Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(9):2407-2423. doi:10.7150/thno.23632 This issue Cite
Research Paper
Imaging, myeloid precursor immortalization, and genome editing for defining mechanisms of leukocyte recruitment in vivo
Sandra Gran1,2*, Lisa Honold2,3*, Olesja Fehler1, Stefanie Zenker1, Sarah Eligehausen3, Michael T. Kuhlmann3, Edwin Geven4, Martijn van den Bosch4, Peter van Lent4, Christoph Spiekermann1, Sven Hermann2,3,5, Thomas Vogl1,5, Michael Schäfers2,3,5,6, Johannes Roth1,2,5 ![]()
1. Institute of Immunology, University of Münster, 48149 Münster, Germany
2. DFG EXC 1003 Cluster of Excellence 'Cells in Motion', University of Münster, Germany
3. European Institute for Molecular Imaging, University of Münster, 48149 Münster, Germany
4. Department of Rheumatology, Radboud University Medical Center, 6525 GA Nijmegen, Netherlands
5. Interdisciplinary Center for Clinical Research, University of Münster, 48149 Münster, Germany
6. Department of Nuclear Medicine, University Hospital Münster, 48149 Münster, Germany
*Both authors contributed equally to this work.
Received 2017-11-2; Accepted 2018-1-18; Published 2018-3-23
Abstract

Recruitment of leukocytes from the blood to sites of inflammation poses a promising target for new diagnostic and therapeutic approaches. We aimed to develop a novel method to non-invasively analyze molecular mechanisms of leukocyte migration in pre-clinical models of inflammation in vivo.
Methods: We used the ER-HoxB8 system to transiently immortalize murine myeloid precursors from wildtype and CD18- as well as MRP14-deficient mice. A VLA4α-/- cell line was generated by CRISPR/Cas9-mediated gene editing. We analyzed the migration of wildtype and knockout leukocytes in vivo by optical and nuclear imaging in mice with irritant contact dermatitis, cutaneous granuloma, experimental arthritis and myocardial infarction.
Results: Transient immortalization, gene editing and in vivo imaging can be combined to analyze migratory mechanisms of murine leukocytes, even for gene deletions resulting in lethal phenotypes in mice. We reliably confirmed known migratory defects of leukocytes deficient for the adhesion molecules CD18 or VLA4α. Also, using our new method we identified a new role of the most abundant calcium-binding proteins in phagocytes and major alarmins in many inflammatory diseases, MRP8 and MRP14, for transmigration in vivo.
Conclusion: We provide a combinatorial approach to rapidly characterize molecular mechanisms of leukocyte recruitment in vivo, with the potential to aid in identification of diagnostic and therapeutic targets in inflammatory pathologies.
Keywords: Cell migration, Genetic engineering, molecular imaging, leukocytes, MRP8 and MRP14
Introduction
Inflammation is the first defense mechanism to cope with tissue injury or invading pathogens. Uncontrolled inflammation, on the other hand, may lead to chronic disorders [1, 2]. Characteristically, neutrophils are the first cells to accumulate in inflamed tissues [1, 3, 4]. Subsequently, monocytes and macrophages play a pivotal regulatory role in the promotion but also in the resolution of inflammation [1, 3, 5, 6]. Recruitment of phagocytes appears also in post-ischemic scenarios such as myocardial infarction (MI). While the initial recruitment of monocytes is necessary to remove dead cells and initiate the wound healing process, numbers of recruited monocytes in the infarct area are frequently too high, impeding the resolution of inflammation and thereby worsening post-MI prognosis [7-9]. Thus, the sequence and dynamics of extravasation of specific phagocyte populations to the site of inflammation is a key process not only for progression but also resolution of inflammation, defining this step as a promising therapeutic target to modulate inflammation [10-12].
Extravasation of phagocytes occurs via an adhesion cascade that involves rolling, arrest, crawling and transmigration of cells through the endothelial cell (EC) layer to the site of inflammation [13]. The relevance of specific adhesion receptors (e.g., the integrin β2 (CD18), a subunit of LFA-1 or MAC-1 binding to ICAM, or integrin α4 (CD49d), a subunit of VLA4 binding to VCAM1 [13-15]) has been shown by gene mutations and by blocking adhesion molecules in mice and man. Although the principal process of transmigration is widely accepted, key questions remain regarding recruitment of distinct phagocyte populations in vivo at specific stages of inflammation and in an organ-specific manner. However, understanding these mechanisms is a prerequisite for the development of innovative diagnostic or therapeutic approaches in many clinically relevant inflammatory diseases. Addressing these questions is difficult due to a wide heterogeneity of ECs in different tissues (e.g., blood brain barrier, high post-capillary venules). A major obstacle is the lack of experimental in vitro setups reliably modeling the ample heterogeneity of phagocytes and ECs in different organs in vivo. Even sophisticated transmigration approaches in vitro cannot reflect the biological complexity in vivo. Analysis of genetically modified mice is often restricted by lethal or complex phenotypes. Still, a reliable and flexible technology allowing monitoring of genetically modified leukocytes in clinically relevant models of inflammation in mice is lacking.
Magnetic resonance (MR), nuclear and optical imaging provide the possibility to non-invasively track the migration of injected cells in mouse models in vivo over a longer period of time but depend on purification of high numbers of primary leukocytes [16-19]. Only limited numbers of monocytes and neutrophils are available as circulating blood cells. Bone marrow cells, on the other hand, represent a very inhomogeneous cell population and purification of a specific cell type generally leads to activation or differentiation of cells. In addition, genetic manipulation of primary phagocytes is not effective and is associated with their activation or damage.
To overcome these challenges, we introduce a novel method combining the estrogen-regulated ER-HoxB8 system for transient immortalization and genetic engineering of murine myeloid precursor cells with in vivo imaging. These precursors can easily be differentiated to monocytes or neutrophils in high quantities [20]. Through the development of fluorescence reflectance imaging (FRI) and single photon emission tomography (SPECT)-based ER-HoxB8 cell labeling protocols we are able to quantitatively analyze the migration of specific phagocyte populations into different organs in the whole body of inflammatory animal models. Establishing precursors of knockout mice (e.g., CD18-/-) or performing targeted genetic modification of ER-HoxB8 cells via CRISPR/Cas9 technology (VLA4-/-) uniquely allows the analysis of leukocyte migration in genetic backgrounds even associated with lethal phenotypes. In addition, we describe a new role of the most abundant calcium-binding proteins, myeloid related protein 8 (MRP8, S100A8) and MRP14 (S100A9), in phagocyte transmigration in vivo. MRP8 and MRP14 represent up to 5-40% of the detergent-soluble cytosolic proteins of monocytes and granulocytes, respectively [21], but their intracellular actions were not well understood so far.
Our novel technology allows a comparative analysis of the migration of wildtype and mutated phagocytes in parallel within the same animal. We thus describe a method for rapid and almost unlimited analysis of migratory properties of genetically modified phagocytes in pre-clinically relevant settings in vivo for identification and confirmation of potential therapeutic anti-inflammatory targets in leukocytes. Our approach is an easy, quick and reliable alternative for establishing genetically modified mouse strains linked with the risk of complex or even lethal phenotypes.
Results
ER-HoxB8 cell labeling and functional analysis
For FRI, differentiated ER-HoxB8 monocytes or neutrophils were labeled with the fluorescent membrane-incorporating dyes DIR or DID. Labeling rates were close to 100% (Figure S1C, D) and viability was not affected by DIR/DID labeling (more than 90% viable cells; Figure S1A, B).
ER-HoxB8 monocytes were labeled with 1.06 ± 0.2 Bq 111In-Oxine per cell for SPECT experiments. Retention of 111In-Oxine in vitro dropped to 74.4% ± 7.2% after 6 h, 28.3% ± 9.1% after 24 h and 24.8% ± 3.5% after 48 h (Figure S1E, F). Labeling with 111In-Oxine did not affect cellular viability (number of dead cells below 2%).
Firstly, ER-HoxB8-derived neutrophils and monocytes were confirmed to express typical differentiation markers and exhibit central phagocytic functions of the primary counterparts, as described previously (Figure S2) [20, 22-24]. In addition, in ER-HoxB8 monocytes and neutrophils neither adhesion properties (Figure S2B) nor spontaneous and chemotactic migration (Figure S2C) nor ROS production and phagocytosis (Figure S2D, E) were altered due to labeling with DIR or DID. Also 111In-Oxine-labeled ER-HoxB8 cells did not show altered migration rates as compared to unlabeled controls (Figure S2F).
In vivo optical imaging of the migration of differentiated ER-HoxB8 cells
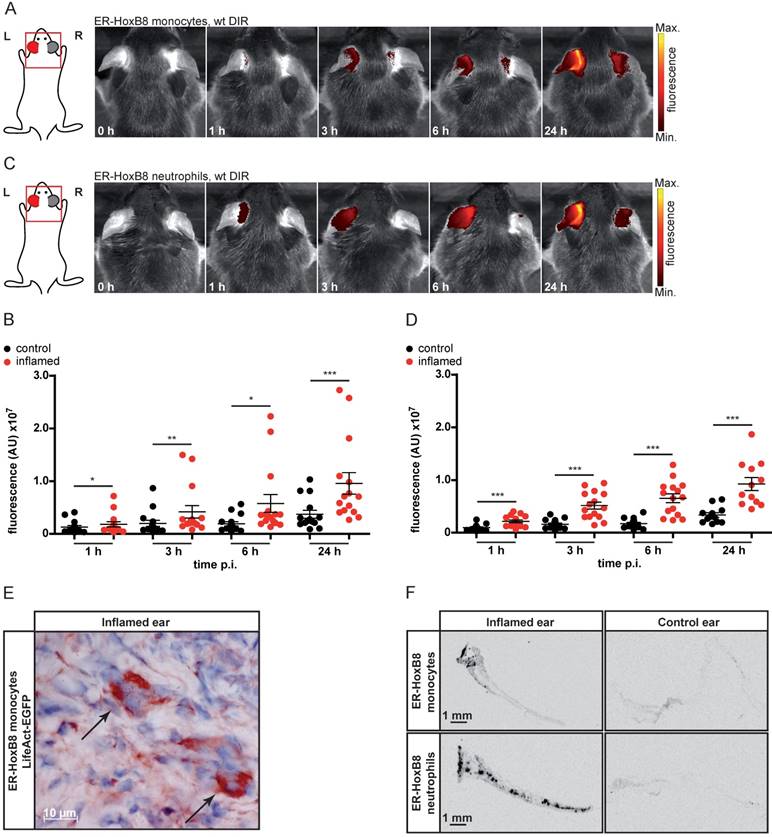
We used irritant contact dermatitis (ICD) as a model of innate immune activation by a non-specific toxic stimulus (left ear: application of croton oil, right ear: control). DIR-labeled ER-HoxB8 monocytes or neutrophils were injected and FRI images were taken 0-24 h post injection (p.i.). We detected strong and significantly higher fluorescence signals in the inflamed ear (left) compared to controls (right) for monocytes (Figure 1A, B) and neutrophils (Figure 1C, D). Due to itching and scratching induced by croton oil treatment, mice distribute minor amounts of croton oil to the right control ears, inducing a weak inflammatory reaction that results in a low background signal caused by some infiltrating immune cells. Injecting ER-HoxB8 LifeAct-EGFP monocytes, we confirmed infiltration of viable monocytes in situ by anti-GFP staining in histological sections (Figure 1E). Immigrated labeled ER-HoxB8 neutrophils and monocytes could also clearly be detected in histological sections of inflamed ears using the LI-COR® Odyssey® Imaging system (Figure 1F).
In vivo optical imaging of monocyte and neutrophil migration in an ear inflammation model (irritant contact dermatitis, ICD). ER-HoxB8 wildtype (wt) monocytes/neutrophils were labeled with DIR and injected i.v. in a mouse showing inflammation at the left ear (right ear served as internal control) due to ICD induction 24 h before. FRI images were taken 0 h, 1 h, 3 h, 6 h and 24 h p.i. (A) Representative imaging series 0-24 h p.i. showing DIR-labeled monocyte infiltration into inflamed tissue by fluorescence signal accumulation. (B) Statistical analysis of immigration of labeled ER-HoxB8 monocytes into inflamed tissue (left ear) as compared to control (right ear) (n=15 mice, 5 independent experiments). (C) Representative imaging series 0-24 h p.i. showing DIR-labeled neutrophil infiltration into inflamed tissue. (D) Statistical analysis of immigration of labeled ER-HoxB8 neutrophils into inflamed tissue (left ear) as compared to control (right ear) (n=15 mice, 5 independent experiments). (E) ER-HoxB8 EGFP monocytes were used for cell tracking and immunolocalized on frozen sections of inflamed tissue by α-GFP/peroxidase staining. Arrows indicate cells positive for α-GFP staining. (F) Frozen sections of inflamed/control ears. Black spots indicate immigrated DIR/DID labeled cells. Images orientation: L = left, R = right, fluorescence = fluorescence intensity (AU). Data are shown as dotplots with mean ± SEM, corrected to baseline and labeling efficiency. Statistical significance was calculated using paired Student's t-test: *p < 0.05, **p < 0.01, ***p < 0.001.
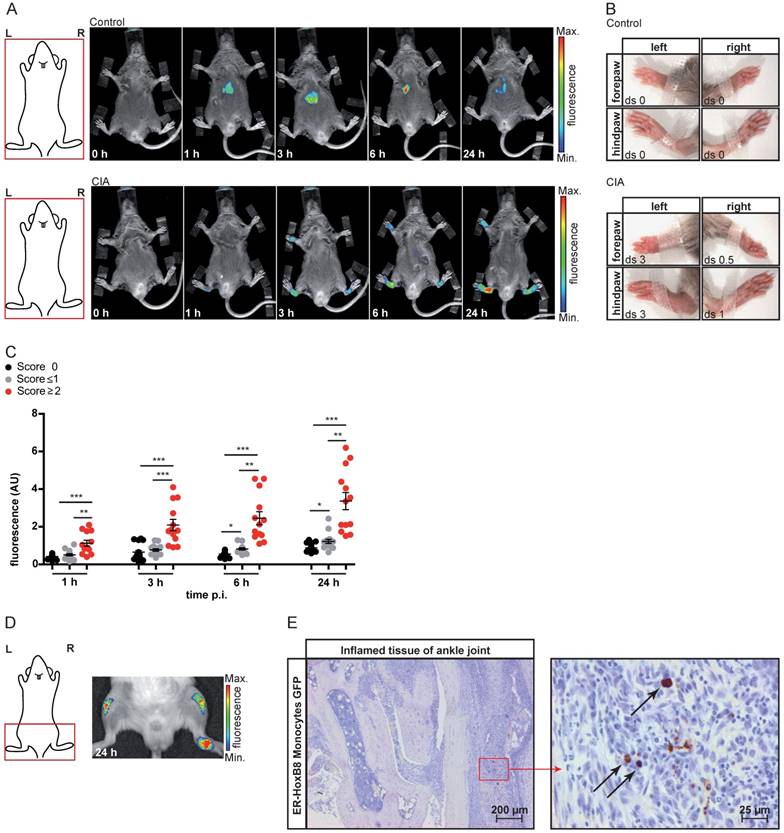
We also analyzed ER-HoxB8 cell migration in experimental arthritis as a clinically relevant model of autoimmunity. In collagen-induced arthritis (CIA), we could detect infiltrating DIR-labeled monocytes in inflamed paws (CIA disease score ≥2). Control animals showed nearly no signal (Figure 2A-C). A significant association of individual fluorescence signals (immigrating ER-HoxB8 cells) and disease scores (“clinical” degree of inflammation) could be found. Also, in IL1-Ra-/- mice, which spontaneously develop arthritis due to overwhelming innate immune activation, labeled ER-HoxB8 GFP monocytes infiltrated inflamed joints (Figure 2D). α-GFP staining of histological sections confirmed infiltration of viable ER-HoxB8 cells (Figure 2E).
In vivo optical imaging of monocyte migration in experimental arthritis. ER-HoxB8 wildtype monocytes were labeled with DIR and injected i.v. in (A-C) a CIA mouse showing joint inflammation with severe disease score and a healthy control mouse. FRI images were taken 0 h, 1 h, 3 h, 6 h and 24 h p.i. (D-E) Labeled ER-HoxB8 monocytes were injected i.v. in an IL-1Ra-/- mouse with severely inflamed joints. A FRI image was taken 24 h p.i. (A) Representative imaging series of control and CIA mouse (0-24 h) indicating monocyte infiltration into inflamed tissue. (B) Stereomicroscopic images of paws corresponding to (A). (C) Statistical analysis of immigration of ER-HoxB8 monocytes into uninflamed or mildly inflamed (score 0 and ≤ 1) paws as compared to severely inflamed (score >2) paws (n=48 paws, 3 independent experiments). (D) Representative image of IL-1Ra-/- mouse (24 h) indicating monocyte infiltration by fluorescence signal accumulation. (E) ER-HoxB8 GFP monocytes were used for cell tracking and immunohistological localization of GFP positive cells was performed on frozen sections of inflamed ankle joints by α-GFP staining. Arrows indicate cells positive for α-GFP staining. Images orientation: L = left, R = right, fluorescence = fluorescence intensity (AU), ds = disease score. Data are shown as dotplots with mean ± SEM, corrected to baseline and labeling efficiency. Statistical significance was calculated using Kruskal-Wallis analysis and Dunn's Multiple Comparison Test: *p < 0.05, **p < 0.01, ***p < 0.001.
In vivo imaging of ER-HoxB8 cells by combined nuclear and optical imaging
In order to analyze defined inflammatory triggers, we employed a cutaneous granuloma model (CG), in which two biogel “plugs” were injected into the right (control) and the left flank (addition of LPS), triggering Toll-like receptor 4-dependent inflammation. In vivo FRI showed highly significant differences between cellular infiltration into control and LPS plugs between 0-48 h after injection of DIR-labeled ER-HoxB8 monocytes (Figure 3A, B). After re-isolation of immigrated cells out of the plugs, we found a viable population of positive cells (DIR/DID+) by flow cytometry representing about 2-10% of the total amount of immigrated cells (Figure 3C).
In vivo imaging of monocyte migration in a cutaneous granuloma (CG) model combining optical (FRI) and nuclear (SPECT) imaging technique. (A-C) ER-HoxB8 DIR-labeled wildtype monocytes were injected i.v. in a CG mouse model (left 20 µg/mL LPS, right plug served as control). FRI images were taken 0-48 h p.i. (A) Representative imaging series showing monocyte immigration. (B) Statistical analysis of migration of ER-HoxB8 monocytes to LPS plug as compared to control plug (n=14 mice, 5 independent experiments). (C) Representative flow cytometry analysis of re-isolated cells from plugs; FL4-H corresponds to DIR/DID-labeling. Representative pseudocolor plots of viable cells (dead cells discriminated by FSC/SSC gating) depicted. (D-G) ER-HoxB8 monocytes were labeled with DIR or 111In-Oxine and were simultaneously injected i.v. in the same animal. FRI and SPECT measurements were taken 6-48 h p.i. (D) Representative imaging series showing DIR-labeled monocyte migration to LPS plug and (E) corresponding SPECT images of the same animal showing surface signals from 111In-Oxine-labeled monocytes (arrowhead) and additional deep tissue signals (arrow). (F) Statistical analysis of migration of DIR-labeled monocytes to LPS plug as compared to control plug (n=6 mice, 2 independent experiments). (G) Statistical analysis of migration of 111In-Oxine-labeled monocytes to LPS plug as compared to control plug (n=6 mice, 2 independent experiments) corresponding to (F). Images orientation: L = left, R = right, fluorescence = fluorescence intensity (AU). Data are shown as dotplots with mean ± SEM, corrected to baseline and labeling efficiency for DIR labeling and displayed as SUV (standardized uptake units) for 111In-Oxine labeling. Statistical significance was calculated using paired Student's t-test: *p < 0.05, **p < 0.01, ***p < 0.001.
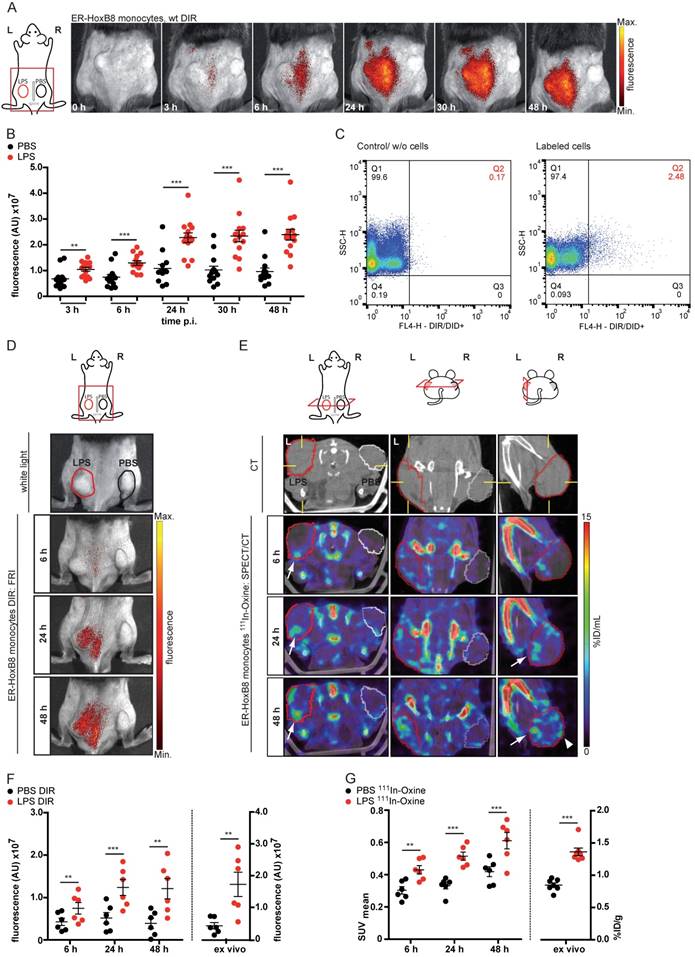
Though FRI is a relatively easy, fast and catchy method for investigating cell migration, it nevertheless has limitations. Regarding the penetration depth of tissue or 3D resolution, FRI is clearly outnumbered by SPECT or MRI. Additionally, SPECT allows the non-invasive, body-wide assessment of injected cells at early time points. We labeled ER-HoxB8 monocytes radioactively with 111In-Oxine and tracked their biodistribution by serial SPECT imaging in healthy mice. Labeled cells accumulated in the lungs for the first 90 min, before being redistributed to spleen, liver and bone. Non-retained radioactivity was eliminated via the kidneys (Figure S3).
To confirm the applicability of 111In-Oxine-labeled ER-HoxB8 monocytes for in vivo tracking in inflammation FRI as well as SPECT measurements were carried out in parallel in the same animals. Finally, cell migration into the biogel plugs was determined by ex vivo scintillation counting and optical imaging. For both approaches, immigrated ER-HoxB8 monocytes could be detected to a significantly higher extent in LPS plugs compared to controls (Figure 3D, E) and time courses of signal accumulation in both techniques fit very well to each other (Figure 3F, G). In contrast to FRI, three-dimensional SPECT acquisitions allowed detection of immigrated cells not only at the surface of the plug, but in deeper tissues, specifically at the inner border of the biogel plug (Figure 3E). The higher in vivo signal from 111In-Oxine-labeled cells in control PBS plugs reveals the higher sensitivity of SPECT compared to FRI.
Simultaneous in vivo FRI analysis of the migration of different cell populations
In the next step, we simultaneously injected ER-HoxB8 wildtype (wt) monocytes labeled with DIR and ER-HoxB8 CD18-/- monocytes labeled with DID, two spectrally distinct fluorophores, into the CG model. This approach allows direct comparison of the migration behaviors of two monocyte populations in the same animal in vivo, at the same time reducing the number and experimental variations between individual animals per experimental setup. FRI measurements were adjusted for differences in labeling efficiencies and corrected for spectral overlap of DIR and DID. CD18-/- was chosen as an example based on its important role in phagocyte migration [13, 25]. Compared to DIR-labeled ER-HoxB8 wildtype monocytes, DID-labeled ER-HoxB8 CD18-/- monocytes exhibited significantly decreased infiltration rates reflected by low FRI signals. Overall recruitment of CD18-/- monocytes into the inflammation site was reduced by about 75% (Figures 4C, D).
In vivo optical imaging of wt and CD18-/- monocyte migration in a CG model. ER-HoxB8 wildtype and CD18-/- monocytes were labeled with DIR and each cell population was injected in an individual animal (A-B). Alternatively, ER-HoxB8 wildtype and CD18-/- monocytes were differentially labeled with DIR and DID and injected in the same mouse (C-D). FRI images were taken 0 h, 3 h, 6 h, 24 h, 30 h and 48 h p.i. (A) Representative imaging series of the one color imaging approach in two individual mice 3-48 h p.i. Upper panel shows LPS plug infiltration of wt monocytes (DIR). Lower panel shows LPS plug infiltration of CD18-/- monocytes (DIR). (B) Statistical analysis of imaging of wt and CD18-/- cell migration to LPS plug corresponding to (A) (n=8 mice, 3 independent experiments). (C) Representative imaging series of the two color simultaneous imaging approach in a single mouse 3-48 h p.i. Upper panel shows LPS plug infiltration of wt monocytes (DIR). Lower panel shows LPS plug infiltration of CD18-/- monocytes (DID). (D) Statistical analysis of simultaneous imaging of wt and CD18-/- cell migration to LPS plug corresponding to (C) (n=9 mice, 3 independent experiments). (E-F) Verification of CD18 knockout in ER-HoxB8 CD18-/- monocytes. Representative results are shown. (E) Result of qRT-PCR of wt (day 0 and day 3) compared to CD18-/- monocytes for CD18 mRNA. (F) Flow cytometry analysis of α-CD18 staining of wt and CD18-/- monocytes (day 3). Expression of CD18 was detected as FL2-H+. Open graphs show isotype control; grey graphs show α-CD18 staining. Images orientation: L = left, R = right, fluorescence = fluorescence intensity (AU). Data are shown as dotplots with mean ± SEM, corrected to baseline and labeling efficiency. Statistical significance was calculated using 2-way ANOVA and Bonferroni post-tests comparing ER-HoxB8 wildtype and CD18-/- cells: *p < 0.05, **p < 0.01, ***p < 0.001.
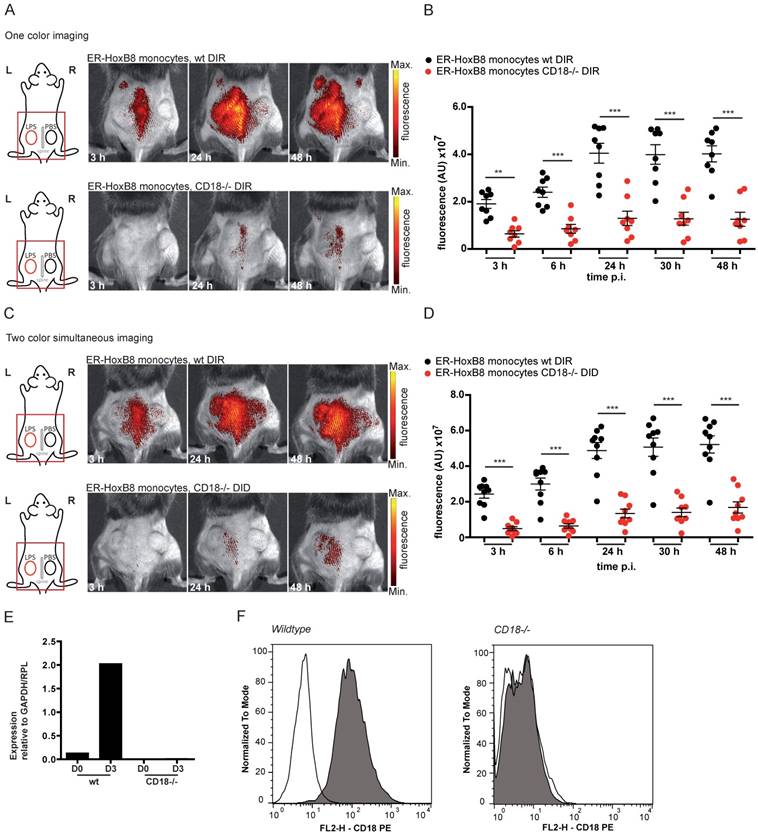
To confirm the accuracy of the simultaneous injection protocol of two labeled monocyte populations, we studied single cell injections of DIR-labeled wildtype or CD18-/- monocytes separately, which tightly reproduced the results of the experiments with simultaneous injection of two monocyte populations (Figures 4A, B). Knockout of CD18-/- cells was confirmed by qRT PCR of mRNA and flow cytometry analysis (Figure 4E, F).
In order to test our method for knockouts resulting in complex or lethal phenotypes in vivo we combined our approach of ER-HoxB8 cell tracking with innovative gene editing techniques, like the clustered regularly interspaced short palindromic repeats/caspase 9 (CRISPR/Cas9) system, allowing for specific and efficient editing of genes [26]. We therefore employed CRISPR/Cas9 in ER-HoxB8 cells to delete the integrin α4 subunit of VLA4, which is known to play a role in phagocyte recruitment but creates a lethal phenotype in mice [13, 25, 27]. VLA4 knockout was confirmed by qRT PCR of mRNA, flow cytometry and DNA sequencing (Figure 5E-G). We simultaneously analyzed and tracked DIR-labeled ER-HoxB8 wildtype and DID-labeled ER-HoxB8 VLA4α-/- monocytes in the CG model. ER-HoxB8 VLA4α-/- monocytes exhibited a major migratory defect. In “two color simultaneous imaging” experiments we found a significant reduction of about 50% for ER-HoxB8 VLA4α-/- monocytes compared to ER-HoxB8 wildtype monocytes (Figure 5A, B). Using DID-labeled ER-HoxB8 wildtype and DIR-labeled ER-HoxB8 VLA4α-/- monocytes in this setting yielded comparable results (data not shown). Applying one color imaging we observed a reduction of cell migration by about 55% for ER-HoxB8 VLA4α-/- compared to ER-HoxB8 wildtype monocytes (Figure S4A, B).
In vivo optical imaging of VLA4α-/- and MRP14-/- monocyte migration in a CG model. ER-HoxB8 wildtype and VLA4α-/- monocytes (A-B) or wildtype and MRP14-/- monocytes (C-D) were differentially labeled with DIR or DID and injected in the same mouse. FRI images were taken 0 h, 3 h, 6 h, 24 h, 30 h and 48 h p.i. (A and C) Representative imaging series (3-48 h) of two color simultaneous imaging of LPS plug infiltration in a single mouse is shown for (A) wt monocytes (DIR, upper panel) and VLA4α-/- monocytes (DID, lower panel) and (C) wt monocytes (DIR, upper panel) and MRP14-/- monocytes (DID, lower panel). (B and D) Statistical analysis of simultaneous imaging of cell migration to LPS plug of (B) wt and VLA4α-/- monocytes corresponding to (A) (n=8 mice, 3 independent experiments) and (D) of wt and MRP14-/- monocytes corresponding to (C) (n=12 mice, 5 independent experiments). (E-G) Verification of VLA4α (CD49d) knockout in ER-HoxB8 VLA4α-/- monocytes. Representative results are shown. (E) qRT-PCR of wt and VLA4α-/- monocytes (day 3) for CD49d (VLA4α) mRNA. (F) Flow cytometry analysis of α-CD49d staining of wt and VLA4α-/- monocytes (day 3). CD49d expression was detected as FL2-H+. Open graphs show isotype control; grey graphs α-CD49d/VLA4α staining. (G) DNA-sequencing of the CRISPR/Cas9 target region of VLA4α-/- monocytes. (H-I) Verification of MRP14 knockout in ER-HoxB8 MRP14-/- monocytes. Representative results are shown. (H) qRT-PCR of wt and MRP14-/- monocytes (day 0 and day 3) for MRP14 mRNA. (I) WB analysis of wt and MRP14-/- monocytes (day 3) by α-MRP14 staining. Images orientation: L = left, R = right, fluorescence = fluorescence intensity (AU). Data are shown as dotplots with mean ± SEM, corrected to baseline and labeling efficiency. Statistical significance was calculated using 2-way ANOVA and Bonferroni post-tests comparing ER-HoxB8 wildtype and VLA4α-/- or MRP14-/- cells: *p < 0.05, **p < 0.01, ***p < 0.001.
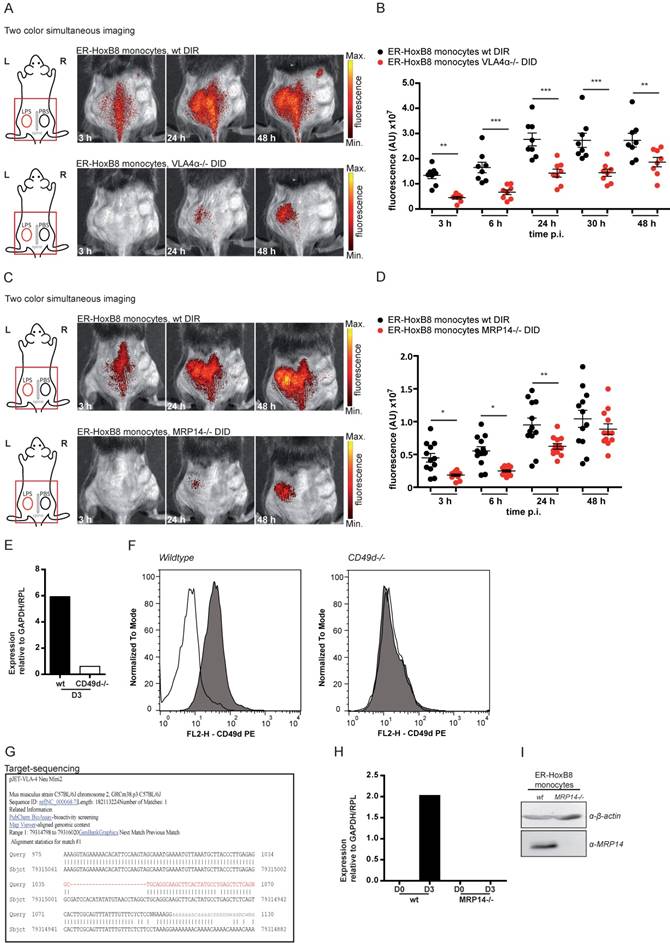
Calcium binding proteins MRP8 and MRP14 were previously found to interact with tubulin under inflammatory conditions, thereby regulating cytoskeletal dynamics of phagocytes in vitro [28, 29]. However, biological relevance of these findings could not be confirmed so far in vivo. We used DIR-labeled ER-HoxB8 wildtype monocytes and DID-labeled ER-HoxB8 MRP14-/- monocytes and compared the migratory capacities of both cell types by “two color simultaneous imaging” in the CG mouse model. We found MRP14-/- cell migration decreased by about 50% at 6 h p.i. and by 40% at 24 h p.i., indicating a significant delay in MRP14-/- monocyte recruitment as compared to wildtype monocytes (Figure 5C, D). MRP14 knockout in MRP14-/- cells was confirmed by qRT PCR of mRNA and Western Blot (WB) analysis (Figure 5H, I).
In vivo nuclear imaging of monocyte migration in myocardial infarction (MI)
We applied SPECT to assess the migration of monocytes in the clinically relevant disease scenario of MI. This model comprises major challenges for in vivo imaging, i.e., detection of small numbers of infiltrating monocytes in a deep and rapidly moving organ (450 beats per minute). 111In-Oxine-labeled monocytes were intravenously administered to mice with permanent MI one day after coronary artery ligation and their migration was followed by serial SPECT imaging. Areas at risk were determined on the day of the surgery by 99mTc-Tetrofosmin SPECT to define the extension of perfusion defects and signal from 111In-Oxine-labeled monocytes was quantified one and two days p.i. (Figure 6A). The SPECT signal within the infarct zone (I) was significantly increased compared to remote myocardium (R) on both days (I: 0.60 ± 0.07 SUV vs. R: 0.37 ± 0.02 SUV on day 1 and I: 0.47 ± 0.04 SUV vs. R: 0.25 ± 0.03 SUV on day 2).
In vivo nuclear imaging of monocyte migration in murine myocardial infarction. ER-HoxB8 wildtype monocytes were labeled with 111In-Oxine and injected 1 day after permanent coronary artery ligation. (A) Representative images showing myocardial perfusion deficit in 99mTc-Tetrofosmin SPECT acquisitions (red VOI) below the coronary artery ligation site (arrowhead). Accumulation of 111In-Oxine-labeled monocytes 1 and 2 days post cell injection in the infarct (arrow) and at sites of surgery-induced thorax inflammation (#). (B) Verification of monocyte migration by ex vivo autoradiography, detecting 111In-Oxine-labeled monocytes in the infarct zone (dotted line), which presents with high numbers of infiltrated macrophages, visualized by Mac-3 staining. (C) Statistical analysis of monocyte migration detected by in vivo SPECT (n= 5 mice, 3 independent experiments). Images orientation: L = left. Data are shown as dotplots with mean ± SEM. SUV: standardized uptake units; %ID/mL: % injected dose per volume. Statistical significance was calculated using 2-way ANOVA and Bonferroni post-test: *p < 0.05, **p < 0.01, ***p < 0.001.
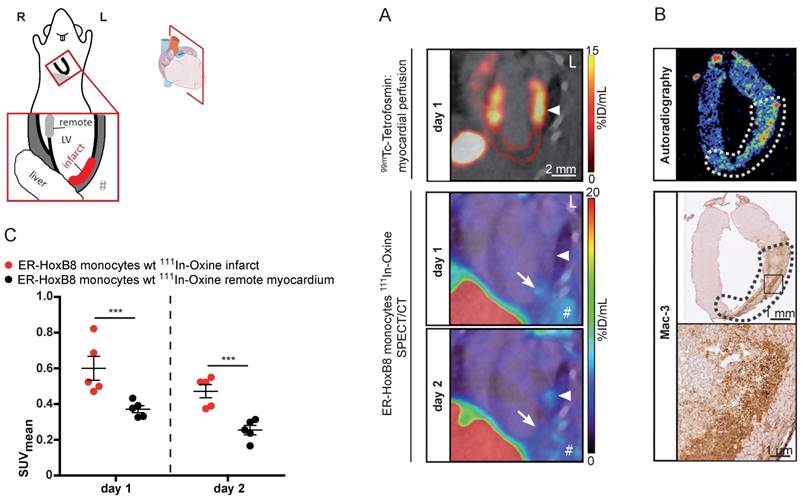
Ex vivo autoradiography detected 111In-Oxine-labeled cells in areas showing large amounts of infiltrated monocytes by immunohistochemistry (Figure 6B). We confirmed signal specificity by application of free 111In-Oxine in MI mice or application of 111In-Oxine-labeled monocytes in sham operated animals (Figure S5).
Discussion
The ability to investigate phagocyte migration under clinically relevant inflammatory conditions in vivo poses an unmet challenge in translational biomedical research for the development of novel diagnostic assays or therapeutic targets. Current experimental approaches exhibit major limitations for modeling the complex processes of leukocyte recruitment due to the high heterogeneity of leukocytes and ECs. Combining reversible immortalization of myeloid progenitors with modern gene editing techniques and in vivo imaging technologies, we now present a novel approach that allows quantitative analysis of leukocyte transmigration combined with genetic interventions in complex preclinical inflamematory models in mice.
We established an efficient labeling method for ER-HoxB8-derived neutrophils and monocytes resulting in high numbers of labeled cells, which is currently not achievable with other methods for the generation of murine phagocytes. Exogenous cell labels, as used here for both imaging strategies, comprise the advantage of flexibility as they can easily be applied to all cell types. Endogenous labels, in contrast, require time-consuming transfection with genetic imaging reporters or even complex and labor-intensive generation of genetically modified mice. On the other hand, cells expressing reporter constructs can be repeatedly followed up in vivo over a longer period of time. Applying our combination of genome editing and HoxB8-induced immortalization on a genetic background expressing a reporter construct may finally combine all advantages of these individual methods [18, 30].
Pure populations of monocytes or neutrophils are not available from primary mouse sources in quantities sufficient for in vivo imaging. Bone marrow cells, which have been used alternatively, comprise a broad spectrum of cells and cell subtypes. Furthermore, bone marrow-derived or peritoneal macrophages represent later differentiation stages not relevant for transendothelial migration studies. As previously described, the ER-HoxB8 cell system allows for the generation of the required quantities of myeloid cells while resembling major characteristics of primary monocytes or neutrophils as shown by morphology, differentiation markers, functional assays and even genome-wide gene expression analysis [20, 22-24, 31-33]. Still, even after withdrawal of estrogen, ER-HoxB8-derived phagocytes are retrovirally transduced cells despite their behavior, which seems similar to primary immune cells. Our labeled cells showed a homogenous differentiation, a normal functionality and a high viability and migratory capacity in vitro. We demonstrate for the first time that this cell system is a feasible technique for monitoring murine neutrophil and monocyte trafficking during inflammation in vivo, independent of the underlying immunological pathomechanism. We got reliable results in a variety of pre-clinical models reflecting toxic and TLR4-mediated innate immune responses in the skin, complex autoimmune diseases like arthritis involving phagocytes, T- and B-lymphocytes or ischemia-induced tissue injury. We confirmed infiltration of viable labeled monocytes and neutrophils by immunohistochemistry and ex vivo analysis. In our arthritis model we could even show a close correlation between clinical disease scores and monocyte immigration, demonstrating that our method is a powerful tool for monitoring leukocyte recruitment and severity of inflammation in preclinical murine models of arthritis.
Using two dyes with different excitation/ emission spectra we further succeeded in direct and quantitative comparison of the migration characterristics of cells with two different genetic backgrounds. We found CD18-/- monocytes exhibiting highly decreased migration rates compared to wildtype controls. Since ER-HoxB8 cells can easily be established for any existing knockout mouse line, our method now allows functional analysis of migratory capacities of a myriad of genetically modified leukocytes. To further overcome restrictions like lack of an appropriate knockout mouse strain or lethal knockout phenotypes, we combined our approach with the CRISPR/Cas9 system for modifying ER-HoxB8 cells [20, 26]. We have chosen VLA4α (CD49d), the integrin α4 subunit of VLA4, which is involved in the leukocyte adhesion cascade where it primarily mediates arrest of phagocytes by binding to VCAM [13]. Knockout of integrin α4 in mice leads to a lethal phenotype impeding generation of ER-HoxB8 VLA4α-/- cells [27]. However, using CRISPR/Cas9 we demonstrate that ER-HoxB8 VLA4α-/- monocytes revealed significantly decreased migration rates compared to controls. Addressing two central adhesion molecules of the integrin family, CD18 (vital phenotype) and CD49d (lethal phenotype) we clearly demonstrate the feasibility of our novel approach for functional analysis of leukocyte migration in vivo [13, 25, 27]. Thus, we have an easy and feasible technique to model any clinically relevant mutation found in patients with innate immune deficiencies for mechanistic studies and testing therapeutic approaches.
Finally, we analyzed the migratory capacity of ER-HoxB8 MRP14-/- monocytes. In contrast to CD18 or CD49d, which were extensively described as part of the migratory network before, evidence for an intracellular role of MRP8/14 during phagocyte migration in vivo was lacking [28]. We could now demonstrate that knockout of the major calcium-binding protein in phagocytes, MRP14, led to a significant reduction of cell migration. MRP14 and its binding partner MRP8 represent the major calcium-binding capacity in granulocytes and monocytes [34]. They play a dominant role in many clinically relevant inflammatory diseases like rheumatoid arthritis, inflammatory bowel disease, lung disease, infections, sepsis, allergies or autoimmune diseases [35-41]. Hence, this is a prototypic example for identification of the relevance of complex and non-obvious phenotypes in vivo that could serve as a blueprint for analysis of almost any molecular system in phagocytes using this new approach in preclinical models in vivo.
Hence, the combination of ER-HoxB8 cells and genetic engineering via CRISPR/Cas9 in principle facilitates the generation of any conceivable knock-out in monocytes or neutrophils, which altogether takes only weeks or a few months before in vivo experiments can be started. Establishment of a conditional knock-out strain for complex or lethal phenotypes, on the other hand, crossing with cell-specific or inducible Cre-mice and final back-cross with adequate reporter mouse strains will take at least one or two years. With a few modifications, this approach may also be feasible for other stem cell models. Introduction of specific gain-of-function mutations is another option of the CRISPR/Cas9 technology. The combination of genetically targeted ER-HoxB8 neutrophils or monocytes with the cutaneous granuloma model now allows molecular analysis of any combination of a genetic mutation in the leukocyte (e.g., adhesion or chemokine receptor, signaling molecule, transcription factor) and any inflammatory trigger in the granuloma (microbial products, cytokines, chemokines or alarmins) in vivo. In addition, differences regarding the biological relevance of specific adhesion and signaling pathways can be analyzed for different tissues and mechanisms of inflammation.
A remaining issue one may quote is the low tissue penetration depth of FRI, excluding organs like lung and heart from being analyzed [18]. Concerning this matter, we successfully used a SPECT-based imaging approach and showed an excellent correlation between results obtained by FRI and SPECT. We thus expanded our approach for quantitative analysis of cell migration in complex disease models affecting deeper tissues and proved this by successfully tracking 111In-Oxine-labeled monocytes in myocardial infarction.
The huge heterogeneity of endothelial cells in different tissues requires new approaches addressing molecular mechanisms of phagocyte transmigration in vivo. Recent advances in intravital high-resolution microscopy have allowed direct visualization of phagocyte recruitment to sites of infections or injury in vivo and revealed differences in phagocyte recruitment between different tissues [30, 42-47].
Intravital microscopy studies have revealed novel insights into self-organized neutrophil recruitment to wounds or infectious sites, and how danger molecules guide phagocytes to sites of sterile inflammation [42, 45].
These techniques are, however, restricted to a small field-of-view, are very sensitive to tissue motion (e.g., heartbeat, respiration), and need invasive preparation of tissues, which may be associated with the risk of experimental artefacts. However, combining these innovative microscopic imaging methods with the genome-editing technique of HoxB8-derived phagocytes described here may also be a promising approach to study molecular mechanisms of phagocyte recruitment. Whole body imaging approaches like FRI or SPECT do not have nearly the spatial resolution of microscopic techniques but have the potential for quantification of dynamic leukocyte recruitment not only in a specific inflammatory site but at the same time in all other organs of an organism in complex inflammatory scenarios [18, 44].
In summary, we present a novel, reliable but nevertheless rapid and simple method that opens almost unlimited possibilities for analysis of phagocyte migration in pre-clinical models in vivo, including targeted modification of phagocyte-specific molecular pathways during adhesion and transendothelial migration. We present an easy, quick and reliable alternative for the time consuming establishment of genetically modified mouse strains for analysis of phagocyte recruitment in vivo. Our approach opens multiple capabilities for testing diagnostic or therapeutic hypotheses at the molecular level in inflammatory processes under clinically relevant conditions.
Methods
Animals
C57 BL/6 mice were obtained from Harlan laboratories, Borchen, Germany, or Charles River, Erkrath, Germany, and DBA/jdba1/j mice from Janvier, Le Genest-Saint-Isle, France. IL-1Ra-/- mice on BALB/c background were kindly provided by Dr. M. Nicklin (Sheffield, UK) and were generated as described previously [48]. Animals were used at the age of 8-12 weeks and were sex matched for each set of experiments. All experiments with mice were performed with the approval of the State Review Board of Nordrhein-Westfalen (Germany) according to the German law for animal welfare or by the Ethics Committee of University Hospital Nijmegen. Animal studies were not randomized or blinded.
Generation and cell culture of ER-HoxB8 cells
ER-HoxB8 cells were generated as previously described [20]. In summary, bone marrow cells were isolated from wildtype as well as CD18-/- and MRP14-/- C57 BL/6 mice by density gradient centrifugation, and cultured for 2-3 days in DMEM with 15% FCSi, 1% penicillin/streptomycin, 1% L-glutamine (Biochrom, Berlin, Germany) and containing IL-3 (10 ng/mL; PeproTech Rocky Hills, USA), IL-6 (20 ng/mL; PeproTech, Rocky Hills, USA) and SCF (25 ng/mL; supernatant SN' from SCF-producing CHO cells, Institute of Microbiology and Hygiene, Freiburg). Subsequently, cells were transfected with ER-HoxB8 Retrovirus (1 mL of virus suspension/1x106 cells, kindly provided by the Häcker lab) and exposed to spinoculation in the presence of lipofectamine (Lipofectin® Transfection reagent, Invitrogen, Darmstadt, Germany). Selection of transduced, i.e., immortalized, progenitor cells was carried out by cultivation of cells in either Optimem Glutamax (Gibco Invitrogen, Darmstadt, Germany) with 10% FCSi, 1% pen/strep, 30 µM ß-mercaptoethanol, 1% SN' SCF, and 1 µM ß-estradiol (Sigma Aldrich, Munich, Germany) for neutrophilic progenitor cells or RPMI1640 (Merck Millipore, Darmstadt, Germany) with 10% FCSi, 1% pen/strep, 1% L-glutamine, 1% SN GM-CSF (obtained from B16 cells, Institute of Microbiology and Hygiene, Freiburg), and 1 µM ß-estradiol for monocytic cells. Cells were routinely tested for mycoplasma contamination.
For differentiation of precursors to neutrophils or monocytes, cells were harvested, washed twice with PBS containing 10% FCSi and 1% SN' SCF or SN' GM-CSF to remove ß-estradiol and seeded at densities of 1x106 cells/mL in either Optimem Glutamax with 10% FCSi, 1% pen/strep, 30 µM ß-mercaptoethanol, and 1% SN' SCF to obtain neutrophils or RPMI1640 with 10% FCSi, 1% pen/strep, 1% L-glutamine, and 0.04 µg/mL rm GM-CSF (Immunotools, Friesoythe, Germany) for monocytes, respectively. After 3 days of differentiation, cells were harvested and assigned to experiments. Differentiation to neutrophils and monocytes was confirmed by expression of differentiation markers CD11b, CD14 and GR-1 by flow cytometry. Knockout of CD18-/- cells was routinely tested by flow cytometry using α-mouse-CD18 (#101401, Biolegend, San Diego, USA) and goat-α-rat IgG PE (#112-116-143, Dianova, Hamburg, Germany) and by qRT PCR (Primer fw: 5'-tctcctactccatgcttgatgacc-3', rv: 5'-aaacgacccaaagccgatg -3'). Knockout of MRP14-/- cells was tested by qRT PCR (Primer fw: 5'-ggcaaaggctgtgggaagt-3', rv: 5'-ccattgagtaagccattcccttta-3') and Western blot analysis using α-mouse-MRP14 [49] and goat-α-rabbit IgG HRP (#P044801-2, Agilent, Santa Clara, USA).
Generation of ER-HoxB8 GFP+ cells
A lentiviral vector for GFP was produced as described previously [50]. Virus concentrations were determined with the INNOTEST HIV antigen mAb kit (DiaSorin, Saluggia, Italy) and expressed as nanograms of p24GAG per microliter. ER-HoxB8 cells were transduced with 50 ng virus/5x104 cells in complete medium (+ 8 µg/mL Polybrene (Sigma-Aldrich, Munich, Germany)). Afterwards, ER-HoxB8 cells were cultured with standard culture medium. Transduction efficiency was around 70% as determined by flow cytometry analysis.
Generation of ER-HoxB8 VLA4α-/- cells via CRISPR/Cas9
ER-HoxB8 VLA4α-/- cells were generated via the CRISPR/Cas9 system of genome editing as described earlier [26, 51, 52].
Briefly, a guide RNA (gRNA) was determined, containing the target sequence for VLA4α (CD49d) and the cutting sequence for BsmBI (fw: 5´-caccgatccacatatatgtaacct-3´, rv: 5´-aaacaggttacatatatgtggatc-3´) allowing for the insertion of gRNA into lentiCRISPRv2 plasmid (Addgene, Cambridge, USA, Nr. 52961) after hybridization and phosphorylation of oligos. Plasmids were then transformed in DH5α-competent bacteria (ThermoFisher Scientific, Waltham, USA), incubated o/n and a mini-prep was inoculated the next day and prepared 24 h later. DNA was subsequently assigned for sequencing.
For virus propagation, HEK cells were split to 1.5x105 cells/mL and transfected the next day at 70% confluency with the lentiCRISPRv2gRNA transfer plasmid + packaging plasmid (psPAX2) + envelope plasmid (pCMV-VSV-G) in a 2:2:1 proportion. 48 h after transfection, the virus-containing supernatants were harvested and used for transduction of ER-Hoxb8 cells.
ER-HoxB8 monocytic wildtype cells were seeded at 3x105 cells/mL, infected and exposed to spinoculation in the presence of polybrene (8 µg/mL). Cells were then cultivated for 2 weeks as described. Selection of transduced cells started 3 weeks post infection by the addition of puromycin (InvivoGen, San Diego, USA) to the culture medium as the lentiCRISPRv2 plasmid comprises puromycin resistance. Lethal concentration of puromycin for wildtype cells was determined beforehand.
After two weeks of continuous selection, cells were tested for knockout of VLA4α by flow cytometry analysis using α-mouse-CD49d (#01271D, Pharmingen, BD Biosciences, San Jose, USA) and goat-α-rat IgG PE (#112-116-143, Dianova, Hamburg, Germany), qRT-PCR (Primer fw: 5'-accatcagcttgctacttgga-3', rv: 5'-acctgagtttttcagttctctcagt-3') and DNA sequencing of CRISPR/Cas9 target region.
Labeling
Differentiated ER-HoxB8 cells (day 3) were harvested and labeled with DIR or DID, both available from ThermoFisher Scientific, Waltham, USA. 1x106 cells/mL were labeled with 98.7 µM DIR/EtOH or 47.5 µM DID/EtOH for 5 min and washed three times. Labeling efficiency was checked by flow cytometry (FL4-H), and viability by trypan blue or 7AAD staining. For in vivo FRI experiments, 5x106 labeled cells/200 µL PBS were i.v. injected into the lateral tail vein of isoflurane-anesthetized mice. For MI experiments, harvested cells were incubated with 111In-Oxine (Mallinckrodt Deutschland, Hennef, Germany) at a concentration of approximately 1.2 MBq/1x106 cells for 20 min at 37°C. Cells were washed once, resuspended in 100 µL saline/1x106 cells and the amount of radioactivity was determined by scintillation counting (Wizard2, PerkinElmer, Waltham, USA). 10x106 labeled cells were i.v. injected 1 day post coronary artery ligation. In some animals, the initial distribution of labeled cells was analyzed by dynamic SPECT imaging. For this purpose, mice were put under isoflurane anesthesia and a tail vein catheter was placed. Immediately after the scan was started, 5x106 cells in 100 µL saline were slowly injected over the course of 60 s.
Viability
Viability of cells was assessed by trypan blue staining or 7-aminoactinomycin D staining (BioLegend, Fell, Germany) and subsequent flow cytometry analysis (FL3-H).
Adhesion assay
Cell adhesion was tested either on plastic surface or fibronectin coated surface. Differentiated (day 3) ER-HoxB8 cells were seeded (5x105 cells/well) onto untreated or fibronectin (FN; 25 µg/mL, Roche, Penzberg, Germany)-coated wells of a 24-well plate and allowed to adhere for 2 h at 37°C. After washing, remaining adherent cells were fixed with 2% glutaraldehyde, washed again and stained with 0.5% crystal violet (in 200 mM boric acid, pH 8). Cells were then lysed with 10% acetic acid and optical density was measured at 560 nm.
Transmigration assay
Spontaneous and chemokine-stimulated transmigration of differentiated ER-HoxB8 monocytes through a transwell filter was analyzed. 1x106 cells were applied to the upper chamber and no or 12 nM LTB4 (Biomol, Hamburg, Germany) was added to the lower chamber of the transwell filter system (5 µm pore size, ThermoFisher Scientific, Waltham, USA). Cells were allowed to migrate for 1 h at 37°C and cells migrated to the lower chamber were subsequently counted by flow cytometry (FSC/SSC).
ROS production assay
5x105 cells were stimulated with 100 nM phorbol myristate acetate (PMA, Abcam, Cambridge, UK) for 1 h or left untreated. 15 µM dihydrorhodamine 123 (DHR123, Sigma Aldrich, Munich, Germany) was added 15 min before the end of incubation. A subsequent washing step was followed by flow cytometry analysis (FL1-H).
Phagocytosis assay
The number of phagocytosing cells was determined by phagocytosis assay with fluorescently labeled beads. 5x105 cells were exposed to 5x106 beads (Fluospheres®Polystyrene Microspheres 1 µm, 530/ 30 nm, ThermoFisher Scientific, Waltham, USA) for 4 h at 37°C. After washing, cells were analyzed for fluorescent phagocytosed particles by flow cytometry (FL1-H).
111In-Oxine retention
111In-Oxine-labeled monocytes were incubated for 6, 24 or 48 h in 12-well plates (1×106 cells, labeled with approximately 1 MBq, in 2 mL medium per well). At the respective time points, cells were harvested as described above and the amount of radioactivity in the cell pellet and supernatant was assessed by gamma counter measurement.
Irritant contact dermatitis (ICD)
Contact dermatitis [53] was induced by the application of 3% croton oil (Sigma Aldrich, Munich, Germany)/olive oil-acetone mixture (1:4) at the left ear (seen from the dorsal surface); the right ear served as control and was exposed to olive oil-acetone mixture (1:4) only. Ears were shaved beforehand. FRI measurements took place the next day, baseline, and 1 h, 3 h, 6 h, 24 h after injection of labeled ER-HoxB8 cells. At the end of the experiments, ears were processed as frozen sections (Tissue Tek, O.C.T.™ Compound, Cryomold®, Sakura, Alphen aan den Rijn, Netherlands).
Cutaneous granuloma model (CG)
As similarly described before [54], mice where shaved and 200 µL of Biogel (P-100, Bio Rad, Hercules, USA) was injected as a “plug” subcutaneously at both the right and the left flank, with the left one (seen from the dorsal surface) containing 20 µg/200 µL lipopolysaccharide LPS (from Salmonella enterica serotype enteritidis, Sigma Aldrich, Munich, Germany) representing the inflammatory site and the right plug serving as control. FRI measurements were performed the next day, baseline, and 3 h, 6 h, 24 h, 30 h, and 48 h after injection of labeled ER-HoxB8 cells. Optionally, plugs were isolated, transferred to PBS and homogenized through a 70 µm cell strainer (Corning®, Sigma Aldrich, Munich, Germany). Purified cells were analyzed by flow cytometry.
Collagen-induced arthritis model (CIA)
CIA was procured as previously described [49]. In short, mice were immunized by subcutaneous injection of 100 µg bovine collagen type II (bCII, MD Biosciences)/0.05 M acetic acid emulsified in Complete Freund's Adjuvant (CFA; Sigma Aldrich) containing HKMT (4 mg/mL; InvivoGen) at the tail base of DBA/jdba1/j mice. 21 days later mice were optionally boosted i.p. with 100 µg bCII in IFA or left untreated. Animals were checked and scored for clinical symptoms at last 3 times a week, with disease scores ranging from 0-3/paw (S0 = no swelling; S1 = slight swelling of entire paw or one joint swollen; S2 = mild swelling of entire paw or more than one joint swollen; S3 = pronounced edematous swelling of entire paw or several joints or ankylosis). First signs of disease occurred around day 25 p.i. with animals reaching high disease scores (≥2) only a few days later. FRI measurements took place as mice showed clear clinical symptoms and were performed at baseline, and 1 h, 3 h, 6 h, and 24 h post injection of labeled ER-HoxB8 cells.
Arthritis in IL1 Ra-/- mice
Interleukin-1 receptor antagonist deficient (IL-1Ra-/-) mice will spontaneously develop an autoimmune T cell-mediated arthritis in the hind paws due to excessive IL-1 signaling [55], TNF-α [56] and IL-17 signaling [57]. It is associated with increased infiltration of neutrophils and monocytes in the arthritic ankle joint [58].
Myocardial infarction
Permanent ligation of the left anterior descending coronary artery was performed as described previously [59, 60]. Briefly, mice were i.p. injected with a mixture of Midazolam (4 mg/g bodyweight), Fentanyl (0.04 mg/g bodyweight) and Rimadyl (0.5 µL/g bodyweight) and kept under isoflurane anaesthesia and on a heating pad during surgery. Mice were intubated and ventilated using a Minivent ventilator. The left thorax was opened and the coronary artery permanently ligated with a suture (prolene 7.0). In sham operated animals, the suture was loosely placed without ligating the artery. The thorax (vicryl rapide 4.0) and skin (prolene 6.0) were closed with sutures before the animal was removed from the ventilator and allowed to recover on a heating pad. Food was provided on the cage floor for the first 48 h after surgery.
FRI measurements
In vivo imaging of labeled ER-HoxB8 cells by FRI in the ICD, the CG and the IL1RA-/- models was carried out using the IVIS Spectrum system (epi-illumination) by PerkinElmer, Waltham, USA (710 nm excitation/780 nm emission for DIR and 605 nm/680 nm for DID). For imaging the CIA model, we employed the In-vivo MS FX PRO system (trans-illumination) by Bruker, Billerica, USA (730 nm/790 nm for DIR). In both cases, mice were kept under anesthesia (DRÄGER Isofluran Vapor, Lübeck, Germany) and warm during the imaging process.
For ICD and CG experiments, imaging analysis and quantification was carried out using Living Image®Software (PerkinElmer) and for CIA experiments using Bruker MI (Bruker). Therefore, ROIs (region of interest) were drawn around the ears, the plug zone and paws based on anatomical context (by white light images) to determine average radiant efficiency [p/s/cm2/sr]/[µW/cm2] (IVIS) and mean intensity AU (FX Pro), respectively. Data was then corrected for autofluorescence, labeling efficiency and, in the case of parallel injection of DIR and DID labeled cells (two color simultaneous imaging approach), corrected for spectral overlap (compensation). Therefore, a dilution series of DIR and DID labeled cells alone or in combination was included in every imaging experiment, allowing recalculation of received data. Fluorescence intensity AU (arbitrary units) was calculated as fluorescence intensity = [S(t) - S(0)]/ Fcolour (one color imaging approach) and as fluorescence intensity = [(S(t) - S(0)) x Fcompensation]/ Fcolour (two color simultaneous imaging approach),with S(t) being the fluorescence signal within a ROI at a time t post injection, S(0) the fluorescence signal within the ROI at baseline, Fcolor the correction factor for the labeling efficiency of cells and Fcompensation the correction factor for spectral overlap.
SPECT Imaging
SPECT experiments were carried out using a small-animal SPECT/CT scanner (nanoScan, Mediso, Budapest, Hungary). During all in vivo scans, mice were kept under isoflurane anesthesia and on a heated bed to maintain body temperature. The initial biodistribution of 111In-Oxine-labeled monocytes was assessed by dynamic whole-body SPECT scans (9x10 min, FOV 100 mm) and static SPECT scans at approximately 3 h, 6 h, 24 h and 48 h post injection of cells (1×30 min, FOV 100 mm).
Myocardial perfusion imaging was performed on the day of surgery to assess the area at risk. 60 MBq 99mTc-Tetrofosmin in 100 µL was i.v. injected directly after closure of the thorax. Animals were then allowed to recover for one hour before undergoing static SPECT scanning of the thorax (1x30 min, FOV 26 mm) followed by native CT. On day 1 and day 2 p.i., the accumulation of monocytes in the infarct area was assessed by static thorax scans (1×60 min, FOV 26 mm). Each scan was followed by a CT scan of the respective region. On day 2, 5 µL ExiTron nano 6000 (Miltenyi Biotec, Bergisch Gladbach, Germany) per gram bodyweight was injected via a tail vein catheter 2 min before the CT was started to better delineate the heart.
SPECT images from cell tracking experiments were analyzed using in-house software MEDgical.
To quantify cell migration in the CG model, volumes of interest (VOI) were drawn over the Biogel pellet based on CT images.
For analysis of MI experiments, signal in the infarct zone was quantified by defining a VOI for the LV myocardium in contrast-enhanced CT. Area of infarction was defined as the part of the LV myocardium with < 25% maximum cardiac 99mTc-Tetrofosmin uptake. Areas affected by spill-over artefacts induced by strong 99mTc-Tetrofosmin accumulation in the adjacent liver were excluded from analysis. Signal in the remote myocardium was quantified by placing an oval VOI in the septum, as this is a region spared from damage in coronary artery ligation in C57BL/6 mice [60]. Biodistribution of injected cells to other organs was analyzed by placing representative oval VOIs within the respective organs.
All SPECT data from cell tracking experiments were corrected for radioactive decay, labeling efficiency and bodyweight by calculating mean standardized uptake units (SUVs) as SUV = [cimg(t) x BW]/ID, with cimg(t) being the concentration of radioactivity within a VOI at a time t post injection, decay corrected to t=0, and ID being the injected dose, i.e., the amount of radioactivity taken up by the injected cells.
Autoradiography
MI mice were sacrificed directly after the 48 h SPECT scan for ex vivo analysis. Following FRI measurements, the hearts were embedded in TissueTek® O.C.T.™ Compound (Sakura Finetek, Staufen, Germany) and snap-frozen at -20°C. 20 µm frozen sections were made and placed in a microimager (Micro-Imager V2, Biospace Lab, Nesles la Vallee, France) for 6 h to obtain autoradiographic images. Adjacent sections (10 µm) were collected for histological analysis.
Scintillation counting
Ex vivo radioactivity biodistribution was analyzed by scintillation counting (Wizard2 gamma counter, Perkin Elmer, Waltham, USA) and the radioactivity in respective organs was decay-corrected and expressed as percentage of injected dose per gram tissue (%ID/g).
Histology
Immunofluorescence staining
Frozen sections of isolated mice ears were fixed in acetone and permeabilized with TBS/Triton x-100. Samples were washed, blocked and incubated with primary antibody α-GFP Alexa Fluor® 647 coupled (1:100, #A-31852, Thermo Fisher Scienific, Waltham, USA). After washing, an HRP-coupled secondary antibody was applied (goat-α-rabbit IgG, #111-036-045, Jackson Immuno Research, West Grove, USA). Samples were incubated in acetate buffer + 10% AEC (3-Amino-9-Ethylcarbazol) + 0.05% H2O2 to stain for peroxidase activity. Sections were counterstained with hematoxylin.
For analysis by LI-COR® Odyssey® Imaging system, frozen sections were mounted with mounting medium (DAKO Fluorescence Mounting Medium, Real Carpinteria, USA). Sample analysis was carried out with LI-COR® Odyssey® Imaging system (LI-COR® Biosciences, Nebraska, USA) using the filter set for 700 nm, 21 µm resolution.
Tissue sections (7 µm) from formalin-fixed, paraffin-embedded ankle joints of IL-1Ra-/- mice were incubated for 2 h at RT in 10 mM citrate buffer (pH 6.0) for antigen retrieval. Tissue sections were incubated overnight at 4°C with rabbit α-GFP (1:800, #2525, Cell Signaling, Danvers, MA, USA) for staining of infiltrated GFP+ ER-HoxB8 cells. Sections were incubated with biotinylated goat anti-rabbit IgG as a second antibody followed by incubation with avidin-streptavidin-peroxidase (VECTASTAIN, Elite ABC HRP kit, Vector, Burlingame, USA). Peroxidase activity was stained with 3,3'-diaminobenzidine (DAB, Powervision DAB, Immunologic, Duiven, the Netherlands) in the presence of H2O2 and all sections were counterstained with hematoxylin.
Mac-3 immunohistochemistry on MI hearts was performed by first demasking in citrate buffer and blocking with peroxidase blocking solution. Slides were washed and incubated with primary antibody (1:50, Mac-3 Rat IgG1, #550292 BD Biosciences, Franklin Lakes, USA), followed by washing and incubation with secondary antibody (biotinylated mouse anti-rat, #550325 BD Biosciences, Franklin Lakes, US). After washing, slides were incubated with streptavidin (DAKO, Hamburg, Germany), followed by DAB solution (Sigma-Aldrich, St. Louis, USA, 49 mL PBS, 1 mL DAB, 20 mL H2O2) and counterstaining with hematoxylin. Slides were then dehydrated and covered with entellan.
Flow cytometry analysis
Analyzed cells were gated for viability, i.e., dead cells were discriminated by FSC/SSC.
Statistical analysis
Statistical significance was analyzed by paired or unpaired two-sided Student's t-test or Kruskal-Wallis Analysis and Dunn's Multiple Comparison or 2-way ANOVA and Bonferroni post-tests. P-values of p*<0.05, p**<0.01 and p***<0.001 were considered to be significant. Size of experimental groups was estimated according to the method Cohen's D for detection of medium effect sizes.
Abbreviations
AEC: 3-amino-9-ethylcarbazol; bCII: bovine collagen II; Bq: Becquerel; CFA: complete Freund's adjuvant; CG: cutaneous granuloma; CIA: collagen induced arthritis; CRISPR/Cas9: clustered regularly interspaced short palindromic repeats/ caspase9; CT: computed tomography; DHR123: dihydrorhodamine 123; EC: endothelial cell; EGFP: enhanced green fluorescent protein; ER: estrogen-regulated; FCS: fetal calf serum; FOV: field of view; FRI: fluorescence reflectance imaging; FSC: forward scatter; GFP: green fluorescent protein; GM-CSF: granulocyte-macrophage colony-stimulating factor; h: hour; HRP: horseradish peroxidase; i.p.: intraperitoneal; i.v.: intravenous; ICD: irritant contact dermatitis; IFA: incomplete Freund's adjuvant; IgG: immunoglobulin G; IL: interleukin; LPS: lipopolysaccharide; LV: left ventricle; MI: myocardial infarction; MR: magnetic resonance; mRNA: messenger ribonucleic acid; MRP: myeloid related protein; o/n: overnight; p.i.: post injection; PBS: phosphate buffered saline; pen/strep: penicillin/streptavidin; PMA: phorbol myristate acetate; qRT PCR: quantitative real-time polymerase chain reaction; ROS: reactive oxygen species; SCF: stem cell factor; SN: supernatant; SPECT: single photon emission computed tomography; SSC: side scatter; SUV: standardized uptake units; TLR4: toll-like receptor 4; TNF: tumor necrosis factor; VCAM: vascular cell adhesion molecule; VOI: volume of interest; WB: western blot; wt: wildtype.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors thank Ina Winkler, Sarah Köster, Christine Bätza, Roman Priebe, Stefanie Bouma, Wiebke Gottschlich, Irmgard Hoppe, Christa Möllmann, Heike Hater, Claudia Terwesten-Solé, Eva Nattkemper for excellent technical assistance. This study was partly funded by the German Research Foundation (DFG) Cluster-of-excellence EXC 1003 'Cells in Motion', Münster, Germany (FF project -2013-24), DFG CRC 656 'Cardiovascular molecular imaging', Münster, Germany (projects C06 and C10), DFG CRC 1009 'Breaking barriers' (projects B8 and B9), the Interdisciplinary Center of Clinical Research at the University of Münster (Vo2/014/09, Ro2/003/15 and core unit PIX) and by the Federal Ministry of Education and Research (BMBF), project AID-NET. L.H. was funded by the CiM-IMPRS Graduate School, Münster, Germany.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author Contributions
SG, LH, MS and JR conceived and designed the experiments; SG, LH, EG, MvdB performed the experiments; SG, LH, SH, MS, OF, SZ, SE, MK, TV, PvL, CS, and JR analyzed the data; SG, LH, MS and JR wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kantari C, Pederzoli-Ribeil M, Witko-Sarsat V. The role of neutrophils and monocytes in innate immunity. Contrib Microbiol. 2008;15:118-146
2. Freire MO, Van Dyke TE. Natural resolution of inflammation. Periodontol. 2000. 2013;63:149-164
3. Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10:427-439
4. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159-175
5. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762-774
6. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14:392-404
7. Nahrendorf M, Swirski FK, Aikawa E. et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037-3047
8. Panizzi P, Swirski FK, Figueiredo JL. et al. Impaired infarct healing in atherosclerotic mice with Ly-6Chi monocytosis. J Am Coll Cardiol. 2010;55:1629-1638
9. Siti HN, Kamisah Y, Kamsiah J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vascul Pharmacol. 2015;71:40-56
10. Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182-1190
11. Zhao G, Wang S, Wang Z. et al. CXCR6 deficiency ameliorated myocardial ischemia/reperfusion injury by inhibiting infiltration of monocytes and IFN-γ-dependent autophagy. Int J Cardiol. 2013;168:853-862
12. Sager HB, Dutta P, Dahlman JE. et al. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci Transl Med. 2016;8:342ra80-342ra80
13. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol. 2015;15:692-704
14. Ulbrich H, Eriksson EE, Lindbom L. Leukocyte and endothelial cell adhesion molecules as targets for therapeutic interventions in inflammatory disease. Trends Pharmacol Sci. 2003;24:640-647
15. Cox D, Brennan M, Moran N. Integrins as therapeutic targets: lessons and opportunities. Nat Rev. 2010;9:804-820
16. Franc BL, Acton PD, Mari C. et al. Small-animal SPECT and SPECT/CT: important tools for preclinical investigation. J Nucl Med. 2008;49:1651-1663
17. Hilderbrand SA, Weissleder R. Near-infrared fluorescence: application to in vivo molecular imaging. Curr Opin Chem Biol. 2010;14:71-79
18. Studwell AJ, Kotton DN. A shift from cell cultures to creatures: In vivo imaging of small animals in experimental regenerative medicine. Mol Ther. 2011;19:1933-1941
19. Ahrens ET, Bulte JWM. Tracking immune cells in vivo using magnetic resonance imaging. Nat Rev Immunol. 2013;13:755-763
20. Wang GG, Calvo KR, Pasillas MP. et al. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat Methods. 2006;3:287-293
21. Edgeworth J, Gorman M, Bennett R. et al. Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J Biol Chem. 1991;266:7706-7713
22. McDonald JU, Cortini A, Rosas M. et al. In vivo functional analysis and genetic modification of in vitro-derived mouse neutrophils. FASEB J. 2011;25:1972-1982
23. Rosas M, Osorio F, Robinson MJ. et al. Hoxb8 conditionally immortalised macrophage lines model inflammatory monocytic cells with important similarity to dendritic cells. Eur J Immunol. 2011;41:356-365
24. Redecke V, Wu R, Zhou J. et al. Hematopoietic progenitor cell lines with myeloid and lymphoid potential. Nat Methods. 2013;10:795-803
25. Ley K, Laudanna C, Cybulsky MI. et al. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678-689
26. Cong L, Ann Ran F, Cox D. et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819-823
27. Yang JT, Rayburn H, Hynes RO. Cell adhesion events mediated by alpha 4 integrins are essential in placental and cardiac development. Development. 1995;121:549-560
28. Vogl T, Ludwig S, Goebeler M. et al. MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood. 2004;104:4260-4268
29. Pruenster M, Vogl T, Roth J. et al. S100A8/A9: From basic science to clinical application. Pharmacol Ther. 2016;167:120-131
30. Germain RN, Miller MJ, Dustin ML. et al. Dynamic imaging of the immune system: Progress, pitfalls and promise. Nat Rev Immunol. 2006 p. 497-507
31. Chen C-W, Sowden M, Zhao Q. et al. Nuclear phospholipid scramblase 1 prolongs the mitotic expansion of granulocyte precursors during G-CSF-induced granulopoiesis. J Leukoc Biol. 2011;90:221-233
32. Friesenhagen J, Viemann D, Börgeling Y. et al. Highly pathogenic influenza viruses inhibit inflammatory response in monocytes via activation of Rar-related Orphan Receptor RORa. J Innate Immun. 2013;5:505-518
33. Fassl SK, Austermann J, Papantonopoulou O. et al. Transcriptome assessment reveals a dominant role for TLR4 in the activation of human monocytes by the alarmin MRP8. J Immunol. 2015;194:575-583
34. Nacken W, Roth J, Sorg C. et al. S100A9/S100A8: Myeloid representatives of the S100 protein family as prominent players in innate immunity. Microsc Res Tech. 2003;60:569-580
35. Tibble JA, Bjarnason I. Non-invasive investigation of inflammatory bowel disease. World J Gastroenterol. 2001;7:460-465
36. Soyfoo MS, Roth J, Vogl T. et al. Phagocyte-specific S100A8 / A9 protein levels during disease exacerbations and infections in systemic lupus erythematosus. J Rheumatol. 2009;36:2190-2194
37. Van Zoelen MAD, Vogl T, Foell D. et al. Expression and role of myeloid-related protein-14 in clinical and experimental sepsis. Am J Respir Crit Care Med. 2009;180:1098-1106
38. Gopal R, Monin L, Torres D. et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med. 2013;188:1137-1146
39. Kuipers MT, Vogl T, Aslami H. et al. High levels of S100A8/A9 proteins aggravate ventilator-induced lung injury via TLR4 signaling. PLoS One. 2013;8:e68694
40. Petersen B, Wolf M, Austermann J. et al. The alarmin Mrp8/14 as regulator of the adaptive immune response during allergic contact dermatitis. EMBO J. 2013;32:100-111
41. Austermann J, Friesenhagen J, Vogl T. et al. Alarmins MRP8 and MRP14 induce stress tolerance in phagocytes under sterile inflammatory conditions. Cell Reports. 2014;9:2112-2123
42. McDonald B, Pittman K, Menezes GB. et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362-366
43. Pittet MJ, Weissleder R. Intravital Imaging. Cell. 2011;147:983-991
44. Germain RN, Robey EA, Cahalan MD. A decade of imaging cellular motility and interaction dynamics in the immune system. Science. 2012;336:1676-1681
45. Lämmermann T, Afonso P V, Angermann BR. et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 2013;498:371-375
46. Choi M, Kwok SJJ, Yun SH. In vivo fluorescence microscopy: Lessons from observing cell behavior in their native environment. Physiology. 2015;30:40-49
47. Wang J, Hossain M, Thanabalasuriar A. et al. Visualizing the function and fate of neutrophils in sterile injury and repair. Science. 2017;358:111-116
48. Nicklin MJH, Hughes DE, Barton JL. et al. Arterial inflammation in mice lacking the Interleukin 1 receptor antagonist gene. J Exp Med. 2000:191
49. Vogl T, Eisenblätter M, Völler T. et al. Alarmin S100A8/S100A9 as a biomarker for molecular imaging of local inflammatory activity. Nat Commun. 2014;5:4593
50. Geurts J, Joosten L a B, Takahashi N. et al. Computational design and application of endogenous promoters for transcriptionally targeted gene therapy for rheumatoid arthritis. Mol Ther. 2009;17:1877-1887
51. Mali P, Yang L, Esvelt KM. et al. RNA-Guided Human Genome Engineering via Cas9. Science. 2013;339:823-826
52. Platt RJ, Chen S, Zhou Y. et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440-455
53. Tonelli G, Thibault L, Ringler I. A Bio-assay for the concomitant assessment of the antiphlogistic and thymolytic activities of topically applied corticoids. Endocrinology. 1965;77:625-634
54. von Stebut E, Metz M, Milon G. et al. Early macrophage influx to sites of cutaneous granuloma formation is dependent on MIP-1alpha /beta released from neutrophils recruited by mast cell-derived TNFalpha. Blood. 2003;101:210-215
55. Horai R, Saijo S, Tanioka H. et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000;191:313-320
56. Horai R, Nakajima A, Habiro K. et al. TNF-α is crucial for the development of autoimmune arthritis in IL-1 receptor antagonist-deficient mice. J Clin Invest. 2004;114:1603-1611
57. Nakae S, Saijo S, Horai R. et al. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. PNAS. 2003;100:5986-5990
58. Geven EJW, van den Bosch MHJ, Di Ceglie I. et al. S100A8/A9, a potent serum and molecular imaging biomarker for synovial inflammation and joint destruction in seronegative experimental arthritis. Arthritis Res Ther. 2016;18:247
59. Fishbein MC, Maclean D, Maroko PR. Experimental myocardial infarction in the rat; qualitative and quantitative changes during pathological evolution. Am J Pathol. 1978;90:57-70
60. Kumar D, Hacker TA, Buck J. et al. Distinct mouse coronary anatomy and myocardial infarction consequent to ligation. Coron Artery Dis. 2005;16:41-44
Author contact
![]() Corresponding author: rothjde
Corresponding author: rothjde