Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2016; 6(7):1023-1030. doi:10.7150/thno.14741 This issue Cite
Research Paper
Solid Tumor Therapy Using a Cannon and Pawn Combination Strategy
Wantong Song1, Zhaohui Tang1 ![]() , Dawei Zhang1, Xue Wen2, Shixian Lv1,3, Zhilin Liu1,3, Mingxiao Deng2, Xuesi Chen1
, Dawei Zhang1, Xue Wen2, Shixian Lv1,3, Zhilin Liu1,3, Mingxiao Deng2, Xuesi Chen1 ![]()
1. Key Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, PR China;
2. Department of Chemistry, Northeast Normal University, Changchun, 130021, PR China;
3. University of Chinese Academy of Sciences, Beijing, 100039, PR China.
Received 2015-12-17; Accepted 2016-2-15; Published 2016-4-28
Abstract

Nanocarrier-based anti-tumor drugs hold great promise for reducing side effects and improving tumor-site drug retention in the treatment of solid tumors. However, therapeutic outcomes are still limited, primarily due to a lack of drug penetration within most tumor tissues. Herein, we propose a strategy using a nanocarrier-based combination of vascular disrupting agents (VDAs) and cytotoxic drugs for solid tumor therapy. Specifically, combretastatin A-4 (CA4) serves as a “cannon” by eradicating tumor cells at a distance from blood vessels; concomitantly, doxorubicin (DOX) serves as a “pawn” by killing tumor cells in close proximity to blood vessels. This “cannon and pawn” combination strategy acts without a need to penetrate every tumor cell and is expected to eliminate all tumor cells in a solid tumor. In a murine C26 colon tumor model, this strategy proved effective in eradicating greater than 94% of tumor cells and efficiently inhibited tumor growth with a weekly injection. In large solid tumor models (C26 and 4T1 tumors with volumes of approximately 250 mm3), this strategy also proved effective for inhibiting tumor growth. These results showing remarkable inhibition of tumor growth provide a valuable therapeutic choice for solid tumor therapy.
Keywords: nanocarrier, vascular disrupting agents, cytotoxic drugs, combination, tumor therapy.
Introduction
Cytotoxic drugs are the bedrock of chemotherapy. However, dose-related side effects limit their use. Nanocarriers have repeatedly been used to successfully improve the body's tolerance to cytotoxic drugs-allowing for the use of increased dosages [1-4] and retention of the drug within tumor sites by virtue of the “enhanced permeability and retention” (EPR) effect [5-7]. However, nanocarrier-delivered chemotherapeutics are still limited by less than 100% kill efficiency within tumors.
One key point behind the poor performance of nanocarrier delivery systems lies in that the delivered cytotoxic drugs require “contact” with tumor cells in order to be effective chemotherapeutics. After delivery to the tumor site, cytotoxic drugs must also be internalized by tumor cells, interact with DNA or other cellular organelles, and induce apoptosis [8-10]. Therefore, delivery of drugs to tumor cells is the initial step in effective tumor therapies. However, hindered by intra-tumor diffusion, nanocarrier-loaded drugs are not delivered to all tumor cells even when a sufficient concentration has been maintained in the tumor site [11-14]. It's commonly observed that nanocarrier-loaded drugs are primarily distributed around tumor vessels, leaving large untreated areas at a distance from the vessels [15-17]. As a result, untreated tumor cells continue to grow, resulting in control rather than eradication of the tumor.
Vascular disrupting agents (VDAs) are tumor vascular targeting drugs that kill tumor cells without the need for “contact”. Studies have shown that VDAs selectively act on endothelial cells in immature or abnormal vessels that lack a full complement of smooth muscle cells or pericyte support and are abundant in tumor tissues [18-20]. The action of VDAs on endothelial cells induces endothelial cell dysfunction, partial vessel occlusion, and diminished blood flow. These effects result in oxygen and nutrient deprivation of tumor cells at a distance from the vessels and widespread tumor cell necrosis far from the endothelium [21-24]. For tumor cells in proximity to vessels or at the tumor rim, the decrease in blood flow may not be fatal because oxygen and nutrients can be supplied by diffusion from the surrounding normal tissues; therefore, these cells typically survive and continue to grow [25, 26]. As a consequence, VDAs serve as efficient non-contacting agents for solid tumor therapy that commonly induce strong necrosis, particularly in regions far from vessels.
It has become recognized that the combination of VDAs and nanocarrier-loaded cytotoxic drugs shows promise for solid tumor therapy. Previously, we reported eradication of tumor cells in both the peripheral and central regions of a solid tumor by coadministration of combretastatin A4 phosphate (CA4P) and cisplatin-loaded nanoparticles [27]. However, the endothelial cell dysfunction induced by CA4P is reversible upon drug removal and a single administration of CA4P lasts for only a very short period of time due to its rapid clearance from the plasma and tissues; therefore, repeated injections are necessary for substantial inhibition of tumor growth [28-30].
Here, we propose a nanocarrier-based combination of VDAs and cytotoxic drugs. The benefits of using nanocarriers as a delivery system for this drug combination are due to selective drug delivery to tumor tissues and reduced side effects to normal organs. Furthermore, the drugs remain within the tumor environment at a high concentration and for a long period of time due to the retention effect and constant release. Previously, S. Sengupta and Y. Wang used a nanocell and micelle system to deliver a CA4 and DOX drug combination, improving tumor therapeutic outcomes by targeting both tumor vasculature and tumor cells [31, 32]. In this paper, we emphasize the synergism of the VDA and DOX cytotoxic drug combination strategy for solid tumor therapy. We suggest that this strategy can be described as a combination of “cannon” and “pawn”, with the VDA acting as a “cannon” by killing tumor cells at a distance from blood vessels, while the cytotoxic drug acts as a “pawn” by killing the residual tumor cells in close proximity to the blood vessels. This combination has the potential to completely eradicate tumors.
We prepared the nanocarrier-based drug combination (PLG-CA4/DOX) by grafting CA4 onto the methoxy poly(ethylene glycol)-block-poly(L-glutamic acid) (mPEG-PLG) copolymer and then loading DOX inside using a nanoprecipitation method. The PLG-CA4/DOX was administered through tail vein injections and the two loaded drugs were delivered to tumor tissues and distributed around the blood vessels. Release of CA4 from PLG-CA4/DOX results in diminished blood flow and large areas of tumor cell necrosis far from the vasculature; release of DOX directly kills the residual tumor cells near the vessels (Scheme 1). We determined the therapeutic efficacy of injected PLG-CA4/DOX for the treatment of solid tumors in in vivo solid tumor models.
Mechanism of the “cannon” and “pawn” combination strategy for solid tumor therapy. After tail vein injection, nanocarriers loaded with CA4 and DOX (PLG-CA4/DOX) arrive around the tumor vessels. DOX is released and acts directly on tumor cells in proximity to the vessels, whereas CA4 causes endothelial cell dysfunction, diminished blood flow, oxygen and nutrient deprivation and widespread tumor cell necrosis at a distance from the vessels.
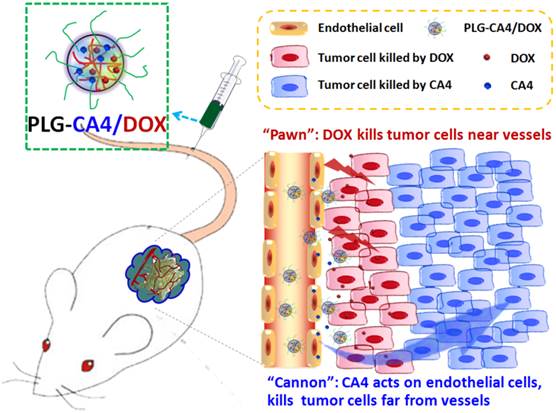
Results
Preparation of PLG-CA4/DOX
Poly(L-glutamic acid) (PLG)-based polymeric-drug conjugates have consistently been used during drug development. PLG is biocompatible, biodegradable, and easily modified [33-35]. As such, PLG is grafted to various small molecule drugs such as paclitaxel (PTX) and camptothecin (CPT) to improve their longevity and bioavailability [36-38]. Herein, CA4 was conjugated to mPEG-PLG using a one-step esterification reaction catalyzed by dicyclohexylcarbodiimide (DIC) and 4-dimethylaminopyridine (DMAP) (Fig. 1A). We used 1H NMR and FT-IR to confirm the chemical structure of the resulting mPEG-PLG-g-CA4 (Fig. S1). Resonance peaks at 6.57 (k), 6.44 (j), 6.29 (i), 3.62 (h), 4.02 (g), and 2.29 (e) ppm were attributed to the presence of CA4. An average of 10 CA4 molecules was grafted to the mPEG-PLG copolymer based on calculations of i, j, k, and b intensity ratios (3.57 ppm). Stretching vibration peaks in FT-IR spectra at 1500 and 1600 cm-1 were attributed to the benzene ring of CA4. Gel permeation chromatography (GPC) results using dimethylformamide as a solvent confirmed that both polymers are unimodal (Fig. S2). The number average molecular weight (Mn) of mPEG-PLG was 7.4 × 103 Da, with a polydispersity index (PDI) of 1.2; Mn of mPEG-PLG-g-CA4 was 9.5 × 103 Da, with a PDI of 1.4.
Preparation of PLG-CA4/DOX: (A) mPEG-PLG-g-CA4 was synthesized by conjugation of CA4 onto mPEG-PLG. (B) DOX was loaded into mPEG-PLG-g-CA4 by nanoprecipitation.

Due to the presence of hydrophobic CA4, mPEG-PLG-g-CA4 formed nano-sized micelles in pH 7.4 phosphate buffered saline, with a hydrodynamic radius (Rh) of 26.5 ± 6.3 nm and zeta potential of -13.3 ± 1.3 mV. DOX was enwrapped inside micelles that formed through self-assembly in water (Fig. 1B). The formed PLG-CA4/DOX had a slightly larger Rh (32.4 ± 5.1 nm) and higher zeta potential (-10.3 ± 1.9 mV). The uniform spherical structures were confirmed by transmission electron microscopy (TEM) as shown in Fig. S3. The static micelle size under dried conditions was 28.8 ± 5.1 nm for mPEG-PLG-g-CA4 and 32.5 ± 4.3 nm for PLG-CA4/DOX. The critical micelle concentration (CMC) was 0.016 mg/mL for mPEG-PLG-g-CA4 micelles and 0.012 mg/mL for PLG-CA4/DOX micelles, as determined by the pyrene probe method (Fig. S4). The CMC values reported here are comparable to related poly(amino acid) polymeric micelle CMC values reported in the literature [39, 40]. The loaded DOX was stabilized inside the micelles by hydrophobic accumulation and electronic interactions between the amine in DOX and the carboxylate in PLG. These dual interactions have been shown to ensure both high drug loading efficacy and high drug loading stability [41]. The final PLG-CA4/DOX micelles had a drug loading content (DLC%) of 25.1% for CA4 and 2.5% for DOX, as confirmed by UV-Vis spectrometry (Fig. S5).
In vitro release and cytotoxicity tests
In vitro release of DOX and CA4 from PLG-CA4/DOX was tested at a pH of 7.4 and 5.5 in phosphate buffered saline (Fig. S6). DOX release was obviously pH-dependent, with a much faster release rate at pH 5.5 than pH 7.4. CA4 was gradually released from the conjugates, while the ester hydrolysis rate was slightly faster at neutral compared to acidic conditions. Unlike many other drug conjugates linked via ester bonds, the phenolic ester breaks relatively efficiently. This understanding of the release rate is essential for polymer-drug conjugates to act in vivo [42, 43]. We also measured the time-dependent size changes of the PLG-CA4/DOX micelles in pH 7.4 and 5.5 phosphate buffered saline (Fig. S7). Consistent with the release results, the average Rh increased over time, suggesting that the structure loosened as the hydrophobic components were released.
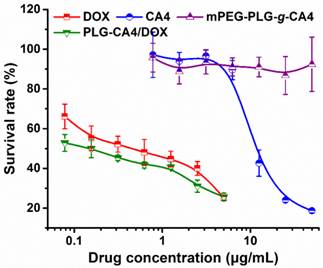
An in vitro tumor cell inhibition test was conducted in murine colon carcinoma C26 cells by MTT assay. As show in Fig. 2, DOX strongly inhibited C26 cell growth, with an IC50 value of 0.44 μg/mL at 48 h. However, CA4 did not dramatically inhibit C26 cell growth, with an IC50 value of 11.8 μg/mL. For mPEG-PLG-g-CA4, the IC50 value was over 50.0 μg/mL. The MTT results reflect the different mechanisms of these two drugs. In particular, DOX is a representative anthracycline, inhibiting DNA or RNA synthesis by intercalating between base pairs of the DNA/RNA strand, thus preventing the replication of rapidly growing cancer cells. In contrast, CA4 is a tubulin-binding agent that induces reversible cell shape changes with a weak growth inhibitory effect. Obviously, the in vitro cytotoxicity of PLG-CA4/DOX in C26 cells is primarily attributable to the presence of DOX.
In vitro survival rates of C26 tumor cells after incubation with DOX, CA4, mPEG-PLG-g-CA4, and PLG-CA4/DOX for 48 h (n = 4).
Intra-tumor distribution of PLG-CA4/DOX
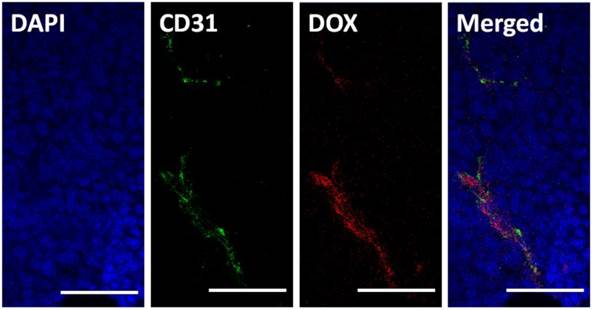
It has frequently been noted that nanoparticles entering tumor tissues are mainly confined to the areas surrounding blood vessels, with only moderate diffusion. Herein, we tested the intra-tumor distribution of prepared micelles. PLG-CA4/DOX or mPEG-PLG-g-CA4 labeled with RhoB was injected into mice via tail vein, and 4 h later tumors were collected and frozen until further analysis. The blood vessels and nuclei within the frozen tissue slices were stained with CD31 and DAPI, respectively. As shown in Fig. 3, DOX was mainly located around the blood vessels. For RhoB labeled mPEG-PLG-g-CA4, the RhoB signal was also mainly detected around the blood vessels (Fig. S8). These results confirmed that penetration of the prepared nano-micelles was restricted inside tumor tissues, and the delivered DOX and CA4 would be released around these blood vessels.
Histopathochemical analysis of tumor tissues 4 h after injection of PLG-CA4/DOX (dose: CA4 50.0 mg/kg and DOX 5.0 mg/kg). Scale bar = 50 μm.
Pathological analysis of PLG-CA4/DOX therapy
Therapeutic effects of PLG-CA4/DOX on C26 tumors were tested after a single injection (CA4 at 50.0 mg/kg and DOX at 5.0 mg/kg). For comparison purposes, mPEG-PLG-g-CA4 and DOX loaded in mPEG-PLG (mPEG-PLG/DOX) were also administered at the same doses. There were significant differences in the pathology of tumors treated with the various formulations after 24 h, as determined by hematoxylin and eosin (H & E) staining (Fig. 4).
H & E results of tumor tissues 24 h after injection of (A) mPEG-PLG/DOX, (B) mPEG-PLG-g-CA4, and (C) PLG-CA4/DOX at a dose of DOX 5.0 mg/kg and CA4 50.0 mg/kg. L and N indicates the live regions and necrotic regions, white arrows in (B) indicate the blood vessels. (D) Summarized percentage of live areas, n = 5, *** p<0.001.
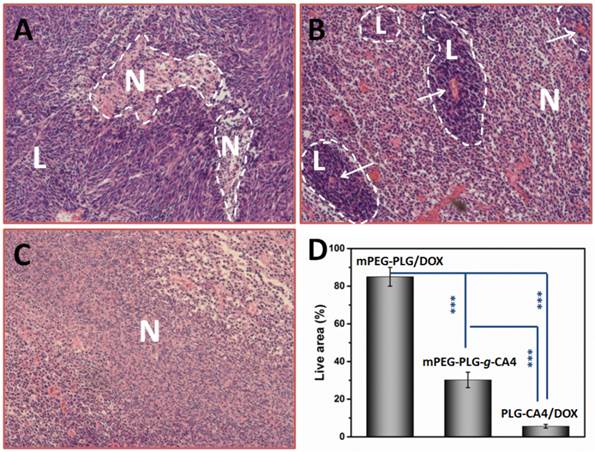
In mice administered mPEG-PLG/DOX, only regional necrosis was observed; while in large tumor regions, no necrosis was observed. For mPEG-PLG-g-CA4, large areas of tumor degeneration occurred, while some tumor cell “islands” were left around the blood vessels. These results support the fact that diminished blood flow caused by CA4 treatment results in necrotic tumor cells located at a distance from the blood vessels, while the cells in proximity to the vessels can still get sufficient amounts of nutrition to survive. These tumor cell “islands” will continue to grow and result in later tumor relapses. For PLG-CA4/DOX, tumor degeneration occurred in the entire tumor, suggesting that the combination of these two drugs works to eradicate cells in the entire tumor. Fig. 4D shows percentage of areas in tumor still containing viable cells. For the PLG-CA4/DOX treatment group, there was only 5.6 ± 1.0% of the total area still containing live cells after treatment. In comparison, the percentage of tumor areas containing live cells after treatment with mPEG-PLG/DOX and mPEG-PLG-g-CA4 were 85.0 ± 5.0% and 30.2 ± 4.1%, respectively.
In the PLG-CA4/DOX treatment group, benefits continued to be noted in later observations. After 72 h, regional necrosis was observed in the mPEG-PLG/DOX group, whereas large areas of relapse occurred in the mPEG-PLG-g-CA4 group. However, in the PLG-CA4/DOX group, there were no relapses and some completely fibrotic regions appeared (Fig. S9). We also tested the intra-tumor drug concentrations of DOX and CA4 in tumors treated with PLG-CA4/DOX and compared them with those treated with free DOX or free CA4. As shown in Fig. S10, administration of free DOX and CA4 resulted in rapid clearance from the tumors. In contrast, DOX and CA4 in PLG-CA4/DOX treated tumors peaked at 24 h and high amounts were maintained throughout the 72 h period. This phenomenon also contributed to the persistent inhibitory effect of the PLG-CA4/DOX delivery system.
In vivo tumor therapy test
We initially conducted the tumor therapy tests in a C26 xenograft tumor model. Once the tumor volumes reached approximately 100 mm3, saline, DOX, mPEG-PLG/DOX, CA4, mPEG-PLG-g-CA4, DOX+CA4, and PLG-CA4/DOX were administered at a relative dose of DOX 5.0 mg/kg and CA4 50.0 mg/kg, which was designated as day 1. A second injection was given on day 8, with additional injections of DOX and mPEG-PLG/DOX given on days 3 and 10. As shown in Fig. 5A, DOX and mPEG-PLG/DOX at 5.0 mg/kg were ineffective treatments in these fast growing murine tumors and a second injection resulted in an obvious loss of bodyweight (Fig. 5B). Treatment with free CA4 was also an ineffective treatment for controlling tumor volume, which can be explained by the short-lived and reversible activity of free CA4. Treatment with mPEG-PLG-g-CA4 resulted in improved inhibition of tumor growth compared to free CA4-confirming that a high and constant CA4 concentration in tumors is necessary for controlling tumor growth. The combination of free DOX and CA4 was also effective in inhibiting tumor growth; however, there was rapid re-growth of the tumor 2 days after drug administration, confirming the short action time of the free drugs. PLG-CA4/DOX obviously inhibited C26 tumor growth in a once per week injection, with a tumor suppression rate (TSR%) of 83.8% on day 15. Differences in C26 tumor inhibition were also observed in photos of the tumors taken on day 15 (Fig. 5C).
In vivo tumor therapy results. (A) C26 tumor volumes after injection with saline, CA4, mPEG-PLG-g-CA4, DOX+CA4, and PLG-CA4/DOX on days 1 and 8, and DOX and mPEG-PLG/DOX on days 1, 3, 8, and 10. (B) Bodyweight changes. (C) Photos of C26 tumors on day 15. (D) C26 tumor volumes after injection with saline or PLG-CA4/DOX on days 1 and 5. Treatment started when tumor volume reached approximately 250 mm3. (E) 4T1 tumor volumes after injection with saline, mPEG-PLG/DOX, mPEG-PLG-g-CA4, and PLG-CA4/DOX on days 1 and 14. (F) Photos of 4T1 tumors on day 21. Dose: DOX 5.0 mg/kg and CA4 50.0 mg/kg. **p < 0.01, ***p < 0.001.
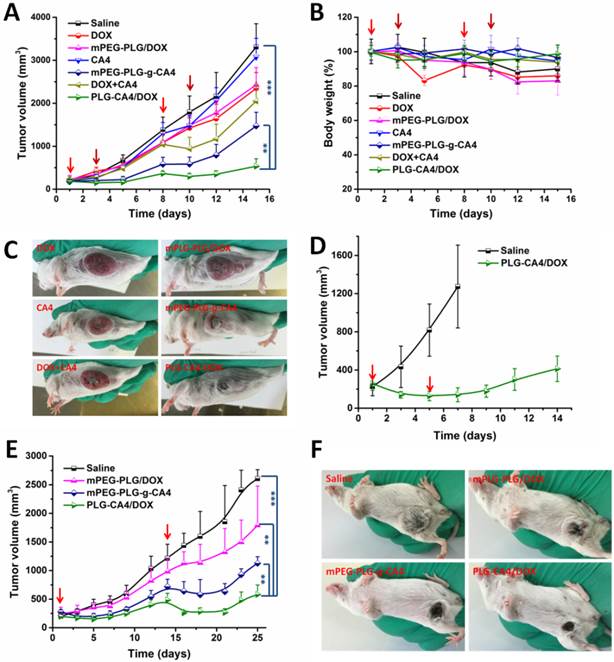
In order to determine the efficacy of PLG-CA4/DOX treatment in large tumors, PLG-CA4/DOX was administered to C26 tumor bearing mice with tumor volumes of approximately 250 mm3. The injections were carried out on days 1 and 5, at a relative dose of DOX 5.0 mg/kg and CA4 50.0 mg/kg. As shown in Fig. 5D, PLG-CA4/DOX was also effective for inhibiting growth in these large tumors, and the tumor inhibition effect lasted for over two weeks.
The in vivo tumor therapy tests were further studied in an orthotopic murine breast 4T1 tumor model. We began treatment when tumor volume reached approximately 250 mm3. Saline, mPEG-PLG/DOX, mPEG-PLG-g-CA4, and PLG-CA4/DOX were administered at a dose of DOX 5.0 mg/kg and CA4 50.0 mg/kg. Injections were given on days 1 and 14. As shown in Fig. 5E, PLG-CA4/DOX was the most effective formulation for inhibiting the growth of large 4T1 tumors. After the first injection, the inhibition continued for over a week. The injection on day 14 resulted in tumor shrinkage that lasted for another week. Photos of the tumors taken on day 21 clearly show the tremendous therapeutic effect of the combination system (Fig. 5F).
Discussion
Nanocarriers hold great promise for solid tumor therapy by reducing the side effects and improving drug accumulation in the tumor tissue. However, due to heterogeneity and diffusion hindrances within tumors, nanocarriers are commonly arrested around tumor vessels, preventing penetration to cells at a distance from the vasculature. As a result, large areas of tumor cells can be left unaffected and continue to grow.
Significant efforts have been made to improve nanoparticle penetration into tumor microenvironments. For example, in many systems small nanoparticles (< 30 nm) have been shown to penetrate much deeper than large nanoparticles (> 100 nm) [44-46]. Nanocarrier systems have also been designed to break into smaller nanoparticles in response to the tumor microenvironment [47]. Positively charged nanoparticles have been shown to penetrate tumors better than neutral or negatively charged nanoparticles, resulting in the design of charge-reversal nanocarriers showing good tumor inhibition properties [48, 49].
Herein, we propose a strategy for overcoming penetration issues by combining VDAs and cytotoxic drugs within one nanocarrier. Although insufficient penetration still exists, this combination is expected to eradicate all the tumor cells inside a solid tumor, both at a distance from and in proximity to the tumor vasculature. Mechanistically, VDAs induce endothelial cell dysfunction, which diminishes blood flow and results in tumor cell necrosis at a distance from the tumor vasculature; the cytotoxic drugs directly kill tumor cells in proximity to the blood vessels. We have termed this approach a “cannon” and “pawn” combination for eradicating tumor cells both at a distance from and in proximity to the tumor vasculature.
We prepared PLG-CA4/DOX by synthesizing mPEG-PLG-g-CA4 and then loading DOX by hydrophobic accumulation and electronic interactions. Using immunofluorescent analysis, we observed that the PLG-CA4/DOX micelles delivered to tumor tissues were arrested near the blood vessels. However, release of the two drugs induced tumor necrosis in greater than 94% of the tumor area as shown in the pathological analysis. The results from this combination approach at drug delivery were much better compared to treatment with either drug alone. In addition, the inhibitory effects lasted over 72 h due to the retention properties of nanocarriers.
Fast growing murine colon C26 tumors were effectively inhibited by a single injection of PLG-CA4/DOX per week, in vivo. Noticeably, large solid tumors (C26 and 4T1 tumor volumes approximately 250 mm3 in size) were also effectively inhibited by PLG-CA4/DOX treatment. Commonly, large tumors are hard to treat by chemotherapy because of limited distribution and vascular degeneration [50-52]. Here, effective inhibition of large solid tumor growth suggests that a combination treatment with CA4 and DOX through the administration of PLG-CA4/DOX is a viable option, despite poor penetration.
Conclusion
Here, we propose a nanocarrier-based combination of VDAs and cytotoxic drugs for solid tumor therapy, in which eradication of tumor cells was expected both at a distance from and in proximity to blood vessels. In practice, the prepared PLG-CA4/DOX resulted in necrosis of greater than 94% of the tumor area, and the effect lasted for over 72 h. In in vivo tumor inhibition tests, PLG-CA4/DOX showed constant inhibition of C26 tumor growth in a once per week injection and effectively inhibited large C26 and 4T1 solid tumor growth. These results provide an effective strategy for solid tumor therapy.
Supplementary Material
Supplementary materials and methods, supplementary figures.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Projects 51173184, 51233004, 51373168, 51390484, 51473029, 81430087 and 51403204), and Jilin province science and technology development program (20130206058GX and 20130521011JH).
Competing Interests
The authors declare no competing interests.
References
1. Alexis F, Pridgen EM, Langer R, Farokhzad OC. Nanoparticle technologies for cancer therapy. Handb Exp Pharmacol. 2010;197:55-86
2. Schutz CA, Juillerat-Jeanneret L, Mueller H, Lynch I, Riediker M, Consortium N. Therapeutic nanoparticles in clinics and under clinical evaluation. Nanomedicine (Lond). 2013;8:449-67
3. Hrkach J, Von Hoff D, Ali MM, Andrianova E, Auer J, Campbell T. et al. Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med. 2012;4:128ra39
4. Yu H, Guo C, Feng B, Liu J, Chen X, Wang D. et al. Triple-layered pH-responsive micelleplexes loaded with siRNA and cisplatin prodrug for NF-Kappa B targeted treatment of metastatic breast cancer. Theranostics. 2016;6:14-27
5. Peer D, Karp JM, Hong S, FaroKHzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751-60
6. Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliver Rev. 2013;65:71-9
7. Moon JH, Moxley JW Jr, Zhang P, Cui H. Nanoparticle approaches to combating drug resistance. Future Med Chem. 2015;7:1503-10
8. Momparler RL, Karon M, Siegel SE, Avila F. Effect of adriamycin on DNA, RNA, and protein-synthesis in cell-free systems and intact-cells. Cancer Res. 1976;36:2891-5
9. Dasari S, Tchounwou PB. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur J Pharmacol. 2014;740:364-78
10. Horwitz SB. Taxol (paclitaxel): mechanisms of action. Ann Oncol. 1994;5(Suppl 6):S3-6
11. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653-64
12. Lee H, Fonge H, Hoang B, Reilly RM, Allen C. The effects of particle size and molecular targeting on the intratumoral and subcellular distribution of polymeric nanoparticles. Mol Pharmaceut. 2010;7:1195-208
13. Huo S, Ma H, Huang K, Liu J, Wei T, Jin S. et al. Superior penetration and retention behavior of 50 nm gold nanoparticles in tumors. Cancer Res. 2013;73:319-30
14. Wu W, Driessen W, Jiang X. Oligo(ethylene glycol)-based thermosensitive dendrimers and their tumor accumulation and penetration. J Am Chem Soc. 2014;136:3145-55
15. Sykes EA, Chen J, Zheng G, Chan WC. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. Acs Nano. 2014;8:5696-706
16. Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK. Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994;54:3352-56
17. Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2013;4:81-9
18. Dark GG, Hill SA, Prise VE, Tozer GM, Pettit GR, Chaplin DJ. Combretastatin A-4, an agent that displays potent and selective toxicity toward tumor vasculature. Cancer Res. 1997;57:1829-34
19. Vincent L, Kermani P, Young LM, Cheng J, Zhang F, Shido K. et al. Combretastatin A4 phosphate induces rapid regression of tumor neovessels and growth through interference with vascular endothelial-cadherin signaling. J Clin Invest. 2005;115:2992-3006
20. Song W, Tang Z, Zhang D, Li M, Gu J, Chen X. A cooperative polymeric platform for tumor-targeted drug delivery. Chem Sci. 2016;7:728-736
21. Siemann DW, Chaplin DJ, Horsman MR. Vascular-targeting therapies for treatment of malignant disease. Cancer. 2004;100:2491-99
22. Baguley BC, Siemann DW. Temporal aspects of the action of ASA404 (vadimezan; DMXAA). Expert Opin Inv Drug. 2010;19:1413-25
23. Tozer GM, Prise VE, Wilson J, Cemazar M, Shan SQ, Dewhirst MW. et al. Mechanisms associated with tumor vascular shut-down induced by combretastatin A-4 phosphate: Intravital microscopy and measurement of vascular permeability. Cancer Res. 2001;61:6413-22
24. Gaya AM, Rustin GJS. Vascular disrupting agents: A new class of drug in cancer therapy. Clin Oncol (R Coll Radiol). 2005;17:277-90
25. Chaplin DJ, Hill SA. The development of combretastatin A4 phosphate as a vascular targeting agent. Int J Radiat Oncol. 2002;54:1491-96
26. Siemann DW, Chaplin DJ, Walicke PA. A review and update of the current status of the vasculature-disabling agent combretastatin-A4 phosphate (CA4P). Expert Opin Inv Drug. 2009;18:189-97
27. Song W, Tang Z, Zhang D, Yu H, Chen X. Coadministration of vascular disrupting agents and nanomedicines to eradicate tumors from peripheral and central regions. Small. 2015;11:3755-61
28. Galbraith SM, Chaplin DJ, Lee F, Stratford MRL, Locke RJ, Vojnovic B. et al. Effects of combretastatin A4 phosphate on endothelial cell morphology in vitro and relationship to tumour vascular targeting activity in vivo. Anticancer Res. 2001;21:93-102
29. Grosios K, Holwell SE, McGown AT, Pettit GR, Bibby MC. In vivo and in vitro evaluation of combretastatin A-4 and its sodium phosphate prodrug. Br J Cancer. 1999;81:1318-27
30. Li J, Cona MM, Chen F, Feng Y, Zhou L, Zhang G. et al. Sequential systemic administrations of combretastatin A4 Phosphate and radioiodinated hypericin exert synergistic targeted theranostic effects with prolonged survival on SCID mice carrying bifocal tumor xenografts. Theranostics. 2013;3:127-37
31. Sengupta S, Eavarone D, Capila I, Zhao GL, Watson N, Kiziltepe T. et al. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature. 2005;436:568-72
32. Wang Y, Yang T, Wang X, Dai W, Wang J, Zhang X. et al. Materializing sequential killing of tumor vasculature and tumor cells via targeted polymeric micelle system. J Control Release. 2011;149:299-306
33. Nair LS, Laurencin CT. Biodegradable polymers as biomaterials. Prog Poly Sci. 2007;32:762-798
34. Tian H, Tang Z, Zhuang X, Chen X, Jing X. Biodegradable synthetic polymers: Preparation, functionalization and biomedical application. Prog Poly Sci. 2012;37:237-280
35. He C, Zhuang X, Tang Z, Tian H, Chen X. Stimuli-sensitive synthetic polypeptide-based materials for drug and gene delivery. Adv Healthc Mater. 2012;1:48-78
36. Li C. Poly(L-glutamic acid) - anticancer drug conjugates. Adv Drug Deliver Rev. 2002;54:695-713
37. Nemunaitis J, Cunningham C, Senzer N, Gray M, Oldham F, Pippen J. et al. A. Phase I study of CT-2103, a polymer-conjugated paclitaxel, and carboplatin in patients with advanced solid tumors. Cancer Invest. 2005;23:671-76
38. Koizumi F, Kitagawa M, Negishi T, Onda T, Matsumoto S.-I, Hamaguchi T. et al. Novel SN-38-incorporating polymeric micelles, NK012, eradicate vascular endothelial growth factor-secreting bulky tumors. Cancer Res. 2006;66:10048-56
39. Prompruk K, Govender T, Zhang S, Xiong CD, Stolnik S. Synthesis of a novel PEG-block-poly(aspartic acid-stat-phenylalanine) copolymer shows potential for formation of a micellar drug carrier. Int J Pharm. 2005;297:242-53
40. Deng J, Gao N, Wang Y, Yi H, Fang S, Ma Y. et al. Self-assembled cationic micelles based on PEG-PLL-PLLeu hybrid polypeptides as highly effective gene vectors. Biomacromolecules. 2012;13:3795-804
41. Lv S, Li M, Tang Z, Song W, Sun H, Liu H. et al. Doxorubicin-loaded amphiphilic polypeptide-based nanoparticles as an efficient drug delivery system for cancer therapy. Acta Biomater. 2013;9:9330-42
42. Xie Z, Guan H, Chen X, Lu C, Chen L, Hu X. et al. A novel polymer-paclitaxel conjugate based on amphiphilic triblock copolymer. J Control Release. 2007;117:210-16
43. Choe YH, Conover CD, Wu DC, Royzen M, Gervacio Y, Borowski V. et al. Anticancer drug delivery systems: multi-loaded N-4-acyl poly(ethylene glycol) prodrugs of ara-C. II. Efficacy in ascites and solid tumors. J Control Release. 2002;79:55-70
44. Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M. et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815-23
45. Tang L, Yang XJ, Yin Q, Cai KM, Wang H, Chaudhury I. et al. Investigating the optimal size of anticancer nanomedicine. P Natl Acad Sci USA. 2014;111:15344-49
46. Wang J, Mao W, Lock LL, Tang J, Sui M, Sun W. et al. The role of micelle size in tumor accumulation, penetration, and treatment. ACS Nano. 2015;9:7195-206
47. Sun Q, Sun X, Ma X, Zhou Z, Jin E, Zhang B. et al. Integration of nanoassembly functions for an effective delivery cascade for cancer drugs. Adv Mater. 2014;26:7615-21
48. Du JZ, Sun TM, Song WJ, Wu J, Wang J. A tumor-acidity-activated charge-conversional nanogel as an intelligent vehicle for promoted tumoral-cell uptake and drug delivery. Angew Chem Int Ed Engl. 2010;49:3621-26
49. Zhou Z, Shen Y, Tang J, Fan M, Van Kirk EA, Murdoch WJ. et al. Charge-reversal drug conjugate for targeted cancer cell nuclear drug delivery. Adv Funct Mater. 2009;19:3580-89
50. Wu J, Akaike T, Maeda H. Modulation of enhanced vascular permeability in tumors by a bradykinin antagonist, a cyclooxygenase inhibitor, and a nitric oxide scavenger. Cancer Res. 1998;58:159-65
51. Holash J, Wiegand SJ, Yancopoulos GD. New model of tumor angiogenesis: dynamic balance between vessel regression and growth mediated by angiopoietins and VEGF. Oncogene. 1999;18:5356-62
52. Graff BA, Benjaminsen IC, Brurberg KG, Ruud EBM, Rofstad EK. Comparison of tumor blood perfusion assessed by dynamic contrast-enhanced MRI with tumor blood supply assessed by invasive imaging. J Magn Reson Imaging. 2005;21:272-81
Author contact
![]() Corresponding author: Zhaohui Tang & Xuesi Chen
Corresponding author: Zhaohui Tang & Xuesi Chen