Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Material & Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(11):1214-1224. doi:10.7150/thno.12403 This issue Cite
Research Paper
Identification of 7 Proteins in Sera of RA Patients with Potential to Predict ETA/MTX Treatment Response
Antoine Obry1,2,3,4, Julie Hardouin2,3,4, Thierry Lequerré1,4,5,6, Frédérique Jarnier2,3,4, Olivier Boyer1,4,7, Patrice Fardellone8, Peggy Philippe9, Christian Marcelli10, Xavier Le Loët1,4,5,6, Olivier Vittecoq1,4,5,6, ![]() , Pascal Cosette2,3,4
, Pascal Cosette2,3,4
1. INSERM, U905, Rouen, France
2. CNRS, UMR6270, Mont Saint Aignan, France
3. PISSARO Proteomic Platform, Mont Saint Aignan, France
4. Normandy University, IRIB, Rouen, France
5. Department of Rheumatology, Rouen University Hospital, Rouen, France
6. CIC/CRB1404, Rouen University, Rouen, France
7. Department of Immunology, Rouen University Hospital, Rouen, France
8. Department of Rheumatology, Amiens University Hospital, Amiens, France
9. Department of Rheumatology, Lille University Hospital, Lille, France
10. Department of Rheumatology, Caen University Hospital, Caen, France
Received 2015-4-14; Accepted 2015-7-8; Published 2015-8-9
Abstract

Objective: The recent growth of innovating biologics has opened fascinating avenues for the management of patients. In rheumatoid arthritis, many biologics are currently available, the choice of which being mostly determined empirically. Importantly, a given biologic may not be active in a fraction of patients and may even provoke side effects. Here, we conducted a comparative proteomics study in attempt to identify a predictive theranostic signature of non-response in patients with rheumatoid arthritis treated by etanercept/methotrexate combination.
Methods: A serum sample was collected prior to treatment exposure from a cohort of 22 patients with active RA. A proteomic “label free” approach was then designed to quantitate protein biomarkers using mass spectrometry. To verify these results, a relative quantification followed by an absolute quantification of interesting protein candidates were performed on a second cohort. The criterion of judgment was the response to etanercept/methotrexate combination according to the EULAR criteria assessed at 6 months of treatment.
Results: These investigations led to the identification of a set of 12 biomarkers with capacity to predict treatment response. A targeted quantitative analysis allowed to confirm the potential of 7 proteins from the latter combination on a new cohort of 16 patients. Two highly discriminating proteins, PROS and CO7, were further evaluated by ELISA on this second cohort. By combining the concentration threshold of each protein associated to a right classification (responders vs non-responders), the sensitivity and specificity reached 88.9 % and 100 %, respectively.
Conclusion: Prior to methotrexate/etanercept treatment, abundance of several sera proteins, notably PROS and CO7, were associated to response status of RA patients 6 month after treatment initiation.
Keywords: biomarkers, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is a chronic, autoimmune and inflammatory disease that results in progressive joint damage and disability. RA prevalence is estimated at 0.3 % in the French population [1]. This disease mostly involves 40-60 years-old women [2]. It affects small joints of hands and feet and is characterized by pain, swelling and joint deformity.
RA is a multi-factorial disease which combines hormonal, environmental, immunological and genetic factors. Nowadays, RA patients, whose conventional disease modifying anti-rheumatic drugs (DMARDs) such as methotrexate (MTX) are not sufficiently effective, require additional treatment by biologic agents. Most of these drugs target the pro-inflammatory cytokine tumor necrosis factor α (TNFα) since it is a key mediator in the pathogenesis of RA [3]. Up to now, five TNFα blocking agents (TBAs) are routinely prescribed for RA patients: a recombinant soluble form of the TNF receptor TNFRSF1B (etanercept, ETA) and four anti-TNFα monoclonal antibodies: infliximab, adalimumab, certolizumab and golimumab. These TBAs are often combined to MTX since the association has proved its effectiveness compared to monotherapy.
However, despite overall improvement of quality of life [4], around 30% of patients do not respond to a given TBA [5], a situation leading to both increased risk of undesired side-effects and over costs [6]. Because of increasing numbers of available molecules, variability of treatment response, risk of serious adverse events and cost, one of the major current challenges in RA therapy is being able to predict drug responsiveness prior to treatment initiation.
Some parameters have been suggested to influence the response to TBAs such as tobacco use [7], high titers of rheumatoid factors of IgA isotype [8] or presence of anti-CCP antibodies [9]. Other studies investigating soluble biomarkers (metalloproteinases, osteocartilaginous markers…) [10] or genetic factors (particularly TNFα-308 G/A polymorphism) [11] have not been conclusive, yet. Several candidate gene polymorphisms have been also associated with differential response to TBAs in RA but the replication of these genetic predictors is lacking [12] except for the G allele of the PTPRC gene [13]. It results that prediction of drug responsiveness by theranostic biomarkers may benefit from more innovative approaches such as transcriptomics [14] and proteomics [15].
To our knowledge, only 2 studies using such an approach have tempted to identify theranostic biomarkers to infliximab, either after surface enhanced laser desorption ionisation (SELDI) investigations [15] or using isobaric tags for relative and absolute quantitation (iTRAQ) labeling [16]. Thus, up to now, no study has investigated the identification of serum proteomic biomarkers for prediction of response to other TBAs than infliximab. The above-mentioned successes were made possible by recent advances in proteomics, and in particular by the developments in mass spectrometry (MS). Previously restricted to protein identification, the improvement of MS instruments in terms of resolution and accuracy associated with a better sensitivity has rendered more accessible the quantification of proteins within complex matrices [17-20]. As a new gold standard, the so-called "label free" approach enables protein relative quantification in several samples by comparing intensities of ion currents extracted from thousands peptides during LC separations [20]. To our point of view, label-free quantification appears to constitute an optimal approach for biomarker discovery by allowing accurate and robust enough measurements of a large number of samples while reducing costs as well as handling. However, despite its demonstrated capability in numerous pathologies [21-25], this technology was not developed in the context of inflammatory rheumatic diseases.
In this study, a label-free proteomics approach was designed to discover protein biomarkers for prediction of response in serum from RA patients whose status has been evaluated after several months of treatment with etanercept (ETA)/ MTX combination. Firstly, relative quantitation with mass spectrometry-based proteomic approach was performed between serum samples of responders (R) and non-responders (NR) patients from a first cohort of 22 patients. This led to the identification of several proteins differentially expressed between the R and NR groups. To confirm these results, a relative quantification by mass spectrometry mass and an absolute quantification by ELISA of two selected biomarker candidates was carried out on serum samples from a second cohort of 16 patients.
Material & Methods
Patients
Two cohorts of RA patients with active disease (defined by a disease activity score (DAS) 28 > 3.2), who required a biologic agent after failure of at least one non-biologic DMARD have been studied. All patients fulfilled the American College of Rheumatology (ACR) criteria [26], were biologic DMARD naïve and received MTX at stable dose since 1 month and for some of them glucocorticoids at a dose ≤ 10 mg per day. The first cohort (population 1) that included 22 RA patients was devoted to identification of proteins associated to drug responsiveness while the second one (population 2) that comprised 16 RA patients was used for verification of the combination of biomarkers identified from population 1. In both cohorts, each patient has received the MTX/ETA combination at stable doses. During the 12 months follow-up period, several clinical and biological parameters were collected at different time-points (i.e, 6, 12, 24 and 52 weeks) as follows: patient's assessment of pain and disease activity (using 0 to 100 mm visual analog scale), duration of morning stiffness, number of tender joints, number of swollen joints, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP) level, disease activity score (DAS28) and the physical function scored with the French version of the Health Assessment Questionnaire (HAQ) for RA. The clinical efficacy of the MTX/ETA association was evaluated at 6 months according to the European league against rheumatism (EULAR) response criteria [27]. So, the patients were categorized into good, moderate or non-responders based on the degree of change in the DAS28 and the level of DAS28 reached at 6 months. Good responders (R) were defined as patients who had a decrease in DAS28 from baseline (ΔDAS28) > 1.2 and a DAS28 at sixth month < 3.2. Moderate responders (MR) had either ΔDAS28 > 1.2 and a DAS28 at sixth month > 3.2 or ΔDAS28 from 0.6 to 1.2 and a DAS28 at sixth month < 5.1. Non-responders (NR) were those who had either ΔDAS28 < 0.6 or a DAS28 at the sixth month > 5.1. Only R and NR patients were included herein.
Sera were collected from patients included in the SATRAPE study (no. 2005/06; ClinicalTrials.gov identifier: NCT 00234234) and another study (no. 2004/120). Both studies were approved by the regional ethics committee (CPP Nord Ouest 1, France) and all participants gave written informed consent at the time of enrollment.
Serum samples
For the present study, serum samples obtained before treatment initiation were investigated by proteomic analysis. Thus, a blood sample was collected, allowed to coagulate and then centrifuged at 2000 g for 10 min. Serum samples were immediately stored at -80 °C until analyzed. In each sample, the protein concentration was determined using the Bradford method. Thereafter, 25 µg of samples were loaded on a stacking polyacrylamide gel (7 %) and after a short migration and staining, the proteins were cut out from the SDS PAGE gel right after they penetrated into the gel. The proteins contained in the gel plugs were submitted to a reduction step (10 mM dithiothreitol) and cysteines were then irreversibly alkylated in 25 mM iodoacetamide. After washing, gel bands were submitted to trypsin digestion (0.5 µg per band, Promega). After peptide extraction by using H20/CH3CN mixtures (50%/50%) acidified with TFA, the peptide fractions were combined and evaporated.
Liquid nano-chromatography and mass spectrometry
For mass spectrometry analyses, peptides were dissolved in 0.1% formic acid in water. All experiments were carried out with a hybrid linear ion trap / orbitrap mass spectrometer (LTQ Orbitrap Elite, Thermo Scientific) equipped with a nano-ESI source and coupled to a liquid nano-chromatography system (Easy-nLC II, Thermo Scientific). Briefly, Samples were loaded onto an enrichment column (C18 PepMap100, Thermo Scientific) and the separation was performed on a reverse phase C18 column (NTCC-360/100-5-153, Nikkyo Technos, Japan). Tryptic peptides were eluted from the reverse phase column into the mass spectrometer, using a linear gradient from 2% to 40% of B in 105 min, followed by a rapid rising to 80% in 4 min and a final 15 min isocratic period at 80% (mobile phase A: H2O/0.1% FA; mobile phase B: CH3CN/0.1% FA). The mass spectrometer was operated in the data-dependent mode with survey full scan mass spectra acquired from 400 to 2000 m/z. The mass spectrometer selected the 20 most intense ions for CID fragmentation within the trap.
Relative quantification by Progenesis LC-MS
Raw data were imported in Progenesis LC-MS software (V4.0.4441.29989, Nonlinear Dynamics). First, a LC-MS run was set as a reference and the retention times of all other samples within the experiment were aligned. After alignment and feature exclusion, raw abundances of all features were normalized. Statistical analysis was performed using normalized abundances for one-way analysis of variance (ANOVA) calculations. Features presenting p-value < 0.05 and q-value < 0.05 were selected. Corresponding extracted peak lists were searched using the MASCOT search engine (version 2.2, Matrix Science) against the SwissProt database (V55.6; 390696 sequences) restricted to the taxonomy Homo sapiens (20009 sequences). All resulting identified peptides were included when they presented an identification score superior to identity threshold (leading to a false discovery rate of 0.45%). The total cumulative abundance of the protein was then calculated by summing the abundances of all retained peptides.
Determination of serum levels of CO7 and PROS by ELISA
Serum levels of the two proteins presenting the best discriminating capacity from the label-free investigations, namely complement component C7 (CO7) and vitamin K-dependent protein S (PROS), were measured in 16 RA patients at baseline in sera, using enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (USCNK, USA and EIAAB, China).
Statistical analyses
The Kolmogorov-Smirnov test was used to evaluate the data distributions. Accordingly, Mann-Whitney non-parametric tests were used to compare median levels of proteins from label free experiments and ELISA. The latter were also used to compare at baseline the differences of clinical and demographic data between responders versus non-responders
To establish wheter a relationship exists between clinical parameters (ESR, CRP …) measured prior to treatment initiation and the candidate protein levels, univariate analyses were performed using the Spearman's rank correlation (GraphPad Prism 5, GraphPad Software). A p-value < 0.05 was considered statistically significant. To evaluate the capacity of the biomarker combination, principal component analyses (PCA) were performed first with the 11 biological, clinical and demographic monitored parameters and also secondly with all the identified protein biomarkers. This statistical part was realized with R software (R Development Core Team 2011) by using consecutively the missMDA and FactoMineR package. The R software was also used for unsupervised hierarchical clustering analysis, using Pearson and Ward linkage options, to separate R and NR patients after MTX/ETA combination exposure.
To evaluate the theranostic value of these potential biomarkers, the areas under curve (AUC) of receiver operating characteristic (ROC) curves were calculated with R software by using ROCR package. The standard error of the area under the ROC curve, as well as the 95% confidence interval were also reported. For cross verification by absolute quantification, the calculated thresholds resulting from ROC curves analyses were combined. So, the patients were categorized into good or non-responders based on concentrations of CO7 and PROS proteins. Good responders (R) were defined as patients who had both concentrations above calculated threshold.
Results
Classification of RA patients
Demographic, clinical and biological data for cohort referred to as “Population 1” are given at the time of treatment initiation (Table 1). This first cohort, in which 12 patients were classified as R and 10 as NR after six months of ETA/MTX treatment (according to the EULAR criteria) was used to discover protein biomarkers. The DAS28 was significantly improved at 6 months in the R group (ΔDAS28 = -2.57 ± 0.18), whereas it was unchanged in NR patients (ΔDAS28 = 0.17 ± 0.17). Prior to treatment initiation, four parameters (CRP, DAS, ESR, HAQ) were a little higher in R compared to NR but the difference was not statistically significant (all p-value > 0.05). Only two parameters (morning stiffness and pain) were higher in the NR group but the difference between these groups was once again not significant. Even if the number of men was higher in R patients, the ratio remained largely in favor of women for each subgroup of patients. Thus, except for gender, R and NR groups were similar in terms of clinical and biological characteristics before beginning of the treatment.
Demographic, clinical and biological data of RA patients from population 1 at baseline.
| Population 1 (serum) | |||||||
|---|---|---|---|---|---|---|---|
| Responders (n=12) | Non responders (n=10) | p-value | |||||
| Mean ± SEM | Median | min_max | Mean ± SEM | Median | min_max | ||
| Age (years) | 51 ± 4 | 50.89 | 22.5_69.5 | 59 ± 5 | 63.81 | 26.69_74.85 | 0.12 |
| Sex (f/m) | 04/08 | _ | 01/09 | _ | _ | ||
| Methotrexate (mg/week) | 15.0 ± 1.4 | 15.00 | 10.0_20.0 | 13.3 ± 1.9 | 15.00 | 7.50_15.0 | 0.51 |
| Corticoids (mg/day) | 5.6 ± 1.7 | 5.00 | 0_15 | 3.6 ± 1.4 | 2.50 | 0.0_10.0 | 0.46 |
| Morning stiffness (minutes) | 47 ± 17 | 30.00 | 0_180 | 62 ± 28 | 30.00 | 10.0_240 | 0.62 |
| Pain (0-100 mm VAS) | 62 ± 5 | 65.00 | 40.0_90.0 | 64 ± 6 | 67.50 | 40.0_80.0 | 0.77 |
| ESR (mm/hour) | 27 ± 6 | 22.00 | 3.0_76.0 | 22 ± 5 | 18.00 | 11.0_56.0 | 0.51 |
| CRP (mg/l) | 20.6 ± 7.9 | 11.00 | 2.0_71.2 | 9.5 ± 5.8 | 5.50 | 1.0_44.0 | 0.07 |
| FR (IU/ml) | 137 ± 52 | 51.00 | 0_621 | 370 ± 250 | 81.50 | 0_2520 | 0.92 |
| Anti_CCP2 (AU/ml) | 84 ± 32 | 45.00 | 0_400 | 89 ± 48 | 7.50 | 0_400 | 0.31 |
| HAQ score (0_3) | 1.4 ± 0.3 | 1.13 | 1.0_2.0 | 1.1 ± 0.2 | 1.13 | 0.75_1.38 | 0.60 |
| DAS28 | 4.17± 0.26 | 4.23 | 2.55_5.69 | 3.41 ± 0.34 | 3.58 | 1.45_4.72 | 0.12 |
| DAS28 6 month | 1.61 ± 0.16 | 1.58 | 0.69_2.48 | 3.59 ± 0.32 | 3.90 | 1.98_4.64 | 0.0004 |
| ΔDAS28 | -2.57 ± 0.18 | -2.65 | -3.27_-1.27 | 0.17 ± 0.17 | -0.052 | -0.33_1.6 | < 0.0001 |
All differences between responders versus non-responders at baseline of the first cohort were non-significant (p-values > 0.05; Mann-Whitney non-parametric test). Only DAS28 at 6 month and ΔDAS28 showed a significant difference between R and NR patients. VAS: Visual Analogue Scale (patient assessment of pain); ESR: Erythrocyte Sedimentation Rate; CRP: C-reactive protein; RF: level of rheumatoid factors; Anti_CCP2: level of antibodies against cyclic citrullinated peptides; HAQ: Health Assessment Questionnaire; DAS28: Disease Activity Score at initiation of treatment; ΔDAS28: DAS28 difference between 6 months and baseline. Values are mean ± SEM.
General traits from proteomic analysis of R and NR patients
All samples from the first cohort were analyzed by mass spectrometry and in these assays 213 proteins were identified. This number is relatively low in agreement with analytical challenges related to the dynamic range of protein concentrations in serum samples. This is also due to the high level of filtering when validating protein identification from MSMS data. To get an overall idea on the sample characteristics and the depth of analysis, main features are presented in Figure S1. As much as 72.8% of proteins were identified in both R and NR populations, and associated with a mean coverage close to 41% and a mean number of unique peptides of around 14 per protein. Proteins identified only in R or NR presented a weaker mean sequence coverage, as well as a reduced number of peptide spectrum match (PSM) (Figure S1).
In order to evaluate the reproducibility of the experiments, different linear regressions were performed by plotting the logarithm of the number of PSMs for 2 replicates of the same sample (Ri1 vs Ri2), for different samples of the same group (Ri vs Ri) and for samples from different groups (Ri vs NRi) (Figure S1). The regression coefficients measured to evaluate the robustness of the technical scenario between replicates (Ri1 vs Ri2) of the same sample were found around 0.975.
Identification of a biomarker combination segregating R and NR samples
Within the first cohort, a differential proteomic analysis was undertaken on R and NR samples collected before treatment (Figure 1). First, peptides were kept when they displayed a statistically differential abundance between the two populations. When MSMS data were produced, this was followed by the identification of these peptides. A typical workflow for this scenario is presented on Figure 1 with the example of a peptide belonging to the Ceruloplasmin sequence. According to this strategy, 101 peptides were retained. For the second step, at the protein level, only proteins identified by at least two proteotypic peptides were conserved. After a similar statistical filtering as the one realized at the peptide level, 12 proteins finally proposed a significant differential expression between the two populations.
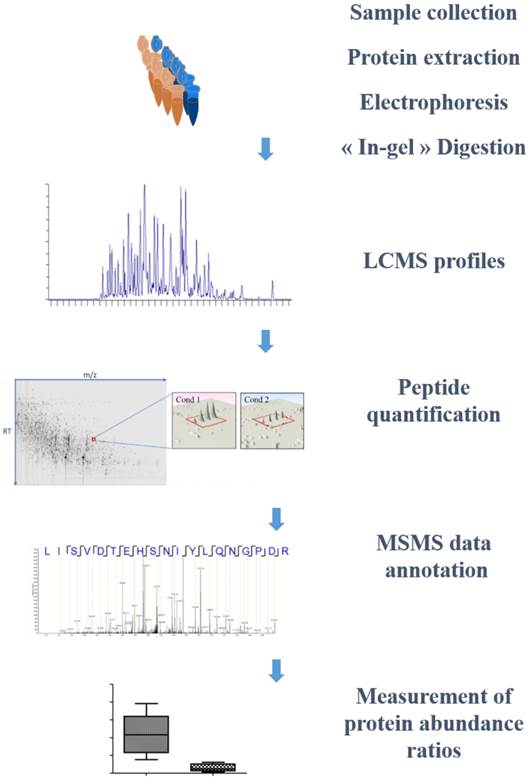
Workflow for biomarker identification. Flowchart representing the different stages of label free quantitative proteomics applied to biomarker discovery. After sample collection, each sample is biochemically processed for protein extraction in adequate buffer. The protein extract is run on a 1D SDS-PAGE stacking gel, allowing concentration of the protein extract and elimination of interfering compounds. The excised gel plug containing the whole protein content is then submitted to protein digestion prior to LCMS analysis. These runs are then converted to 2D LCMC maps (coordinates: m/z ratio; retention time) and aligned. On these maps, the isotopic profile of each peptide is integrated over the different samples; the fragmentation profile (MSMS data) allowing the annotation of the corresponding peptide sequence. Finally, the abundances of each contributing peptide for a given protein are summed to yield to the measurement of the protein relative abundance ratios.
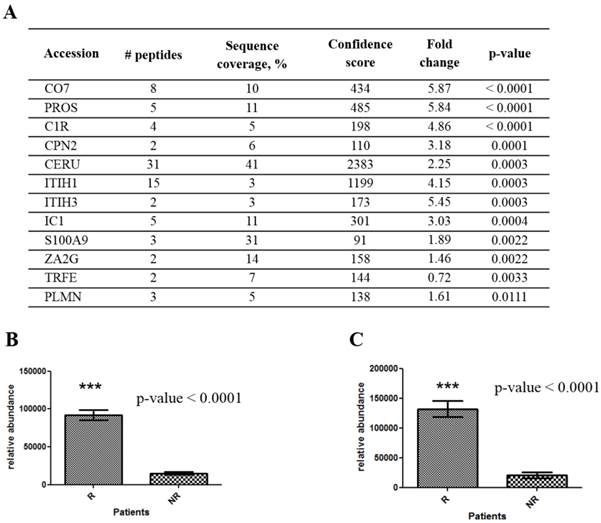
Indeed, CO7 and PROS proteins were significantly over-expressed in R patients with p-value below 0.001 (Figure 2). In contrast, transferrin (TRFE) was the only over-expressed protein in NR patients, all other being over-expressed in R samples. From these results, the 3 most discriminating proteins appeared to be CO7, PROS and complement C1r subcomponent (C1R). However, ceruloplasmin (CERU) whose quantitation relies on a large number of peptides and inter-alpha-trypsin inhibitor heavy chain 1 and 3 (ITIH1, ITIH3) associated with high fold change (~5) were also interesting biomarker candidates.
Serum proteins showing differential abundance between ETA/MTX responder and non-responder RA patients prior to treatment initiation. (A) Table shows the 11 proteins over-expressed in R serum samples and the unique protein (transferrin, TRFE) over-expressed in NR serum samples identified by mass spectrometry. Np is the number of peptides above significance threshold allowing protein quantitation and the associated protein sequence coverages. Confident score is the value extracted from Progenesis LCMS calculating after importing Mascot search results and restricted to the peptides used for quantitation. Fold change refers to the ratio of protein abundance in responders samples divided by the one of non-responders samples. The associated p-value, given in the last column is significant if p-value is < 0.05 (Mann-Whitney test). Illustration of relative quantification for proteins PROS (B) and CO7 (C) at baseline between R and NR samples. The results are presented as mean ± SEM. Ceruloplasmin : CERU; complement C1r subcomponent : C1R; complement component C7: CO7; inter-alpha-trypsin inhibitor heavy chain ITIH, C1-inhibitor : IC1, plasminogen : PLMN, vitamin K-dependent protein S: PROS, protein S100A9 : S100A9, transferrin : TRFE; zinc-alpha2-glycoprotein : ZA2G.
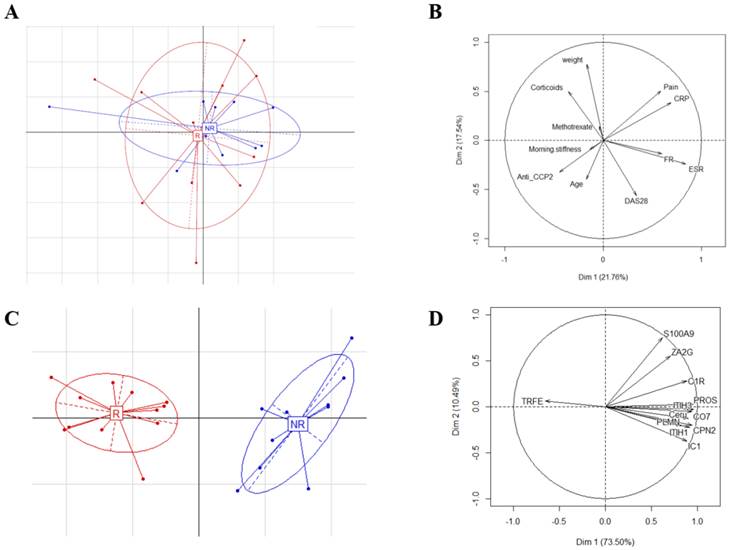
Thereafter, PCA was used to evaluate the discriminating capacity of the whole measurements for the 2 panels of patients. With the demographic, clinical and biological parameters (Figure 3A and B), the first two axes of the PCA account for 39.29% of the total variability. These results show the presence of a low variability between populations and indicate that these parameters do not allow to separate the R and NR patients groups. A similar analysis was then performed after identification of 12 potential theranostic biomarkers (Figure 3C and D). The results of the PCA show that the first two axes account for 83.99% of the total variability. The first component, which alone accounts for 73.5% of the total diversity is positively correlated with the level of TRFE and negatively correlated with PROS, CO7, and ITIH3 levels (Figure 3D). Here, in contrast, these results demonstrate that protein abundances of these 12 biomarkers allow to separate the R and NR groups before the initiation of treatment (Figure 3C).
Compared principal component analyses between consensual monitored parameters and identified biomarkers. Graphical representations of the PCA results (principal components 1 and 2) from demographic, clinical and biological parameters (A) and from protein abundances of the 12 biomarker candidates (C) for R and NR patients groups (R patients in red and NR in blue). Correlation circle of the projection of demographic, clinical and biological variables (B) and correlations of protein abundances (D) with the 2 first principal components.
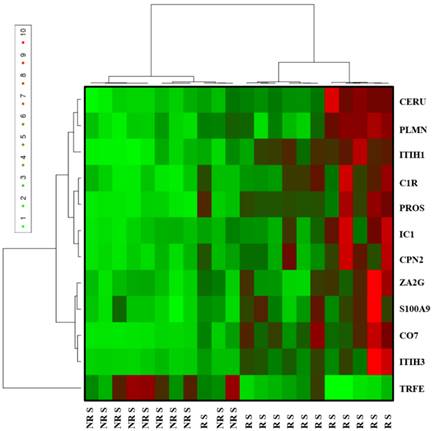
Furthermore, an unsupervised classification of patients was achieved by using the relative abundance values extracted for each protein (Figure 4). This combination of 12 proteins allows to split into two distinct R and NR groups with a specificity of 100 % and a sensitivity of 91.67 %. In this analysis, only one R patient was misclassified.
Analysis by hierarchical clustering of proteins differentially expressed in sera of R patients (n = 12) compared to NR patients (n = 10) from the population 1 prior to MTX/ETA initiation. The heat map was built from the 12 proteins that were differentially expressed between both groups (R and NR). Each row represents a protein and each column represents an individual patient serum sample (the name below the cluster indicates patient response status to ETA/MTX combination at six months according to the EULAR criteria: NR, non-responder and R: responder). Relative abundance levels are colored green for lower intensities and red for higher intensities in R patient samples.
After this clustering step, the potential correlation between the abundance of each protein and the various clinical parameters was tested. These results showed no correlation between individual biomarker and DAS28, age, CRP, ESR or pain. The unique relevant correlation concerned the relation between treatment response and biomarker abundance (p-value < 0.05; Table S1).
Cross-validation by relative quantification and absolute quantification
To independently corroborate these results, we used a second cohort of RA patients to specifically study the expression of the highlighted proteins during the discovery phase. Table S2 shows the clinical profiles for “population 2” in which 9 patients were classified as R and 7 as NR after six months of ETA/MTX exposure (in line with the EULAR criteria). As with the first cohort, these parameters showed no significant difference between the R and NR groups before the beginning of treatment.
At first, a targeted relative quantification focusing on these biomarkers was carried out by mass spectrometry. Seven proteins from the 12 potential biomarkers were identified as differently expressed (p-value < 0.05) between R and NR from population 2 (Figure S2A and S2B). In order to characterize these potential biomarkers of patients' response, ROC analysis was performed. Table 2 shows the detailed sensitivity and specificity levels with 95% confidence interval of the 7 proteins. When considered as a single biomarker in sera, S100A9 had a sensitivity of 100% and a specificity of 100%. Interestingly, the 6 other potential biomarkers provided an AUC value above 0.80. All combinations with S100A9 allowed to identify R with a sensitivity and specificity of 100%.
Cross-verification by targeted relative quantification in patients' sera from the second cohort.
| Responders vs no Responders | |||||
|---|---|---|---|---|---|
| Protein biomarkers | Area | Std. Error a | 95% confidence interval | Sensitivity | Specificity |
| CERU | 0.92 | 0.07 | 0.78-1.00 | 87.50% | 87.50% |
| CO7 | 0.89 | 0.09 | 0.72-1.00 | 87.50% | 87.50% |
| ITIH1 | 0.86 | 0.10 | 0.67-1.00 | 75.00% | 62.50% |
| PLMN | 0.88 | 0.09 | 0.69-1.00 | 87.50% | 87.50% |
| PROS | 1.00 | 0.00 | 1.00-1.00 | 100.00% | 100.00% |
| S100A9 | 0.98 | 0.02 | 0.94-1.00 | 100.00% | 87.50% |
| ZA2G | 0.91 | 0.08 | 0.76-1.00 | 75.00% | 100.00% |
a. Under the nonparametric assumption
Receiver Operating Characteristic (ROC) curve analysis of potential theranostic biomarkers in population 2 (from targeted label free analysis on the proteins CERU, CO7, ITIH1, PLMN, PROS, S100A9 and ZA2G). The AUCs and associated standard errors, the 95% confidence interval as well as sensitivities and specificities are reported.
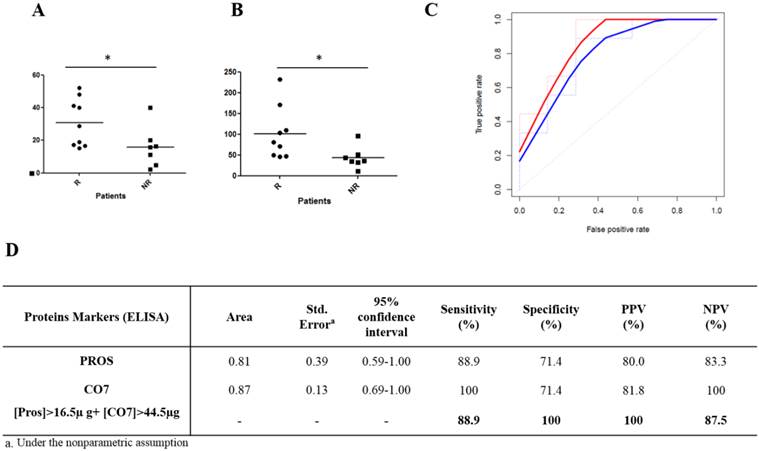
Thereafter, an absolute quantification was performed to confirm the theranostic interest of these potential biomarkers using a complementary approach. The two most discriminant proteins identified within the population 1 (CO7 and PROS) and validated by relative quantification in population 2 were tested in population 2 by ELISA. As illustrated in Figure 5, CO7 and PROS proteins were significantly over-expressed in responders (respectively p-value: 0.015 and 0.042). This absolute quantification for these 2 proteins allowed to determine a concentration threshold associated to a right classification by the construction of a ROC curve. This threshold was 16.5 µg/ml for PROS and could discriminate R from NR with a sensitivity of 88.9 % and a specificity of 71.4 %. For CO7, a threshold of 44.5 µg/ml could discriminate R and NR with a sensitivity of 100 % and a specificity of 71.4%. To improve the identification of R and NR, combining the thresholds of these 2 biomarkers allowed to discriminate R from NR with a sensitivity and a specificity that reached 88.9 % and 100 %, respectively.
Cross-verification by absolute quantification in patient sera from the second cohort prior to MTX/ETA initiation. Absolute quantification by ELISA of serum proteins PROS (A) and CO7 (B) at baseline in responders versus non-responders. Significant difference is noted by asterisk (p < 0.05, Mann- Whitney test). The horizontal bars correspond to the means. (C) Receiver Operating Characteristic (ROC) curves averaging of CO7 (red line), PROS (blue line) were built. (D) Table showing the different parameters resulting from ROC curve analysis from each individual protein and for their combination.
Discussion
Traditional biomarkers such as CRP, RF and anti-CCP are valuable tools for RA diagnosis or prognosis. In contrast, theranostic biomarkers are lacking despite their potential value for personalized medicine. In the field of biologic agents, the ability to anticipate the patient response to a given molecule before the beginning of treatment will allow to prescribe the most appropriate drug.
To identify predictive factors of drug responsiveness, most studies focused on a well-defined marker usually derived from RA pathophysiology. Whatever their nature (immunological, inflammatory, genetic….), none of them was sufficiently relevant to guide clinicians in the choice of a given TBA, up to now. In particular, results obtained from studies focused on the target of TBA appeared disagreeing. In a first attempt, serum TNFα level, evaluated by ELISA, was not correlated to the clinical response to adalimumab [28]. However, in another study, high level of TNF-α was associated with a good response to infliximab by direct estimation of the bioactivity of its circulating form [29].
Studies based on multiplex approaches sound more promising. So far, five main studies were conducted using different tools (SELDI, ELISA, antigen or cytokine microarrays, protein chips, iTRAQ labeling) for screening of sera from patients with RA treated with infliximab, rituximab or etanercept [15-16, 30-32]. After a discovery stage, no combination has been yet validated. However, serum levels of proteins such as MCP-1 and EGF, or ApoA1 appeared to be related to a good response to etanercept or infliximab respectively. Here, we used the label-free approach based on relative quantification of the serum proteome of patients treated with etanercept before treatment initiation. This approach allows the simultaneous evaluation of hundreds of potential biomarkers in biological samples of different nature.
The present study, using an approach based on relative quantification of proteins by label free proteomics, has highlighted a combination of 12 serum proteins able to discriminate two groups of patients by predicting drug responsiveness before the initiation of ETA treatment in patients suffering from active RA refractory to MTX. Importantly, the expression of these proteins is not influenced by clinical or demographic parameters. Using an additional set of patients, a cross validation was carried out by two complementary quantitative approaches. Results of relative quantification by mass spectrometry confirmed the potential theranostic value of 7 of the 12 protein biomarkers identified from the first cohort (table 2). None of them is common to those identified by previous studies dedicated to prediction of response to ETA [30, 32].
Previously, we have reported the potential theranostic interest of S100A9 protein, known as a marker of inflammation [33]. Acute inflammation seems to be at the heart of the prediction of response since, among the seven proteins identified in the two cohorts, 5 of them belong to the class of acute phase proteins (APPs). The latter are synthesized in the liver, and their production is stimulated (positive AAPs) or inhibited (negative AAPs) in response to inflammation. From the combination identified herein, CERU, ITIH1, PLMN, CO7, PROS are positive APPs [34]. Accordingly, the observed modified levels of these proteins appear to be connected to acute inflammation. However, the measurement of the two usual parameters, CRP and ESR, did not reveal any significant difference between the two populations, although CRP tends to be more informative (Table 1 and Table S2). This situation is not exceptional since a discordance between CRP and the serum concentrations of the different proteins of the acute phase has already been described [34]. Moreover, the usual markers of inflammation have never made it possible yet to predict the response to treatment [35]. Since etanercept is a soluble form of TNFα receptor, it is not surprising that TNF-induced proteins may play an important role in prediction of response.
Proteins over-expressed in the acute phase response of the R patients seem to exert an anti-inflammatory effect in two processes: complement system and coagulation. Interestingly, ITIH1 is a protein that inhibits the early stages of complement activation by different pathways [36]. Besides, ITIH1 and ITIH3 are also protease inhibitors forming complexes with hyaluronan (HA) and thus generating a serum-derived hyaluronan-associated proteins (SHAP)-HA complex [37]. ITIH1 may act as a carrier of hyaluronan in serum to regulate its localization, synthesis and degradation. Noteworthy, the SHAP-HA complex is found at very high concentration in RA synovial fluid suggesting its implication in the inflammatory response.
Inflammation initiates clotting, reduces the activity of natural anticoagulant mechanisms and down-regulates the fibrinolytic system [38]. However, some proteins of acute inflammation inhibit the clotting process. PROS exists in two forms: a free form and a form bound to a complement protein, C4b-binding protein (C4BP) [39]. However, C4BP did not exhibit a differential expression between R and NR patients (data not shown) which indicates that only the free form is over-expressed. The latter functions as a cofactor to protein C associated to the inactivation of factors Va and VIIIa. The combined action of these anticoagulant proteins would limit the conversion of PLMN into plasmin, which would explain the greater amount of PLMN in R patients. Noteworthy, PLMN could bind to CO7, also over-expressed in R patients [40].
In addition to understanding the reasons related to the abundance of these proteins in inflammation, we tried to find the best combination able to predict the non-response to ETA/MTX treatment prior to its initiation. In this respect, we have quantified by ELISA two highly relevant proteins. Individually, we obtained satisfactory results but using the combination of these two proteins, we identified 100% of non-responders.
Taken together, those data are robust since the discovery phase was followed by a subsequent confirmation step by targeted proteomics and ELISA in an independent population. These results obtained from 2 well-documented cohorts, suggest that the abundance of serum proteins might be informative for the prediction of response to a given biologic agent such as ETA. Nevertheless, those data remain preliminary due to the limited size of both cohorts and need to be confirmed in a large population of RA treated by ETA/MTX. When validated in another independent cohort, these findings should pave the route for future development of a simple non-invasive test to predict patient non-response to this targeted biotherapy, optimizing the rheumatologist prescription of an alternative biologic agent for a given patient.
Supplementary Material
Figures S1-S2 and Tables S1-S2.
Abbreviations
APPs: Acute phase proteins; ANOVA: ANalysis Of VAriance; anti-CCP2: anti-Cyclic Citrullinated Peptide antibodies; AUC: areas under curve; C4BP: C4b-binding protein; CERU: ceruloplasmin; C1R: complement C1r subcomponent; CO7: Complement component; CRP level: C-Reactive Protein; DAS28: Disease Activity Score 28; DMARD: Disease Modifying Anti-Rheumatic Drugs; ESI: ElectroSpray Ionization; ELISA: Enzyme-Linked Immuno-Sorbent Assay; DMARDs: Disease modifying anti-rheumatic drugs; ESR: Erythrocyte Sedimentation Rate; EULAR: EUropean League Against Rheumatism; HAQ: Health Assessment Questionnaire; HA: hyaluronan; ITIH: inter-alpha-trypsin inhibitor heavy chain; iTRAQ: Isobaric Tags for Relative and Absolue Quantitation; LTQ: Linear Trap Quadrupole; MS: Mass Spectrometry; MTX/ETA: Methotrexate/Etanercept; MR: Moderate Responder; NR: Non Responder; CPN2: N2 carboxypeptidase; PSM: peptide spectrum match; PLMN: plasminogen; PCA: Principal Component Analysis; PROS: Protein S Vitamin K-dependent protein; S100A9: protein S100A9; ROC: receiver operating characteristic; R: Responder; RA: Rheumatoid Arthritis; RF: Rheumatoid Factors; SELDI: surface enhanced laser desorption ionisation; TBAs: TNF Blocking Agents; TRFE: transferrin; TNF-a: Tumor Necrosis Factor alpha; ZA2: Gzinc -alpha2 - glycoprotein.
Acknowledgements
This study was supported by the European Regional Development Fund (FEDER program 33267) and the French Ministry of Health. The authors gratefully acknowledge the assistance of A. Ben Abdelkrim and C. Bérard for discussions in biostatistics.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Roux CH, Saraux A, Le Bihan E, Fardellone P, Guggenbuhl P, Fautrel B. et al. Rheumatoid arthritis and spondyloarthropathies: geographical variations in prevalence in France. J Rheumatol. 2007;34:117-22
2. Carbonell J, Cobo T, Balsa A, Descalzo MA, Carmona L. The incidence of rheumatoid arthritis in Spain: results from a nationwide primary care registry. Rheumatology (Oxford). 2008;47:1088-92
3. Brennan FM, Maini RN, Feldmann M. Role of pro-inflammatory cytokines in rheumatoid arthritis. Springer Semin Immunopathol. 1998;20:133-47
4. Bathon JM, Martin RW, Fleischmann RM, Tesser JR, Schiff MH, Keystone EC. et al. A comparison of etanercept and methotrexate in patients with early rheumatoid arthritis. N Engl J Med. 2000;343:1586-93
5. Keystone EC, Schiff MH, Kremer JM, Kafka S, Lovy M, DeVries T. et al. Once-weekly administration of 50 mg etanercept in patients with active rheumatoid arthritis: results of a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2004;50:353-63
6. Fautrel B, Woronoff-Lemsi MC, Ethgen M, Fein E, Monnet P, Sibilia J. et al. Impact of medical practices on the costs of management of rheumatoid arthritis by anti-TNFalpha biological therapy in France. Joint Bone Spine. 2005;72:550-6
7. Saevarsdottir S, Wedren S, Seddighzadeh M, Bengtsson C, Wesley A, Lindblad S. et al. Patients with early rheumatoid arthritis who smoke are less likely to respond to treatment with methotrexate and tumor necrosis factor inhibitors: observations from the Epidemiological Investigation of Rheumatoid Arthritis and the Swedish Rheumatology Register cohorts. Arthritis Rheum. 2011;63:26-36
8. Bobbio-Pallavicini F, Caporali R, Alpini C, Avalle S, Epis OM, Klersy C. et al. High IgA rheumatoid factor levels are associated with poor clinical response to tumour necrosis factor alpha inhibitors in rheumatoid arthritis. Ann Rheum Dis. 2007;66:302-7
9. Braun-Moscovici Y, Markovits D, Zinder O, Schapira D, Rozin A, Ehrenburg M. et al. Anti-cyclic citrullinated protein antibodies as a predictor of response to anti-tumor necrosis factor-alpha therapy in patients with rheumatoid arthritis. J Rheumatol. 2006;33:497-500
10. Lequerre T, Jouen F, Brazier M, Clayssens S, Klemmer N, Menard JF. et al. Autoantibodies, metalloproteinases and bone markers in rheumatoid arthritis patients are unable to predict their responses to infliximab. Rheumatology (Oxford). 2007;46:446-53
11. Pavy S, Toonen EJ, Miceli-Richard C, Barrera P, van Riel PL, Criswell LA. et al. Tumour necrosis factor alpha -308G->A polymorphism is not associated with response to TNFalpha blockers in Caucasian patients with rheumatoid arthritis: systematic review and meta-analysis. Ann Rheum Dis. 2010;69:1022-8
12. Suarez-Gestal M, Perez-Pampin E, Calaza M, Gomez-Reino JJ, Gonzalez A. Lack of replication of genetic predictors for the rheumatoid arthritis response to anti-TNF treatments: a prospective case-only study. Arthritis Res Ther. 2010;12:R72
13. Plant D, Bowes J, Potter C, Hyrich KL, Morgan AW, Wilson AG. et al. Genome-wide association study of genetic predictors of anti-tumor necrosis factor treatment efficacy in rheumatoid arthritis identifies associations with polymorphisms at seven loci. Arthritis Rheum. 2011;63:645-53
14. Lequerre T, Gauthier-Jauneau AC, Bansard C, Derambure C, Hiron M, Vittecoq O. et al. Gene profiling in white blood cells predicts infliximab responsiveness in rheumatoid arthritis. Arthritis Res Ther. 2006;8:R105
15. Trocme C, Marotte H, Baillet A, Pallot-Prades B, Garin J, Grange L. et al. Apolipoprotein A-I and platelet factor 4 are biomarkers for infliximab response in rheumatoid arthritis. Ann Rheum Dis. 2009;68:1328-33
16. Ortea I, Roschitzki B, Ovalles JG, Longo JL, de la Torre I, Gonzalez I. et al. Discovery of serum proteomic biomarkers for prediction of response to infliximab (a monoclonal anti-TNF antibody) treatment in rheumatoid arthritis: an exploratory analysis. J Proteomics. 2012;77:372-82
17. Cho H, Smalley DM, Theodorescu D, Ley K, Lee JK. Statistical identification of differentially labeled peptides from liquid chromatography tandem mass spectrometry. Proteomics. 2007;7:3681-92
18. Higgs RE, Knierman MD, Gelfanova V, Butler JP, Hale JE. Comprehensive label-free method for the relative quantification of proteins from biological samples. J Proteome Res. 2005;4:1442-50
19. Silva JC, Denny R, Dorschel CA, Gorenstein M, Kass IJ, Li GZ. et al. Quantitative proteomic analysis by accurate mass retention time pairs. Anal Chem. 2005;77:2187-200
20. Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR. et al. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol Cell Proteomics. 2005;4:1487-502
21. Fischer R, Trudgian DC, Wright C, Thomas G, Bradbury LA, Brown MA. et al. Discovery of candidate serum proteomic and metabolomic biomarkers in ankylosing spondylitis. Mol Cell Proteomics. 2012;11:M111 013904
22. Stoop MP, Singh V, Stingl C, Martin R, Khademi M, Olsson T. et al. Effects of natalizumab treatment on the cerebrospinal fluid proteome of multiple sclerosis patients. J Proteome Res. 2013;12:1101-7
23. Bostanci N, Ramberg P, Wahlander A, Grossman J, Jonsson D, Barnes VM. et al. Label-free quantitative proteomics reveals differentially regulated proteins in experimental gingivitis. J Proteome Res. 2013;12:657-78
24. Dagley LF, Emili A, Purcell AW. Application of quantitative proteomics technologies to the biomarker discovery pipeline for multiple sclerosis. Proteomics Clin Appl. 2013;7:91-108
25. Bleijerveld OB, Wijten P, Cappadona S, McClellan EA, Polat AN, Raijmakers R. et al. Deep proteome profiling of circulating granulocytes reveals bactericidal/permeability-increasing protein as a biomarker for severe atherosclerotic coronary stenosis. J Proteome Res. 2012;11:5235-44
26. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS. et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315-24
27. van Gestel AM, Prevoo ML, van 't Hof MA, van Rijswijk MH, van de Putte LB, van Riel PL. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism Criteria. Arthritis Rheum. 1996;39:34-40
28. Cuchacovich M, Soto L, Edwardes M, Gutierrez M, Llanos C, Pacheco D. et al. Tumour necrosis factor (TNF)alpha -308 G/G promoter polymorphism and TNFalpha levels correlate with a better response to adalimumab in patients with rheumatoid arthritis. Scand J Rheumatol. 2006;35:435-40
29. Marotte H, Arnaud B, Diasparra J, Zrioual S, Miossec P. Association between the level of circulating bioactive tumor necrosis factor alpha and the tumor necrosis factor alpha gene polymorphism at -308 in patients with rheumatoid arthritis treated with a tumor necrosis factor alpha inhibitor. Arthritis Rheum. 2008;58:1258-63
30. Hueber W, Tomooka BH, Batliwalla F, Li W, Monach PA, Tibshirani RJ. et al. Blood autoantibody and cytokine profiles predict response to anti-tumor necrosis factor therapy in rheumatoid arthritis. Arthritis Res Ther. 2009;11:R76
31. Fabre S, Guisset C, Tatem L, Dossat N, Dupuy AM, Cohen JD. et al. Protein biochip array technology to monitor rituximab in rheumatoid arthritis. Clin Exp Immunol. 2009;155:395-402
32. Fabre S, Dupuy AM, Dossat N, Guisset C, Cohen JD, Cristol JP. et al. Protein biochip array technology for cytokine profiling predicts etanercept responsiveness in rheumatoid arthritis. Clin Exp Immunol. 2008;153:188-95
33. Obry A, Lequerre T, Hardouin J, Boyer O, Fardellone P, Philippe P. et al. Identification of S100A9 as biomarker of responsiveness to the methotrexate/etanercept combination in rheumatoid arthritis using a proteomic approach. PLoS One. 2014;9:e115800
34. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448-54
35. Emery P, Dorner T. Optimising treatment in rheumatoid arthritis: a review of potential biological markers of response. Ann Rheum Dis. 2011;70:2063-70
36. Okroj M, Holmquist E, Sjolander J, Corrales L, Saxne T, Wisniewski HG. et al. Heavy chains of inter alpha inhibitor (IalphaI) inhibit the human complement system at early stages of the cascade. J Biol Chem. 2012;287:20100-10
37. Zhuo L, Hascall VC, Kimata K. Inter-alpha-trypsin inhibitor, a covalent protein-glycosaminoglycan-protein complex. J Biol Chem. 2004;279:38079-82
38. Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;131:417-30
39. Castoldi E, Hackeng TM. Regulation of coagulation by protein S. Curr Opin Hematol. 2008;15:529-36
40. Reinartz J, Hansch GM, Kramer MD. Complement component C7 is a plasminogen-binding protein. J Immunol. 1995;154:844-50
Author contact
![]() Corresponding author: O. Vittecoq MD, PhD: Department of Rheumatology, Rouen University Hospital & INSERM U905, Rouen, Normandy, France, Tel.: +33 2 32 88 90 19; fax: +33 2 32 88 91 10, E-mail address: olivier.vittecoqfr
Corresponding author: O. Vittecoq MD, PhD: Department of Rheumatology, Rouen University Hospital & INSERM U905, Rouen, Normandy, France, Tel.: +33 2 32 88 90 19; fax: +33 2 32 88 91 10, E-mail address: olivier.vittecoqfr