Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results and Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(8):2204-2219. doi:10.7150/thno.19571 This issue Cite
Research Paper
E-selectin-targeted Sialic Acid-PEG-dexamethasone Micelles for Enhanced Anti-Inflammatory Efficacy for Acute Kidney Injury
Jing-Bo Hu1, Xu-Qi Kang1, Jing Liang2, Xiao-Juan Wang1, Xiao-Ling Xu1, Ping Yang3, Xiao-Ying Ying1, Sai-Ping Jiang3 ![]() , Yong-Zhong Du1
, Yong-Zhong Du1 ![]()
1. Institute of Pharmaceutics, College of Pharmaceutical Sciences, Zhejiang University, 866 Yuhangtang Road, Hangzhou 310058, China;
2. Department of Pharmacy, Zhejiang Hospital, Hangzhou 310013, China;
3. Department of Pharmacy, The First Affiliated Hospital, College of Medicine, Zhejiang University, 79 Qingchun Road, Hangzhou 310003, China.
Received 2017-2-8; Accepted 2017-3-16; Published 2017-6-1
Abstract

The effective treatment for acute kidney injury (AKI) is currently limited, and care is primarily supportive. Sialic acid (SA) is main component of Sialyl Lewisx antigen on the mammalian cell surface, which participates in E-selectin binding. Therefore, dexamethasone(DXM)-loaded E-selectin-targeting sialic acid-polyethylene glycol-dexamethasone (SA-PEG-DXM/DXM) conjugate micelles are designed for ameliorating AKI. The conjugates are synthesized via the esterification reaction between PEG and SA or DXM, and can spontaneously form micelles in an aqueous solution with a 65.6 µg/mL critical micelle concentration. Free DXM is incorporated into the micelles with 6.28 ± 0.21% drug loading content. In vitro DXM release from SA-PEG-DXM/DXM micelles can be prolonged to 48h. Much more SA-PEG-DXM micelles can be internalized by lipopolysaccharide (LPS)-activated human umbilical vein endothelial cells (HUVECs) in comparison to PEG-DXM micelles due to specific interaction between SA and E-selectin expressed on HUVECs, and consequently more SA-PEG-DXM micelles are accumulated in the kidney of AKI murine model. Furthermore, SA in SA-PEG-DXM conjugates can significantly ameliorate LPS-induced production of pro-inflammatory cytokines via suppressing LPS-activated Beclin-1/Atg5-Atg12-mediated autophagy to attenuate toxicity. Compared with free DXM and PEG-DXM/DXM micelles, SA-PEG-DXM/DXM micelles show better therapeutical effects, as reflected by the improved renal function, histopathological changes, pro-inflammatory cytokines, oxidative stress and expression of apoptotic related proteins.
Keywords: Acute kidney injury, Sialic acid, Dexamethasone, Micelles, Anti-inflammatory effect.
Introduction
Acute kidney injury (AKI) occurs during endotoxemia or ischemia/reperfusion, where endotoxin binds to endothelium and leukocytes, and then causes the production and release of pro-inflammatory cytokines, along with increased oxidative stress and apoptosis [1, 2]. There is strong evidence that patients with AKI are associated with a higher increased mortality rate [3, 4]. Glucocorticosteroids have been extensively studied as renoprotective agents due to their potent anti-inflammatory and immunosuppressive efficacy [5, 6]. But meanwhile, glucocorticoid systemic therapy is inevitably associated with several serious side effects in a dose-dependent manner [7, 8]. Therefore, identifying more effective therapeutic approach with less systemic toxicity for AKI remains a primary aim.
In the last years, the use of carrier systems has been proposed in the renal drug delivery adopting different targeting strategies for the diverse target sites [9-11]. Drug-carrier conjugates were designed to delivery drugs to the proximal tubular cells, based on their role in the reabsorption and secretion of substances, to achieve kidney targeting [9]. For example, chitosan-drug conjugates were developed renal targeting therapy via specific accumulation in the proximal tubular cells by means of megalin and cubilin receptor-mediated endocytosis [11]. However, recent study indicated that acute endotoxemia would down-regulate megalin and cubilin receptor, two internalizing receptors on the proximal tubular cells responsible for reabsorption [12]. With the progressive deterioration of disease, the reabsorption function of proximal tubular cells weaken due to the down-regulation of megalin and cubilin receptors, and thus the uptake of drugs in these cells are potentially reduced. Antibody mediated liposomes were another therapeutic strategy to site-selectively delivery anti-inflammatory agents to the kidney to sustain efficacy and prevent drug-induced side effects [10, 13-15], but this design thought is hampered by the potential immunogenicity, increased size of drug delivery system and high cost of antibody production. In addition, the restricted species specificity between animals and humans must be given sufficient consideration.
Encouraged by these inadequacies, we attempt to identify a small molecule ligand to achieve efficient and safe cell-specific drug delivery to the kidney. Vascular endothelial cells (VECs), lining the entire circulatory system (from the heart to the smallest capillaries), have unique vascular biology functions, including fluid filtration (e.g. the glomerulus), blood vessel tone, neutrophil recruitment and hormone trafficking [16]. VECs are activated by pro-inflammatory cytokines and endothelial expression of E-selectin increases during inflammation, which affect the interaction of leukocytes with activated VECs through process of tethering and rolling [17]. Next, the leukocytes adhere to the vascular endothelium through interaction between integrins of the leukocytes and adhesion molecules (e.g. and E-selectin) on the VECs, and subsequently transmigrate into the inflamed tissue to cause inflammation-related organ impairment [18]. Therefore, VECs play a key role in the renal damage, and they are also potential therapeutic target for the treatment of AKI.
Sialic acid (SA), a family of 9-carbon carboxylated monosaccharides, was found as the termini of mammalian cell surface glycoproteins and glycolipids, and was a component of sialyl Lewisx antigen participating in E-selectin binding [19]. Previous studies demonstrated that SA was a well-known ligand for selectin responsible for tumor metastases through modulating cell-cell interactions among leukocytes, platelets, endothelial cells and tumor cells [20-23]. Therefore, we speculate that SA has the potential to specifically bind to E-selectin expressed on activated VECs during inflammation. If this is true, SA potentially provides many advantages over sialyl Lewisx antigen. First, unlike the macromolecular antibody, SA can achieve further development in clinical practice benefiting from its homogeneous and well-defined molecular structure. Second, as an endogenous substance, SA possesses a higher biological safety over macromolecular antibody. Third, SA has been found to ameliorate acute hepatic and renal failure via neutralizing toxicity [24].
In this study, dexamethasone loaded sialic acid-PEG-dexamethasone (SA-PEG-DXM/DXM) micelles were designed for the treatment of AKI. SA-PEG-DXM conjugates were firstly synthesized via an esterification reaction, and then SA-PEG-DXM/DXM micelles were developed via the solvent diffusion method. The characteristics of SA-PEG-DXM/DXM micelles as a drug delivery carrier, including drug-loading ability, size, potential and in vitro drug release behavior, were examined in detail. As a potential ligand for E-selectin, the target function of SA-PEG-DXM micelles was investigated in vitro and vivo. The therapeutic potential of SA-PEG-DXM/DXM micelles was assessed in LPS-induced AKI murine model in comparison to PEG-DXM/DXM micelles and free DXM (equal DXM), both of which was used as control. The evaluation index included renal function, histopathological changes, production of pro-inflammatory cytokines, oxidative stress levels and expression of apoptotic related proteins.
Materials and Methods
Materials
Dexamethasone and sialic acid were purchased from Aladdin Bio-chem Technology Co. Limited (Shanghai, China). HOOC-PEG-COOH (Mw = 2.0 kDa) was purchased from Sigmae-Aldrich Inc., USA. Fluorescein isothiocyanate (FITC), indocyanine green (ICG) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies, including Anti-Bax, Anti-Bcl-2, Anti-Caspase-3, Anti-CD62E(E-selectin), Anti-Beclin 1, Anti-Atg5 and Anti-Atg5/12 antibodies were purchased from Abcom. A TUNEL assay kit was obtained from Roche (Nutley, NJ, USA). All other solvents were of analytical or chromatographic grade.
Human umbilical vein endothelial cells (HUVECs) were purchased from the Thermo Fisher. HUVECs were maintained in RPMI-1640 containing 10% fetal bovine serum (Gibco) as well as 1% penicillin and streptomycin (Sigma) and were cultured at 37°C in a humidified atmosphere of 5% CO2.
ICR mice (body weight: 18-20 g) were purposed from the Zhejiang Medical Animal Centre. All animal experiments were carried out in accordance with the National Institutes of Health (NIH, USA) guidelines for the care and use of laboratory animals in research. The surgical procedures and experiment protocols were approved by the Committee for Animal Experiments of Zhejiang University.
Synthesis of sialic acid-PEG-dexamethasone conjugates
Synthesis of sialic acid-g-PEG-g-dexamethasone (SA-PEG-DXM) was accomplished in two steps: (1) esterification reaction between the carboxyl groups of PEG (HOOC-PEG-COOH) and the hydroxyl group of DXM, (2) esterifying sialic acid to PEG-DXM (HOOC-PEG-DXM). Briefly, PEG (5.00 g, 2.50 mmol, 1 equiv), DCC (1.55 g, 7.50 mmol, 3 equiv) and DMAP (0.10 g, 0.82 mmol, 0.3 equiv) were dissolved in anhydrous dimethyl formamide (DMF). The solution was stirred under the protection of nitrogen for 2h to activate the carboxylic acid of PEG. Then, DXM (0.98 g, 2.50 mmol, 1 equiv) was added in the solution and the reaction was carried out for 48h in nitrogen gas at 60℃ under stirring at 250 rpm. The reaction solution was dialyzed (MWCO 7.0 kDa) against pure water for 48 h with frequent exchange (4h) of pure water to remove DMF and water-soluble byproducts, and the dialyzed solution was centrifugated at 15000 rpm to remove water-insoluble byproducts. After the supernatants were lyophilized, the crude product was purified by cold diethyl ether to remove the unreacted DXM, and thus the resulting PEG-DXM was obtained (4.31 g, 72% yield).
Next, SA-PEG-DXM was synthesized via the esterification reaction between carboxyl group of PEG-DXM and hydroxy group of SA in the presence of DCC and DMAP. Similarly, PEG-DXM DCC and DMAP (PEG-PLGA conjugates:DCC:DMAP = 1:3:0.3, mol:mol:mol) were dissolved in anhydrous DMF. The solution was stirred at 60℃ for 1 h to activate the carboxylic acid of PEG-PLGA conjugates under the protection of nitrogen. After SA was added, the reaction was stirred 60℃ under the protection of nitrogen for 48 h at 250 rpm. The reaction solution was then dialyzed (MWCO 7.0 kDa) to against pure water for 48 h with frequent exchanges of pure water. The dialyzed solution was centrifugated at 15000 rpm to remove water-insoluble byproducts, and lyophilized to achieve SA-PEG-DXM
Characterization of sialic acid-PEG-dexamethasone conjugates
The composition of obtained SA-PEG-DXM conjugates was confirmed using a NMR spectrometer (AC-80, Bruker Bios pin. Germany). Samples were dissolved at 10 mg·mL-1 in dimethylsulfoxide-d6 for detection.
The CMC of SA-PEG-DXM was determined using pyrene as a fluorescence probe [25]. The concentration of SA-PEG-DXM solution was varied from 3.0 × 10-3 to 1.0 mg·mL-1 and the concentration of pyrene was fixed at 6.0 × 10-7 M. The fluorescence spectra were recorded using a fluorescence spectrophotometer (F-2500, Hitachi Co., Japan) with the excitation wavelength of 337 nm. The emission fluorescence at a wavelength range of 300-470 nm was monitored. The CMC was determined via calculating the emission intensity ratio of the first peak (I1, 374 nm) to the third peak (I3, 385 nm) [25].
Preparation and characterization of sialic acid-PEG-dexamethasone micelles
DXM-loaded SA-PEG-DXM micelles were prepared through solvent diffusion method. Briefly, 10.0 mg SA-PEG-DXM conjugates and free DXM were dissolved in 0.2 mL ethanol, and then the mixture solution was rapidly injected into 10.0 mL of deionized water with constant stirring for 10 min. Afterwards, the mixture solution was dialyzed against pure water (MWCO 7.0 kDa) for 6h at room temperature with frequent change of dialysis media to remove ethanol. Finally, the suspension was centrifuged at 4 000 rpm for 10 min to remove unencapsulated DXM. The supernatant was lyophilized, and ultimately SA-PEG-DXM/DXM micelles were obtained. PEG-DXM/DXM micelles were prepared by the same method.
The micellar size, size distribution and zeta potential were measured by dynamic light scattering after suitable dilution (Zetasizer, Malvern Co., UK). The morphological examinations were checked by transmission electron microscopy (TEM) (JEOL JEM-1230, Japan).
To determine the encapsulated DXM in micelles, SA-PEG-DXM/DXM micelles were dispersed in 0.1M hydrochloric acid solution and stirred at 37 ℃ for 90 min to produce free DXM. The total DXM content (including encapsulated DXM and conjugated DXM) was determined by HPLC with C18 column (250 mm × 4.6 mm, 5 μm). Methanol and water (v/v = 60:40) was used as the mobile phase. The column temperature and the detection wavelength were set as 25 ℃ and 240 nm with flow rate at 1.0 mL·min-1. The DXM-loading content (DL%) was calculated using equation below:
DL (%) = ((Total DXM - encapsulated DXM)/Weight of the micelle) × 100%
In vitro drug release studies
The dialysis membrane diffusion technique was used to investigate drug release characteristics. The content of ATO was measured using HPLC method. DXM release from micelles is performed in the phosphate buffer solution (PBS) at pH 7.4 for a period of 48 h. A known amount of free DXM, SA-PEG-DXM/DXM micelles, PEG-DXM/DXM solution, SA-PEG-DXM conjugates and PEG-DXM conjugates (equal DXM) were respectively sealed in a dialysis membrane (MWCO 7.0 kDa), and then immersed into 30 mL release medium. This experiment was carried out in an incubator shaker (HZ-8812S, Scientific and Educational Equipment Plant, Taicang, China) maintained at 37℃ and shaken horizontally at 60 rpm. At predefined time intervals, the release medium was withdrawn and replaced with fresh medium. Released DXM was qualified by HPLC from the standard DXM curve.
Cytotoxicity studies
For assessing the cytotoxicity of SA-PEG-DXM conjugates, MTT assay was performed on HUVECs [35-25]. They were seeded on a 96-well plate with 1×104 cells/well and incubated for 24 h. After preincubation, cells were exposed to SA-PEG-DXM and PEG-DXM conjugates with serial concentrations (including 100, 200, 400, 600 and 800 µg·mL-1) for 48 h. 10 µL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (C18H16BrN5S, Sigma Chemical Co.) was added and incubated for another 4 h at 37 °C. Then, the medium was withdrawn and MTT formazan was dissolved in 200 μL of DMSO. After shaken for 20 min, the absorbance at a wavelength of 570 nm of the formazan product was measured using a microplate reader (Bio-Rad, model 680, U.S.A.). Relative cell viability was calculated with the following equation:
Cell viability = (Asample-Ablank)/(Anegative control-Ablank) × 100%
Immunofluorescence of E-selectin
HUVECs were seeded on sterile round coverslips at a density of 1 × 105 cells/well into 6-well plates and cultured for 48 h before they were activated with LPS (100 ng·mL-1) for 4 hours. Then, LPS-activated HUVECs were incubated with octadecanoic acid(ODA)-FITC-loaded SA-PEG-DXM micelles for 1 hour, PEG-DXM micelles as control. After rinsing with PBS, HUVECs were fixed with cold methanol (-20℃) and kept in the refrigerator for 3 min. HUVECs were washed with PBS at room temperature for 3 times, 5min for each. Nonspecific sites were blocked with blocking buffer (5% fetal bovine serum in PBS) at room temperature for 1hour. Next, HUVECs were incubated with monoclonal anti-human E-selectin antibody (10 µg·mL-1 in blocking buffer) overnight at 4℃. After washing with PBS at room temperature for 5 times, 10 min for each, HUVECs were incubated with secondary antibody (anti-rabbit, 1:200 dilution, HRP-labeled Goat Anti-Mouse IgG) blocking buffer at room temperature for 2 h. After washing with PBS, DAPI was prepared in PBS at 1:1000 dilution and stained at room temperature for 10 min. Imaging was performed with the laser scanning confocal microscope.
Cellular uptake
HUVECs were seeded in a 12-well plate (5 × 105 cells per well) and cultured for 24 hours, and then exposed to LPS (50 ng·mL-1). After 4 hours, LPS-activated HUVECs were incubated with ODA-FITC-loaded SA-PEG-DXM micelles for 0.5, 1 and 2 hours in 37℃, ODA-FITC-loaded PEG-DXM micelles as control. After washing the cells with PBS 3 times, the cellular uptake was observed using laser scanning confocal microscope (Zeiss LSM 510 META, Carl Zeiss, Germany). For the competitive experiment for E-selectin, HUVECs were pretreated with different concentration SA solution (1.0, 2.0 and 4.0 mg·mL-1) for 1 hour and then incubated with SA-PEG-DXM micelles for 1 hour. The cells were resuspended in PBS and fluorescence intensity was determined by flow cytometry (FC 500 MCL; Beckman Coulter, USA), and blanked by untreated cells.
In vitro cell culture and treatment
HUVECs, human umbilical vein endothelial cells (Thermo Fisher), were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin solution at 37°C in a humidified atmosphere of 5% CO2. HUVECs were seeded in a 96-well plate (5 × 105 cells per well) and cultured for 24 hours. Cells were treated with LPS (100 ng·mL-1) and different interventions (SA, DXM, SA+DXM, PEG-DXM, SAP-PEG-DXM) for 24h. The levels of TNF-α and IL-6 in the supernatant were determined using commercial kits (Thermo Scientific™) according to the manufacturer's protocol. All samples were measured triplicate.
Renal injury induction and treatment
For all of the mouse studies, ICR male mice (Zhejiang Medical Animal Centre, Hangzhou, China) were used. Age- and weight-matched mice (8 to 10 wk old, 20 to 25 g) were anesthetized with an intraperitoneal injection of pentobarbital sodium (1%, 50 mg·kg-1). This experiment was performed on a preheated to maintain body temperature at 37°C. A midline incision was made, and the kidney was directly injected with LPS (preheated to 37°C, 10 µL, 1 mg·mL-1) or saline. After 12h, animals were assigned to receive treatment. After 2 and 7d, animals were killed, and blood samples and kidneys were obtained for measurements.
Mice were randomly allocated into the following groups: (1) sham + saline group (n = 6); (2) AKI + saline group (n = 6); (3) AKI + free DXM (n = 6); (4) AKI + PEG-DXM/DXM (n = 6); (5) AKI + SA-PEG-DXM/DXM (n = 6). The dose of DXM was determined as 1.2 mg/kg body weight because it was consistent with stress-dose of DXM in the clinical practice [26].
Biodistribution of ICG-labelled micelles
To investigate the in vivo distribution, the mice with AKI or without AKI were injected intravenously with ICG-tetrabutylammonium iodide complex-loaded SA-PEG-DXM micelles or PEG-DXM micelles at a dose of 5.0 mg·kg-1. The mice were sacrificed at predetermined time (2, 4, 6, 12, 24, 36 and 48h) and organs were harvested, including the heart, liver, spleen, lung and kidney. Fluorescence signal in collected tissues was analyzed using the Maestro in vivo imaging system (Cambridge Research & Instrumentation, Inc., Woburn, MA, USA).
Histological analysis and immunohistochemistry
Renal tissues were fixed in 4.5% buffered formalin and embedded in paraffin. Five micrometer-thick sections were stained using hematoxylin and eosin (H&E) and periodic acid-Schiff (PAS) for morphology analysis of kidneys with light microscopy. For immunohistochemical staining, primary antibody E-selectin (1:100) was applied to the sections. Examination and semi-quantitative score were carried out in a blinded fashion by an experienced pathologist. The following parameters were used to indicate morphological damage to the kidney: epithelial necrosis, luminal necrotic debris, and tubular dilation. For each mouse, at least 3 sections per kidney and 10 fields per section were scored as follows: none = 0; <10% = 1; 11-25% = 2; 26-75% = 3; and >75% = 4.
TUNEL
Apoptotic cells were determined by the changes showed by widespread terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay (Milipore, MA, USA) according to the manufacturer's protocol. The mount of apoptosis cells were counted at ×400 magnification and 10 fields per slide for each samples were randomly selected.
Assessment of renal function
Renal function was assayed for serum creatinine (SCR) and blood urea nitrogen (BUN) levels. Serum samples were measuring by using an automated Beckman Analyzer (Beckman Instruments GmbH, Munich, Germany).
Detection of pro-inflammatory cytokines and oxidative stress
The levels of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) as pro-inflammatory cytokines were measured using ELISA kits according to the protocol provided by the manufacturer (Boster Co., Ltd., Wuhan, China). The levels of NO, SOD, MPO and MDA were measured using commercial kits according to the manufacturer's protocol (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). All samples were measured triplicate.
Western blot
Kidney tissues were homogenized and lysed in ice-cold modified RIPA lysis buffer. Then, the protein concentrations were determined by a bicinchoninic acid assay (Beyotime Biotechnology, Shanghai, China) according to the manufacturer's instructions. Samples were matched for protein, separated by SDS-PAGE, and transferred to PVDF membranes (Sigma-Aldrich). After blocked at room temperature for at least 1 h in blocking buffer, the membranes were blotted with anti-Bax (1:1000, Abcam), anti-Bcl (1:1000, Abcam), anti-caspase 3 (1:1000, Abcam) and anti-β-actin (1:2000, Abcam) antibodies overnight at 4 ℃. The blots were washed and incubated with the secondary ant-rabbit immunoglobulin G (IgG) horseradish peroxidase (HRP) (1:1000, Beyotime Biotechnology, Shanghai, China) for 2 h at room temperature. The membrane was exposed to Fuji Medical X-ray film (Fujifilm) for various periods in a film cassette. The target bonds were selected and the average density was calculated. To facilitate comparisons, densitometry values of target protein were standardized with that of β-actin.
Statistical Analysis
All values are reported as mean ± SEM unless otherwise stated. All statistical analysis was performed using SPSS 14.0 statistical software. The significant differences between the groups were analyzed by a Student's t test, and P < 0.05 was considered significant.
Results and Discussion
Synthesis and characterization of SA-PEG-DXM conjugates
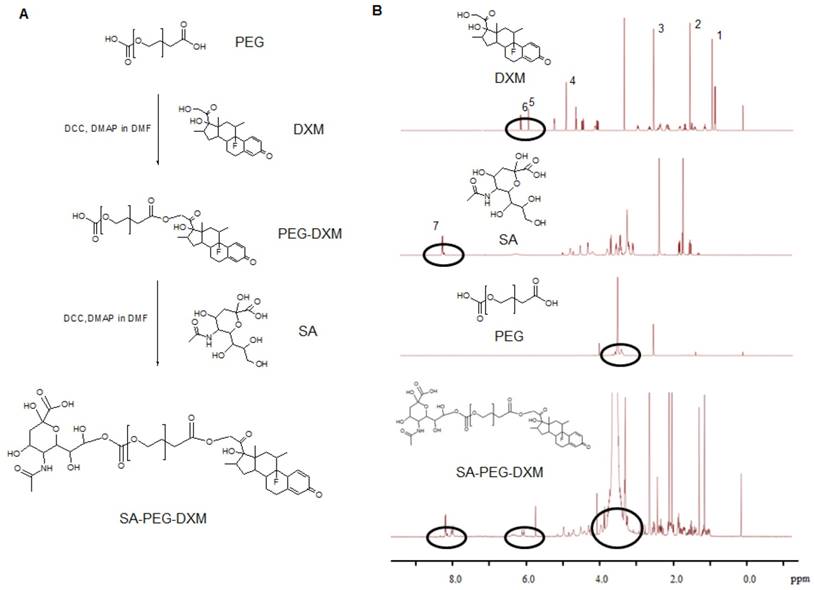
Sialic acid-g-PEG-g-dexamethasone (SA-PEG-DXM; 3.78 g, 76.82% yield) conjugates were successfully synthesized via the esterification reaction between the carboxyl groups of PEG (HOOC-PEG-COOH) and the hydroxyl groups of SA and DXM in the presence of 4-dimethylaminopyridine (DMAP) and dicyclohexylcarbodiimide (DCC). The detailed synthesis route is presented in Figure 1A. 1H NMR spectrum of SA-PEG-DXM taken in DMSO-d6, a solvent in which micelles did not form, was used to confirm the structure of SA-PEG-DXM conjugate, and the results are presented in Figure 1B. The proton peak of amino in SA (at about 8.1 ppm) and olefin in DXM (at about 6.2 ppm) were observed in 1H NMR spectrum of SA-PEG-DXM, which indicated that SA and DXM were successfully conjugated upon PEG. During the synthesis of SA-PEG-DXM conjugates, by-product (DXM-PEG-DXM) was inevitably obtained. The conjugated DXM content in SA-PEG-DXM conjugates was 15.9% (HPLC analysis for DXM as shown in Figure S1), which was slightly higher than theoretical content of DXM (15.4%, SA: PEG: DXM = 1:1:1). This result showed that the proportion of DXM-PEG-DXM in end-product was far less than SA-PEG-DXM. The conjugated DXM content in PEG-DXM conjugates, as control, was 16.4%.
Synthesis and characterization of sialic acid-g-PEG-g-dexamethasone conjugates. (A) The synthesis route of SA-PEG-DXM conjugates. (B) 1H NMR spectrum of DXM, SA, PEG and SA-PEG-DXM conjugates.
The inherent amphiphilicity property of SA-PEG-DXM and PEG-DXM conjugates leads to spontaneously forming micelles in aqueous medium. The hydrophobic DXM became the core to store hydrophobic drugs and the hydrophilic PEG formed the shell to enhance micellar stability in aqueous medium. The critical micelle concentration (CMC), an important characteristic of amphiphilic materials, was determined using pyrene as a fluorescence probe. The CMC values of SA-PEG-DXM and PEG-DXM conjugates were 63.9 and 60.4 µg/mL (Figure 2A), respectively. The low CMC values could ensure conjugates to form micelles with a core-shell structure in aqueous medium, and keep integrity and stability during systemic circulation [27].
Preparation and characterization of SA-PEG-DXM micelles. (A) The relationship of variation of fluorescence intensity ration (I1/I3) for pyrene and concentration of SA-PEG-DXM and PEG-DXM conjugates. (B) Size distribution obtained by DLS and TEM images: Blank SA-PEG-DXM Micelles; Blank PEG-DXM Micelles; SA-PEG-DXM/DXM Micelles; PEG-DXM/DXM Micelles. Scale bar = 50nm. (C) In vitro drug release profiles of free DXM, SA-PEG-DXM/DXM, PEG-DXM/DXM micelles, SA-PEG-DXM conjugates and PEG-DXM conjugates in pH 7.4 PBS at 37 ℃.
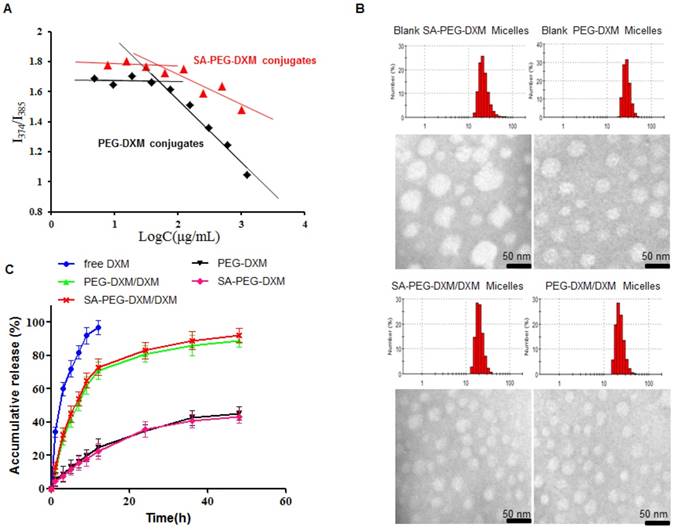
Preparation and characterization of DXM-loaded SA-PEG-DXM micelles
The SA-PEG-DXM/DXM and PEG-DXM/DXM micelles were successfully prepared via the solvent diffusion method. The micelle solution was dialyzed (MWCO 7.0 kDa) against pure water with frequent change of dialysis media to remove ethanol. Table 1 presents the characteristics of SA-PEG-DXM/DXM and PEG-DXM/DXM micelles. Figure 2B presents the size distribution and morphology measured by dynamic light scattering (DLS) and transmission electron microscopy (TEM) of blank and DXM-loaded micelles. It was found that both SA-PEG-DXM/DXM and PEG-DXM/DXM micelles presented had a uniform spherical shape and were well dispersed as individual nanoparticles in comparison to blank micelles. The average diameters of SA-PEG-DXM/DXM and PEG-DXM/DXM micelles both were around 20 nm, while the blank micelles were about 50 and 30 nm, respectively. The size difference between DXM-loaded micelles and blank micelles was due to the hydrophobic interaction between the hydrophobic chains (DXM) of conjugates and free DXM becoming stronger after DXM loading.
Characteristics of DXM-loaded micelles
| Type of micelles | dn (nm) | PDI | DL% | ζ Potential |
|---|---|---|---|---|
| SA-PEG-DXM | 45.3 ± 4.6 | 0.158 ± 0.028 | 6.28 ± 0.21 | -13.3 ± 4.8 |
| PEG-DXM | 42.7 ± 3.7 | 0.126 ± 0.023 | 9.82 ± 0.26 | -7.2 ± 3.1 |
dn: hydrodynamic diameter, PDI: polydispersity index, DL: drug-loading
Data are presented as mean ± S.E.M. (n = 3).
The drug-loading (DL%) of SA-PEG-DXM and PEG-DXM micelles were 6.28 ± 0.21 and 9.82 ± 0.26 during 15% DXM-feeding amount (Table 1), which was exclusive of the conjugated DXM. In vitro drug release profiles of SA-PEG-DXM/DXM in pH 7.4 phosphate buffer solution (PBS) are presented in Figure 2C. Free DXM was released rapidly, more than 90% within 12h. In contrast, sustained release of DXM from micelles could be maintained for more than 48 h, despite fast release in the initial 12 h. In addition, the release profiles of DXM from SA-PEG-DXM/DXM and PEG-DXM/M micelles were mostly similar, which indicated that SA had no effects on drug-release behaviors. The in vitro drug release profiles of SA-PEG-DXM and PEG-DXM conjugates were also presented in Figure 2C, and the results showed that the conjugated DXM could slowly release from SA-PEG-DXM and PEG-DXM. Therefore, the release of DXM includes two parts: the encapsulated DXM from micelles and conjugated DXM from conjugates. The release of DXM from SA-PEG-DXM/DXM micelles could be sustained for more than 48h, which suggested that administration frequency might be reduced and therapeutic effects could be maintained for a prolonged time.
In vitro cytotoxicity assay of SA-PEG-DXM micelles
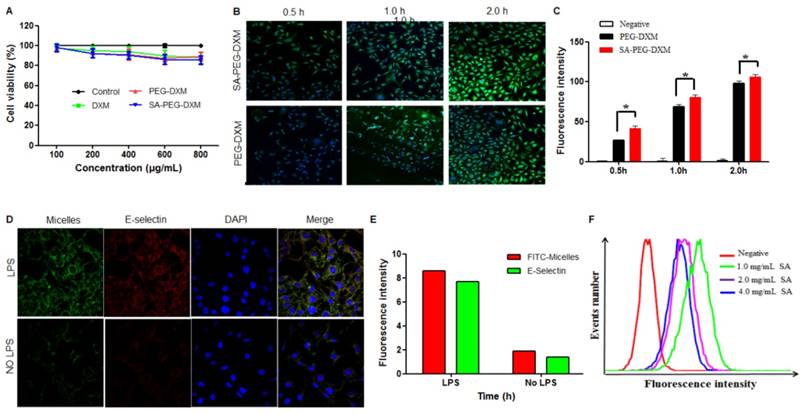
The cytotoxicity of SA-PEG-DXM and PEG-DXM micelles was assessed in HUVECs for a range of concentrations via the MTT assay. As shown in Figure 3A, SA-PEG-DXM micelles displays minimum cytotoxicity with over 80% HUVECs remaining viable at the highest concentration of 800 µg·mL-1, which indicated that SA-PEG-DXM micelles had a good biocompatibility and low toxicity.
Assessments on targeting function of SA-PEG-DXM micelles in vitro. (A) In vitro cytotoxicity assay. (B) Imaging of LPS-activated HUVECs was incubated with FITC-loaded SA-PEG-DXM and PEG-DXM micelles for 0.5, 1 and 2 h, respectively. (C) Fluorescence intensity inside cells measured by flow cytometry when HUVECs pre-treated with LPS and then incubated with FITC-loaded PEG-DXM and SA-PEG-DXM micelles for 0.5, 1 and 2 h, respectively (* p < 0.05 vs PEG-DXM micelles, n = 3). (D) Imaging of LPS-activated HUVECs (upper panels) and non-activated ones (lower panels) exposure to FITC-loaded SA-PEG-DXM micelles. The red staining shows E-selectin receptor localization. The green channel shows FITC-loaded SA-PEG-DXM micelles. (E) The semi-quantitative values of fluorescence intensity of (D). (F) Fluorescence intensity measured by flow cytometry when LPS-activated HUVECs pre-treated with different concentration of free SA and then treated with FITC-loaded SA-PEG-DXM micelles.
Cellular uptake study
In this experiment, HUVECs were used as the model cell of VECs due to their human origin. They are well characterized and widely used in vascular experiments in vitro [28]. HUVECs, under the treatment of LPS or TNF-α, can be used to simulate an in vitro model of activated endothelium. It was reported that E-selectin expression reached a maximum after incubation with LPS for 4 hours [29]. To investigate the potential targeting ability of SA-PEG-DXM micelles, cellular uptake test for SA-PEG-DXM and PEG-DXM micelles on LPS-activated HUVECs at different times (0.5, 1.0 and 2.0 h) was performed and observed by confocal microscopy. Figure 3B shows the green fluorescence images after LPS-activated HUVECs exposure to FITC-loaded SA-PEG-DXM micelles or PEG-DXM micelles for 0.5, 1 and 2 h. It was found that both SA-PEG-DXM and PEG-DXM micelles could be internalized into HUVECs in a time-dependent manner. However, much higher fluorescence intensity was observed in cells exposure to SA-PEG-DXM micelles than those incubated with PEG-DXM micelles at three point-in-times (0.5, 1.0 and 2.0h), which was consistent with the results of cellular uptake measured by flow cytometry (Figure 3B). The significant difference (p < 0.05) in fluorescence intensity between SA-PEG-DXM and PEG-DXM micelles was observed at 0.5 and 1.0 hour (p < 0.05, Figure 3B). Initially, more SA-PEG-DXM micelles were internalized in LPS-activated HUVECs than PEG-DXM micelles. With the prolonged exposure to micelles (at 2 hours), the fluorescence intensities in HUVECs incubated with SA-PEG-DXM and PEG-DXM micelles were similar (Figure 3C). Therefore, it is supposed that the more internalization of SA-PEG-DXM micelles into LPS-activated HUVECs at initial time was due to the conjugated SA.
To investigate whether the cellular uptake mechanism was associated with the specific binding between SA and E-selectin expressed on LPS-activated VECs, HUVECs with or without LPS-pretreatment were incubated with FITC-loaded SA-PEG-DXM micelles (Green) for 1 hour. The result showed that the fluorescence intensity (Green) in LPS-activated HUVECs (Figure 3D, upper panels) was higher than that in non-activated HUVECs (Figure 3D, lower panels). The lipophilic dye (Red) was used to localize and stain E-selectin receptor. The expression of E-selectin (Red) in LPS-activated HUVECs was also higher than that in non-activated HUVECs, the result of which was consistent with the difference of micellar fluorescence intensity (Green), indicating a predominantly selective uptake mediated by E-selectin receptor (Figure 3E). Moreover, competitive experiment was performed to further testify this E-selectin receptor-involved internalization process. Different content free SA (1.0, 2.0 and 4.0 mg·mL-1) was used to block E-selectin receptor beforehand, and the results (Figure 3F) showed that the uptake of SA-PEG-DXM micelles in LPS-activated HUVECs gradually diminished with the increased doses of free SA.
In vitro anti-inflammatory activity
The in vitro anti-inflammatory capacity of SA-PEG-DXM/DXM micelles was investigated in LPS-induced HUVECs. LPS-activated HUVECs were treated with SA-PEG-DXM micelles or other appropriate pharmacological intervention, including SA, SA-PEG, free DXM, free SA + DXM and PEG-DXM. The supernatants were determined for TNF-α and IL-6 by enzyme-linked immunosorbent assay (ELISA). It was found that SA-PEG-DXM micelles could significantly ameliorate LPS-induced production of cytokines TNF-α and IL-6 in comparison to LPS (both p < 0.05, Figure 4A and 4B). The reduced TNF-α and IL-6 levels were also observed in HUVECs treated with free DXM, free DXM + SA and PEG-DXM, which was mainly attributed to the anti-inflammatory activity of DXM [5,30]. The anti-inflammatory capacity of free DXM and free DXM + SA was higher than SA-PEG-DXM and SA-PEG-DXM, which was attributed to the delayed anti-inflammatory effects induced by conjugated DXM dissociating from conjugates.
Assessment on the anti-inflammatory activity in vitro. (A) Effect of SA-PEG-DXM micelles on the production of TNF-α (*p < 0.05, n = 3). (B) Effect of SA-PEG-DXM micelles on the production of IL-6 (*p < 0.05, n = 3). (C, D and E) Effect of SA or PEG-SA treatment on LPS-induced autophagy-related proteins expression, β-actin was exploited as control. (F, G and H) Densitometric analysis of the western blot bands of Beclin-1, Atg5 and Atg5/12 (*p < 0.05, n = 3).
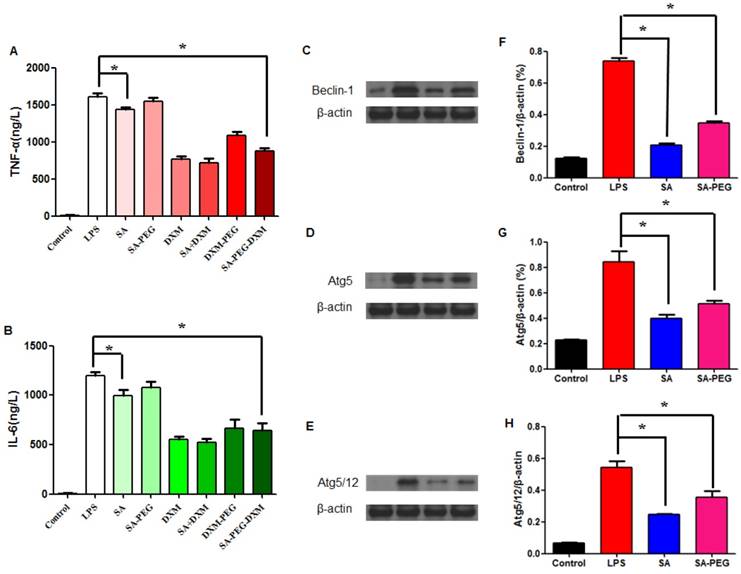
In addition to anti-inflammatory action of DXM, free SA could modestly attenuate LPS-driven cytokines production. The significant reduction in TNF-α and IL-6 levels was observed in cells treated with free SA in comparison to those with LPS (both p < 0.01, Figure 4A and 4B), despite the reduction with SA-PEG was no statistical significance in comparison to LPS, which is probably due to the delayed release of SA dissociating from SA-PEG. Previous study reported that SA could attenuate LPS-induced kidney injury through suppressing LPS-activated Beclin-1/Atg5-Atg12-mediated autophagy [24]. We next examined the expression of related proteins. The results showed that both SA and SA-PEG could significantly reduce LPS-induced Beclin-1, truncated Atg5 and Atg5/12 expression (both p < 0.01, Figure 4C, 4E and 4G), which suggested that both free SA and SA-PEG were activated in depressing LPS-enhanced renal autophagy despite different expression. The quantitative values of bands were presented in Figure 4F, 4G and 4H.
Biodistribution of SA-PEG-DXM micelles
To investigate the kidney-targeting efficacy of SA-PEG-DXM micelles in vivo, AKI murine models were built and then injected intravenously with indocyanine green (ICG)-tetrabutylammonium iodide complex-loaded SA-PEG-DXM (SA-PEG-DXM/ICG) or PEG-DXM (PEG-DXM/ICG) micelles. At 24 h after administration, mice were sacrificed, and their organs (heart, liver, spleen, lung and kidney) were collected. Compared with mice without AKI, the higher renal fluorescence signal was observed in AKI model mice treated with SA-PEG-DXM micelles (Figure 5A vs 5B). Moreover, the renal fluorescence intensity in AKI model mice treated with SA-PEG-DXM micelles was higher than that treated with PEG-DXM micelles (Figure 5A vs 5C). Therefore, much more SA-PEG-DXM micelles were accumulated in the renal tissue of mice with AKI due to SA-PEG-DXM micelles specifically binding to E-selectin expressed on VECs in the renal tissue, which provided strong evidence for the increased accumulation of SA-PEG-DXM micelles in the kidney benefiting from the specific interaction between SA and E-selectin receptor. The distributional difference of SA-PEG-DXM micelles in mice with or without AKI was consistent with the expression of E-selectin in vivo that highly expressed during inflammation, but only a small amount of E-selectin expressed under normal physiological condition (Figure S2). The quantitative values of fluorescence intensity in all three mice were analyzed and presented in Figure 5D.
Assessment on targeting function of SA-PEG-DXM micelles in vivo. (A) Mice with AKI injected intravenously with SA-PEG-DXM/ICG micelles. (B) Mice without AKI injected intravenously with SA-PEG-DXM/ICG micelles. (C) Mice with AKI injected intravenously with PEG-DXM/ICG micelles. (D) The semi-quantitative values of fluorescence intensity. (E) The renal fluorescence signal in AKI murine model treated with SA-PEG-DXM and PEG-DXM micelles at different point-in-times. (F) The semi-quantitative values of fluorescence intensity.
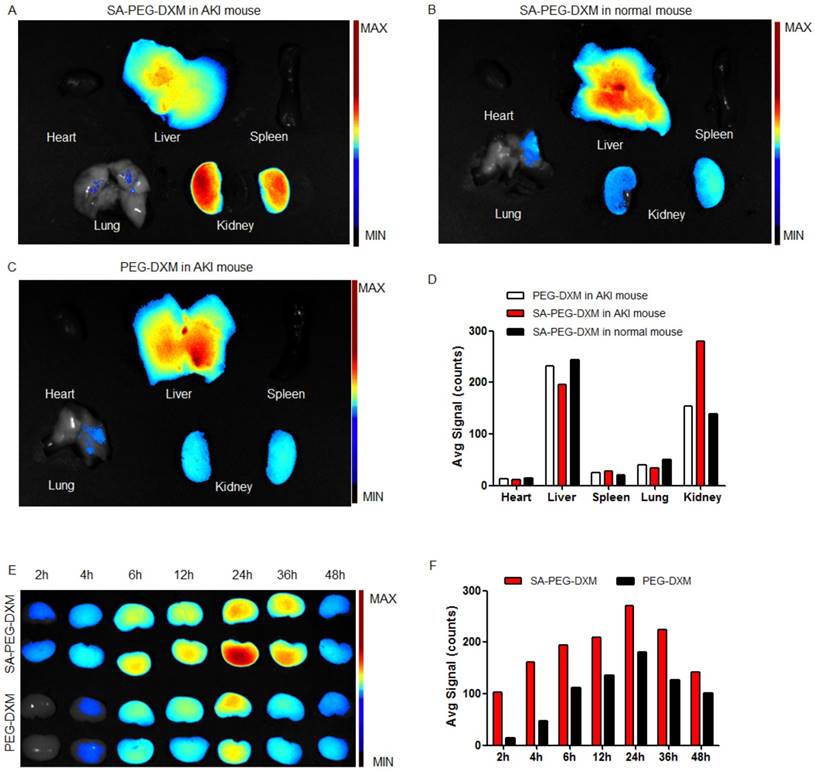
The renal accumulation of SA-PEG-DXM and PEG-DXM micelles in AKI murine models at different point-in-time (2, 4, 6, 8, 12, 24, 36 and 48h) was presented in Figure 5E. AKI mice injected intravenously with SA-PEG-DXM/ICG micelles showed ICG-exciting fluorescence signal in the kidney as early as 2 h after injection, and fluorescence signal maintained more than 48 hours (Figure 4E, upper panels). In contrast, renal fluorescence signal began to appear at 4 hours in mice treated with PEG-DXM/ICG micelles and the fluorescence signal observed at all monitoring points was obviously lower than that treated with SA-PEG-DXM/ICG micelles (Figure 4E, lower panels). The quantitative values of fluorescence intensity in mice treated with SA-PEG-DXM/ICG micelles and PEG-DXM/ICG micelles presented in Figure 4F. A larger amount of SA-PEG-DXM micelles were accumulated in the kidney than PEG-DXM micelles, despite both fluorescence signals reached their maximum at 24h. Based on the obtained data, we demonstrated that both micelles showed long persistence in circulation, but SA-mediated micelles are easier to be accumulated in the kidney due to the specific interaction between SA and E-selectin receptor expressed on renal VECs during AKI.
In vivo efficacy of SA-PEG-DXM/DXM Micelles in AKI murine model
The therapeutic efficacy of SA-PEG-DXM/DXM micelles was examined in the AKI murine model. AKI mice were induced with LPS (20 µL, 1mg·mL-1) and treated with SA-PEG-DXM/DXM micelles, free DXM and PEG-DXM/DXM micelles as control. In comparison with sham animals, animals with AKI caused significant renal dysfunction at 2 days after LPS-activation as showed by significant increase in serum creatinine (Scr, go up ≥ 0.5mg/dl) and BUN (Figure 6A and 6B), which confirmed the successful induction of AKI. The reduced levels of Scr and BUN were observed in AKI mice treated with free DXM, PEG-DXM/DXM and SA-PEG-DXM/DXM micelles. As shown in Figure 6A and 6B, SA-PEG-DXM/DXM micelles significantly reduced Scr (0.32 ± 0.11 vs 0.56 ± 0.08 mg·dl-1, *P < 0.05) and BUN (28.3 ± 7.27 vs 46.9 ± 7.54 mg·dl-1, *P < 0.05) level compared to free DXM at 7 days, despite the protection was no statistical difference in Scr (0.29 ± 0.11 vs 0.38 ± 0.12 mg·dl-1, **P > 0.05) and BUN (33.57 ± 7.75 vs 38.1 ± 10.20 mg·dl-1, **P > 0.05) between SA-PEG-DXM/DXM micelles and free DXM at 2 days. This time-dependent therapeutic efficacy is due to the sustained release behavior of SA-PEG-DXM/DXM micelles and the needed time for physiological functional recovery. In addition, the significant reduction of Scr and BUN levels were observed in AKI mice treated with SA-PEG-DXM/DXM micelles than those treated with PEG-DXM/DXM micelles both at day 2 and day 7, which was due to the higher accumulation of SA-PEG-DXM/DXM micelles in the renal tissue.
Assessment on kidney function and histopathological changes. (A and B) The increase in Scr and BUN at 2 and 7 days in AKI mice is abrogated by SA-PEG-DXM/DXM micelles (*p < 0.05, **p > 0.05, n=3). (C) Hematoxylin and eosin (HE)-stained sections are from the outer medulla (at day 7). (D) Periodic acid-Schiff (PAS) staining from the outer medulla (at day 7). (E and F) Quantitative scores of tubular injury in the outer medulla. The analysis was performed under the same magnification for respective groups (*p < 0.05).
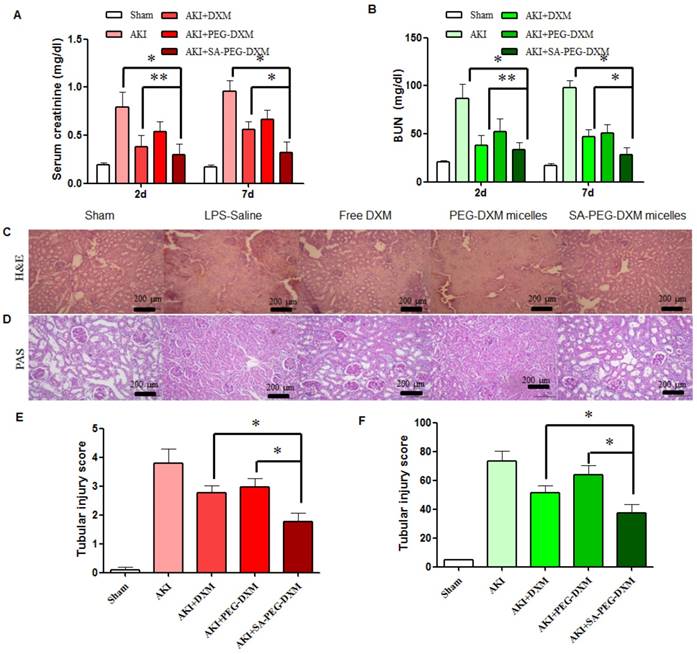
Histopathological change, a direct indication of renal injury, showed that the renal tissue from AKI group had severe tubular damage in the outer medulla, as evidenced widespread by tubular necrosis, glomerular atrophy, and luminal congestion in comparison to normal kidney at day 7 (Figure 6C). Pharmacological intervention (free DXM, PEG-DXM/DXM and SA-PEG-DXM/DXM micelles) could markedly improve LPS-induced kidney injury to varying degrees. Quantification of kidney pathological damage in the outer medulla showed that a significant reduction of mean tubular injury score in animals treated with SA-PEG-DXM/DXM micelles than that with free DXM and PEG-DXM/DXM micelles (Figure 6E, both *p < 0.05). Periodic acid-Schiff (PAS) staining also reached the similar conclusion that SA-PEG-DXM/DXM micelles significantly ameliorated LPS-induced kidney injury in comparison to free DXM and PEG-DXM/DXM micelles at day 7 (Figure 5D and 5F, *p < 0.05).
The anti-inflammatory efficacy of SA-PEG-DXM/DXM micelles was investigated in AKI murine model. As expected, LPS caused a sharp increase of pro-inflammatory cytokines, such as TNF-α and IL-6. The cytokine levels in animals treated with SA-PEG-DXM/DXM micelles were significantly reduced in comparison to those treated with free DXM and PEG-DXM/DXM micelles at day 7 (Figure 7A and 7B, both *p < 0.05). SA-PEG-DXM/DXM micelles showed better anti-inflammatory efficacy due to increased drug accumulation in the kidney and longer action duration benefiting from sustained release behavior of micelles. In this study, DXM was selected as a model drug due to its extensive use as anti-inflammatory agents in clinical practice. It mainly inhibits the NF-κB signaling pathway and decreases expression of the inflammatory cytokines, ultimately suppressing the inflammatory response [31, 32]. Several studies demonstrated it was a good candidate for kidney disease caused by sepsis, ischemia-reperfusion and contrast via its Inflammation inhibition [6, 33].
Pro-inflammatory cytokines and oxidative stress changes after PEG-DXM/DXM micelles therapy. (A and B) Renal cytokine responses in the sham mice and AKI mice treated with saline, free DXM, PEG-DXM/DXM and SA-PEG-DXM/DXM micelles (*p < 0.05). (C, D and E) Oxidative stress changes in the sham mice and AKI mice treated with saline, free DXM, PEG-DXM/DXM and SA-PEG-DXM/DXM micelles (*p < 0.05, **p > 0.05, n=3).
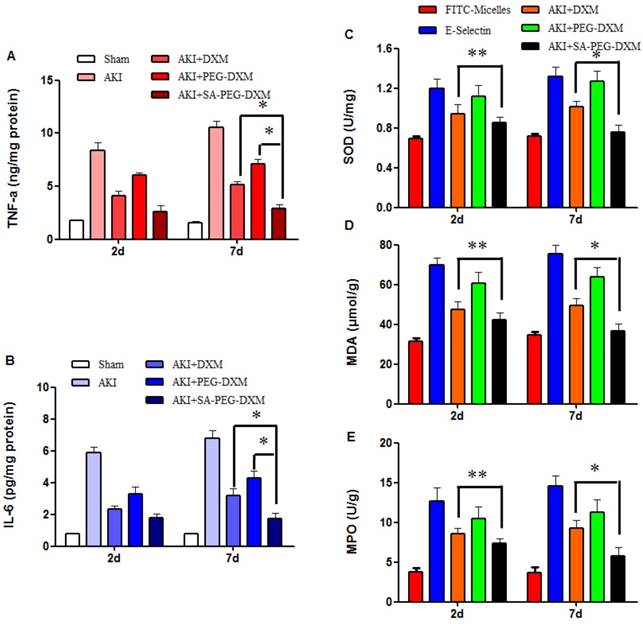
To assess the effects of SA-PEG-DXM/DXM micelles on oxidative stress, the activities of superoxide dismutase (SOD), malondialdehyde (MDA) and myeloperoxidase (MPO) were detected at day 2 and 7, respectively. The results showed that the reduction of SOD, MDA and MPO levels in SA-PEG-DXM/DXM micelles group was slightly higher than that in free DXM group (Figure 7C, 7D and 7E **p > 0.05 versus free DXM), but significantly higher than that in PEG-DXM/DXM micelles group (*p < 0.05 versus free DXM) at day 2. The SOD, MDA and MPO levels were significantly reduced in SA-PEG-DXM/DXM micelles group compared with free DXM and PEG-DXM/DXM micelles group (both *p < 0.05).
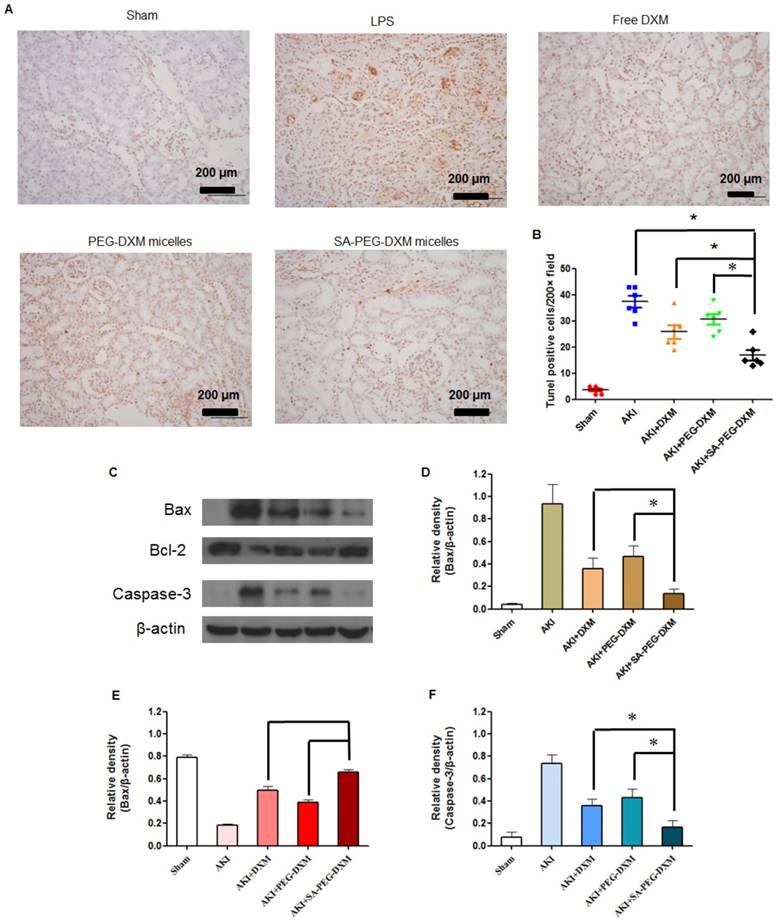
Apoptosis is a major pathological process in AKI, and is triggered by certain factors in vivo. Both infiltration of inflammatory cells and oxidative stress can cause apoptosis in AKI. Therefore, inhibition of apoptosis effectively decreases kidney injury during AKI. Previous study has indicated that the increased cytokine levels can cause renal tissue damage via inducing apoptosis [34]. TUNEL staining was used to examine the apoptosis in kidney (Figure 8A). Kidney from animals with AKI showed extensive nuclear changes in line with apoptotic cell death as shown by TUNEL staining. In comparison, no TUNEL-positive cells were observed in kidney sections from sham group. SA-PEG-DXM/DXM micelles could significantly reduce the percentage of TUNEL-positive cells in comparison to free DXM and PEG-DXM/DXM micelles treatment at day 7 (both p < 0.05, Figure 8B), which suggests a better anti-apoptotic effect in vivo benefiting from more DXM accumulated in the renal tissues with SA-PEG-DXM/DXM micelles. Kidney injury also causes the abnormal expression of apoptotic related proteins, such as Bax, Bcl-2 and Caspase-3. Bax is a pro-apoptosis protein but Bcl-2 is a pro-apoptosis protein acts the opposite effect. The changes of Bax and Bcl-2 expression potentially initiate mitochondrial outer membrane permeabilization, and then caspase cascade is activated and apoptosis occurs. The Caspase-3 protein is a key member of caspase family, and sequential activation of caspases plays a critical role in the execution-phase of cell apoptosis. Lee et al. indicated that Caspase-3 activity positively correlated with renal dysfunction in animals with AKI [35]. In this study, the immunoreactivity of Bax and Caspase-3 significantly increased, but Bcl-2 lowered at day 7 in LPS-activated animals. SA-PEG-DXM/DXM micelles significantly attenuated the expression of Bax and Caspase-3, and enhanced Bcl-2 expression compared with free DXM and PEG-DXM/DXM micelles (Figure 8C, 8D, 8E and 8F, *p < 0.05).
Anti-apoptotic efficacy of SA-PEG-DXM/DXM micelles. (A) TUNEL assay. The experiments were conducted using tissues harvested at day 7 from the sham mice and AKI mice treated with saline, free DXM, PEG-DXM/DXM and SA-PEG-DXM/DXM micelles. (B) The number of TUNEL-positive cells was quantified (*p < 0.05). (C) Western blot analysis of Bax, Bcl-2 and Caspase-3 in the kidney at day 7 for the cases of the sham mice and AKI mice treated with saline, free DXM, PEG-DXM/DXM and SA-PEG-DXM/DXM micelles, β-actin was exploited as control. (D, E and F) Densitometric analysis of the western blot bands of Bax, Bcl-2, and Caspase-3 (*p < 0.05, n = 3).
Consequently, we demonstrated that targeting delivery of DXM into vascular endothelial cells in the kidney by micelles, modified with SA as a homing device, potently suppressed the expression of pro-inflammatory cytokines and improved renal function for the treatment of AKI. As drug carrier for encapsulation of DXM, SA-PEG-DXM micelles could prolong the circulation time and reduce the clearance rate compared with free DXM. Moreover, specific binding between SA-mediated micelles and E-selectin receptor expressed on VECs in the kidney during AKI contributed to the increased accumulation of SA-PEG-DXM/DXM micelles in renal tissue to achieve strong local pharmacological efficacy, as showed by our current data. In addition to AKI, elevated E-selectin expression was detected in patients with IgA nephropathy, lupus nephritis, and diabetic nephropathy, which supports the clinical relevance of the development of E-selectin-based drug-targeting strategies and represents an important added value for future clinical therapy. SA-PEG-DXM micelles can also encapsulate ultra-small particles of iron oxide (USPIO) used for magnetic resonance angiography to the diagnosis of AKI.
Conclusion
In summary, SA-PEG-DXM/DXM micelles were successfully prepared for ameliorating AKI through E-selectin-targeting strategy, which is distinct from other reported strategies. Here, we have showed that micelle surface display of SA binds to E-selectin receptor and then increases the uptake by LPS-activated HUVECs. We have examined this effect to elicit therapeutically useful effects in AKI murine model, as reflected by reduced Scr and BUN levels, oxidative stress, pro-inflammatory cytokines production and expression of apoptotic related proteins. Hence, micelles modified with SA delivering potent drugs into inflamed VECs represent a new therapeutic strategy for the treatment of AKI.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81573362, 81373345, 81301611, 81671889), the Nature Science Foundation of Zhejiang province (LZ13H300001, Q17H050002) and the Scientific Research Fund of Ministry of Health-Medical Science Major Technology Fund Project of Zhejiang Province.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Basile DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney Int. 2007;72:151-156
2. Molitoris BA, Sutton TA. Endothelial injury and dysfunction: role in the extension phase of acute renal failure. Kidney Int. 2004;66:496-499
3. Oppert M, Engel C, Brunkhorst FM. et al. Acute renal failure in patients with severe sepsis and septic shock-a significant independent risk factor for mortality: results from the German Prevalence Study. Nephrol Dial Transplant. 2008;23:904-909
4. Plataki M, Kashani K, Cabello-Garza J. et al. Predictors of acute kidney injury in septic shock patients: an observational cohort study. Clin J Am Soc Nephrol. 2011;6:1744-1751
5. Choi HM, Jo SK, Kim SH. et al. Glucocorticoids attenuate septic acute kidney injury. Biochem Biophys Res Commun. 2013;435:678-684
6. Kumar S, Allen DA, Kieswich JE. et al. Dexamethasone ameliorates renal ischemia-reperfusion injury. J Am Soc Nephrol. 2009;20:2412-2425
7. Perez A, Jansen-Chaparro S, Saigi I, Bernal-Lopez MR, Miñambres I, Gomez-Huelgas R. Glucocorticoid-induced hyperglycemia. J Diabetes. 2014;6:9-20
8. Bultink IE, Baden M, Lems WF. Glucocorticoid-induced osteoporosis: an update on current pharmacotherapy and future directions. Expert Opin Pharmacother. 2013;14:185-197
9. Lin Y, Li Y, Wang X, Gong T, Zhang L, Sun X. Targeted drug delivery to renal proximal tubule epithelial cells mediated by 2-glucosamine. J Control Release. 2013;167:148-156
10. Asgeirsdóttir SA, Kamps JA, Bakker HI, Zwiers PJ, Heeringa P, van der Weide K. et al. Site-specific inhibition of glomerulonephritis progression by targeted delivery of dexamethasone to glomerular endothelium. Mol Pharmacol. 2007;72:121-131
11. He XK, Yuan ZX, Wu XJ, Xu CQ, Li WY. Low molecular weight hydroxyethyl chitosan-prednisolone conjugate for renal targeting therapy: synthesis, characterization and in vivo studies. Theranostics. 2012;2:1054-1063
12. Schreiber A, Theilig F, Schweda F. et al. Acute endotoxemia in mice induces downregulation of megalin and cubilin in the kidney. Kidney Int. 2012;82:53-59
13. Asgeirsdóttir SA, Zwiers PJ, Morselt HW. et al. Inhibition of proinflammatory genes in anti-GBM glomerulonephritis by targeted dexamethasone-loaded AbEsel liposomes. Am J Physiol Renal Physiol. 2008;294:554-561
14. Kim YK, Kwon JT, Jiang HL. et al. Kidney-specific peptide-conjugated poly(ester amine) for the treatment of kidney fibrosis. J Nanosci Nanotechnol. 2012;12:5149-5154
15. Geng Q, Sun X, Gong T, Zhang ZR. Peptide-drug conjugate linked via a disulfide bond for kidney targeted drug delivery. Bioconjug Chem. 2012;23:1200-1210
16. Daiber A, Steven S, Weber A. et al. Targeting vascular (endothelial) dysfunction. Br J Pharmacol. 2016 doi: 10.1111/bph.13517. [Epub ahead of print]
17. Auvinen K, Jalkanen S, Salmi M. Expression and function of endothelial selectins during human development. Immunology. 2014;143:406-415
18. Lerolle N, Nochy D, Guérot E, Bruneval P, Fagon JY, Diehl JL, Hill G. Histopathology of septic shock induced acute kidney injury: apoptosis and leukocytic infiltration. Intensive Care Med. 2010;36:471-478
19. Varki A. Sialic acids as ligands in recognition phenomena. FASEB J. 1997;11:248-255
20. Kneuer C, Ehrhardt C, Radomski MW. et al. Selectins-potential pharmacological targets? Drug Discov Today. 2006;11:1034-1040
21. Barthel SR, Gavino JD, Descheny L. et al. Targeting selectins and selectin ligands in inflammation and cancer. Expert Opin Ther Targets. 2007;11:1473-1491
22. Ehrhardt C, Kneuer C, Bakowsky U. Selectinsdan emerging target for drug delivery. Adv Drug Deliv Rev. 2004;56:527-549
23. Zeisig R, Stahn R, Wenzel K. et al. Effect of sialyl Lewis Xglycoliposomes on the inhibition of E-selectin-mediated tumour cell adhesion in vitro. Biochim Biophys Acta. 2004;1660:31-40
24. Yang CC, Yao CA, Yang JC. et al. Sialic acid rescues repurified lipopolysaccharide-induced acute renal failure via inhibiting TLR4/PKC/gp91-mediated endoplasmic reticulum stress, apoptosis, autophagy, and pyroptosis signaling. Toxicol Sci. 2014;141:155-165
25. Du YZ, Weng Q, Yuan H, Hu FQ. Synthesis and antitumor activity of stearate-g-dextran micelles for intracellular doxorubicin delivery. ACS Nano. 2010;4:6894-6902
26. Yang J, Wu DW, Tang LN, Zhang F, Guo HP, Huang SY. The protective effect of different doses of dexamethasone on acute kidney injury induced by sepsis in mice. Chin Crit Care Med. 2013;25:424-428
27. Liu P, Situ JQ, Li WS, Shan CL, You J, Yuan H, Hu FQ, Du YZ. High tolerated paclitaxel nano-formulation delivered by poly (lactic-co-glycolic acid)-g-dextran micelles to efficient cancer therapy. Nanomedicine. 2015;11:855-866
28. Park HJ, Zhang Y, Georgescu SP. et al. Human umbilical vein endothelial cells and human dermal microvascular endothelial cells offer new insights into the relationship between lipid metabolism and angiogenesis. Stem Cell Rev. 2006;2:93-102
29. Wu J, Li X, Huang L. et al. HSPA12B inhibits lipopolysaccharide-induced inflammatory response in human umbilical vein endothelial cells. J Cell Mol Med. 2015;19:544-554
30. Xu C, Chang A, Hack BK. et al. TNF-mediated damage to glomerular endothelium is an important determinant of acute kidney injury in sepsis. Kidney Int. 2014;85:72-81
31. NCT00065611. Steroid treatment for kidney disease. https://clinicaltrials.gov/ct2/show/NCT00065611. Accessed 25 Sep. 2016
32. NCT00004990. Once-A-Month steroid treatment for patients with focal segmental glomerulosclerosis. https://clinicaltrials.gov/ct2/show/NCT00004990. Accessed 25 Sep. 2016
33. Schmidt C, Höcherl K, Schweda F. et al. Regulation of renal sodium transporters during severe inflammation. J Am Soc Nephrol. 2007;18:1072-1083
34. Ortiz A, Justo P, Catalán MP. et al. Apoptotic cell death in renal injury: the rationale for intervention. Curr Drug Targets Immune Endocr Metabol Disord. 2002;2:181-192
35. Distinct pathophysiologic mechanisms of septic acute kidney injury. role of immune suppression and renal tubular cell apoptosis in murine model of septic acute kidney injury, Crit. Care Med. 2012;40:2997-3006
Author contact
![]() Corresponding authors: Yong-Zhong Du, Institute of Pharmaceutics, College of Pharmaceutical Sciences, Zhejiang University, 866 Yuhangtang Road, Hangzhou 310058, China, E-mail: duyongzhongedu.cn. Sai-Ping Jiang, Department of Pharmacy, The First Affiliated Hospital, College of Medicine, Zhejiang University, 79 Qingchun Road, Hangzhou 310003, China, E-mail: j5145com
Corresponding authors: Yong-Zhong Du, Institute of Pharmaceutics, College of Pharmaceutical Sciences, Zhejiang University, 866 Yuhangtang Road, Hangzhou 310058, China, E-mail: duyongzhongedu.cn. Sai-Ping Jiang, Department of Pharmacy, The First Affiliated Hospital, College of Medicine, Zhejiang University, 79 Qingchun Road, Hangzhou 310003, China, E-mail: j5145com