Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Variation of tunable...
Modulating physiological state...
Hitchhiking-based delivery of...
Conclusions and prospects
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(9):1007-1020. doi:10.7150/thno.11742 This issue Cite
Review
Current Approaches for Improving Intratumoral Accumulation and Distribution of Nanomedicines
Mikhail O Durymanov1, Andrey A Rosenkranz1,2, Alexander S Sobolev1,2 ![]()
1. Department of Molecular Genetics of Intracellular Transport, Institute of Gene Biology, Russian Academy of Sciences, 34/5, Vavilov St., 199334, Moscow, Russia
2. Department of Biophysics, Faculty of Biology, Moscow State University, 1-12, Leninskie Gory, 119991, Moscow, Russia
Received 2015-1-31; Accepted 2015-5-9; Published 2015-6-8
Abstract

The ability of nanoparticles and macromolecules to passively accumulate in solid tumors and enhance therapeutic effects in comparison with conventional anticancer agents has resulted in the development of various multifunctional nanomedicines including liposomes, polymeric micelles, and magnetic nanoparticles. Further modifications of these nanoparticles have improved their characteristics in terms of tumor selectivity, circulation time in blood, enhanced uptake by cancer cells, and sensitivity to tumor microenvironment. These “smart” systems have enabled highly effective delivery of drugs, genes, shRNA, radioisotopes, and other therapeutic molecules. However, the resulting therapeutically relevant local concentrations of anticancer agents are often insufficient to cause tumor regression and complete elimination. Poor perfusion of inner regions of solid tumors as well as vascular barrier, high interstitial fluid pressure, and dense intercellular matrix are the main intratumoral barriers that impair drug delivery and impede uniform distribution of nanomedicines throughout a tumor. Here we review existing methods and approaches for improving tumoral uptake and distribution of nano-scaled therapeutic particles and macromolecules (i.e. nanomedicines). Briefly, these strategies include tuning physicochemical characteristics of nanomedicines, modulating physiological state of tumors with physical impacts or physiologically active agents, and active delivery of nanomedicines using cellular hitchhiking.
Keywords: nanoparticles, macromolecules, nanomedicines
Introduction
The development of effective and highly selective drugs against cancer as a part of Ehrlich's “magic bullet” concept is one of the main challenges of contemporary medicine. Macromolecule-based and nanoparticle-based delivery systems have turned out to be a significant breakthrough. Due to large fenestrations in disorganized tumor endothelium [1], slowing of blood flow [2], and lack of lymphatic drainage [3], intravenously administered nanomedicines can extravasate and delay in tumor interstitium. On the contrary, their penetration through the tight endothelial junctions of normal blood vessels is strongly limited. As a result, local tumor concentrations of nano-scaled anticancer agents can be several-fold higher than in most normal tissues. This phenomenon was firstly described in 1986 and called the “enhanced permeability and retention” (EPR) effect [4]. A significant contribution of the EPR effect in accumulation of nano-scaled therapeutics in solid tumors has been noted in numerous studies [5-10]. However, the above-mentioned morphological and physiological abnormalities of tumors are the source of transport barriers that limit intratumoral accumulation and uniform distribution of nanomedicines [11]. First, tumors have a poorly organized and leaky vasculature network that in combination with increased hematocrit and viscosity of tumor blood diminishes blood flow rate [2, 11]. Reduction of tumor blood flow also originates from growth-induced solid stress because active proliferation of cancer cells leads to compression of blood and lymphatic vessels, especially in its inner regions [12]. This fact causes poor perfusion and collapse of lymphatic drainage in the core of a tumor, which is usually accompanied by a decrease in extracellular pH and an increase in necrosis. Lack of lymphatic drainage along with leaky blood vessels is the source of elevated interstitial fluid pressure [13], which results in almost complete absence of fluid pressure gradient between interstitium of tumor tissue and intravascular space. Furthermore, cellular hyperplasia reduces intercellular spacing and promotes increase in extracellular matrix density along with cross-linking of matrix components including collagen fibers, fibronectin, and glycosaminoglycans [14-17]. Thus, reduction of perfusion and blood flow rate significantly impedes tumor supply with anticancer agents, whereas vascular barrier, interstitial hypertension, as well as high density of extracellular matrix limit transvascular permeability and intratumoral diffusion of nanomedicines [18].
It is important to emphasize that nanotherapeutics are unable to cross biological barriers actively. Convection and diffusion are the main driving forces of nanomedicine transport, but their contribution varies depending on the transport step. Vascular transport of nanomedicines to the site of a tumor occurs substantially by convection with blood flow. Subsequently, nanoparticles extravasate and penetrate into the tumor interstitium due to diffusion affected by convection. The convective constituent at this step is determined by interstitial fluid flux towards lymphatic capillaries and takes place only at the tumor margins where lymphatic drainage remains, in contrast to the core of the tumor [12]. Another determinant of effective tumoral uptake of nanomedicines is combination of their “passive” features like size, surface charge, shape, biocompatibility, susceptibility to microenvironment, targeting functionality, and biodegradability [19]. In this connection, there are three general strategies to improve delivery of nano-scaled particles and macromolecules to a tumor. Because lifetime in the bloodstream as well as crossing of vascular and stromal barriers in tumor depends on physicochemical characteristics of nanoparticles, the first approach is based on chemical modification and variation of tunable features of nanomedicines resulting in prolonged circulation in blood and enhanced passive accumulation in tumor. The next category of strategies is founded on modulating physiological state of tumors including changing of tumor blood flow, interstitial fluid pressure, vascular and stromal permeability using external actions which can have physical (temperature, magnetic field, ultrasound, radiation) or chemical (hormones, cytokines, matrix-modifying agents and other physiologically active agents) nature followed by administration of nanotherapeutics. Hence, reducing the impact of transport barriers results in the EPR effect and increases tumor accumulation of nanomedicines. At last, the third approach to improve delivery of nanomedicines, termed “cellular hitchhiking” [20], does not depend on passive accumulation in tumor at all. Harnessing of live cells with natural tumor-homing ability enables to impart “active” features to nanomedicines in terms of crossing transvascular barrier and reaching of inner poorly perfused areas of tumor.
In this review, we briefly describe the basic aspects of tumor-targeted delivery of intravenously injected nanomedicines and discuss current approaches including tuning “passive” properties of nanomedicines, modulating physiological state of tumors, and cellular hitchhiking in order to reduce the influence of transport barriers and improve intratumoral uptake and distribution.
Variation of tunable physicochemical properties of nanomedicines by means of chemical modifications
Generally, the vast majority of nanomedicines after intravenous injection accumulate mainly in liver, spleen, and to a lesser extent in lungs [21]. As for tumor, the EPR effect significantly determines selectivity of nanomedicine deposition there, although its contribution may be different because of heterogeneity of tumor morphology between patients [22] and different tumor models. On the other hand, extravasation through abnormal openings between endothelial cells, penetration into the interstitium, and retention of nanomedicines in tumors are affected by the combination of tunable “passive” characteristics of nanoparticles including their hydrophilicity, size, shape, surface charge, targeting functionality, susceptibility to external stimuli, and tumor microenvironment. Changing of these properties in the context of effective tumor deposition as well as possible pitfalls and limitations are discussed in this chapter and summarized briefly in Table 1.
Functionalization and tuning physicochemical characteristics of nanotherapeutics.
| Characteristics of nanomedicines | Advantages and means of optimizations | Disadvantages and limitations |
|---|---|---|
| PEGylation (hydrophilicity) | PEGylation leads to extended circulation time and improved biocompatibility of nanoformulations [28-31] | “PEG-dilemma” (see 2.1 for details) [33] Accelerated blood clearance due to production of PEG-specific antibodies [34,35] |
| Size | The optimal size of nanomedicines for improved tumor accumulation should be 30-60 nm [40-42] | Nanoparticles with diameters > 200 nm are trapped by lung, spleen, and liver macrophages [23,25] Fast renal clearance of small nanoparticles (less than 8 nm) [25] |
| Surface charge | Neutral and slightly negatively charged nanoparticles have extended lifetime in the bloodstream [49,50] The optimal surface charge of nanomedicines for improved tumoral uptake should be close to neutral or slightly negative [46, 47, 49, 50] | Nanoparticles with highly positive surface charge can promote platelet aggregation and hemolysis [45] Highly positively or negatively charged nanoparticles usually display faster clearance than neutral or slightly charged ones [27,49,50] Suspensions of nanoparticles with zeta-potential between -25 mV and + 25 mV are considered unstable after long-term storage [51] |
| Shape of nanoparticles | Elongated nanoparticles sometimes display extended lifetime in blood as compared with spherical ones [53,54] | Influence of shape on extravasation ability is multifaceted depending on morphological characteristics of the tumor [57] |
| Stimulus-responsive delivery systems | Accumulation of nanomedicines in tumors can be improved using incorporation of magnetically responsive nanoparticles in combination with magnetic field [60, 61] or thermosensitive polymeric delivery systems and local hyperthermia [63, 64] Triggering systems sensitive to tumor microenvironment can help to overcome the “PEG-dilemma” [66] and minimize particle size | Feasibility of triggering systems, sensitive to tumor microenvironment, must be validated for a particular tumor |
| Modification with a target moiety | Targeting with a ligand causes strong binding and/or internalization of nanomedicines into cancer cells in tumor [72] Targeting prevents washout of nanoparticles from the extravascular compartment back to the blood circulation [42,73] | Modification with a ligand moiety does not always cause augmentation in tumoral uptake of nanomedicines [10, 42, 71-73, 78] “Binding site barrier” (see 2.6 for details) [42,80,81] Targeted nanoparticles can lose their specificity after placing in biological fluids due to shielding with protein corona [79] |
Hydrophilicity and PEGylation
In the context of tumor uptake enhancement, circulation time of nanotherapeutics in the bloodstream becomes one of the most important parameters because it increases the amount of nanomedicine that passes through the tumor and the possibility of extravasation. The key role in recognition and elimination of intravenously injected nanoparticles, which determines their circulation time, is played by the reticuloendothelial system (RES), which is comprised of resident macrophages and dendritic cells mainly in the liver and spleen [23]. Normally, these cells participate in recognition and phagocytosis of pathogens, production of inflammatory cytokines, and antigen presentation [24]. In the case of nanoparticle application, their fast elimination from the bloodstream by the RES may cause off-target side effects and decrease in tumoral uptake [25]. Circulation time as well as tumor accumulation depends on a combination of such characteristics of nanomedicines as hydrophilicity/hydrophobicity, size, surface charge, and shape [26]. As a rule, hydrophobic nanoformulations as well as nanoparticles with large size or high positive or negative surface charge usually have short circulation times (seconds to minutes) [25, 27]. The most appropriate and simplest modification of nanomedicines to minimize their unwanted interactions with plasma proteins and macrophages is attachment of polyethylene glycol (PEG) onto the nanoparticle surface, termed “PEGylation”. It has been shown that PEGylation significantly enhances circulation time and improves biocompatibility and tumor accumulation of gold nanorods [28], polymeric-based nanoparticles [29, 30], therapeutic proteins, and liposomes [31]. In general, higher density of a PEG shell or longer PEG length is more effective in avoiding opsonization and subsequent nonspecific uptake by phagocytic cells [32]. However, issues about appropriate PEG length and density are still under discussion. The matter is that high surface PEG coverage may hinder interaction of nanovehicles with target cell surface and diminish therapeutic effect of nanomedicines whose mechanisms implicate obligatory internalization into cancer cells (for example, nanoparticles for nucleic acid delivery). This contradictory effect is known as the “PEG-dilemma” [33]. Another obstacle of PEGylation is producing of PEG-specific IgM antibodies by splenic B cells, which have been observed in a few animal species [34]. As a result, it causes accelerated blood clearance of PEG-modified nanomedicines, including polymeric micelles [35], PEGylated proteins [36], etc., upon repeated doses. In recent years, this phenomenon has been attracting increasing attention among researchers and clinicians, but still there is no well-defined approach to avoid this problem completely [34]. Application of alternative hydrophilic polymers instead of PEG seems to be the most perspective approach to abrogate the accelerated blood clearance phenomenon. Some steps forward in this direction have been made. Use of poly(hydroxyethyl-L-asparagine) [37] and poly(N-vinyl-2-pyrrolidone) for coating of nanomedicines decreases the accelerated blood clearance phenomenon upon repeated administration [38].
Size
The size of nanotherapeutics significantly affects transvascular transport and accumulation in tumor. Because of high interstitial fluid pressure and low contribution of convection into traffic through vascular barrier, the diffusive constituent remains the main driving force of extravasation and penetration of nanoparticles into tumor. Therefore, minimization of size enhances uniform distribution of nanomedicines, but it is also important to take into account possibility of fast renal clearance (cutoff size < 8 nm) [25] and extravasation in normal tissues with upper pore size in endothelium between 5 and 12 nm [39]. Thus, tuning size of nanoparticles may help to minimize side effects and to find the optimal variants with effective tumor-targeted accumulation. It has been shown using hydrophilic dextrans of various weights (from 3.3 kDa to 2 MDa) that increase in the molecular weight (and hydrodynamic diameter) statistically significantly reduces its vascular permeability [40]. Despite better penetration, low molecular weight 3.3-kDa dextran (2 nm) displays high rate of tumor washout because of renal clearance and low plasma half-life. The highest extent of tumor accumulation was achieved by dextrans with molecular weights between 40 and 70 kDa (5-7 nm), though its high concentration was observed approximately 15 μm from the vessel wall after a 30-minute period. Analysis of transvascular transport of quantum dot-based nanoparticles (ζ-potential range was from -9 to +5 mV) has shown that 12-nm nanoparticles extravasate easily and penetrate up to 100 μm in 60 min, 60-nm nanoparticles also extravasate but do not leave the immediate perivascular space, while the 125-nm particles do not appreciably extravasate [41]. This trend was also exhibited in the case of neutral polymeric micelles: 25-nm nanoparticles displayed faster clearance, greater penetration, but lower total accumulation in tumor versus 60-nm counterparts [42]. Penetration of nanomedicines with different sizes significantly depends on viscosity of tumor stroma, which has been shown on neutral polymeric micelles of different diameters (from 30 to 100 nm) in both highly and poorly permeable tumors. Whereas all nanoparticles extravasated and penetrated into tumor tissue more than 100 nm in highly permeable tumors, only 30-nm nanoparticles could extravasate into poorly permeable tumors and cause distinct therapeutic effect [43]. In addition, nanoparticle size also affects cellular uptake, which should be taken into consideration when nanomedicine internalization into cancer cells is necessary. It was found that gold nanoparticles with diameters of 40-50 nm coated with antibodies against ErbB2 receptors displayed maximal level of internalization in ErbB2-overexpressing human breast cancer cells [44].
In terms of long circulation in the bloodstream, the diameter of nanomedicines should be between 20 nm and 150 nm to prevent fast renal clearance and to attenuate off-target uptake by liver, respectively. With respect to enhanced uptake and penetration into tumor tissue, the size of 30-60 nm seems to be preferred.
Surface charge
Surface charge (or electrophoretic mobility) of nanoparticles also predetermines their transvascular transport, penetration, and safety [45-48]. It is evident that surface charge of nanoformulations directly depends on chemical composition. Modification of nanoparticles with negatively or positively charged groups can be exploited as a tool for driving their transvascular transport and selectivity. It has been shown that cationized IgG antibodies (approximate size ≈ 11 nm) extravasate faster in solid tumors compared to the same macromolecules with neutral or negative charges [46]. At the same time, positively charged antibodies exhibited faster clearance, which indicates increase in their delivery to normal tissues. As for total accumulation in tumor tissue, unmodified antibodies turned out to be more selective due to relatively fast extravasation and slow plasma clearance [46]. Analysis of tumoral accumulation and microdistribution of 150-nm fluorescently labeled liposomes with different surface charge showed that by 6 hours post-injection, cationic liposomes (zeta-potential, ζ = +55 mV) accumulated in tumor 1.5-fold and 7-fold more effectively than their slightly anionic (ζ = -30 mV) and anionic (ζ = -75 mV) counterparts, respectively. In spite of higher selectivity of cationic liposomes to tumor site, they accumulated on the vascular endothelium without following penetration into the extravascular compartment of the tumor [47, 48], whereas slightly anionic and anionic counterparts accumulate within tumor tissue [47]. Some other groups investigated the influence of surface charge on pharmacokinetics and total tumor accumulation of polymeric nanoparticles [49, 50]. They showed that nanoparticles with neutral or slightly negative surface charge tended to accumulate in tumor more efficiently, with minimal uptake in lungs and liver. At the same time, this category of nanoparticles showed prolonged circulation in blood, whereas nanoparticles with high surface charge, either positive or negative, were taken up more efficiently by murine macrophages [49, 50]. Therefore, the optimal surface charge of nanomedicines should be close to neutral or slightly negative. Such surface charge extends lifetime of nanomedicines in the bloodstream, attenuates their uptake by macrophages, and gives them the ability to penetrate into tumor tissue as deeply as possible without electrostatic binding with positively or negatively charged components of the extracellular matrix. On the other hand, one needs to keep in mind that suspensions of neutral or slightly charged nanoparticles (-25 mV ≤ ζ ≤ +25 mV) are not stable, with a propensity to coagulate, flocculate, and/or agglomerate after long-term storage [51]. This requires the use of such nanomedicines immediately after their preparation.
Shape of nanoparticles
The next important “passive” property of nanomedicines that affects tumor accumulation and extravasation is the shape of the particles. First, the shape of nanoparticles influences phagocytosis by macrophages. It has been shown that elongated nanoparticles with high aspect (length/diameter) ratios have little chance to be internalized by phagocytic cells in comparison with spherical counterparts [52]. Therefore, worm-like particles must have extended lifetime in the bloodstream, which was confirmed in the case of filomicelles and gold nanorods versus their spherical analogs [53, 54]. Besides prolonged circulation, high aspect ratio and contact surface area of elongated nanoparticles might also contribute to their superior tumor deposition. The matter is that worm-like nanoparticles moving along a fluid have internal momentum. Thus, drastic change in blood flow direction in tortuous tumor vessels can cause tumbling and collision with the endothelial cell walls if the momentum force of the particles exceeds the capability of the fluid force to change direction [32, 55]. Tumbling effects as well as higher contact surface area of elongated nanoparticles increase their binding probability and strong adhesion in comparison with spherical ones [32, 55], especially at lower shear rates [55, 56].
Although use of elongated nanoparticles could increase their binding with tumor endothelium and total accumulation, it does not mean that they possess superior extravasation. For example, high-aspect-ratio carbon nanotubes exhibited extravasation different from spherical quantum dots for three different tumor models, although the nanoparticles had similar surface coatings, area, and charge [57]. The observed discrepancies in extravasation behavior were due to different morphology of tumor vasculature [57]. Therefore, it seems very complicated to predict which nanoparticle geometry is more appropriate for effective delivery to a certain tumor.
Triggering systems
Improvement of therapeutic properties of nanomedicines can be achieved due to development of stimulus-responsive, or triggering systems. These delivery systems are able to accumulate in tumors passively and change their properties under the influence of external impacts (e.g., temperature, magnetic field) or tumor microenvironment (e.g., tumor matrix metalloproteinases (MMPs), low pH) [58]. Such transformations of nanomedicines enable them to overcome transport barriers, problems of controlled release, and safety. Herein, we dwell on strategies aimed at improvement of nanoparticle accumulation and distribution in tumor.
Tumoral uptake of nanomedicines can be improved by incorporation of a magnetically responsive moiety, like magnetite (Fe3O4 or γ-Fe2O3) or magnetized metal particles [58], and external magnetic field. As a result, magnetic field-driven nanoparticles collide with vessel wall in tumor more intensively, extravasate, and move towards the magnet despite the elevated interstitial fluid pressure, showing magnetically enhanced retention. As likely as not, magnetic-enhanced extravasation of magnetically responsive nanoparticles can occur via transendothelial transport [59]. Anyway, some of these nanopreparations display enhanced delivery of drugs [60] and nucleic acids [61] to tumors. Improved deposition of nanomedicines in superficial tumors can also be achieved using thermosensitive polymeric delivery systems that can change conformation from water-soluble to hydrophobic at phase transition temperature (about 40 °C that corresponds to hyperthermal conditions) [62]. This hydrophilic-hydrophobic transition enables accumulation of nanomedicines in locally heated tumor tissues, preventing their washout. Nanovehicles based on thermosensitive poly(N-isopropylacrylamide) have been reported to have superior DNA [63] and drug [64] delivery to hyperthermia-treated tumors compared to non-treated ones. It should be noted that triggering systems responsive to external physical stimuli are applicable to treatment of tumors with well-defined localization. However, these systems, especially magnetically responsive ones, can be used clinically in case of nonresectable tumors, because this approach enables substantially enhance tumor permeability and displays small dependence on individual tumor properties.
Creation of triggering systems sensitive to tumor microenvironment aims to overcome the PEG-dilemma and minimize particle size. One of the most common and highly favorable triggering mechanisms relies on harnessing of enzymatic activity of tumor-specific proteases, mainly MMP-2 and -9. These MMPs are gelatinases overexpressed in various tumors and implicated in angiogenesis, epithelial-mesenchymal transition, and invasion [65]. Use of PEG-grafted polymeric micelles with MMP-degradable peptide linkers between the core-associated part of copolymer and surface PEG provides enzyme-mediated deshielding of these nanomedicines in a tumor and their stronger interaction with target cells, resulting in improved intratumor localization [66]. A similar strategy to achieve tumor-specific disassembly of aggregated nanoparticles was applied to a model delivery system composed of gelatin core covered with 10-nm quantum dots [67]. These 100-nm nanoparticles, administered intravenously, accumulate in tumor due to the EPR effect and undergo subsequent MMP-mediated cleavage into 10-nm particles able to diffuse deeply into tumor interstitium [67]. It should be taken into account that feasibility of triggering systems sensitive to tumor microenvironment must be validated for a particular tumor.
Modification with a target moiety
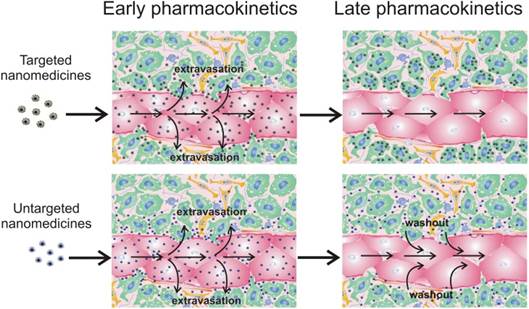
For targeted delivery of nanomedicines to cancer cells, molecular recognition processes, like ligand-receptor interactions, can be exploited. Usually, this approach aims for i) strong binding of nanotherapeutics to target cells and prolonged retention in tumor site, or ii) receptor-mediated internalization into cancer cells that is necessary for therapeutic nanovehicles containing nucleic acids (plasmid DNA, shRNA, mRNA) and locally acting molecules like Auger electron emitters or photosensitizers. In any event, modification of nanoparticles with a ligand often leads to their enhanced accumulation in tumor as compared with untargeted counterparts [42, 68-70]. However, this approach does not always augment nanomedicine deposition in tumors, which has been shown for targeted gold nanoparticles [71], liposomes [72,73], polymeric gene delivery vehicles (polyplexes) [10], and polymeric micelles [42] in comparison with their untargeted analogs. Most likely, discrepancies in the data can be explained by the fact that total accumulation is the integral characteristic of nanoparticle delivery. It is clear that the process of nanomedicine transport subdivides into some steps including vascular transport, extravasation, and interstitial transport. Detailed analysis of nanoparticle microdistribution revealed that the major part of “in tumor accumulated” nanoparticles reside on the vessel wall surface or in close proximity to the blood vessels [10, 40-42, 47, 48], which does not exclude their washout back to the blood circulation (Fig. 1). This means that strong binding with cancer cells is one of the main causes of superior tumor accumulation rate of targeted nanomedicines (Fig. 1). It should be noted that targeting mainly prevents the washout of the smaller nanoparticles from the extravascular compartment [42, 73], especially in slow flow tumor regions [73]. Therefore, high expression of target receptors in tumor and highly effective nanoparticle targeting are crucial conditions for prolonged retention of targeted nanomedicines in a tumor. The influence of expression level of target EGF receptors (EGFR) was evaluated in two breast murine tumor models with high (106 EGFR per cell) and low (104 EGFR per cell) level of EGFR expression. As a result, the greater accumulation of EGF-modified polymeric micelles compared with unmodified ones was observed only in the case of an EGFR-overexpressing model [42, 74]. The importance of the degree of nanoparticle targeting has been demonstrated for magnetic nanoparticles with spherical and elongated shape. Unlike spherical, worm-like shape provides a more effective scaffold to generate multivalent interactions of ligand moieties and receptors, resulting in enhancement of targeted efficiency and decrease in washout from a tumor [75]. Thus, low expression of target receptors and/or insufficient targeting of nanomedicines will lead to the same tumor deposition of targeted and untargeted nanoparticles. Even so, the benefit of ligand targeting can be illustrated by our own investigations of gene delivery into Cloudman S91 melanoma tumors (4,500 melanocortin receptors per cell) using polyplex nanoparticles. We found no differences in tumor accumulation and microdistribution of targeted polyplexes containing melanocortin receptor-1-specific peptide and untargeted ones. However, targeted polyplexes cause superior transfection in tumor [10], because the faster cellular uptake for polyplexes, provided by inclusion of a ligand into the polymeric part, is preferred for successful gene transfer [76, 77]. A similar effect has been demonstrated in case of 100-nm siRNA nanoparticles [78]. There is also a noteworthy fact that ligand targeting of PEGylated gene delivery vehicles makes it possible to overcome the above-mentioned “PEG-dilemma” by an increase in receptor-mediated internalization resulting in more effective gene therapy.
Influence of nanomedicine functionalization with target moieties on tumor accumulation. Targeted nanomedicines extravasate into tumor interstitium and bind/internalize into cancer cells (green) due to antigen-antibody/ligand-receptor interactions. Untargeted nanomedicines can bind and internalize into cancer (green) and non-cancer (yellow and blue) cells due to unspecific surface adsorption; endotheliocytes are shown in red. However, a significant part can be washed out from the extravascular compartment back to the blood circulation, resulting in lower tumor accumulation.
It is very important to reckon with some obstacles related to functionalization of nanomedicines with target moieties. In fact, such nanoparticles can lose their specificity after placing them in biological fluids. It has been shown that PEGylated silica nanoparticles modified with transferrin became shielded with a protein corona after exposure to serum. As a result, these nanoparticles lost targeting ability [79]. Apart from affinity, design of targeted nanomedicines should also consider size and charge of a ligand moiety. For instance, attachment of the highly cationic and large peptidic F-3 ligand to magnetic nanoparticles caused their fast elimination from the blood circulation mainly by the liver and lack of tumor homing [75]. Functionalization of macromolecules or nanoparticles with a ligand can also limit their penetration in tumor because of so-called “binding-site barrier”. This phenomenon is because penetration of ligand-containing macromolecules into tumors can be hampered by their overall binding to target cells, possessing receptors/determinants for the ligand, in close proximity to blood vessels. The effect of binding-site barrier has been revealed for antibodies [80, 81] as well as for targeted polymeric particles [42].
Thus, modification of nanoparticles with a ligand in designing of nanomedicines should be very careful and take into account possible limitations connected with targeting.
Modulating physiological state of tumors
Superior accumulation of nanomedicines in tumor site can be achieved not only by tuning their characteristics, but also via modulating tumor blood flow, vasculature leakiness, and extracellular matrix penetration prior to administration of nanomedicines. Applied strategies subdivide into physical methods and treatment with physiologically active agents. Because many were applied only for low molecular weight chemotherapeutics [82], we highlight here only ones that were tested for nanomedicines and found to have high clinical potential in terms of safety and therapeutic outcome.
Physical methods for increasing nanomedicine accumulation
Hyperthermia
Local heating of a tumor site often leads to enhanced accumulation of nano-scaled agents, yielding more effective outcome of gene [83], drug [84], and radioisotope [85] delivery. To achieve the maximal uptake of nanomedicines by tumor, the regimen of heating up to 42 °C is usually exploited, because further heating led to hemorrhage and stasis in tumor vessels [86]. The effect of hyperthermia can be enhanced by performing alternate cooling and heating on a tumor and its vasculature [87]. The phenomenon of improved nanomedicine delivery after hyperthermal exposure is based on increase in tumor blood flow as a result of vasodilation [88] and vascular permeability [89] owing to increase in intercellular gaps between endotheliocytes [90].
In spite of high potential in clinical use, it should be noted that the effect of hyperthermia on macromolecular and nanoparticle accumulation might depend on the chosen tumor model [84]. Another limitation of this approach is ability to treat only tumors with well-defined localization.
Local ionizing radiation treatment of tumor
Tumor-targeted delivery of nanomedicines can be improved by combining with local X- or γ-irradiation of the tumor, which was been demonstrated for polymeric [84, 91] and liposomal [92] drug delivery systems and antibodies [93, 94]. Some possible mechanisms underlie enhanced tumor uptake. The first is based on induction of inflammatory response after irradiation, which increases vascular permeability. It has been reported that irradiation of tumor-bearing mice causes 2-fold upregulation of proinflammatory cytokines like IFNγ and TNFα [95], which leads to vascular barrier dysfunction [96]. Tumor irradiation may also upregulate VEGF expression [97], which stimulates endothelial cells and cause disruption of adherens junction complexes between endotheliocytes [98]. Lastly, radiation treatment of tumor induces apoptotic changes in morphology of well-oxygenated tumor cells surrounding the microvessels but not in endothelial cells [91], resulting in decrease in interstitial fluid pressure [99].
The obvious advantage of ionizing radiation treatment is the reliability of this strategy for different tumor models [84]. However, this approach is suitable only for treatment of localized tumors.
Treatment with ultrasound
Ultrasound exposure at frequencies of 0.5-5 MHz can be used to magnify accumulation and extravasation of nanomedicines in localized tumors due to cavitational, thermal, and acoustic radiation force effects whose occurrence depends on exposure conditions [100]. Cavitation is a rapid formation of volumetrically oscillating microbubbles caused by ultrasound. These gas-filled microbubbles can be spontaneously nucleated by ultrasound in the tumor tissue or/and can be injected intravenously together with nanomedicines [101]. Collapsing of these bubbles can create transient pores in blood vessel walls and cell membranes (sonoporation), through which nanomedicines of different nature can extravasate and enter into neighboring cells [102,103]. Ultrasound exposure of tumors can also cause local hyperthermia (see 3.1.1) in an acoustic intensity and time dependent manner [104]. Finally, acoustic radiation force promotes flow of fluid in the direction of the sound wave propagation, enhancing convection of nanomedicines through tumor extracellular matrix [105].
Ultrasound exposure is also appropriate for treatment of deep-lying tumors. Moreover, multiple therapeutic mechanisms make this approach very attractive for augmentation of nanomedicine accumulation in tumor. Another advantage of ultrasound treatment is that it almost does not depend on the intensity of the EPR effect in the particular tumor [103].
Physiologically active modulators
Mediators of vascular permeability
Permeability of tumor vasculature can be enhanced after application of inflammatory molecules including TNFα [106], interleukin 2 [107], prostaglandin analogs [108], VEGF [109], histamine [110], nitric oxide (NO) donors [111], and other agents. Besides enhancing vascular permeability, these mediators also induce vasodilatation and increased blood flow. Although inflammatory mediators significantly increase accumulation of different nanomedicines, there are some serious obstacles for clinical application of these agents. First, some of them (inflammatory cytokines, histamine) have strong adverse effects after systemic application [112]. Therefore, the use of such agents requires local application [106,111] or targeted delivery to tumor site [107, 113]. Another obstacle is involvement of several inflammatory mediators in cancer progression [114]. In this connection, application of nitroglycerin ointment on the skin at the tumor site seems to be a safer and simpler method [111]. Under hypoxic and acidic conditions, nitroglycerin generates NO, which affects vascular permeability. Efficacy of this approach was found comparable with TNFα [106] or prostaglandin analogs [108], and it caused 2-fold augmentation in nanomedicine accumulation [111].
An alternative approach to attenuate barrier function of vessels is low-dose TGFβ type I receptor inhibition by small-molecule drugs, which results in decreased pericyte coverage of the endothelium in tumor neovasculature and promotes superior accumulation of nanomedicines in tumors [115]. The main challenge of this strategy is accurate selection of acting dose because total TGF-β signaling inhibition can have either positive or negative influence on cancer progression [116].
Matrix-modifying agents
The treatment of tumors with matrix-modifying agents aims to reduce extracellular matrix density, thus resulting in enhanced permeability of tumor interstitium, alleviation of solid stress, and, therefore, improvement of tumor perfusion. This category of drugs consists of matrix-degrading enzymes, small-molecule inhibitors, and hormones.
It has been shown that hyaluronan and collagen are the main components of tumor interstitium, being responsible for solid stress, increasing interstitial fluid pressure, and causing poor penetration of nanomedicines [17]. Consistent with this fact, direct degradation of these components with intratumoral injections of hyaluronidase and bacterial collagenases significantly improved distribution of liposomal doxorubicin [117] and oncolytic herpes simplex virus vectors [118], respectively. The same effect on 2 MDa dextran and IgG delivery in tumor caused by upregulation of MMPs by treatment of tumor-bearing mice with relaxin, a nontoxic hormone secreted by women during pregnancy [119]. Hence, extracellular matrix seems to be the critical barrier for deep penetration of nanomedicines into tumor tissue. However, hormone-mediated upregulation of MMPs or direct application of matrix-degrading enzymes can increase the risk of metastasis [118]. In this connection, the safest and most appropriate strategy is inactivation of cancer-associated fibroblasts (CAFs), which can also reduce solid stress and improve perfusion. It has been shown that inhibition of angiotensin receptors-1 with the clinically approved small-molecule losartan or other antagonists impairs activation of CAFs, leading to decreased CAF density and CAF-mediated production of hyaluronan and collagen [120], which enhances the penetration and efficacy of nanotherapeutics [121]. It is also noteworthy that inhibition of angiotensin receptors-1 signaling in itself attenuates growth of different types of tumors [122-124]. It does matter that application of losartan or analogs might cause synergetic contribution to anticancer therapy with nanomedicines.
Mild hypertension
Hypertension induced by angiotensin II infusion significantly increases accumulation of IgGs [125], polymeric drug delivery systems [126], and liposomes [127] in tumors. Angiotensin-II-induced hypertension shifts the fluid pressure gradient towards tumor interstitium and improves convective influx of nanomedicines into the tumor compartment out of the blood capillary. Moreover, application of vasoconstrictors increases tumor blood flow, which might also contribute to tumor uptake of nanomedicines.
Recently, this method was clinically tested resulting in improved delivery and therapeutic effects of polymeric drug conjugate for various advanced solid tumors [128].
Normalization of tumor vasculature
As a perspective strategy to obtain superior accumulation rate of nanomedicines in tumor and uniform microdistribution of anticancer agents, the concept of “normalization” of tumor vessels was developed by Jain's laboratory [129-132]. This approach aims vessel remodeling to a more “normal” morphological state with recovered basal membrane and pericyte coverage that makes tumor vessels less leaky and decreases interstitial fluid pressure and solid stress, resulting in improved perfusion and penetration of anticancer agents [129]. It has been found that transient “normalization” of tumor vasculature arose after antiangiogenic cancer therapy with small doses of anti-VEGF and VEGFR antibodies or small-molecule inhibitors of VEGFR tyrosine kinase activity, and this was accompanied by improved accumulation of both chemotherapeutics [129-131] and quantum dot-based nanoparticles [132]. Regarding nanoparticles, improvement of their accumulation in tumor interstitium occurs in a size-dependent manner and is preferential for 12-nm nanoparticles compared with 125-nm ones because recovery of basal membrane and pericyte coverage creates steric barrier for larger particles [132]. Therefore, “normalization” of tumor vasculature has great potential for cancer therapy with small nanomedicines.
Hitchhiking-based delivery of nanomedicines
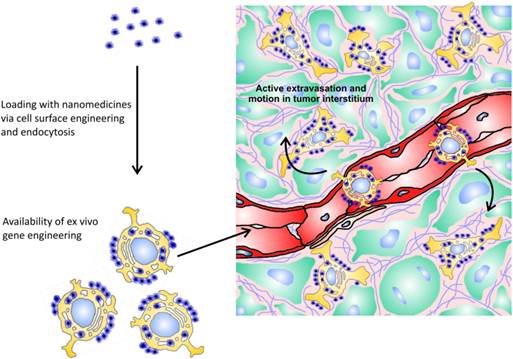
Use of cells with tumor-homing capacity for delivery of nanomedicines is a relatively new and appealing approach in cancer therapy (Fig. 2). Being natural “moving gears” per se, these cells confer their own “active” properties to attached nanoparticles, making them capable of tumor targeting, of crossing endothelial barrier, and of penetrating to poorly perfused areas of a tumor. These cells can be isolated from bone marrow and other sources, genetically engineered to impart additional capabilities, loaded with nanoparticles, and injected intravenously back into the patient. Nanoparticles can be attached to tumor-tropic cells due to internalization, nonspecific surface adsorption, ligand-receptor interactions, and covalent binding to amine (—NH2) or thiol (—SH) groups intrinsic to cell membrane proteins, or to other reactive groups introduced exogenously into plasma membrane [133].
The principle of cellular hitchhiking. Tumor-tropic cells can be loaded with nanomedicines via internalization, nonspecific surface adsorption, ligand-receptor interactions, and covalent binding. Furthermore, hitchhiked cells can be genetically engineered to impart additional capabilities to them. Injected intravenously, these cells can actively extravasate in tumor and relatively freely move in tumor interstitium.
Currently, there are three main classes of hitchhiked cells possessing innate tumor-homing ability and relative ease of isolation: mesenchymal stem cells (MSC), T-cells, and monocytes/macrophages.
Mesenchymal stem cells (MSCs)
MSCs are considered promising candidates for tumor-targeted delivery of nanomedicines. These cells can be harvested from the bone marrow, adipose tissue, umbilical cord blood, liver, placenta, muscles, and amniotic fluid [134,135]. When activated, MSCs migrate to sites of tissue damage and inflammation (including cancer) upon stimulation with CXCL12 and CXCL8 chemokines and growth factors [136, 137], and then extravasate via both leukocyte-like and other mechanisms [138]. Although MSCs are known to secrete antiinflammatory cytokines and factors stimulating both angiogenesis and tissue regeneration [139], their role in tumors is still unclear [140].
Intravenously injected MSCs initially accumulate in lungs and to a lesser extent in liver followed by gradual increase in their number in tumors [137], reaching the maximum at 14 days post-injection [140]. Only about 2-5% of intravenously administered MSCs accumulated in tumors, and almost the same fraction was found in lymph nodes and bone marrow [141], which might potentiate side effects there if MSCs are used for delivery of cytotoxic drug-loaded nanovehicles. Nevertheless, MSCs can be applied for photothermal therapy that involves delivery of gold nanoparticles followed by local irradiation of the tumor with near infrared light, which leads to its absorption by gold nanoparticles, dissipation into heat, and tumor necrosis [142]. A similar approach of MSC application is hitchhiking-based delivery of magnetic nanoparticles to tumor aiming at hyperthermal therapy by local treatment under alternating magnetic field [140]. In the latter case, harnessing of MSCs as delivery vehicles led to 4-fold increase in tumor accumulation and apparent inhibition of tumor growth compared with intravenous application of 50-nm magnetic nanoparticles alone [140].
Low efficacy of tumor targeting after intravenous administration is the main challenge of MSC application. However, generation of MSCs expressing key ligands that are required for homing (P-selectin glycoprotein ligand-1 and Sialyl-Lewisx) is a potential way to reduce their off-target accumulation and hasten migration to tumor. The advantage of this approach has been shown for targeted delivery of MSCs to sites of inflammation [143]. Another possible limitation of clinical application of MSCs is their ability to promote metastasis [141, 144, 145]. Perhaps, incorporation of a suicide gene into MCSs can help to overcome this problem.
It is worth noting that MSCs can be exploited for gene transfer to tumors after ex vivo transfection [146, 147]. To prevent off-target expression of a therapeutic gene, gene-specific promoters sensitive to tumor microenvironment can be exploited [147]. Thus, it does matter that anticancer effect of nanotherapeutics delivered by MSCs might be heightened by combining with gene therapy.
T cells
Cytotoxic T-cells (CTLs) possess innate ability to accumulate in a tumor and recognize and kill cancer cells with MHC restriction and antigen specificity, although their activity can be suppressed in tumor via CTLA-4 signaling and other mechanisms [148]. However, inactive tumor-infiltrating lymphocytes can be isolated from the tumor mass and incubated with IL-2 to activate and expand tumor-specific T cell population. Additionally, these cells can be engineered ex vivo to express tumor-specific T-cell receptors. Infusion of modified CTLs into a patient, known as adoptive T cell therapy, became the first attempt to exploit natural tumor-infiltrating capacity of T lymphocytes [149]. It was found that exogenously injected T cells preferentially accumulate in antigen-positive tumors, reaching a plateau at 7-8 days post-injection [150]. Infiltration of tissues with CTLs was observed in liver, lymph nodes, lungs, and spleen [151]. Therapeutic outcome of adoptive T cell therapy might be improved by exploiting antigen-specific T cells as delivery vehicles for nanomedicines, aiming to eliminate tumor cells directly or maintain activity and proliferation of CTLs. To accomplish the first task, T cells can be loaded with retroviral vectors encoding Herpes Simplex Virus thymidine kinase (HSVtk) due to nonspecific adsorption. Subsequently, viral nanoparticles can be delivered and released at the tumor site, causing expression of the cytotoxic HSVtk gene in the infected tumor cells and improving therapeutic effect [152]. The maintenance of anticancer activity and expansion of T cells can be substantially enhanced using the same retroviral system for transfer of the gene encoding proinflammatory cytokine IL-12 [152]. CTL-mediated hitchhiking of multilamellar lipid nanoparticles loaded with adjuvant drugs like cytokines IL-15 and IL-21 also aims for the achievement of T-cell expansion and enhancement of cancer immunotherapy [153].
Activated T-cells also may be exploited for delivery of cytotoxic gold nanoparticles aiming at mentioned above photothermal therapy. The use of T cell-mediated hitchhiking causes more than 4-fold augmentation in tumor deposition of 45-nm gold nanoparticles compared with their clinically approved intravenous application alone [154].
Monocytes/macrophages
Application of monocytes/macrophages as active tumor-targeted carriers is based on their significant capacity to infiltrate tumors, reaching up to 50 % of tumor mass [155]. Migration of monocytes to tumor occurs in response to a spectrum of cytokines released by cancer cells in hypoxic conditions [156]. In-tumor macrophages acquire alternatively activated M2 phenotype, promoting angiogenesis and cancer development [157].
Regarding pharmacokinetics of exogenously injected monocytes, initially they reside in lungs temporary followed by accumulation in liver and spleen, which in consistent with their natural behavior [158]. The maximal degree of tumor infiltration with systemically administered macrophages labeled with micrometer-sized particles of iron oxide was observed 3-5 days after injection [159]. Macrophages can be easily loaded with anticancer nanotherapeutics due to their unique property, phagocytosis. Generally, monocytes/macrophages act as “Trojan Horses” for delivery of cytotoxic nanomedicines, like gold nanoshells [157] or liposomal doxorubicin [160], to tumors. It should be emphasized that accumulated in tumor macrophages loaded with doxorubicin-containing liposomes inhibit tumor growth to greater extent than clinically approved intravenous injection of liposomal doxorubicin [160]. Moreover, macrophage-mediated hitchhiking is considered a perspective strategy for delivery of contrast agents to tumors aiming at MRI diagnostics of cancer [160]. The ability of engineered monocyte/macrophage-like cells to transfer both a therapeutic gene encoding activating enzyme and dextran-conjugated prodrug to tumor for gene-directed enzyme/prodrug therapy has also been reported [161].
Hence, the diversity of monocytes/macrophage application as well as demonstrated superior therapeutic effects in comparison with clinically approved nanomedicines should be motivating for development of this approach, especially for enhancement of tumor homing ability. Besides incorporation of magnetic nanoparticles followed by application of local magnetic field, gene engineering of macrophages to express homing machinery might be used.
Conclusions and prospects
Efficient cancer therapy with nanomedicines implies their tumor-selective delivery, accumulation, and uniform distribution in the tumor. Exploiting the EPR effect remains the main basis for targeted delivery of intravenously injected nanomedicines to tumors. Combining of tunable “passive” features of nanoparticles governing their pharmacokinetics has made it possible to yield in general 2-3-fold augmentation in tumor accumulation rate of nanomedicines in experiments on animals. However, in spite of numerous variants of nanomedicines for more than 25 years, there are only a few clinically approved ones and some under preclinical development [162]. Such limited progress in this field can be attributed to intratumoral barriers, poor intensity of the EPR effect in humans as compared with rodents [163], and its heterogeneity [21]. Actually, the extent of tumor vascularization, degree of vessel maturation, value of interstitial fluid pressure, and intensity of interstitial matrix density, which govern the EPR effect, can substantially vary between tumor types and depend on tumor size [11, 164-167]. This means that application of intravenously injected nanomedicines should take into account individual characteristics of tumors. Therefore, the high efficiency of nanomedicine uptake by the particular patient's tumor have to be confirmed using for example combining of nano-scaled contrast or labeled agent injection with an image-guided technique like MRI, PET, SPECT, or CT. Some strategies based on modulating physiological state of tumors can attenuate the impact of transport barriers and increase tumoral uptake of nanomedicines, but the clinical outcome of these methods is also to some extent affected by morphological characteristics of a tumor. In this context, it is very important to develop delivery approaches that do not rely on passive accumulation in the tumor. These strategies should imply active crossing of transport barriers, which may be possible due to application of magnetically responsive nanomedicines and local magnetic field (although small dependence on tumor morphology is retained), or cellular hitchhiking. The first strategy is relevant only for treatment of localized tumors, whereas hitchhiking-based delivery is appropriate for targeted delivery to metastatic tumors too, although there are some challenges for its clinical application so far. Meanwhile, harnessing of cells with tumor-tropic features enables delivery of nanoparticles almost regardless of their passive features and the individual characteristics of a tumor. Moreover, cellular hitchhiking may serve as a platform for both i) diagnostics and ii) combined therapy including cytotoxic therapy, immunotherapy, and gene therapy. Another possible way of nanomedicine design development is incorporation of propulsion and navigation capabilities in order to deliver nanoformulations deep into the tumor tissue and minimize off-target delivery. Although the concept of using nanomachines that are able to convert chemical or external energy into mechanical motion for drug delivery purpose seems rather futuristic now, some initial steps in this direction have been already done [168]. Perhaps, the use of bio-inspired nanomotors sensitive to the tumor microenvironment (e.g., enzyme and acidity) will help to achieve further progress in this field.
Abbreviations
EPR: enhanced permeability and retention; RES: reticuloendothelial system; PEG: polyethylene glycol; ErbB2: human epidermal growth factor receptor 2; IgG: Immunoglobulin G; MMP: matrix metalloproteinase; EGF: epidermal growth factor; EGFR: epidermal growth factor receptor; VEGF: vascular endothelial growth factor; VEGFR: receptor for vascular endothelial growth factor; IFNγ: interferon gamma; TNFα: tumor necrosis factor alpha; NO: nitric oxide; TGFβ: transforming growth factor beta; CAF: cancer-associated fibroblast; MSC: mesenchymal stem cell; CTL: cytotoxic T-lymphocytes; CTLA-4: cytotoxic T-lymphocyte-associated protein 4; HSVtk: herpes simplex virus thymidine kinase; IL: interleukin; MRI: magnetic resonance imaging; PET: positron emission tomography; SPECT: single-photon emission computed tomography; CT: computed tomography.
Acknowledgements
This research was supported by the Russian Foundation for Basic Research grant no. 13-04-01282-a and by the Program of Presidium of the Russian Academy of Sciences "Basic Sciences for Development of Biomedical Technologies".
Competing Interests
The authors declare that no competing interest exists.
References
1. Hashizume H, Baluk P, Morikawa S. et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156:1363-80
2. Sevick EM, Jain RK. Geometric resistance to blood flow in solid tumors perfused ex vivo: effects of tumor size and perfusion pressure. Cancer Res. 1989;49:3506-12
3. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27-31
4. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent SMANCS. Cancer Res. 1986;46:6387-92
5. Malik N, Evagorou EG, Duncan R. Dendrimer-platinate: a novel approach to cancer chemotherapy. Anticancer Drugs. 1999;10:767-76
6. Ambruosi A, Khalansky AS, Yamamoto H. et al. Biodistribution of polysorbate 80-coated doxorubicin-loaded [14C]-poly(butyl cyanoacrylate) nanoparticles after intravenous administration to glioblastoma-bearing rats. J Drug Target. 2006;14:97-105
7. Okuda T, Kawakami S, Akimoto N. et al. PEGylated lysine dendrimers for tumor-selective targeting after intravenous injection in tumor-bearing mice. J Control Release. 2006;116:330-6
8. Kim JH, Kim YS, Park K. et al. Antitumor efficacy of cisplatin-loaded glycol chitosan nanoparticles in tumor-bearing mice. J Control Release. 2008;127:41-9
9. Morton JG, Day ES, Halas NJ. et al. Nanoshells for photothermal cancer therapy. Methods Mol Biol. 2010;624:101-17
10. Durymanov MO, Slastnikova TA, Kuzmich AI. et al. Microdistribution of MC1R-targeted polyplexes in murine melanoma tumor tissue. Biomaterials. 2013;34:10209-16
11. Chauhan VP, Stylianopoulos T, Boucher Y. et al. Delivery of molecular and nanoscale medicine to tumors: transport barriers and strategies. Annu Rev Chem Biomol Eng. 2011;2:281-98
12. Leu AJ, Berk DA, Lymboussaki A. et al. Absence of functional lymphatics within a murine sarcoma: a molecular and functional evaluation. Cancer Res. 2000;60:4324-7
13. Fukumura D, Jain RK. Tumor microenvironment abnormalities: causes, consequences, and strategies to normalize. J Cell Biochem. 2007;101:937-49
14. Levental KR, Yu H, Kass L. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;193:891-906
15. Laurich C, Wheeler MA, Iida J. et al. Hyaluronan mediates adhesion of metastatic colon carcinoma cells. J Surg Res. 2004;122:70-4
16. Payne SL, Hendrix MJ, Kirschmann DA. Paradoxical roles for lysyl oxidases in cancer - a prospect. J Cell Biochem. 2007;101:1338-54
17. Alexandrakis G, Brown EB, Tong RT. et al. Two-photon fluorescence correlation microscopy reveals the two-phase nature of transport in tumors. Nat Med. 2004;10:203-7
18. Torosean S, Flynn B, Axelsson J. et al. Nanoparticle uptake in tumors is mediated by the interplay of vascular and collagen density with interstitial pressure. Nanomedicine. 2013;9:151-8
19. Rabanel JM, Aoun V, Elkin I. et al. Drug-loaded nanocarriers: passive targeting and crossing of biological barriers. Curr Med Chem. 2012;19:3070-102
20. Anselmo AC, Mitragotri S. Cell-mediated delivery of nanoparticles: Taking advantage of circulatory cells to target nanoparticles. J Control. Release. 2014;190:531-41
21. Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153:198-205
22. Harrington KJ, Mohammadtaghi S, Uster PS. et al. Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylated liposomes. Clin Cancer Res. 2001;7:243-54
23. Bhadra D, Bhadra S, Jain P. et al. Pegnology: a review of PEG-ylated systems. Pharmazie. 2002;57:5-29
24. Sankari SL, Babu NA, Masthan MK. et al. The mononuclear phagocyte system in health and disease. Indian J Dent Sci. 2013;1:126-128
25. Nel AE, Mädler L, Velegol D. et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8:543-57
26. Wang B, He X, Zhang Z. et al. Metabolism of nanomaterials in vivo: blood circulation and organ clearance. Acc Chem Res. 2013;46:761-9
27. Alexis F, Pridgen E, Molnar LK. et al. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505-15
28. Akiyama Y, Mori T, Katayama Y. et al. The effects of PEG grafting level and injection dose on gold nanorod biodistribution in the tumor-bearing mice. J Control Release. 2009;139:81-4
29. Fang C, Shi B, Pei YY. et al. In vivo tumor targeting of tumor necrosis factor-alpha-loaded stealth nanoparticles: effect of MePEG molecular weight and particle size. Eur J Pharm Sci. 2006;27:27-36
30. Kano A, Moriyama K, Yamano T. et al. Grafting of poly(ethylene glycol) to poly-lysine augments its lifetime in blood circulation and accumulation in tumors without loss of the ability to associate with siRNA. J Control Release. 2011;149:2-7
31. Milla P, Dosio F, Cattel L. PEGylation of proteins and liposomes: a powerful and flexible strategy to improve the drug delivery. Curr Drug Metab. 2012;13:105-19
32. Duan X, Li Y. Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small. 2013;9:1521-32
33. Hatakeyama H, Akita H, Harashima H. The polyethyleneglycol dilemma: advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol Pharm Bull. 2013;36:892-9
34. Abu Lila AS, Kiwada H, Ishida T. The accelerated blood clearance (ABC) phenomenon: clinical challenge and approaches to manage. J Control Release. 2013;172:38-47
35. Shiraishi K, Yokoyama M. Polymeric micelles possessing polyethyleneglycol as outer shell and their unique behaviors in accelerated blood clearance phenomenon. Biol Pharm Bull. 2013;36:878-82
36. Lu W, Wan J, She Z. et al. Brain delivery property and accelerated blood clearance of cationic albumin conjugated pegylated nanoparticle. J Control Release. 2007;118:38-53
37. Romberg B, Oussoren C, Snel CJ. et al. Pharmacokinetics of poly(hydroxyethyl-l-asparagine)-coated liposomes is superior over that of PEG-coated liposomes at low lipid dose and upon repeated administration. Biochim Biophys Acta. 2007;1768:737-43
38. Ishihara T, Maeda T, Sakamoto H. et al. Evasion of the accelerated blood clearance phenomenon by coating of nanoparticles with various hydrophilic polymers. Biomacromolecules. 2010;11:2700-6
39. Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res. 2010;2:14
40. Dreher MR, Liu W, Michelich CR. et al. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J Natl Cancer Inst. 2006;98:335-44
41. Popovic Z, Liu W, Chauhan VP. et al. A nanoparticle size series for in vivo fluorescence imaging. Angew Chem Int Ed Engl. 2010;49:8649-52
42. Lee H, Fonge H, Hoang B. et al. The effects of particle size and molecular targeting on the intratumoral and subcellular distribution of polymeric nanoparticles. Mol Pharm. 2010;7:1195-208
43. Cabral H, Matsumoto Y, Mizuno K. et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815-23
44. Jiang W, Kim BY, Rutka JT. et al. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol. 2008;3:145-50
45. Goodman CM, McCusker CD, Yilmaz T. et al. Toxicity of gold nanoparticles functionalized with cationic and anionic side chains. Bioconjug Chem. 2004;15:897-900
46. Dellian M, Yuan F, Trubetskoy VS. et al. Vascular permeability in a human tumour xenograft: molecular charge dependence. Br J Cancer. 2000;82:1513-8
47. Krasnici S, Werner A, Eichhorn ME. et al. Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels. Int J Cancer. 2003;105:561-7
48. Campbell RB, Fukumura D, Brown EB. et al. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002;62:6831-6
49. He C, Hu Y, Yin L. et al. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31:3657-66
50. Xiao K, Li Y, Luo J. et al. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. 2011;32:3435-46
51. Dair BJ, Tyner K, Sapsford KE. Techniques for the Characterization of Nanoparticle-Bioconjugates. In: (ed.) Rege K, Medintz IL. Methods in Bioengineering. Nanoscale Bioengineering and Nanomedicine, 1st ed. London: Artech House. 2009:293-331
52. Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145:182-95
53. Geng Y, Dalhaimer P, Cai S. et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol. 2007;2:249-55
54. Arnida M.M. Janát-Amsbury, A. Ray, et al. Geometry and surface characteristics of gold nanoparticles influence their biodistribution and uptake by macrophages. Eur J Pharm Biopharm. 2011;77:417-23
55. Shah S, Liu Y, Hu W. et al. Modeling particle shape-dependent dynamics in nanomedicine. J Nanosci Nanotechnol. 2011;11:919-28
56. Doshi N, Prabhakarpandian B, Rea-Ramsey A. et al. Flow and adhesion of drug carriers in blood vessels depend on their shape: a study using model synthetic microvascular networks. J Control Release. 2010;146:196-200
57. Smith BR, Kempen P, Bouley D. et al. Shape matters: intravital microscopy reveals surprising geometrical dependence for nanoparticles in tumor models of extravasation. Nano Lett. 2012;12:3369-77
58. Zhu L, Torchilin VP. Stimulus-responsive nanopreparations for tumor targeting. Integr Biol (Camb). 2013;5:96-107
59. Mondalek FG, Zhang YY, Kropp B. et al. The permeability of SPION over an artificial three-layer membrane is enhanced by external magnetic field. J Nanobiotechnology. 2006;4:4
60. Klostergaard J, Seeney CE. Magnetic nanovectors for drug delivery. Nanomedicine. 2012;8(Suppl 1):S37-50
61. Plank C, Zelphati O, Mykhaylyk O. Magnetically enhanced nucleic acid delivery. Ten years of magnetofection - progress and prospects. Adv Drug Deliv Rev. 2011;63:1300-31
62. de Las Heras Alarcon C, Pennadam S, Alexander C. Stimuli responsive polymers for biomedical applications. Chem Soc Rev. 2005;34:276-85
63. Schwerdt A, Zintchenko A, Concia M. et al. Hyperthermia-induced targeting of thermosensitive gene carriers to tumors. Hum Gene Ther. 2008;19:1283-92
64. Chilkoti A, Dreher MR, Meyer DE. et al. Targeted drug delivery by thermally responsive polymers. Adv Drug Deliv Rev. 2002;54:613-30
65. Turpeenniemi-Hujanen T. Gelatinases (MMP-2 and -9) and their natural inhibitors as prognostic indicators in solid cancers. Biochimie. 2005;87:287-97
66. Zhu L, Wang T, Perche F. et al. Enhanced anticancer activity of nanopreparation containing an MMP2-sensitive PEG-drug conjugate and cell-penetrating moiety. Proc Natl Acad Sci U S A. 2013;110:17047-52
67. Wong C, Stylianopoulos T, Cui J. et al. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc Natl Acad Sci U S A. 2011;108:2426-31
68. Liu J, Liu J, Xu H. et al. Novel tumor-targeting, self-assembling peptide nanofiber as a carrier for effective curcumin delivery. Int J Nanomedicine. 2014;9:197-207
69. Gao H, Yang Z, Zhang S. et al. Ligand modified nanoparticles increases cell uptake, alters endocytosis and elevates glioma distribution and internalization. Sci Rep. 2013;3:2534
70. Chen F, Hong H, Zhang Y. et al. In vivo tumor targeting and image-guided drug delivery with antibody-conjugated, radiolabeled mesoporous silica nanoparticles. ACS Nano. 2013;7:9027-39
71. Choi CH, Alabi CA, Webster P. et al. Mechanism of active targeting in solid tumors with transferrin-containing gold nanoparticles. Proc Natl Acad Sci U S A. 2010;107:1235-40
72. Kirpotin DB, Drummond DC, Shao Y. et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66:6732-40
73. Toy R, Hayden E, Camann A. et al. Multimodal in vivo imaging exposes the voyage of nanoparticles in tumor microcirculation. ACS Nano. 2013;7:3118-29
74. Lee H, Hoang B, Fonge H. et al. In vivo distribution of polymeric nanoparticles at the whole-body, tumor, and cellular levels. Pharm Res. 2010;27:2343-55
75. Park JH, von Maltzahn G, Zhang L. et al. Systematic surface engineering of magnetic nanoworms for in vivo tumor targeting. Small. 2009;5:694-700
76. Durymanov MO, Beletkaia EA, Ulasov AV. et al. Subcellular trafficking and transfection efficacy of polyethylenimine-polyethylene glycol polyplex nanoparticles with a ligand to melanocortin receptor-1. J Control Release. 2012;163:211-9
77. von Gersdorff K, Ogris M, Wagner E. Cryoconserved shielded and EGF receptor targeted DNA polyplexes: cellular mechanisms. Eur J Pharm Biopharm. 2005;60:279-85
78. Bartlett DW, Su H, Hildebrandt IJ. et al. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci U S A. 2007;104:15549-54
79. Salvati A, Pitek AS, Monopoli MP. et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat Nanotechnol. 2013;8:137-43
80. Juweid M, Neumann R, Paik C. et al. Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Res. 1992;52:5144-53
81. Adams GP, Schier R, McCall AM. et al. High affinity restricts the localization and tumor penetration of single-chain Fv antibody molecules. Cancer Res. 2001;61:4750-5
82. Marcucci F, Corti A. How to improve exposure of tumor cells to drugs — Promoter drugs increase tumor uptake and penetration of effector drugs. Adv Drud Deliv Rev. 2012;64:53-68
83. Chang E, Chalikonda S, Friedl J. et al. Targeting vaccinia to solid tumors with local hyperthermia. Hum Gene Ther. 2005;16:435-44
84. Lammers T, Peschke P, Kühnlein R. et al. Effect of radiotherapy and hyperthermia on the tumor accumulation of HPMA copolymer-based drug delivery systems. J Control Release. 2007;117:333-41
85. Hauck ML, Dewhirst MW, Bigner DD. et al. Local hyperthermia improves uptake of a chimeric monoclonal antibody in a subcutaneous xenograft model. Clin Cancer Res. 1997;3:63-70
86. Kong G, Braun RD, Dewhirst MW. Characterization of the effect of hyperthermia on nanoparticle extravasation from tumor vasculature. Cancer Res. 2001;61:3027-32
87. Shen Y, Liu P, Zhang A. et al. Study on tumor microvasculature damage induced by alternate cooling and heating. Ann Biomed Eng. 2008;36:1409-19
88. Dudar TE, Jain RK. Differential response of normal and tumor microcirculation to hyperthermia. Cancer Res. 1984;44:605-12
89. Kong G, Braun RD, Dewhirst MW. Hyperthermia enables tumor-specific nanoparticle delivery: effect of particle size. Cancer Res. 2000;60:4440-5
90. Chen BG, Zhou MJ, Xu LX. Study of vascular endothelial cell morphology during hyperthermia. J Therm Biol. 2005;30:111-7
91. Kobayashi H, Reijnders K, English S. et al. Application of a macromolecular contrast agent for detection of alterations of tumor vessel permeability induced by radiation. Clin Cancer Res. 2004;10:7712-20
92. Davies C de L, Lundstrøm LM, Frengen J. et al. Radiation improves the distribution and uptake of liposomal doxorubicin (caelyx) in human osteosarcoma xenografts. Cancer Res. 2004;64:547-53
93. Kalofonos H, Rowlinson G, Epenetos AA. Enhancement of monoclonal antibody uptake in human colon tumor xenografts following irradiation. Cancer Res. 1990;50:159-63
94. Buchegger F, Rojas A, Delaloye AB. et al. Combined radioimmunotherapy and radiotherapy of human colon carcinoma grafted in nude mice. Cancer Res. 1995;5:83-9
95. Ganss R, Ryschich E, Klar E. et al. Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. 2002;62:1462-70
96. Goldblum SE, Ding X, Campbell-Washington J. TNF-alpha induces endothelial cell F-actin depolymerization, new actin synthesis, and barrier dysfunction. Am J Physiol. 1993;264:894-905
97. Park JS, Qiao L, Su ZZ. et al. Ionizing radiation modulates vascular endothelial growth factor expression through multiple mitogen activated protein kinase dependent pathways. Oncogene. 2001;25:3266-80
98. Claesson-Welsh L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med. 2013;273:114-27
99. Znati CA, Rosenstein M, Boucher Y. et al. Effect of radiation on interstitial fluid pressure and oxygenation in a human tumor xenograft. Cancer Res. 1996;56:964-68
100. Mo S, Coussios CC, Seymour L, Carlisle R. Ultrasound-enhanced drug delivery for cancer. Expert Opin Drug Deliv. 2012;9:1525-38
101. Ferrara KW. Driving delivery vehicles with ultrasound. Adv Drug Deliv Rev. 2008;60:1097-102
102. Thakkar D, Gupta R, Monson K. et al. Effect of ultrasound on the permeability of vascular wall to nano-emulsion droplets. Ultrasound Med Biol. 2013;39:1804-11
103. Sirsi SR, Hernandez SL, Zielinski L. et al. Polyplex-microbubble hybrids for ultrasound-guided plasmid DNA delivery to solid tumors. J Control Release. 2012;157:224-34
104. Wei H, Huang J, Yang J. et al. Ultrasound exposure improves the targeted therapy effects of galactosylated docetaxel nanoparticles on hepatocellular carcinoma xenografts. PLoS One. 2013;8:e58133
105. Arvanitis CD, Bazan-Peregrino M, Rifai B. et al. Cavitation-enhanced extravasation for drug delivery. Ultrasound Med Biol. 2011;37:1838-52
106. Seki T, Carroll F, Illingworth S. et al. Tumour necrosis factor-alpha increases extravasation of virus particles into tumour tissue by activating the RhoA/Rho kinase pathway. J Control Release. 2011;156:381-9
107. Epstein AL, Mizokami MM, Li J. et al. Identification of a protein fragment of interleukin 2 responsible for vasopermeability. J Natl Cancer Inst. 2003;95:741-9
108. Tanaka S, Akaike T, Wu J. et al. Modulation of tumor-selective vascular blood flow and extravasation by the stable prostaglandin I2 analogue beraprost sodium. J Drug Target. 2003;11:45-52
109. Monsky WL, Fukumura D, Gohongi T. et al. Augmentation of transvascular transport of macromolecules and nanoparticles in tumors using vascular endothelial growth factor. Cancer Res. 1999;59:4129-35
110. Hennigan TW, Begent RH, Allen-Mersh TG. Histamine, leukotriene C4 and interleukin-2 increase antibody uptake into a human carcinoma xenograft model. Br J Cancer. 1991;64:872-4
111. Seki T, Fang J, Maeda H. Enhanced delivery of macromolecular antitumor drugs to tumors by nitroglycerin application. Cancer Sci. 2009;100:2426-30
112. Schiller JH, Storer BE, Witt PL. et al. Biological and clinical effects of intravenous tumor necrosis factor-alpha administered three times weekly. Cancer Res. 1991;51:1651-8
113. van Laarhoven HW, Gambarota G, Heerschap A. et al. Effects of the tumor vasculature targeting agent NGR-TNF on the tumor microenvironment in murine lymphomas. Invest New Drugs. 2006;24:27-36
114. Atsumi T. Singh R, Sabharwal L, et al. Inflammation amplifier, a new paradigm in cancer biology. Cancer Res. 2014;74:8-14
115. Kano MR, Bae Y, Iwata C. et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc Natl Acad Sci U S A. 2007;104:3460-5
116. Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov. 2004;3:1011-22
117. Eikenes L, Tari M, Tufto I. et al. Hyaluronidase induces a transcapillary pressure gradient and improves the distribution and uptake of liposomal doxorubicin (Caelyx) in human osteosarcoma xenografts. Br J Cancer. 2005;93:81-8
118. McKee TD, Grandi P, Mok W. et al. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006;66:2509-13
119. Brown E, McKee T, di Tomaso E. et al. Dynamic imaging of collagen and its modulation in tumors in vivo using second-harmonic generation. Nat Med. 2003;9:796-800
120. Chauhan VP, Martin JD, Liu H. et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013;4:2516
121. Diop-Frimpong B, Chauhan VP, Krane S. et al. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci U S A. 2011;108:2909-14
122. Arnold SA, Rivera LB, Carbon JG. et al. Losartan slows pancreatic tumor progression and extends survival of SPARC-null mice by abrogating aberrant TGFβ activation. PLoS One. 2012;7:e31384
123. Chen X, Meng Q, Zhao Y. et al. Angiotensin II type 1 receptor antagonists inhibit cell proliferation and angiogenesis in breast cancer. Cancer Lett. 2013;328:318-24
124. Otake AH, Mattar AL, Freitas HC. et al. Inhibition of angiotensin II receptor 1 limits tumor-associated angiogenesis and attenuates growth of murine melanoma. Cancer Chemother Pharmacol. 2010;66:79-87
125. Kinuya S, Yokoyama K, Konishi S. et al. Effect of induced hypertension with angiotensin II infusion on biodistribution of 111In-labeled monoclonal antibody. Nucl Med Biol. 1996;23:137-40
126. Li CJ, Miyamoto Y, Kojima Y. et al. Augmentation of tumour delivery of macromolecular drugs with reduced bone marrow delivery by elevating blood pressure. Br J Cancer. 1993;67:975-80
127. Hattori Y, Ubukata H, Kawano K. et al. Angiotensin II-induced hypertension enhanced therapeutic efficacy of liposomal doxorubicin in tumor-bearingmice. Int J Pharm. 2011;403:178-84
128. Nagamitsu A, Greish K, Maeda H. Elevating blood pressure as a strategy to increase tumor-targeted delivery of macromolecular drug SMANCS: cases of advanced solid tumors. Jpn J Clin Oncol. 2009;39:756-66
129. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58-62
130. Goel S, Duda DG, Xu L. et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071-121
131. Stylianopoulos T, Jain RK. Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc Natl Acad Sci U S A. 2013;110:18632-7
132. Chauhan VP, Stylianopoulos T, Martin JD. et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. 2012;7:383-8
133. Stephan MT, Irvine DJ. Enhancing cell therapies from the outside in: cell surface engineering using synthetic nanomaterials. Nano Today. 2011;6:309-25
134. Cihova M, Altanerova V, Altaner C. Stem cell based cancer gene therapy. Mol Pharm. 2011;8:1480-7
135. Salem HK, Thiemermann C. Mesenchymal stromal cells: current understanding and clinical status. Stem Cells. 2010;28:585-96
136. Ringe J, Strassburg S, Neumann K. et al. Towards in situ tissue repair: human mesenchymal stem cells express chemokine receptors CXCR1, CXCR2 and CCR2, and migrate upon stimulation with CXCL8 but not CCL2. J Cell Biochem. 2007;101:135-46
137. Gao Z, Zhang L, Hu J. et al. Mesenchymal stem cells: a potential targeted-delivery vehicle for anti-cancer drug, loaded nanoparticles. Nanomedicine. 2013;9:174-84
138. Teo GS, Ankrum JA, Martinelli R. et al. Mesenchymal stem cells transmigrate between and directly through tumor necrosis factor-α-activated endothelial cells via both leukocyte-like and novel mechanisms. Stem Cells. 2012;30:2472-86
139. Marquez-Curtis LA, Janowska-Wieczorek A. Enhancing the migration ability of mesenchymal stromal cells by targeting the SDF-1/CXCR4 axis. Biomed Res Int. 2013;2013:561098
140. Ruan J, Ji J, Song H. et al. Fluorescent magnetic nanoparticle-labeled mesenchymal stem cells for targeted imaging and hyperthermia therapy of in vivo gastric cancer. Nanoscale Res Lett. 2012;7:309
141. Albarenque SM, Zwacka RM, Mohr A. Both human and mouse mesenchymal stem cells promote breast cancer metastasis. Stem Cell Res. 2011;7:163-71
142. Zhang YS, Wang Y, Wang L. et al. Labeling human mesenchymal stem cells with gold nanocages for in vitro and in vivo tracking by two-photon microscopy and photoacoustic microscopy. Theranostics. 2013;3:532-43
143. Levy O, Zhao W, Mortensen LJ. et al. mRNA-engineered mesenchymal stem cells for targeted delivery of interleukin-10 to sites of inflammation. Blood. 2013;122:e23-32
144. Xu WT, Bian ZY, Fan QM. et al. Human mesenchymal stem cells (hMSCs) target osteosarcoma and promote its growth and pulmonary metastasis. Cancer Lett. 2009;281:32-41
145. Karnoub AE, Dash AB, Vo AP. et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557-63
146. Martinez-Quintanilla J, Bhere D, Heidari P. et al. Therapeutic efficacy and fate of bimodal engineered stem cells in malignant brain tumors. Stem Cells. 2013;31:1706-14
147. Knoop K, Schwenk N, Dolp P. et al. Stromal targeting of sodium iodide symporter using mesenchymal stem cells allows enhanced imaging and therapy of hepatocellular carcinoma. Hum Gene Ther. 2013;24:306-16
148. Drake CG. Combination immunotherapy approaches. Ann Oncol. 2012;23(Suppl 8):S41-6
149. Liu Z, Li Z. Molecular imaging in tracking tumor-specific cytotoxic T lymphocytes (CTLs). Theranostics. 2014;4:990-1001
150. Charo J, Perez C, Buschow C. et al. Visualizing the dynamic of adoptively transferred T cells during the rejection of large established tumors. Eur J Immunol. 2011;41:3187-97
151. Palmer DC, Balasubramaniam S, Hanada K. et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J Immunol. 2004;173:7209-16
152. Cole C, Qiao J, Kottke T. et al. Tumor-targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific T cells. Nat Med. 2005;11:1073-81
153. Stephan MT, Moon JJ, Um SH. et al. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat Med. 2010;16:1035-41
154. Kennedy LC, Bear AS, Young JK. et al. T cells enhance gold nanoparticle delivery to tumors in vivo. Nanoscale Res Lett. 2011;6:283
155. Solinas G, Germano G, Mantovani A. et al. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065-73
156. Tripathi C, Tewari BN, Kanchan RK. et al. Macrophages are recruited to hypoxic tumor areas and acquire a Pro-Angiogenic M2-Polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget. 2014;14:5350-68
157. Lal S, Clare SE, Halas NJ. Nanoshell-enabled photothermal cancer therapy: impending clinical impact. Acc Chem Res. 2008;41:1842-51
158. Choi MR, Bardhan R, Stanton-Maxey KJ. et al. Delivery of nanoparticles to brain metastases of breast cancer using a cellular Trojan horse. Cancer Nanotechnol. 2012;3:47-54
159. Valable S, Barbier EL, Bernaudin M. et al. In vivo MRI tracking of exogenous monocytes/macrophages targeting brain tumors in a rat model of glioma. Neuroimage. 2008;40:973-83
160. Choi J, Kim HY, Ju EJ. et al. Use of macrophages to deliver therapeutic and imaging contrast agents to tumors. Biomaterials. 2012;33:4195-203
161. Basel MT, Balivada S, Shrestha TB. et al. A cell-delivered and cell-activated SN38-dextran prodrug increases survival in a murine disseminated pancreatic cancer model. Small. 2012;8:913-20
162. Lammers T, Kiessling F, Hennink WE. et al. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release. 2012;161:175-87
163. Nichols JW, Bae YH. EPR: Evidence and fallacy. J Control Release. 2014;190:451-64
164. Raju B, Haug SR, Ibrahim SO. et al. High interstitial fluid pressure in rat tongue cancer is related to increased lymph vessel area, tumor size, invasiveness and decreased body weight. J Oral Pathol Med. 2008;37:137-44
165. Zätterström UK, Brun E, Willén R. et al. Tumor angiogenesis and prognosis in squamous cell carcinoma of the head and neck. Head Neck. 1995;17:312-18
166. Eberhard A, Kahlert S, Goede V. et al. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res. 2000;60:1388-93
167. Line SR, Torloni H, Junqueira LC. Diversity of collagen expression in the pleomorphic adenoma of the parotid gland. Virchows Arch A Pathol Anat Histopathol. 1989;414:477-83
168. Guix M, Mayorga-Martinez CC, Merkoçi A. Nano/micromotors in (bio)chemical science applications. Chem Rev. 2014;114:6285-322
Author contact
![]() Corresponding author: Dr. Alexander S. Sobolev, Department of Molecular Genetics of Intracellular Transport, Institute of Gene Biology, 34/5, Vavilov St., 199334, Moscow, Russia. Phone number: +7 (499) 135-31-00; Fax number: +7 (499) 135-41-05; E-mail: sobolevac.ru
Corresponding author: Dr. Alexander S. Sobolev, Department of Molecular Genetics of Intracellular Transport, Institute of Gene Biology, 34/5, Vavilov St., 199334, Moscow, Russia. Phone number: +7 (499) 135-31-00; Fax number: +7 (499) 135-41-05; E-mail: sobolevac.ru