Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results and discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(7):1012-1022. doi:10.7150/thno.15095 This issue Cite
Research Paper
A Facile Approach to Functionalize Cell Membrane-Coated Nanoparticles
Hao Zhou1, Zhiyuan Fan1, Pelin K. Lemons1, Hao Cheng1,2 ![]()
1. Department of Materials Science and Engineering, Drexel University, Philadelphia, Pennsylvania, USA;
2. School of Biomedical Engineering, Science and Health Systems, Drexel University, Philadelphia, Pennsylvania, USA.
Received 2016-1-26; Accepted 2016-3-9; Published 2016-4-28
Abstract

Convenient strategies to provide cell membrane-coated nanoparticles (CM-NPs) with multi-functionalities beyond the natural function of cell membranes would dramatically expand the application of this emerging class of nanomaterials. We have developed a facile approach to functionalize CM-NPs by chemically modifying live cell membranes prior to CM-NP fabrication using a bifunctional linker, succinimidyl-[(N-maleimidopropionamido)-polyethyleneglycol] ester (NHS-PEG-Maleimide). This method is particularly suitable to conjugate large bioactive molecules such as proteins on cell membranes as it establishes a strong anchorage and enable the control of linker length, a critical parameter for maximizing the function of anchored proteins. As a proof of concept, we show the conjugation of human recombinant hyaluronidase, PH20 (rHuPH20) on red blood cell (RBC) membranes and demonstrate that long linker (MW: 3400) is superior to short linker (MW: 425) for maintaining enzyme activity, while minimizing the changes to cell membranes. When the modified membranes were fabricated into RBC membrane-coated nanoparticles (RBCM-NPs), the conjugated rHuPH20 can assist NP diffusion more efficiently than free rHuPH20 in matrix-mimicking gels and the pericellular hyaluronic acid matrix of PC3 prostate cancer cells. After quenching the unreacted chemical groups with polyethylene glycol, we demonstrated that the rHuPH20 modification does not reduce the ultra-long blood circulation time of RBCM-NPs. Therefore, this surface engineering approach provides a platform to functionlize CM-NPs without sacrificing the natural function of cell membranes.
Keywords: Tumor penetration, extracellular matrix, biomimetics, lymph nodes, drug delivery.
Introduction
Cell membrane-coated nanoparticles (CM-NPs) are drawing increasing attention because their surfaces preserve the natural structures of cell membranes, making them a unique class of biomimetic materials that combine both natural and synthetic components. Due to the easy availability of red blood cells (RBCs), RBC membrane-coated nanoparticles (RBCM-NPs) have been extensively studied [1-8]. RBC membrane coating resists non-specific protein adsorption and provides nanoparticles (NPs) with membrane protein CD47, a “self” signal that prevents NP clearance by phagocytic cells [1, 9]. Because of these merits, RBCM-NPs have an ultra-long blood circulation time [1]. RBCM-NPs have also been applied to absorb membrane-damaging toxins for detoxification and deliver antibiotics [2, 4]. To harness the natural binding and targeting ability of membrane proteins, NPs coated with membranes from leukocytes and platelets were used for active targeting of inflammatory or injured tissues [10-12]. In addition, CM-NPs have been utilized as an emerging platform of vaccines [3, 5, 13-15]. Cancer cell and bacteria membrane-coated NPs present entire surface antigens to host dendritic cells, which is ideal to generate a robust immune response when coupled with adjuvants and immunomodulators [13-15].
The emerging demand for multitasking applications in complex biological systems requires multi-functionality of CM-NPs, involving active targeting, signal stimulation, immunomodulation etc. However, cells may not naturally exhibit all the desired functional molecules on their membranes. Genetic engineering is a common strategy to functionalize cells with new membrane proteins [16, 17]. However, the most commonly used cells in CM-NP fabrication, such as RBCs and platelets lack nuclei and cannot be directly modified using genetic engineering. Therefore, non-genetic approaches that allow modification of CM-NPs or live cell membranes prior to CM-NP fabrication are desired to functionalize CM-NPs for expanding their applications. Non-genetic approaches also enable the use of bioactive molecules that cannot be produced by cells [18].
Compared to the direct modification of CM-PNs, modifying membranes of live cells before membrane isolation has its advantages. 1) It is convenient to separate cells and biomolecules that are not anchored on cell membranes after the step of biomolecule conjugation, while the separation of CM-NPs with free biomolecules requires time-consuming processes and may disrupt the membrane coating on NPs. 2) A fraction of CM-NPs exhibit an inside-out membrane orientation [6, 19]. Modification of live cells ensures that newly anchored biomolecules are on the same side as the extracellular domain of membrane proteins, important for achieving multifunctionality from both natural membrane proteins and conjugated biomolecules. 3) The anchored molecules may even increase the right-side-out membrane orientation on NPs if the molecule-NP interaction increases energy. Several non-genetic engineering techniques have been reported to modify cell surfaces [18]. Physical attachment largely relies on the insertion of the hydrophobic alkyl chain or lipid portion of biomolecules into lipid bilayers of membranes [20, 21]. This method has been applied to render CM-NPs with targeting ability and present tumor antigens [15, 22]. Although convenient, this physical interaction strategy cannot secure a strong connection when forces are exerted on the attached molecules. Free proteins in biological systems also reduce the energy required to pull out alkyl chain or lipid anchors from cell membranes due to hydrophobic interactions between free proteins and hydrophobic anchors. Chemical modifications, which mostly conjugate molecules onto primary amines of cell membrane proteins, provide more stable anchorage as it takes more energy to uproot a transmembrane protein than a lipid or alkyl chain from cell membranes [23]. Biotin-avidin binding is widely-used to anchor molecules onto biotinylated cell membranes. However the method is not optimal for clinical applications because of the immunogenicity of avidin and streptavidin [24]. The use of 1-ethyl-3-(3'-dimethyl aminopropyl)-carbodiimide (EDC) and N-hydroxysuccinimide (NHS) or N-hydroxysulfosuccinimide (sulfo-NHS) is another popular strategy, linking molecules and cell membranes via amide bonds [25]. We recently developed a cell membrane modification method using succinimidyl-[(N-maleimidopropionamido)-diethyleneglycol] ester (NHS-(PEG)2-Maleimide), a cell membrane impermeable linker molecule, which reacts with amine groups of cell membrane proteins and conjugate thiolated molecules via thiol-maleimide reaction [6, 26]. This conjugation strategy is superior to the commonly used EDC/NHS approach as it avoids protein aggregation due to random carboxyl-amine reaction and EDC-induced cell apoptosis [25, 27], which can cause the exposure of phosphatidylserine to the outer leaflet of cell membranes [28], leading to fast CM-NPs clearance in the blood. This strategy also allows the control of linker molecule length, critical for maintaining enzyme activity.
Herein, we report a facile approach of direct modification of live cell membranes with proteins to functionalize CM-NPs and demonstrate that the anchored proteins maintain their function and do not diminish the original advantages of CM-NPs. RBCM-NPs are selected for this study because they are the most representative CM-NPs. As a proof of concept, we anchored recombinant human hyaluronidase, PH20 (rHuPH20) on RBCM-NPs as anchored exogenous enzymes may significantly expand CM-NP functions. rHuPH20 degrades hyaluronic acid (HA), which is a major component of extracellular matrix (ECM) [29]. Moderate to high levels of HA expression has been observed in almost 100% pancreatic ductal adenocarcinomas[30], about 80% prostate cancer[31] and more than 50% breast cancer [32]. The elevated tumor HA causes high interstitial fluid pressure and reduces therapeutic diffusion [33]. Anchoring rHuPH20 on CM-NPs can potentially improve the NP diffusion in tumors, increasing the access of CM-NPs to tumor cells. In addition, it may be used for imaging atherosclerotic plaques and treating the cardiovascular disease as HA is also abundant in atherosclerotic plaques and actively involved in the development of atherosclerosis [34, 35]. Another potential application is to improve the interstitial transport of CM-NPs for entering lymphatic capillaries and their draining lymph nodes through intradermal or subcutaneous injection to stimulate or induce tolerogenic immune responses.
Materials and methods
Materials
Recombinant human hyaluronidase, PH20 (rHuPH20) in pH=6.5, 10 mM sodium phosphate, 150 mM NaCl buffer was obtained from Halozyme, Inc.. Poly(D,L-lactide-co-glycolide) with PLA:PGA ratio of 50:50 (Mw = 47.5 kDa, inherent viscosity = 0.66 dL/g) was purchased from DURECT Corporation. PC3 prostate cancer cell line was purchased from ATCC Inc. 1,2-dimyristoyl-sn-glycero-3-phospho-ethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (Rhod-PE) was acquired from Avanti Polar Lipids Inc. Long linker, succinimidyl-[(N-maleimidopropionamido)-polyethyleneglycol] ester (NHS-PEG-Maleimide, Mw = 3400) was purchased from NANOCS, whereas short linker, succinimidyl-[(N-maleimidopropionamido)-diethyleneglycol] ester (NHS-PEG2-Maleimide, Mw = 425), o-Phenylene-diamine (OPD), 1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindodicarbocyanine Perchlorate (DiD), horseradish peroxidase conjugated streptavidin (HRP-streptavidin), 2-Iminothiolane•HCl (Traut's Reagent), N-hydroxysulfosuccinimide (Sulfo-NHS), 1-ethyl-3-(3-dimethylamin-opropyl) carbidodiimide (EDC) and (biotinyl)hydrazide (Biotin hydrazide) were purchased from ThermoFisher Scientific. Bovine hyaluronidase was purchased form Sigma-Aldrich. Additional salts, solvents and buffers were purchased from Fisher Scientific.
Conjugation of rHuPH20 on RBCs
Traut's reaction was employed to thiolate rHuPH20. Traut's reagent was first dissolved in PBS at 2 mg/mL and added into 1 mg/mL of rHuPH20 solution at 5:1 molar ratio and reacted for 1 h at room temperature under stirring. After the reaction, the thiolated rHuPH20 was purified by filtering through a desalting column (Thermo Scientific.). Fresh RBCs were collected from mice through a submandibular blood collection method and kept in a blood collecting tube containing 0.04 mL EDTAK3 7.5% solution. The cells were washed three times in PBS via centrifuge at 600×g for 5 min. After washing, RBCs from 0.5 mL of blood were resuspended in 50 mL PBS. Long or short linker molecules were dissolved in DMSO before adding to cell solution at a final concentration of 100 µM. The reaction was kept at room temperature for 20 min and 1 h for short and long linkers, respectively. The maleimide-modified cells were washed three times with PBS and resuspended in 10 mL PBS and reacted with thiolated rHuPH20 at a concentration of 20 µg/mL. After gently shaking cells at room temperature for 20 min and 1 h for short and long linkers, respectively, RBCs were washed three times with PBS to remove all unconjugated rHuPH20. To quench the unreacted thiol groups on rHuPH20, modified cells were first reacted with PEG2K-maleimide (PEG2K-MAL) (10-fold) for 2 hours at room temperature, and then were incubated with PEG2K-thiol (PEG2K-SH) (10-fold) for another 2 hours at room temperature to quench unreacted maleimide groups. The resulting RBCs were spun down and washed three times in PBS.
To image the hyaluronidase on RBC membranes, bovine hyaluronidase was first labeled with NHS-Fluorescein (8-fold) at a concentration of 2 mg/mL by incubation in PBS for 2 h at room temperature. Then the FITC-hyaluronidase was purified through the desalting column followed with a Traut's reaction. The thiolated FITC-hyaluronidase was added to cells modified with long linkers and short linkers respectively at a final concentration of 1 mg/mL. After reaction, the RBCs were washed three times with PBS before imaging under a confocal microscope.
Fabrication of RBCM-NPs with and without PH20 modification
Poly(lactic-co-glycolic acid) nanoparticles (PLGA-NPs) were prepared through a modified nanoprecipitation method [36]. PLGA was first dissolved in acetone at various concentrations before being added swiftly to 4 times of volumes of DI H2O. The mixture was placed under vaccum overnight to remove the acetone. PLGA-NPs encapsulating DiD (DiD-PLGA-NPs) were prepared by dissolving 0.05% (w%) of DiD in acetone together with PLGA before being added to water. The resulting PLGA-NPs were then subsequently filtered through 450 nm and 200 nm syringe filters to remove any aggregates.
To rupture RBCs with osmotic pressure, 950 μL of 0.2 mM EDTA in H2O was used to resuspend the washed RBCs from 0.1 mL of blood followed by adding 50 μL of 20X PBS. The membranes were centrifuged at 17000×g for 7 min. The same process was repeated twice before a pink pellet was collected and resuspended in 2 mL DI H2O. The membranes were first sonicated in a bath sonicator for 10 mins, and then were extruded seven times back and forth through 100 nm extrusion membranes for fabricating small NPs or through 200 nm extrusion membranes for fabricating large NPs using an Avanti mini extruder. To coat PLGA-NPs with RBC membranes, 0.5 mL of RBC vesicles from 75 μL of blood were mixed with 0.5 mL of 2 mg/mL of 105 nm and 1 mg/mL of 65 nm PLGA-NPs solutions respectively. At these compositions, the ratios of cell membrane and PLGA-NP surface area are approximately 1:1. The mixture was then extruded again for 11 times through 400 nm extrusion membrane before collecting the samples. To fabricate rHuPH20-modified RBCM-NPs (PH20-RBCM-NPs), the same ratio of rHuPH20-modified RBC membranes and PLGA-NPs were used according to the above-described method of RBCM-NP fabrication. PLGA-NPs and DiD-PLGA-NPs were used in fabrication for rHuPH20 activity assay, and gel diffusion and cell uptake respectively.
Characterization of NPs
Sizes and zeta potentials of the prepared NPs were detected by dynamic light scattering (DLS, Zetasizer Nano ZS90). For transmission electron microscopy (TEM), 10 μL of 0.2 mg/mL of sample was added onto the carbon coated grid after being cleaned by plasma. The grid was then rinsed with DI water. 5 μL of 2% uranyl acetate water solution was dropped on the grid and removed instantly via filter paper. The staining step was repeated 3 times prior to sample imaging via TEM (JEOL JEM2100) at 200KV.
Enzyme activity assay
The activity of rHuPH20 on cells and on NPs were detected with a modified microtiter-based assay [37]. Briefly, the plate was coated with biotin-labeled HA via an EDC-mediated reaction between amine groups on well substrates and carboxyl groups on HA. The samples were diluted five times in PBS before assaying at 100 μL/well in triplicate. After 50 min of incubation at 37 oC, the reaction was terminated and the non-degraded HA were quantified with a HRP-OPD color reaction. Absorbance for each well was read at 492 nm (TECAN, Infinite M200). The rHuPH20 activity of each sample was determined by comparing with the standard curve that was generated by diluting 1 mg/mL of rHuPH20 solution with original activity of 116000 U/mg to a series of solutions with rHuPH20 activity ranging from 1 U/mL to 1000 U/mL.
Diffusion of NPs in ECM-mimicking gels
To prepare the ECM-mimicking gels composed of collagen and HA, the following components were mixed in order on ice: 48 μL of 10X PBS, 12.8 μL of 1N NaOH, 24.3 μL of DI H2O, 160 μL at 5 mg/mL of HA (MW=1.5 ~ 2.0 × 106) in 2X PBS and 555 μL at 9.37 mg/mL of rat tail collagen type I (Corning, Bedford, MA). The final concentration of collagen was 6.5 mg/mL and HA was 1 mg/mL. After being vortexed and vacuumed on ice to remove bubbles, the mixture was added at 60 μL into each capillary tube (0.4×4.00 mm ID, Vitrocom, Mountain Lakes, NJ), the tubes were then incubated overnight at 37 °C. PLGA-NP core were labeled with DiD, while the RBC membranes were labeled with Rhodamine by inserting Rhod-PE into the lipid bilayers after isolation of RBC membranes. In the diffusion tests, 10 μL of equalized 1 mg/mL of RBCM-NPs, RBCM-NPs + 50 U/mL of free rHuPH20, and 1 mg/mL of PH20-RBCM-NP solution were slowly added onto the top of ECM-mimic gels. The tubes were sealed and left at 37 oC for 1 hour then imaged using a confocal laser scanning microscope. Image analysis was performed via ImageJ. Diffusion coefficient D in the ECM-mimicking gel was obtained by fitting diffusion profiles of the NPs to the following one-dimensional diffusion model with relative intensity (C) and the diffusion distance (x) via fminsearch in MATLAB:
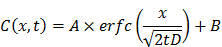
where erfc is the complementary error function and A, B are the constant for the function.
Cell culture
PC3 cells were maintained in F-12K medium (ATCC) containing 10% fetal bovine serum in 5% CO2 incubator at 37 oC and sub-cultured every 2-3 days when reached 90% confluence.
RBC exclusion assay
RBCs were fixed in 2% formaldehyde in PBS overnight. PC3 cells were cultured in a 12-well plate at 5000 cells/well two days before the study. The control PC3 cells were pretreated in medium with 1000 U/mL of free rHuPH20. After 2 h incubation at 37 oC, all the wells were washed with PBS three times. 0.5 mL of fixed RBC solution (5×108 RBCs/mL in medium) was added into each well after the removal of PBS. After 20 min, phase contrast images of the cells were taken using Optic microscopy (Lexia). Areas of exclusion were quantified using ImageJ.
NP internalization by PC3 cells
The amount of NPs were equalized using the fluorescence intensity of encapsulated DiD. PC3 cells were cultured at 5000 cells/well on round glass coverslips in a 12-well plate for two days to reach 70% confluence before treatment. NP solutions were diluted five times in cell culture medium before being added to each well at 1 mL/well. The cells were treated at 37 oC for different time points for the kinetic study and 2 h for the rest of studies. The cells were then fixed and observed under a confocal microscope (Olympus IX81, 60 ×, C.A. = 80 μm) with 405 nm and 635 nm lasers for DAPI and DiD respectively. The HV and gain were set high enough to just avoid saturation and % off was set slightly higher than the threshold to avoid background signals. Ten images were taken for each sample at random locations and focused at the focal plane where the largest nuclei showed. The fluorescence intensity of DiD was quantified via ImageJ and averaged by the number of cells.
NP blood circulation
DiD-labeled RBCM-NPs and PH20-RBCM-NPs were fabricated following the above mentioned method except replacing DI H2O with PBS in the steps of PLGA-NP formation and membrane coating. Each female BALB/c mouse was systemically administered with 100 µL of 8 mg/mL NP solution via tail vein injection. A small volume of 15 µL of blood was collected at 2 min, 15 min, 0.5 h, 1 h, 2 h, 4 h, 8 h, 24 h, 48 h, and 72 h post i.v. injection. The blood was diluted in 200 μL of PBS containing 16 U/mL heparin as an anti-coagulant. Blood cells were removed by spinning at 300 g for 5 min, and 180 µL of the supernatant was used for fluorescence intensity measurement with TECAN. All animal procedures were conducted according to the protocols of the Committee on Animal Care of Drexel in compliance with NIH guidelines.
Results and discussion
Fabrication and characterization of PH20-RBCM-NPs
RBCs were modified with rHuPH20 using our recently developed membrane engineering technique, which does not affect the viability, proliferation or multipotency of stem cells [26]. The RBCs were first treated with NHS-PEG-maleimide, a cell membrane impermeable linker molecule, and then coupled with thiolated rHuPH20 through thiol-maleimide reaction. rHuPH20 was thiolated using Traut's reagent (Figure 1A). The sequential conjugations of NHS-PEG-maleimide and thiolated rHuPH20 to cell membranes are necessary to avoid the reaction between NHS groups in the linker molecules and amine groups in the rHuPH20. When bovine hyaluronidase was labeled with FITC, the surface-modified RBC showed concentric rings in confocal microscopy images (Figure 1B,C) because of the biconcave shape of RBCs, indicating a successful conjugation of enzymes on RBC membranes. To acquire sufficient fluorescence signals for imaging, a high concentration of hyaluronidase is needed. Therefore, inexpensive bovine hyaluronidase was employed for this study, while rHuPH20 was used for the rest of studies involving hyaluronidase. No fluorescence signal was observed when hyaluronidase had not been thiolated or linker molecules were not added during the membrane modifications (data not shown), demonstrating that enzymes were conjugated on cells through the above discussed reactions.
Preparation and characterization of rHuPH20-modified red blood cell membrane-coated nanoparticles (PH20-RBCM-NPs). A) Schematic illustration of the fabrication of PH20-RBCM-NPs and the diffusion of PH20-RBCM-NPs in extracellular matrix. The efficient interstitial diffusion can be achieved through the extracellular matrix (ECM) degradation by the ECM degrading enzymes on RBCM-NP surfaces. B,C) Confocal microscopy images of RBCs conjugated with FITC-labeled bovine hyaluronidase (green). (B): Hyaluronidase conjugated on RBCs with short linkers, (C): Hyaluronidase conjugated on RBCs with long linkers. (Scale bar = 20 μm; Inset scale bar = 5 μm). D) Representative transmission electron microscopy (TEM) image of PH20-RBCM-NPs. Cell membrane coating is marked by arrows. (Scale bar: 50 nm) E) Quantification of the enzymatic activity of PH20-modified cells and PH20-RBCM-NPs with long and short linkers. Values indicate mean ± s.d. (n = 3).
By adjusting the PLGA concentration in acetone, PLGA-NPs ranging from 50 nm to 120 nm can be generated (Figure S1A). The particle polydispersity indexes were all lower than 0.2. PLGA-NPs with sizes 65 nm and 105 nm were prepared using 7.5 mg/mL and 15 mg/mL of PLGA acetone solution respectively. Coating of RBC membranes on PLGA-NP surfaces is demonstrated by TEM images (Figure 1D) as well as the changes in particle size and zeta potential (Figure S1B,C). The coated RBC membranes on PLGA-NPs have a thickness around 8 nm as shown in between two arrows in the TEM image and around 10 nm according to the zeta average size, consistent with previous studies [1]. Incorporating 0.05% DiD in PLGA-NPs increased particle size by around 5 nm for all the NPs. Modification of RBC membranes with rHuPH20 did not change the sizes and zeta potential of the vesicles and NPs. Therefore the small RBCM-NPs and PH20-RBCM-NPs are 85 nm, and their large counterparts are both 125 nm for the enzyme activity test, whereas the small and large NPs are 90 nm and 130 nm, respectively, for the rest of studies that require fluorescence labeling.
Effect of linker length on enzyme activity
Two linkers with different numbers of repeating PEG units were used in the conjugation of FITC-labeled hyaluronidase onto RBC membranes. Though the reactions took place at the same initial molar concentration, the resulting fluorescence intensities of hyaluronidase modified-RBCs (Figure 1B,C) showed that less amount of hyaluronidase were conjugated on cells via the long linkers compared to the short linker-mediated conjugation. It is likely because that the efficiencies of end-group reactions are lower for long chains compared to short linker molecules, resulting in relatively low densities of anchored long linkers and hyaluronidase. The enzyme activity maintains throughout the conjugation and fabrication steps (Figure 1D). Although rHuPH20 conjugated via long linkers should have a lower density on RBC membranes according to the study using bovine hyaluronidase, the cells showed more than twice the enzymatic activity compared to the cells modified using short linkers. This is likely because the long linkers provide the conjugated rHuPH20 with better flexibility to access and degrade HA than the short linkers. The overall effective activities of rHuPH20 per unit area on NPs were dramatically higher than that on cells even though the membranes were originally isolated from the same batch of modified cells. NPs with sizes much smaller than cells are more likely to penetrate into the HA layer of substrates, providing more contact area between rHuPH20 and HA in the enzyme activity assay. Although the density of total rHuPH20 on long linker-modified NPs is lower than that on short linker-modified NPs, these two kinds of NPs showed a comparable effective enzyme activity. Similarly, it is because that long linker provides the freedom to conjugated rHuPH20 to access HA and allows it to adopt a correct orientation to degrade HA. In order to exclude the influence of non-covalently attached rHuPH20 during the modification steps, controls of non-thiolated rHuPH20, cells without linker molecule treatment and a simple mixture of cells and rHuPH20 were also analyzed by the activity assay (Figure S2). The enzyme activity only remained on RBC membranes when rHuPH20 was covalently attached, indicating that the cell washing steps are sufficient to remove non-covalently attached rHuPH20.
Based on this result, the long linker (MW: 3400) was selected for PH20-RBCM-NPs fabrication since its lower conjugation density on RBCs better maintains the surface properties of RBC membranes while still exhibiting comparable effective activities of rHuPH20 to NPs fabricated using short linkers. For each mg of PH20-RBCM-NPs, there were 271 ± 8 U effective rHuPH20 on small NPs and 53 ± 2 U effective rHuPH20 on large NPs. It is estimated that there were about 8 and 14 effective rHuPH20 molecules on 85 and 125 nm PH20-RBCM-NPs respectively under the fabrication condition (Figure S3), giving a high local concentration of rHuPH20.
Nanoparticle diffusion through ECM-mimicking gels
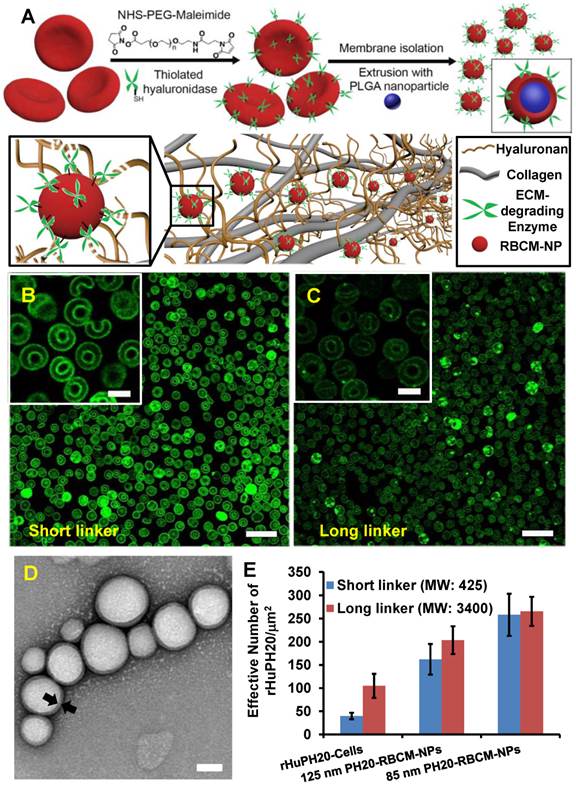
Gel matrix was generated by mixing collagen and HA, mimicking the natural structure of ECM, in which collagen fibers compose the scaffold, while HA fills the space of collagen scaffolds. The concentration of collagen and HA were also selected in consistent with physiological conditions [38]. When 130 nm RBCM-NPs with and without conjugated rHuPH20 or free rHuPH20 were added on top of the ECM-mimicking gels, RBCM-NPs alone barely diffuse into the gels, while the NPs were able to penetrate into the gels at different depth with the assistance of rHuPH20 (Figure 2 A-D). More interestingly, we found that the surface-conjugated rHuPH20 were more efficient in facilitating RBCM-NPs to diffuse through the gels than free rHuPH20 with the same enzyme activity. The diffusion coefficient is more than doubled (Figure 2D). This is because when rHuPH20 were conjugated on NP surfaces, the enzymatic degradation of HA took place on the pathway of NP diffusion, which is more efficient for enhanced NP diffusion than random HA degradation by free rHuPH20. The co-localization of fluorescence signals from the PLGA-NPs (green) and RBC membranes (red) indicates that RBCM-NPs maintained their structure during diffusion in matrix.
Nanoparticle diffusion in ECM-mimicking gels. (Scale bar: 200 μm). (A-C) The gels were prepared with 6.5 mg/mL of rat collagen I and 1 mg/mL of HA in capillary tubes. Ten μL of 1 mg/mL of nanoparticles (PLGA-green; RBC membrane-red) were added on the top of gels and incubated at 37ºC for 1 h before sample imaging. The separate labeling of membranes and nanoparticle cores, and the co-localization of two colors illustrated that the membranes did not separate from the particles. The activity of free or conjugated rHuPH20 was 50 U/mL. (D) Normalized NP fluorescence with diffusion distance in gels. Images were analyzed via ImageJ. Diffusion coefficients were obtained by fitting the data to the one-dimension diffusion model in MATLAB. Lines display theoretical intensity profiles (from top to bottom) for particles with diffusion coefficients of 4.11 × 10-8 cm2·s-1, 2.08 × 10-8 cm2·s-1 and 2.18 × 10-9 cm2·s-1.
Nanoparticle internalization by PC3 cells
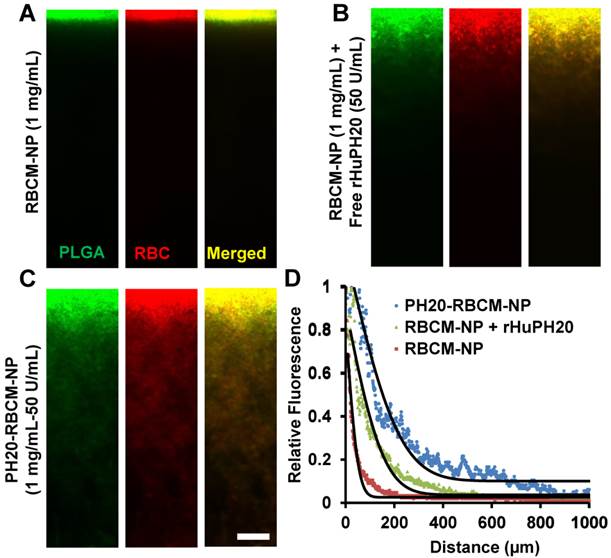
Some cancer cells such as PC3 cells have pericellular HA matrix that protect cancer cells from therapeutics and even inhibit immune cells to access cancer cells [39]. Pericellular HA matrix around PC3 cells was demonstrated through a RBC exclusion assay where the matrix blocked the access of RBCs (Figure S4). The calculated pericellular matrix is approximately 128 μm2 (Figure S4), which is smaller than the previously reported value because extra aggrecan was added in the previous study to physically crosslink HA matrix [29, 40]. The pericellular HA matrix can be depleted by a high concentration of rHuPH20 (Figure S3). The diffusion of DiD-labeled NPs was studied using confocal microscopy and analyzed with ImageJ. The signals were from both internalized NPs and membrane-bound NPs. Flow cytometry was not used in this study because high concentrations of trypsin and EDTA are required to trypsinize PC3 cells, depleting membrane-bound NPs. Therefore, imaging analysis, which is also a widely used method, was employed (Figure 3 A-C). PC3 cells were treated with small and large NPs equalized with the fluorescence intensity from PLGA cores. Each well contained 1 mL medium with 70 μg/mL of NPs, giving approximately 19 U and 3.5 U enzyme activity/well for cells that were treated with 90 nm and 130 nm DiD-labeled PH20-RBCM-NPs, respectively. It was shown that more PH20-RBCM-NPs were internalized by or bound on PC3 cells than RBCM-NPs without rHuPH20 modification for both large and small NPs. The enhanced diffusion, which is indicated by the derivative with respect to time of fluorescence intensity difference between PH20-RBCM-NPs and RBCM-NPs-treated cells, gradually disappeared after about 4 h of treatment (Figure 3C). Since the counted signals included both internalized and membrane-bound NPs, the process of NP targeting of cells can be divided into two steps, matrix penetration and binding on membranes. Cells may only uptake limited amounts of NPs within a certain timeframe. Increasing amounts of NPs diffused through the HA matrix layer, but did not bind to cell surfaces, building up the NP concentration close to the cells. Gradually, NP concentrations in both sides of HA matrix reached equilibrium, and NPs lost the driving force for further net diffusion toward cells. This explains the disappearance of the enhanced diffusion after 4 h treatment. The enhanced diffusion is less significant for smaller sized NPs as counted by the percentage of increased fluorescence signals (Figure 3B and C). This can be understood by the relatively easier diffusion of smaller sized NPs. Due to the denser and thicker matrix in tumors, the enhanced diffusion is expected to be critical for better therapeutic efficacy.
Kinetic study of nanoparticle internalization by PC3 cells. The cells have a hyaluronic acid pericellular matrix. Nanoparticles need to overcome this barrier before binding on or being internalized by PC3 cells. A) Confocal microscopy images of PC3 cells after treatment with either 130 nm PH20-RBCM-NPs, 130 nm RBCM-NPs, 90 nm PH20-RBCM-NPs, 90 nm RBCM-NPs for 1 h, 2 h, 4 h and 8 h. (Scale bar: 50 μm) Images were adjusted by enhancing green signals for presentation. Original images with unsaturated green signals were analyzed with ImageJ. B,C) Quantitative analysis of NPs internalized and bound on PC3 cells. B) Mean fluorescence intensity/cell was obtained by ImageJ analysis of DiD signal that was averaged by the number of cells. Normalized value was compared with the value of 130 nm PH20-RBCM-NPs at 8 h treatment. Values indicate mean ± s.d. (n = 10). C) Fluorescence intensity difference between PH20-RBCM-NP and RBCM-NP-treated PC3 cells with respect to time. Fluorescence intensities of cells treated with 130 nm and 90 nm NPs for 8 h were used as references. Data represent mean ± s.d. (n = 10).
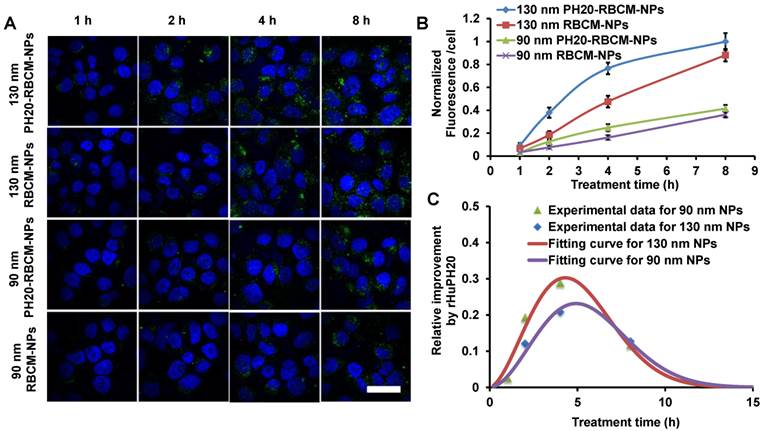
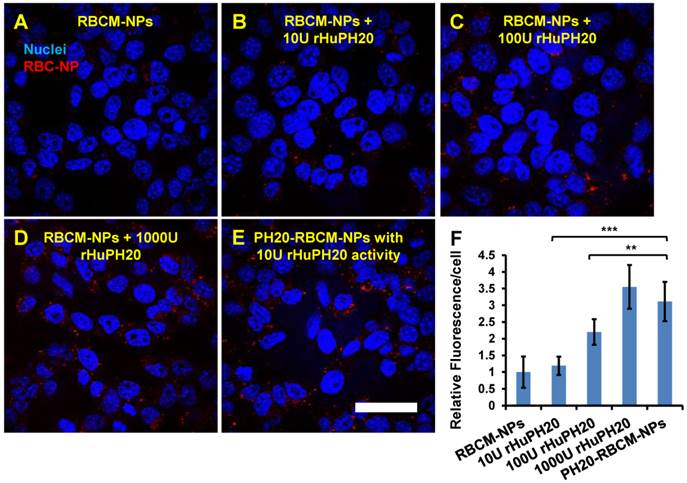
The effect of conjugated rHuPH20 were also compared with free rHuPH20 in NP diffusion through the pericellular HA matrix. In this study, PC3 cells were treated with 1 mL of 0.2 mg/mL of 130 nm PH20-RBCM-NPs, which contained 10 U rHuPH20 activity or a same concentration of RBCM-NPs with 10, 100, 1000 U of free rHuPH20 (Figure 4 A-F). In 2 h, the conjugated rHuPH20 tripled the amount of NPs internalized by or bound to PC3 cells compared to that of RBC-NPs with 10 U free rHuPH20. Moreover, the amount of internalized and membrane-bound PH20-RBCM-NPs was also significantly higher than the RBCM-NPs with 100 U of free rHuPH20. This result is consistent with the NP diffusion data in ECM-mimicking gels, showing that conjugated rHuPH20 is more efficient than free rHuPH20 in facilitating NP diffusion.
Quantitative analysis of NP fluorescence intensity in PC3 cells. The cells were treated for 2 h with RBCM-NPs supplemented with free rHuPH20 at 0 U/mL (A), 10 U/mL (B), 100 U/mL (C) or 1000 U/mL (D) or treated with PH20-RBCM-NPs with 10 U/mL of enzyme activity (E). (Scale bar = 50 μm). F) Quantitative analysis of NP internalization and binding on PC3 cells. The conjugated rHuPH20 on NPs (10 U activity) was significantly more efficient than free rHuPH20 (10 U and 100 U) in increasing NP internalization or bound to PC3 cells through assisting NP penetration of the pericellular HA matrix of PC3 cells. (A-E) Images were adjusted by enhancing red signals for presentation. Original images with unsaturated red signals were analyzed with ImageJ. Values indicate mean ± s.d. (n = 10). **P < 0.01, ***P < 0.001. (one-way ANOVA followed by Tukey post hoc test).
Blood circulation of PH20-RBCM-NPs
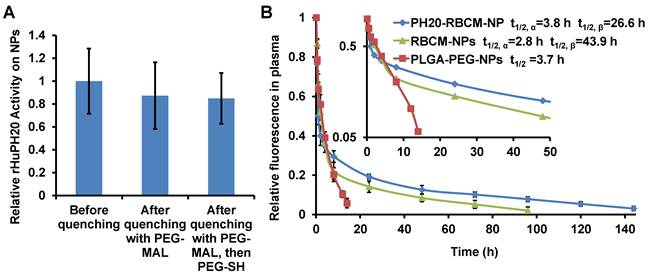
RBCM-NPs were previously shown to possess a long blood circulation half-life. To validate our facile approach to functionalize CM-NPs, it is critical to demonstrate that PH20-RBCM-NPs at least maintain the circulation half-life of RBCM-NPs. We have found that RBCM-NPs prepared in PBS are more stable in the blood than those prepared in DI H2O. Therefore, DiD-labeled RBCM-NPs or PH20-RBCM-NPs were fabricated following the method used for in vitro studies except replacing DI H2O with PBS in the steps of PLGA-NP formation and membrane coating. We first tested the effect of membrane-to-NP ratio on the blood circulation of RBCM-NPs. It was found that increasing the membrane/PLGA-NP surface area ratio prolongs NP blood circulation probably due to an improved stability of membrane coating on PLGA-NPs (Figure S5). It is known that local membrane compositions are heterogeneous on individual cells [41]. Some compositions may not provide stable membrane coating on PLGA-NPs. Therefore, a high membrane/PLGA-NP surface area ratio is necessary to achieve an ultra-long blood circulation time for RBCM-NPs. To study the effect of conjugated rHuPH20 on RBCM-NP blood circulation, NPs have been prepared by mixing 0.2 mL of RBC membranes from 1 mL of blood to 0.2 mL of 15 mg/mL of 105 nm PLGA-NPs in the NP coating step. The unreacted extra thiol groups on the rHuPH20 and maleimide groups on the linkers after modification were quenched with PEG2K-MAL and PEG2K-SH sequentially, and more than 50% of the enzyme activity of rHuPH20 on RBCs was maintained after quenching unreacted thiol and maleimide groups (Figure S6). When the RBC membranes were isolated and coated on NPs, there was no significant difference of rHuPH20 activity on NPs before and after quenching the unreacted groups with PEG molecules (Figure 5A). This can be explained by the increased surface-to-volume ratio, making NPs easier to access and degrade HA after membrane coating on NPs. Similar to the previous work, the RBCM-NPs showed blood retention superior to poly(lactic-co-glycolic acid)-b-polyethylene glycol nanoparticles (PLGA-PEG-NPs) (Figure 5B). Interestingly, when fitted into a two-compartment model, PH20-RBCM-NPs have an elimination half-life (t1/2,β ) of 43.9 h, which is longer than the 26.6 h half-life of RBCM-NPs. The t1/2,β of PH20-RBCM-NPs is also comparable to the 39.6 h half-life of a previous work, in which a higher membrane and NP ratio was used [1]. The relative short circulation half-life in the distribution phase is due to the clearance of RBCM-NPs with defects in coating or with an inside-out membrane coating. Protein adsorption on NPs is the major reason for NP clearance in the blood by phagocytic cells. The improved circulation half-life of PH20-RBCM-NPs may be because PEG molecules interfere with protein adsorption on membrane coating defects, thereby providing additional protective effects that complement those offered by the self-signal on the membranes. The reason may also be that conjugated PH20-RBCM-NPs and PEG reduced the fraction of RBCM-NPs with an inside-out membrane coating as the conjugated molecules prevent access to the outer leaflet of cell membranes to PLGA-NPs in the coating step. For other applications, the unreacted groups may also be quenched by small molecules to eliminate the effect of PEG on protein interactions. In spite of the advantages of our facile approach in CM-NP functionalization, there may be a limitation on the number of enzymes that we can conjugate on cells because extensive chemical modification of cell membranes affects the natural function of cell membranes.
Effect of rHuPH20 modification on RBCM-NP blood circulation. A) The rHuPH20 activity on PH20-RBCM-NPs remained after quenching unreacted thiol and maleimide groups on modified RBCs with PEG2K-maleimide and PEG2K-thiol sequentially in NP fabrication. B) In vivo blood circulation of DiD-labeled PH20-RBCM-NPs and RBCM-NPs via measuring the fluorescence intensity of NPs. PLGA-PEG-NPs were tested as a control. Value indicates mean ± s.d. (n = 6 from two independent experiments). Inset: Semi-log scale NP signals with time in which the slope illustrates circulation half-life. The circulation half-lives for RBCM-NPs and PH20-RBCM-NPs were fitted into a two-compartment model, while the half-life for PLGA-PEG-NPs was fitted into a one-compartment model.
Conclusion
We have developed a non-genetic cell membrane modification approach to functionalize CM-NPs. The strategy enables a stable anchorage of functional molecules on the extracellular domain of cell membrane proteins via a cell impermeable linker, NHS-PEG-Maleimide. Through a two-step conjugation, rHuPH20 can be chemically anchored on RBC membranes while still maintains its enzymatic activity. Long linkers were found to be superior to short linkers in enzyme anchorage and were selected for CM-NP functionalization. Under the assistance of the conjugated rHuPH20, PH20-RBCM-NPs can diffuse through ECM-mimicking gels as well as the pericellular HA matrix of PC3 cells faster than RBCM-NPs in combination with free rHuPH20 thanks to a high local rHuPH20 concentration around NPs. RBCM-NPs are known for their long blood circulation. Surprisingly, the functionalization strategy even improved the circulation time of RBCM-NPs. This may be because of a combined effect of the “self” signals on NP surfaces and the conjugated PEGs that were used in quenching unreacted thiol and maleimide groups. Unlike the steric repulsion of a high density PEG layer to proteins, these membrane associated PEGs may interfere with protein attachment to the defect area of membrane coating, and along with the conjugated rHuPH20 reduce the inside-out membrane orientation on NPs.
In summary, our functionalization approach maintained the original properties of the natural cell membranes while adding desired functions. This platform strategy may expand CM-NP applications, especially when multi-functions are required.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by a faculty startup fund from Drexel University and seed funding from the Clinical & Translational Research Institute (CTRI). We thank Halozyme Therapeutics for providing rHuPH20 and Mr. David Kang and Dr. Tzung Yang for the helpful discussion about hyaluronidase. We also thank Dr. Yichao Lu for his help on analyzing the data of nanoparticle diffusion in matrix. TEM imaging was performed in the Centralized Research Facilities (CRF) at Drexel University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hu CMJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang LF. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:10980-5
2. Hu CMJ, Fang RH, Copp J, Luk BT, Zhang LF. A biomimetic nanosponge that absorbs pore-forming toxins. Nat Nanotechnol. 2013;8:336-40
3. Hu CMJ, Fang RH, Luk BT, Zhang LF. Nanoparticle-detained toxins for safe and effective vaccination. Nat Nanotechnol. 2013;8:933-8
4. Li LL, Xu JH, Qi GB, Zhao XZ, Yu FQ, Wang H. Core-Shell Supramolecular Gelatin Nanoparticles for Adaptive and "On-Demand" Antibiotic Delivery. ACS Nano. 2014;8:4975-83
5. Guo YY, Wang D, Song QL, Wu TT, Zhuang XT, Bao YL. et al. Erythrocyte Membrane-Enveloped Polymeric Nanoparticles as Nanovaccine for Induction of Antitumor Immunity against Melanoma. ACS Nano. 2015;9:6918-33
6. Fan ZY, Zhou H, Li PY, Speer JE, Cheng H. Structural elucidation of cell membrane-derived nanoparticles using molecular probes. Journal of Materials Chemistry B. 2014;2:8231-8
7. Tan SW, Wu TT, Zhang D, Zhang ZP. Cell or cell membrane-based drug delivery systems. Theranostics. 2015;5:863-81
8. Gao WW, Zhang LF. Engineering red-blood-cell-membrane-coated nanoparticles for broad biomedical applications. Aiche Journal. 2015;61:738-46
9. Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051-4
10. Parodi A, Quattrocchi N, van de Ven AL, Chiappini C, Evangelopoulos M, Martinez JO. et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8:61-8
11. Hu C-MJ, Fang RH, Wang K-C, Luk BT, Thamphiwatana S, Dehaini D. et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526:118-21
12. Hu Q, Sun W, Qian C, Wang C, Bomba HN, Gu Z. Anticancer Platelet-Mimicking Nanovehicles. Advanced Materials. 2015;27:7043-50
13. Fang RH, Hu CMJ, Luk BT, Gao WW, Copp JA, Tai YY. et al. Cancer Cell Membrane-Coated Nanoparticles for Anticancer Vaccination and Drug Delivery. Nano Lett. 2014;14:2181-8
14. Gao WW, Fang RH, Thamphiwatana S, Luk BT, Li JM, Angsantikul P. et al. Modulating Antibacterial Immunity via Bacterial Membrane-Coated Nanoparticles. Nano Lett. 2015;15:1403-9
15. Patel JM, Vartabedian VF, Bozeman EN, Caoyonan BE, Srivatsan S, Pack CD. et al. Plasma membrane vesicles decorated with glycolipid-anchored antigens and adjuvants via protein transfer as an antigen delivery platform for inhibition of tumor growth. Biomaterials. 2016;74:231-44
16. Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nature Reviews Genetics. 2003;4:346-58
17. Hwang WY, Fu YF, Reyon D, Maeder ML, Tsai SQ, Sander JD. et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31:227-9
18. Wang Q, Cheng H, Peng H, Zhou H, Li PY, Langer R. Non-genetic engineering of cells for drug delivery and cell-based therapy. Advanced Drug Delivery Reviews. 2015;91:125-40
19. Luk BT, Hu CMJ, Fang RNH, Dehaini D, Carpenter C, Gao WW. et al. Interfacial interactions between natural RBC membranes and synthetic polymeric nanoparticles. Nanoscale. 2014;6:2730-7
20. Chung HA, Kato K, Itoh C, Ohhashi S, Nagamune T. Casual cell surface remodeling using biocompatible lipid-poly(ethylene glycol)(n): Development of stealth cells and monitoring of cell membrane behavior in serum-supplemented conditions. Journal of Biomedical Materials Research Part A. 2004;70A:179-85
21. Hymel D, Peterson BR. Synthetic cell surface receptors for delivery of therapeutics and probes. Advanced Drug Delivery Reviews. 2012;64:797-810
22. Fang RNH, Hu CMJ, Chen KNH, Luk BT, Carpenter CW, Gao WW. et al. Lipid-insertion enables targeting functionalization of erythrocyte membrane-cloaked nanoparticles. Nanoscale. 2013;5:8884-8
23. Bell GI. Models for the specific adhesion of cells to cells. Science (New York, NY). 1978;200:618-27
24. Chinol M, Casalini P, Maggiolo M, Canevari S, Omodeo ES, Caliceti P. et al. Biochemical modifications of avidin improve pharmacokinetics and biodistribution, and reduce immunogenicity. Br J Cancer. 1998;78:189-97
25. Smarr CB, Hsu CL, Byrne AJ, Miller SD, Bryce PJ. Antigen-Fixed Leukocytes Tolerize Th2 Responses in Mouse Models of Allergy. J Immunol. 2011;187:5090-8
26. Cheng H, Byrska-Bishop M, Zhang CT, Kastrup CJ, Hwang NS, Tai AK. et al. Stem cell membrane engineering for cell rolling using peptide conjugation and tuning of cell-selectin interaction kinetics. Biomaterials. 2012;33:5004-12
27. Turley DM, Miller SD. Peripheral tolerance induction using ethylenecarbodiimide-fixed APCs uses both direct and indirect mechanisms of antigen presentation for prevention of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:2212-20
28. Standley SM, Toft DJ, Cheng H, Soukasene S, Chen J, Raja SM. et al. Induction of Cancer Cell Death by Self-assembling Nanostructures Incorporating a Cytotoxic Peptide. Cancer Res. 2010;70:3020-6
29. Jiang P, Li XM, Thompson CB, Huang ZD, Araiza F, Osgood R. et al. Effective Targeting of the Tumor Microenvironment for Cancer Therapy. Anticancer Res. 2012;32:1203-12
30. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2012;21:418-29
31. Aaltomaa S, Lipponen P, Tammi R, Tammi M, Viitanen J, Kankkunen JP. et al. Strong stromal hyaluronan expression is associated with PSA recurrence in local prostate cancer. Urologia Internationalis. 2002;69:266-72
32. Auvinen P, Tammi R, Kosma VM, Sironen R, Soini Y, Mannermaa A. et al. Increased hyaluronan content and stromal cell CD44 associate with HER2 positivity and poor prognosis in human breast cancer. International Journal of Cancer. 2013;132:531-9
33. Whatcott CJ, Han H, Posner RG, Hostetter G, Von Hoff DD. Targeting the Tumor Microenvironment in Cancer: why Hyaluronidase Deserves a second Look. Cancer Discovery. 2011;1:291-6
34. Bot PT, Hoefer IE, Piek JJ, Pasterkamp G. Hyaluronic acid: Targeting immune modulatory components of the extracellular matrix in atherosclerosis. Curr Med Chem. 2008;15:786-91
35. Sadowitz B, Seymour K, Gahtan V, Maier KG. The Role of Hyaluronic Acid in Atherosclerosis and Intimal Hyperplasia. J Surg Res. 2012;173:E63-E72
36. Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX. et al. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28:869-76
37. Frost GI, Stern R. A microtiter-based assay for hyaluronidase activity not requiring specialized reagents. Analytical Biochemistry. 1997;251:263-9
38. Wong C, Stylianopoulos T, Cui JA, Martin J, Chauhan VP, Jiang W. et al. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:2426-31
39. Singha NC, Nekoroski T, Zhao CM, Symons R, Jiang P, Frost GI. et al. Tumor-Associated Hyaluronan Limits Efficacy of Monoclonal Antibody Therapy. Mol Cancer Ther. 2015;14:523-32
40. Thompson CB, Shepard HM, O'Connor PM, Kadhim S, Jiang P, Osgood RJ. et al. Enzymatic Depletion of Tumor Hyaluronan Induces Antitumor Responses in Preclinical Animal Models. Mol Cancer Ther. 2010;9:3052-64
41. Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F. et al. Ceramide triggers budding of exosome vesicles into multivesicular Endosomes. Science. 2008;319:1244-7
Author contact
![]() Corresponding author: hchengedu.
Corresponding author: hchengedu.