Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(9):1403-1414. doi:10.7150/thno.15394 This issue Cite
Research Paper
Serum Stability and Affinity Optimization of an M2 Macrophage-Targeting Peptide (M2pep)
Chayanon Ngambenjawong, Heather H. Gustafson, Julio M. Pineda, Nataly A. Kacherovsky, Maryelise Cieslewicz, Suzie H. Pun ![]()
Department of Bioengineering and Molecular Engineering and Sciences Institute, University of Washington, Seattle, WA 98195, United States.
Received 2016-2-27; Accepted 2016-4-14; Published 2016-6-15
Abstract

Tumor associated macrophages (TAMs) are a major stromal component of the tumor microenvironment in several cancers. TAMs are a potential target for adjuvant cancer therapies due to their established roles in promoting proliferation of cancer cells, angiogenesis, and metastasis. We previously discovered an M2 macrophage-targeting peptide (M2pep) which was successfully used to target and deliver a pro-apoptotic KLA peptide to M2-like TAMs in a CT-26 colon carcinoma model. However, the effectiveness of in vivo TAM-targeting using M2pep is limited by its poor serum stability and low binding affinity. In this study, we synthesized M2pep derivatives with the goals of increasing serum stability and binding affinity. Serum stability evaluation of M2pepBiotin confirmed its rapid degradation attributed to exolytic cleavage from the N-terminus and endolytic cleavages at the W10/W11 and S16/K17 sites. N-terminal acetylation of M2pepBiotin protected the peptide against the exolytic degradation while W10w and K(17,18,19)k substitutions were able to effectively protect endolytic degradation at their respective cleavage sites. However, no tested amino acid changes at the W10 position resulted in both protease resistance at that site and retention of binding activity. Therefore, cyclization of M2pep was investigated. Cyclized M2pep better resisted serum degradation without compromising binding activity to M2 macrophages. During the serum stability optimization process, we also discovered that K9R and W10Y substitutions significantly enhanced binding affinity of M2pep. In an in vitro binding study of different M2pep analogs pre-incubated in mouse serum, cyclic M2pep with K9R and W10Y modifications (cyclic M2pep(RY)) retained the highest binding activity to M2 macrophages over time due to its improved serum stability. Finally, we evaluated the in vivo accumulation of sulfo-Cy5-labeled M2pep and cyclic M2pep(RY) in both the CT-26 and 4T1 breast carcinoma models. Cyclic M2pep(RY) outperformed M2pep in both tumor localization and selective accumulation in M2-like TAMs. In conclusion, we report cyclic M2pep(RY) as our lead M2pep analog with improved serum stability and M2 macrophage-binding activity. Its enhanced utility as an in vivo M2-like-TAM-targeting agent was demonstrated in two tumor models, and is expected to be applicable for other tumor models or in models of M2 macrophage-related diseases.
Keywords: targeted drug delivery, M2 macrophages, M2pep, serum stability, binding affinity, immunomodulation.
Introduction
Despite advancement in medical research, cancer remains one of the leading causes of death in the United States [1]. One of the main limitations in cancer therapies is non-specific side effects, which significantly compromise patients' quality of life and restrict the effective dosage that can be administered [2]. Improvement in nanotechnology and molecular biology enables development of therapeutics that can more precisely target the diseased organs or cells, thus alleviating the side effects [3]. Ligand-mediated active targeting is a powerful strategy used to enhance cellular internalization, and hence potency, of drug-loaded nanoparticles or biomacromolecular therapeutics to cells overexpressing the corresponding receptors [2-4]. This active targeting strategy is used in ligand-decorated nanoparticles that are progressing in different stages of clinical trials as well as the recent FDA approvals of antibody-drug conjugates; Brentuximab vedotin and Trastuzumab emtansine [3,5,6].
The increased use of active targeting strategies for drug delivery or imaging applications especially in cancer is in part due to advances in molecular engineering and combinatorial screening techniques that have led to new targeting ligands for different cell targets [7,8]. Antibodies, proteins, oligonucleotides, peptides, and small molecules have all been successfully used as targeting moieties in several targeted drug delivery systems. The inherent advantages and disadvantages of each of these ligand classes has been reviewed in recent literature [3,8-10]. For tumor targeting applications, peptide targeting ligands offer significant advantages over antibodies due to their smaller size which can enhance tumor penetration, in addition to their lower immunogenicity and higher scalability. Small molecule screening is usually more tedious and less successful compared to the discovery of targeting peptides, which is greatly accelerated by robust phage display screening technology. Moreover, diverse functional groups of amino acids also allow for more flexible conjugation chemistry to cargo of interest. Nonetheless, using peptides as targeting moiety may have disadvantages from lower target affinity and serum stability, which in several cases have been successfully improved via multivalent display of the peptides and rational modifications respectively [8,11-13].
We have been interested in targeted drug delivery for local immunomodulation in solid tumors [13,14]. The immune-suppressive microenvironment of tumors has been reported to contribute toward immune evasion and tumor progression [15,16]. One strategy for tumor immunomodulation is selective depletion of tumor-associated macrophages (TAMs), which are known to mostly express an anti-inflammatory “M2” phenotype and contribute to tumor progression, angiogenesis, and metastasis [17]. We have previously performed phage display biopanning to identify an M2 macrophage-targeting peptide (M2pep) that binds preferentially to murine anti-inflammatory M2 macrophages/M2-like TAMs over other leukocytes including pro-inflammatory M1 macrophages/M1-like TAMs, which are known to promote anti-tumor immunity [14]. A fusion peptide comprised of M2pep with a pro-apoptotic peptide KLA was shown to successfully mediate selective depletion of M2-TAMs in a syngeneic CT-26 colon carcinoma model [14]. However, poor serum half-life and low target binding affinity (KD ~ 90 µM) of M2pep are two major obstacles limiting its therapeutic efficacy.
In this study, we aim to improve serum stability and binding affinity of M2pep through rational peptide engineering. Following successful optimization of M2pep, we demonstrate its enhanced performance in in vivo targeting of M2-like TAMs in both the syngeneic CT-26 colon carcinoma model as well as in the 4T1 breast carcinoma model.
Materials and Methods
Materials
Protected amino acids and 2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU) were purchased from AAPPTec (Louisvulle, KY) and AnaSpec (Fremont, CA). NovaPEG Rink Amide was purchased from Merck Millipore (Billerica, MA). Collagenase (C0130), dispase II, triisopropylsilane (TIPS), 1,2-ethanedithiol (EDT), 1,3-dimethoxybenzene (DMB), tris(2-carboxyethyl)phosphine hydrochloride (TCEP), copper(II) sulfate pentahydrate (CuSO4Ÿ5H2O), tris(3-hydroxypropyltriazolylmethyl)amine (THPTA), and sodium ascorbate were purchased from Sigma-Aldrich (St. Louis, MO). Sulfo-Cy5-alkyne was purchased from Lumiprobe (Hallandale Beach, FL). Streptavidin FITC was purchased from eBioscience (San Diego, CA). Pacific blue anti-mouse F4/80 antibody (Clone BM8) was purchased from Life Technologies (Grand Island, NY). PerCP anti-mouse/human CD11b antibody (Clone M1/70) and anti-mouse CD16/32 antibody (Fc receptor block, clone 93) were purchased from BioLegend (San Diego, CA). FITC anti-mouse Ly-6G antibody (Clone 1A8) was purchased from BD Pharmingen (San Diego, CA). Mouse macrophage colony-stimulating factor (M-CSF), interleukin-4 (IL-4), and interferon-γ (IFN-γ) were purchased from R&D Systems (Minneapolis, MN). Lipopolysaccharide (LPS) was purchased from InvivoGen (San Diego, CA). Normal mouse serum (Catalog number 10410) and other reagents were purchased from Thermo Fisher Scientific (Waltham, MA).
Peptide synthesis
Biotinylated peptides
Peptide synthesis was performed on an automated PS3 peptide synthesizer (Protein Technologies, Phoenix, AZ) following the standard Fmoc solid phase peptide synthesis chemistry. When needed, amino acids were manually coupled by incubation in a solution of amino acid and HCTU dissolved in 0.4 M N-methylmorpholine in DMF for 3 h. The coupling reaction was checked for completion by Kaiser Test as previously described [18]. Fmoc protecting groups were removed by two 30 min incubations in 20% (v/v) piperidine in DMF. Biotinylated peptides were synthesized from the biotinylated resin manually as follows: Fmoc-Lys(Mtt)-OH was first coupled to the NovaPEG rink amide resin. The Mtt-protecting group was removed by 3-min incubations in 1.8% (v/v) TFA in DCM until completion (15 - 20 times) as indicated by the disappearance of the yellow color (Mtt cation) in the drained deprotection solution. Biotin was then coupled to the resin, and the subsequent amino acid extension was continued on the peptide synthesizer. Peptides were acetylated at the N-terminus in acetic anhydride/triethylamine/DCM (1:1:5 v/v/v) for 2 h. Peptides were cleaved in TFA/TIPS/EDT/DMB (90:2.5:2.5:5 v/v/v/v) for 2.5 h. EDT was included in the cleavage solution only for the cysteine-containing peptides. The cleaved peptides were precipitated in cold ether twice and purified by RP-HPLC (Agilent 1200, Santa Clara, CA) using Phenomenex Fusion-RP C18 semi-preparative column (Torrance, CA) in H2O (0.1% TFA) as a mobile phase A and ACN (0.1% TFA) as a mobile phase B. Disulfide cyclization of the purified peptides was performed by incubation in deaerated 0.1 M ammonium bicarbonate buffer (4 mg/mL peptide concentration) for 2 days. The peptides were then desalted using the HyperSepTM C18 cartridge and confirmed for purity with RP-HPLC. Molecular weights of the purified peptides were confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS, Bruker Daltonics, Billerica, MA).
Sulfo-Cy5-labeled peptides
Azide-functionalized peptides were synthesized following the procedure described above replacing Fmoc-Lys(Mtt)-OH with Fmoc-D-Lys(N3)-OH at the C-terminus. Sulfo-Cy5 labeling via copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) was performed by reacting 1 eq. of peptide with 1.3 eq. of sulfo-Cy5-alkyne in the presence of 5 eq. of CuSO4Ÿ5H2O, THPTA, and sodium ascorbate. CuSO4Ÿ5H2O and THPTA were pre-mixed for a few minutes before adding to the peptide solution, and sodium ascorbate was added last. The final peptide concentration was 1 mM in H2O/DMF (1:1 v/v). The conjugation reaction was maintained at 37 °C for 1.5 h after which any solid precipitate was centrifuged. The supernatant was initially cleaned up by eluting through Sep-Pak C18 cartridge, concentrated by rotary evaporator, and then purified by RP-HPLC. Peptide was cyclized after sulfo-Cy5 labeling.
Serum stability study
The peptide of interest (30 µL of 10 mg/mL stock solution in dH2O) was added to normal mouse serum (300 µL) and incubated at 37 °C in an incubator. At each time point, an aliquot of 40 µL was drawn and an equal volume of ACN was added to the aliquot to precipitate serum proteins. The cloudy mixture was centrifuged at 10,000 rpm for 5 min and then supernatant removed. A solution of 1:1 H2O/ACN (80 µL) was added to the pellet and sonicated for 10 min to further extract the remaining peptides. This resuspended mixture was then centrifuged, and the supernatant was pooled with the former and vacuum-dried on a Speedvac machine. The dried pellet was solubilized in H2O (50 µL) by sonication for 10 min. The mixture was then centrifuged, and the supernatant was drawn and analyzed by MALDI-TOF MS.
Bone marrow harvest
All animal handling protocols were approved by the University of Washington Institutional Animal Care and Use Committee. Bone marrow harvest was performed following the previously reported protocol [14]. Briefly, femur and tibia were excised from 6-8 week-old female c57bl6/027 mice. Cuts were made on the ends of each bone, and RPMI 1640 medium was used to flush out bone marrow cells via 18G needle. The cells were cultured on petri dishes in RPMI 1640 medium supplemented with 20% donor horse serum, 1% antibiotic-antimycotic (AbAm), and 20 ng/mL M-CSF. After 7 d of culture, macrophages were activated by replacing M-CSF with 25 ng/mL IFN-γ and 100 ng/mL LPS for M1 macrophage or 25 ng/mL IL-4 for M2 macrophage. The macrophages were activated for 2 d and then scraped off the petri dishes with cell lifter for binding studies.
Binding study
Peptide solutions at different concentrations for binding study were prepared by dilutions from the stock solution (10 mg/mL in dH2O) with PBS containing 1% BSA (PBSA). M1 and M2 macrophages (50,000 cells/well) were seeded on a black 96-well plate and incubated in the peptide solutions on ice for 20 min. Unbound peptides were washed off with PBSA twice. The macrophages were subsequently incubated with streptavidin-FITC for 15 min on ice to probe for the bound peptides. Excess streptavidin-FITC was washed off with PBSA twice, and the macrophages were then resuspended in PBS for analysis by MACSQuant Flow Cytometer (Miltenyi Biotec, San Diego, CA). Propidium iodide was added to the samples before the data acquisition to discriminate dead cells. Flow cytometry data were analyzed with FlowJo Analysis Software (Tree Star, Ashland, OR). Binding study of peptides post serum incubation was similarly performed with the following additions: The peptides (5 mM stock solution in H2O) were diluted in serum to a final concentration of 0.5 mM and incubated at 37 °C in an incubator. At each time point, an aliquot of the peptides was drawn and diluted in PBS to the final concentration for the binding study. The diluted aliquot was heated at 80 °C for 30 min to inactivate serum proteins, stored in a -20 °C freezer, and thawed for use in the binding study after aliquots at all time points were collected.
In vivo biodistribution study in CT-26 and 4T1 tumor models
CT-26 colon carcinoma cells and 4T1 mammary carcinoma cells were maintained in DMEM medium supplemented with 10% fetal bovine serum (FBS) and 1% Penicillin Streptomycin. Subcutaneous tumors were formed by the injection of 106 CT-26 or 4T1 cells into the right flank or mammary fat pad, respectively, of 6 week-old female BALB/c mice. Tumor-bearing mice were used 2 weeks post inoculation at a maximum tumor diameter of 1.5 cm. The mice were injected retro-orbitally with 150 µL of PBS control or sulfo-Cy5-labeled peptides (2.24 nmol in PBS with 10% DMSO). At 20 min after peptide injection, mice were anesthetized with a ketamine/xylazine cocktail and perfused with PBS. Organs were harvested and imaged on the Caliper Xenogen IVIS (PerkinElmer, Hopkinton, MA). The fluorescence intensity emitted from each organ was subsequently quantified utilizing Xenogen's Living Image software. Following imaging, each tumor was then processed into single cell suspension by mincing into small pieces, homogenizing on gentleMACS Dissociator (Miltenyi Biotec, San Diego, CA), and incubating in 5 mL RPMI 1640 medium with 100 μL collagenase (10,000 CDU/mL stock solution) and 100 μL dispase II (32 mg/mL stock solution) for 40 min in the TC incubator. The dissociated single cells were collected through 70 μm cell strainer and plated on a black round bottom 96-well plate at the density of 200,000 cells/well. Incubation of the cells with Fc receptor block was performed prior to staining with CD11b, Ly-6G, and F4/80 antibodies. Finally, the cells were fixed with 4% paraformaldehyde and analyzed with flow cytometer.
Statistical analysis
Normalized median fluorescent intensity values in the binding studies were presented as mean ± SD of triplicate in the same experiment. Statistical significance in binding between M1 and M2 macrophages or binding between M2pep and its modified analogs was evaluated by Student's unpaired t-test. For clarity of presentation, only statistical significance at 50 µM concentration was labeled to elucidate the general trend in binding activity. Analysis of the binding of M2pep analogs post-serum incubation at the 24-h time point was performed by One-way ANOVA with Tukey's post-hoc tests. Accumulation of M2pep analogs in different organs were compared by Student's unpaired t-test. Finally, intratumoral biodistribution of each M2pep analog in different tumor subpopulations as well as comparison of accumulation in M2-like TAMs of the M2pep analogs were analyzed by one-way ANOVA with Tukey's post-hoc tests. All data analysis was processed on GraphPad Prism 6 (GraphPad Software Inc., La Jolla, CA). P < 0.5 was considered as statistically significant.
Results and Discussion
The effect of N-terminal acetylation on serum stability and binding activity of M2pepBiotin
In vivo applications of peptides as therapeutic or imaging agents are usually limited by their short serum half-life, typically on the order of a few minutes, due to degradation by serum proteases as well as clearance via kidneys and liver [19-21]. In regard to serum stability, several strategies including N-terminal acetylation, C-terminal amidation, D-amino acid substitution, and peptide stapling/macrocyclization have been utilized to impart peptide resistance against peptidase degradation [19,22]. In this study, serum stability of M2pep was first evaluated to determine peptidase-susceptible sites. A series of rationally-engineered peptides were then synthesized and evaluated for their serum stability and binding activity (Table 1).
Amino acid sequences and molecular weights of M2pep analogs.
| Name | Sequence | Mw (Calculated) | Mw (Observed) | Note |
|---|---|---|---|---|
| Linear M2pepBiotin analogs | ||||
| M2pepBiotin | YEQDPWGVKWWYGGGSKK(K-Biotin) | 2,524.86 | 2,524.21 | Original sequence |
| AcM2pepBiotin | Ac-YEQDPWGVKWWYGGGSKKK(K-Biotin) | 2,694.77 | 2,693.32 | |
| W10w | Ac-YEQDPWGVKwWYGGGSKKK(K-Biotin) | 2,694.77 | 2,694.04 | |
| W(10,11)w | Ac-YEQDPWGVKwwYGGGSKKK(K-Biotin) | 2,694.77 | 2,693.83 | |
| W10P | Ac-YEQDPWGVKPWYGGGSkkk(K-Biotin) | 2,605.67 | 2,605.47 | |
| W10Y | Ac-YEQDPWGVKYWYGGGSkkk(K-Biotin) | 2,671.73 | 2,671.02 | |
| K9R | Ac-YEQDPWGVRWWYGGGSKKK(K-Biotin) | 2,722.78 | 2,721.22 | |
| AcM2pep(RY)Biotin | Ac-YEQDPWGVRYWYGGGSkkk(K-Biotin) | 2,699.74 | 2,699.45 | Supplementary info. |
| W10(P,D,T,R,H) (Crude mixture) | Ac-YEQDPWGVK(P,D,T,R,H)WYGGGSkkk(K-Biotin) | P: 2,605.67 D: 2,623.64 T: 2,609.66 R: 2,664.74 H: 2,645.70 | P:2,604.96 D:2,624.78 T:not detected R:2,664.17 H:2,646.17 | Supplementary info. |
| Y12y | Ac-YEQDPWGVRYWyGGGSkkk(K-Biotin) | 2,699.74 | 2,698.65 | . |
| P5Hyp | Ac-YEQD(Hyp)WGVRYWyGGGSkkk(K-Biotin) | 2,715.74 | 2,715.00 | |
| R0 | Ac-RYEQDPWGVRYWyGGGSkkk(K-Biotin) | 2,855.93 | 2,856.18 | |
| Cyclic M2pepBiotin analogs | ||||
| Cyclic M2pep(RY)Biotin | CGYEQDPWGVRYWYGCkkk(K-Biotin) | 2,718.16 | 2,717.59 | GGGS spacer to GC |
| cRYWY | CGDPWGVRYWYGCkkk(K-Biotin) | 2,297.74 | 2,297.25 | GGGS spacer to GC Deletion of Y1E2Q3 |
| cRYW | CGDPWGVRYWGCkkk(K-Biotin) | 2,134.56 | 2,134.00 | GGGS spacer to GC Deletion of Y1E2Q3, Y12 |
| Sulfo-Cy5-labeled peptides | ||||
| M2pep-sulfoCy5 | YEQDPWGVKWWYGGGSKKK(k(N3)-sulfoCy5) | 3,132.74 | 3,132.17 | |
| Cyclic M2pep(RY)-sulfoCy5 | CGYEQDPWGVRYWYGCkkk(k(N3)-sulfoCy5) | 3,197.86 | 3,198.13 | |
All peptides are amidated at the C terminus. Ac denotes N-terminal acetylation. Small letters denote d-amino acids.
Amino acid positions are in reference to the original M2pepBiotin sequence.
Modifications to the original M2pepBiotin are highlighted in red.
Mw observed from MALDI-TOF MS is reported as [M+H]+.
To evaluate serum stability of M2pep, the peptide was incubated in mouse serum, and aliquots of the serum at various time points were withdrawn for analysis of degradation patterns by MALDI-TOF MS. M2pepBiotin was quickly degraded in mouse serum and was no longer detected after 4 h of serum incubation (Fig. 1A). Instead, a serum peptide (652.15 Da) became the predominant signal detected from this time point onwards. Analysis of the degraded peptide fragments revealed that the peptide degradation is both exolytic from the N-terminus and endolytic within the peptide (Fig. 1B). Poor serum stability of this peptide therefore warranted investigation into more serum-resistant M2pep analogs.
(A) MALDI-TOF MS spectra of M2pepBiotin at different serum incubation times. (B) Zoomed-in MALDI-TOF MS spectra of M2pepBiotin at 1 and 2 h. (C) MALDI-TOF MS spectra of AcM2pepBiotin at different serum incubation times. Adjacent to the primary peaks are their respective Na+ adducts. (D) Binding of AcM2pepBiotin versus M2pepBiotin to M1 and M2 macrophages. Stars denote statistical significance between M1 and M2 macrophages in the same treatment group. * P < 0.05, ns = not statistically significant.

N-terminal acetylation of peptides is a common peptide modification strategy shown to effectively prevent N-terminal peptide degradation by exopeptidases [23,24]. Hence, M2pep was acetylated at the N-terminus by reaction with acetic anhydride before cleavage from resin. The acetylated peptide, AcM2pepBiotin, was indeed resistant to N-terminal degradation (Fig. 1C). However, this modification did not improve the overall serum stability of the peptide since the peptide remained susceptible to endolytic degradation. With the N-terminal protection, the peptide degradation patterns became clearer showing two distinct peptide fragments corresponding to endolytic cleavages at W10/W11 and S16/K17 sites with the latter occurring with relatively faster kinetics.
Next, the binding specificity of AcM2pepBiotin to M2 versus M1 macrophage was evaluated using primary bone marrow-derived macrophages polarized to the M1 or M2 phenotypes as we previously described [14]. AcM2pepBiotin retained binding selectivity to M2 macrophages over M1 macrophages although with slight, but not statistically significant, reduction in binding activity compared to M2pep at the concentrations tested (Fig. 1D). Hence, we decided to acetylate all our subsequent peptides to protect against exolytic degradation and focused our next modifications at the W10/W11 and S16/K17 endolytic cleavage sites.
The effect of D-amino acid substitutions on serum stability and binding activity of AcM2pepBiotin
As reported previously, our high throughput sequencing of phage that bound to M2 macrophages showed a consensus motif of DPWXXXXW where X denoted other amino acids with less or no consensus [25]. Hence, we deduced that the motif may be essential for binding to the M2pep receptor and targeted our next peptide modification to the W10 position which lies in the non-consensus region. Since D-amino acids are not normally present in nature and substitution with D-amino acids into peptide sequences often confer serum stability [11], we explored the effect of D-tryptophan substitution at the W10 position (W10w). Despite its effective protection against endolytic cleavage at the W10/W11 site (Fig 2A), the W10w substitution abrogated binding activity of the peptide (Fig. 2B). In addition, D-tryptophan substitutions to both W10 and W11 (W(10,11)w) also did not restore the binding activity of M2pep.
(A) MALDI-TOF MS spectra of W10w analog at different serum incubation times. Adjacent to the primary peaks are their respective Na+ adducts. (B) Binding of M2pepBiotin compared to W10w and W(10,11)w analogs to M1 and M2 macrophages. Unless labeled in pairs, stars denote statistical significance between M1 and M2 macrophages in the same treatment group. * P < 0.05.

The effect of L-amino acid substitutions on serum stability and binding activity of AcM2pepBiotin
Next, we investigated how substitutions of W10 with other ʟ-amino acids may help protect against degradation at the W10/W11 site. We first synthesized a W10P analog due to the apparent serum stability at the P5W6 site in AcM2pepBiotin and also a W10Y analog since tyrosine, like tryptophan, is an amino acid with a bulky aromatic side chain [26]. For both analogs, the trilysine spacer was also replaced with D-lysines to evaluate if this replacement would improve serum stability at the S16/K17 site. In addition, we also studied a K9R analog. K9 is the only lysine in the targeting region and being able to replace this lysine with arginine would allow us to synthesize AcM2pep with C-terminal lysine for facile and site-selective conjugation to drug cargos with activated NHS ester. D-lysine substitution at the trilysine spacer improved serum stability at S16/K17 site while W10P, W10Y, and K9R substitutions did not confer protection at the W10/W11 site (Fig. 3A). Interestingly, binding studies showed that both K9 and W10 amino acid positions are essential for binding activity of AcM2pepBiotin (Fig. 3B). In this case, K9R and W10Y substitutions significantly increased binding activity of AcM2pepBiotin while a W10P substitution abrogated the binding activity. Combining K9R and W10Y substitutions together (AcM2pep(RY)Biotin) did not improve the binding activity any further compared to the K9R analog and also did not improve serum stability at the W10/W11 site (Fig. S1). Given high natural abundance of pi-cation interactions between Lys/Arg and Phe/Tyr/Trp in many proteins [27], K9 may be involved in the interaction with the aromatic amino acids either on the M2pep itself or on the target receptor, and the higher binding affinity of the K9R analog may thus be attributed to the stronger pi-cation interaction of the relatively more hydrophobic arginine with the corresponding aromatic amino acid [28,29]. Additional investigation into W10(D,R,H) substitutions also did not give a positive result in term of improving serum stability (Fig. S2).
MALDI-TOF MS spectra of M2pep analogs: (A) W10P, (B) W10Y, and (C) K9R at different serum incubation times. Adjacent to the primary peaks are their respective Na+ adducts. (D) Binding of the M2pep analogs to M1 and M2 macrophages. Unless labeled in pairs, stars denote statistical significance between M1 and M2 macrophages in the same treatment group. * P < 0.05.

The effect of cyclization on serum stability and binding activity of M2pepBiotin
Head-to-tail cyclization of peptides may impose structural rigidity to the peptides and in several cases, leads to improvement in bioactivity and serum stability [30]. Hence, we modified linear M2pep(RY)Biotin with two flanking cysteines to enable disulfide cyclization of the peptide. The synthesized peptide (cyclic M2pep(RY)Biotin) had markedly improved serum stability over the linear AcM2pep(RY)Biotin (Fig. 4A). Unlike the linear, acetylated M2pep derivatives which were degraded within 2 to 8 hrs post-serum incubation, cyclic M2pep(RY)Biotin remained detectable by MALDI-TOF MS even after serum incubation for 48 h. No endolytic cleavage was observed at the W10/W11 site in cyclic M2pep(RY)Biotin for up to at least 8 h. At 24 and 48 h, however, signals corresponding to N-terminal degradation and W10/W11 endolytic cleavage were observed. This degradation might be attributed to reduction of the disulfide bond over time, which converts the peptide to the protease-accessible linear form (Fig. 4B). Interestingly, cleavage of biotin from the peptide was also observed at later time points and was not appreciated previously due to poor serum stability of the other analogs. Furthermore, cyclic M2pep(RY)Biotin has comparable binding activity to the linear AcM2pep(RY)Biotin, the highest affinity linear M2pep analog (Fig. 4C). The binding curve of cyclic M2pep(RY)Biotin was shown in Fig. S3. Hence, in this study, we showed that cyclization of M2pep(RY)Biotin significantly imparted serum stability over all linear AcM2pep analogs while retaining high binding activity comparable to AcM2pep(RY)Biotin. In the future, more stable cyclization strategies, such as incorporation of azide- and alkyne-functionalized amino acids for cyclization via CuAAC, may be investigated to further improve serum stability of this peptide.
(A) MALDI-TOF MS spectra of cyclic M2pep(RY)Biotin at different serum incubation times. (B) Zoomed-in MALDI-TOF MS spectra of cyclic M2pep(RY)Biotin at 48 h. (C) Binding of cyclic M2pep(RY)Biotin and AcM2pep(RY)Biotin to M1 and M2 macrophages. (D) Binding of cyclic M2pep(RY)Biotin, cRYWY, and cRYW to M1 and M2 macrophages. Unless labeled in pairs, stars denote statistical significance between M1 and M2 macrophages in the same treatment group. * P < 0.05, ns = not statistically significant.

In an effort to truncate the cyclic M2pep(RY)Biotin sequence, we investigated the effect of deleting amino acids that lie outside the DPWXXXXW motif by synthesizing two cyclic M2pep(RY)Biotin analogs; 1) cRYWY having deletion of Y1, E2, and Q3 and 2) cRYW containing deletions of Y1, E2, Q3, and also Y12. Both cRYWY and cRYW analogs have significantly lower binding activity compared to cyclic M2pep(RY)Biotin implying that Y1, E2, and Q3 may be important for binding to M2 macrophages (Fig 4D). However, Y12 may not be a critical residue since cRYWY and cRYW analogs have similar binding activity.
The effect of Y12y on serum stability and binding activity of AcM2pep(RY)Biotin
Since Y12 is not required for binding activity, we next synthesized linear AcM2pep(RY)Biotin with a D-tyrosine substitution at the Y12 position (Y12y) to evaluate if this substitution confers protection to the nearby W10/W11 cleavage site. However, the Y12y analog did not improve serum stability at the W10/W11 site (Fig. 5A) and also unexpectedly reduced binding activity of the peptide (Fig. 5B). Thus, while deletion of Y12 does not affect binding, substitution with a D-amino acid reduces binding affinity. Since AcM2pepBiotin shows trends of slightly reduced binding compared with M2pepBiotin (Fig 1D), we investigated the effect of introducing an arginine (R0) at the start of the AcM2pep(RY)Biotin sequence to offset for loss in amine from acetylation of M2pep. In addition, we also substituted P5 with hydroxyproline (HyP), which has been shown previously to improve binding activity of another proline-containing peptide [31]. Both analogs were synthesized with the Y12y substitution, and binding study was evaluated in comparison to the Y12y analog. The R0 analog did not improve binding activity any further while P5Hyp significantly ablated the binding activity (Fig. 5C).
(A) MALDI-TOF MS spectra of Y12y analog at different serum incubation times. (B) Binding of AcM2pep(RY) and Y12y to M1 and M2 macrophages. (C) Binding study Y12y, P5Hyp, and R0 analogs to M1 and M2 macrophages. Stars denote statistical significance between M1 and M2 macrophages in the same treatment group. * P < 0.05, ns = not statistically significant.

Binding of M2pep analogs post serum incubation
For TAM targeting applications, M2pep analogs will be exposed to serum during systemic circulation. Therefore, we next studied how the serum stability of M2pep analogs affects subsequent binding to target cells. Peptides were incubated with mouse serum at 37 °C to mimic peptidase exposure in systemic circulation, and aliquots of the serum were withdrawn at various time points for binding studies with polarized macrophages. In general, binding activity of the M2pep analogs corresponded with the peptides' serum stability (Fig. 6). Binding studies were performed at 150 µM of M2pepBiotin and 50 µM of AcM2pep(RY)Biotin and cyclic M2pep(RY)Biotin such that all the analogs had similar extent of binding at 0 h. M2pepBiotin had the fastest and greatest decline in binding activity while cyclic M2pep(RY)Biotin had the least decline which was also partially accounted by the cleavage of biotin. At 24 h, cyclic M2pep(RY)Biotin retained the highest binding activity compared to M2pepBiotin and AcM2pep(RY)Biotin reaffirming the highest serum stability of this M2pep analog observed in the previous study.
M2 macrophage-binding study of M2pepBiotin, AcM2pepRY)Biotin, and cyclic M2pep(RY)Biotin pre-incubated in serum for different durations. * P < 0.05.

Biodistribution of M2pep analogs in CT-26 and 4T1 tumor models
Finally, we assessed the effectiveness of cyclic M2pep(RY) as an in vivo TAM-targeting ligand in a syngeneic CT-26 colon carcinoma and a 4T1 breast carcinoma tumor model. The biodistribution study was first performed in the CT-26 tumor model which we previously used to evaluate the original M2pep [14]. CT-26 tumor-bearing mice were intravenously administered with PBS control, M2pep-sulfoCy5, or cyclic M2pep(RY)-sulfoCy5 and perfused 20 min after. Organs were then harvested and imaged under the Xenogen IVIS. Subsequently, the tumors were processed into cell suspension, stained with antibodies, and analyzed for intratumoral distribution by flow cytometry. The gating strategy is depicted in Fig. S4. In general, higher accumulation of cyclicM2pep(RY)-sulfoCy5 than M2pep-sulfoCy5 in tumors and other organs was observed with an exception of liver and kidneys (Fig. 7A(i-ii)). The highest fluorescent signal was observed in kidneys where the peptides were filtered and excreted. Intratumorally, cyclic M2pep(RY)-sulfoCy5 promoted significantly higher uptake in M2-like TAMs compared to M2pep-sulfoCy5 while selectivity to M2-like TAMs over M1-like TAMs and non-TAM CD11b- cells was observed in both peptide treatments (Fig. 7A(iii)).
In vivo biodistribution study of M2pep-sulfoCy5 versus cyclic M2pep(RY)-sulfoCy5 in (A) CT-26 tumor model (n = 5) and (B) 4T1 tumor model (n = 3); (i) Representative xenogen images of the harvested organs (∆ indicates the corresponding pair of the tumor images and the scale bar), (ii) Quantified fluorescence intensity in each organ (Stars denote statistical significance relative to PBS and M2pep-sulfoCy5-treated groups. P < 0.05), and (iii) Intratumoral accumulation in CD11b- cells, M1-like TAMs, and M2-like TAMs. Unless labeled in pairs, stars denote statistical significance relative to both CD11b- cells and M1-like TAMs in the same treatment group. * P < 0.05.
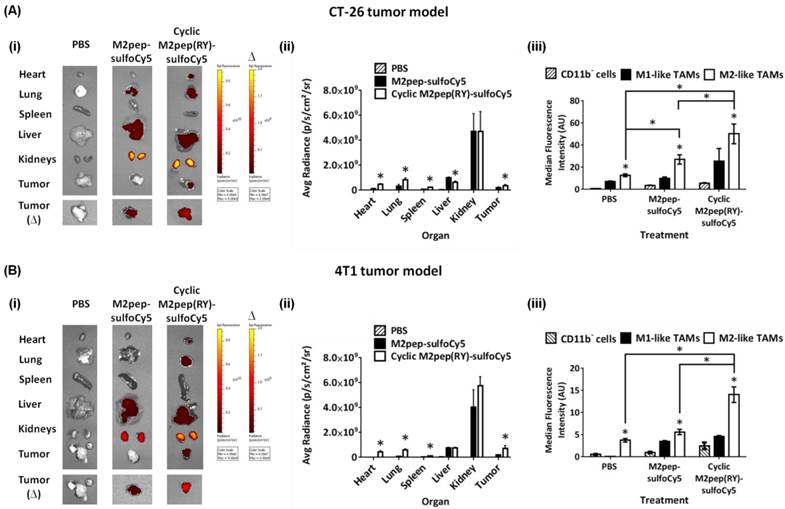
Following successful application of cyclic M2pep(RY) for targeting M2-like TAMs in CT-26 model, we further evaluated its effectiveness as an in vivo targeting ligand in the 4T1 breast tumor model since high accumulation of M2-like TAMs in breast cancer has been correlated to poor disease prognosis [32]. As observed in the CT-26 tumor model, cyclic M2pep(RY)-sulfoCy5 localized to 4T1 tumors to a greater extent compared to M2pep-sulfoCy5 (Fig. 7B(i-ii)), and also accumulated preferentially in M2-like TAMs (Fig. 7B(iii)). Hence, we have demonstrated through the studies of two tumor models that cyclic M2pep(RY) can be effectively used as an in vivo M2-TAM-targeting ligand due to its superior serum stability and enhanced binding affinity over the original M2pep. Its utility is expected to extend across multiple tumor models. In perspective, cyclic M2pep(RY)-mediated delivery of therapeutic cargos or imaging agents to M2-like TAMs could enable scientists to develop a more potent and selective M2-like TAM immunomodulation regimen or to better monitor their behavior over the course of tumor development.
Conclusions
M2pep was successfully optimized for serum stability via cyclization of the peptide as well as replacement of trilysine spacer to D-lysines. Binding activity of M2pep was further enhanced via K9R and W10Y substitutions. The optimized cyclic M2pep(RY) performed significantly better than M2pep at tumor localization and M2-like TAM accumulation in both CT-26 and 4T1 tumor models confirming its superior utility as an in vivo M2-like TAM-targeting ligand.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by NIH 1R01CA177272. Chayanon Ngambenjawong was supported by an Anandamahidol Foundation Fellowship. Heather H. Gustafson was supported by the Cardiovascular Pathology Training Grant (5 T32 HL 007312-37). Julio M. Pineda was supported by a Mary Gates Research Scholarship. Maryelise Cieslewicz was supported by a National Science Foundation Graduate Fellowship. We thank David S.H. Chu for consultation on serum stability study of peptides.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2015. CA Cancer J Clin. 2015;65:5-29
2. Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2002;2:750-63
3. Sanna V, Pala N, Sechi M. Targeted therapy using nanotechnology: Focus on cancer. Int J Nanomedicine. 2014;9:467-83
4. Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci U S A. 2007;104:15549-54
5. Chari RVJ, Miller ML, Widdison WC. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew Chemie - Int Ed. 2014;53:3796-827
6. Srinivasarao M, Galliford CV, Low PS. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat Rev Drug Discov. 2015;14:203-19
7. Khoury GA, Smadbeck J, Kieslich CA, Floudas CA. Protein folding and de novo protein design for biotechnological applications. Trends Biotechnol. 2014;32:99-109
8. Brown KC. Peptidic tumor targeting agents: the road from phage display peptide selections to clinical applications. Curr Pharm Des. 2010;16:1040-54
9. Yu MK, Park J, Jon S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics. 2012;2:3-44
10. Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2-25
11. Kim J-W, Kim T-D, Hong BS, Kim OY, Yoon W-H, Chae C-B. et al. A serum-stable branched dimeric anti-VEGF peptide blocks tumor growth via anti-angiogenic activity. Exp Mol Med. 2010;42:514-23
12. Akcan M, Stroud MR, Hansen SJ, Clark RJ, Daly NL, Craik DJ. et al. Chemical re-engineering of chlorotoxin improves bioconjugation properties for tumor imaging and targeted therapy. J Med Chem. 2011;54:782-7
13. Ngambenjawong C, Cieslewicz M, Schellinger JG, Pun SH. Synthesis and evaluation of multivalent M2pep peptides for targeting alternatively activated M2 macrophages. J Control Release. 2016;224:103-11
14. Cieslewicz M, Tang J, Yu JL, Cao H, Zavaljevski M, Motoyama K. et al. Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc Natl Acad Sci U S A. 2013;110:15919-24
15. Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014-22
16. Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904-12
17. Lamagna C, Aurrand-Lions M, Imhof B a. Dual role of macrophages in tumor growth and angiogenesis. J Leukoc Biol. 2006;80:705-13
18. Zheng J-S, Tang S, Qi Y-K, Wang Z-P, Liu L. Chemical synthesis of proteins using peptide hydrazides as thioester surrogates. Nat Protoc. 2013;8:2483-95
19. Cochran R, Cochran F. Phage display and molecular imaging: expanding fields of vision in living subjects. Biotechnol Genet Eng Rev. 2010;27:57-94
20. McGregor DP. Discovering and improving novel peptide therapeutics. Curr Opin Pharmacol. 2008;8:616-9
21. Pollaro L, Heinis C. Strategies to prolong the plasma residence time of peptide drugs. Medchemcomm. 2010;1:319-24
22. Walensky LD, Bird GH. Hydrocarbon-Stapled Peptides: Principles, Practice, and Progress. J Med Chem. 2014;57:6275-88
23. Sahu A, Soulika AM, Morikis D, Spruce L, Moore WT, Lambris JD. Binding kinetics, structure-activity relationship, and biotransformation of the complement inhibitor compstatin. J Immunol. 2000;165:2491-9
24. Nguyen LT, Chau JK, Perry NA, de Boer L, Zaat SAJ, Vogel HJ. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS One. 2010;5:1-8
25. Liu GW, Livesay BR, Kacherovsky NA, Cieslewicz M, Lutz E, Waalkes A. et al. Efficient Identification of Murine M2 Macrophage Peptide Targeting Ligands by Phage Display and Next-Generation Sequencing. Bioconjug Chem. 2015;26:1811-7
26. Bordo D, Argos P. Suggestions for “safe” residue substitutions in site-directed mutagenesis. J Mol Biol. 1991;217:721-9
27. Gallivan JP, Dougherty DA. Cation-pi interactions in structural biology. Proc Natl Acad Sci U S A. 1999;96:9459-64
28. Dougherty DA. Cation-pi Interactions Involving Aromatic Amino Acids. J Nutr. 2007;137:1504S-1508
29. Hughes RM, Wiggins KR, Khorasanizadeh S, Waters ML. Recognition of trimethyllysine by a chromodomain is not driven by the hydrophobic effect. Proc Natl Acad Sci U S A. 2007;104:11184-8
30. Clark RJ, Fischer H, Dempster L, Daly NL, Rosengren KJ, Nevin ST. et al. Engineering stable peptide toxins by means of backbone cyclization: stabilization of the alpha-conotoxin MII. Proc Natl Acad Sci U S A. 2005;102:13767-72
31. Kolodziej AF, Zhang Z, Overoye-Chan K, Jacques V, Caravan P. Peptide optimization and conjugation strategies in the development of molecularly targeted magnetic resonance imaging contrast agents. Methods Mol Biol. 2014;1088:185-211
32. Laoui D, Movahedi K, Van Overmeire E, Van den Bossche J, Schouppe E, Mommer C. et al. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. Int J Dev Biol. 2011;55:861-7
Author contact
![]() Corresponding author: Email address: spunedu, 3720 15th Ave NE, Foege N530P, Box 355061, Seattle, WA 98195
Corresponding author: Email address: spunedu, 3720 15th Ave NE, Foege N530P, Box 355061, Seattle, WA 98195