Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2016; 6(9):1415-1424. doi:10.7150/thno.15250 This issue Cite
Research Paper
Colorimetric TMPRSS2-ERG Gene Fusion Detection in Prostate Cancer Urinary Samples via Recombinase Polymerase Amplification
Kevin M. Koo1, Eugene J.H. Wee1 ![]() , Matt Trau1, 2
, Matt Trau1, 2 ![]()
1. Centre for Personalized Nanomedicine, Australian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, QLD 4072, Australia;
2. School of Chemistry and Molecular Biosciences, The University of Queensland, QLD 4072, Australia.
Received 2016-2-12; Accepted 2016-4-27; Published 2016-6-15
Abstract

TMPRSS2 (Exon 1)-ERG (Exon 4) is the most frequent gene fusion event in prostate cancer (PC), and is highly PC-specific unlike the current serum prostate specific antigen (PSA) biomarker. However, TMPRSS2-ERG levels are currently measured with quantitative reverse-transcription PCR (RT-qPCR) which is time-consuming and requires costly equipment, thus limiting its use in clinical diagnostics. Herein, we report a novel rapid, cost-efficient and minimal-equipment assay named “FusBLU” for detecting TMPRSS2-ERG gene fusions from urine. TMPRSS2-ERG mRNA was amplified by isothermal reverse transcription-recombinase polymerase amplification (RT-RPA), magnetically-isolated, and detected through horseradish peroxidase (HRP)-catalyzed colorimetric reaction. FusBLU was specific for TMPRSS2-ERG mRNA with a low visual detection limit of 105 copies. We also demonstrated assay readout versatility on 3 potentially useful platforms. The colorimetric readout was detectable by naked eye for a quick yes/no evaluation of gene fusion presence. On the other hand, a more quantitative TMPRSS2-ERG detection was achievable by absorbance/electrochemical measurements. FusBLU was successfully applied to 12 urinary samples and results were validated by gold-standard RT-qPCR. We also showed that sediment RNA was likely the main source of TMPRSS2-ERG mRNA in urinary samples. We believe that our assay is a potential clinical screening tool for PC and could also have wide applications for other disease-related fusion genes.
Keywords: TMPRSS2-ERG, FusBLU
Introduction
Prostate cancer (PC) is the most common form of cancer and the second leading cause of cancer death in males of developed countries [1]. The customary screening approach for prostate biopsies is mainly based on high serum prostate specific antigen (PSA) levels and/or abnormal digital rectal examination (DRE). PSA is the primary PC biomarker used by clinical practitioners and has led to a significant increase in PC diagnosis since its introduction [2]. However, due to its low PC-specificity, the majority of PSA-diagnosed cases are actually benign and do not require medical intervention. This has in turn led to redundant medical expenses as well as negative effects on patients' overall well-being [3]. Thus, there is a pressing need for better PC diagnostic biomarkers, particularly one with high PC-specificity for discriminating benign cases from aggressive PC cases.
In 2005, Tomlins and co-workers identified recurrent PC fusion genes between the promoter of transmembrane protease, serine 2 (TMPRSS2) and the coding sequence of erythroblastosis virus E26 (ETS) family members [4]. These chromosomal rearrangements-induced fusions resulted in androgen-dependent overexpression of oncogenic ETS transcription factors [5]. The fusion of TMPRSS2 exon 1 to ERG exon 4 (TMPRSS2-ERG) is the most frequent subtype of these gene fusions, appearing in about 50% of PC patients and 90% of all PC gene fusions [6]. More importantly, TMPRSS2-ERG is highly PC-specific and absent in non-PC samples. Furthermore, TMPRSS2-ERG may correlate with PC aggression and metastatic potential [7-9]; potentially making them more attractive as diagnostic and prognostic biomarkers than serum PSA.
TMPRSS2-ERG mRNA is commonly detected from the cell sediment fraction of urine [10, 11]. Recently, Nilsson and co-workers had reported TMPRSS2-ERG detection in urinary exosomes [12]. Additionally, it is also highly plausible that TMPRSS2-ERG mRNA is freely circulating within the urine sample. Given that TMPRSS2-ERG is detectable from multiple sources within urine [13], it could be worth investigating the primary source (circulating free mRNA in whole urine, cellular RNA or exosomal RNA) of the fusion mRNA. This could potentially lead to a more in-depth understanding of the cancer biology and aid in future assay development.
Currently, quantitative reverse transcription PCR (RT-qPCR) is mainly used to measure TMPRSS2-ERG mRNA levels in urinary samples [4, 10, 14]. However, this approach is too time-consuming and laborious for use in a clinical setting. Therefore, the development of newer, faster and convenient assays for quantifying TMPRSS2-ERG mRNA in clinical samples may aid in improved PC screening and treatment monitoring.
Colorimetric assays have been developed for a wide range of target analytes and offer benefits such as low cost, short assay time, visual readout, and quantitative detection via absorbance measurements [15]. In particular, the reaction between tetramethyl benzidine (TMB) and horseradish peroxidase (HRP) in presence of hydrogen peroxide (H2O2) to produce a blue-colored product [16] has been employed in many colorimetric assays, with ELISA as a well-known example. Additionally, it has been demonstrated by previous studies [16-18] that TMB could serve as an electrochemical substrate. Given the excellent simplicity, sensitivity and speed of electrochemical biosensors [19-21], it may be ideal to adapt TMB-based colorimetric assays for alternative electrochemical readouts to exploit such advantages. Therefore, we hypothesized that a TMB-based assay which displays a quantitative colorimetric/electrochemical signal change in TMPRSS2-ERG presence could be potentially useful for diagnostic purposes.
Herein, we describe the FusBLU assay, a novel approach to detect and quantify TMPRSS2-ERG fusion mRNA. By the innovative merger of isothermal reverse-transcription recombinase polymerase reaction (RT-RPA) and HRP-catalyzed colorimetric readout, TMPRSS2-ERG mRNA in urine could be specifically amplified and detected by naked-eye or by quantitative absorbance measurements. The versatility of the assay readout was further shown by conveniently performing FusBLU on a portable potentiostat for electrochemical detection. The practical application of our assay was demonstrated using PC cell lines and patient urinary samples. We also applied our assay to detect TMPRSS2-ERG mRNA in whole urine, urinary sediments and exosomes of PC urinary specimens in order to investigate the primary source of the mRNA.
Materials and Methods
RNA extraction from cell lines
DuCap and LnCap cells were kindly donated by Matthias Nees (VTT, Finland); Gregor Tevz (APCRC, Australia); and Michelle Hill (UQDI, Australia). DuCap and LnCap cells were cultured in RPMI-1640 growth media (Life Technologies, Australia) supplemented with 10% fetal bovine serum (Life Technologies, Australia) in a humidified incubator (Sanyo MCO-19AIC, Japan) containing 5% CO2 at 37 ºC. Total RNA was extracted by lysing cells with Trizol® reagent (Life Technologies, Australia) before adding chloroform to separate RNA into a clear upper aqueous layer. Then, RNA was precipitated from the aqueous layer and resuspended in 50 µL of RNase-free water. RNA purity and integrity were checked using a NanoDrop Spectrophotometer (Thermo Fisher Scientific, Australia)
RNA extraction from clinical urinary samples
Ethics approval was obtained from The University of Queensland Institutional Human Research Ethics Committee (Approval No. 2004000047). Informed consent was obtained from all subjects prior to sample collection and methods pertaining to clinical samples were carried out in accordance with approved guidelines.
For whole urine RNA extraction, 1 mL of fresh urine sample was briefly mixed with 500 µL of lysis buffer [22] (150 mM Tris-HCI pH 8.0, 4.5 M guanidium-HCI, 3% v/v Triton-X and 1.5 mM EDTA). Total RNA was purified from 25 µL of cell lysate using the Agencourt AMPure XP kit (Beckman Coulter, Australia). Briefly, two volumes of Solid Phase Reversible Immobilization (SPRI) reagent was incubated with the cell lysate for 10 minutes. The RNA-bound magnetic beads were then separated from the lysate using a magnet and washed twice with 80% ethanol. Finally, total RNA was eluted in 25 µL of RNase-free water.
For urinary cell sediment RNA extraction, urinary samples (30-50 mL) were centrifuged (Beckman Coulter AllegraTM X-22R, Australia) at 700 g, 4ºC for 10 min and urinary sediments (supernatant was kept for exosomal RNA extraction) were washed with ice-cold 10 mM PBS buffer before being centrifuged again at the same conditions. Then, 25 µL of lysis buffer was added to a 50 µL fraction of PBS-suspended urinary sediments with vigorous mixing to release total RNA. Next, total RNA was purified from 25 µL of cell lysate using the procedure as described for whole urine RNA extraction.
For urinary exosomal RNA extraction, microvesicles in the supernatant (after urinary sediment centrifugation) was firstly pelleted by ultracentrifugation (Beckman Coulter OptimaTM XL-100 K, Australia) at 100 000 g, 4ºC for 90 min and resuspended in 200 µL of 10 mM PBS buffer. Then, the Exo-spinTM kit (Cell Guidance Systems, USA) was used accordingly to manufacturer's instructions to isolate exosomes from the pellet. To ensure that extracted RNA was of exosomal origin, the isolated urinary exosomes were treated with RNase A (New England Biolabs, Australia) to degrade all non-exosomal RNA before RNA extraction. Exosomal total RNA was extracted by Trizol® reagent (Life Technologies, Australia) using the procedure as described for RNA extraction from cells.
Colorimetric FusBLU assay
Synthetic oligonucleotide and primer sequences used in our experiments were obtained commercially (IDT, Singapore). RT-RPA primer sequences were designed based on the fusion junction of the most common TMPRSS2 (Exon 1):ERG (Exon 4) mRNA isoform reported in literature [4]. The RT-qPCR primers were also designed to amplify the same target TMPRSS2:ERG isoform.
For cell line experiments, the TwistAmp Basic RPA kit (Twist-DX, UK) was used with slight modifications to manufacturer's instructions. Briefly, 1 µL of extracted RNA, 50 units of MMuLV reverse transcriptase (New England Biolabs, Australia), 375 nM of each primer (Table 1) and 20 nM of biotinylated dUTPs (Thermo Fisher Scientific, Australia) were added to make a 12.5 µL reaction volume prior to incubation at 43°C for 20 min. For all other experiments, the TwistAmp Basic RT-RPA kit (Twist-DX, UK) with pre-included reverse transcriptase was used with the same modifications to manufacturer's instructions. The 43ºC amplification temperature, which is above the manufacturer's recommended optimum temperature, was required for higher amplification specificity.
Oligonucleotide sequences used in experiments.
| Oligos | 5'-Sequence-3' |
|---|---|
| RT-RPA TMPRSS2-ERG Fwd Primer Sequence | CCTGGAGCGCGGCAGGAAGCCTTATCAGTTG |
| RT-RPA TMPRSS2-ERG Rev Primer Sequence | GCTAGGGTTACATTCCATTTTGATGGTGAC |
| RT-RPA Housekeeping Fwd Primer Sequence | GCTATGCCGATCGGGTGTCCGCACTAAGTT |
| RT-RPA Housekeeping Rev Primer Sequence | GACGGGGTCTCGCTATGTTGCCCAGGCTGG |
| RT-qPCR Fwd Primer Sequence | CCTGGAGCGCGGCAGGAAGCCTTATCAGTTG |
| RT-qPCR Rev Primer Sequence | TCCTGCTGAGGGACGCGTGGGCTCATCTTG |
For colorimetric detection of RT-RPA products, 2 µL of RT-RPA products was incubated with 1 µL of 1:1000 diluted streptavidin (SA)-HRP (BD Biosciences, Australia), 1 µL of SA-magnetic beads (New England Biolabs, Australia) and 10 µL of wash buffer (10 mM PBS buffer, 0.5% triton-X) for 5 min. Then, a magnet was used to separate the beads and beads were washed three times with wash buffer (10 mM PBS buffer, 0.1% Tween20). Lastly, 25 µL of 1-Step™ TMB substrate solution (Thermo Fisher Scientific, Australia) was added to the beads and the mixture was observed for color change after 5 min. For each sample, two separate RT-RPA assays were performed for independent detection of TMPRSS2-ERG and housekeeping (RN7SL1) RNA. For quantitative measurements of color change, absorbance readings were recorded at 650 nm with a NanoDrop Spectrophotometer (Thermo Fisher Scientific, Australia). For each sample, relative TMPRSS2-ERG level was calculated by normalizing the absorbance value of TMPRSS2-ERG to the respective housekeeping RN7SL1 as follows:
Relative TMPRSS2-ERG Level = XTmprss2-ERG / XRN7SL1 (1)
where XTmprss2:ERG and XRN7SL1 are the average absorbance readings at 650 nm for TMPRSS2-ERG and housekeeping RN7SL1 RNA respectively.
Alternative electrochemical readout
For electrochemical detection of RT-RPA products, total RNA was isolated from patient urinary sediments and after which, the same steps as described previously for colorimetric FusBLU assay were performed. At 5 min after TMB-H2O2 addition, 500 mM H2SO4 was added to stop the reaction and activate TMB for electrochemical detection. The electrodes used in our experiments were screen-printed electrodes (DRP110, Dropsens, Spain) with carbon as working and counter electrodes, and silver as reference electrode. Briefly, 45 µL of the resulting mixture after H2SO4 addition was pipetted onto the electrode surface, and ensured that the solution was in contact with all three electrode types (i.e. working, counter, and reference). Amperometry measurements were carried out using a portable µSTAT 400 bipotentiostat/galvanostat (Dropsens, Spain) at 150 mV, 30 s. Each sample's electrochemical measurement was scored for relative TMPRSS2-ERG level in the same manner as described previously for absorbance readings. All measurements were performed at room temperature.
RT-qPCR
The KAPA SYBR® FAST One-Step qRT-PCR kit (KAPA Biosystems, Australia) was used to set up a single reaction volume of 10 µl for each sample. Each reaction volume consist of 1X KAPA SYBR® FAST qPCR Master Mix, 200 nM of each forward and reverse primer (Table 1), 1X KAPA RT Mix, 50 nM ROX dye and 30 ng of cell line total RNA template. RT-qPCR was performed using the Applied Biosystems® 7500 Real-Time PCR System (Thermo Fisher Scientific, Australia). The cycling protocol was: 42°C for 10 min to synthesize cDNA, followed by 95°C for 5 min to deactivate RT before cycling 35 times (95°C for 30 s, 50°C for 30 s and 72°C for 1 min) and finished with 72°C for 10 min.
Results
FusBLU assay for TMPRSS2-ERG detection
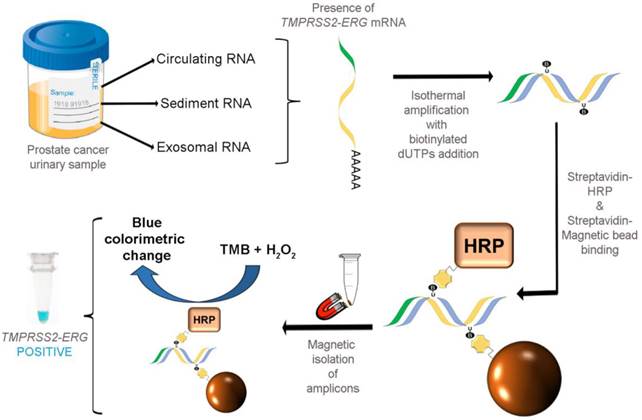
As illustrated in Fig. 1, our colorimetric assay utilized a combination of isothermal RT-RPA and TMB-based colorimetric readout for detecting TMPRSS2-ERG mRNA. Total RNA was firstly isolated from various sources used in our study and specific primers were used to amplify the TMPRSS2-ERG mRNA region at a constant temperature of 43ºC. During the isothermal RT-RPA, biotinylated dUTPs were randomly incorporated into the newly-synthesized strands. Then, SA-magnetic beads and SA-HRP were added to select for and label biotinylated RT-RPA products through biotin-streptavidin interactions. Lastly, TMB-H2O2 was added for color change reaction to test for the presence of HRP which in turn signifies the presence of RT-RPA products (i.e. TMPRSS2-ERG positive).
FusBLU assay for rapid TMPRSS2-ERG detection. Total RNA is isolated from urine and TMPRSS2-ERG mRNA is specifically amplified by RT-RPA isothermally. During strand polymerization, biotinylated dUTP bases are randomly incorporated and subsequently, SA-magnetic beads and SA-HRP are added to RT-RPA products for coupling through biotin-SA interactions. After magnetic isolation of RT-RPA products, TMB is added and a blue color change indicates the presence of TMPRSS2-ERG.
Assay specificity
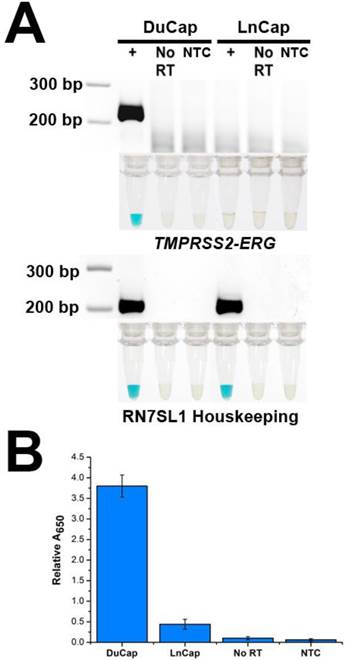
To demonstrate the specificity of FusBLU in detecting TMPRSS2-ERG, we applied our assay to total RNA from two different PC cell lines. DuCap and LnCap are well-studied cell lines with the presence and absence of TMPRSS2-ERG respectively [4]. As shown in Fig. 2A, our assay was able to specifically amplify TMPRSS2-ERG mRNA from DuCap RNA and subsequently generated a blue color change. On the other hand, when the assay was performed on LnCap RNA, RT-RPA products were not generated at a detectable level colorimetrically (Fig. 2A). The specific colorimetric detection of TMPRSS2-ERG could be followed by naked-eye observation of color change or more quantitatively by absorbance measurements (Fig. 2B). Furthermore, gel electrophoresis was also used to verify that the RT-RPA primers used in our experiments were specific and generated 216 bp products only in TMPRSS2-ERG positive DuCap samples. RN7SL1 housekeeping RNA was used as a loading control to validate successful RNA isolation from the cell lines (Fig. 2A). In addition, control experiments without the reverse transcriptase did not generate any RPA products, thus demonstrating that amplification was dependent on TMPRSS2-ERG mRNA, not DNA targets (Fig. 2).
Specificity of FusBLU assay. (A) HRP-catalyzed colorimetric results for DuCap (fusion positive) and LnCap (fusion negative) cell lines with corresponding gel electrophoresis images after RT-RPA. Top panel: TMPRSS2-ERG. Bottom panel: RN7SL1. No reverse transcriptase (No RT) and no-template control (NTC) were included. (B) TMPRSS2-ERG levels normalized to RN7SL1. Error bars represent standard deviation of three independent experiments.
Assay sensitivity
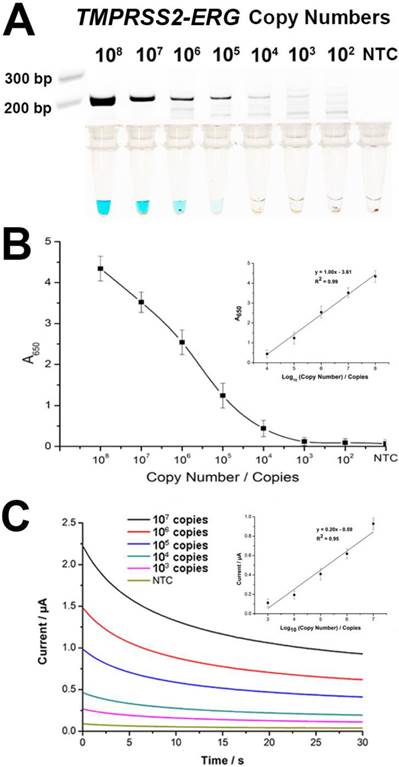
To evaluate assay sensitivity, we prepared a titration of synthetic TMPRSS2-ERG mRNA (102-108 copies) into a background of LnCap total RNA to test the limit of detection (LOD). As expected, the color change intensity increased with increasing amounts of input targets (Fig. 3A). By visual detection, the assay LOD was 105 copies of TMPRSS2-ERG mRNA at which a blue color change in solution was still observable. The determination of assay sensitivity could also be followed quantitatively by absorbance measurements and assay LOD was further improved by 10-fold to 104 TMPRSS2-ERG mRNA copies with a linear range of 104-108 copies (Fig. 3B). However, it is worthy to note that the LOD of 105 copies by visual detection is approximately equivalent to single cell level of detection [23] and thus may potentially be adequate for TMPRSS2-ERG screening purpose in a clinical setting. FusBLU also showed good assay reproducibility with intra- and inter-assay variability of 12.5% and 10.5% respectively (n = 3).
Sensitivity of FusBLU assay. (A) HRP-catalyzed colorimetric change generated from 102 - 108 copies of synthetic TMPRSS2-ERG mRNA, and corresponding gel electrophoresis images after RT-RPA. (B) Calibration plot of the average absorbance measurements at different initial RT-RPA input amounts. Inset shows the analogous linear calibration plot. (C) HRP-catalyzed current change generated from 103 - 107 copies of synthetic TMPRSS2-ERG mRNA. Inset shows the analogous linear calibration plot. Error bars represent standard deviation of three independent experiments.
Colorimetric assay performance on clinical specimens
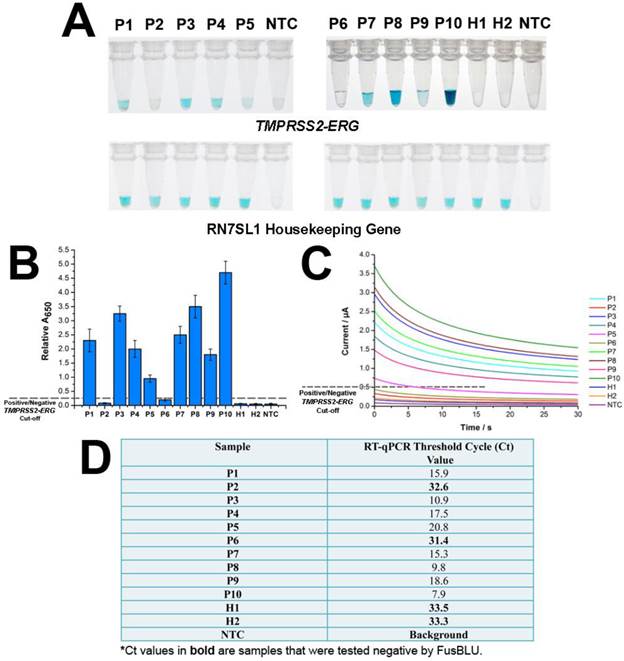
After establishing assay specificity and sensitivity, we challenged FusBLU with urine specimens to demonstrate potential clinical utility. To this end, we isolated sediment RNA from the urinary samples of 12 men and applied FusBLU for TMPRSS2-ERG detection. The magnetic SPRI method provided a minimal yield of 15 ng total RNA; an amount which was sufficient for our analysis using FusBLU. Of the 12 clinical urinary samples used in our experiments: 5 (P1-P5) were metastatic hormone-refractory PC patients undergoing treatment; 5 (P6-P10) were diagnosed with PC after biopsies; and the remaining 2 (H1 and H2) were healthy controls. As shown on Fig. 4A, we could visually detect TMPRSS2-ERG in 8 of the patient samples (P1, P3, P4, P5, P7, P8, P9, and P10), while the remaining samples (P2, P6, H1, and H2) showed no observable color change as similar to the no-target control (NTC). For quantitative detection, the relative absorbance measurements (Eqn. 1) provided additional information of TMPRSS2-ERG levels in different patients (Fig. 4B). In addition, we also performed RT-qPCR, the current gold standard gene fusion detection approach on the same 12 urinary samples as a benchmark comparison. The RT-qPCR threshold cycle (Ct) values (Fig. 4D) showed that higher Ct values (i.e. lower amount of targets in samples) were generated for samples with low FusBLU-detected TMPRSS2-ERG levels. We found that our FusBLU results were in excellent agreement with the RT-qPCR results, thus validating the TMPRSS2-ERG status of the patient samples.
FusBlu assay on clinical urinary samples. (A) HRP-catalyzed colorimetric results of 12 urinary samples over two different assay runs (1st run (Left): P1-P5; 2nd run (Right): P6-H2). Top panel: TMPRSS2-ERG. Bottom panel: RN7SL1. (B) TMPRSS2-ERG levels normalized to RN7SL1. (C) TMB-derived currents of 12 urinary samples compared to no template-control (NTC). (D) RT-qPCR threshold cycle (Ct) values for each sample as validation for FusBLU results. Error bars represent standard deviation of three independent experiments.
Alternative electrochemical readout
Since TMB is electrochemically active and considering the multiple advantages of an electrochemical biosensor [20, 24, 25], we integrated the use of a portable potentiostat as an alternative FusBLU readout strategy to further demonstrate the potential for clinical screening applications. We firstly established the LOD of the electrochemical readout by a titration assay, and observed that 103 TMPRSS2-ERG copies were detectable with a linear range of 103-107 copies (Fig. 3C).
We next used the same collection of 12 urinary samples to demonstrate the electrochemical readout. Fig. 4C showed the TMPRSS2-ERG levels detected electrochemically were similar to that of spectrophotometry (Fig. 4B) and RT-qPCR (Fig. 4D). Taken together, these results indicated that FusBLU was a robust method for detecting TMPRSS2-ERG levels in urine and could be readily adapted for naked-eye, spectrophotometrical or electrochemical readout platforms.
Investigating TMPRSS2-ERG source in urine
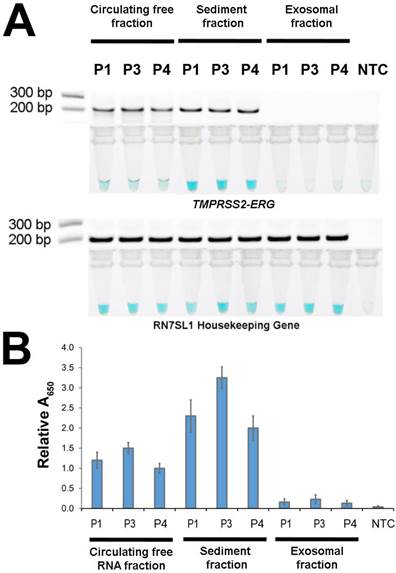
After demonstrating FusBLU's potential for clinical applications, we used our assay to investigate the primary source of TMPRSS2-ERG mRNA in urine. The potential sources included cell-free circulating RNA, cell sediment RNA and exosomal RNA. To this end, 3 TMPRSS2-ERG-positive PC patients (P1, P3, and P4) were first selected (Fig. 4) and total RNA was then isolated from the three candidate sources and assayed with FusBLU. As shown on Fig. 5A, the sediment fraction gave the strongest color intensity change (i.e. highest amount of TMPRSS2-ERG mRNA), followed by circulating free fraction, while the exosomal fraction contain the least amount of TMPRSS2-ERG mRNA. In addition, quantitative absorbance measurements showed that TMPRSS2-ERG mRNA in the sediment fraction was 2-fold and 12-fold higher than in the circulating free and exosomal fractions respectively (Fig. 5B). Absorbance results were also validated by gel electrophoresis following RT-RPA of the isolated total RNA from the various candidate sources (Fig. 5A). Our results therefore suggested that the sediment fraction is likely the main source of TMPRSS2-ERG mRNA in urine.
Source of TMPRSS2-ERG mRNA in urine. Total RNA isolated from circulating free, sediment, and exosomal fractions of 3 patient urine samples were studied (A) HRP-catalyzed colorimetric changes and corresponding gel electrophoresis images after RT-RPA. Top panel: for TMPRSS2-ERG. Bottom panel: RN7SL1. (B) TMPRSS2-ERG levels normalized to RN7SL1. Error bars represent standard deviation of three independent experiments.
Discussion
Recurrent gene fusions between TMPRSS2 exon 1 and ERG exon 4 is one of the emerging PC biomarker alternative to serum PSA. TMPRSS2-ERG offers higher PC specificity, potential prognostic value and is detectable in urine as a non-invasive biomarker [10, 11]. However, the conventional RT-qPCR approach for measuring TMPRSS2-ERG levels is not ideal for routine clinical screening due to long assay time and the need for specialized instrument [4, 14]. Therefore, we have developed FusBLU, an assay (Fig. 1) which combined isothermal RT-RPA and HRP-based colorimetric readout for quick and quantitative TMPRSS2-ERG detection in 75 min without requiring expensive readout instruments. With FusBLU, we demonstrated (i) potential TMPRSS2-ERG detection in clinical urinary samples with a quick and convenient visual readout; (ii) quantitative TMPRSS2-ERG detection by alternative absorbance/electrochemical readout platforms; (iii) cell sediment RNA as potentially the main source of TMPRSS2-ERG mRNA in patient urinary samples.
In order for the assay to be used for TMPRSS2-ERG detection in real biological samples such as urine specimens, assay specificity and sensitivity are of paramount importance. High assay specificity is required to avoid false-positive/negative results due to detection of non-target mRNA and high assay sensitivity is required to detect trace amounts of TMPRSS2-ERG mRNA from a complex background of other biological molecules. FusBLU was highly specific for TMPRSS2-ERG mRNA in fusion-positive DuCap cells but not fusion-negative LnCap cells (Fig. 2). This high assay specificity was largely enabled by designing RT-RPA forward primer across the fusion junction to selectively amplify only gene fusion events. In future, this flexibility of primer design may also allow specific detection of other TMPRSS2-ERG isoforms for multiplexed FusBLU gene fusion detection. Our assay visual detection limit of 105 copies of TMPRSS2-ERG mRNA (Fig. 3A); which was approximately single-cell level [26], may be especially useful for non-invasively detecting the low levels of TMPRSS2-ERG in urine. Furthermore, this visual LOD could be further improved up to 100-fold with easily-adaptable absorbance/electrochemical readouts. In comparison to other standard urinary TMPRSS2-ERG detection methodologies such as RT-qPCR, FusBLU required lower sample input (10 ng total RNA vs. 50 ng total RNA for RT-qPCR in our experiments) and was able to produce results within a shorter timeframe (75 min vs. 150 min for RT-qPCR in our experiments).
To demonstrate the potential clinical utility of FusBLU, we applied our assay to 12 urinary samples (Fig. 4) and found excellent agreement between our assay results and current gold-standard RT-qPCR results (Fig. 4D). Importantly, the healthy and NTC samples generated negligible background signals, and varying TMPRSS2-ERG levels were detected for the other samples with good reproducibility over independent runs. These data therefore suggested the reliability and functional applicability of our assay for routine diagnostics. Furthermore, the presence of TMPRSS2-ERG in the urine samples of the late-stage metastatic, hormone refractory samples was consistent with previous reports in literature [27, 28]. Hence, our results suggested that FusBLU may be a useful screening tool for the TMPRSS2-ERG positive metastatic subtype of aggressive PC. Considering the good assay sensitivity as well as speed and simple naked-eye evaluation of color change detection; FusBLU may also be a rapid and convenient preliminary PC screening tool for guiding clinical decisions. In addition, quantitative absorbance measurements of the color intensity can also provide additional information that could be beneficial for tracking patient response to treatment or for relapse monitoring. Lastly, electrochemical approaches have high potential for rapid, sensitive, inexpensive and portable biosensing [29-31] and thus have attracted great interest for clinical diagnostic developments. To this end, we exploited TMB's compatibility as an electrochemical substrate and adapted FusBLU into an electrochemical assay with a portable potentiostat to highlight the versatility of the method (Fig. 4C). Despite the use of a portable potentiostat, the FusBLU electrochemical readout provided the highest detection sensitivity (103 copies) out of the three different readout platforms. In short, FusBLU is potentially a flexible methodology that could be tailored to specific diagnostic needs.
While it is known that TMPRSS2-ERG mRNA is present in urine, it is not previously clear where the fusion mRNA originated from. Hence, identifying the primary source of TMPRSS2-ERG mRNA in urine might be useful for future assay development and may help better understand the cancer biology of TMPRSS2-ERG subtypes. Using FusBLU, we found that TMPRSS2-ERG mRNA was most abundant in the sediment fraction of urine (Fig. 5). TMPRSS2-ERG mRNA in circulating free fraction was the second most abundant source while minimal fusion mRNA was detected in the exosomal fraction. A possible explanation for our observation could be the shedding of tumor cells from the primary (and metastastic) tumors in the prostate into the urine through the prostatic urethra [32, 33]. It could also be from ingested tumor cells that are present in urine within white blood cells of the immune system [34]. The presence of the TMPRSS2-ERG mRNA in circulating free fraction was likely from the spontaneous lysis of cells since minimal RNA was detected in the exosomal fraction. Nevertheless, the data is still preliminary and warrants further investigation. Notwithstanding, the data did suggested that although TMPRSS2-ERG was detectable in whole urine, the sediment fraction is most likely the best choice for sampling TMPRSS2-ERG mRNA due to ease of preparation and high abundance. Additionally, the use of our assay as a rapid and convenient research tool for biological studies was also demonstrated through this set of successful experiments to investigate the source of TMPRSS2-ERG mRNA in urine.
FusBLU shows promise as a faster, simpler, and more cost-effective solution for gene fusion biomarkers detection as compared to current techniques. Nevertheless, there remain some aspects of the assay which could be further investigated in future studies. Firstly, our main aim in this report is to demonstrate FusBLU as a successful proof-of-concept technology which could be applied to real patient samples. While FusBLU detected TMPRSS2-ERG levels in 12 clinical urinary samples as a demonstration, it would be ideal to apply the method to a larger pool of patients and also of samples at various stages of PC. This would allow a better evaluation of the clinical utility of the assay for PC diagnosis. Secondly, we envisaged an improved version of FusBLU assay with multiplexing capabilities to detection multiple biomarkers. It has been suggested that detecting a combination of gene fusion isoforms or other PC biomarkers could improve PC diagnostic and predictive accuracy [14]. In this regard, we feel that an electrochemical readout would be better suited for this purpose as performing the assay on an array of miniaturized electrodes could greatly increase throughput.
In summary, we have developed FusBLU; a quantitative TMPRSS2-ERG assay with single-cell level detection sensitivity and highly specific for the most common gene fusion isoform in PC. This assay was achieved by the novel combination of isothermal RT-RPA and HRP-catalyzed colorimetric readout of RT-RPA products. Our assay was successfully applied to cell lines and patient urine samples with minimal equipment and at a low cost in approximately 75 minutes. Through the use of this assay, we also determined that total RNA isolated from urinary sediments as the main source for TMPRSS2-ERG mRNA detection from urinary samples. We believe that our colorimetric assay is potentially useful for POC detection of TMPRSS2-ERG, or other oncogenic fusion genes.
Abbreviations
PC: prostate cancer; PSA: prostate specific antigen; RT-qPCR: quantitative reverse-transcription PCR; HRP: horseradish peroxidase; DRE: digital rectal examination; TMB: tetramethyl benzidine; H2O2: hydrogen peroxide; RT-RPA: reverse-transcription recombinase polymerase reaction; SPRI: Solid Phase Reversible Immobilization; SA: streptavidin; LOD: limit-of-detection; NTC: no-template control; Ct: threshold cycle.
Acknowledgements
We thank Matthias Nees, Gregor Tevz, and Michelle Hill for providing the cell lines used in our experiments.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA A Cancer J Clinicians. 2012;62:10-29
2. Welch HG, Albertsen PC. Prostate cancer diagnosis and treatment after the introduction of prostate-specific antigen screening: 1986-2005. J Natl Cancer Inst. 2009;101:1325-9
3. Loeb S, Catalona WJ. Prostate-specific antigen in clinical practice. Cancer Lett. 2007;249:30-9
4. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW. et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644-8
5. White NM, Feng FY, Maher CA. Recurrent rearrangements in prostate cancer: Causes and therapeutic potential. Curr Drug Targets. 2013;14:450-9
6. Prensner JR, Chinnaiyan AM. Oncogenic gene fusions in epithelial carcinomas. Curr Opin Genet Dev. 2009;19:82-91
7. Tomlins SA, Aubin SMJ, Siddiqui J, Lonigro RJ, Sefton-Miller L, Miick S. et al. Urine TMPRSS2:ERG fusion transcript stratifies prostate cancer risk in men with elevated serum PSA. Science Transl Med. 2011;3:94ra72
8. Tomlins SA, Bjartell A, Chinnaiyan AM, Jenster G, Nam RK, Rubin MA. et al. ETS gene fusions in prostate cancer: From discovery to daily clinical practice. Eur Urol. 2009;56:275-86
9. Yoshimoto M, Joshua AM, Cunha IW, Coudry RA, Fonseca FP, Ludkovski O. et al. Absence of TMPRSS2:ERG fusions and PTEN losses in prostate cancer is associated with a favorable outcome. Mod Pathol. 2008;21:1451-60
10. Hessels D, Smit FP, Verhaegh GW, Witjes JA, Cornel EB, Schalken JA. Detection of TMPRSS2-ERG fusion transcripts and prostate cancer antigen 3 in urinary sediments may improve diagnosis of prostate cancer. Clin Cancer Res. 2007;13:5103-8
11. Laxman B, Tomlins SA, Mehra R, Morris DS, Wang L, Helgeson BE. et al. Noninvasive detection of TMPRSS2: ERG fusion transcripts in the urine of men with prostate cancer. Neoplasia. 2006;8:885-8
12. Nilsson J, Skog J, Nordstrand A, Baranov V, Mincheva-Nilsson L, Breakefield XO. et al. Prostate cancer-derived urine exosomes: a novel approach to biomarkers for prostate cancer. Br J Cancer. 2009;100:1603-7
13. Dijkstra S, Mulders PFA, Schalken JA. Clinical use of novel urine and blood based prostate cancer biomarkers: A review. Clin Biochem. 2014;47:889-96
14. Wang JH, Cai Y, Ren CX, Ittmann M. Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res. 2006;66:8347-51
15. Li HX, Rothberg L. Colorimetric detection of DNA sequences based on electrostatic interactions with unmodified gold nanoparticles. Proc Natl Acad Sci U S A. 2004;101:14036-9
16. Wee EJH, Ngo TH, Trau M. Colorimetric detection of both total genomic and loci-specific DNA methylation from limited DNA inputs. Clin Epigenet. 2015;7:65
17. Volpe G, Compagnone D, Draisci R, Palleschi G. 3,3 ',5,5 '-tetramethylbenzidine as electrochemical substrate for horseradish peroxidase based enzyme immunoassays. A comparative study. Analyst. 1998;123:1303-7
18. Ng BYC, Wee EJH, West NP, Trau M. Naked-eye colorimetric and electrochemical detection of Mycobacterium tuberculosis-towards rapid screening for active case finding. ACS Sens. 2015;1:173-8
19. Gasparac R, Taft BJ, Lapierre-Devlin MA, Lazareck AD, Xu JM, Kelley SO. Ultrasensitive electrocatalytic DNA detection at two- and three-dimensional nanoelectrodes. J Am Chem Soc. 2004;126:12270-1
20. Ronkainen NJ, Halsall HB, Heineman WR. Electrochemical biosensors. Chem Soc Rev. 2010;39:1747-63
21. Koo KM, Sina AAI, Carrascosa LG, Shiddiky MJA, Trau M. eMethylsorb: rapid quantification of DNA methylation in cancer cells on screen-printed gold electrodes. Analyst. 2014;139:6178-84
22. Wee EJH, Lau HY, Botella JR, Trau M. Re-purposing bridging flocculation for on-site, rapid, qualitative DNA detection in resource-poor settings. Chem Commun. 2015;51:5828-31
23. Velculescu VE, Madden SL, Zhang L, Lash AE, Yu J, Rago C. et al. Analysis of human transcriptomes. Nat Genet. 1999;23:387-8
24. Das J, Ivanov I, Montermini L, Rak J, Sargent EH, Kelley SO. An electrochemical clamp assay for direct, rapid analysis of circulating nucleic acids in serum. Nat Chem. 2015;7:569-75
25. Sadik OA, Aluoch AO, Zhou A. Status of biomolecular recognition using electrochemical techniques. Biosens Bioelectron. 2009;24:2749-65
26. Matthew T, Yang B, Jarrard DF. Toward the detection of prostate cancer in urine: A critical analysis. J Urol. 2013;189:422-9
27. Mehra R, Tomlins SA, Yu J, Cao X, Wang L, Menon A. et al. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res. 2008;68:3584-90
28. Attard G, Swermenhuis JF, Olmos D, Reid AHM, Vickers E, A'Hern R. et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 2009;69:2912-8
29. Nie Z, Nijhuis CA, Gong J, Chen X, Kumachev A, Martinez AW. et al. Electrochemical sensing in paper-based microfluidic devices. Lab Chip. 2010;10:477-83
30. Hsieh K, Patterson AS, Ferguson BS, Plaxco KW, Soh HT. Rapid, sensitive, and quantitative detection of pathogenic DNA at the point of care through microfluidic electrochemical quantitative loop-mediated isothermal amplification. Angew Chem, Int Ed. 2012;51:4896-900
31. Ferguson BS, Buchsbaum SF, Wu T-T, Hsieh K, Xiao Y, Sun R. et al. Genetic analysis of H1N1 influenza virus from throat swab samples in a microfluidic system for point-of-care diagnostics. J Am Chem Soc. 2011;133:9129-35
32. Fujita K, Pavlovich CP, Netto GJ, Konishi Y, Isaacs WB, Ali S. et al. Specific detection of prostate cancer cells in urine by multiplex immunofluorescence cytology. Hum Pathol. 2009;40:924-33
33. Hessels D, Gunnewiek J, van Oort I, Karthaus HFM, van Leenders GJL, van Balken B. et al. DD3(PCA3)-based molecular urine analysis for the diagnosis of prostate cancer. Eur Urol. 2003;44:8-15
34. Kato T, Suzuki H, Komiya A, Imamoto T, Naya Y, Tobe T. et al. Clinical significance of urinary white blood cell count and serum C-reactive protein level for detection of non-palpable prostate cancer. Int J Urol. 2006;13:915-9
Author contact
![]() Corresponding authors: j.weeedu.au; m.trauedu.au Tel: +61733464173 Fax: +61733463973.
Corresponding authors: j.weeedu.au; m.trauedu.au Tel: +61733464173 Fax: +61733463973.