Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(2):549-562. doi:10.7150/thno.20047 This issue Cite
Research Paper
A cell-penetrating whole molecule antibody targeting intracellular HBx suppresses hepatitis B virus via TRIM21-dependent pathway
Jun-Fang Zhang1,2,3*, Hua-Long Xiong1,2*, Jia-Li Cao1,2, Shao-Juan Wang1,2, Xue-Ran Guo1,2, Bi-Yun Lin4, Ying Zhang1,2, Jing-Hua Zhao1,2, Ying-Bin Wang1,2, Tian-Ying Zhang1,2, ![]() , Quan Yuan1,2,
, Quan Yuan1,2, ![]() , Jun Zhang1,2, Ning-Shao Xia1,2
, Jun Zhang1,2, Ning-Shao Xia1,2
1. State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics, School of Public Health, Xiamen University, Xiamen 361102, China;
2. National Institute of Diagnostics and Vaccine Development in Infectious Diseases, School of Life Science, Xiamen University, Xiamen 361102, China;
3. School of Medicine, Shenzhen University, Shenzhen, 518060, China;
4. Department of Clinical Pathology, Affiliated Hospital of Guangdong Medical College, Zhanjiang, 524001, China.
* These authors contributed equally to this work
Received 2017-3-13; Accepted 2017-9-27; Published 2018-1-1
Abstract

Rationale: Monoclonal antibodies (mAbs) mostly targeting extracellular or cell surface molecules have been widely used in the treatment of various diseases. However, mAbs cannot pass through the cell membrane as efficiently as small compounds, thus limiting their use against intracellular targets. Methods to shuttle antibodies into living cells may largely expand research and application in areas based on mAbs. Hepatitis B virus X protein (HBx) is an important intracellular multi-functional viral protein in the life cycle of hepatitis B virus (HBV). HBx plays essential roles in virus infection and replication and is strongly associated with HBV-related carcinogenesis.
Methods: In this study, we developed a cell-penetrating whole molecule antibody targeting HBx (9D11-Tat) by the fusion of a cell penetrating peptide (CPP) on the C-terminus of the heavy chain of a potent mAb specific to HBx (9D11). The anti-HBV effect and mechanism of 9D11-Tat were investigated in cell and mouse models mimicking chronic HBV infection.
Results: Our results demonstrated that the recombinant 9D11-Tat antibody could efficiently internalize into living cells and significantly suppress viral transcription, replication, and protein production both in vitro and in vivo. Further analyses suggested the internalized 9D11-Tat antibody could greatly reduce intracellular HBx via Fc binding receptor TRIM21-mediated protein degradation. This process simultaneously stimulated the activations of NF-κB, AP-1, and IFN-β, which promoted an antiviral state of the host cell.
Conclusion: In summary, our study offers a new approach to target intracellular pathogenesis-related protein by engineered cell-penetrating mAb expanding their potential for therapeutic applications. Moreover, the 9D11-Tat antibody may provide a novel therapeutic agent against human chronic HBV infection.
Keywords: antibodies for intracellular targets, cell-penetrating peptide, antibody-mediated intracellular immunity, hepatitis B virus, Hepatitis B virus X protein
Introduction
Monoclonal antibodies (mAbs) have a well-established therapeutic role for cancer, autoimmune diseases, inflammations and acute infectious diseases [1-3]. MAbs are highly specific and potent binders, capable of labeling cells for immunologic attack and modulating signaling pathways to inhibit cell proliferation and/or induce cell death. However, unlike small molecule drugs which easily cross cellular membranes, mAbs cannot directly access intracellular targets due to their large molecular size [4]. Currently approved therapeutic mAbs all target secreted or cell-surface antigens, whereas many oncogenic proteins and pathogens are intracellular and therefore not accessable to mAbs. Previous studies have shown the usefulness of mAbs in visualizing or functionally blocking the target proteins in living cells by microinjection or protein transduction [5-7]. It has also been demonstrated that antibodies can be carried into cells when attached to non-enveloped viral particles during virus infection, and subsequently induce proteasomal targeting and rapid degradation of intracellular antibody-bound pathogens [8, 9]. These studies provided evidence for the stability and function of mature antibodies in the cytoplasm of cells. Therefore, cell-penetrating mAbs directed against intracellular disease targets may provide novel therapeutic agents, in particular for cancers and persistent viral infections [10-13].
As one of the most important pathogens worldwide, the hepatitis B virus (HBV) causes a chronic, even life-long infection. Chronic HBV infection (CHB) causes chronic hepatitis and places patients at high risk of death due to liver cirrhosis and hepatocellular carcinoma (HCC) [14]. Worldwide, more than78,000 people die each year because of the acute or chronic consequences of HBV infection [14, 15]. Despite the successful development of preventive hepatitis B vaccine that have effectively reduced new cases of HBV infection globally, there are still hundreds of millions of people who are infected with HBV and require more effective therapies [14]. The approved anti-HBV drugs, which are either interferon or nucleos(t)ide analogues, can only induce disease remission, but cannot eradicate the virus or completely block HCC development [16]. The ideal end point for anti-HBV therapy is hepatitis B surface antigen (HBsAg) loss; however, this goal is achieved infrequently with the currently available drugs (in <5% of cases [17]). Development of more effective anti-HBV drugs or treatment strategies is urgently required. Hepatitis B virus X protein (HBx, 154 aa) is a multi-functional non-structural protein of HBV. For viral life cycle, HBx is essentially required to initiate and maintain HBV replication after infection [18]. HBx-defective HBV mutants showed significantly reduced replication efficiency both in vitro and in vivo [19, 20]. Moreover, several studies have suggested that HBx is a promiscuous transactivator and plays multiple roles in HBV-related hepatocarcinogenesis. Transgenic expression of HBx in mouse liver resulted in animals at high risk of HCC, whereas knockdown of HBx via RNA interference (RNAi) suppressed tumor growth and enhanced cisplatin chemo sensitivity in HBV-related hepatoma cells [21, 22]. Recent studies revealed that HBx could destroy the “structural maintenance of chromosomes” (Smc) complex Smc5/6, which is a host restriction factor for HBV replication, thereby enabling the virus to evade host antiviral defense [23, 24]. Because of the critical roles of HBx in promoting viral replication and HBV-related hepatocarcinogenesis, we propose HBx as a potential target for therapeutic intervention against HBV infection. However, due to the absence of a crystal structure for full-length HBx protein, development of pharmacological inhibitors targeting HBx has not been possible [25].
In this study, we investigated the feasibility of intracellular delivery of HBx mAb as a treatment against HBV infection. A cell-penetrating antibody targeting HBx (9D11-Tat) was developed via the recombinant fusion expression of a cell-penetrating peptide (CPP) on the C-terminus of the heavy chain of a potent mAb specific to HBx (9D11). Therapeutic roles and mechanisms of 9D11-Tat were systematically investigated in cell culture and mouse models.
Results
Characterization of the 9D11 mAb specific for HBx protein
Based on the binding activity evaluations of several anti-HBx mAbs generated by mouse immunization and hybridoma technology, the 9D11 mAb was selected for genetic engineering modifications and further anti-HBV tests. The 9D11 mAb is an IgG1 subtype antibody and recognizes the aa 26-43 region of HBx. Immunofluorescence (Figure 1A) and Western blot (Figure 1B) assays to detect HBx expressed by transient transfection with plasmids encoding HBx of various genotypes revealed the 9D11 mAb could bind to HBx proteins of HBV genotypes A, B, C and D. It should be noted that the transfection efficiency of Huh7 cells was ~30% in our hand; therefore, not all cells presented immunofluorescence signals in Figure 1A. Both cytoplasmic and nuclear HBx staining signals by 9D11 could be observed in cells expressing HBx of various genotypes, consistent with previous reports [19, 26]. To test whether the 9D11 mAb recognizes natural HBx in liver tissues of CHB patients, we performed immunohistochemical assays for 3 matched pair tissue sections (both adjacent non-tumor and tumor tissue derived from the same patient) of CHB related HCC patients (patient 1-3) by using 9D11 and 83H12 (an anti-HBsAg mAb previously described) [27]. As the results show in Figure 1C, the expressions of HBx and HBsAg were correlated in the 3 patients. Strong HBx staining signals (by 9D11) were observed in non-tumor hepatocytes of all 3 patients, whereas less HBx staining was noted in paired tumor sections, particularly in that of patient 2 and 3 (Figure 1C). Previous studies suggested that the random integration of HBV genome may led to the C-terminal truncation of HBx (HBx-ΔC, usually loss the C-terminal 27-30 aa) [28]. The HBx-ΔC frequently expressed in HBV-associated liver tumors, and play a important role in facilitating hepatocarcinogenesis [29, 30]. Therefore, we further tested the reactivity of 9D11 in detections of natural HBx mutants via Western blot analyses of cell lysates of matched pair tissues derived from 5 additional CHB related HCC patients (patient 4 to 8). As the 9D11 binds an N-terminal epitope (aa 26-43) of HBx, both full-length (wt-HBx) and C-terminal truncated HBx (HBx-ΔC) were recognized by the mAb as expected (Figure 1D). Among 4 of 5 patients (except patient 8), the levels of wt-HBx were significantly higher in non-tumor samples than that in tumors (Figure 1D), which was consistent with a previous report [29]. Notably, in tumour samples of patient 4 and 7, only the HBx-ΔC band was observed whereas the wt-HBx band was absent. Overall, these results demonstrated the 9D11 mAb reacts well with natural HBx protein and its C-terminal truncated mutant in human hepatocytes with HBV infection.
Characterization of the 9D11 mAb specific for HBx protein. Immunofluorescence (A) and Western blot (B) detection of intracellular HBx of genotypes A, B, C, and D in Huh7 cells transfected with HBx-expressing plasmids using the 9D11 mAb. For Western blots, GFP (derived from co-transfected plasmid of pcDNA3.1-GFP) served as a transfection control and β-tubulin served as a sample loading control. Relative HBx-expressing levels of various plasmids were determined by ImageJ and normalized with β-tubulin. The scale bar is 20 µm for the immunofluorescence images. (C) Immunohistochemical analyses of HBx (by 9D11) and HBsAg (by 83H12) in liver tissue samples from patients with CHB related HCC. A total of 3 matched pair tissue sections (both adjacent paracancerous and tumor tissue derived from the same patient) of HBV-positive HCC patients were analyzed. The scale bar is 50 µm. Red arrows indicate representative HBx-stained cells. (D) Western blot analyses of intracellular HBx (by 9D11) and HBsAg (by 83H12) in 5 human CHB-related liver tumor samples and their surrounding non-tumor liver samples. T represents tumor samples; NT represents their surrounding non-tumor samples. Wt-HBx represents the full-length HBx; HBx-ΔC represents C-terminal truncated HBx. The unglycosylated small-HBsAg is p24 and its N-glycosylated form is gp27. (E) Western blot analyses of antibody expressions of Huh7 cells transfected with 9D11 and Ctr-Ab expressing plasmids. Relative expressing levels of antibody H-Chain and L-Chain of the two plasmids were determined by ImageJ and normalized with β-tubulin. (F) Analyses of HBV suppression effect of plasmid transfection-mediated intracellular expression of 9D11 mAb in HBV-transfected Huh7 cells. The levels of various viral antigens (HBsAg, HBeAg, and HBcAg) in supernatants and cell lysates were quantitatively measured by commercial assays. The results represented the fold changes of viral antigen levels from the mAb expressing plasmids-transfected cells compared with those from the vector-transfected cells. The data represented mean ± SD from three independent experiments.
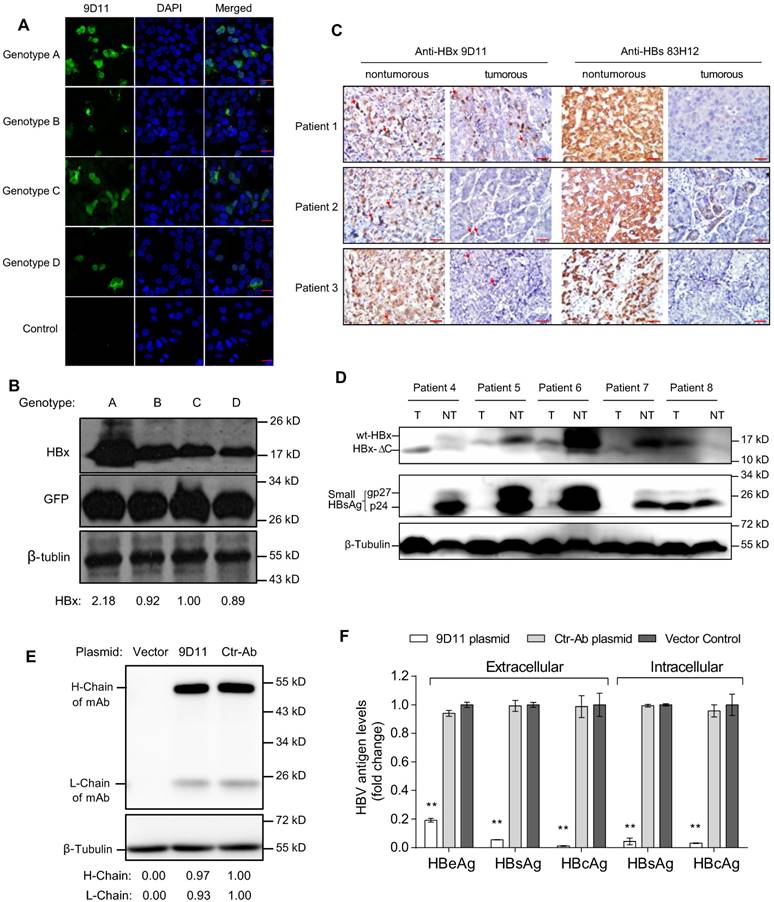
To investigate the potential anti-HBV effect of intracellular expression of 9D11 mAb, we constructed plasmids containing full-length 9D11 and control mAb (Ctr-Ab) expression cassettes. The two plasmids expressed mAbs well in Huh7 cells (Figure 1E). Compared with that of Ctr-Ab plasmid and vector plasmid, co-transfection of the 9D11 expression plasmid and replication-competent HBV genome (HBV48-WT) containing plasmid exhibited significantly reduced viral antigen levels (HBsAg, HBeAg, and HBcAg) both in culture medium and cell lysates (Figure 1F).
Development of a cell-penetrating 9D11 mAb
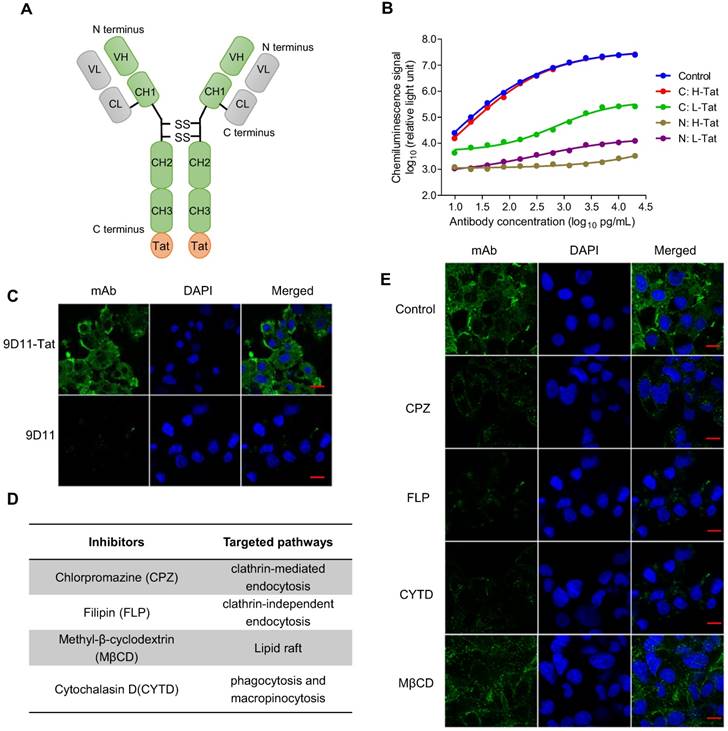
The results from a proof of concept study based on plasmid transfection (Figure 1) suggested intracellular 9D11 mAb may have a potent HBV suppression effect. However, native 9D11 mAb protein could not be internalized into live cells effectively. To enable its cell-penetrating capability, we modified the mAb via fusion expression of a cell penetrating peptide (Tat) on the N- or C-terminus of the heavy chain or light chain of 9D11 (Figure 2A). Four 9D11 variants were produced in CHO cells by transient transfection and purified by affinity chromatography. As is evident from binding tests of the 9D11 variants shown in Figure 2B, fusion of Tat on the C-terminus of the 9D11 heavy chain (hereafter referred to as 9D11-Tat) could completely preserve its HBx-binding activity. On the contrary, its HBx-binding activity was impaired by Tat fusions on the light chain or N-terminus of the heavy chain (Figure 2B). As expected, the 9D11-Tat mAb could successfully enter Huh7 cells, whereas the native 9D11 could not (Figure 2C). We further investigated the uptake pathway of 9D11-Tat by using endocytosis inhibitors (Figure 2D), including CPZ, FLP, CYTD and MβCD [31]. As displayed in Figure 2E, treatment of cells with CPZ, FLP and CYTD nearly completely blocked 9D11-Tat internalization while treating cells with MβCD showed a slight inhibition on the intracellular delivery of 9D11-Tat. These results suggested that 9D11-Tat enters cells through an endocytic pathway.
Tat-mediated efficient intracellular delivery of HBx mAb via the endocytic pathway. (A) Schematic structure of Tat-modified antibody. (B) HBx-binding activities of various Tat-modified recombinant 9D11 mAbs. A total of 4 Tat-modified mAbs were generated via fusion expression of the Tat sequence (12 aa) on the N- or C-terminus of the heavy chain (H) or light chain (L) of 9D11. The HBx-binding activities to recombinant HBx were measured by chemiluminescence enzyme immunoassay (CLEIA). The data represent the mean values from three independent experiments. Native 9D11 mAb was used as the control. (C) Immunofluorescence images of intracellular antibodies of Huh7 cells treated with 9D11-Tat and 9D11 at a concentration of 200 µg/mL. The scale bar is 20 µm. (D) Compounds that inhibit various endocytic pathways, including clathrin-mediated endocytosis inhibited by CPZ; lipid raft inhibited by MβCD; clathrin-independent endocytosis inhibited by FLP; and phagocytosis and micropinocytosis inhibited by CYTD. (E) Inhibition of intracellular delivery of 9D11-Tat by CPZ (10 µg/mL), MβCD (1.0 µg/mL), FLP (5.0 mM) and CYTD (4.0 µg/mL) treatments in Huh7 cells. The scale bar is 20 µm.
In vitro suppression of HBV by 9D11-Tat
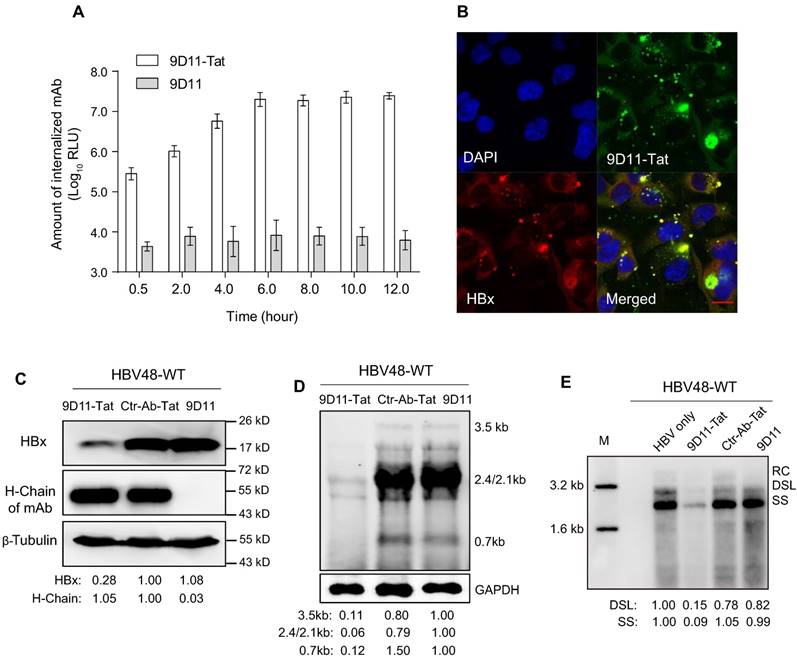
When 9D11-Tat was incubated with cells in culture medium, the internalized antibody could be detected 30-min after incubation, with maximum internalization occurring ~6 h after incubation (Figure 3A). The co-localization (Figure 3B) of the antibody and HBx in 9D11-Tat-treated cells demonstrated the well-preserved HBx binding activity of the internalized mAb. Treatment of HBV48-WT-transfected Huh7 cells with 9D11-Tat could significantly reduce the intracellular HBx protein and HBV RNA, and HBV DNA replicative intermediates as shown by Western, Northern, and Sourthern blotting, respectively (Figure 3C, 3D, and 3E). On the other hand, the levels of either HBx or HBV RNA/DNA did not change when cells were treated with Ctr-Ab-Tat (an isotype control mAb with Tat fusion on its heavy chain C-terminus) or native 9D11 mAb. A dose-effect analysis indicated that the maximum suppression effect of 9D11-Tat on HBsAg can be achieved at a dose of ≥200 µg/mL (Supplemental Figure S1A). However, a slight viral inhibition (~30%) was still observable when HBV-transfected cells were treated with 9D11-Tat at a dose as low as 25 µg/mL. No significant cytotoxicity was observed when cells were treated with 9D11-Tat at 400 µg/mL, which was 2-fold higher that that used in anti-HBV tests (Supplemental Figure S1B).
Intracellular delivery of 9D11-Tat mAb suppressed HBV transcription and replication. (A) Time-course monitoring of internalization of 9D11-Tat and 9D11 in Huh7 cells. Cells were incubated with 9D11-Tat or 9D11 at 200 µg/mL for 0.5, 2.0, 4.0, 6.0, 8.0, 10.0, and 12.0 h. At the end of incubation, cells were washed to remove residual mAbs and collected by trypsinization. The intracellular mAb levels of cell lysates were determined by CLEIA. The data represent mean ± SD from three independent experiments. (B) Immunofluorescence confocal laser scanning microscopy to assess the intracellular localizations of internalized 9D11-Tat and HBx. Huh7 cells transfected with HBV48-WT plasmid were incubated with 9D11-Tat mAb. 6 h after the incubation, the cells were fixed and immunofluorescence detections were performed. The internalized 9D11-Tat was detected by using an Alexa Fluor® 488 labelled goat anti-human antibody, whereas HBx was detected using Alexa Fluor® 594 conjugated anti-HBx antibody 20F3, which targets a different HBx epitope. (C) Intracellular HBx levels of HBV48-WT-transfected Huh7 cells after treatments of 9D11-Tat, Ctr-Ab-Tat, and 9D11 mAbs (200 µg/mL). The internalized mAbs were indicated by the heavy chains. β-tubulin served as a sample loading control. Relative levels of HBx and antibody H-Chain were determined by ImageJ and normalized with β-tubulin. (D) Northern blot and (E) Southern blot assays to assess the effect of mAb treatments on HBV RNA transcription and DNA replication. Relative levels of HBV RNAs and DNAs were determined by ImageJ and normalized with GAPDH. RC: relaxed-circular DNA; DSL: double-strand linear DNA; SS: single-stranded DNA.
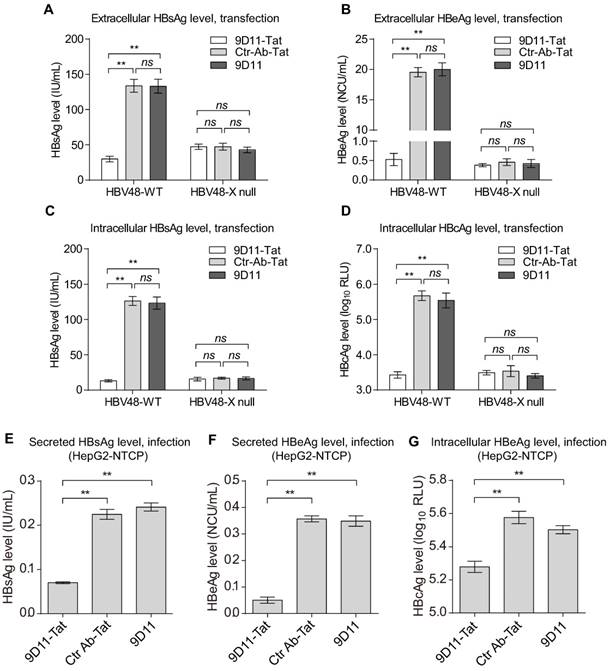
To examine whether the 9D11-Tat-mediated anti-HBV effect is indeed dependent on the presence of HBx target, we constructed an HBx-defective HBV mutant (HBV48-X null). The mutant could not produce HBx protein and possessed significantly decreased capability for HBV biosynthesis (Figure 4). Compared with Ctr-Ab-Tat mAb and native 9D11 mAb, HBV48-WT transfected Huh7 cells treated with 9D11-Tat produced substantially decreased viral antigens both in culture medium (HBsAg and HBeAg, Figure 4A and 4B) and cell lysates (HBsAg and HBcAg, Figure 4C and 4D). In contrast, for HBV48-X null transfected cells, 9D11-Tat treatment didn't change either extracellular or intracellular viral antigens compared with Ctr-Ab-Tat mAb or native 9D11 treatment. These results suggested that 9D11-Tat suppresses HBV in an HBx-dependent manner. We further used HepG2 cells expressing the HBV receptor sodium taurocholate cotransporting polypeptide (NTCP) to evalute the activity of 9D11-Tat in decreasing viral antigen levels in HBV-infected cells. The HepG2-NTCP cell line supports in vitro HBV infection, thus providing a more physiologically relavant model to test the effects of 9D11-Tat in the context of the entire virus life cycle. In HBV-infected HepG2-NTCP cells, HBx was expressed at levels too low for detection due to the low HBV infection efficiency in this system [32]. However, treatment of the cells with 9D11-Tat still exhibited significant inhibition on the levels of HBsAg (Figure 4E), HBeAg (Figure 4F), and HBcAg (Figure 4G).
Intracellular delivery of 9D11-Tat mAb suppressed the expression of HBV proteins. Quantitative analyses for extracellular HBsAg (A), extracellular HBeAg (B), intracellular HBsAg (C) and HBcAg (D) of the HBV plasmid-transfected Huh7 cells treated with 9D11-Tat, Ctr-Ab-Tat, and 9D11 mAbs (200 µg/mL). HBV48-X null is an HBx-defective HBV mutant. The suppression effects of the mAbs were also investigated in HepG2-NTCP cells that support in vitro HBV infection. The secreted HBsAg (E), secreted HBeAg (F), and intracellular HBcAg (G) of HBV-infected HepG2-NTCP cells treated with 9D11-Tat, Ctr-Ab-Tat, and 9D11 mAbs (200 µg/mL) were analyzed at day 8 post infection (2 days after treatment). The data represented mean± SD from three independent experiments. p values were calculated using a two-sided unpaired t test, ** indicates p<0.01 and ns indicates p>0.05.
The role of TRIM21 in 9D11-Tat-mediated HBV suppression
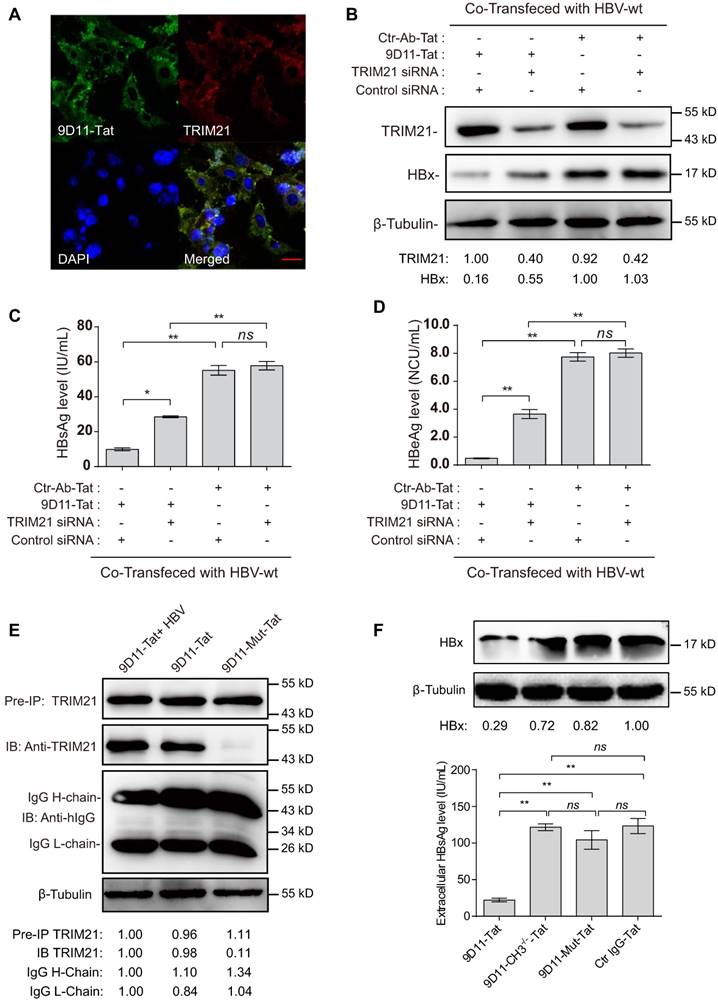
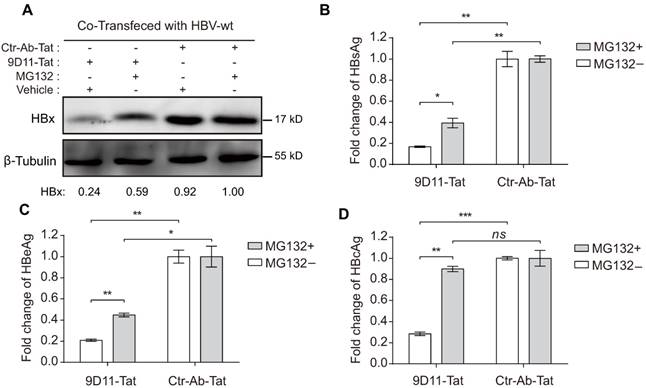
Previous studies suggested that the Fc receptor TRIM21 is essential to initiate clearance of intracellular antibody-bound pathogens [9]. Therefore, we investigated the role of TRIM21 in 9D11-Tat-mediated HBV suppression. In 9D11-Tat treated cells, the 9D11-Tat colocalized with endogenous TRIM21, suggesting an association between them in the cytoplasm (Figure 5A). To test the requirement of endogenous TRIM21 for 9D11-Tat-mediated HBV suppression, we first examined the effect of TRIM21 gene silencing in HBV transfection assay. Huh7 cells were co-transfected with TRIM21-specific or a control small inferfering RNA (siRNA) and HBV48-WT. When TRIM21 protein level was reduced to ~30% in TRIM21 siRNA-tranfected cells (Figure 5B), the 9D11-Tat-mediated suppression effects on the levels of HBx (Figure 5B), HBsAg (Figure 5C), and HBeAg (Figure 5D), were partially reversed in these cells compared to those transfected with control siRNA. As 9D11 recognizes an epitope in the HBx aa 26-43 region, which is located in the regulatory domain (aa 1-50) of HBx [33], but not in its trans-activation domain (aa 51-154) [34], it was reasonable that intracellular binding of HBx with 9D11 may not impaire its HBV promoting function in the absence of TRIM21. On the other hand, in the presence of Ctr-Ab-Tat treatment, TRIM21 knockdown did not show any inhibition on viral protein levels (Figure 5B, 5C, and 5D), suggesting that that TRIM21 does not inhibit HBV at its physiological level when intracellular anti-HBV anibody is absent. Coimmunoprecipitation experiments (Figure 5E) revealed that 9D11-Tat interacts with TRIM21, regardless of the presence of HBV. The interaction between 9D11-Tat and TRIM21 could be abolished by introducing the H433A, N434A and H435A mutations [35] into the CH3 domain of the 9D11 Fc region (denoted 9D11-Mut-Tat) (Figure 5E). Functional assays demonstrated that the introduction of the triple-alanine CH3 mutation or CH3 deletion mutation into 9D11-Tat resulted in the loss of HBV suppression effects both to the levels of HBx (Figure 5F, upper panel) and HBsAg (Figure 5F, lower panel). We also performed promoter luciferase reporter assays to test whether 9D11-Tat treatment activated intracellular immune signaling pathways. The results suggested the promoter activities of NF-kB, AP-1, and IFN-β were significantly stimulated in HBV-transfected cells after 9D11-Tat treatment compared to the cells treated by control mAbs (Supplemental Figure S2). Although the 9D11-Tat-stimulated IFN-β was too low to be detected at the protein level, a neutralizing anti-IFN-β antibody could partially block the suppression effects of 9D11-Tat on viral proteins (Supplemental Figure S3), suggesting that the activation of IFN-β in this process may contribute contribute to the anti-HBV effects of 9D11-Tat. Various reports suggested that TRIM21 mediates antibody coated viruses for degradation in the proteasome, which is essential for the process of TRIM21-mediated antibody-dependent intracellular neutralization (ADIN) [8, 9, 36]. We therefore investigated the association between 9D11-Tat mediated HBV suppression effect and proteasome activity. As expected, addition of proteasome inhibitor MG132 prevented HBx degradation (Figure 6A) and significantly restored the levels of HBsAg (Figure 6A), HBeAg (Figure 6B), and HBcAg (Figure 6C) in 9D11-Tat-treated cells. These results showed that TRIM21-mediated ADIN and activations of innate antiviral pathways may both contribute to 9D11-Tat-mediated HBV suppression.
9D11-Tat suppressed HBV by TRIM21-dependent pathway (A) Immunofluorescence confocal laser scanning microscopy to assess the intracellular localizations of internalized 9D11-Tat and TRIM21. (B) 9D11-Tat-mediated inhibitions on HBx (B), HBsAg (C), and HBeAg (D) were reduced by TRIM21 knockdown. (C) The interaction between 9D11-Tat and TRIM21 was HBV-independent, and could be abolished via introduction of mutations in the CH3 domain of the 9D11 Fc region (9D11-Mut-Tat). (D) TRIM21 binding-defected 9D11-Tat mutants (9D11-Mut-Tat and 9D11-CH3-/--Tat) lost HBV suppression effects. For Western blots, relative levels of all detection targets were determined by ImageJ and normalized with β-tubulin. For (C), (D), and (F), the data represent mean ± SD from three independent experiments. p values were calculated using a two-sided unpaired t test, ** indicates p<0.01, * indicates p<0.05 and ns indicated p>0.05.
MG132 decreased the HBV suppression effects of 9D11-Tat. Proteasome inhibitor (MG132) treatment (2 µM) reduced the HBV suppression effects of 9D11-Tat in HBV-transfected Huh7 cells on the levels of intracellular HBx (A), secreted HBsAg (B), secreted HBeAg (C), and intracellular HBcAg (D). Relative HBx levels in (A) were determined by ImageJ and normalized with β-tubulin. For (B), (C), and (D), the data represent mean ± SD from three independent experiments. p values were calculated using a two-sided unpaired t test, ** indicates p<0.01, * indicates p<0.05, and ns indicates p>0.05.
In vivo suppression of HBV by 9D11-Tat
We evaluated the in vivo therapeutic effects of 9D11-Tat in a hydrodynamic injection (HDI)-based HBV mouse model. For this, mice received a single intravenous infusion of 9D11-Tat or Ctr-Ab-Tat at a dose of 10 mg/kg at 6 h post hydrodynamic injection of HBV plasmids (HBV48-WT or HBV48-X null). Serum and liver samples of mice were analyzed for viral markers at 4 days post injection (dpi 4). Similar to the observations from in vitro studies, infusion of 9D11-Tat significantly reduced serum levels of HBsAg (average decline by 90%) and HBeAg (average decline by 75%) in mice that received HBV48-WT plasmid compared with those treated with Ctr-Ab-Tat (Figure 7A and 7B). In mice that received HBV48-X null plasmid, no significant difference in serum HBV antigen levels was observed between groups treated with 9D11-Tat and Ctr-Ab-Tat. Viral DNA from liver samples of mice that received HBV48-WT plasmid was isolated and subjected to Southern blot analysis. The average levels of replicative HBV DNA intermediates (including RC/DSL DNA and SS DNA) in livers of mice receiving infusion of 9D11-Tat were reduced to 20-50% of that of mice receiving infusion of Ctr-Ab-Tat (Figure 7C and 7D).
9D11-Tat suppressed HBV in mice. Evaluation of the HBV suppression effects of 9D11-Tat in mice on the levels of serum HBsAg (A) and HBeAg (B) at 4 days after hydrodynamic injections of HBV48-WT and HBV-X null plasmids. Antibodies were used at a dosage of 10 mg/kg. The data are expressed as the mean ± SD (n=4 per group). (C) Southern blot assays for intrahepatic levels of HBV DNA after hydrodynamic injection of HBV48-WT; mice received mAb treatments (4 days after mAb infusion). (D) Semi-quantitative analyses of HBV replicative intermediates from data in (C), normalized by the input DNA band. (E-I) Effects of mAb treatment in HBV transgenic mice (n=4 per group). Serum HBV DNA (E), HBsAg (F), creatinine (G), and ALT (H) profiles of HBV-Tg mice after a single infusion of mAbs (9D11-Tat and Ctr-Ab-Tat, 10 mg/kg) or daily oral regimen of entecavir (3.2 mg/kg). The data are expressed as the mean± SD.
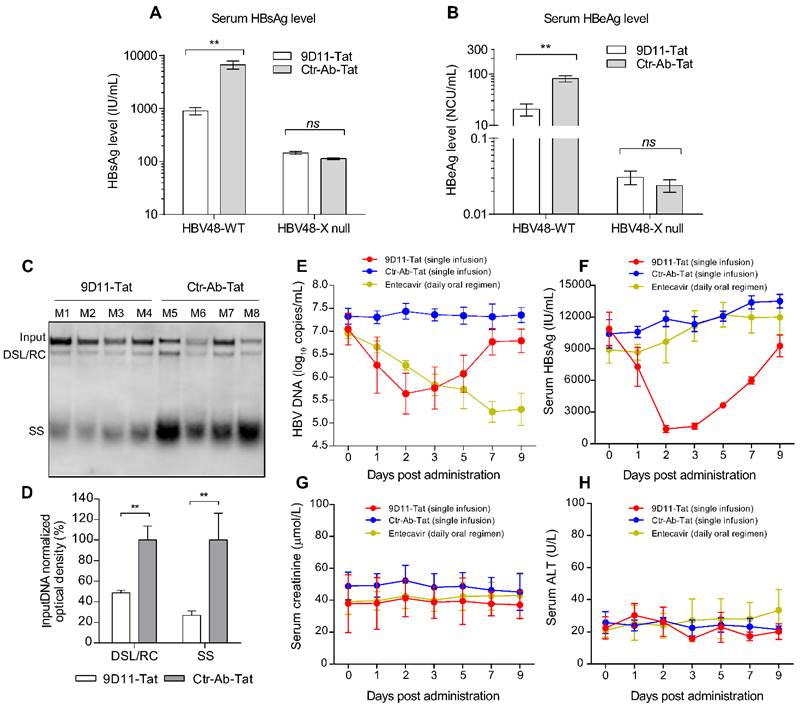
To further validate the therapeutic potential of 9D11-Tat, we performed treatment experiments in HBV-transgenic (HBV-Tg) mice that persistently expressed high serum levels of HBV DNA and HBsAg. Significant decreases in serum HBV DNA (Figure 7D) and HBsAg (Figure 7E) were observed in mice that received a single infusion of 9D11-Tat since dpi 2, whereas no viremia suppression was detected in mice that received Ctr-Ab-Tat. However, a serum virological rebound occurred in most 9D11-Tat-treated mice between dpi 7 and dpi 9. We also compared the anti-HBV effects by single 9D11-Tat infusion with a daily regimen of oral entecavir. The advantage of 9D11-Tat was evident by its parallel suppression effects on HBsAg and HBV DNA, whereas entecavir did not decrease the HBsAg level in HBV-Tg mice (Figure 7E and 7F). No significant elevation was observed in monitoring of the serum levels of creatinine (Figure 7G) and ALT (Figure 7H) of mice during treatment, suggesting a low risk of 9D11-Tat treatment for causing kidney or liver injury. Furthermore, we investigated the bio-distribution profiles of intravenously-administered 9D11-Tat in HBV-Tg mice. The mAbs of 9D11-Tat and 9D11 were labeled with DyLight 680 fluorophore, infused at 5 mg/kg, and visualized at 24 h after injection with an in vivo imaging system to reveal their distribution in both living mice and isolated organs. As the results show in Supplemental Figure S4A, the two mAbs mainly accumulated in the liver and lung. Compared to mice that received 9D11-fluorophore, the animals that received 9D11-Tat-fluorophore showed slightly higher fluorescence signals both for whole-body and isolated organs (Supplemental Figure S4B and S4C), possibly attributed to the cell-penetrating capacity of 9D11-Tat that may increase tissue retention time of the molecule.
Discussion
During the past several years, numerous methods have been developed for the intracellular delivery of antibody fragments with binding activities (Fab and ScFv). These approaches include direct intracellular expression by viral vectors carrying antibody genes, liposome or polymer-based protein transfection systems, and chemical conjugation or genetic fusion of antibodies with cell-penetrating peptides (CPPs) [37]. However, efficient intracellular delivery of a whole molecule antibody is still a challenge. Recent studies have suggested the importance of the Fc region of antibodies in immunomodulation, particularly in recruiting intracellular Fc receptor (TRIM21) to mediate ADIN by proteasome degradation and activate innate immune signaling [8, 9, 36]. Therefore, development of technologies that can efficiently deliver the Fc-containing whole molecule antibody into living cells may greatly expand the potential uses of antibody-based therapeutics.
In this study, we investigated the feasibility of genetically engineering whole molecule monoclonal antibody with efficiently cell-penetrating capability. We engineered anti-HBx 9D11 mAb by recombinant fusion expression of Tat CPP on the C-terminus of the heavy chain, which enabled 9D11 to be shuttled into living cells without affecting its antigen binding activity and Fc-mediated function. Based on an identical strategy, we had successfully generated another cell-penetrating anti-HBs mAb (data not shown). Chemical conjugation of CPP to antibodies, usually results in a heterogeneous mixture containing species with both less and more CPP loading than desired, and requires further purification [38]. Recombinant expression of the genetically modified antibody would be ideal for large-scale production of homogeneous products with more reproducible pharmacological properties. Therefore, our approach represents a potential universal strategy for producing engineered cell-penetrating whole molecule antibodies with little influence on their functional characteristics.
The cell-penetrating mAbs provided robust tools to achieve specific functional depletion of targeted intracellular proteins, and, therefore presented an alternative approach to develop therapeutic antibodies against intracellular targets. The feasibility of this strategy was investigated in our study by the therapeutic use of 9D11-Tat mAb against HBV infection in both cell culture and mouse models. Despite the successful development and clinical application of current antiviral treatments based on pegylated interferon, or nucleos(t)ide analogues, which have demonstrated clinical benefit in chronic hepatitis B patients resulting in liver disease remission, persistent HBV infection remains a difficult-to-cure disease [39, 40]. As the HBx protein plays an essential role in promoting viral transcription, replication, and evasion from host innate antiviral defense, specific depletion of intracellular HBx may facilitate HBV eradication and reduce the risk of HBV-related hepatocarcinogenesis. Although HBx-deficient HBV was still lowly replicative in the HBV-plasmid transfection-based system (Figure 3 and Figure 7), several studies have demonstrated that HBx is indispensably required to initiate and maintain HBV replication after infection in HepaRG cells [18] or in human hepatocyte chimeric mice [41], and highlight HBx as the key regulator during the natural HBV infection process. Given the low expression level but indispensable role of HBx in the context of HBV infection, we believed that HBx is a potential cost-effective antiviral drug target. However, the development of HBx specific inhibitor was limited because of the absence of high-resolution protein structure. Unlike small molecule protein inhibitors, whose development is largely dependent on the presence of a protein structure, the mAbs against defined proteins are easier to produce via hybridoma or phage display techniques when the structure data is absent. Our studies revealed that a cell-penetrating HBx antibody (9D11-Tat) could reduce intracellular HBx via Fc binding receptor TRIM21-mediated protein degradation (Figure 5), and therefore significantly suppressed HBV transcription, replication, and viral antigen production both in vitro (Figure 3 and 4) and in vivo (Figure 7). The activation of TRIM21 by antibody-antigen immune complex simultaneously stimulated the immune signaling pathways of NF-κB and AP-1 and production of IFN-β, promoting an antiviral state of the host cells and further expanding the anti-HBV effects (Supplemental Figure S2 and S3) [9]. To our knowledge, this is the first proof-of-concept study for treatment of HBV infection via antibody-mediated specific targeting of intracellular HBx, and may provide a new avenue for development of novel therapeutic strategies.
It should be noted that several previous studies suggested Tat peptide can deliver various protein cargos into practically all cells [42, 43]. In vivo intracellular delivery of 9D11-Tat may not be hepatocyte-specific. However, our in vivo imaging results suggested 9D11-Tat predominantly accumulated in the liver and lung in HBV-Tg mice received the intravenous mAb infusion (Supplemental Figure S4), which may facilitate its in vivo HBV suppression effect. Possible strategies to develope tissue and/or cell-specific targeting of cell-penetrating antibodies may be achieved in future via CPP modification on a bispecific antibody with one arm binds to a cell-specific surface marker and the other arm binds to an intracellular target (such as HBx).
In summary, our study provided a novel strategy for developing a cell-penetrating whole molecule antibody. Degradation of the intracellular HBx protein by the engineered 9D11-Tat mAb exhibited potent antiviral activity against HBV infection both in vitro and in vivo. Our results suggest that the 9D11-Tat is an excellent therapeutic antibody candidate for further improving CHB treatment. Further studies should focus on advancing cell- and/or tissue- specific intracellular mAb delivery with high efficiency, which will further promote the therapeutic application of cell-penetrating mAb in humans.
Materials and Methods
Antibodies and plasmids
The 9D11 mAb against HBx protein was generated using standard hybridoma technology via immunization of Balb/C mice using E.Coli derived recombinant HBx. To construct recombinant mAb expression plasmids, the variable genes of the cell clones of 9D11 and 16G12 (an isotype control mAb specific to the HIV-1 p24 protein, denoted Ctr-Ab) were obtained from the hybridoma by reverse-PCR using a mouse Ig primer set (Merck millipore, Darmstadt, Germany). The heavy chain and kappa chain of the two mAbs were cloned into a pTT5 vector containing the constant region of mouse IgG1. To construct Tat-modified recombinant mAbs, the coding sequence of the cell-penetrating peptide of HIV Tat-1 protein (aa48-60) [44] was fused on the N- or C-terminus of the heavy chain or light chain of 9D11 with a (GGGGS)2 linker. The 9D11-Mut-Tat variant was constructed through introducing H433A, N434A and H435A mutations in the Fc region of the mAb using site-directed mutagenesis. The 9D11-CH3-/--Tat variant was constructed through deletion of the CH3 domain coding sequence of Fc using PCR overlap extension. The detailed amino acid sequences of 9D11 mAb and its variants are listed in Supplemental Table S1. The recombinant antibodies were produced in CHO cells and purified using Mabselect Xtra affinity chromatography (Amersham GE Health, Uppsala, Sweden) from culture media after transient transfection, as previously described [45]. The anti-TRIM21 antibody was purchased from Abcam. The plasmids of HBV48-WT and HBV48-X null plasmids that contained 1.2 copies of the HBV genome of genotype Ae with wildtype X expression cassette (HBV48-WT) or X-deletion mutation (HBV48-X null) were provided by professor Pei-Jer Chen (National Taiwan University) [46]. HBx-expressing plasmids of HBV genotypes A, B, C, and D were constructed through cloning of the entire HBx cassette of various HBV strains into pcDNA3.1 vector [47]. Luciferase reporter plasmids for activities of NF-κB, AP-1, and IFN-β were purchased from Genomeditech.
Virological and immunological assays
To determine the HBx-binding activities of 9D11 mAbs, a chemiluminescence enzyme immunoassay (CLEIA) was used. Briefly, wells were coated with recombinant HBx at a concentration of 100 ng per well, and nonspecific binding was blocked with PBS containing 2% (vol/vol) bovine serum albumin (BSA) and 10% sucrose. A series of 2-fold dilutions that ranged from 10 to 20,000 pg/mL of mAbs were prepared. 100 μL of specimens were added to the reaction wells and incubated for 1 h at 37°C, followed by washing and reaction with horseradish peroxidase (HRP)-conjugated anti-mouse pAb (Thermo Scientific, Rockford, USA). The relative light unit (RLU) was evaluated using an Orion II Microplate Luminometer (Berthold, Germany) following the addition of SuperSignal ELISA Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, USA). Immunofluorescence and Western blot analyses for intracellular antibodies, HBx, and TRIM21 were performed according to standard procedures. Quantitative analyses of HBV DNA, HBsAg, HBeAg and HBcAg were detected using commercial kits (Wantai, Beijing, China). For detection of HBcAg in secreted virions in cell culture medium, supernatants (50μL) were treated with a suitable lysis buffer (50μL, 20 mM HEPES, 0.5% (vol/vol) Triton X-114, 100 mM NaCl, pH 8.0) at room temperature for 30 min, then the mixture was subjected for immunoassay quantification. Southern and Northern blots were performed according to the methods described previously using DIG-labelled DNA fragments from the X gene as a probe [45]. Coimmunoprecipitation experiments were performed using a rabbit anti-TRIM21 antibody (Abcam) and Protein G Sepharose beads (GE Healthcare) according to the protocol described previously. To test the reactivity of 9D11 to HBx in HBV-infected human liver samples, 8 matched pair tissue sections (both adjacent non-tumorous and tumorous tissues derived from same patient) from HBV-positive HCC patients (patient 1-8) were collected from Zhongshan Hospital of Xiamen University. The liver tissue samples from patient 1, 2, and 3, were fixed in 10% (vol/vol) buffered formalin and embedded in paraffin. Immunohistochemical analyses were performed using 9D11 (for HBx, 4 µg/mL) and 83H12 (for HBsAg, 1 µg/mL) according the staining protocol previously described [48]. The liver tissue samples from other patients (patient 4-8) were lysed in RIPA buffer (Sigma). Western blot assays were performed using 9D11 (for HBx, 1 µg/mL) and 83H12 (for HBsAg, 0.5 µg/mL). The experiments conformed to the 1975 Declaration of Helsinki and were approved by the Ethics Committee of Zhongshan Hospital of Xiamen University. Written informed consent was obtained from all subjects.
Cell and animal studies
Huh7 cells were grown in DMEM supplemented with 10% (vol/vol) FBS (Thermo Scientific, Rockford, USA). Plasmid transfections were performed using X-tremeGENE HP DNA Transfection Reagent (Roche Applied Science, Mannheim, Germany). Inhibitors for endocytosis including Chlorpromazine (CPZ), Filipin (FLP), Cytochalasin D (CYTD) and Methyl-beta-cyclodextrin (MβCD) [31] were purchased from Sigma (Sigma, St. Louis, MO). For transient siRNA knockdown experiments, cells (2 × 105 cells per well) in 6-well plates were transfected with siRNA oligonucleotides of T21siRNA1 and T21siRNA2 (150 pmol each, the sequences of two siRNA are listed in Supplemental Table 1) or 300 pmol of control oligo by using Oligofectamine transfection reagent (Thermo Scientific, Rockford, USA). For mAb treatments in HBV-transfection system, HBV plasmid-transfected Huh7 cells were incubated with 9D11-Tat or control mAbs at 200 µg/mL in culture medium for 48 h, then the culture medium and cell lysates were collected for analyses. NTCP-overexpressing HepG2 cells (HepG2-NTCP) that support in vitro HBV infection were developed as reported in our previous study [49]. HepAD38 cell line with tetracycline-regulated HBV DNA replication was used as the source of HBV [50]. Briefly, HepAD38 cells were induced to produce HBV virions by tetracycline removal from the culture medium. The HBV-containing culture supernatants were collected and concentrated by precipitation with 5% PEG. For infection assay, HepG2-NTCP cells were incubated overnight with HBV at a multiplicity of 100 genome equivalents per cell in the presence of 4% PEG8000 (Sigma), followed by a wash. The culture medium was then refreshed every 2 days. At day 6 post infection, the cells were incubated with 9D11-Tat or control mAbs at 200 µg/mL in culture medium. Two days after mAb treatments, the supernatants and cell lysates were collected for analyses.
All mice were maintained under specific pathogen-free conditions in the Laboratory Animal Center of Xiamen University. HBV-Tg mice were provide by professor Pei-Jer Chen (National Taiwan University). The characteristics of HBV-Tg mice had been described in our previous studies [45]. To generate hydrodynamic injection-based HBV mice, 25 μg of HBV plasmid was injected into the tail veins of C57BL/6 mice within 5 s in a volume of PBS equivalent to 10% of the mouse body weight. For anti-HBV treatment of HBV mice, antibody (10 mg/kg) was intravenously injected into each animal. The mice were bled via orbital punctures at the baseline and indicated time points after treatment. Mouse serum specimens taken from different time points were kept at -80°C until use. For in vivo fluorescence imaging, HBV-Tg mice were intravenously injected with 9D11-Tat-DyLight 680 or 9D11-DyLight 680 (5mg/kg). One day after injection, the fluorescence signal of DyLight 680 was recorded by IVIS Lumina II system (Caliper Life Sciences, USA) in animals under isoflurane anaesthesia. Then the mice were sacrificed, and the major organs (heart, liver, spleen, lung, and kidney) were isolated and examined. All animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 7.0 (San Diego, CA, USA). The unpaired t-test was used to compare continuous variables. Differences were considered significant at a a two-sided p<0.05.
Abbreviations
HBV: hepatitis B virus; CHB: chronic HBV infection; HCC: hepatocellular carcinoma; HBx: hepatitis B x protein; HBsAg: hepatitis B surface antigen; HBeAg: hepatitis B e antigen; HBcAg: hepatitis B core antigen; HBV-Tg: HBV transgenic mouse; ADIN: antibody-dependent intracellular neutralization; CPP: cell penetrating peptide.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
National Natural Science Foundation of China (81672023, 81502685), Excellent Youth Foundation of Fujian Scientific Committee (2015J06018), Natural Science Foundation of Guangdong Province (2016A030310179) and China Postdoctoral Science Foundation (2016M602596) supported this work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Marasco WA, Sui J. The growth and potential of human antiviral monoclonal antibody therapeutics. Nat Biotechnol. 2007;25:1421-34
2. Leavy O. Therapeutic antibodies: past, present and future. Nature reviews Immunology. 2010;10:297
3. Weiner GJ. Building better monoclonal antibody-based therapeutics. Nature reviews Cancer. 2015;15:361-70
4. Gura T. Therapeutic antibodies: magic bullets hit the target. Nature. 2002;417:584-6
5. Kaiser PD, Maier J, Traenkle B, Emele F, Rothbauer U. Recent progress in generating intracellular functional antibody fragments to target and trace cellular components in living cells. Biochim Biophys Acta. 2014;1844:1933-42
6. Rinaldi AS, Freund G, Desplancq D, Sibler AP, Baltzinger M, Rochel N. et al. The use of fluorescent intrabodies to detect endogenous gankyrin in living cancer cells. Exp Cell Res. 2013;319:838-49
7. Lin JJ, Feramisco JR. Disruption of the in vivo distribution of the intermediate filaments in fibroblasts through the microinjection of a specific monoclonal antibody. Cell. 1981;24:185-93
8. Bai Y, Ye L, Tesar DB, Song H, Zhao D, Bjorkman PJ. et al. Intracellular neutralization of viral infection in polarized epithelial cells by neonatal Fc receptor (FcRn)-mediated IgG transport. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18406-11
9. McEwan WA, Tam JC, Watkinson RE, Bidgood SR, Mallery DL, James LC. Intracellular antibody-bound pathogens stimulate immune signaling via the Fc receptor TRIM21. Nature immunology. 2013;14:327-36
10. Song Z, Liu L, Wang X, Deng Y, Nian Q, Wang G. et al. Intracellular delivery of biomineralized monoclonal antibodies to combat viral infection. Chem Commun (Camb). 2016;52:1879-82
11. Veomett N, Dao T, Scheinberg DA. Therapeutic antibodies to intracellular targets in cancer therapy. Expert Opin Biol Ther. 2013;13:1485-8
12. Guo K, Li J, Tang JP, Tan CP, Hong CW, Al-Aidaroos AQ. et al. Targeting intracellular oncoproteins with antibody therapy or vaccination. Science translational medicine. 2011;3:99ra85
13. Dao T, Yan S, Veomett N, Pankov D, Zhou L, Korontsvit T. et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Science translational medicine. 2013;5:176ra33
14. Schweitzer A, Horn J, Mikolajczyk RT, Krause G, Ott JJ. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet (London, England). 2015;386:1546-55
15. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V. et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet (London, England). 2012;380:2095-128
16. Kwon H, Lok AS. Hepatitis B therapy. Nat Rev Gastroenterol Hepatol. 2011;8:275-84
17. Trepo C, Chan HL, Lok A. Hepatitis B virus infection. Lancet (London, England). 2014;384:2053-63
18. Lucifora J, Arzberger S, Durantel D, Belloni L, Strubin M, Levrero M. et al. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. Journal of hepatology. 2011;55:996-1003
19. Cha MY, Ryu DK, Jung HS, Chang HE, Ryu WS. Stimulation of hepatitis B virus genome replication by HBx is linked to both nuclear and cytoplasmic HBx expression. J Gen Virol. 2009;90:978-86
20. Geng X, Huang C, Qin Y, McCombs JE, Yuan Q, Harry BL. et al. Hepatitis B virus X protein targets Bcl-2 proteins to increase intracellular calcium, required for virus replication and cell death induction. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:18471-6
21. Cheng AS, Wong N, Tse AM, Chan KY, Chan KK, Sung JJ. et al. RNA interference targeting HBx suppresses tumor growth and enhances cisplatin chemosensitivity in human hepatocellular carcinoma. Cancer Lett. 2007;253:43-52
22. Lin WH, Yeh SH, Yang WJ, Yeh KH, Fujiwara T, Nii A. et al. Telomerase-specific oncolytic adenoviral therapy for orthotopic hepatocellular carcinoma in HBx transgenic mice. Int J Cancer. 2013;132:1451-62
23. Decorsiere A, Mueller H, van Breugel PC, Abdul F, Gerossier L, Beran RK. et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature. 2016;531:386-9
24. Murphy CM, Xu Y, Li F, Nio K, Reszka-Blanco N, Li X. et al. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep. 2016;16:2846-54
25. Slagle BL, Andrisani OM, Bouchard MJ, Lee CG, Ou JH, Siddiqui A. Technical standards for hepatitis B virus X protein (HBx) research. Hepatology. 2015;61:1416-24
26. Forgues M, Marrogi AJ, Spillare EA, Wu CG, Yang Q, Yoshida M. et al. Interaction of the hepatitis B virus X protein with the Crm1-dependent nuclear export pathway. J Biol Chem. 2001;276:22797-803
27. Huang CH, Yuan Q, Chen PJ, Zhang YL, Chen CR, Zheng QB. et al. Influence of mutations in hepatitis B virus surface protein on viral antigenicity and phenotype in occult HBV strains from blood donors. Journal of hepatology. 2012;57:720-9
28. Ma NF, Lau SH, Hu L, Xie D, Wu J, Yang J. et al. COOH-terminal truncated HBV X protein plays key role in hepatocarcinogenesis. Clin Cancer Res. 2008;14:5061-8
29. Li W, Li M, Liao D, Lu X, Gu X, Zhang Q. et al. Carboxyl-terminal truncated HBx contributes to invasion and metastasis via deregulating metastasis suppressors in hepatocellular carcinoma. Oncotarget. 2016;7:55110-27
30. Quetier I, Brezillon N, Revaud J, Ahodantin J, DaSilva L, Soussan P. et al. C-terminal-truncated hepatitis B virus X protein enhances the development of diethylnitrosamine-induced hepatocellular carcinogenesis. J Gen Virol. 2015;96:614-25
31. Dutta D, Donaldson JG. Search for inhibitors of endocytosis: Intended specificity and unintended consequences. Cellular logistics. 2012;2:203-8
32. Li J, Zong L, Sureau C, Barker L, Wands JR, Tong S. Unusual Features of Sodium Taurocholate Cotransporting Polypeptide as a Hepatitis B Virus Receptor. J Virol. 2016;90:8302-13
33. Murakami S, Cheong JH, Kaneko S. Human hepatitis virus X gene encodes a regulatory domain that represses transactivation of X protein. J Biol Chem. 1994;269:15118-23
34. Tang H, Delgermaa L, Huang F, Oishi N, Liu L, He F. et al. The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J Virol. 2005;79:5548-56
35. James LC, Keeble AH, Khan Z, Rhodes DA, Trowsdale J. Structural basis for PRYSPRY-mediated tripartite motif (TRIM) protein function. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6200-5
36. Mallery DL, McEwan WA, Bidgood SR, Towers GJ, Johnson CM, James LC. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19985-90
37. D'Astolfo DS, Pagliero RJ, Pras A, Karthaus WR, Clevers H, Prasad V. et al. Efficient intracellular delivery of native proteins. Cell. 2015;161:674-90
38. Marschall AL, Frenzel A, Schirrmann T, Schungel M, Dubel S. Targeting antibodies to the cytoplasm. MAbs. 2011;3:3-16
39. Chen YC, Liaw YF. Pharmacotherapeutic options for hepatitis B. Expert Opin Pharmacother. 2016;17:355-67
40. Brahmania M, Feld J, Arif A, Janssen HL. New therapeutic agents for chronic hepatitis B. Lancet Infect Dis. 2016;16:e10-21
41. Tsuge M, Hiraga N, Akiyama R, Tanaka S, Matsushita M, Mitsui F. et al. HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J Gen Virol. 2010;91:1854-64
42. Toro A, Grunebaum E. TAT-mediated intracellular delivery of purine nucleoside phosphorylase corrects its deficiency in mice. J Clin Invest. 2006;116:2717-26
43. Bolhassani A, Jafarzade BS, Mardani G. In vitro and in vivo delivery of therapeutic proteins using cell penetrating peptides. Peptides. 2017;87:50-63
44. Schwarze SR, Hruska KA, Dowdy SF. Protein transduction: unrestricted delivery into all cells? Trends Cell Biol. 2000;10:290-5
45. Zhang TY, Yuan Q, Zhao JH, Zhang YL, Yuan LZ, Lan Y. et al. Prolonged suppression of HBV in mice by a novel antibody that targets a unique epitope on hepatitis B surface antigen. Gut. 2016;65:658-71
46. Wang SH, Yeh SH, Lin WH, Wang HY, Chen DS, Chen PJ. Identification of androgen response elements in the enhancer I of hepatitis B virus: a mechanism for sex disparity in chronic hepatitis B. Hepatology. 2009;50:1392-402
47. Wu Y, Zhang TY, Fang LL, Chen ZX, Song LW, Cao JL. et al. Sleeping Beauty transposon-based system for rapid generation of HBV-replicating stable cell lines. J Virol Methods. 2016;234:96-100
48. Wu HC, Tsai HW, Teng CF, Hsieh WC, Lin YJ, Wang LH. et al. Ground-glass hepatocytes co-expressing hepatitis B virus X protein and surface antigens exhibit enhanced oncogenic effects and tumorigenesis. Hum Pathol. 2014;45:1294-301
49. Li H, Zhuang Q, Wang Y, Zhang T, Zhao J, Zhang Y. et al. HBV life cycle is restricted in mouse hepatocytes expressing human NTCP. Cell Mol Immunol. 2014;11:175-83
50. Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT. et al. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother. 1997;41:1715-20
Author contact
![]() Corresponding authors: Quan Yuan (yuanquanedu.cn), or Tian-Ying Zhang (tyzhang1003com), State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics, Xiamen University, Xiamen 361102, People's Republic of China. Fax: (86)-05922181258.
Corresponding authors: Quan Yuan (yuanquanedu.cn), or Tian-Ying Zhang (tyzhang1003com), State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics, Xiamen University, Xiamen 361102, People's Republic of China. Fax: (86)-05922181258.