Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2016; 6(7):918-929. doi:10.7150/thno.14689 This issue Cite
Review
Current and Future Theranostic Applications of the Lipid-Calcium-Phosphate Nanoparticle Platform
Andrew B. Satterlee1,2, Leaf Huang1,2 ![]()
1. Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7571, USA;
2. UNC and NCSU Joint Department of Biomedical Engineering, Chapel Hill, NC 27599.
Received 2015-12-14; Accepted 2016-3-15; Published 2016-4-27
Abstract

Over the last four years, the Lipid-Calcium-Phosphate (LCP) nanoparticle platform has shown success in a wide range of treatment strategies, recently including theranostics. The high specific drug loading of radiometals into LCP, coupled with its ability to efficiently encapsulate many types of cytotoxic agents, allows a broad range of theranostic applications, many of which are yet unexplored. In addition to providing an overview of current medical imaging modalities, this review highlights the current theranostic applications for LCP using SPECT and PET, and discusses potential future uses of the platform by comparing it with both systemically and locally delivered clinical radiotherapy options as well as introducing its applications as an MRI contrast agent. Strengths and weaknesses of LCP and of nanoparticles in general are discussed, as well as caveats regarding the use of fluorescence to determine the accumulation or biodistribution of a probe.
Keywords: Cancer, Theranostic, SPECT, PET, SIRT, Nanoparticle, LCP.
Introduction
The fields of tumor imaging and cancer therapy have been steadily sliding closer together since the field recognized a need for targeted therapeutics. Targeted delivery must be coupled with verification of payload deposition in order to effectively monitor and analyze therapeutic outcomes. A wealth of theranostic agents have been developed to address the growing desire for this dual-purpose paradigm, and nanoparticle formulations have recently come to the forefront of theranostic research1, 2.
Calcium phosphate precipitates have been used as in vitro transfection agents since the 1970s3, when it was discovered that DNA could be co-precipitated in a solution containing calcium chloride and phosphate-buffered saline. Since that time, there has been a substantial effort to control particle size and aggregation in order to increase transfection efficiency and allow delivery in vivo. Some formulation strategies gained moderate success in vitro4-6, but it was not until 2010 that calcium phosphate nanoparticles were successfully coated with a lipid bilayer7, and not until 2012 that these so-called Lipid-Calcium-Phosphate (LCP) nanoparticles showed efficient gene silencing in vivo8. Since that time, the two seminal papers have garnered nearly 300 citations, inspiring many other laboratories to use LCP themselves or innovate on its design9-14. The Huang lab has published nearly 20 original research articles using LCP, due to its versatility in encapsulating many types of therapeutic compounds, such as small molecule drugs15-17, siRNA18-22, DNA23, peptides24, and radionuclides25, 26. The principles behind LCP formulation have also allowed the Huang lab to formulate lipid-coated nanoparticles with cores entirely made of cisplatin27, 28. Over the last six years, LCP has transitioned from a novel formulation to a powerful tool used to probe for answers regarding PEGylation kinetics29, the tumor microenvironment15, 18, 24, 25, cancer vaccines18, 24, and gene therapy23. The Huang lab has now begun using LCP as a theranostic agent, using imaging modalities such as SPECT and PET.
LCP is formulated using a reverse (water-in-oil) microemulsion system. In separate round-bottom flasks, water droplets containing either calcium or phosphate are stirred in an oil phase containing cyclohexane and the surfactant igepal. The surfactant maintains the oil-water interface and stabilizes nano-sized water droplets in the oil. When the two emulsions are mixed and the separate droplets collide, the soluble calcium and phosphate react to form nanoprecipitates, still suspended in the water droplets. Addition of the phospholipid DOPA soon after mixing allows the formation of a single lipid layer at the oil-water interface in which the hydrophilic head of DOPA inserts inside the water droplet and its hydrophobic tail remains in the oil phase. The phosphate head group of the DOPA co-precipitates into the calcium-phosphate nanoprecipitate, positioning the hydrophobic tail to remain in the oil. Over time, the co-precipitated DOPA replaces more and more igepal in the water-oil interface, and eventually the calcium phosphate precipitates are so well coated with DOPA that they are able to be dissolved in organic solvents such as chloroform or dichloromethane after washing with ethanol. The final LCP nanoparticle, shown in Figure 1, is generated by adding free lipids such as DOTAP, cholesterol, and DSPE-PEG2000, also in organic solvent, to the cores. The solvent is then evaporated to leave a thin film of lipids and cores on the edge of the vial. Addition of warm water/sucrose solution and brief sonication is all that is needed to resuspend the final particles for testing or injection into mice.
Schematic of LCP nanoparticle synthesis. LCP is capable of encapsulating siRNA, mRNA, DNA, phosphorylated peptides, nucleotide mimics, and radioactive isotopes in its calcium phosphate core. Source: Adapted from Satterlee et al25. Copyright © 2015 Elsevier B.V. License number: 3815560646760
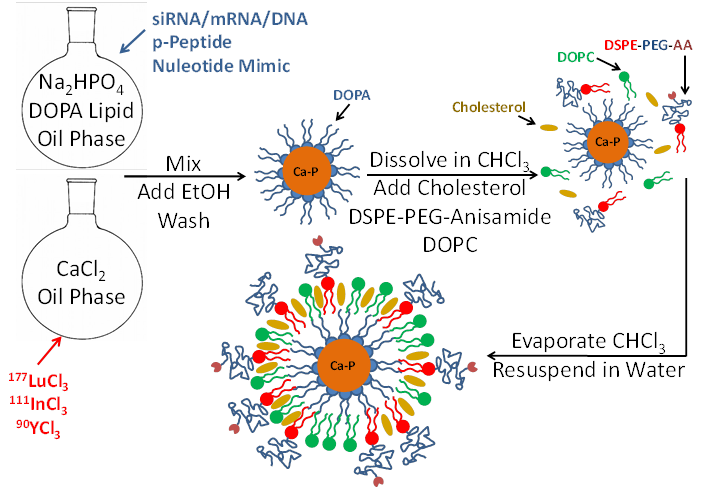
Encapsulation of therapeutics into LCP occurs during the calcium-phosphate nanoprecipitation portion of the procedure. Phosphorylated prodrugs, nucleotide mimics, siRNA, and DNA are all ideal candidates for encapsulation into LCP because the phosphate groups on all of these molecules will co-precipitate in the calcium-phosphate LCP core. Radionuclides such as indium and lutetium can similarly be encapsulated via their low solubility with phosphate. Because calcium-phosphate is acid-sensitive, after the LCP is delivered to the cellular endosome, the late endosome's acidic environment will dissolve the calcium-phosphate core7. The increase in endosomal salt concentration increases the osmolarity in the endosome, destabilizing it and releasing the therapeutics into the cytosol. The cell avoids calcium-induced toxicity by quickly lowering cytosolic calcium via the mitochondrial calcium uniporter and then by more slowly removing the excess intracellular calcium via the plasma membrane calcium ATPase30.
Herein we aim to review current imaging techniques and compare the theranostic capabilities of LCP to clinical practices that combine therapy and imaging. This review will focus its discussion on the nuclear imaging modalities SPECT and PET.
Single Photon Emission Computed Tomography (SPECT)
SPECT imaging detects γ photon emissions from a decaying radioactive source inside the body31. Radionuclides with a high abundance of γ decay at energies around 100-200 keV32, such as 111In, 99mTc, and 123I, are ideal for detection using SPECT, as their γ energies are high enough to avoid attenuation in tissue but low enough to be adequately captured by the SPECT detectors. Because the source emits photons in all directions, in order to maximize spatial resolution, a collimator must be used to block all photons that are not approaching the detector from the region or angle of interest. More selective collimators increase the resolution of the image but also reduce signal to the detectors, requiring longer detection times or higher doses of radionuclide to maintain adequate image reconstruction33. SPECT is often run in combination with a computed tomography (CT) scan to provide anatomical contrast.
Positron Emission Tomography (PET)
PET imaging is similar to SPECT imaging, as the PET detectors also detect γ photons from a radioactive source inside the body32. In contrast to SPECT, the photons detected in PET are generated from an annihilation reaction between a positron emitted by the source and a nearby electron present in the body. The annihilation reaction converts the electron-positron pair into 511 keV photons that travel in opposite directions toward the PET detector. By only counting photon pairs that are detected at a 180o angle from each other, all other noise not attributed to positron emission can be disregarded, eliminating the need for collimation. By also calculating the small differences in detector arrival time of the photon pair—referred to as time-of-flight—, more accurate spatial localization of the radionuclide can be determined34. The unique imaging properties of positron emitters allow PET imaging to be more sensitive than SPECT, but PET must also be coupled with an imaging modality like CT or MRI for anatomical contrast.
Comparing two common PET agents, copper-64 (64Cu) and gallium-68 (68Ga), will demonstrate the additional considerations that must be made when planning a PET imaging study. Half-life must first be considered, as many PET agents have very short half-lives and must be produced and purified on-site. 64Cu has a relatively long half-life of 12.7 h, allowing it to be shipped overnight without decaying prohibitively, unlike 68Ga which has a half-life of just 68 minutes. Fortunately, 68Ga is generated by the slow decay of Germanium-68 (68Ge) and can be quickly purified from its parent on-site35, unlike 64Cu which must be generated by neutron or proton bombardment in a nuclear reactor or cyclotron36. Half-life also affects the total dose of nuclide that must be given to achieve the same decay rate during imaging. It is desirable to inject a low total dose with a high rate of decay such that a large portion of the decay will occur during the imaging experiment. In a one-hour PET imaging session using 68Ga, half of the remaining 68Ga atoms will decay during the imaging experiment, but if 64Cu is used, less than 10% of the 64Cu will decay during the experiment, leaving the rest to decay over the next few days. In order to produce the same quality image, both 64Cu and 68Ga need to be dosed at similar positron decay rates, and since 68Ga decays more quickly, fewer total atoms also need to be given, further lowering the required dose.
The energy with which a positron is emitted, β+max = 653 keV for 64Cu and β+max = 1899 keV for 68Ga, also affects the resolution of the image. A positron must be at rest energy before annihilating with an electron, and any extra energy is released as the positron proceeds away from its parent nucleus and collides with other matter37. Positrons ejected at higher energies must release more energy before annihilation and therefore must travel further away from the nucleus before the resulting two γ photons are generated. Because the PET detectors can only detect where in space the photons were generated, and not where the positron was emitted, a longer positron range will generate larger areas between the positron emitter and the photon generation, thus decreasing the spatial resolution of the image. The average positron ranges in water for 64Cu and 68Ga are 0.55 mm and 2.8 mm, respectively38, which is not a negligible difference and should be considered when planning an experiment.
Clinical Nuclear Imaging
In the clinic, SPECT and PET contrast agents are often used diagnostically for blood vessel perfusion imaging or tumor diagnosis. Radiometals have recently seen more use in the clinic, and are often chelated to small molecules or targeted proteins. A huge amount of effort has gone into discovering bifunctional chelators that stably bind both the radiometal and the tumor-targeting moiety39-43. Because each radiometal ion can differ in atomic size, oxidation state, and electron configuration, a chelator that stably binds one metal may not be suitable for another. Stability tests must include exposing the chelated metal to serum conditions, as an inadequate chelator may have higher affinity for an endogenous metal ion or allow reduction of its metal into a less stable state44. The expected circulation time of the radionuclide must also be considered, as a nuclide chelated to a protein with a circulation half-life on the order of days would need a much more stable chelator than one on a particle with a half-life of an hour or less. If an unsuitable chelator is chosen to bind a radionuclide, the nuclide may prematurely shed in circulation and skew the biodistribution data, as highly charged heavy metals such as 111In(III) or 177Lu(III) prefer to accumulate in the bone, and actively metabolized metals such as copper may be filtered through the liver and into the intestines.
There are several typical radioisotopes used diagnostically as SPECT and PET contrast agents. Current clinical uses involve chelation or conjugation of the isotopes to small molecules for blood vessel perfusion imaging or tumor diagnosis. Classically, the γ emitter 99mTc has been injected intravenously into patients after chelation to one of several small molecules such as exametazime and sestamibi45. The lipophilic conjugate formed can accumulate in tumors and allow detection of many types of cancer via SPECT. In several clinical trials, 99mTc has also been conjugated to the apoptosis marker Annexin V to gauge a patient's response to treatment46. A similar agent exists for PET imaging. Incorporation of the positron emitter 18F as the 2' group on glucose molecules yields 18Fluoro-deoxyglucose (18FDG), which can accumulate in tumors due to a tumor's high glucose uptake and the inability for cells to metabolize FDG after uptake. 111In is also currently used as a diagnostic agent to measure tumor burden or metastasis by chelating it to an antibody targeted to an overexpressed tumor receptor47. The clinically used ProstaScint® is an 111In-conjugated monoclonal antibody against prostate specific membrane antigen used to diagnose prostate cancer. In some cases, such as in the phase II clinical trial for a doxorubicin-linked polymeric micelle termed PK1, pharmacokinetics and biodistribution of the micelle were determined by conjugating the γ emitter 123I onto a small amount of PK1 analog during one treatment cycle48. The imaging analog contained an additional methacryloyltyrosinamide group whose tyrosine moiety was labeled with 123I via the Iodogen® labeling procedure49. These imaging strategies have laid the groundwork for those therapeutics that can simultaneously image and treat the tumor.
Radioimmunotherapy
Another recent effort has been placed on formulations that can accumulate in the tumor to provide diagnosis while simultaneously treating the tumor mass, and so far the most successful radio-theranostics in the clinic seem to be those used in radioimmunotherapy (RIT). RIT is a treatment strategy in which a therapeutic radioisotope such as 90Y or 177Lu is chelated and covalently bound to an antibody that will selectively target the tumor. 90Y has a higher β energy and a longer β range in tissue, while 177Lu decays via both β and γ emission, allowing SPECT imaging with simultaneous therapy. The radioimmunotherapeutic Zevalin®, which uses 90Y, was the first in its class to receive FDA approval and is currently seeing success in the clinic50. Zevalin® is used against Non-Hodgkin's Lymphoma (NHL) and uses the murine anti-CD20 antibody Ibritumomab against B-cells as a targeting and therapeutic agent. Unfortunately, the antibodies used in this therapy are known to cause sometimes serious toxicity via complement activation and subsequent cytokine release51-53. Antibodies used in RIT are also very expensive, especially those that have been humanized or otherwise modified to reduce toxicity.
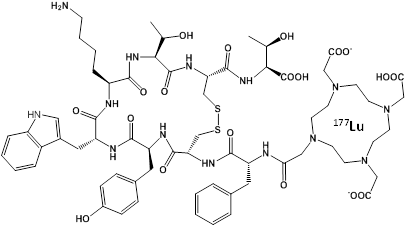
An effective alternative to RIT may be to use a peptide instead of an antibody to target the chelated radionuclide to the tumor, which would be less toxic and less expensive. This approach has been termed Peptide Receptor Radionuclide Therapy (PRRT) and the most successful designs have used a somatostatin analog called octreotate to target the somatostatin receptor overexpressed in neuroendocrine tumors54-56. The novel molecule DOTA-TATE (Figure 2) was generated by covalently linking the chelator DOTA to octreotate, allowing chelation of radionuclides such as 177Lu, 90Y, 64Cu, and 111In for imaging and therapy. PRRT with octreotate was recently approved by the FDA for use in patients with neuroendocrine cancer. The dual therapy (β) and imaging (γ) capabilities of 177Lu allow simultaneous treatment and dosimetry of patients receiving 177Lu-DOTA-TATE57. The absorbed dose to healthy organs, especially the kidneys and bone marrow, was calculated from γ-scintigraphy and SPECT/CT images in order to provide the maximum therapeutic dose without exceeding the maximum acceptable dose to healthy tissue.
Chemical Structure of the PRRT agent 177Lu-DOTA-TATE
SPECT Imaging with LCP
The LCP formulation procedure allows efficient encapsulation of radiometals without the use of a chelator, and as of 2015, two articles have been published using LCP for SPECT imaging. The first encapsulated the γ emitter 111In3+ into the core of LCP as an imaging agent to measure lymph node accumulation of LCP, initiating research on treating lymph node metastasis26. The second article encapsulated the γ and β emitter 177Lu3+ as both an imaging and therapeutic agent25. It was discovered that the 3+ charge on radiometals like 177Lu enabled very selective encapsulation in the LCP core, as their solubility product with phosphate are many orders of magnitude lower than that of calcium and phosphate (ksp lutetium-phosphate = 2x10-25; ksp calcium-phosphate = 1x10-7). Consequently, when 177Lu was co-precipitated into the LCP core, a 70% encapsulation efficiency of 177Lu was maintained as the feed ratio of lutetium to calcium was increased by four orders of magnitude, allowing a tunable loading of radionuclide in a single-sized batch of LCP. Because only a trace amount of 177Lu is required to provide therapeutic effect, small-scale batches are still sufficient to produce 177Lu-LCP particles at human-level therapeutic dosing. Even at the highest stable feed ratio of Lu:Ca, 177Lu-LCP maintains its encapsulation efficiency and morphology in particles that are still overwhelmingly composed of calcium phosphate (1:1000 Lu:Ca input ratio). This eliminates the risk of heavy metal toxicity that would be presented in neutron-activatable theranostic nanoparticles, which can be loaded with as much as 20 wt% of the heavy metal ion58. In this case, the nanoparticles are bombarded with neutrons in a nuclear reactor to produce radioactive 166Ho from stable 165Ho. Only a small fraction of the 165Ho is converted, increasing the dose of nanoparticles and heavy metal that must be given, but this method does decrease radioactive handling during fabrication.
177Lu has a max β energy of ~500 keV and γ energies of 113 and 208 keV. The γ energies are in the ideal range for efficient SPECT imaging and allow the biodistribution and tumor accumulation of 177Lu-LCP to be measured using SPECT/CT (Figure 3A) and γ scintillation25. The therapeutic β emission from 177Lu-LCP was found to cause significant tumor inhibition via DNA damage-induced H2AX phosphorylation, followed by cellular apoptosis. A change in the tumor microenvironment's stromal structure was also detected, resulting in a decrease in fibroblast and collagen organization. Furthermore, the cytotoxic β emission from 177Lu also induced photons in the visible spectrum from the surrounding media. This Cerenkov radiation is generated from the β electron moving faster than the speed of light in tissue and was detected using optical imaging in a live mouse (Figure 3B). Although Cerenkov imaging does not provide a 3D reconstruction of biodistribution as SPECT does, it does allow less expensive and faster image acquisition that would be especially useful in determining early biodistribution kinetics using multiple scans in quick succession. Cerenkov radiation also allows imaging of other β-emitting nuclides such as 90Y that would otherwise not be available for imaging studies.
(A) SPECT/CT and (B) Cerenkov imaging of a live athymic nude mouse bearing a subcutaneous UMUC3/3T3 stroma-rich bladder cancer tumor at t = 24 h after i.v. injection of 2.5 mCi of 177Lu-LCP. Tumor accumulation of 177Lu is clearly visible above background, although not as high as liver and spleen accumulation. SPECT and Cerenkov agree on the overall biodistribution of 177Lu. Source: Adapted from Satterlee et al25. Copyright © 2015 Elsevier B.V. License number: 3815560646760

Local Delivery of Radiation Therapy
When delivered systemically, nanoparticle-based therapeutics are generally too large to clear from the body through the kidneys, and therefore tend to accumulate in the body's other clearing organs, which are primarily the liver and spleen. Splenocytes and the liver's Kupffer cells will internalize these nanoparticles and attempt to metabolize the contents59, so when delivering therapeutic siRNA or DNA, this off-target delivery is not of much consequence. Additionally, since siRNA is generally chosen to knock down upregulated mRNA that is not abundant in healthy cells, and gene therapy often seeks to reintroduce a lost or downregulated gene that is present in healthy cells, off-target effects resulting from delivery to hepatocytes or other healthy cells are dampened. In contrast, when radiation is delivered, the cytotoxic decay products can reach several millimeters away from the delivered source, indiscriminately acting on healthy and cancerous cells alike. Although cancer cells are more susceptible to DNA damage due to their increased proliferation rate and decreased DNA repair capacity, care must be taken to minimize the dose to off-target organs. Thus, current internal radiation therapies focus on local delivery of the radiation in order to increase the absorbed dose to the tumor and limit dose to healthy tissues.
Brachytherapy
Brachytherapy is a type of clinical radiotherapy in which the therapeutic radioactive source is permanently or temporarily placed in or near the tumor site. The majority of brachytherapy procedures are performed in prostate cancer, and placement of the source(s) is pre-planned using transrectal ultrasound to maximize dose to the tumor while minimizing dose to healthy tissue, allowing safe dose escalation to nearly double that for external beam radiation therapy60. In high dose rate (HDR) brachytherapy, sealed sources of Iridium-192 are temporarily placed within the prostate via intraperineal catheterization, while in low dose rate (LDR) brachytherapy, sealed radioactive seeds measuring ~0.8 mm x 4.5 mm are permanently deposited at precise intervals in the prostate. The radioisotopes Cesium-131, Iodine-125, and Palladium-103 are preferred for LDR brachytherapy because their low-energy emissions provide rapid dose fall-off with increasing distance from the source, minimizing off-target dose61. To decrease incidence of seed movement or loss, each row of seeds can be sutured together to create a strand of several seeds62. LDR seeds are typically placed 1 cm apart63, which allows a tunable dose to different areas of the prostate based on the activity encapsulated in each seed.
The new concept of nanobrachytherapy is currently limited to simple intratumoral injection of radioactive nanoparticles64, but there may be some value in delivering nanoparticles that can evenly diffuse within the tumor. Major difficulties would be in reproducible dosimetry and clearance of the nanoparticles out of the tumor, as clearance would affect dosimetry as well as convey off-target toxicity to the clearing organ. In order to control diffusion and minimize clearance, nanoparticle penetration through tissue could be tuned by modifying their charge and PEGylation density. Highly loaded 177Lu-LCP may be a good probe to quantify nanoparticle diffusion and clearance in the tumor, both by live animal imaging and in frozen tissue sections. In its current formulation, LCP may be too acid-sensitive to ensure minimal clearance of the radioisotope over the initial decay of the delivered isotope, but a similar precipitate with a lower ksp such as Cu(PO4) may more effectively resist dissolution while maintaining the preferential encapsulation of trivalent radiometals. Interestingly, excess cellular copper is known to generate reactive oxygen-dependent DNA damage, which could itself provide an effective combination therapy with the delivered radionuclide.
Selective Internal Radiation Therapy (SIRT)
SIRT is another clinically approved local delivery method for internal radiation therapy. This approach delivers radioactive microspheres loaded with 90Y to the liver via a surgically implanted catheter that is fed up the hepatic artery. It is known that while the portal vein provides the majority of the blood supply to healthy hepatocytes in the liver, liver tumors are fed by the hepatic artery. Thus, introduction of microspheres through the hepatic artery selectively target the tumor regions. Commercially available microsphere formulations such as Sir-Spheres by Sirtex are approximately 30 μm in diameter, which prevent cellular uptake and deep tumor penetration, but also help retain the microspheres around the tumor site while minimizing clearance through the hepatic capillaries. SIRT is currently in several phase II and III clinical trials for use in combination with front-line chemotherapeutics against hepatocellular carcinoma (HCC) and advanced metastatic colorectal cancer65, 66. The data from recent clinical trials show that 90Y-SIRT + systemic chemotherapy provides significantly more hepatic progression-free survival than chemotherapy alone while maintaining tolerable liver and lung toxicity.
Although local hepatic delivery using 90Y-microspheres for SIRT has shown success, there are areas in which it can be improved. Most notably, the lack of tumor penetration by the microspheres requires a high-energy β emitter with a long depth of penetration to reach the deeper tumor tissue. To maximize the absorbed dose to deep tumor cells, a large dose must be provided at the tumor periphery, which increases toxicity to healthy liver tissue. A future 177Lu-LCP-SIRT therapy may be able to address these issues. With a diameter of just 40-60 nm, the much smaller 177Lu-LCP could penetrate much deeper into the tumor mass, and the addition of an appropriate targeting ligand would allow selective uptake of the particles into the tumor cells themselves, bringing the radioactive payload closer to the target DNA in the cell nucleus. The lower energy and shorter depth of penetration of 177Lu's β emissions may then be adequate to treat the entire tumor at a lower total dose and with less toxicity to nearby healthy tissue, although the efficacy of 177Lu as the encapsulated radionuclide should be compared with 90Y. If 177Lu-LCP is effective, the γ decay from 177Lu could then allow simultaneous confirmation of 177Lu-LCP accumulation in the tumor mass via SPECT imaging. Additionally, 177Lu-LCP could be easily co-loaded with a phosphorylated chemotherapeutic for localized chemoradiation therapy.
The much smaller size of LCP compared to Sir-Spheres also presents new challenges regarding the clearance of 177Lu away from the tumor site, not only from those particles that are injected into the hepatic artery and do not enter the tumor mass, but also from any radiation that initially enters the tumor and exits at a later time. To ensure maximal tumor accumulation and minimal clearance of 177Lu-LCP through the vena cava, simple changes to the surface of the particle should be made to encourage interaction with the tumor cell membrane and increase receptor-mediated endocytosis, such as modifying the outer leaflet with cationic lipids, lowering the surface PEG density, and adding a high affinity targeting ligand. Peptide targeting ligands may be the most successful class of ligand for this purpose, as they generally bind with higher affinity than small molecule ligands, and are less bulky, expensive, and immunostimulatory than antibodies. Peptide ligands against the α6β1 receptor overexpressed in human colon cancer cells may be successful against liver metastasis67, 68, and similarly, overexpression of glypican-3 (GPC-3) in HCC has allowed synthesis and use of targeted peptides69, 70. Changes in the nanoparticle injection speed should also be considered, as a slower infusion of particles into the tumor will avoid saturation of the surrounding blood vessels and tumor cell receptors. The introduction of novel nanodelivery approaches does indeed present new challenges in local delivery of radioisotopes, but the advantages posed by these new methods should make overcoming these challenges a worthwhile venture.
Original Research: PET Imaging with LCP
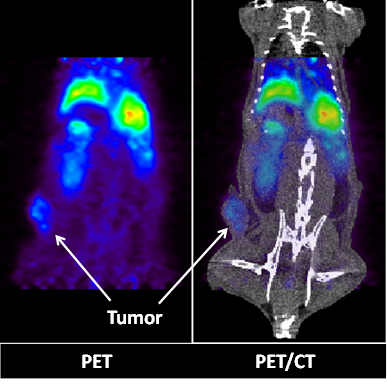
The successful encapsulation of 177Lu and 111In in the LCP core25, 26 prompted experiments to encapsulate a radionuclide into an LCP formulation already containing chemotherapeutics in order to better study its kinetics and biodistribution. The LCP formulation that was chosen contained the cytotoxic nucleotide analog gemcitabine monophosphate, and had previously been published as a monotherapy and in combination with siRNA16, 19, 20. In previously unpublished experiments from this lab, trace 64Cu was encapsulated in the GMP-LCP core via co-precipitation and achieved a moderate encapsulation efficiency of ~50% because of its slightly lower solubility with phosphate when compared to calcium (a difference in Ksp of 3-4 orders of magnitude). Using PET/CT, we were able to record a continuous dynamic scan documenting the first two hours after intravenous injection into mice (Supplementary Video S1), followed by additional time points at 16, 22, and 48 h after injection. As shown in Figures 4 and 5A, tumor accumulation of 64Cu-GMP-LCP reached a maximum at t = 22 h and was clearly visible in the PET image. Figure 5A shows that while max tumor uptake was sustained throughout the study, liver uptake peaked just 20 min after injection and showed a continuous decrease over time. This time-dependent decrease in liver accumulation was not consistent with our experiments using 177Lu and 111In, in which liver accumulation of the probes remained high after 24 h.
PET (left) and PET/CT (right) images of a live athymic nude mouse bearing a subcutaneous A549 lung cancer tumor. Images show 2-D coronal sections at t = 22h after 64Cu-GMP-LCP administration. Liver and tumor accumulation of 64Cu are both clearly visible. Dose of 64Cu = 500 μCi; dose of GMP = 5 mg/kg.
Biodistribution kinetics in a live athymic nude mouse bearing a subcutaneous A549 lung cancer tumor: (A) Quantification of 64Cu from 64Cu-GMP-LCP in liver and tumor, calculated from PET reconstruction ROIs; (B) PET images of 2-D transverse liver sections at different time points after injection; (C) PET images of 2-D coronal tumor sections at different time points after injection; (D) 3-D reconstruction of 64Cu biodistribution from 64Cu-GMP-LCP at different time points after injection. Arrows at 16 h and 22 h time points show accumulation of 64Cu in the intestines. Dose of 64Cu = 500 μCi; dose of GMP = 5 mg/kg.
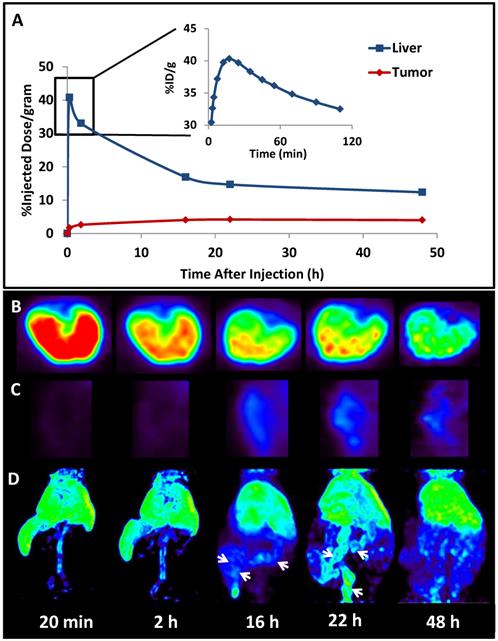
The difference in 177Lu/111In and 64Cu biodistribution is likely because of downstream action after the radionuclide cargoes are released from LCP via acid-sensitive dissolution in the cellular endosome. Because trivalent radiometals like 177Lu are not metabolically active, they are not actively excreted in the bile and intestines by the liver. When free `177LuCl3 is injected intravenously, intestinal accumulation of 177Lu is not observed25. In contrast, copper is a micronutrient that is actively metabolized in the liver, secreted into the bile, and excreted into the intestines71. Previously published time-dependent PET data of free 64CuCl2 injected intravenously shows high initial liver uptake, followed by a decrease in liver accumulation via washout through the intestines72. Figure 5C similarly shows that as 64Cu is excreted from the liver, the intestines begin to show 64Cu accumulation. Because free 64Cu can only be excreted after release from LCP, this may suggest that LCP releases its payload within just a few hours of deposition.
Quick copper clearance from off-target organs can be desirable if one wants to decrease background signal or use copper itself as a theranostic agent72, but when using 64Cu as a probe for nanoparticle biodistribution, it is important that 64Cu pharmacokinetics accurately represent those of the nanoparticle. Problematic copper metabolism after delivery can be remedied by chelating the copper to render it metabolically inactive. For many years, the most effective copper chelators were the tetraazamacrocyclics DOTA and TETA, which admittedly had well-documented stability issues in vivo, allowing 64Cu to transchelate onto ceruloplasmin, the main copper-binding enzyme in the liver73, 41. The chelator TE2A has shown better 64Cu stability than DOTA and TETA, allowing faster and more complete off-target clearance in vivo74, 75, and recently, a class of bifunctional, bicyclic chelators called sarcophagines has shown excellent ability to stably chelate copper for tumor imaging76, 77. Sarcophagines could potentially be covalently attached to the distal ends of PEG or outer leaflet lipids on nanoparticles in order to provide stable PET imaging. To achieve reproducible results, chelator density on the nanoparticle must be optimized, excess chelators must be removed before addition of 64Cu, and unincorporated 64Cu—if any—must be purified away before injection. Alternatively, it may be possible to incorporate chelated 64Cu directly into the nanoparticle core. A chelator that remains highly charged after 64Cu incorporation may co-precipitate with calcium phosphate in the LCP core, eliminating the need for surface chelation and accurately representing the biodistribution of the LCP core while also maintaining the metabolic inertness of the isotope.
Future Directions: Magnetic Resonance Imaging (MRI) with LCP
MRI uses the concept of nuclear magnetic resonance to reconstruct anatomical images. By using a large magnet to manipulate the spin and magnetic moment of hydrogen atoms in the body, and then measuring their relaxation times, the distribution and structural characteristics of the hydrogen atoms can be measured78. MRI contrast agents such as iron oxide or gadolinium can themselves be magnetized when under the influence of an external magnetic field, which shortens both the T1 and T2 relaxation times of nearby hydrogen atoms79. Thus, to localize the contrast agents, the affected hydrogen atoms are measured, and not the contrast agents themselves. This requires that a baseline reading be completed before adding the contrast agents.
LCP has not yet been utilized in MRI for any purpose, but there are definitely some interesting theranostic applications for this imaging modality, since gadolinium is another trivalent cation that should be easily incorporated into the LCP core. One advantage that MRI has over SPECT and PET is that it does not use radioactivity to generate an image, which makes formulation and optimization of a theranostic nanoparticle formulation safer and less expensive. What functionally separates gadolinium and MRI from SPECT and PET imaging, however, is the fact that gadolinium must interact with the hydrogen atoms found mostly in water or tissue to generate its contrast, unlike radioisotopes that decay independent of their surroundings. The magnetic moment generated by gadolinium on water decreases with distance to the sixth power, which means that gadolinium encapsulated in LCP and surrounded by calcium phosphate and lipid membrane will provide a much lower signal than free or released gadolinium. Our previous experiments with 64Cu have provided some insight into the release profile of LCP in the liver, but using MRI to quantify gadolinium release from LCP could show relative release rates in all organs, determine if any release occurs in circulation, and measure whether the low pH of the tumor does indeed cause more rapid LCP dissolution. This type of information would not be available if gadolinium were chelated to the outside of the particle, as it has been repeatedly shown that chelated gadolinium retains its ability to magnetize water80, 81.
This approach can be taken one step further. By encapsulating or chelating 64Cu to LCP as described above and also encapsulating gadolinium in the core, combined PET/MRI could be used to simultaneously measure the LCP biodistribution and release, showing which organ regions contain both released and unreleased LCP over time. This type of information could provide insight into what kind of dose the tumor and other organs are receiving, as faster release of a small molecule drug or siRNA from LCP should cause more acute toxicity than slow, sustained release.
Optical Imaging: Use Caution
Optical imaging generally uses fluorescent or luminescent contrast agents with emission spectrums in or near the visible light spectrum. Fluorescent compounds require excitation by light before they are able to emit light of their own, while other luminescent compounds can utilize chemical reactions or biological processes to emit light without an incident excitation wavelength. The wide range of available fluorescent probes and the low cost of optical imaging make fluorescent theranostics a very desirable modality for both in vitro and in vivo work, although attenuation and quenching in vivo can be problematic.
A discussion on fluorescent theranostics has been conspicuously missing from this review, in part because of a recent article published by the Huang lab that has detailed several caveats when dealing with in vivo and ex vivo quantification of fluorescent signal82. Because of high tissue autofluorescence at lower wavelengths in the visible spectrum83, red and near-infrared (NIR) probes are extensively used to quantify nanoparticle biodistribution. In order to determine if these fluorescent probes accurately measure nanoparticle accumulation, the referenced article compared the biodistribution of LCP using three different quantification methods: Texas red oligonucleotide fluorescence, 3H-oligonucleotide β emission, and 111In γ emission. For consistency, each contrast agent was loaded into the LCP core. While the biodistributions of 3H and 111In agreed, the high amounts of blood in the liver, spleen, and other organs quenched and attenuated the fluorescent signal in those organs, inaccurately causing measurements of higher relative fluorescence in the tumor. Even after perfusion to remove circulating blood from the liver, fluorescence quenching was not completely eliminated. This phenomenon also occurred when using a near-infrared dye. The quenching occurs at least in part because the secondary absorption peaks of hemoglobin and oxyhemoglobin present around the red and NIR emission spectra absorb the light emitted from the fluorescent probes, effectively lowering their signals in blood-rich organs such as the liver. Signal quenching could also occur because of greater tissue thickness of liver vs tumor, but the referenced article also compared tissue homogenates to show that equal amounts of different tissues will yield different fluorescence readings after adding equal concentrations of a fluorescent probe. These results suggest that while comparing fluorescent signals in the same type of organ can provide qualitative results, comparing relative fluorescence across different organ types, especially when comparing blood-rich organs like the liver to sparsely vascularized tumors, is an inaccurate quantitative method and should not be used unless adequate standards and controls are generated.
Conclusions
The versatile LCP platform has proven itself to be a successful theranostic agent and it is clear that there are many more future opportunities for LCP to combine simultaneous therapy and imaging in vivo. Our lab has been primarily known as a formulation lab, but we have recently focused on innovating upon LCP in order to broaden therapeutic approaches and learn more about tumor biology. LCP's success has not been based on changes in formulation as much as on innovative applications in cancer vaccine therapy, gene therapy, and theranostics that provide solutions in areas where therapeutic options are lacking. In 2015, nearly 3,000 peer-reviewed articles were published in the field of cancer nanotechnology, and many had imaging or theranostic components to aid in tumor detection and treatment. It is surely an exciting time in this field, as hundreds of minds collaborate against cancer, but as competition increases, the focus must not be on simply publishing novel papers, but on broadening our knowledge of cancer and improving treatment options for others.
Supplementary Material
Supplementary video S1.
Abbreviations
DOPA: 1,2-dioleoyl-sn-glycero-3-phosphate
DOTAP: 1,2-dioleoyl-3-trimethylammonium-propane
DSPE-PEG2000: N-(Carbonyl-methoxypolyethyleneglycol 2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine, sodium salt
Ksp: Solubility Product Constant
LCP: Lipid-Calcium-Phosphate
Lu: Lutetium
NIR: Near-Infrared
SPECT: Single Photon Emission Computed Tomography
PET: Positron Emission Tomography
MRI: Magnetic Resonance Imaging
SIRT: Selective Internal Radiation Therapy
Acknowledgements
ABS is supported by the National Science Foundation's Graduate Research Fellowship Program and by the Graduate School at the University of North Carolina at Chapel Hill. The work in this lab is supported by NIH grants DK100664, CA194067, and CA149387.
We thank the Small Animal Imaging facility at the UNC Biomedical Research Imaging Center, especially staff Jon Frank and Joseph Merrill, for providing the PET/CT imaging service.
Original Animal Studies
Animal Studies used to produce original research in Figures 4 and 5 utilized 6-8 week old female athymic nude mice. These mice were purchased from the National Cancer Institute (Bethesda, MD) and bred at the Division of Laboratory Animal Medicine at the University of North Carolina-Chapel Hill. All work performed on these animals was approved by the Institutional Animal Care and Use Committee at the University of North Carolina-Chapel Hill.
Permissions
Figure 1 and Figure 3 reproduced with permission from: http://www.journals.elsevier.com/journal-of-controlled-release/. License number: 3815560646760
Competing Interests
The authors have declared that no competing interest exists.
References
1. Xie J, Lee S, Chen X. Nanoparticle-based theranostic agents. Adv Drug Deliv Rev. 2010;62(11):1064-79
2. Duncan R, Gaspar R. Nanomedicine(s) under the microscope. Mol Pharm. 2011;8(6):2101-41
3. Graham FL, van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52(2):456-67
4. Maitra A. Calcium phosphate nanoparticles: second-generation nonviral vectors in gene therapy. Expert Rev Mol Diagn. 2005;5(6):893-905
5. Sokolova VV, Radtke I, Heumann R, Epple M. Effective transfection of cells with multi-shell calcium phosphate-DNA nanoparticles. Biomaterials. 2006;27(16):3147-53
6. Liu T, Tang A, Zhang G. et al. Calcium phosphate nanoparticles as a novel nonviral vector for efficient transfection of DNA in cancer gene therapy. Cancer Biother Radiopharm. 2005;20(2):141-9
7. Li J, Chen YC, Tseng YC, Mozumdar S, Huang L. Biodegradable calcium phosphate nanoparticle with lipid coating for systemic siRNA delivery. J Control Release. 2010;142(3):416-21
8. Li J, Yang Y, Huang L. Calcium phosphate nanoparticles with an asymmetric lipid bilayer coating for siRNA delivery to the tumor. J Control Release. 2012;158(1):108-14
9. Giger EV, Puigmarti-Luis J, Schlatter R, Castagner B, Dittrich PS, Leroux JC. Gene delivery with bisphosphonate-stabilized calcium phosphate nanoparticles. J Control Release. 2011;150(1):87-93
10. Lee MS, Lee JE, Byun E. et al. Target-specific delivery of siRNA by stabilized calcium phosphate nanoparticles using dopa-hyaluronic acid conjugate. Journal of Controlled Release. 2014;192:122-30
11. Chen WH, Lecaros RL, Tseng YC, Huang L, Hsu YC. Nanoparticle delivery of HIF1alpha siRNA combined with photodynamic therapy as a potential treatment strategy for head-and-neck cancer. Cancer Lett. 2015;359(1):65-74
12. Tang J, Li L, Howard CB, Mahler SM, Huang L, Xu ZP. Preparation of optimized lipid-coated calcium phosphate nanoparticles for enhanced in vitro gene delivery to breast cancer cells. Journal of Materials Chemistry B. 2015;3(33):6805-12
13. Jung H, Kim SA, Yang YG, Yoo H, Lim SJ, Mok H. Long chain microRNA conjugates in calcium phosphate nanoparticles for efficient formulation and delivery. Arch Pharm Res. 2015;38(5):705-15
14. Au KM, Satterlee A, Min Y. et al. Folate-targeted pH-responsive calcium zoledronate nanoscale metal-organic frameworks: Turning a bone antiresorptive agent into an anticancer therapeutic. Biomaterials. 2016;82:178-93
15. Zhang J, Miao L, Guo S. et al. Synergistic anti-tumor effects of combined gemcitabine and cisplatin nanoparticles in a stroma-rich bladder carcinoma model. J Control Release. 2014;182:90-6
16. Zhang Y, Kim WY, Huang L. Systemic delivery of gemcitabine triphosphate via LCP nanoparticles for NSCLC and pancreatic cancer therapy. Biomaterials. 2013;34(13):3447-58
17. Yao J, Zhang Y, Ramishetti S, Wang Y, Huang L. Turning an antiviral into an anticancer drug: nanoparticle delivery of acyclovir monophosphate. J Control Release. 2013;170(3):414-20
18. Xu Z, Wang Y, Zhang L, Huang L. Nanoparticle-delivered transforming growth factor-beta siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano. 2014;8(4):3636-45
19. Zhang Y, Schwerbrock NM, Rogers AB, Kim WY, Huang L. Codelivery of VEGF siRNA and gemcitabine monophosphate in a single nanoparticle formulation for effective treatment of NSCLC. Mol Ther. 2013;21(8):1559-69
20. Zhang Y, Peng L, Mumper RJ, Huang L. Combinational delivery of c-myc siRNA and nucleoside analogs in a single, synthetic nanocarrier for targeted cancer therapy. Biomaterials. 2013;34(33):8459-68
21. Yang Y, Li J, Liu F, Huang L. Systemic delivery of siRNA via LCP nanoparticle efficiently inhibits lung metastasis. Mol Ther. 2012;20(3):609-15
22. Yang Y, Hu Y, Wang Y, Li J, Liu F, Huang L. Nanoparticle delivery of pooled siRNA for effective treatment of non-small cell lung cancer. Mol Pharm. 2012;9(8):2280-9
23. Hu Y, Haynes MT, Wang Y, Liu F, Huang L. A highly efficient synthetic vector: nonhydrodynamic delivery of DNA to hepatocyte nuclei in vivo. ACS Nano. 2013;7(6):5376-84
24. Xu Z, Ramishetti S, Tseng YC, Guo S, Wang Y, Huang L. Multifunctional nanoparticles co-delivering Trp2 peptide and CpG adjuvant induce potent cytotoxic T-lymphocyte response against melanoma and its lung metastasis. J Control Release. 2013;172(1):259-65
25. Satterlee AB, Yuan H, Huang L. A radio-theranostic nanoparticle with high specific drug loading for cancer therapy and imaging. J Control Release. 2015;217:170-82
26. Tseng YC, Xu Z, Guley K, Yuan H, Huang L. Lipid-calcium phosphate nanoparticles for delivery to the lymphatic system and SPECT/CT imaging of lymph node metastases. Biomaterials. 2014;35(16):4688-98
27. Guo ST, Miao L, Wang YH, Huang L. Unmodified drug used as a material to construct nanoparticles: delivery of cisplatin for enhanced anti-cancer therapy. Journal of Controlled Release. 2014;174:137-42
28. Guo S, Wang Y, Miao L. et al. Lipid-coated Cisplatin nanoparticles induce neighboring effect and exhibit enhanced anticancer efficacy. ACS Nano. 2013;7(11):9896-904
29. Liu Y, Hu Y, Huang L. Influence of polyethylene glycol density and surface lipid on pharmacokinetics and biodistribution of lipid-calcium-phosphate nanoparticles. Biomaterials. 2014;35(9):3027-34
30. Tseng YC, Yang A, Huang L. How Does the Cell Overcome LCP Nanoparticle-Induced Calcium Toxicity? Mol Pharm. 2013;10(11):4391-95
31. Khalil MM, Tremoleda JL, Bayomy TB, Gsell W. Molecular SPECT Imaging: An Overview. Int J Mol Imaging. 2011;2011:796025
32. Lu Z-R. Modern Pharmaceutics, Ch 6: In Vivo Imaging of Dosage Forms. 5 ed. New York: Informa Healthcare. 2009
33. Van Audenhaege K, Van Holen R, Vandenberghe S, Vanhove C, Metzler SD, Moore SC. Review of SPECT collimator selection, optimization, and fabrication for clinical and preclinical imaging. Med Phys. 2015;42(8):4796-813
34. Karp JS, Surti S, Daube-Witherspoon ME, Muehllehner G. Benefit of time-of-flight in PET: experimental and clinical results. J Nucl Med. 2008;49(3):462-70
35. Banerjee SR, Pomper MG. Clinical applications of Gallium-68. Appl Radiat Isot. 2013;76:2-13
36. McCarthy DW, Shefer RE, Klinkowstein RE. et al. Efficient production of high specific activity 64Cu using a biomedical cyclotron. Nucl Med Biol. 1997;24(1):35-43
37. Bednarczyk EM. Radiopharmaceuticals in Nuclear Pharmacy and Nuclear Medicine, 3rd Edition. Ann Pharmacother. 2012
38. Khalil M. Basic Sciences of Nuclear Medicine. Springer. 2011
39. Wadas TJ, Wong EH, Weisman GR, Anderson CJ. Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for PET and SPECT imaging of disease. Chem Rev. 2010;110(5):2858-902
40. Bartholoma MD. Recent developments in the design of bifunctional chelators for metal-based radiopharmaceuticals used in Positron Emission Tomography. Inorganica Chimica Acta. 2012;389:36-51
41. Wadas TJ, Wong EH, Weisman GR, Anderson CJ. Copper chelation chemistry and its role in copper radiopharmaceuticals. Curr Pharm Des. 2007;13(1):3-16
42. Pandya DN, Bhatt N, Dale AV. et al. New bifunctional chelator for 64Cu-immuno-positron emission tomography. Bioconjug Chem. 2013;24(8):1356-66
43. Dale AV, An GI, Pandya DN. et al. Synthesis and Evaluation of New Generation Cross-Bridged Bifunctional Chelator for (64)Cu Radiotracers. Inorg Chem. 2015;54(17):8177-86
44. Woodin KS, Heroux KJ, Boswell CA. et al. Kinetic inertness and electrochemical behavior of copper(II) tetraazamacrocyclic complexes: Possible implications for in vivo stability. European Journal of Inorganic Chemistry. 2005;23:4829-33
45. Wilczek B, Svensson L, Danielsson R, Celebiouglu F, Larsson SA, Jacobsson H. 99mTc-exametazime as a breast tumor-seeking agent: comparison with 99mTc-sestamibi. J Nucl Med. 2004;45(12):2040-4
46. Belhocine TZ, Blankenberg FG, Kartachova MS. et al. (99m)Tc-Annexin A5 quantification of apoptotic tumor response: a systematic review and meta-analysis of clinical imaging trials. Eur J Nucl Med Mol Imaging. 2015;42(13):2083-97
47. Rizvi T, Deng C, Rehm PK. Indium-111 Capromab Pendetide (ProstaScint((R))) Demonstrates Renal Cell Carcinoma and Aortocaval Nodal Metastases from Prostate Adenocarcinoma. World J Nucl Med. 2015;14(3):209-11
48. Seymour LW, Ferry DR, Kerr DJ. et al. Phase II studies of polymer-doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal cancer. Int J Oncol. 2009;34(6):1629-36
49. Pimm MV, Perkins AC, Strohalm J, Ulbrich K, Duncan R. Gamma scintigraphy of a 123I-labelled N-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin conjugate containing galactosamine following intravenous administration to nude mice bearing hepatic human colon carcinoma. J Drug Target. 1996;3(5):385-90
50. Grillo-Lopez AJ. Zevalin: the first radioimmunotherapy approved for the treatment of lymphoma. Expert Rev Anticancer Ther. 2002;2(5):485-93
51. Rezvani AR, Maloney DG. Rituximab resistance. Best Pract Res Clin Haematol. 2011;24(2):203-16
52. van der Kolk LE, Grillo-Lopez AJ, Baars JW, Hack CE, van Oers MH. Complement activation plays a key role in the side-effects of rituximab treatment. Br J Haematol. 2001;115(4):807-11
53. Kulkarni HS, Kasi PM. Rituximab and cytokine release syndrome. Case Rep Oncol. 2012;5(1):134-41
54. Kwekkeboom DJ, de Herder WW, Kam BL. et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0,Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26(13):2124-30
55. Garske U, Sandstrom M, Johansson S. et al. Lessons on Tumour Response: Imaging during Therapy with (177)Lu-DOTA-octreotate. A Case Report on a Patient with a Large Volume of Poorly Differentiated Neuroendocrine Carcinoma. Theranostics. 2012;2(5):459-71
56. Delpassand ES, Samarghandi A, Zamanian S. et al. Peptide receptor radionuclide therapy with 177Lu-DOTATATE for patients with somatostatin receptor-expressing neuroendocrine tumors: the first US phase 2 experience. Pancreas. 2014;43(4):518-25
57. Sandstrom M, Garske-Roman U, Granberg D. et al. Individualized dosimetry of kidney and bone marrow in patients undergoing 177Lu-DOTA-octreotate treatment. J Nucl Med. 2013;54(1):33-41
58. Di Pasqua AJ, Yuan H, Chung Y. et al. Neutron-activatable holmium-containing mesoporous silica nanoparticles as a potential radionuclide therapeutic agent for ovarian cancer. J Nucl Med. 2013;54(1):111-6
59. Sadauskas E, Wallin H, Stoltenberg M. et al. Kupffer cells are central in the removal of nanoparticles from the organism. Part Fibre Toxicol. 2007;4:10
60. Challapalli A, Jones E, Harvey C, Hellawell GO, Mangar SA. High dose rate prostate brachytherapy: an overview of the rationale, experience and emerging applications in the treatment of prostate cancer. Br J Radiol. 2012;85(Spec No 1):S18-27
61. Yaparpalvi R, Rossinow J, Cavanagh WA. et al. Is Cs-131 or I-125 or Pd-103 the "Ideal" isotope for prostate boost brachytherapy? A dosimetric view point. International Journal of Radiation Oncology Biology Physics. 2007;69(3):S677-S78
62. Reed DR, Wallner KE, Merrick GS. et al. A prospective randomized comparison of stranded vs. loose 125I seeds for prostate brachytherapy. Brachytherapy. 2007;6(2):129-34
63. Yu Y, Anderson LL, Li Z. et al. Permanent prostate seed implant brachytherapy: report of the American Association of Physicists in Medicine Task Group No. 64. Med Phys. 1999;26(10):2054-76
64. Khan MK, Minc LD, Nigavekar SS. et al. Fabrication of {Au-198(0)} radioactive composite nanodevices and their use for nanobrachytherapy. Nanomedicine-Nanotechnology Biology and Medicine. 2008;4(1):57-69
65. Gulec SA, Pennington K, Wheeler J. et al. Yttrium-90 microsphere-selective internal radiation therapy with chemotherapy (chemo-SIRT) for colorectal cancer liver metastases: an in vivo double-arm-controlled phase II trial. Am J Clin Oncol. 2013;36(5):455-60
66. Gibbs P, Gebski V, Van Buskirk M, Thurston K, Cade DN, Van Hazel GA. Selective Internal Radiation Therapy (SIRT) with yttrium-90 resin microspheres plus standard systemic chemotherapy regimen of FOLFOX versus FOLFOX alone as first-line treatment of non-resectable liver metastases from colorectal cancer: the SIRFLOX study. BMC Cancer. 2014;14:897
67. Ren Y, Mu Y, Song Y. et al. A new peptide ligand for colon cancer targeted delivery of micelles. Drug Deliv. 2015:1-10
68. Zhang J, Gao J, Tan X, Wang M, Qin R. Effects of down-regulation of integrin-beta1 expression on migration and hepatic metastasis of human colon carcinoma. J Huazhong Univ Sci Technolog Med Sci. 2010;30(4):464-9
69. Filmus J, Capurro M. Glypican-3: a marker and a therapeutic target in hepatocellular carcinoma. FEBS J. 2013;280(10):2471-6
70. Lee YL, Ahn BC, Lee Y, Lee SW, Cho JY, Lee J. Targeting of hepatocellular carcinoma with glypican-3-targeting peptide ligand. J Pept Sci. 2011;17(11):763-9
71. Roberts EA, Sarkar B. Liver as a key organ in the supply, storage, and excretion of copper. Am J Clin Nutr. 2008;88(3):851S-4S
72. Ferrari C, Asabella AN, Villano C. et al. Copper-64 Dichloride as Theranostic Agent for Glioblastoma Multiforme: A Preclinical Study. Biomed Research International. 2015
73. Hellman NE, Gitlin JD. Ceruloplasmin metabolism and function. Annu Rev Nutr. 2002;22:439-58
74. Pandya DN, Kim JY, Park JC. et al. Revival of TE2A; a better chelate for Cu(II) ions than TETA? Chem Commun (Camb). 2010;46(20):3517-9
75. Garrison JC, Rold TL, Sieckman GL. et al. In vivo evaluation and small-animal PET/CT of a prostate cancer mouse model using Cu-64 bombesin analogs: Side-by-side comparison of the CB-TE2A and DOTA chelation systems. Journal of Nuclear Medicine. 2007;48(8):1327-37
76. Liu S, Li D, Huang CW. et al. The efficient synthesis and biological evaluation of novel bi-functionalized sarcophagine for (64)cu radiopharmaceuticals. Theranostics. 2012;2(6):589-96
77. Gourni E, Del Pozzo L, Kheirallah E. et al. Copper-64 Labeled Macrobicyclic Sarcophagine Coupled to a GRP Receptor Antagonist Shows Great Promise for PET Imaging of Prostate Cancer. Mol Pharm. 2015;12(8):2781-90
78. Edelman RR, Warach S. Magnetic resonance imaging (1). N Engl J Med. 1993;328(10):708-16
79. Fries PaB, Elie. The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging. West Sussex, United Kingdom: Wiley. 2013
80. Carr DH, Brown J, Bydder GM. et al. Intravenous Chelated Gadolinium as a Contrast Agent in Nmr Imaging of Cerebral-Tumors. Lancet. 1984;1(8375):484-86
81. Jenjob R, Kun N, Ghee JY. et al. Enhanced conjugation stability and blood circulation time of macromolecular gadolinium-DTPA contrast agent. Mater Sci Eng C Mater Biol Appl. 2016;61:659-64
82. Liu Y, Tseng YC, Huang L. Biodistribution studies of nanoparticles using fluorescence imaging: a qualitative or quantitative method? Pharm Res. 2012;29(12):3273-7
83. Masilamani V. et al. Cancer diagnosis by autofluorescence of blood components. Journal of Luminescence. 2004;109:143-54
Author contact
![]() Corresponding author: Tel.: 919 843 0736 E-mail address: leafhedu.
Corresponding author: Tel.: 919 843 0736 E-mail address: leafhedu.