Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(9):2350-2362. doi:10.7150/thno.19119 This issue Cite
Research Paper
[177Lu]pentixather: Comprehensive Preclinical Characterization of a First CXCR4-directed Endoradiotherapeutic Agent
Margret Schottelius1 ![]() , Theresa Osl1, Andreas Poschenrieder1, Frauke Hoffmann1, Seval Beykan4, Heribert Hänscheid4, Andreas Schirbel4, Andreas K. Buck4, Saskia Kropf5, Markus Schwaiger3, 6, Ulrich Keller2, 6, Michael Lassmann4, Hans-Jürgen Wester1
, Theresa Osl1, Andreas Poschenrieder1, Frauke Hoffmann1, Seval Beykan4, Heribert Hänscheid4, Andreas Schirbel4, Andreas K. Buck4, Saskia Kropf5, Markus Schwaiger3, 6, Ulrich Keller2, 6, Michael Lassmann4, Hans-Jürgen Wester1
1. Chair for Pharmaceutical Radiochemistry, Technische Universität München, Walther-Meissner-Strasse 3, 85748 Garching, Germany;
2. III. Medical Department, Klinikum rechts der Isar, Technische Universität München, Ismaningerstr. 22, 81675 Munich, Germany;
3. Department of Nuclear Medicine, Klinikum rechts der Isar, Technische Universität München, Ismaningerstr. 22, 81675 Munich, Germany;
4. Department of Nuclear Medicine, Universität Würzburg, Oberdürrbacher Str. 6, 97080 Würzburg, Germany;
5. Scintomics GmbH, Lindach 4, 82256 Fürstenfeldbruck, Germany;
6. Deutsches Konsortium für translationale Krebsforschung (DKTK) and Deutsches Krebsforschungszentrum (DKFZ), Im Neuenheimer Feld 280, 69120 Heidelberg, Germany.
Received 2017-1-9; Accepted 2017-3-11; Published 2017-6-11
Abstract

Purpose: Based on the clinical relevance of the chemokine receptor 4 (CXCR4) as a molecular target in cancer and on the success of [68Ga]pentixafor as an imaging probe for high-contrast visualization of CXCR4-expression, the spectrum of clinical CXCR4-targeting was expanded towards peptide receptor radionuclide therapy (PRRT) by the development of [177Lu]pentixather.
Experimental design: CXCR4 affinity, binding specificity, hCXCR4 selectivity and internalization efficiency of [177Lu]pentixather were evaluated using different human and murine cancer cell lines. Biodistribution studies (1, 6, 48, 96h and 7d p.i.) and in vivo metabolite analyses were performed using Daudi-lymphoma bearing SCID mice. Extrapolated organ doses were cross-validated with human dosimetry (pre-therapeutic and during [177Lu]pentixather PRRT) in a patient with multiple myeloma (MM).
Results: [177Lu]pentixather binds with high affinity, specificity and selectivity to hCXCR4 and shows excellent in vivo stability. Consequently, and supported by >96% plasma protein binding and a logP=-1.76, delaying whole-body clearance of [177Lu]pentixather, tumor accumulation was high and persistent, both in the Daudi model and the MM patient. Tumor/background ratios (7d p.i.) in mice were 499±202, 33±7, 4.0±0.8 and 116±22 for blood, intestine, kidney and muscle, respectively. In the patient, high tumor/kidney and tumor/liver dose ratios of 3.1 and 6.4 were observed during [177Lu]pentixather PRRT (7.8 GBq), with the kidneys being the dose-limiting organs.
Conclusions: [177Lu]pentixather shows excellent in vivo CXCR4-targeting characteristics and a suitable pharmacokinetic profile, leading to high tumor uptake and retention and thus high radiation doses to tumor tissue during PRRT, suggesting high clinical potential of this [68Ga]pentixafor/[177Lu]pentixather based CXCR4-targeted theranostic concept.
Keywords: CXCR4, pentixafor, pentixather, PRRT, endoradiotherapy.
Introduction
More than 20 years of research have provided detailed insight into the multifaceted and oftentimes pivotal roles of the interplay between the chemokine receptor 4 (CXCR4) and its endogenous ligand CXCL12, both in physiology and in a variety of pathological conditions [1, 2]. The central functional element in these processes is the migration of CXCR4-expressing cells towards tissues with high local CXCL12 expression and their retention, homing and differentiation at these sites. Physiologically, this includes the recruitment of stem and progenitor cells during embryogenesis, hematopoiesis and neoangiogenesis as well as leukocyte trafficking during immune response [2]. In cancer, chemotactic CXCR4/CXCL12 interactions fulfil a variety of tumor-growth supporting functions, including direct autocrine stimulation of proliferation and angiogenesis, promotion of tumor invasiveness as well as metastasis of CXCR4-expressing tumor cells to mesenchymal stromal tissues with high CXCL12 expression (liver, bone marrow, lung, lymph nodes) [3-5]. An increasing number of preclinical studies underline the relevance of the multiple CXCR4/CXCL12 mediated interactions between cancer cells and the tumor microenvironment (fibroblasts, endothelial cells, immune cells), which provides a protective and supportive niche for tumor growth [6].
CXCR4 overexpression is documented for more than twenty human tumor types, ranging from hematological malignancies to solid tumors [7]. Furthermore, high CXCR4 expression has been found to be significantly associated with distant metastasis and with poor overall and disease-free survival in patients with e.g. breast, prostate, pancreatic and lung cancer, lymphoma and leukemia and thus represents an important independent prognostic factor in these diseases [8]. The accurate in vivo assessment of the level and kinetics of CXCR4 expression in a given tumor entity may therefore not only provide relevant information on tumor biology and tumor grade as well as on its metastatic potential, but may also allow selection of patients eligible for CXCR4-targeted therapies.
Intense efforts have therefore been directed towards the development of a suitable CXCR4-targeted molecular imaging agent in recent years [9, 10]. The only compound that has entered clinical application so far is the small cyclic pentapeptide [68Ga]pentixafor [11]. With its high affinity and selectivity for human CXCR4, rapid renal excretion and very low non-specific background accumulation, it allows sensitive and high-contrast imaging of CXCR4-expressing tissues in vivo using positron emission tomography (PET). Besides its successful application in patients with lymphoma [12], multiple myeloma [13], AML [14], SCLC [15], glioblastoma [16] or other solid tumors [17] such as non-small cell lung cancer [18] or adrenocortical cancer [19], [68Ga]pentixafor-PET has recently been shown to be a valuable tool for the in vivo detection of inflammatory processes e.g. after myocardial infarction [20-22] or ischemic stroke [23] as well as in atherosclerosis [24] or urinary tract infection [25]. The possibility to non-invasively quantify CXCR4 expression using [68Ga]pentixafor-PET is anticipated to further strengthen the emerging role of CXCR4 as a diagnostic marker, matching its already established importance as a therapeutic target.
Currently, several potent CXCR4 antagonists are being investigated in clinical trials for CXCR4-directed anticancer therapy [4, 26, 27]. Their therapeutic effect, both in hematological and solid cancers, has been shown to be primarily based on an effective prevention of distant organ metastasis [28]. Furthermore, by mobilizing tumor cells from their chemoprotective microenvironment into the peripheral blood, CXCR4 antagonists also act as potent chemo/radiosensitizers [4]. Combining CXCR4 inhibition with chemo- or radiotherapy has therefore been shown to provide a significant therapeutic advantage relative to the respective conventional monotherapy alone in preclinical mouse models [29-31]. In contrast, CXCR4 targeting alone, although effective in preventing metastasis, only has negligible effect on primary tumor growth. In this context, CXCR4-targeted PRRT might represent a highly promising alternative treatment concept in cancer therapy [32].
By the direct administration of therapeutic radionuclides conjugated to a CXCR4 targeting vector, high doses of ionizing radiation may be specifically deposited in CXCR4-overexpressing tissues with high precision, with significantly reduced systemic side effects for the patient. Since PRRT requires very low mass amounts of the CXCR4-targeted probe, unwanted pharmacological side effects are highly improbable to occur. As opposed to “conventional” CXCR4-directed therapeutic approaches, the efficacy of PRRT also relies on the so-called “crossfire effect” and is thus not restricted to cells expressing the target epitope. In contrast, even in the case of pronounced intratumoral heterogeneity in CXCR4 expression [12] or insufficient vascularization, the choice of a radionuclide with an adequate particle range in tissue allows delivery of therapeutic radiation doses to areas of the tumor with low or no target expression or limited target accessibility.
To exploit the potential of such an approach, the current study was directed towards implementing a CXCR4-targeted theranostic concept based on the pentixafor scaffold as a targeting vector.
The concept of using a peptide-receptor-targeted “theranostic agent” for radiolabeling with both diagnostic (PET/SPECT) and therapeutic radionuclides has been pioneered by the introduction of radiolabeled somatostatin analogs, e.g. [68Ga]DOTATOC/TATE and [177Lu/90Y]DOTATOC/TATE for the diagnosis and PRRT of neuroendocrine tumors [33]. The success of this concept has recently been paralleled by the use of [68Ga/177Lu]PSMA-I&T [34] and [68Ga/177Lu]PSMA-617 [35] for theranostics of metastatic castration resistant prostate cancer. It is important to note, that these approaches invariably use one single precursor molecule for both diagnostic and therapeutic applications. This has certain practical advantages, but does not fully account for the fundamentally different pharmacokinetic requirements which need to be met by a diagnostic agent and the corresponding therapeutic analog. Sensitive high-contrast diagnostic imaging relies on the fast and high accumulation of the respective tracer in the target tissue as well as rapid background clearance, usually within ≤2h p.i.. In contrast, therapeutic targeting in PRRT primarily aims at achieving maximal activity uptake and retention in the tumor lesions, all the while keeping radiation doses to non-target and excretion organs to a minimum. Thus, longer circulation times and delayed whole-body clearance, which in the case of small bioactive peptides are primarily the result of efficient plasma protein binding as a consequence of a suitably balanced lipophilicity, are desirable features for PRRT applications, leading to prolonged availability of the therapeutic agent for target binding.
These considerations as well as our previous finding, that the highly optimized structure of Ga-pentixafor does not tolerate an exchange of Ga3+ by other M3+ ions (e.g. Lu or Y) without a significant loss in CXCR4 affinity [36, 37], led towards the development of pentixather (3-iodo-D-Tyr1-pentixafor, Figure 1) as a dedicated probe for CXCR4-targeted PRRT and as the companion therapeutic to [68Ga]pentixafor within a CXCR4-directed theranostic concept. Pentixather was selected from a series of D-Tyr1-substituted pentixather analogs [unpublished data] as the most promising candidate for the intended application due to the particularly high affinity of its Lu3+, Y3+ and Bi3+ complexes to human CXCR4 and its suitable physicochemical characteristics.
Chemical structures of [68Ga]pentixafor and [177Lu]pentixather
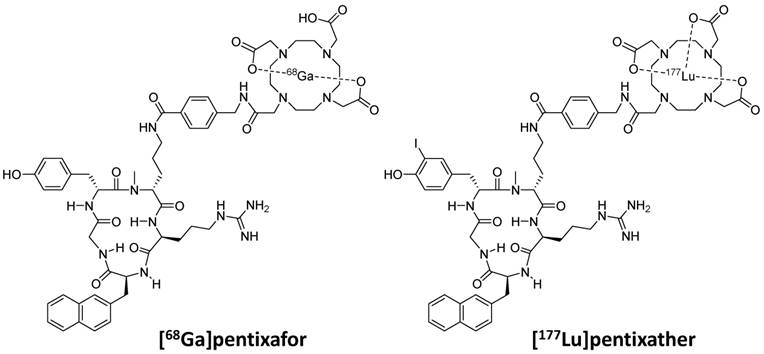
It is important to note at this point that very much in contrast to the usual sequence of operations in radiopharmaceutical development, the in-depth preclinical evaluation of [177Lu]pentixather, which is summarized in this manuscript, was conducted in parallel to the first human application of [177Lu]pentixather [38, 39]. Driven by an urgent clinical need and supported by German legislation, which explicitly allows application of non-approved radiopharmaceuticals on a compassionate-use basis (German Drug Act, §13,2b), [177Lu]pentixather PRRT was offered to very few and selected patients with relapsed multiple myeloma as a last therapy option on the basis of the very promising initial data even before the entire preclinical dataset had been completed.
The complete preclinical characterization of [177Lu]pentixather as a prerequisite for further human studies and the initiation of clinical studies is now provided in this study, including biodistribution studies in Daudi-lymphoma xenograft bearing mice and the extrapolation of organ dose estimates. To cross-validate mouse biokinetic and dosimetry data and to assess their predictive value for the human situation, patient dosimetry was also performed in an exemplary patient with multiple myeloma, both before and during [177Lu]pentixather PRRT.
Materials and Methods
Peptide synthesis
Pentixather (cyclo(D-3-iodo-Tyr1-[NMe]-D-Orn2(AMBS-DOTA)-Arg3-2-Nal4-Gly5), Figure 1) was prepared via solid phase peptide synthesis [37] and consecutive solution phase functionalization with 4-aminomethyl benzoic acid (AMBS) and DOTA [40], followed by direct iodination of Tyr1 in the unprotected peptide using N-iodosuccinimide [41]. Analytical data on pentixather and its natLu-, natY- and natBi-complexes are provided in the supplementary data.
Radiolabeling
For 177Lu-labeling, pentixather was dissolved in water to yield a 1 mM solution. Of this solution, the required volume was added to 177LuCl3 in 0.04 M HCl (ITG/ITM, Garching, Germany; activity concentration: 370 MBq/500 µl) to achieve a peptide-to-177Lu-activity ratio of 1 nmol peptide per 225 MBq 177LuCl3. To this mixture, 1 M NH4OAc was added (calculated to be 10% of total reaction volume), and the mixture was heated to 95°C for 20 min. After cooling and determination of the radiochemical purity (usually >98%), the reaction mixture was diluted with PBS to the desired activity concentration and used as such for in vitro and in vivo studies.
For patient application, [177Lu]pentixather was prepared using a Scintomics GRP synthesis module (Scintomics, Fürstenfeldbruck, Germany). Briefly, a solution of 150 µg pentixather and 7 mg gentisic acid in 600 µl sodium acetate buffer (pH = 4-5) was added to a solution of 300 MBq [177Lu]Cl3 in 0.04 M hydrochloric acid (ITG/ITM) and heated for 35 minutes at 95°C. The product was diluted with saline and passed through a sterile filter (0.22 µm) into a sterile vial.
Radiochemical purity was determined using radio-TLC and analytical radio-HPLC. Radio-TLC was carried out using Agilent iTLC silica gel impregnated chromatography paper (Agilent, Waldbronn, Germany) and two different mobile phases, i.e. A) 0.1 M aq. sodium citrate and B) a 1:1 (v/v) mixture of 1 M aq. NH4OAc and MeOH. TLC-strips were analyzed using a Bioscan TLC analyzer.
Lipophilicity
The lipophilicity of [177Lu]pentixather was determined via a modified shake-flask method as described [42].
In vitro studies
CXCR4 affinity
Affinities for human CXCR4 (hCXCR4) were determined in competitive binding assays (IC50) [11] using Jurkat lymphoma cells (4x105 cells/sample) in Hank's buffered salt solution (1% BSA) and [125I]FC-131 as radioligand. Detailed cell culture conditions are provided in the supplementary data. To allow data normalization, natGa-pentixafor and FC-131 were included as references in this study. Experiments were performed in triplicate with n=3 per concentration in each experiment. IC50 values were calculated using GraphPad Prism 6.01 (Graph Pad Software, San Diego, USA).
Receptor selectivity
The receptor selectivity of [177Lu]pentixather was investigated using CHO-K1 cells transiently transfected with h(HA)CXCR4, m(HA)CXCR4, hCXCR7 and mCXCR7 (negative control: untransfected CHO-K1 cells) as described [12].
Comparative binding studies using various cell lines
CXCR4-mediated cellular uptake of [177Lu]pentixather was investigated in various lymphoma (Daudi), leukemia (MV4-11, THP-1, Molm-13) and multiple myeloma (MM.1S, RPMI, OPM-2) cell lines with different CXCR4 expression levels. Samples containing 2 × 105 cells in assay medium were incubated with [177Lu]pentixather (1 nM) at 37 °C for different time points up to 60 min in the presence (non-specific binding) or absence (control) of 100 µM AMD3100 (n=3 per concentration). After incubation, the tubes were centrifuged (3 min, 1300 rcf, Megafuge 1.0, Heraeus Thermo Scientific) and the supernatant was carefully removed. After washing twice with 200 µL of cold HBSS, the amount of bound radioligand in the cell pellet was quantified using a γ-counter.
Binding and internalization kinetics
Daudi cells (2 × 105 cells/sample) in assay medium were incubated with [177Lu]pentixafor (1 nM) at 37 °C for different time points up to 60 min in the presence (non-specific binding) or absence (control) of 100 µM AMD3100 (n=3 per concentration). Samples were then centrifuged, the supernatant (free radioligand) was removed and combined with 250 µL HBSS used for washing the cells. To differentiate between receptor bound (acid releasable) and internalized radioactivity, cells were then washed twice with 250 µL ice cold acid wash buffer (0.02 M NaOAc buffered with AcOH to pH = 5). The amount of free, acid releasable and internalized radioligand in the cell pellet, respectively, was quantified using a γ-counter.
Binding to human blood cells and plasma protein binding
To 225 µL samples of heparinized whole blood from a healthy donor, 25 µL of PBS (n=3, total binding) or 25 µL of 1 mM AMD3100 in PBS (n=3, non-specific binding) were added, immediately followed by the addition of [177Lu]pentixather in PBS (20 µL). After 15 min at RT, erythrocytes were spun down at 700·g for 5 min, and washed twice with 200 µL PBS. The combined supernatants of each sample were then centrifuged at 6200·g for 5 min, and the pellet (leukocytes and platelets) was washed once with PBS. The amount of activity in the three fractions containing erythrocytes, leukocytes and cell-free plasma, respectively, was quantified using a γ-Counter.
For the determination of plasma protein binding, fresh human plasma (400 µL) was incubated with [177Lu]pentixather for 15 min at RT. Then, a 300 µL sample was transferred into an ultracentrifugation device (Amicon Ultra 0.5 mL, 30K, regenerated cellulose, Merck Millipore, Darmstadt, Germany). Upon ultrafiltration at 14.000·g for 6 min, the activity in 100 µL aliquots of the ultrafiltrate and of unfiltered plasma was quantified using a γ-Counter. Additionally, binding of [177Lu]pentixather to human serum albumin (HSA) was quantified via a previously described gradient HPLC method [43] with minor modifications using a Chiralpak HSA column (50x3 mm, 5µm, Chiral Technologies Europe) equipped with a Chiralpak HSA Guard Cartridge (10x3 mm, 5µm). For elution (flow rate 0.5 mL/min), 50 mM ammonium acetate (pH=6.9, solvent A) and 2-propanol (solvent B) and a linear gradient (0-3 min: 0% B, 3-15 min: 0-20% B) were used.
Flow Cytometry
Membrane expression levels of CXCR4 were determined by flow cytometry (Cyan 9C, Beckman Coulter, Krefeld, Germany) using a PE-conjugated anti-CXCR4 antibody (hCD184; clone 12G5; BD Pharming, Becton Dickinson, Bergisch-Gladbach, Germany) and its isotype control (mouse IgG 2a,κ) according to the manufacturer's instructions. Data were analyzed using FlowJo software.
In vivo animal studies
Tumor model
For the induction of tumor growth, female CB-17 SCID mice (6-8 weeks, Charles River, Sulzfeld, Germany) were injected subcutaneously with app. 5×106 Daudi cells suspended in 100 µL of a 1:1 (v/v) mixture of serum free culture medium and Matrigel (BD Biosciences, Heidelberg, Germany). The Daudi xenograft model was chosen due to the well characterized endogenous CXCR4 expression on these cells, which is comparable to that of the non-tumorigenic Jurkat cells used for the in vitro assays [12]. Within 10-21 days, solid palpable tumors had grown (100-800 mg), and the animals were used for the experiments. All animal experiments were performed in accordance with current animal welfare regulations in Germany (approval #55.2-1-54-2532-71-13).
Biodistribution studies
For biodistribution studies, animals (n=4-5 per group) were injected intravenously with ≈1.5-4 MBq (depending on the specific activity of the preparation; the injected peptide amount was kept constant at 0.16 nmol peptide per mouse) of [177Lu]pentixather in PBS (100 µL) into the tail vein under isoflurane anesthesia. CXCR4-specificity of binding was determined via coinjection of 50 µg AMD3100 per mouse. The animals were sacrificed 1, 6, 48, 96h and 7d post injection (p.i.), and the organs of interest were dissected. The radioactivity was measured in weighted tissue samples using a γ-counter. Data are expressed in % ID/g tissue (mean ± SD).
Investigation of in vitro/in vivo stability
To determine the stability of [177Lu]pentixather in human plasma, 1-2 MBq of the tracer were incubated in 500 µL fresh human plasma (37° C, 1 h). Plasma proteins were removed by precipitation with acetonitrile, centrifugation and ultrafiltration using an Amicon Ultra 30K centrifugal filter (Merck Millipore, Darmstadt, Germany). In vivo stability of [177Lu]pentixather was investigated by injection of 30-40 MBq into the tail vein of anesthetized CB-17 SCID mice (n=3). Animals were sacrificed at 1h p.i., and blood and urine were collected. After centrifugation of the blood samples and isolation of cell-free plasma, sample preparation was performed as described above. All samples were analyzed via Radio-TLC and Radio-RP-HPLC using a Chromolith RP-18e performance column (4.6x150 mm) and isocratic elution with 3% acetonitrile (0.1% TFA) in 0.1% TFA for 3 min, followed by a linear gradient of 3-95% acetonitrile (0.1% TFA) in 0.1% TFA within 6 min at a flow rate of 3 mL/min.
Dosimetry and patient imaging
Based on the tissue uptake data obtained from the biodistribution study in mice, an extrapolation of the absorbed doses to humans was performed. This involved the scaling of the time-integrated activity coefficients (TIACs) and the subsequent calculation of the absorbed doses from the animal biodistribution data using two different methods. Mouse TIACs were calculated using the software solution NUKFIT [44]. The dose calculation was performed for a selected group of organs using OLINDA/EXM V1.1 [45]. Details on the extrapolation methodology used are provided in the supplementary data.
Human biokinetics and dosimetry of [177Lu]pentixather were assessed by SPECT/CT and planar imaging in a patient with kappa light-chain multiple myeloma (f, 54 y). [177Lu]pentixather administration on a compassionate-use basis (German Drug Act, §13,2b) was in compliance with §37 of the Declaration of Helsinki and had been approved by the clinical ethics committee of Würzburg University. The patient gave written informed consent prior to receiving therapy.
Maximum tolerable treatment activity was determined in a pre-therapeutic dose assessment from scintigraphic images consecutively acquired within 4d after intravenous injection of 197 MBq [177Lu]pentixather. Absorbed doses in organs at risk were deduced from the highest activity concentration measured by SPECT/CT in a contiguous 1 ml volume. Based on a dose limit of 23 Gy for the kidneys, the maximum safe activity was estimated to be 9.1 GBq [177Lu]pentixather (if combined with subsequent stem cell transplantation). After treatment with 7.8 GBq [177Lu]pentixather, tracer kinetics were measured by SPECT/CT and serial planar imaging up to 13d p.i.. All scintigraphic images were acquired with a 20% energy window at 208 keV using dual-head gamma-cameras (Siemens Symbia E for planar imaging, Symbia T2 for SPECT/CT) equipped with medium-energy collimators and calibrated by phantom measurements with 177Lu activity standards. Pharmacokinetic data were fitted by bi-exponential decay functions and normalized to activity concentrations measured quantitatively by SPECT/CT. SPECT/CT data were reconstructed using 3-dimensional ordered-subsets expectation maximization (6 subsets, 6 iterations, 6-mm gaussian filter) with corrections for scatter and attenuation to obtain absolute activity quantification in 4.8 mm voxels (0.11 cm³).
Results
CXCR4 affinity and selectivity
The hCXCR4 affinities of pentixather and its natLu- and natY-complexes are summarized in Table 1. Since the experimental setup of the competitive binding assay was modified compared to previous studies [11], the reference compounds natGa-pentixafor and FC-131 were re-assayed under the novel conditions (increased cell number per sample, higher BSA content in assay medium) for comparison and data normalization.
Binding affinities (IC50 in nM) of pentixather and its natLu-, natY- and natBi-complexes to hCXCR4. Data for the reference compounds Ga-pentixafor and FC-131 are also included.
| Peptide | IC50 [nM] |
|---|---|
| natGa-pentixafor | 24.8 ± 2.5 |
| FC-131 | 10.2 ± 2.4 |
| pentixather (uncomplexed) | 35.8 ± 13 |
| natLu-pentixather | 14.6 ± 1.0 |
| natY-pentixather | 20.4 ± 0.3 |
| natBi-pentixather | 4.4 ± 1.3 |
Affinities to hCXCR4 were determined using Jurkat human T-cell leukemia cells (400.000 cells/sample) and [125I]FC-131 as radioligand. Each experiment was performed in triplicate, and results are means ± SD from a minimum of three separate experiments.
Compared to natGa-pentixafor, both natLu-pentixather and natY-pentixather show improved hCXCR4 affinities. As previously observed for pentixafor and its natGa- and natIn-analogs, metal chelation leads to an enhanced hCXCR4 affinity of natLu-pentixather and natY-pentixather compared to non-complexed pentixather (Table 1). However, this effect is by far less pronounced than in the case of pentixafor/natGa-pentixafor, where Ga-complexation lead to a 30-fold increase in receptor affinity [37].
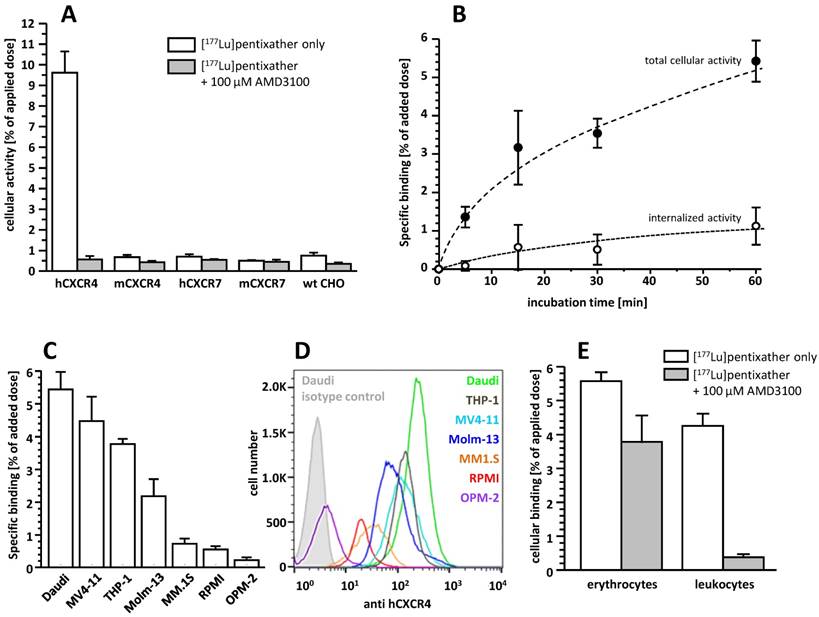
[177Lu]pentixather exhibits very high selectivity and specificity for hCXCR4 (Figure 2A) and only low specific binding to hCXCR7, an alternative receptor for CXCL12, and murine CXCR4 (mCXCR4), while virtually no binding to mCXCR7 is observed.
A: Binding of [177Lu]pentixather to wild type (wt) CHO-K1 cells and CHO-K1 cells transiently transfected with HA-tagged hCXCR4, mCXCR4, hCXCR7 and mCXCR7. Cells were incubated with [177Lu]pentixather (1 nM) at 37°C for 60 min in the absence (white bars) and presence (grey bars) of competitor (100 µM AMD3100). Experiments were carried out in triplicate, and data are means±SD. B: Binding and internalization kinetics of [177Lu]pentixather (1 nM) in Daudi lymphoma cells (37°C). Data are corrected for non-specific binding/internalization in the presence of 100 µM AMD3100. Experiments were performed in triplicate, and data are means±SD. C: Binding of [177Lu]pentixather to different lymphoma (Daudi), AML (MV4-11, THP-1, Molm-13) and multiple myeloma (MM.1S, RPMI, OPM-2) cell lines. Cells were incubated with [177Lu]pentixather (1 nM) at 37°C for 60 min. Data are corrected for non-specific binding in the presence of 100 µM AMD3100. Experiments were performed in triplicate, and data are means±SD. D: CXCR4 expression levels on the assayed cell lines determined via flow cytometry. E: Binding of [177Lu]pentixather to human blood cells. Whole blood from a healthy donor was incubated with [177Lu]pentixather at RT for 15 min in the absence (white bars) and presence (grey bars) of 100 µM AMD3100. Experiments were performed in triplicate, and data are means±SD.
Binding and internalization
CXCR4-mediated cellular binding of [177Lu]pentixather was investigated in a variety of human lymphoma (Daudi), multiple myeloma (MM1.S, RPMI, OPM-2) and acute myeloid leukemia cell lines (THP-1, MV4-11, Molm-13) (Figure 2C). Tracer uptake in all cell lines closely correlated with the respective CXCR4-expression levels determined via flow cytometry (Figure 2D), with the lymphoma and AML cell lines showing significantly higher CXCR4 expression than the MM cell lines, resulting in correspondingly enhanced cellular [177Lu]pentixather uptake.
Based on the particularly high uptake of [177Lu]pentixather in Daudi cells, this cell line was used to investigate its binding and internalization kinetics in more detail (Figure 2B). [177Lu]pentixather shows rapid cellular uptake kinetics. CXCR4-specific total cellular tracer uptake (binding + internalization) is improved compared to the reference [68Ga]pentixafor (6.5±1.1% of added dose for [177Lu]pentixather vs 4.6±0.6% of added dose for [68Ga]pentixafor after 60 min [12]) and does not yet reach a plateau within the 60 min observation period. Interestingly, however, only 16.6±1.9% of the total cellular [177Lu]pentixather activity are internalized at time points ≥ 15 min, whereas 41.2±14.4% of the total cellular activity were found to be internalized for [68Ga]pentixafor [12].
Binding to human blood cells and plasma protein binding
Incubation of whole blood of a healthy donor with [177Lu]pentixather revealed significant radioligand association with blood cells (Figure 2E). While activity association to erythrocytes was found to be primarily non-specific, [177Lu]pentixather binding to leukocytes and platelets was almost exclusively CXCR4-mediated, amounting to app. 4% of the added dose.
Quantification of the plasma protein (HSA) binding of [177Lu]pentixather via two alternative methods (ultrafiltration and HPLC) revealed consistent values of 96.8 and 96.6%, respectively. Thus, in contrast to [68Ga]pentixafor, which showed a lower extent of plasma protein binding (68% (determined by ultrafiltration [11]) and 86.0% (determined by HPLC), respectively), almost all of the circulating [177Lu]pentixather activity is protein associated.
In vivo biodistribution studies and metabolite analysis
The biodistribution of [177Lu]pentixather in Daudi human B-cell lymphoma xenograft bearing CB-17 SCID mice is summarized in Table 2. Compared to the reference [68Ga]pentixafor (logP= -2.90) [12] (Figure 3A), [177Lu]pentixather (logP= -1.76) exhibits slightly increased blood activity levels at 1h p.i., accompanied by enhanced overall background accumulation. Furthermore, the 177Lu-labeled peptide shows comparably high initial uptake in all excretion organs. However, while activity is rapidly cleared from stomach, intestines and the kidneys, hepatic activity levels remain persistently high up to 7d p.i., suggesting tracer retention in the liver rather than slow and continuous hepatobiliary excretion of the tracer. However, as demonstrated by coinjection of an excess of unlabeled competitor (2mg/kg AMD3100) at 6h p.i., hepatic [177Lu]pentixather uptake is at least partially blockable, which hints towards a certain contribution of CXCR4-mediated liver accumulation.
Biodistribution of [177Lu]pentixather in Daudi (human B cell lymphoma) xenograft bearing CB-17 SCID mice at 1h, 6h, 2d, 4d and 7d p.i. (groups of n=4-5 animals).
| organ | 1 h | 6 h | 6 h block | 48 h | 96 h | 7 d |
|---|---|---|---|---|---|---|
| blood | 1.3 ± 0.4 | 0.09 ± 0.01 | 0.06 ± 0.01 | 0.02 ± 0.001 | 0.01 ± 0.001 | 0.004 ± 0.002 |
| heart | 0.9 ± 0.3 | 0.18 ± 0.02 | 0.17 ± 0.02 | 0.11 ± 0.01 | 0.10 ± 0.01 | 0.07 ± 0.003 |
| lung | 2.5 ± 0.7 | 0.42 ± 0.06 | 0.39 ± 0.03 | 0.21 ± 0.06 | 0.17 ± 0.01 | 0.11 ± 0.02 |
| liver | 10.3 ± 0.8 | 9.59 ± 0.69 | 5.85 ± 0.24 | 8.25 ± 2.23 | 6.29 ± 0.36 | 5.44 ± 0.48 |
| stomach | 1.2 ± 0.3 | 0.21 ± 0.01 | 0.21 ± 0.04 | 0.12 ± 0.02 | 0.10 ± 0.01 | 0.06 ± 0.02 |
| small intestine | 0.9 ± 0.2 | 0.22 ± 0.04 | 0.17 ± 0.004 | 0.11 ± 0.02 | 0.09 ± 0.02 | 0.04 ± 0.01 |
| large intestine | 1.3 ± 1.2 | 0.50 ± 0.16 | 0.43 ± 0.12 | 0.25 ± 0.10 | 0.15 ± 0.01 | 0.06 ± 0.006 |
| kidney | 3.4 ± 0.6 | 2.52 ± 0.33 | 2.76 ± 0.18 | 1.20 ± 0.13 | 0.85 ± 0.15 | 0.51 ± 0.04 |
| pancreas | 0.4 ± 0.1 | 0.09 ± 0.01 | 0.10 ± 0.002 | 0.08 ± 0.01 | 0.07 ± 0.01 | 0.04 ± 0.01 |
| adrenals | 1.5 ± 0.6 | 0.64 ± 0.13 | 0.51 ± 0.01 | 0.56 ± 0.08 | 0.48 ± 0.03 | 0.46 ± 0.25 |
| spleen | 1.8 ± 0.3 | 1.35 ± 0.41 | 0.64 ± 0.55 | 1.58 ± 0.33 | 1.27 ± 0.43 | 1.10 ± 0.23 |
| femur | 0.6 ± 0.2 | 0.23 ± 0.02 | 0.15 ± 0.001 | 0.22 ± 0.05 | 0.21 ± 0.01 | 0.31 ± 0.02 |
| uterus | 1.1 ± 0.2 | 0.31 ± 0.05 | 0.39 ± 0.07 | 0.23 ± 0.05 | 0.23 ± 0.06 | 0.21 ± 0.09 |
| muscle | 0.3 ± 0.1 | 0.06 ± 0.01 | 0.06 ± 0.001 | 0.04 ± 0.005 | 0.04 ± 0.01 | 0.02 ± 0.001 |
| skin | 0.9 ± 0.5 | 0.19 ± 0.02 | 0.22 ± 0.01 | 0.11 ± 0.02 | 0.095 ± 0.008 | 0.03 ± 0.003 |
| brain | 0.05 ± 0.01 | 0.02 ± 0.005 | 0.02 ± 0.002 | 0.007 ± 0.002 | 0.006 ± 0.001 | 0.004 ± 0.001 |
| tumor | 12.4 ± 3.7 | 6.79 ± 0.68 | 0.43 ± 0.02 | 3.27 ± 0.41 | 2.07 ± 0.12 | 2.06 ± 0.37 |
To determine CXCR4 specificity of tracer uptake (6h p.i.), 50 µg AMD3100 (2 mg/kg) were coinjected. Data are given in %iD/g and are means ± SD.
A: Comparative biodistribution of [68Ga]pentixafor [12] and [177Lu]pentixather in Daudi (human B cell lymphoma) xenograft bearing CB17-SCID mice (groups of n=5) at 90 and 60 min p.i., respectively. Uptake values are given in %iD/g and are means±SD. B: Comparison of the tracer kinetics of [177Lu]pentixather in kidneys and liver of the multiple myeloma patient (f, 54y) in this study with mouse biodistribution data. Total organ uptake is given in % of the injected activity (%iD). Patient data are depicted in green (pre-therapeutic injection, 197 MBq [177Lu]pentixather) and black ([177Lu]pentixather therapy, 7.8 GBq), mouse data are depicted in red. Open circles represent data for kidneys, filled dots data for liver. C: Whole body planar imaging of the multiple myeloma patient in this study at 22h after [177Lu]pentixather therapy (7.8 GBq). D: Corresponding SPECT/CT images at 23h after [177Lu]pentixather therapy (7.8 GBq).
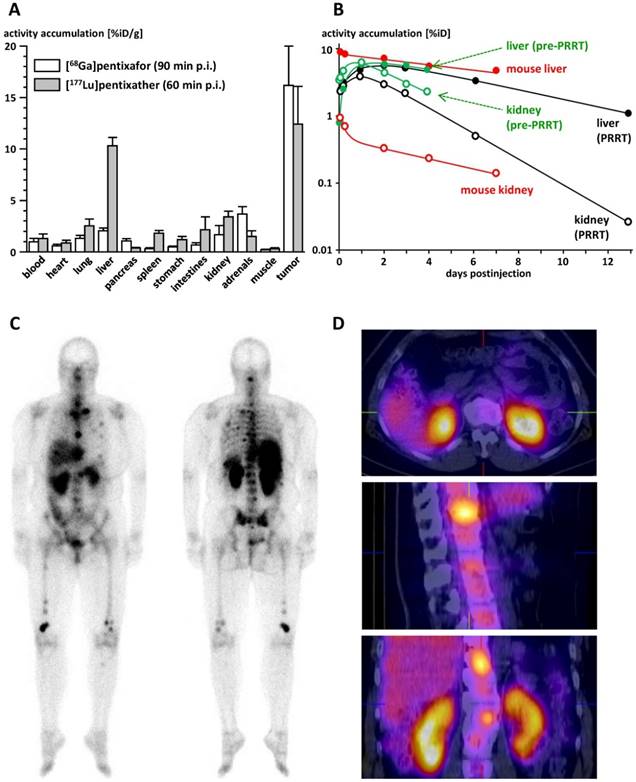
Since, as opposed to the highly hCXCR4-selective ligand [68Ga]pentixafor, [177Lu]pentixather seems to exhibit some affinity to mCXCR4 (Figure 2A), CXCR4-specificity of tracer uptake was not only observed in the Daudi xenografts, but also in mouse tissues with endogenous CXCR4 expression, i.e. liver, spleen and the bone marrow (femur). Overall, [177Lu]pentixather showed prolonged retention or even increasing accumulation (femur) in all CXCR4-positive tissues up to 7 d p.i., while the tracer was effectively cleared from the background within this period. This leads to increasing tumor/background ratios over time, reaching 499±202, 0.4±0.1, 33±7, 4.0±0.8 and 116±22 for blood, liver, large intestine, kidney and muscle, respectively, at 7d p.i..
The metabolic stability of [177Lu]pentixather was investigated both by incubation in human serum and by an in vivo metabolite study in CB-17 SCID mice at 1h p.i. (n=3). Both in human serum and in mice, virtually no tracer degradation was observed (see supplementary data, Fig. 1), indicating excellent metabolic stability of [177Lu]pentixather.
Dosimetry
TIACs and absorbed dose coefficients for [177Lu]pentixather in humans were extrapolated from mouse biodistribution data using two alternative extrapolation methods. Details on the methodology, a list of the TIACs for organs of relevance for dosimetry, a full list of the corresponding absorbed doses, and information on the absorbed dose by β-particles and photons are supplied in the supplementary data.
The highest TIAC was calculated for the liver (method 1: 20.4 h; method 2: 11.2 h), with values for all other tissues, including the kidneys, being considerably lower. Consequently, the highest extrapolated dose coefficients were found for the liver (0.95 mGy/MBq (method 1) and 0.52 mGy/MBq (method 2), respectively), with estimated values for other organs at risk (kidneys, spleen, red bone marrow) being by a factor of 5-10 lower, depending on the extrapolation method used (Table 3).
Estimates of the total absorbed doses for [177Lu]pentixather in humans based on mouse biokinetic data.
| Target Organ | Total absorbed dose [mGy/MBq] | |
|---|---|---|
| method 1 | method 2 | |
| Adrenals | 8.4E-02 | 3.2E-02 |
| Brain | 7.9E-04 | 7.2E-04 |
| Breasts | 1.8E-03 | 1.2E-03 |
| Gallbladder Wall | 1.9E-02 | 1.1E-02 |
| Lower Large Intestine Wall | 4.4E-02 | 2.2E-02 |
| Small Intestine | 4.2E-02 | 1.7E-02 |
| Stomach Wall | 1.1E-02 | 4.9E-03 |
| Upper Large Intestine Wall | 4.0E-02 | 1.7E-01 |
| Heart Wall | 8.5E-03 | 6.1E-03 |
| Kidneys | 2.1E-01 | 5.8E-02 |
| Liver | 9.5E-01 | 5.2E-01 |
| Lungs | 1.1E-02 | 1.6E-02 |
| Muscle | 4.1E-03 | 3.4E-03 |
| Ovaries | 6.1E-02 | 1.2E-02 |
| Pancreas | 3.0E-02 | 9.7E-03 |
| Red Marrow | 9.9E-02 | 9.9E-02 |
| Osteogenic Cells | 4.9E-02 | 4.8E-02 |
| Skin | 1.1E-03 | 7.9E-04 |
| Spleen | 3.0E-02 | 6.8E-02 |
| Testes | 4.1E-04 | 3.9E-04 |
| Thymus | 1.7E-03 | 1.2E-03 |
| Thyroid | 6.5E-04 | 5.7E-04 |
| Urinary Bladder Wall | 1.0E-03 | 8.6E-04 |
| Uterus | 6.3E-02 | 1.4E-02 |
Data were calculated using OLINDA/EXM using two different scaling methods (for details see supplementary data).
Extrapolated liver absorbed doses (Table 3) are in good agreement with the human data obtained in the multiple myeloma patient in this study (Figure 3B), i.e. 0.95 mGy/MBq in the pre-therapeutic dose assessment and 0.75 mGy/MBq during [177Lu]pentixather therapy. For the kidneys, however, the absorbed dose observed in the pre-therapeutic dosimetry assessment of the patient (2.51 mGy/MBq) was substantially higher than the values extrapolated from the mouse biodistribution data (Table 3; 0.21 and 0.058 mGy/MBq, respectively), necessitating nephroprotective treatment [46] during [177Lu]pentixather therapy.
As shown in Figure 3C, uptake of [177Lu]pentixather was higher in tumor lesions than in all other organs, leading to tumor/kidney and tumor/liver dose ratios of 3.1 and 6.4, respectively. Substantial [177Lu]pentixather accumulation was also observed in the spleen and bone marrow of the patient, resulting in organ doses of 0.81 and 0.97 mGy/MBq to spleen and bone marrow, respectively, and thus to myeloablation by [177Lu]pentixather PRRT, which represents a therapy objective in patients with multiple myeloma. Again, extrapolated doses from the mouse biodistribution data (Table 3) have limited predictive value for the human situation, which in this case, however, is the result of the low affinity of [177Lu]pentixather to mCXCR4 and thus an underproportionally low tracer accumulation in mouse spleen and bone marrow as compared to humans.
Discussion
Given the importance of CXCR4 as an oncological target, the objective of this study was the development of a suitable therapeutic counterpart to the already established PET imaging probe [68Ga]pentixafor [11-17] and to thereby provide a first and sustainable CXCR4-targeted theranostic concept.
As shown in Figure 1, the only structural difference between pentixafor and pentixather is the iodine-for-H-substitution in Tyr1 in the peptide backbone. Given the fact that D-Tyr1 iodination is a straightforward one-step synthetic procedure [41], only minimal synthetic effort is required for obtaining the dedicated therapeutic probe, which represents a considerable practical advantage. Despite representing a minimal structural modification compared to pentixafor, however, iodination of D-Tyr1 conveys several characteristics to pentixather that represent essential prerequisites for successful application in CXCR4-targeted PRRT.
Firstly, introduction of 3-iodo-D-Tyr1 not only substantially improves CXCR4 affinity of the unlabeled peptide as such, it also confers much greater flexibility towards chelation of different (radio)metal ions by the DOTA moiety. As opposed to pentixafor, where exchange of Ga3+ for In3+ or Lu3+ had led to an app. tenfold decrease in receptor affinity [37], complexation of Lu3+, Y3+ and Bi3+ by pentixather even has beneficiary effect on CXCR4 affinity, leading to analogs with two- to fivefold higher CXCR4 affinity than Ga-pentixafor in the same assay (Table 1).
The enhanced affinity of the respective pentixather (radio)metal chelates towards hCXCR4 is also evident in the improved in vitro CXCR4 targeting efficiency of [177Lu]pentixather; it not only shows high and specific uptake in CXCR4 expressing human cancer cell lines (Figure 2C), which accurately reflects CXCR4 expression levels, but also improved CXCR4-mediated tracer accumulation in Daudi lymphoma cells compared to the reference [68Ga]pentixafor. Interestingly, however, internalization efficiency of [177Lu]pentixather was found to be considerably lower than that of [68Ga]pentixafor. Most probably, slight differences in the agonistic/antagonistic profile of the compounds may be responsible for this observation, and ongoing studies are aimed at elucidating these relationships.
It is important to note, that recent studies on radiolabeled somatostatin receptor (sst) agonists and antagonists have induced a paradigm shift concerning the relevance of efficient ligand internalization for high in vivo tumor accumulation and retention. As opposed to previous assumptions, the in vivo tumor uptake and retention of non-internalizing sst-antagonists, both in preclinical mouse models and in patients with neuroendocrine tumors, is dramatically increased compared to efficiently internalizing agonists [47]. The present data on [177Lu]pentixather also demonstrate the negligible effect of ligand internalization on the in vivo performance of the compound. Despite different internalization efficiencies, [177Lu]pentixather and [68Ga]pentixafor show comparable uptake in the Daudi lymphoma xenografts at 60 and 90 min p.i., respectively (Figure 3A). Even more importantly, [177Lu]pentixather shows slow late-phase tumor washout kinetics and persistent activity retention in the tumor xenografts up to 7 d p.i. (Table 2), which is the primary prerequisite for delivery of sufficient radiation doses to the tumor during PRRT.
Besides its effect on CXCR4 affinity, CXCR4 targeting efficiency and species selectivity, the iodination of Tyr1 has a pronounced impact on the physicochemical properties of [177Lu]pentixather and consequently its pharmacokinetic behavior. The lipophilicity of [177Lu]pentixather is increased by more than an order of magnitude compared to [68Ga]pentixafor. On the one hand, this leads to enhanced plasma protein binding, which in turn entails a slightly delayed activity clearance from the circulation (Figure 3A) and from the background tissues. As mentioned, this circumstance is expected to have a beneficiary influence on tracer uptake in the target tissues, since it leads to prolonged tracer delivery to the tumor site, given a sufficient metabolic stability of the targeted agent. In the case of [177Lu]pentixather, no significant tracer degradation or deiodination was observed up to 1h p.i. in mice and in human serum (see supplementary Figure 1), indicating sufficient tracer stability for efficiently exploiting the aforementioned effect. In humans, the extended availability of [177Lu]pentixather at the target site by plasma protein binding may even be further amplified by an additional reservoir function, i.e. the binding of [177Lu]pentixather to hCXCR4-expressing blood cells. Given the low affinity of [177Lu]pentixather for mCXCR4, tracer binding to mouse blood cells was not expected and thus not investigated in detail; in vitro studies using human full blood, however, showed that a total of app. 6% of the added activity were specifically bound to both erythrocytes and leukocytes/platelets, with the latter fraction primarily contributing to the effect (Figure 2E). This finding is in accordance with the documented high CXCR4-expression on human leukocytes [48] and may also contribute to maintaining a sufficient [177Lu]pentixather concentration in the blood pool for prolonged tracer delivery to the tumor.
Of course, the increased lipophilicity of [177Lu]pentixather was also expected to be reflected in an enhanced contribution of hepatobiliary clearance to overall tracer elimination. At a first glance, the comparably high liver accumulation of [177Lu]pentixather observed in the mouse biodistribution studies as well as in the patient (Figure 3A, 3C, 3D) supports this assumption. It is important to note, however, that in mice, the hepatic uptake of [177Lu]pentixather is overproportional to what would have been expected on the basis of differences in lipophilicity between structurally closely related peptides as the determining factor alone [42]. Furthermore, both in mice and in humans, virtually none of the initial hepatic activity is cleared into the intestines within the observation period (Table 2 and Figure 3B), indicating effective trapping of the activity in liver tissue by a yet unknown mechanism.
In mice, at least part of this uptake seems CXCR4 mediated, as demonstrated by the reduction of the hepatic [177Lu]pentixather accumulation by coinjection of a high dose of competitor (2 mg/kg AMD3100; Table 2). This observation may be accounted for by the documented expression of functional CXCR4 in the mouse liver [49] in combination with the observed, albeit low affinity of [177Lu]pentixather to mCXCR4 (Table 1). In humans, CXCR4 expression in normal liver has also been described [50]. However, since [68Ga]pentixafor, despite comparably high hCXCR4 affinity, shows virtually no hepatic accumulation/retention in patients [12-17], other factors than CXCR4-mediated tracer accumulation seem to be responsible for the persistent retention of hepatic 177Lu-activity in humans.
Overall, absorbed liver doses calculated from mouse biodistribution data and from patient dosimetry in this study, both pre-therapeutically and during [177Lu]pentixather PRRT, were highly consistent and also in agreement with patient data obtained in the aforementioned parallel patient studies, where liver doses ranging from 0.37 mGy/MBq to 0.85 mGy/MBq were observed [38, 39]. However, due to the substantially lower renal activity retention in mice (Figure 3B), the mouse data failed to accurately predict the human kidney dose, which ranged from 0.48 to 3.07 mGy/MBq in the pre-therapeutic dosimetry assessments (2.51 mGy/MBq in the present patient), but was reduced by app. 40% by nephroprotective treatment during therapy [38, 39].
As mentioned, the primary goal of this study was the comprehensive characterization of [177Lu]pentixather with respect to its in vitro and in vivo CXCR4 targeting efficiency, general pharmacokinetics and dosimetric profile in mice and a cross-validation with exemplary human data. Detailed investigations on the therapeutic efficacy of [177Lu]pentixather PRRT in mouse models of different human cancers are currently ongoing and are anticipated to further substantiate the findings from the aforementioned first human studies [38, 39], which had been conducted in parallel to this work. In the majority of patients with relapsed multiple myeloma, significant tumor response to [177Lu]pentixather PRRT was observed. Of course, given the small number of patients and the fact that all patients received a combination of [177Lu/90Y]pentixather endoradiotherapy with high-dose chemotherapy and consecutive stem cell support, which precludes a dissection of the therapeutic effects of each individual treatment component, these results are preliminary in nature. Nevertheless, they demonstrate the general feasibility and high potential of a [68Ga]pentixafor/[177Lu]pentixather based CXCR4-targeted theranostic concept, which may open new perspectives towards individualized cancer therapies, especially in hematological cancers [51]. The tolerance of the pentixather scaffold towards complexation with different radiometals (Table 1), ranging from high-energy beta-emitters (90Y) to alpha-emitters (213Bi), without biasing CXCR4 affinity provides the option to specifically select the best-suited radionuclide for targeting tumors of a given size, from larger tumor masses down to micrometastases. Recent insights into the regulation of CXCR4 expression and function by specific chemotherapeutics may also contribute to improving the efficacy of CXCR4-directed endoradiotherapy using the pentixather platform. For example, pronounced CXCR4 upregulation was observed by daunomycin treatment of selected AML cell lines [52], leading to more effective chemosensitization of these cells by CXCR4-targeted treatment with AMD3100. This effect may also help to improve uptake of CXCR4-targeted therapeutic vectors such as radiometal-labeled pentixather and thus promote the efficacy of endoradiotherapeutic treatment.
In summary, the detailed preclinical characterization of [177Lu]pentixather demonstrates its excellent CXCR4 targeting characteristics, both in vitro and in vivo, along with a suitable pharmacokinetic profile for efficient tracer delivery to the target tissues as well as favorable overall dosimetry. Ongoing evaluation of [177Lu]pentixather, both preclinically in patient-derived lymphoma and leukemia xenograft models and clinically in patients with hematological cancers will help to further assess its potential role in the endoradiotherapeutic treatment of CXCR4-overexpressing malignancies.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors are indebted to Monika Beschorner and Sven Hintze for their excellent technical assistance during precursor synthesis and in vitro/in vivo evaluations, to Katharina Franke for performing the flow cytometry measurements and to Stephanie Robu for carrying out metabolite analyses. Furthermore, our sincere thanks go to Marcus Schneider-Ludorff for his valuable contribution in proofreading the manuscript.
Grant Support
This study was financially supported by the Deutsche Forschungsgemeinschaft (SFB824; subprojects B5, C3 and Z1) and by an IGSSE (International Graduate School of Science and Engineering, TUM) grant (BioMat03:CXCR4).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jacobson O, Weiss ID. CXCR4 chemokine receptor overview: biology, pathology and applications in imaging and therapy. Theranostics. 2013;3:1-2
2. Nagasawa T. CXC chemokine ligand 12 (CXCL12) and its receptor CXCR4. J Mol Med (Berl). 2014;92:433-9
3. Balkwill FR. The chemokine system and cancer. J Pathol. 2012;226:148-57
4. Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EG. et al. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49:219-30
5. Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res. 2014;124:31-82
6. Guo F, Wang Y, Liu J, Mok SC, Xue F, Zhang W. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene. 2016;35:816-26
7. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540-50
8. Zhao H, Guo L, Zhao H, Zhao J, Weng H, Zhao B. CXCR4 over-expression and survival in cancer: a system review and meta-analysis. Oncotarget. 2015;6:5022-40
9. Weiss ID, Jacobson O. Molecular imaging of chemokine receptor CXCR4. Theranostics. 2013;3:76-84
10. George GPC, Pisaneschi F, Quang-De N, Aboagye EO. Positron Emission Tomographic Imaging of CXCR4 in Cancer: Challenges and Promises. Molecular Imaging. 2015:14
11. Gourni E, Demmer O, Schottelius M, D'Alessandria C, Schulz S, Dijkgraaf I. et al. PET of CXCR4 Expression by a Ga-68-Labeled Highly Specific Targeted Contrast Agent. J Nucl Med. 2011;52:1803-10
12. Wester HJ, Keller U, Schottelius M, Beer A, Philipp-Abbrederis K, Hoffmann F. et al. Disclosing the CXCR4 Expression in Lymphoproliferative Diseases by Targeted Molecular Imaging. Theranostics. 2015;5:618-30
13. Philipp-Abbrederis K, Herrmann K, Knop S, Schottelius M, Eiber M, Luckerath K. et al. In vivo molecular imaging of chemokine receptor CXCR4 expression in patients with advanced multiple myeloma. EMBO Mol Med. 2015;7:477-87
14. Herhaus P, Habringer S, Philipp-Abbrederis K, Vag T, Gerngross C, Schottelius M. et al. Targeted positron emission tomography imaging of CXCR4 expression in patients with acute myeloid leukemia. Haematologica. 2016;101:932-40
15. Lapa C, Luckerath K, Rudelius M, Schmid JS, Schoene A, Schirbel A. et al. [68Ga]Pentixafor-PET/CT for imaging of chemokine receptor 4 expression in small cell lung cancer - initial experience. Oncotarget. 2016;7:9288-95
16. Lapa C, Luckerath K, Kleinlein I, Monoranu CM, Linsenmann T, Kessler AF. et al. (68)Ga-Pentixafor-PET/CT for Imaging of Chemokine Receptor 4 Expression in Glioblastoma. Theranostics. 2016;6:428-34
17. Vag T, Gerngross C, Herhaus P, Eiber M, Philipp-Abbrederis K, Graner FP. et al. First Experience with Chemokine Receptor CXCR4-Targeted PET Imaging of Patients with Solid Cancers. J Nucl Med. 2016;57:741-6
18. Watts A, Singh B, Basher R, Singh H, Bal A, Kapoor R. et al. 68Ga-Pentixafor PET/CT demonstrating higher CXCR4 density in small cell lung carcinoma than in non-small cell variant. Eur J Nucl Med Mol Imaging. 2017
19. Bluemel C, Hahner S, Heinze B, Fassnacht M, Kroiss M, Bley TA. et al. Investigating the Chemokine Receptor 4 as Potential Theranostic Target in Adrenocortical Cancer Patients. Clin Nucl Med. 2017;42:e29-e34
20. Thackeray JT, Derlin T, Haghikia A, Napp LC, Wang Y, Ross TL. et al. Molecular Imaging of the Chemokine Receptor CXCR4 After Acute Myocardial Infarction. JACC Cardiovasc Imaging. 2015;8:1417-26
21. Lapa C, Reiter T, Werner RA, Ertl G, Wester HJ, Buck AK. et al. [(68)Ga]Pentixafor-PET/CT for Imaging of Chemokine Receptor 4 Expression After Myocardial Infarction. JACC Cardiovasc Imaging. 2015;8:1466-8
22. Rischpler C, Nekolla SG, Kossmann H, Dirschinger RJ, Schottelius M, Hyafil F. et al. Upregulated myocardial CXCR4-expression after myocardial infarction assessed by simultaneous GA-68 pentixafor PET/MRI. J Nucl Cardiol. 2016;23:131-3
23. Schmid JS, Schirbel A, Buck AK, Kropf S, Wester HJ, Lapa C. [68Ga]Pentixafor-Positron Emission Tomography/Computed Tomography Detects Chemokine Receptor CXCR4 Expression After Ischemic Stroke. Circ Cardiovasc Imaging. 2016;9:e005217
24. Hyafil F, Pelisek J, Laitinen I, Schottelius M, Mohring M, Doring Y. et al. Imaging the cytokine receptor CXCR4 in atherosclerotic plaques with the radiotracer 68Ga-pentixafor for positron emission tomography. J Nucl Med. 2016
25. Derlin T, Wester HJ, Bengel FM, Hueper K. Visualization of Posttraumatic Splenosis on Chemokine Receptor CXCR4-Targeted PET/CT. Clin Nucl Med. 2017
26. Peled A, Wald O, Burger J. Development of novel CXCR4-based therapeutics. Expert Opin Investig Drugs. 2012;21:341-53
27. Mishan MA, Ahmadiankia N, Bahrami AR. CXCR4 and CCR7: Two eligible targets in targeted cancer therapy. Cell Biol Int. 2016
28. Ramsey DM, McAlpine SR. Halting metastasis through CXCR4 inhibition. Bioorg Med Chem Lett. 2013;23:20-5
29. Domanska UM, Boer JC, Timmer-Bosscha H, van Vugt MA, Hoving HD, Kliphuis NM. et al. CXCR4 inhibition enhances radiosensitivity, while inducing cancer cell mobilization in a prostate cancer mouse model. Clin Exp Metastasis. 2014;31:829-39
30. Domanska UM, Timmer-Bosscha H, Nagengast WB, Oude Munnink TH, Kruizinga RC, Ananias HJ. et al. CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy. Neoplasia. 2012;14:709-18
31. Xiang J, Hurchla MA, Fontana F, Su X, Amend SR, Esser AK. et al. CXCR4 Protein Epitope Mimetic Antagonist POL5551 Disrupts Metastasis and Enhances Chemotherapy Effect in Triple-Negative Breast Cancer. Mol Cancer Ther. 2015;14:2473-85
32. Zoller F, Eisenhut M, Haberkorn U, Mier W. Endoradiotherapy in cancer treatment-basic concepts and future trends. Eur J Pharmacol. 2009;625:55-62
33. Kjaer A, Knigge U. Use of radioactive substances in diagnosis and treatment of neuroendocrine tumors. Scand J Gastroenterol. 2015;50:740-7
34. Weineisen M, Schottelius M, Simecek J, Baum RP, Yildiz A, Beykan S. et al. 68Ga- and 177Lu-Labeled PSMA I&T: Optimization of a PSMA-Targeted Theranostic Concept and First Proof-of-Concept Human Studies. J Nucl Med. 2015;56:1169-76
35. Afshar-Oromieh A, Hetzheim H, Kratochwil C, Benesova M, Eder M, Neels OC. et al. The Theranostic PSMA Ligand PSMA-617 in the Diagnosis of Prostate Cancer by PET/CT: Biodistribution in Humans, Radiation Dosimetry, and First Evaluation of Tumor Lesions. J Nucl Med. 2015;56:1697-705
36. Poschenrieder A, Schottelius M, Schwaiger M, Kessler H, Wester HJ. The influence of different metal-chelate conjugates of pentixafor on the CXCR4 affinity. EJNMMI Res. 2016;6:36
37. Demmer O, Gourni E, Schumacher U, Kessler H, Wester HJ. PET imaging of CXCR4 receptors in cancer by a new optimized ligand. Chemmedchem. 2011;6:1789-91
38. Herrmann K, Schottelius M, Lapa C, Osl T, Poschenrieder A, Hanscheid H. et al. First-in-Human Experience of CXCR4-Directed Endoradiotherapy with 177Lu- and 90Y-Labeled Pentixather in Advanced-Stage Multiple Myeloma with Extensive Intra- and Extramedullary Disease. J Nucl Med. 2016;57:248-51
39. Lapa C, Herrmann K, Schirbel A, Hänscheid H, Lückerath K, Schottelius M. et al. CXCR4-directed endoradiotherapy induces high response rates in extramedullary relapsed Multiple Myeloma. Theranostics. 2017 accepted
40. Schottelius M, Schwaiger M, Wester HJ. Rapid and high-yield solution-phase synthesis of DOTA-Tyr(3)-octreotide and DOTA-Tyr(3)-octreotate using unprotected DOTA. Tetrahedron Lett. 2003;44:2393-6
41. Schottelius M, Konrad M, Osl T, Poschenrieder A, Wester HJ. An optimized strategy for the mild and efficient solution phase iodination of tyrosine residues in bioactive peptides. Tetrahedron Lett. 2015;56:6602-5
42. Schottelius M, Wester HJ, Reubi JC, Senekowitsch-Schmidtke R, Schwaiger M. Improvement of pharmacokinetics of radioiodinated Tyr(3)-octreotide by conjugation with carbohydrates. Bioconjug Chem. 2002;13:1021-30
43. Valko K, Nunhuck S, Bevan C, Abraham MH, Reynolds DP. Fast gradient HPLC method to determine compounds binding to human serum albumin. Relationships with octanol/water and immobilized artificial membrane lipophilicity. J Pharm Sci. 2003;92:2236-48
44. Kletting P, Schimmel S, Kestler HA, Hanscheid H, Luster M, Fernandez M. et al. Molecular radiotherapy: The NUKFIT software for calculating the time-integrated activity coefficient. Med Phys. 2013:40
45. Stabin MG, Sparks RB, Crowe E. OLINDA/EXM: The second-generation personal computer software for internal dose assessment in nuclear medicine. J Nucl Med. 2005;46:1023-7
46. Bodei L, Mueller-Brand J, Baum RP, Pavel ME, Horsch D, O'Dorisio MS. et al. The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2013;40:800-16
47. Wang X, Fani M, Schulz S, Rivier J, Reubi JC, Maecke HR. Comprehensive evaluation of a somatostatin-based radiolabelled antagonist for diagnostic imaging and radionuclide therapy. Eur J Nucl Med Mol Imaging. 2012;39:1876-85
48. Lee B, Sharron M, Montaner LJ, Weissman D, Doms RW. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc Natl Acad Sci U S A. 1999;96:5215-20
49. Mendt M, Cardier JE. Stromal-derived factor-1 and its receptor, CXCR4, are constitutively expressed by mouse liver sinusoidal endothelial cells: implications for the regulation of hematopoietic cell migration to the liver during extramedullary hematopoiesis. Stem Cells Dev. 2012;21:2142-51
50. Shibuta K, Mori M, Shimoda K, Inoue H, Mitra P, Barnard GF. Regional expression of CXCL12/CXCR4 in liver and hepatocellular carcinoma and cell-cycle variation during in vitro differentiation. Jpn J Cancer Res. 2002;93:789-97
51. Mesguich C, Zanotti-Fregonara P, Hindie E. New Perspectives Offered by Nuclear Medicine for the Imaging and Therapy of Multiple Myeloma. Theranostics. 2016;6:287-90
52. Sison EA, McIntyre E, Magoon D, Brown P. Dynamic chemotherapy-induced upregulation of CXCR4 expression: a mechanism of therapeutic resistance in pediatric AML. Mol Cancer Res. 2013;11:1004-16
Author contact
![]() Corresponding author: PD Dr. Margret Schottelius, Chair for Pharmaceutical Radiochemistry, Technische Universität München, Walther-Meissner-Strasse 3, 85748 Garching, Germany m.schotteliusde Phone 0049 89 289 12258 Fax 0049 89 289 12204
Corresponding author: PD Dr. Margret Schottelius, Chair for Pharmaceutical Radiochemistry, Technische Universität München, Walther-Meissner-Strasse 3, 85748 Garching, Germany m.schotteliusde Phone 0049 89 289 12258 Fax 0049 89 289 12204