Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Overview of cell-SELEX procedure
Procedures of cell-SELEX
Characterization of aptamers
Challenges and perspectives
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(9):1440-1452. doi:10.7150/thno.15666 This issue Cite
Review
Generating Cell Targeting Aptamers for Nanotheranostics Using Cell-SELEX
Yifan Lyu*, Guang Chen*, Dihua Shangguan*, Liqin Zhang, Shuo Wan, Yuan Wu, Hui Zhang, Lian Duan, Chao Liu, Mingxu You, Jie Wang ![]() , Weihong Tan4
, Weihong Tan4 ![]()
1. Department of Cardiology, Guang'anmen Hospital, China Academy of Chinese Medical Sciences, Beijing 100053, China
2. Molecular Science and Biomedicine Laboratory, State Key Laboratory of Chemo/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology, Collaborative Innovation Center for Chemistry and Molecular Medicine, Hunan University, Changsha 410082, People's Republic of China.
3. Beijing National Laboratory for Molecular Sciences, Key Laboratory of Analytical Chemistry for Living Biosystems, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China and University of the Chinese Academy of Sciences, Beijing 100049, China
4. Department of Chemistry and Physiology and Functional Genomics, Center for Research at the Bio/Nano Interface, Health Cancer Center, UF Genetics Institute, McKnight Brain Institute, University of Florida, Gainesville, FL 32611-7200, USA Fax: (+1)352-846-2410.
* These authors made equal contributions.
Received 2016-3-27; Accepted 2016-5-12; Published 2016-6-15
Abstract

Detecting and understanding changes in cell conditions on the molecular level is of great importance for the accurate diagnosis and timely therapy of diseases. Cell-based SELEX (Systematic Evolution of Ligands by EXponential enrichment), a foundational technology used to generate highly-specific, cell-targeting aptamers, has been increasingly employed in studies of molecular medicine, including biomarker discovery and early diagnosis/targeting therapy of cancer. In this review, we begin with a mechanical description of the cell-SELEX process, covering aptamer selection, identification and identification, and aptamer characterization; following this introduction is a comprehensive discussion of the potential for aptamers as targeting moieties in the construction of various nanotheranostics. Challenges and prospects for cell-SELEX and aptamer-based nanotheranostic are also discussed.
Keywords: Aptamers, Cell-SELEX, nanotheranostic, aptamer drug conjugates, DNA network.
Introduction
The molecular mechanisms behind the changes in cell conditions are of great significance to understanding and developing treatments for diseases. However, detection in cell behavior reaches a bottleneck due to a lack of molecular probes that recognize features of cellular architecture at the molecular level. Morphological evidence is an important standard commonly used by pathologists to serve as a basis for cancer diagnosis. However, molecular-level signals, e.g., mass-density fluctuations, lie far beyond the detection limit of traditional optical microscopy. Proteomic technologies have renewed our understanding of the interconnections between genes, proteins, and diseases. However, due to their reliance on known biomarkers for the development of corresponding probes, most technologies fail to illuminate particular molecular signatures on the cell surface. Moreover, only a small amount of biomarkers are currently identified and validated for detecting cancerous, viral, bacterial or fungal infection. In other words, the diagnosis of infectious agents is particularly cumbersome. Therefore, new methods to discover unknown molecular features of diseased cells and pathogens are highly desirable.
Aptamers are single-stranded DNA or RNA oligonucleotides that bind their targets with high affinity and selectivity. Aptamers, also known as nucleic acid antibodies, have unique merits, including ease of chemical synthesis, high chemical stability, low molecular weight, lack of immunogenicity, and ease of modification and manipulation[1] compared to their protein counterparts. These characteristics enable aptamers, good candidates for molecular probes, to recognize extracellular matrix signatures of cancer cells and target cell-specific ligands for therapeutic purposes. An aptamer is evolved from a random oligonucleotide library by repetitively selecting oligonucleotides from the oligonucleotide pool based on their binding affinities to target molecules; this process is known as Systematic Evolution of Ligands by EXponential enrichment (SELEX)[2, 3]. Initially, aptamers were only generated to bind with simple targets, such as small molecules and purified proteins.[4] More complicated aptamer selections, such as SELEX against red blood cell membranes[5] and whole cells[6-8], were demonstrated from 2001 to 2003. However, at that time, aptamers involved cancer detection were limited by the absence of aptamers targeting cancer cell membrane proteins.
By 2003, only several cancer biomarkers had been identified. These biomarkers were very expensive or could not be synthesized by commercial vender, preventing their widespread use. Then, the Tan group at the University of Florida conceived the strategy of selecting aptamers specific to whole live cells. Phenotypic variations between cell types, such as between normal and cancer cells or multiple types of cancer cells, represent differences in molecular signature. Therefore, the ability to isolate aptamers based on these molecular signatures would allow for the creation of molecular probes specific to cancer cell types for use in cancer diagnosis and therapy. This process, known as cell-based SELEX, or cell-SELEX, was first developed using a human acute lymphoblastic leukemia cell line, CCRF-CEM (T-cell line), as the target cell, and a human diffuse large cell lymphoma cell line, Ramos (B-cell line), as the control cell.[9] As a result, a panel of aptamers specifically binding target cells, including one able to bind to a membrane protein tyrosine kinase 7, which has been identified as a biomarker for leukemia,[10, 11] was generated. Many aptamers have been developed via cell-SELEX,[12-28] with targets including various cancer cells, virus-infected cells and bacteria. Moreover, these aptamers have shown their advantages in cell detection, cell capture, and in vivo imaging.[1]
The merits of cell-SELEX are as follows:
(1) Aptamers may be generated by cell-SELEX technology that have the ability to not only to specifically recognize cells, but also differentiate between the molecular signatures of diverse cell types without any prior knowledge of their molecular characteristics.
(2) The surfaces of different cell types often display numerous molecular differences, particularly membrane-bound proteins. These molecules are potential targets in cell-SELEX. Therefore, multiple aptamers may be generated against diverse targets through successful selections. Also, probes may be developed based on these aptamers that assist in accurate disease diagnosis, a boon for personalized medicine.
(3) It is possible for aptamer probes to distinguish their cognate targets directly because aptamers bind to target molecules in the native state, creating a true molecular profile of diseased cells. Additionally, bound aptamers and unbound oligonucleotides can be separated easily through washing or centrifugation during the SELEX process, because target molecules are anchored on the surface of cells naturally. Thus, there is no need to purify and fix the target molecules on a solid support.
(4) New biomarkers are discovered with the help of aptamers. Both sophisticated pathological and physiological processes are related to the changes at the molecular level in cells. Although the cause of such changes has not been elucidated, cell-based SELEX makes it possible to generate aptamers that recognize unknown biomarkers. These aptamers, in turn, can act as molecular tools to identify and purify their targets, which have potential to be new biomarkers.
Due to this host of benefits, cell-SELEX technology is now used worldwide and new cell-specific aptamers are reported every year. The use of these aptamers as targeting moieties has led to the development of numerous nanotools for the efficient cancer diagnosis and therapy.[29-34] Here we present an overview of DNA aptamer development against different cell types using cell-SELEX technology, mainly based on the experiments and results of our lab. We believe the following discussion on the valuable experiences gathered in our lab will give readers an “inside look” at the mechanics of the process and the key considerations of each step. Aptamers offer great potential as molecular probes in biomedicine studies. With a benefit of the natural properties of DNA, aptamers can be easily modified by both chemical and enzymatic reactions, which make them good candidates as targeting moieties in the construction of nanotheranostics. In order to demonstrate the great potential of aptamers as powerful nanotools for cancer nanotheranostics, we introduce several strategies with sharply distinct mechanisms. At the end of this review, challenges and prospects of cell-SELEX are discussed.
Overview of cell-SELEX procedure
The SELEX strategy, first described in 1990 by Gold and Szostak,[2, 3] has been modified in different ways.[35] But in general, the process of SELEX involves some basic steps including incubation of targets with an oligonucleotide library, isolation of the oligonucleotide-target complexes from unbound sequences, and amplification of the bound sequences by PCR or RT-PCR to obtain an enriched pool for the next round of selection. The bound sequences are enriched by repeating the process. These DNA or RNA sequences enriched pool is then cloned into bacteria and sequenced to obtain the individual sequences which are further tested for obtaining potential aptamer candidates through chemically synthesizing and labeling with reporters, and the testing against the target.
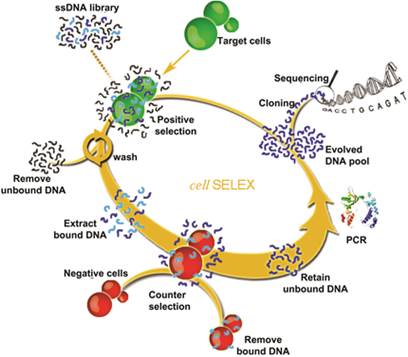
The most important step of SELEX is to distinguish the target-binding sequences from unbinding sequences.[35] But for cell-SELEX, partitioning is relatively simple, essentially because the unbound sequences can be easily removed by centrifugation or washing. A typical cell-SELEX procedure is shown in Figure 1. Cell-SELEX begins with the preparation of a synthesized random oligonucleotide library and the growth of cells of interest. The iterative cycles of cell-SELEX usually follows several steps: incubation of target cells with the DNA pool, collection of bound oligonucleotides via elution from target cells, generation of a new enriched pool through the amplification of eluted oligonucleotides, counter-selection (also known as subtractive selection) to reduce nonspecific binding and common binding to both target and control cells, and evaluation of binding infinity using flow cytometry, to monitor the aptamer enrichment after each round or after several rounds. While the process is iterative, each round represents an increasing selection pressure to ensure the generation of aptamers with high affinity and specificity instead of repeating other rounds. The number of selection rounds can be defined by the progress of enrichment, and 10-20 rounds are usually suggested. Once the binding assay indicates enough affinity and specificity, the last selected pool will be cloned and sequenced to generate candidate sequences. Candidate sequences are selected, synthesized and applied to binding assays, and aptamers with high affinity and specificity to target are obtained and modified for further applications. The efficiency of cell-SELEX can be affected by many factors, such as library construction, growth state of cells, selection conditions, potential targets on the cell surface, and human error. Our previous paper[36] describes a detailed protocol of the whole process of cell-SELEX; therefore, in this paper we only focus on the general principles of each step. A comprehensive discussion of the aptamer selection and identification processes will be presented in the following section.
Schematic representation of cell-SELEX. Reprinted with permission from ref. [9]. Copyright (2006) National Academy of Sciences, USA.
Sequences of aptamers need to be optimized by minimizing length and maximizing binding affinity. Dissociation constant (Kd) is an important parameter to describe the binding affinity of aptamer, and must be measured subsequently. Though aptamers can be selected without knowledge of the target membrane receptor, it is still beneficial to identify the target protein of a certain aptamer after cell-SELEX, because the target could be a new potential biomarker or disease-related protein. Aptamer characterization, including aptamer optimization, Kd determination and target identification, will be discussed in this review.
Apart from traditional cell-SELEX process, we also introduce a noncanonical cell-SELEX method, using artificial expanded genetic information systems (AEGIS)[37-39] with two added artificial nucleotides Z and P, to construct the library. There are three innovations that allow the success of AEGIS-SELEX: (a) artificial nucleotide phosphoramidites Z and P for GACTZP library synthesis, (b) polymerases that can recognize Z and P nucleotides for efficient PCR amplification, and (c) deep-sequence technology of GACTZP DNA survivors.
Procedures of cell-SELEX
Preparation of oligonucleotide library and cells
The SELEX process begins with the design and chemical synthesis of a random oligonucleotide library, with strands containing a random domain (20-80 nucleotides (nt)) in the middle and two constant sequences at either end for primer binding during PCR amplification. Since most cell-SELEX is carried out using a DNA library, due to the superior stability of DNA over RNA, herein we confine our discussions to DNA aptamer selection.
The primer sequence must be specified before library synthesis based on the general rules for primer design in PCR. Fluorescent dye modifications, like fluorescein amidite (FAM), carboxytetramethylrhodamine (TAMRA), or cyanine, are usually conjugated to the 5'-end of the sense primer to monitor selection progress. Biotins are often modified at the 5'-end of the antisense primer so that duplex PCR products may be captured on a streptavidin-coated matrix, followed by separation of the dye-labeled sense strand from the antisense strand using alkaline denaturation.
Generally, the diversity of the DNA library is determined by the length of the randomized domain, which is commonly in the range of 20-80 nt for cell-SELEX. Short libraries are cheaper to synthesize, while longer randomized regions ensure more complex secondary structures with better binding potential.[35] However, since the amount of synthesized DNA in a library used for one cell-SELEX is usually in the range of 20 pmol - 20 nmol, which is equivalent to 1013-1016 random sequences, it is difficult to include all possible sequences when using a longer random sequence pool. For example, if the random domain is longer than 30 nt, there are more than 430 (~1018) possible sequences.
Aptamers can be generated against a large variety of target cells, including those expressing a specific protein of interest,[40] types of cancer cells,[9] cancer stem cells,[17] adipocyte cells,[41] virus-infected cells,[19, 21] and bacteria.[42, 43] Most of these aptamers are selected against live cells, although in some cases target cells may be fixed.[8] It is vital that at least one type of control cell be used for counter-selection to eliminate the sequences that bind to the common molecules present on surface of both cell types.[12, 44] This is especially important because two closely-related cell types are often chosen as target cells and control cells; for example, tumor cells and homologous normal cells, differentiated cells and parental cells, drug-resistant cancer cells and drug-sensitive cancer cells, virus-infected cells and uninfected parental cells, or antibiotic-resistant bacterial strains and antibiotic-sensitive bacterial strains.[17]
Improper cell culture during cell-SELEX may affect enrichment of binding sequences and even lead to failed selection because of alteration in cell morphology and protein expression. Based the same consideration, when using adherent cells for selection, we suggest not doing cell dissociation before incubation with DNA pool, because cells dissociated whether by short-term trypsin treatment, non-enzymatic dissociation buffer, or scraping, may result in change of surface expression, cell death and cell rupture.
Aptamer selection against live cells
The basic principle for a successful SELEX is to enrich specific binding sequences while removing nonspecific binding sequences. Selection conditions, include binding buffer, washing buffer, elution buffer and binding temperature and time, play an important role in cell-SELEX. The pH value, osmolality, and ion concentrations of binding and washing buffers must be compatible with cells to avoid harm during the binding and washing steps. Elution buffer is used to dissociate potential aptamer sequences from cells. Choice of binding temperature depends on the application of the aptamer. Higher temperatures, like 37 °C, can induce endocytosis of oligonucleotides, which may result in the accumulation of nonspecific sequences. Binding performed at 4 °C or on ice is most commonly used, because most aptamers selected at 4 °C also bind very well at 37 °C, especially those with very high affinity.[9, 15, 16, 18]
As shown in Figure 1, each cycle generally includes incubation with target cell, washing, elution, PCR amplification, enriched pool preparation, and counter-selection. Theoretically, the initial library contains only one copy of each sequence. Once some specific sequences are lost in the first round of selection, they can never be recovered. In order to keep as many binding sequences as possible, more target cells, longer incubation times and moderate washing strength are required in the first round compared with subsequent rounds. Counter-selection should be delayed until after the first round. After incubation and elution, all sequences undergo PCR amplification and are converted to an enriched pool for the second round of selection.
From the second round of selection, counter-selection can be carried out before the target cell binding step [16] or after the elution step.[9] In the first case, the selection cycles start with the incubation of control cells with the DNA pool. The supernatant containing unbound sequences is then incubated with target cells as described before. In the latter case, after incubation with target cells, the eluted sequences need to incubate with control cells and the supernatant is applied for PCR amplification. Finally, for both cases, the PCR products are converted to an enriched pool for the next round of selection. In order to obtain aptamers with high affinity and specificity, selection pressure beyond the second round should be gradually enhanced by decreasing the amount of the ssDNA pool, reducing the incubation time for target cell binding and using less target cells, as well as increasing wash times with more wash buffer.
One should choose different processes when using suspension cells or adherent cells in cell-SELEX. For suspension cells, the cells bound with DNA sequences are collected by centrifugation in the binding and washing steps. For adherent cells, DNA-bound cells are collected by removing the supernatant with a pipette and scraping the bottom of the flask or dish. Scraped cells are then collected via centrifugation, followed by the elution of bound DNA. Trypsin should not be used in this case for cell detachment because trypsin can digest the target proteins on the cell surface and affect aptamer binding.[9, 16]
Enrichment and monitoring
PCR amplification is the most attractive advantage of nucleic acid library-based selection. The high complexity of DNA libraries and enriched pools calls for an increase in the efficiency of PCR amplification through the optimization of PCR conditions. After PCR amplification, a gel electrophoresis assay is performed to assess the PCR reaction.[36]
Double stranded PCR products must be transformed into a new ssDNA pool of only sense strands. Several methods have been described in literatures for this purpose, but here we will only describe the highly efficient streptavidin/biotin approach. In this method, bound sequences are amplified by PCR with dye-labeled sense primers and biotin-labeled antisense primers. The PCR products are captured by a streptavidin-coated support, such as sepharose or magnetic beads, and the sense ssDNA strands are then eluted from the beads with NaOH solution. After desalting and quantification, the ssDNA solution is finally dried to obtain the enriched pool for the next round of selection.
The progress of the selection must be monitored through binding assays at the end of every cycle or after several cycles to ensure that the aptamer sequences are enriched. Flow cytometry can be used to monitor the enrichment of bound sequences, which is beneficial due to its high statistical precision and substantial reproducibility. For suspension cells, binding assays may be carried out by incubation of cells with a dye-labeled DNA pool in binding buffer. For adherent cells, before incubation with selected pool, cells should be detached with nonenzymatic cell dissociation solution like EDTA and suspended in binding buffer. Confocal microscopy imaging can provide intuitive information about DNA binding on cells. However, it is difficult to collect the quantitative data necessary to compare binding ability of selected pools from different rounds with statistical significance.
Control experiments should also be performed by using unselected library or nonbinding sequence labeled with the same dye and control cells. A typical flow cytometry result of a successful cell-SELEX is shown in Figure 2.[9] Fluorescence intensity of target cells incubated with the selected pools gradually increases with selection round, while fluorescence intensity of control cells does not.
An example of flow cytometry binding assays of the selected pools from the 3th, 6th, 16th rounds to monitor the progress of cell-SELEX. Left, CEM cells (target); right, Ramos cells (control); dye-labeled ssDNA library as control DNA. Reprinted with permission from ref.[9]. Copyright (2006) National Academy of Sciences, USA.

It is possible for selected pools to bind both target and control cell lines, which may result from the enrichment of sequences that bind to common molecules on both cell lines. To solve this problem, one can choose to strengthen counter-selection by increasing the number of control cells, reducing the amount of DNA pool and decreasing the number of target cells.
Aptamer identification
Aptamer candidates with high affinity and specificity are enriched after each round of selection. The selection cycle is terminated when the binding assay shows that the affinity of the pool to target cells is higher than binding to control cells and cannot be further increased in two or three successive rounds of selection. Then, the final selected pool is PCR-amplified using unmodified primers, cloned into Escherichia coli., and then sequenced to identify individual aptamer candidates.
After removing the primer sequences at both ends, the evolved random domains of potential aptamers are analyzed by sequence alignment through the use of such programs as Clustal X, clustal omega and CLUSTAL W. Usually, the sequence alignment program groups the potential aptamer sequences into several families. Not all the sequences in the final pool can bind with target cells. It is not necessary to synthesize all the obtained sequences to find the effective ones (aptamers), because the sequences in a given family represent nearly identical aptamers. Typically, only a representative sequence from each family is chosen for validation. Fluorescent dyes or other reporters can be attached to the synthesized sequences. Based on the results of binding assays performed via flow cytometry or confocal imaging, strands with high binding affinity and specificity are identified as aptamers for further optimization.
Characterization of aptamers
Optimization and secondary structure of aptamers
Often times, not every nucleotide within the aptamer is required for direct binding or maintenance or formation of the aptamer binding structure.[45, 46] Longer sequences reduce the yield and increase the cost of aptamer synthesis. The presence of extraneous nucleotides can also cause the formation of various secondary structures that destabilize the target-binding conformation of the aptamer. Therefore, most aptamers are abbreviated to a minimal functional sequence following SELEX. Generally, truncated aptamers possess the same binding affinity as, if not better than, the binding affinity of the full-length aptamer. However, it can be difficult to determine which nucleotides are unnecessary. Current methods for determining boundary and binding sites of aptamers include partial hydrolysis and protein footprinting.[47-49] Selection of high affinity fragments from full-length aptamers has been used to determine the minimal sequence of aptamers.[50] In these methods, detection of the aptamer fragments was achieved via radioactive labeling.
In addition to the previously mentioned methods, a new method has been developed to predict the secondary structure of aptamers, as well as the critical sequences for target binding.[45, 46] Secondary structure prediction is generally carried out using the program mfold[51] (http://unafold.rna.albany.edu/?q=DINAMelt) based on an energy-minimizing method. Sequences in the same family generally are specific to the same target with the same secondary structures, but vary in their affinity. Thus, studying the affinity of sequences within the same family, and even different families, may help to find the secondary structures required for aptamer binding and the binding motif of the aptamer.[45, 46] Once the secondary structures and binding motifs have been determined, nucleotides unnecessary to the formation of these structures may be eliminated from the aptamer sequence. The optimized sequences are synthesized and evaluated on their binding ability, which is then compared with that of the full-length aptamer. The process of sequence optimization may be repeated to find the ideal minimized sequence,[46, 52-55] as seen in Figure 3.
(A) Secondary structures of sgc4 and sgc4a. Sgc4a had a shorter stem and lower binding affinity than sgc4. Removing nucleotides outside of the region of sgc4 marked with a box resulted in sgc4c, which had a Kd similar to that of full-length sgc4. (B) Truncated and mutated sequences of sgc4, known as sgc4e and sgc4f. It was clear that the long stem in sgc4 and sgc4c was important for maintaining the binding affinity, and that loop marked in box was not the critical binding site (made apparent by the Kd of sgc4f). Sgc4f can be used as the minimal binding motif of sgc4, with 30 fewer nucleotides than sgc4. Reprinted with permission from ref. [46]. Copyright (2007) WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.

Unfortunately, truncation and optimization based on software-predicted secondary structures is often empirical and sometimes unreliable. Another method to optimize aptamer binding affinity was reported by Kimoto et al, in which a secondary selection is performed using doped libraries.[56] After several rounds of selection and deep sequencing of the isolated library, the conserved bases and co-variations of base pairs are identified. Depending on these co-variation data, optimized and minimized sequences as well as the predicted secondary structures of aptamers can be easily determined.
Determination of dissociation constant (Kd) of aptamer
Once aptamers have been optimized, their properties must be further characterized. These properties include the dissociation constant (Kd), specificity to the target cell line, intensity and location of binding sites, tertiary binding structure, and cellular interactions (e.g. molecular targets on cells, cellular internalization, and biological activities against cells).[57] In cell-SELEX, Kd values of aptamer-cell interaction are usually measured by fitting the dependence of fluorescence intensity of specific binding on the concentration of the aptamers to equation:[9]
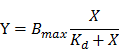
where Y, Bmax, and X are the fluorescence intensity, maximum fluorescence intensity, and aptamer concentration, respectively. The fluorescence intensity of target cells bound to fluorophore-labeled aptamers can be acquired via flow cytometry.[9]
Fluorescence correlation spectroscopy (FCS) can also act as a useful tool for investigation of aptamer binding affinity. By incubating various concentrations of fluorophore-labeled aptamers with cells, the different diffusion times of bound and free aptamers can be used to assess the percentage of bound aptamer, followed by yielding the absolute number of total aptamers inside the confocal volume. A sub-nanomolar dissociation constant (Kd=790 ± 150 pM) of sgc8 and HeLa cells has been determined by using this method, which is in good agreement with the result determined by flow cytometry (Kd = 810 pM).[58] Though both antibody and aptamer have Kd in the range of nM-pM according to the reported comparison, the binding affinity parameters can be controlled on demand during selection of aptamers while it is difficult for antibodies to modify affinity parameters.[59]
Aptamer target identification
The typical method of target protein isolation using aptamers generated by cell-SELEX is depicted in Figure 4. Aptamers used for target identification should be modified with some functional groups to improve their performance as molecular probes. For example, biotin tags allow the isolation and enrichment of aptamer-protein complexes after cell incubation and lysis with the help of streptavidin beads; photocrosslinkable agents enables the covalent linking between aptamers and targets; radioactive tracers either 32P or 3H modified on aptamers can be used for sensitive detection.[60]
General processes of aptamer target identification.

Gel electrophoresis is typically used for the isolation of the aptamer-protein complex after cell lysis. The aptamer-protein complex can be traced by staining of the protein through the use of commercially available protein stains, such as coomassie blue, or via imaging of radioactive tracers, like 32P, modified on the aptamer. Finally, the aptamer-protein complex is digested in situ, and the resulting peptides are analyzed using mass spectrometry. The identification of aptamer binding targets can reveal the binding mechanism and, more importantly, can be used to find potential biomarkers of some diseases. One of the first examples of using an aptamer generated by cell-SELEX to discover a biomarker is the discovery of expression of PTK7 on CCRF cells, as reported by Shangguan et al.[11]
In vitro selection with artificial expanded genetic information systems (AEGISs)
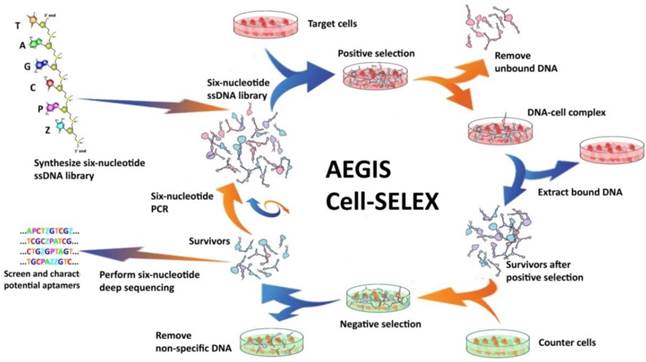
The Artificially Expanded Genetic Information System (AEGIS) is a novel SELEX platform that increases the number of independently replicating nucleotide building blocks.[38, 39] In AEGIS-SELEX, two additional nucleotides (2-amino-8-(1'-β-D-2-deoxyribofuranosyl)-imidazo[1,2-a]-1,3,5-triazin-4(8H)one, hereinafter termed as P, and 6- amino-5-nitro-3-(1'-β-D-2`-deoxyribofuranosyl)-2(1H)-pyridone, hereinafter termed as Z) were exploited. As shown in Figure 5, AEGIS-SELEX begins with the solid-phase synthesis of a GACTZP DNA library by adding Z and P phosphoramidites to the typical 4-nucleotide phosphoramidite mixture. Here, the pool of DNA survivors is collected after each round of selection and amplified by six-letter GACTZP PCR with a mixture of nucleotide triphosphates (dGTP, dATP, dCTP, dTTP, dZTP, and dPTP). It should be noted that Z and P bases are somewhat less sensitive toward polymerase, making it necessary to increase the concentrations of dZTP and dPTP during PCR to maintain the Z and P sites among the enriched GACTZP DNA survivors. Enriched ssDNA libraries obtained from the six-nucleotide PCR are then used for the next round of selection. Similar to SELEX, the selection pressure can be increased by reducing the number of cells and incubation times and increasing the volume of washing buffer and number of washes, as needed. The binding affinity of survivors is also monitored by flow cytometry. Survivors with a high shift are deep sequenced, resynthesized, and characterized. Deep sequencing after AEGIS-SELEX can be done following the “conversion” strategy previously reported.[61] The AEGIS-aptamers show lower dissociation constants toward target cells compared with the ACGT aptamers selected in the same SELEX process, indicating that the addition of artificial bases Z and P helps to improve the binding performance of aptamers. AEGIS-SELEX expands the power and utility of SELEX, and, more importantly, by using the AEGIS pool, AEGIS-based receptors, ligands, and catalysts will be generated with sequence diversities nearer to those displayed by proteins.
Schematic of AEGIS Cell-SELEX with both positive and counter-selections. In AEGIS-SELEX, a six-nucleotide single-stranded DNA library (GACTZP) is used as the DNA pool. After both positive and negative selections, survivors are amplified by GACTZP six-nucleotide PCR. Reprinted (adapted) with permission from ref.[39]. Copyright (2015) American Chemical Society.
Using DNA aptamers to build nanotheranostics
An important application of aptamer is to build nanotheranostics. Chemo- and/or radio-therapy, as conventional anticancer therapies, have obvious drawbacks, such as severe damage to normal tissues, low treatment efficiency, and increased drug/radiation resistance.[62] In order to solve these problems and meet the increasing demand for personalized medicine, numerous nanotheranostic modalities have been reported by integrating real-time diagnostic and therapeutic functions into a single nanosystem.[63] Aptamers, due to their high selective binding affinity towards targets, easy modification and programmable hybridization as well-studied biomolecules, can be easily designed as active targeting groups and integrated into nanotheranostics after sequence optimization.
In order to demonstrate the huge potential of aptamers as efficient and convenient molecular tools in biomedical applications, we will provide some examples of recent advancements in aptamer-based nanotheranostics. Since there have been some reviews about DNA-nanoparticle complexes,[64] here we want to focus our discussions on the examples in which DNA aptamers are used to build targeting DNA nanostructures, aptamer-drug conjugates and smart DNA diagnostic/therapeutic networks, because such applications are seldom summarized.
Programmable hybridization makes DNA an excellent building block to construct different kinds of nanostructures. Through rational design and combinations with aptamers, these nanostructures can be developed as nanotheranostics to solve clinical problems. For instance, widely regarded problems of chemotherapeutic drugs consist of severe side effects, limited maximum tolerated doses (MTD) and reduced therapeutic efficacy. To overcome these problems, Zhu et al reported a hybridization chain reaction (HCR)-based targeting drug delivery system, termed aptamer-nanotrain (aptNTrs).[29] As shown in Figure 6A, a cascading hybridization between hairpin M1 and M2 can be triggered by the aptamer-tethered trigger strand, which leads to the generation of an aptamer-tethered long linear DNA nanostructure. The periodically aligned 'boxcar' segments provide a large number of spatially addressable sites, allowing high-capacity loading of therapeutics (anticancer drug doxorubicin can be inserted into π-π stacking of double-stranded 5'-GC-3' or 5'-CG-3' sequences) or bioimaging agents. In vivo evaluation of this targeted drug transport system using a xenograft mouse tumor model demonstrated potent antitumor efficacy and reduced side effects of drugs delivered via aptNTrs. Other advantages of this drug delivery strategy are its easy design and the cost reduction of DNA preparation because only three short strands are needed in preparing this kind of DNA nanostructure.
A) Schematics of the self-assembly of aptamer-tethered DNA nanotrains (aptNTrs) for transport of molecular drugs in theranostic applications. Reprinted with permission from from ref.[29]. Copyright (2013) National Academy of Sciences, USA. B) RCR-based self-assembly of monodisperse, densely packed, and hierarchical DNA NFs. RCR generates a large amount of elongated non-nicked concatemer DNA by polymerizing uncombined dNTP and dye-modified dUTP on the 3' -terminal end of the primer. The circular template is designed to be complementary with an aptamer sequence so that aptamers are easily generated and integrated during RCR. Reprinted with permission from ref.[65]. Copyright (2015) Macmillan Publishers Limited.

DNA nanostructures can also be created by enzymatic reactions due to the biological properties of DNA molecules. This kind of aptamer-based targeting nanostructure includes DNA nanoflowers (NFs), which are self-assembled from reduplicated DNA strands generated via rolling circle replication (RCR) of a designed template.[30, 65] Different from traditional DNA nanostructures like DNA tetrahedra, DNA dendrimers, DNA origami and hybridization chain reaction (HCR)-based DNA polymers, the assembly of NFs is driven by liquid crystallization and dense packaging of building blocks instead of DNA hybridization. Based on the programmability of DNA strands, aptamers, drug-loading sites and dyes can be easily integrated into the size-tunable and stable structure by rational design of the circular DNA template; NFs are ideal candidates for a variety of applications in biomedicine, like targeted cancer cell recognition, bioimaging, and targeted drug delivery (Figure 6B).
Apart from using DNA structures as carriers to load anticancer drugs, drugs can be directly conjugated with aptamer-based DNA structures via covalent bonds to build targeting aptamer-drug conjugates (ApDCs). Inspired by the mature DNA chemical synthesis technology, Wang et al designed and synthesized a therapeutic module for solid phase synthesis (Figure 7A), which is a phosphoramdite containing an anticancer drug moiety and a photocleavable linker (phosphoramidite D).[31] Multiple drugs can hence be efficiently incorporated into ApDCs at pre-designed positions via a DNA synthesizer. Their results showed that this kind of ApDC not only recognized target cells specifically, but also released drugs in a photo-controllable manner.
A) Automated and modular synthesis of ApDCs from phosphoramidites A, T, C, G and D (top); Structural features of phosphoramidite D (bottom). Reprinted (adapted) with permission from ref. [31]. Copyright (2014) American Chemical Society. B) Schematic illustration of nuclease-resistant synthetic drug-DNA adducts as a simple, yet versatile and programmable, platform for targeted anticancer drug delivery. Adapted by permission from Macmillan Publishers Ltd: [NPG Asia Materials] (ref.[32]), copyright (2015).

The well-developed DNA base chemistry also offers powerful platforms to build aptamer-based nanotheranostics. Zhu et al reported drug-DNA adducts (DDAs) using a formaldehyde linker to site-specifically conjugate drug molecules (e. g. deoxyguanosine) to DNA strands (Figure 7B).[32] The DDAs maintained DNA functionalities, including hybridization-mediated DNA nanoadduct formation, aptamer-mediated target recognition, and targeted drug delivery into cancer cells. The conjugated drugs could be gradually released at physiological temperature and showed a significant inhibitory effect on target tumor growth in a tumor xenograft mouse model.
Countless biochemical circuits exist in biological organisms, and their orderly operation ensures normal organismal functioning. By mimicking the natural biosystem, the cascading of DNA-based switches, or logic gates, has been suggested to result in the generation of artificial biochemical circuits, or even the construction of molecular computers. As a potential repository of disease-related biomarkers, principally proteins, the cell membrane is the most suitable biological unit on which to base the construction of DNA circuits. In this field, smart networks for autonomous cancer targeting and therapy can be created by combining toehold-mediated strand displacements with aptameric structure-switching. For example, You et al. reported a series of Boolean logic gate-based DNA networks to realize multicellular, marker-based cancer analysis.[33, 34] As shown in Figure 8, diagnostic signals and/or targeted photodynamic therapy will be triggered only when multiple biomarkers are recognized on the same cell surface, such as the AND-AND logic gate. Thus, in the most simplistic view, DNA networks can act as “nanodoctors” that test the expression level(s) of one or more cancer-associated biomarkers on the cell surface and then dispense the appropriate therapy according to the diagnostic result. The greatest advantage of such DNA circuits lies in their direct interaction with other biomolecules, leading to subsequent control or regulation of the particular biosystem, not otherwise attainable with silicon-based circuits.
A) The symbols and construction schemes are shown for 2-input trivalent “Y”-shaped Nano-Claw and 3-input tetravalent “X”-shaped Nano-Claw. B) Experimental scheme of aptamer-switch AND gate. Reprinted (adapted) with permission from ref.[33]. Copyright (2013) American Chemical Society.

Challenges and perspectives
Cell-SELEX allows targeting aptamers to be generated without prior knowledge of molecular differences on cell membranes. Large numbers of aptamers against various cell lines, especially different types of cancer cells, have been reported and further exploited in cell capture and detection, selective imaging, drug delivery and biomarker discovery,[66-69] which make cell-SELEX an emerging and promising platform for biomedicine study. However, in spite of this considerable progress, cell-SELEX still faces many challenges.
First, theory and practice often vary greatly. For example, the high abundance of sequences in the final selected pool that bind neither control nor target cells cannot be easily interpreted. Moreover, the entire SELEX process is lengthy, and, as stated above, selection is not so straightforward, nor is the outcome always favorable after many rounds of selection. Besides, sequence discrimination in PCR amplification of the random DNA pools can impede enrichment.
Secondly, the diversity and structural complexity of molecules found on the cell surface make the cell-based SELEX process more sophisticated. Theoretically, hydrogen bond donors/acceptors, target molecules with positive charges, and/or aromatic groups could advance aptamer selection and high-abundance target molecules could advance selection as well. However, if cell-surface molecules with these features are not the desired targets, i.e., molecules common to many cell types, they would interfere with the enrichment of the desired aptamers. The aptamer selection would be hindered by target molecules with negative charges, high hydrophobicity, and low abundance. The abundance of some membrane molecules may change with cell growth and cell passages, which interferes with aptamer selection, underscoring the necessity of maintaining good cell culture.
Thirdly, compared with traditional SELEX against purified targets, a target identification step is of great necessity for cell-SELEX when the desired aptamers are obtained. Nevertheless, the separation and purification of aptamer targets, especially membrane protein targets, still remains a serious limitation that exerts a negative effect on target identification, due to the fact that up to now, only a limited number of aptamer targets have been identified[11, 60, 70, 71], which greatly blocks the further application of selected aptamers.
Lastly, as time consuming and tedious as selection is, it also expensive and requires a high level of technical experience, which prevents the technology from being widely utilized.
Some of these problems can be solved with modified cell-SELEX strategies. FACS-SELEX allows simultaneous removal of dead cells during SELEX process and selects aptamers selectively against cell subpopulations according to flow cytometry and cell sorting.[72, 73] An on-chip Cell-SELEX strategy for automatic selection of aptamers has shortened the selection process to as few as five rounds.[74] 3D Cell-SELEX, which combines the advantages of Cell-SELEX technology and 3D cell culture, has been reported to generate aptamers in mimicked tissue micro-environments.[75]
In the future, continued efforts are needed to determine the principles of aptamer enrichment in cell-SELEX, reveal the binding mechanisms of aptamers to their targets on the cell surface, and develop new methods to enhance selection efficiency, shorten the selection period and lower costs. These improvements may consist of a more automatic selection platform, development of efficient and universal strategies for target identification, and development of a database library of aptamers generated by cell-SELEX, including information on target and control cell lines, initial library, and selection conditions. This database would provide comprehensive information for further understanding of cell-SELEX and cellular biology.
Aptamers are powerful molecules with both biological and chemical properties which allow various modifications and conjugations. It can be predicted that in the near future, aptamers will not only play a more important role in the field of nanotheranostics, aiding in clinical detection and personalized therapies, but also become ubiquitous nanotools in biomedicine studies like cell biology research. Further improvements in aptamer selection by cell-SELEX should lead to an equally improved understanding of the biochemical and molecular basis of changes, including mass-density fluctuations, which manifest in diseased cells, in turn, spurring exciting new technologies for detection, diagnosis, and treatment of cancer.
Acknowledgements
This work is supported by NSFC grants (NSFC 21521063 and NSFC 21327009), and the U.S. National Institutes of Health (GM079359, GM 111386 and CA133086).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tan W, Donovan MJ, Jiang J. Aptamers from cell-based selection for bioanalytical applications. Chem Rev. 2013;113:2842-62
2. Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818-22
3. Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505-10
4. Lee JF, Stovall GM, Ellington AD. Aptamer therapeutics advance. Curr Opin Chem Biol. 2006;10:282-9
5. Morris KN, Jensen KB, Julin CM, Weil M, Gold L. High affinity ligands from in vitro selection: Complex targets. Proc Natl Acad Sci U S A. 1998;95:2902-7
6. Blank M, Weinschenk T, Priemer M, Schluesener H. Systematic Evolution of a DNA Aptamer Binding to Rat Brain Tumor Microvessels: SELECTIVE TARGETING OF ENDOTHELIAL REGULATORY PROTEIN PIGPEN. J Biol Chem. 2001;276:16464-8
7. Daniels DA, Chen H, Hicke BJ, Swiderek KM, Gold L. A tenascin-C aptamer identified by tumor cell SELEX: Systematic evolution of ligands by exponential enrichment. Proc Natl Acad Sci U S A. 2003;100:15416-21
8. Wang C, Zhang M, Yang G, Zhang D, Ding H, Wang H. et al. Single-stranded DNA aptamers that bind differentiated but not parental cells: subtractive systematic evolution of ligands by exponential enrichment. J Biotechnol. 2003;102:15-22
9. Shangguan D, Li Y, Tang Z, Cao ZC, Chen HW, Mallikaratchy P. et al. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc Natl Acad Sci U S A. 2006;103:11838-43
10. Jiang G, Zhang M, Yue B, Yang M, Carter C, Al-Quran SZ. et al. PTK7: a new biomarker for immunophenotypic characterization of maturing T cells and T cell acute lymphoblastic leukemia. Leuk Res. 2012;36:1347-53
11. Shangguan D, Cao Z, Meng L, Mallikaratchy P, Sefah K, Wang H. et al. Cell-Specific Aptamer Probes for Membrane Protein Elucidation in Cancer Cells. J Proteome Res. 2008;7:2133-9
12. Tang Z, Shangguan D, Wang K, Shi H, Sefah K, Mallikratchy P. et al. Selection of Aptamers for Molecular Recognition and Characterization of Cancer Cells. Anal Chem. 2007;79:4900-7
13. Chen HW, Medley CD, Sefah K, Shangguan D, Tang Z, Meng L. et al. Molecular Recognition of Small-Cell Lung Cancer Cells Using Aptamers. ChemMedChem. 2008;3:991-1001
14. Shangguan D, Meng L, Cao ZC, Xiao Z, Fang X, Li Y. et al. Identification of Liver Cancer-Specific Aptamers Using Whole Live Cells. Anal Chem. 2008;80:721-8
15. Li W-M, Bing T, Wei J-Y, Chen Z-Z, Shangguan D-H, Fang J. Cell-SELEX-based selection of aptamers that recognize distinct targets on metastatic colorectal cancer cells. Biomaterials. 2014;35:6998-7007
16. Wang Y, Luo Y, Bing T, Chen Z, Lu M, Zhang N. et al. DNA Aptamer Evolved by Cell-SELEX for Recognition of Prostate Cancer. PLoS One. 2014;9:e100243
17. Sefah K, Bae K-M, Phillips JA, Siemann DW, Su Z, McClellan S. et al. Cell-based selection provides novel molecular probes for cancer stem cells. Int J Cancer. 2013;132:2578-88
18. Sefah K, Tang ZW, Shangguan DH, Chen H, Lopez-Colon D, Li Y. et al. Molecular recognition of acute myeloid leukemia using aptamers. Leukemia. 2009;23:235-44
19. Tang Z, Parekh P, Turner P, Moyer RW, Tan W. Generating Aptamers for Recognition of Virus-Infected Cells. Clin Chem. 2009;55:813-22
20. Zhao Z, Xu L, Shi X, Tan W, Fang X, Shangguan D. Recognition of subtype non-small cell lung cancer by DNA aptamers selected from living cells. Analyst. 2009;134:1808-14
21. Parekh P, Tang Z, Turner PC, Moyer RW, Tan W. Aptamers Recognizing Glycosylated Hemagglutinin Expressed on the Surface of Vaccinia Virus-Infected Cells. Anal Chem. 2010;82:8642-9
22. Sefah K, Meng L, Lopez-Colon D, Jimenez E, Liu C, Tan W. DNA Aptamers as Molecular Probes for Colorectal Cancer Study. PLoS One. 2010;5:e14269
23. Wu X, Zhao Z, Bai H, Fu T, Yang C, Hu X. et al. DNA Aptamer Selected against Pancreatic Ductal Adenocarcinoma for in vivo Imaging and Clinical Tissue Recognition. Theranostics. 2015;5:985-94
24. Wang H, Liang J, Ma Y, Sun B, Li X, Wei Y. et al. Identification of a novel molecular probe for recognition of human osteosarcoma cell using the cell-SELEX method. Int J Clin Exp Med. 2015;8:18151-7
25. Lu M, Zhou L, Zheng X, Quan Y, Wang X, Zhou X. et al. A novel molecular marker of breast cancer stem cells identified by cell-SELEX method. Cancer Biomark. 2015;15:163-70
26. Li X, An Y, Jin J, Zhu Z, Hao L, Liu L. et al. Evolution of DNA Aptamers through in Vitro Metastatic-Cell-Based Systematic Evolution of Ligands by Exponential Enrichment for Metastatic Cancer Recognition and Imaging. Anal Chem. 2015;87:4941-8
27. Benedetto G, Hamp TJ, Wesselman PJ, Richardson C. Identification of Epithelial Ovarian Tumor-Specific Aptamers. Nucleic Acid Ther. 2015;25:162-72
28. Rong Y, Chen H, Zhou X-F, Yin C-Q, Wang B-C, Peng C-W. et al. Identification of an aptamer through whole cell-SELEX for targeting high metastatic liver cancers. Oncotarget. 2016;7:8282-94
29. Zhu G, Zheng J, Song E, Donovan M, Zhang K, Liu C. et al. Self-assembled, aptamer-tethered DNA nanotrains for targeted transport of molecular drugs in cancer theranostics. Proc Natl Acad Sci U S A. 2013;110:7998-8003
30. Zhu G, Hu R, Zhao Z, Chen Z, Zhang X, Tan W. Noncanonical self-assembly of multifunctional DNA nanoflowers for biomedical applications. J Am Chem Soc. 2013;135:16438-45
31. Wang R, Zhu G, Mei L, Xie Y, Ma H, Ye M. et al. Automated modular synthesis of aptamer-drug conjugates for targeted drug delivery. J Am Chem Soc. 2014;136:2731-4
32. Zhu G, Cansiz S, You M, Qiu L, Han D, Zhang L. et al. Nuclease-resistant synthetic drug-DNA adducts: programmable drug-DNA conjugation for targeted anticancer drug delivery. NPG Asia Mater. 2015;7:e169
33. You M, Peng L, Shao N, Zhang L, Qiu L, Cui C. et al. DNA “nano-claw”: logic-based autonomous cancer targeting and therapy. J Am Chem Soc. 2014;136:1256-9
34. You M, Zhu G, Chen T, Donovan MJ, Tan W. Programmable and multiparameter DNA-based logic platform for cancer recognition and targeted therapy. J Am Chem Soc. 2014;137:667-74
35. Stoltenburg R, Reinemann C, Strehlitz B. SELEX—A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol Eng. 2007;24:381-403
36. Sefah K, Shangguan D, Xiong X, O'Donoghue MB, Tan W. Development of DNA aptamers using Cell-SELEX. Nat Protoc. 2010;5:1169-85
37. Benner SA. Understanding nucleic acids using synthetic chemistry. Acc Chem Res. 2004;37:784-97
38. Sefah K, Yang Z, Bradley KM, Hoshika S, Jiménez E, Zhang L. et al. In vitro selection with artificial expanded genetic information systems. Proc Natl Acad Sci U S A. 2014;111:1449-54
39. Zhang L, Yang Z, Sefah K, Bradley KM, Hoshika S, Kim M-J. et al. Evolution of Functional Six-Nucleotide DNA. J Am Chem Soc. 2015;137:6734-7
40. Hicke BJ, Marion C, Chang Y-F, Gould T, Lynott CK, Parma D. et al. Tenascin-C Aptamers Are Generated Using Tumor Cells and Purified Protein. J Biol Chem. 2001;276:48644-54
41. Liu J, Liu H, Sefah K, Liu B, Pu Y, Van Simaeys D. et al. Selection of Aptamers Specific for Adipose Tissue. PLoS One. 2012;7:e37789
42. Cao X, Li S, Chen L, Ding H, Xu H, Huang Y. et al. Combining use of a panel of ssDNA aptamers in the detection of Staphylococcus aureus. Nucleic Acids Res. 2009;37:4621-8
43. Turek D, Van Simaeys D, Johnson J, Ocsoy I, Tan W. Molecular recognition of live methicillin-resistant staphylococcus aureus cells using DNA aptamers. World J Transl Med. 2013;2:67
44. Bayrac AT, Sefah K, Parekh P, Bayrac C, Gulbakan B, Oktem HA. et al. In vitro selection of DNA aptamers to glioblastoma multiforme. ACS Chem Neurosci. 2011;2:175-81
45. Bing T, Yang X, Mei H, Cao Z, Shangguan D. Conservative secondary structure motif of streptavidin-binding aptamers generated by different laboratories. Bioorg Med Chem. 2010;18:1798-805
46. Shangguan D, Tang Z, Mallikaratchy P, Xiao Z, Tan W. Optimization and modifications of aptamers selected from live cancer cell lines. Chembiochem. 2007;8:603-6
47. Legiewicz M, Yarus M. A more complex isoleucine aptamer with a cognate triplet. J Biol Chem. 2005;280:19815-22
48. Manimala JC, Wiskur SL, Ellington AD, Anslyn EV. Tuning the specificity of a synthetic receptor using a selected nucleic acid receptor. J Am Chem Soc. 2004;126:16515-9
49. Sayer NM, Cubin M, Rhie A, Bullock M, Tahiri-Alaoui A, James W. Structural determinants of conformationally selective, prion-binding aptamers. J Biol Chem. 2004;279:13102-9
50. Green LS, Jellinek D, Jenison R, Östman A, Heldin C-H, Janjic N. Inhibitory DNA ligands to platelet-derived growth factor B-chain. Biochemistry. 1996;35:14413-24
51. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406-15
52. Mei H, Bing T, Yang X, Qi C, Chang T, Liu X. et al. Functional-group specific aptamers indirectly recognizing compounds with alkyl amino group. Anal Chem. 2012;84:7323-9
53. Qi C, Bing T, Mei H, Yang X, Liu X, Shangguan D. G-quadruplex DNA aptamers for zeatin recognizing. Biosens Bioelectron. 2013;41:157-62
54. Yang X, Bing T, Mei H, Fang C, Cao Z, Shangguan D. Characterization and application of a DNA aptamer binding to L-tryptophan. Analyst. 2011;136:577-85
55. Bing T, Chang T, Yang X, Mei H, Liu X, Shangguan D. G-quadruplex DNA aptamers generated for systemin. Bioorg Med Chem. 2011;19:4211-9
56. Kimoto M, Yamashige R, Matsunaga K-i, Yokoyama S, Hirao I. Generation of high-affinity DNA aptamers using an expanded genetic alphabet. Nat Biotechnol. 2013;31:453-7
57. Xiao Z, Shangguan D, Cao Z, Fang X, Tan W. Cell-Specific Internalization Study of an Aptamer from Whole Cell Selection. Chem Eur J. 2008;14:1769-75
58. Chen Y, Munteanu AC, Huang YF, Phillips J, Zhu Z, Mavros M. et al. Mapping receptor density on live cells by using fluorescence correlation spectroscopy. Chem Eur J. 2009;15:5327-36
59. Nimjee SM, Rusconi CP, Sullenger BA. Aptamers: an emerging class of therapeutics. Annu Rev Med. 2005;56:555-83
60. Mallikaratchy P, Tang Z, Kwame S, Meng L, Shangguan D, Tan W. Aptamer directly evolved from live cells recognizes membrane bound immunoglobin heavy mu chain in Burkitt's lymphoma cells. Mol Cell Proteomics. 2007;6:2230-8
61. Yang Z, Chen F, Alvarado JB, Benner SA. Amplification, mutation, and sequencing of a six-letter synthetic genetic system. J Am Chem Soc. 2011;133:15105-12
62. O'Connor ML, Xiang D, Shigdar S, Macdonald J, Li Y, Wang T. et al. Cancer stem cells: a contentious hypothesis now moving forward. Cancer Lett. 2014;344:180-7
63. Sun H, Zu Y. Aptamers and their applications in nanomedicine. Small. 2015;11:2352-64
64. Xiang D, Shigdar S, Qiao G, Wang T, Kouzani AZ, Zhou S-F. et al. Nucleic acid aptamer-guided cancer therapeutics and diagnostics: the next generation of cancer medicine. Theranostics. 2015;5:23-42
65. Lv Y, Hu R, Zhu G, Zhang X, Mei L, Liu Q. et al. Preparation and biomedical applications of programmable and multifunctional DNA nanoflowers. Nat Protoc. 2015;10:1508-24
66. Fang X, Tan W. Aptamers generated from cell-SELEX for molecular medicine: a chemical biology approach. Acc Chem Res. 2009;43:48-57
67. Pu Y, Zhu Z, Liu H, Zhang J, Liu J, Tan W. Using aptamers to visualize and capture cancer cells. Anal Bioanal Chem. 2010;397:3225-33
68. Ye M, Hu J, Peng M, Liu J, Liu J, Liu H. et al. Generating aptamers by cell-SELEX for applications in molecular medicine. Int J Mol Sci. 2012;13:3341-53
69. Meyer C, Hahn U, Rentmeister A. Cell-Specific Aptamers as Emerging Therapeutics. J Nucleic Acids. 2011. 2011
70. Van Simaeys D, Turek D, Champanhac C, Vaizer J, Sefah K, Zhen J. et al. Identification of cell membrane protein stress-induced phosphoprotein 1 as a potential ovarian cancer biomarker using aptamers selected by cell systematic evolution of ligands by exponential enrichment. Anal Chem. 2014;86:4521-7
71. Yang M, Jiang G, Li W, Qiu K, Zhang M, Carter CM. et al. Developing aptamer probes for acute myelogenous leukemia detection and surface protein biomarker discovery. J Hematol Oncol. 2014;7:1-14
72. Raddatz MSL, Dolf A, Endl E, Knolle P, Famulok M, Mayer G. Enrichment of Cell-Targeting and Population-Specific Aptamers by Fluorescence-Activated Cell Sorting. Angew Chem Int Ed Engl. 2008;47:5190-3
73. Mayer G, Ahmed M-SL, Dolf A, Endl E, Knolle PA, Famulok M. Fluorescence-activated cell sorting for aptamer SELEX with cell mixtures. Nat Protoc. 2010;5:1993-2004
74. Hung L-Y, Wang C-H, Hsu K-F, Chou C-Y, Lee G-B. An on-chip Cell-SELEX process for automatic selection of high-affinity aptamers specific to different histologically classified ovarian cancer cells. Lab Chip. 2014;14:4017-28
75. Souza AG, Marangoni K, Fujimura PT, Alves PT, Silva MJ, Bastos VAF. et al. 3D Cell-SELEX: Development of RNA aptamers as molecular probes for PC-3 tumor cell line. Exp Cell Res. 2016;341:147-56
Author contact
![]() Corresponding authors: Email: tanufl.edu; wangjie0103com
Corresponding authors: Email: tanufl.edu; wangjie0103com