143837, P41EB015903).
Author contributions
Y.R.K., S.K., S.H.Y designed the study; Y.R.K. performed in vitro and in vivo experiments; S.K. synthetized nanoparticles; S.H.L., H.K. obtained TEM images; Y.R.K., S.K., J.W.C., S.K.H., G.Y.K., S.H.Y. analyzed the data; Y.R.K., S.H.Y. wrote the manuscript with inputs from all authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO. et al. Photodynamic therapy of cancer: an update. CA Cancer J Clin. 2011;61:250-81
2. Brown SB, Brown EA, Walker I. The present and future role of photodynamic therapy in cancer treatment. Lancet Oncol. 2004;5:497-508
3. Castano AP, Mroz P, Hamblin MR. Photodynamic therapy and anti-tumour immunity. Nat Rev Cancer. 2006;6:535-45
4. Huang Y-Y, Chen AC-H, Carroll JD, Hamblin MR. Biphasic dose response in low level light therapy. Dose Response. 2009;7:358-83
5. Wollensak G, Spoerl E, Seiler T. Riboflavin/ultraviolet-A-induced collagen crosslinking for the treatment of keratoconus. Am J Ophthalmol. 2003;135:620-7
6. Ye H, Daoud-El Baba M, Peng R-W, Fussenegger M. A synthetic optogenetic transcription device enhances blood-glucose homeostasis in mice. Science. 2011;332:1565-8
7. Cheong W-F, Prahl SA, Welch AJ. A review of the optical properties of biological tissues. IEEE J Quantum Electron. 1990;26:2166-85
8. Ntziachristos V, Ripoll J, Wang LV, Weissleder R. Looking and listening to light: the evolution of whole-body photonic imaging. Nat Biotechnol. 2005;23:313-20
9. Choi M, Choi JW, Kim S, Nizamoglu S, Hahn SK, Yun SH. Light-guiding hydrogels for cell-based sensing and optogenetic synthesis in vivo. Nat Photonics. 2013;7:987-94
10. Kim T-i, McCall JG, Jung YH, Huang X, Siuda ER, Li Y. et al. Injectable, cellular-scale optoelectronics with applications for wireless optogenetics. Science. 2013;340:211-6
11. Bagley AF, Hill S, Rogers GS, Bhatia SN. Plasmonic Photothermal Heating of Intraperitoneal Tumors through the Use of an Implanted Near-Infrared Source. ACS Nano. 2013;7:8089-97
12. Ran C, Zhang Z, Hooker J, Moore A. In vivo photoactivation without “light”: use of Cherenkov radiation to overcome the penetration limit of light. Mol Imaging Biol. 2012;14:156-62
13. Theodossiou T, Hothersall JS, Woods EA, Okkenhaug K, Jacobson J, MacRobert AJ. Firefly Luciferin-activated Rose Bengal In Vitro Photodynamic Therapy by Intracellular Chemiluminescence in Transgenic NIH 3T3 Cells. Cancer Res. 2003;63:1818-21
14. Schipper ML, Patel MR, Gambhir SS. Evaluation of firefly luciferase bioluminescence mediated photodynamic toxicity in cancer cells. Mol Imaging Biol. 2006;8:218-25
15. Hsu C-Y, Chen C-W, Yu H-P, Lin Y-F, Lai P-S. Bioluminescence resonance energy transfer using luciferase-immobilized quantum dots for self-illuminated photodynamic therapy. Biomaterials. 2013;34:1204-12
16. Huang L, Chen T-C, Lin F-H. Luminol As In Situ Light Source in Meso-Tetraphenylporphyrin-Mediated Photodynamic Therapy. Curr Med Chem. 2013;20:1195-202
17. Berglund K, Birkner E, Augustine GJ, Hochgeschwender U. Light-Emitting Channelrhodopsins for Combined Optogenetic and Chemical-Genetic Control of Neurons. PLoS One. 2013;8:e59759
18. Yuan H, Chong H, Wang B, Zhu C, Liu L, Yang Q. et al. Chemical molecule-induced light-activated system for anticancer and antifungal activities. J Am Chem Soc. 2012;134:13184-7
19. So M-K, Xu C, Loening AM, Gambhir SS, Rao J. Self-illuminating quantum dot conjugates for in vivo imaging. Nat Biotechnol. 2006;24:339-43
20. Angers S, Salahpour A, Joly E, Hilairet S, Chelsky D, Dennis M. et al. Detection of β2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc Natl Acad Sci U S A. 2000;97:3684-9
21. Breitkreutz A, Choi H, Sharom JR, Boucher L, Neduva V, Larsen B. et al. A global protein kinase and phosphatase interaction network in yeast. Science. 2010;328:1043-6
22. Collins BM, Skinner CF, Watson PJ, Seaman MNJ, Owen DJ. Vps29 has a phosphoesterase fold that acts as a protein interaction scaffold for retromer assembly. Nat Struct Mol Biol. 2005;12:594-602
23. Pfleger KD, Eidne KA. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat Methods. 2006;3:165-74
24. Carriba P, Navarro G, Ciruela F, Ferré S, Casadó V, Agnati L. et al. Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat Methods. 2008;5:727-33
25. Yuan H, Bai H, Liu L, Lv F, Wang S. A glucose-powered antimicrobial system using organic-inorganic assembled network materials. Chem Commun. 2015;51:722-4
26. Buturlakin M, Kiseleva V, Shmelev V. Interaction of human serum albumin and luminol. Biofizika. 1975;20:975-7
27. Lee-Chen S, Yu C, Wu D, Jan K. Differential effects of luminol, nickel, and arsenite on the rejoining of ultraviolet light and alkylation-induced DNA breaks. Environ Mol Mutagen. 1994;23:116-20
28. Ikushima T. Bimodal induction of sister-chromatid exchanges by luminol, an inhibitor of poly (ADP-ribose) synthetase, during the S-phase of the cell cycle. Chromosoma. 1990;99:360-4
29. Celli JP, Spring BQ, Rizvi I, Evans CL, Samkoe KS, Verma S. et al. Imaging and photodynamic therapy: mechanisms, monitoring, and optimization. Chem Rev. 2010;110:2795-838
30. Loening AM, Fenn TD, Wu AM, Gambhir SS. Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng Des Sel. 2006;19:391-400
31. So M-K, Loening AM, Gambhir SS, Rao J. Creating self-illuminating quantum dot conjugates. Nat Protoc. 2006;1:1160-4
32. Collins HA, Khurana M, Moriyama EH, Mariampillai A, Dahlstedt E, Balaz M. et al. Blood-vessel closure using photosensitizers engineered for two-photon excitation. Nat Photonics. 2008;2:420-4
33. Lee SJ, Koo H, Jeong H, Huh MS, Choi Y, Jeong SY. et al. Comparative study of photosensitizer loaded and conjugated glycol chitosan nanoparticles for cancer therapy. J Control Release. 2011;152:21-9
34. Douillard S, Olivier D, Patrice T. In vitro and in vivo evaluation of Radachlorin® sensitizer for photodynamic therapy. Photochem Photobiol Sci. 2009;8:405-13
35. Rota C, Chignell CF, Mason RP. Evidence for free radical formation during the oxidation of 2′-7′-dichlorofluorescin to the fluorescent dye 2′-7′-dichlorofluorescein by horseradish peroxidase:: Possible implications for oxidative stress measurements. Free Radic Biol Med. 1999;27:873-81
36. Wang H, Luo K, Tan L-Z, Ren B-G, Gu L-Q, Michalopoulos G. et al. p53-induced gene 3 mediates cell death induced by glutathione peroxidase 3. J Biol Chem. 2012;287:16890-902
37. Kalghatgi S, Spina CS, Costello JC, Liesa M, Morones-Ramirez JR, Slomovic S. et al. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in mammalian cells. Sci Transl Med. 2013;5:192ra85
38. Gorski DH, Beckett MA, Jaskowiak NT, Calvin DP, Mauceri HJ, Salloum RM. et al. Blockade of the vascular endothelial growth factor stress response increases the antitumor effects of ionizing radiation. Cancer Res. 1999;59:3374
39. Mauceri HJ, Hanna NN, Beckett MA, Gorski DH, Staba MJ, Stellato KA. et al. Combined effects of angiostatin and ionizing radiation in antitumour therapy. Nature. 1998;394:287-91
40. Nielsen BS, Lund LR, Christensen IJ, Johnsen M, Usher PA, Wulf-Andersen L. et al. A precise and efficient stereological method for determining murine lung metastasis volumes. Am J Physiol. 2001;158:1997-2003
41. Jackson JD. Classical electrodynamics. Classical Electrodynamics, 3rd Edition, by John David Jackson, pp 832 ISBN 0-471-30932-X Wiley-VCH, July 1998. 1998 1
42. Medintz IL, Hildebrandt N. FRET - Förster resonance energy transfer from theory to applications. In. Wiley-VCH Verlag GmbH. 1999 http://dx.doi.org/10.1002/9783527656028
43. Mitsunaga M, Ogawa M, Kosaka N, Rosenblum LT, Choyke PL, Kobayashi H. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat Med. 2011;17:1685-91
44. Bulina ME, Chudakov DM, Britanova OV, Yanushevich YG, Staroverov DB, Chepurnykh TV. et al. A genetically encoded photosensitizer. Nat Biotechnol. 2005;24:95-9
45. Halliwell B, Chirico S. Lipid peroxidation: its mechanism, measurement, and significance. Am J Clin Nutr. 1993;57:715S-24S
46. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509-17
47. del Carmen MG, Rizvi I, Chang Y, Moor AC, Oliva E, Sherwood M. et al. Synergism of epidermal growth factor receptor-targeted immunotherapy with photodynamic treatment of ovarian cancer in vivo. J Natl Cancer Inst. 2005;97:1516-24
48. Duska LR, Hamblin MR, Miller JL, Hasan T. Combination photoimmunotherapy and cisplatin: Effects on human ovarian cancer ex vivo. J Natl Cancer Inst. 1999;91:1557-63
49. Rizvi I, Dinh TA, Yu WP, Chang YC, Sherwood ME, Hasan T. Photoimmunotherapy and irradiance modulation reduce chemotherapy cycles and toxicity in a murine model for ovarian carcinomatosis: perspective and results. Isr J Chem. 2012;52:776-87
50. Tang P, Zhang D-M, Xuan N, Tsui S, Waye M, Kong S-K. et al. Photodynamic therapy inhibits P-glycoprotein mediated multidrug resistance via JNK activation in human hepatocellular carcinoma using the photosensitizer pheophorbide a. Mol Cancer. 2009;8:56-67
51. Khdair A, Handa H, Mao G, Panyam J. Nanoparticle-mediated combination chemotherapy and photodynamic therapy overcomes tumor drug resistance in vitro. Eur J Pharm Biopharm. 2009;71:214-22
52. Goff BA, Bamberg M, Hasan T. Photoimmunotherapy of human ovarian-carcinoma cells ex vivo. Cancer Res. 1991;51:4762-7
53. Celli JP, Solban N, Liang A, Pereira SP, Hasan T. Verteporfin-based photodynamic therapy overcomes gemcitabine insensitivity in a panel of pancreatic cancer cell lines. Lasers Surg Med. 2011;43:565-74
54. Capella MAM, Capella LS. A light in multidrug resistance: photodynamic treatment of multidrug-resistant tumors. J Biomed Sci. 2003;10:361-6
55. Li W, Zhang WJ, Ohnishi K, Yamada I, Ohno R, Hashimoto K. 5-Aminolaevulinic acid-mediated photodynamic therapy in multidrug resistant leukemia cells. J Photochem Photobiol B. 2001;60:79-86
56. Teiten M-H, Bezdetnaya L, Merlin J-L, Bour-Dill C, Pauly M, Dicato M. et al. Effect of meta-tetra(hydroxyphenyl)chlorin (m THPC)-mediated photodynamic therapy on sensitive and multidrug-resistant human breast cancer cells. J Photochem Photobiol B. 2001;62:146-52
57. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA. et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429-34
58. Čunderlı́ková B, Gangeskar L, Moan J. Acid-base properties of chlorin e 6: relation to cellular uptake. J Photochem Photobiol B. 1999;53:81-90
59. Kessel D, Poretz R. Sites of photodamage induced by photodynamic therapy with a chlorin e6 triacetoxymethyl ester (CAME). Photochem Photobiol. 2000;71:94-6
60. Mojzisova H, Bonneau S, Vever-Bizet C, Brault D. Cellular uptake and subcellular distribution of chlorin e6 as functions of pH and interactions with membranes and lipoproteins. Biochim Biophys Acta. 2007;1768:2748-56
61. Bulina ME, Chudakov DM, Britanova OV, Yanushevich YG, Staroverov DB, Chepurnykh TV. et al. A genetically encoded photosensitizer. Nat Biotechnol. 2006;24:95-9
62. Liao Z-X, Li Y-C, Lu H-M, Sung H-W. A genetically-encoded KillerRed protein as an intrinsically generated photosensitizer for photodynamic therapy. Biomaterials. 2014;35:500-8
63. Watanabe R, Hanaoka H, Sato K, Nagaya T, Harada T, Mitsunaga M. et al. Photoimmunotherapy Targeting Prostate-Specific Membrane Antigen: Are Antibody Fragments as Effective as Antibodies? J Nucl Med. 2015;56:140-4
64. Bulina ME, Lukyanov KA, Britanova OV, Onichtchouk D, Lukyanov S, Chudakov DM. Chromophore-assisted light inactivation (CALI) using the phototoxic fluorescent protein KillerRed. Nat Protoc. 2006;1:947-53
65. Kessel D, Luo Y. Mitochondrial photodamage and PDT-induced apoptosis. J Photochem Photobiol B. 1998;42:89-95
66. Kessel D, Luo Y. Photodynamic therapy: a mitochondrial inducer of apoptosis. Cell Death Differ. 1999;6:28-35
67. Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part three—photosensitizer pharmacokinetics, biodistribution, tumor localization and modes of tumor destruction. Photodiagnosis Photodyn Ther. 2005;2:91-106
68. Loening AM, Wu AM, Gambhir SS. Red-shifted Renilla reniformis luciferase variants for imaging in living subjects. Nat Methods. 2007;4:641-3
69. Boyle RW, Dolphin D. Structure and biodistribution relationships of photodynamic sensitizers. Photochem Photobiol. 1996;64:469-85
70. Hamblin MR, Miller JL, Hasan T. Effect of charge on the interaction of site-specific photoimmunoconjugates with human ovarian cancer cells. Cancer Res. 1996;56:5205-10
71. Spring BQ, Abu-Yousif AO, Palanisami A, Rizvi I, Zheng X, Mai ZM. et al. Selective treatment and monitoring of disseminated cancer micrometastases in vivo using dual-function, activatable immunoconjugates. Proc Natl Acad Sci U S A. 2014;111:E933-E42
72. Ozaslan C, Kuru B. Lymphedema after treatment of breast cancer. Am J Surg. 2004;187:69-72
73. Jenkins DE, Oei Y, Hornig YS, Yu SF, Dusich J, Purchio T. et al. Bioluminescent imaging (BLI) to improve and refine traditional murine models of tumor growth and metastasis. Clin Exp Metastasis. 2003;20:733-44
74. Tammela T, Saaristo A, Holopainen T, Ylä-Herttuala S, Andersson LC, Virolainen S. et al. Photodynamic ablation of lymphatic vessels and intralymphatic cancer cells prevents metastasis. Sci Transl Med. 2011;3:69ra11
Author contact
![]() Corresponding author: S. H. Andy Yun, Ph.D., Associate Professor. Tel: 1-617-768-8704; Email: syunharvard.edu
Corresponding author: S. H. Andy Yun, Ph.D., Associate Professor. Tel: 1-617-768-8704; Email: syunharvard.edu