Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Imaging biomarkers of...
Microglia
Matrix Metalloproteinases (MMPs)
Cell-based therapies and in vivo...
Future perspectives
Discussion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(10):2603-2620. doi:10.7150/thno.24128 This issue Cite
Review
In vivo imaging biomarkers of neuroinflammation in the development and assessment of stroke therapies - towards clinical translation
Bastian Zinnhardt1,2,3,4,6 ![]() , Maximilian Wiesmann5, Lisa Honold1*, Cristina Barca1,4, Michael Schäfers1,3,6, Amanda J Kiliaan5, Andreas H Jacobs1,2,3,4,7
, Maximilian Wiesmann5, Lisa Honold1*, Cristina Barca1,4, Michael Schäfers1,3,6, Amanda J Kiliaan5, Andreas H Jacobs1,2,3,4,7 ![]()
1. European Institute for Molecular Imaging (EIMI), Westfälische Wilhelms University Münster, Münster, Germany
2. EU 7 th FP Programme “Imaging Inflammation in Neurodegenerative Diseases (INMiND)”
3. Cells in Motion (CiM) Cluster of Excellence, University of Münster, Münster, Germany
4. PET Imaging in Drug Design and Development (PET3D)
5. Department of Anatomy, Radboud university medical center, Donders Institute for Brain, Cognition & Behaviour, Nijmegen, The Netherlands
6. Department of Nuclear Medicine, Universitätsklinikum Münster, Münster, Germany
7. Department of Geriatrics, Johanniter Hospital, Evangelische Kliniken, Bonn, Germany
* Current address: Center for Systems Biology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA
Received 2017-12-1; Accepted 2018-1-31; Published 2018-4-3
Abstract

Modulation of the inflammatory microenvironment after stroke opens a new avenue for the development of novel neurorestorative therapies in stroke. Understanding the spatio-temporal profile of (neuro-)inflammatory imaging biomarkers in detail thereby represents a crucial factor in the development and application of immunomodulatory therapies. The early integration of quantitative molecular imaging biomarkers in stroke drug development may provide key information about (i) early diagnosis and follow-up, (ii) spatio-temporal drug-target engagement (pharmacodynamic biomarker), (iii) differentiation of responders and non-responders in the patient cohort (inclusion/exclusion criteria; predictive biomarkers), and (iv) the mechanism of action. The use of targeted imaging biomarkers for may thus allow clinicians to decipher the profile of patient-specific inflammatory activity and the development of patient-tailored strategies for immunomodulatory and neuro-restorative therapies in stroke. Here, we highlight the recent developments in preclinical and clinical molecular imaging biomarkers of neuroinflammation (endothelial markers, microglia, MMPs, cell labeling, future developments) in stroke and outline how imaging biomarkers can be used in overcoming current translational roadblocks and attrition in order to advance new immunomodulatory compounds within the clinical pipeline.
Keywords: Neuroinflammation, Stroke, Imaging, Microglia, TSPO, Matrix Metalloproteinases, Brain-gut axis
Introduction
Cerebral ischemia is one of the major causes of death and disability world-wide due to an overall increase in stroke burden. Ischemic stroke represents the most prevalent form of stroke [1, 2]. Currently available treatment options are limited to tissue plasminogen activator (tPA) and endovascular treatment after stroke solely aiming at recanalization of an occluded vessel [3-5]. Over a thousand neuroprotective candidates, targeting different parameters of the pathophysiological cascade after stroke, have been suggested by preclinical research [3]. However, there is a remarkable gap between the huge number of candidates and their clinically effective translation to stroke patients. The failure of most clinical trials is partially due to the complexity of molecular alterations and their kinetics with regards to the variety of cell types and molecules and their respective functions at different stages after cerebral ischemia [6, 7]. Together with the enormous average costs of clinical development programs of up to 1.4 billion dollars [8], there is a need for a refinement of preclinical and clinical stroke research [9]. Apart from improving quality of research, data acquisition and collaborations, novel non-invasive molecular imaging biomarkers and techniques may help to overcome the translational roadblock in stroke research and to facilitate drug development processes [10].
As current recanalization treatment options are limited to the first couple of hours after stroke, there is an urgent need for molecular imaging techniques employing other markers besides those targeting cerebral blood flow, oxygen and glucose consumption. Therefore, a rapid imaging workup is necessary, and processes like the inflammatory response after stroke remain to be investigated as these represent a variety of attractive novel targets for neurorestorative stroke therapies and extension of the therapeutic window. The multi-factorial and multi-phasic nature of cerebral ischemia, with an initial pro-inflammatory and a delayed repair and regeneration phase, emphasizes the intriguing possibility to shift the inflammatory response towards a pro-resolving, anti-inflammatory phenotype [11, 12]. The detailed understanding of the dynamics of cerebral and peripheral inflammation markers is a key step towards the development of next generation immunomodulatory therapies for ischemic stroke.
Molecular imaging combining positron emission tomography (PET) and single photon computed emission tomography (SPECT) together with functional and anatomical imaging by magnetic resonance imaging (MRI) in hybrid preclinical and clinical scanners represents an important approach to decipher these molecular alterations after stroke in a spatio-temporal manner. Multi-modality imaging bears the advantage of being non-invasive, allowing intra-individual follow-up of multiple disease processes at a time, while at the same time reducing experimental attrition and animal numbers as well as increasing statistical power and translatability [13]. The superb sensitivity of PET (pico-nanomolar range) and SPECT [14, 15] in combination with the fast acquisition time, clinical availability and excellent soft tissue contrast obtained by MRI, allows for the parallel intra-individual determination of various parameters after stroke [16].
In particular, nuclear imaging techniques like PET have been widely used for drug development purposes: (i) direct radiolabeling of a new drug candidate for distribution (microdosing; <µg), (ii) mathematical modeling approaches to decipher drug-receptor interactions and subsequent assessment of pharmacodynamic responses, (iii) lead validation and optimization, (iv) following the (patho-)physiological response to a new drug over time, (v) allowing early diagnosis, and (vi) following longitudinally an imaging biomarker for patient monitoring [13, 17-19]. Ideally these imaging parameters can be used to enhance our understanding of disease pathogenesis and serve as predictive biomarkers for patient selection and stratification to improve clinical study protocols.
This review will highlight innovative preclinical and clinical imaging biomarkers targeting various aspects of neuroinflammation beyond monitoring and re-establishing cerebral blood flow (CBF). We will highlight their potential use in disease modulation and drug development processes towards personalized stroke therapies.
Imaging biomarkers of neuroinflammation
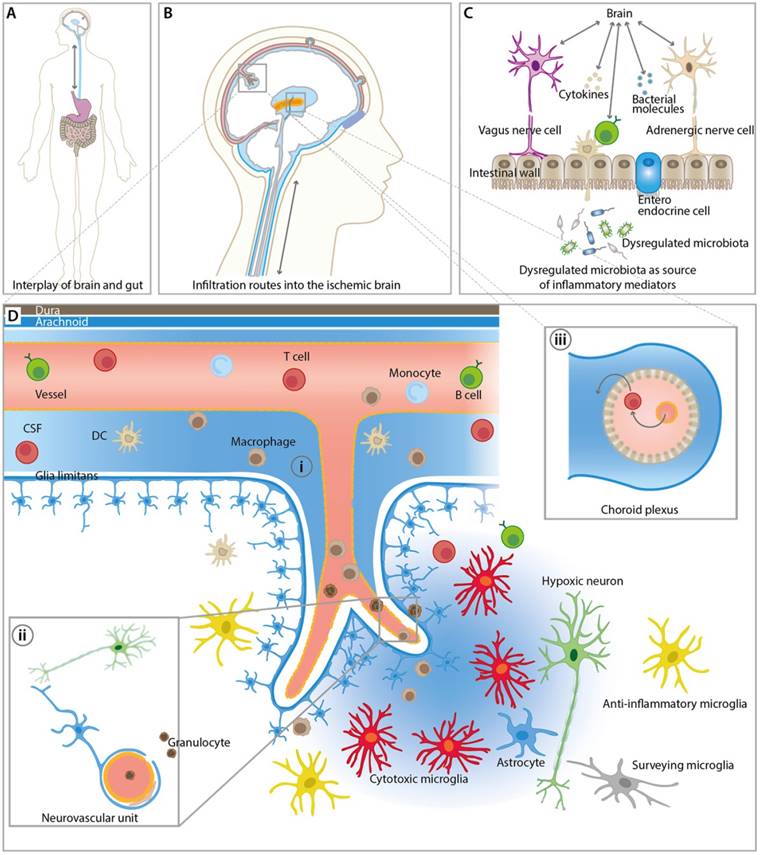
The lack of blood supply to certain brain regions after stroke leads to an orchestrated inflammatory response [12]. Resident and peripheral immune cells become rapidly activated and peripheral immune cells can infiltrate the brain through three major routes including (i) direct crossing of the BBB of (meningeal) blood vessels, (ii) direct crossing over the subarachnoid space into the brain parenchyma and (iii) infiltration via the blood-CSF barrier at the choroid plexus (Figure 1) [20]. The inflammatory response can roughly be divided into an initial damaging period with expression and release of pro-inflammatory mediators and into a late neuroprotective phase of inflammation that is hallmarked by release and expression of anti-inflammatory mediators [12, 21]. However, Hu and colleagues described an increase of anti-inflammatory biomarkers a few days after stroke followed by a progressive increase of pro-inflammatory markers in a mouse stroke model [22]. These contradictory results underline the complex overall dynamics, heterogeneity and multi-phasic nature of the immunological response after stroke making the development and application of immunomodulatory therapies challenging [12, 21]. This heterogeneity is an interesting possibility to extend the therapeutic window after stroke [23]. Given the heterogeneous location and size of infarctions in the clinical setting, the non-invasive assessment of the time course of inflammatory parameters by molecular imaging (Table 1) may help to investigate the pathophysiological response to immunomodulatory therapies in a temporal manner (Table 2). Another possible application of molecular imaging is therapy selection and patient stratification for immunomodulatory treatment, thereby increasing the power of a clinical study.
The following paragraphs will highlight recent advances in imaging of several aspects of the inflammatory cascade and their use in the investigation of treatment responses in preclinical and clinical settings.
Overview of inflammatory pathways and imaging targets in the CNS and periphery. (A) Interplay between gut and the brain as an important route for inflammatory signals from and to the brain influencing stroke outcome. (B) Overview of key infiltration routes of peripheral innate and adaptive immune cells. Peripheral immune cells can enter into the ischemic brain from the (i) subarachnoid space (SAS), (ii) direct crossing of the blood brain barrier, and (iii) the choroid plexus across the blood-cerebrospinal fluid (CSF) barrier. (C) Dysregulated microbiota and resident intestinal immune cells send inflammatory mediators including cytokines via the hypothalamus-pituitary-adrenal glands axis (HPA), the autonomic nervous system, and the enteric nervous system (ENS). (D) Detailed view of the inflammatory events after ischemic stroke at the neurovascular unit (NVU) and SAS. CNS-resident microglia and astrocytes become activated and, depending on their polarization state, promote disease progression and repair. Resident and infiltrating immune cells exert their bi-modal functions via cell contact-dependent mechanisms and through the action of soluble inflammatory mediators. The NVU represents the target structure for functional tissue repair by neuroinflammatory processes. The NVU components may serve as imaging targets for the investigation of novel image-guided therapies targeting the rescue of NVU function.
Selected molecular imaging studies highlighting the dynamics of imaging biomarkers after stroke.
| Marker/Target | Imaging Modality | Compound/Tracer | Species | Occlusion time | Peak signal after stroke | Reference |
|---|---|---|---|---|---|---|
| VCAM-1 | MR | MPIOs-αVCAM-1 | C57BL/6 mice | pMCAo and tMCAo (various) | 24 hours - 5 days | [34] |
| P-Selectin | MR | MPIOs-αP-selectin | Swiss mice | MCAo and TIA (15 min) | 48 hours | [40] |
| TSPO | PET | [11C]PK11195 | Sprague -Dawley rat | tMCAo (1h) | 7 days | [52] |
| TSPO | PET | [18F]DPA-714 | Sprague-Dawley rats | tMCAo (2h) | 11 days | [53] |
| TSPO | PET | [18F]DPA-714 | Sprague-Dawley rats | tMCAo (1.5 h) | 7 days | [72] |
| TSPO | PET | [18F]DPA-714 | C57BL/6 mice | tMCAo (30 min) | 14 days | [60] |
| TSPO | PET | [18F]DPA-714 | Balb/c mice | tMCAo (60 min) | 10 days | [70] |
| TSPO | PET | [11C]PBR28 | Sprague-Dawley rats | M2Cao (1.5 h) | 4-14 days | [71] |
| TSPO | PET | [18F]DPA-714 | human | n.a. | n.a. | [64] |
| cystine-glutamate antiporter (system xc-) | PET | [18F]FSPG | Sprague-Dawley rats | tMCAo (1.5h) | 3-7 days | [72] |
| α4β2 Nicotinic Acetylcholine Receptor | PET | 2-[18F]-fluoro-A85380 | Sprague-Dawley rats | tMCAo (2h) | 7 days | [77] |
| Matrix metallo-proteinases | PET | [18F]BR-351 | C57BL/6 mice | 30 min | 7 days | [60] |
Selected studies of anti-inflammatory treatments in stroke and their respective nuclear imaging readouts.
| Drug | Target | Imaging readout | Effect | Reference |
|---|---|---|---|---|
| Multi-nutrient diet | Broad spectrum | [18F]DPA-714, DTI, rsfMRI | Inflammation ↓, DTI, rsfMRI ↑, functional outcome ↑ | [54] |
| Minocycline | Broad spectrum | [18F]DPA-714 and [18F]PBR06 PET, MRI | Inflammation ↓, functional outcome ↑ | [69, 76] |
| sulfasalazine and S-4-CPG | inhibition of system xc | [18F]DPA-714, [18F]FSPG | Inflammation ↓ | [72] |
| DhβE | α4β2 antagonist | [11C]PK11195 and 2[18F]-fluoro-A85380 | Inflammation ↑ | [77] |
| AMD3100 | CXCR4 antagonist | [18F]DPA-714 | Inflammation ↓ | [70] |
Infiltration of immune cells
Leukocytes are among the first inflammatory cells to enter the ischemic brain within hours after ischemia and, depending on the animal model, infiltration peaks about three days after stroke [6, 7, 24]. The infiltration of leukocytes depends on adhesion markers that are expressed on leukocytes and endothelial cells, including P/E-Selectin, ICAM-1 and vascular cell adhesion molecule 1 (VCAM-1). These adhesion markers represent an early and essential step in the recruitment of leukocytes into the ischemic brain and, therefore, provide an important therapeutic target for modulation of leukocyte infiltration [25].
Beneficial effects of blocking or genetic deletion of these adhesion molecules on functional and molecular parameters in different animal models of ischemic stroke have been studied extensively (reviewed by Yilmaz and Granger [25]).
Several imaging approaches targeting intracellular and cell surface markers have been used to investigate their temporal expression profile after cerebral ischemia (for review: [16, 26, 27]). Among the imaging methods for tracking neutrophil recruitment to the infarction site, intravital imaging is increasingly being used. The applicability of intravital microscopy, however, is limited by the invasiveness of the disease models, as well as the relatively low temporal resolution and translatability into clinical practice [28]. Imaging approaches with PET, SPECT, and MRI represent imaging techniques with a higher translational value due to their non-invasive character.
Most of the imaging paradigms targeting cell adhesion markers are based on molecular MRI. Molecular MRI utilizes contrastophores (either gadolinium- or iron oxide-based) that are linked to a protein of interest using a targeting moiety (pharmacophore: protein, peptide, antibody or enzymatic substrate). Over the last years, micro-sized particles of iron oxide (MPIOs) have been shown to possess superior characteristics in terms of sensitivity and specificity compared to classical ultra-small particles of iron oxide (USPIOs) [16, 29]. However, data on the application of imaging biomarkers for the assessment of treatment efficacy in stroke is still limited.
VCAM-1
Several approaches have been undertaken to image VCAM-1 expression in infarcted areas [30-32]. More recent studies focus on the use of MPIOs targeted to VCAM-1. VCAM-1 activation was visualized within hours after stroke by MRI and the VCAM MPIOs-mediated signal was compared with diffusion weighted imaging (DWI). Interestingly, the spatial extent of VCAM-1 expression was found to be considerably larger than the detectable lesion delineated by DWI. The authors concluded that the VCAM MPIOs might therefore delineate penumbral and core regions of the stroke. Ischemic preconditioning was found to significantly reduce VCAM MPIOs levels and improved functional outcome after stroke [33]. In line with these findings, Gauberti et al. demonstrated spatio-temporal kinetics of VCAM-1 surface expression on endothelial cells after stroke by using MPIOs conjugated to monoclonal VCAM-1 antibodies (MPIOs-αVCAM-1). VCAM-1 expression was found perilesional and peaked after 24 hours for up to 5 days, depending on the severity of the stroke model. The activation of VCAM-1 in the periphery suggests that noninvasive imaging of this inflammatory reaction could be useful to identify the penumbra.
Interestingly, MPIOs-αVCAM-1 may be used to demonstrate the efficacy of immunomodulatory therapies with celecoxib and atorvastatin. Both compounds reduced VCAM-1 expression in the contralateral cortex, but not in the ischemic cortex [34]. Results in other disease models including Alzheimer's disease encourage the use of MPIOs-αVCAM-1 as an imaging biomarker for VCAM-1 expression [35]. To overcome current limitations of contrastophores, another study used liposomes loaded with the neuroprotectant Citicoline. Citicoline has the advantage of inherent chemical exchange saturation transfer (CEST) signal, which can be detected by MRI. In a targeted theranostic approach, Citicoline-loaded liposomes were specifically targeted against VCAM-1, leading to improved imaging-guided drug delivery to the ischemic area in a label-free fashion [36, 37]. However, it should be noted that this signal was detected in areas of BBB damage, potentially indicating the presence of unspecific leakage of liposomes into damaged tissue [36].
Selectins
E- and P-selectin expression, as an important marker of the tether-roll phase of leukocytes, was visualized in vivo by T2-type glyconanoparticle reagent GNP-sLex [38]. In this study, E- and P-selectin could be visualized by cross-linked iron oxide (CLIO) nanoparticles conjugated to anti-human E-selectin (CLIO-F(ab')(2)). Interestingly, this study described a hypointensity in the T2* area only 3 hours after permanent stroke; whereas, this hypointense area could only be detected 24 hours after transient ischemia. These results underline the sensitivity issues related to these techniques. To address this issue, paramagnetic gadolinium diethylenetriaminepentaacetic acid (Gd-DTPA) conjugated to Sialyl Lewis X (Slex) was designed. The authors reported improved targeting of the tracer to E- and P-selectin mediated by Slex, as it mimics P-selectin glycoprotein ligand-1 tetrasaccharide Slex, which mediates the binding of leukocytes and platelets to activated endothelium [39].
Strikingly, antibody-based microparticles of iron oxide targeting P-selectin (MPIOs-αP-selectin), similar to the previously addressed MPIOs-αVCAM-1 [34, 35], could be found in mouse brains 24 hours after stroke induction. In contrast to MPIOs-αVCAM-1, MPIOs-αP-selectin was able to identify and delineate transient ischemic attacks (TIA) 24 hours after induction. Using these microparticles, the authors could discriminate TIAs from epilepsy and migraine-mimicking TIAs in the clinical setting [40], suggesting sensitivity and specificity of the compound for TIAs.
Despite the high translational value of MRI-based assessment of adhesion marker profiles, and first successful theranostic approaches, these techniques still suffer from various drawbacks, including (i) low sensitivity, (ii) low specificity, (iii) quantification issues, and (iv) problems with possible in vivo toxicity of the molecular imaging marker [34, 37, 41]. Drug-loaded micelles/liposomes or activatable probes based on enzymatic substrates [16, 42] in combination with paramagnetic particles may further allow the combination of a therapeutic approach with improved diagnosis (theranostics) [36, 37, 41].
Microglia
Microglia have been implicated as important mediators in a variety of diseases. Under physiological conditions, microglia constantly screen the brain parenchyma for pathophysiological alterations [12, 43]. Under pathological conditions, microglia fulfill a dual role. In the acute phase (days) of ischemic stroke, microglia produce pro-inflammatory mediators such as TNF-α, IL-1β and reactive oxygen species (ROS), i.e., proinflammatory M1-like microglia. Thereafter, under chronic (days-weeks) conditions, microglia obtain an anti-inflammatory M2-like phenotype, i.e., anti-inflammatory M2-like microglia. The neuroprotective function of these M2-like microglia is mediated by the release of IL-10 and TGF-β and other growth factors. However, it should be noted that the concept of M1 and M2 microglia is rather simplified and only represents two aspects of microglia polarization, at the same time neglecting a variety of different sub-phenotypes [44]. However, it has to be pointed out that in chronic post-stroke phases inflammation may spread from focal to remote areas following Wallerian degeneration and may thus not solely be beneficial [45]. Contrarily, it may worsen the outcome and lead to cognitive impairment, again highlighting the transient function of the inflammatory response. (for review: [46]).
One important marker of general microglial activation (i.e., not function of microglia and infiltration of macrophages), is the 18 kDa translocator protein (TSPO) located on the outer mitochondrial membrane. TSPO is part of mitochondrial permeability transition pore (MPTP) and is involved in shuttling cholesterol across the mitochondrial membrane and in steroid synthesis [47-49]. In the past 30 years, TSPO has become an attractive imaging target in neuroinflammation since basal expression levels in the brain cells (microglia, astrocytes, endothelial and smooth muscle cells, subpial glia, intravascular monocytes and ependymal cells) are relatively low under physiological conditions, while it becomes highly upregulated upon neuroinflammatory stimuli in glial cells (microglia and astrocytes) and macrophages in different models of neurological disease [48-55]. Imaging studies in ischemic stroke revealed TSPO to be highly expressed by microglia/macrophages in the infarct core, peri-infarct areas, as well as in remote areas [45, 56-60].
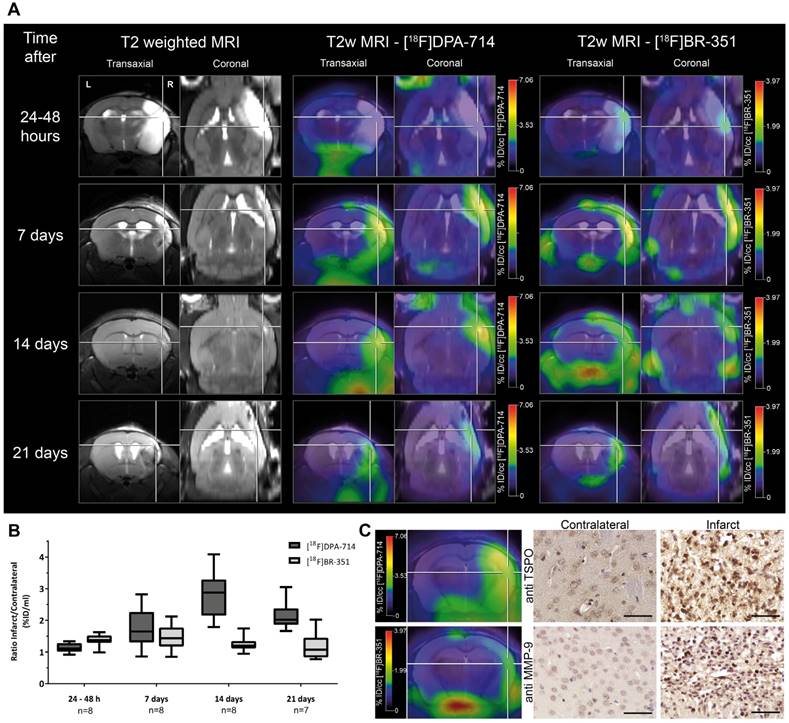
TSPO tracers can be subdivided into first and second generation tracers. The most prominent first generation TSPO ligand is [11C]PK11195. However, it suffers from a poor signal-to-noise ratio, and short half-life of the 11C-label leading to the development of second generation tracers with improved bioavailability, higher specificity, and favorable signal-to-noise ratios. Second generation TSPO tracers like [18F]PBR06, [11C]PBR-28, [125I]CLINDE, [18F]GE-180, [18F]DPA-714 were used in target validation studies in preclinical stroke (Figure 2) [53, 60-63] and clinical stroke [64, 65], allowing an improved understanding of the spatio-temporal profile of TSPO as an imaging biomarker.
Investigation of the temporal dynamics of [18F]DPA-714 (TSPO) and [18F]BR-351 (MMPs) uptake after transient middle cerebral artery occlusion (tMCAo). (A) The inter-individual comparison highlights differential time-dependent radiotracer uptake patterns. (B) [18F]DPA-714 uptake was significantly increased from day 7, peaked at day 14, and was still significantly elevated 21 days after tMCAo. [18F]BR-351 was significantly increased after 24 to 48 hours after tMCAo and 7 days. Data are represented as ratio ± SD. (C) Comparison of in vivo PET data with immunohistochemistry for TSPO and MMP-9. Both markers are increased in the infarct zone. Scale bar: 50 µm. (Adapted with permission from [60], copyright 2015 SAGE Publishing Group).
Recently, a third generation of TSPO tracers was described. Although a general definition of the requirements for a third generation tracer is lacking, [11C]ER176 and [18F]GE180 were recently described as third generation tracers being less susceptible for the rs6971 polymorphism [66-68], However the experimental data supporting this is relatively sparse [68] or do not fulfill the expectations [67].
Application of second generation tracers in stroke allowed detection of increased inflammation in the infarct region from subacute 3 days post-stroke, while the peak of microglia/macrophage response was found around 7-14 days after stroke for up to 7 months post-stroke in remote areas, depending on the stroke model in mice and rats (Table 1) [53, 60, 69-73]. Especially at late time points after stroke, TSPO-positive astrocytes contribute to the amount of TSPO-positive cells [53, 71, 74]. Thus, TSPO is a useful biomarker of inflammation from sub-acute to chronic phases after stroke.
The knowledge about the spatiotemporal profile of TSPO expression was further used to monitor therapy effects in experimental stroke for subsequent translation (Table 2). Minocycline, a well described antibiotic with anti-inflammatory function [75], was applied in a rat model of cerebral ischemia. TSPO levels, indicated by [18F]DPA-714 PET, were reduced in response to minocycline treatment. However, no effect on stroke size was observed and functional parameters were not obtained [76]. This study underlines the still open question of when and how to therapeutically interfere with the post-ischemic neuroinflammatory cascade to serve a beneficial tissue and functional outcome. Another study using the PET ligand [18F]PBR06 showed a correlation between [18F]PBR06 radiotracer uptake and impaired motor function [69]. In contrast to the previous study by Martin et al., this group demonstrated treatment effects of minocycline through improved motor function 22 days after stroke, along with decreased [18F]PBR06 uptake after minocycline treatment. However, this study involved longer observation times and more sophisticated treatment readouts, i.e., motor function by rot-a-rod [69]. Domercq et al. have further shown that the inhibition of Cystine-glutamate antiporter (system xc-) resulted in reduced [18F]DPA-714 uptake and microglial M1 proinflammatory markers 7 days after treatment onset (CCL2, TNF and iNOS) [72]. In line with these results, treatment with an α4β2 antagonist reduced neuroinflammation seven days post stroke in rats, as measured with the first generation TSPO tracer [11C]PK11195 [77]. The anti-inflammatory effect of AMD3100, a CXCR4 antagonist, was evaluated by [18F]DPA-714 in a stroke model for up to 16 days after stroke induction. In line with previous studies, the peak of [18F]DPA-714 uptake was found around 10 days after stroke. A reduction of [18F]DPA-714 after AMD3100 was observed three days after stroke, but was not detectable after seven days. No effects on functional parameters were investigated [70].
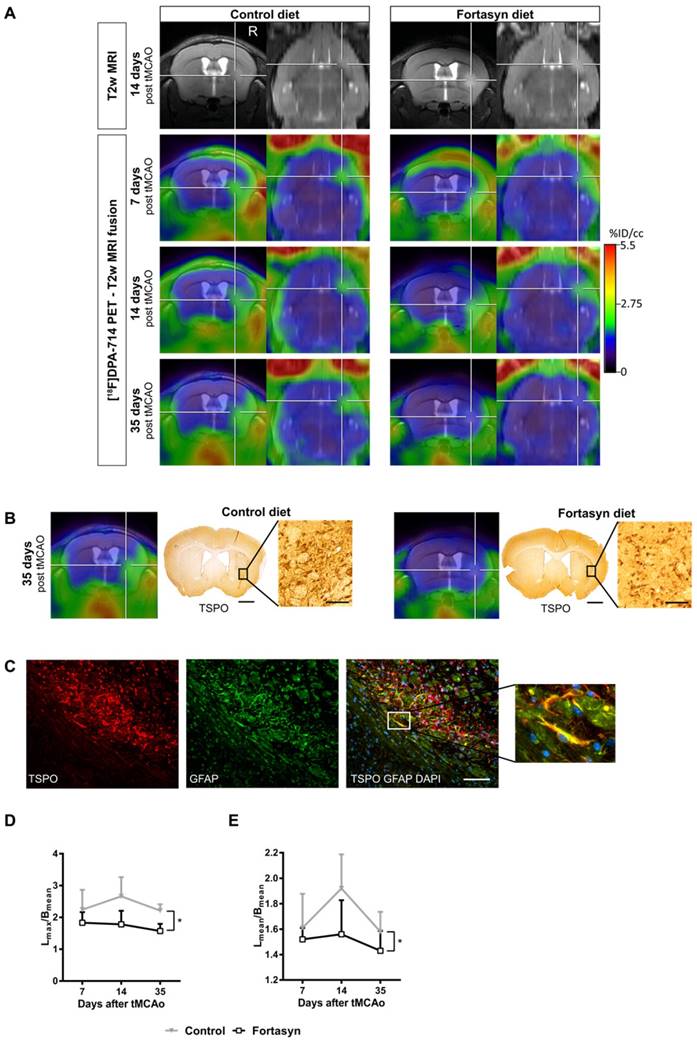
Recent studies highlight the potential of novel diet-based interventions in a variety of disease models [54, 78-83]. In a multicenter study, the potential therapeutic use of a multi-component diet enriched with, e.g., omega-3 long-chain polyunsaturated fatty acids (n3-LCPUFAs), membrane precursors, vitamins, and anti-oxidants on several parameters including microglia were investigated. The same diet is already being applied in the management of patients with early Alzheimer's disease [84, 85]. In order to assess the potential treatment effects of this multi-component diet, a bi-modal translational neuroimaging approach was employed to assess treatment effects on perfusion (arterial spin labeling, ASL), brain structure (diffusion tensor imaging, DTI) and function (resting state functional MRI, rsfMRI), inflammation/microglia ([18F]DPA-714 PET) and functional parameters (behavioral testing). Interestingly, it could be shown that the multi-component diet was able to improve the imaging parameters, as well as functional outcome after stroke (Figure 3) [54]. The beneficial effects of dietary interventions enriched with n3-LCPUFAs may be attributed to different metabolites of n3-LCPUFAs, so called pro-resolving factors (resolvins). These are able to increase phagocytosis and exert anti-inflammatory functions on glial cells via peroxisome proliferator-activated receptor-γ (PPARγ) modulation, ω-3 FA incorporation into the cell membrane, and inhibition of ion currents [for review: 62]. Moreover, in preclinical studies, docosahexaenoic acid (DHA), an n3-LCPUFA, and its oxygenated derivates, docosanoids, have been shown to regulate gene expression of inflammatory mediators like PPARγ; for further review see [87].
These results underline the striking potential of dietary interventions in modulation of stroke pathogenesis and should be considered for clinical translation in the management of stroke patients. Notably, microglia/macrophage imaging in combination with sophisticated MR neuroimaging techniques and ex vivo tissue biomarkers (e.g., polarization markers) has proven a powerful tool for detection of treatment effects and may serve as a blueprint for clinical translation of other neuroprotective compounds.
[18F]DPA-714 PET in the investigation of diet-mediated immunomodulatory effects on microglial activation. (A) Intra-individual follow up with [18F]DPA-714 PET/MRI of mice fed with a control diet (left) versus mice fed with the Fortasyn diet (right). Fortasyn-fed mice showed a reduced uptake of [18F]DPA-714 in the infarction after 35 days. (B) Comparison of [18F]DPA-714 with TSPO immunohistochemistry. TSPO levels were reduced in Fortasyn-fed animals (scale bars: 1000 µm and 50 µm). (C) TSPO (red) was also expressed by astrocytes (green) 35 days after tMCAo (scale bar: 100 µm). (D) Values represent mean ± SD. [18F]DPA-714 mean uptake ratios were significantly reduced from 7 to 35 days after tMCAo in the Fortasyn group when compared to control animals (p<0.029). (E) The maximum [18F]DPA-714 radio uptake ratios show reduced [18F]DPA-714 uptake in the Fortasyn diet group from 7 to 35 days (p<0.016). (Adapted with permission from [54], copyright 2017 Ivyspring International Publisher).
Despite the advances in preclinical microglia imaging, clinical data on TSPO in ischemic stroke is still relatively sparse. Imaging with the first generation TSPO ligand [11C]PK11195 revealed early (72 hours post stroke) changes as well as chronic changes (up to months) in remote areas, indicative of Wallerian degeneration [45, 57, 58, 88, 89]. Besides [11C]PK11195, second generation TSPO ligands have been used in initial clinical studies. Gulyás and colleagues investigated the TSPO signal evolution within the ischemic region and peri-infarct regions using the second generation TSPO tracer [11C]vinpocetine. The authors reported the highest radiotracer uptake in the peri-infarct regions independent of the time after the insult. However, the authors also reported very moderate BPND values for [11C]vinpocetine [90]. In another study, the same group reported no significant improvements of [11C]vinpocetine in comparison to the first generation tracer [11C]PK11195 [91]. In a single case study, Kreisl et al. found increased binding of [11C]PBR28 after subacute infarction [92], but this tracer has not yet been used in further stroke research. The fluorine 18 label of [18F]DPA-714 has the advantage of allowing longer scan times and broader clinical availability, as evaluated in nine stroke patients. Increased [18F]DPA-714 uptake was found within and around the infarct, exceeding the blood brain barrier damage, as indicated by Gd-enhanced T1-weighted MRI [64]. Nevertheless, future studies will have to investigate the relative contribution of the BBB damage for radiotracer uptake in stroke.
Using the SPECT TSPO ligand [125I]CLINDE, Feng et al. investigated the spatial and temporal change of TSPO levels 28 days and 8 months after middle cerebral artery stroke. Increased distribution volume (VT, in mL cm-3) values were observed mostly in peri-infarct areas after 28 days and decreased to baseline values after 8 months. Interestingly, accumulation of [125I]CLINDE was found in the insula of the contralesional hemisphere at this late time point [65], indicating the involvement of the entire brain in ischemia-induced neuroinflammation. Next to TSPO ligands, novel targets for microglia imaging may include the 18F-FSPG /system xc- and 2-18F-fluoro-A85380 (α4β2). Both system xc- and α4β2 have been reported to be overexpressed in microglia after stroke [72, 77].
One of the limitations of second generation TSPO ligands exacerbating clinical translational hurdles is the existence of an rs6971 polymorphism within the gene encoding for TSPO that affects the binding affinity of TSPO ligands [93-97]. This polymorphism results in a non-conservative single amino acid substitution from alanine to threonine at amino acid residue 147 (Ala147Thr) of the TSPO gene causing an inter- and intra-individual variability in binding affinity, hampering quantification of imaging data [98], while at the same time no effect of Ala147Thr on inflammatory and cerebrovascular biomarkers was observed [99].
Based on the expression of high and low affinity sites of TSPO, patients can be subdivided into low-, mixed-, and high-affinity binders, whereas different ligands display a different sensitivity for the rs6971 polymorphism [94]. This implicates that the choice of the TSPO ligand, as well as the TSPO status of the individuals included in a study, may severely influence the outcome of the clinical trial. Thus, statistical methods accounting for the TSPO status and matching of patients should be performed [98].
Newly developed third generation TSPO ligands, being unsusceptible to the rs6971 polymorphism, and new non-TSPO molecular microglial targets are currently under investigation within the EU FP7 program Imaging Inflammation in Neurodegenerative diseases (INMiND consortium; http://www.uni-muenster.de/InMind/) [100, 101].
Nevertheless, current second generation TSPO ligands are under intense clinical investigation in a variety of diseases including Alzheimer's disease, mild cognitive impairment, multiple sclerosis, amyotrophic lateral sclerosis, Parkinson's disease, rheumatoid arthritis, brain tumors and alcoholism.
In conclusion, TSPO imaging provides a valuable tool to study the neuroinflammatory response in stroke and allows for assessment of immunomodulatory treatments targeting inflammation in pre-clinical and clinical settings.
Matrix Metalloproteinases (MMPs)
MMPs belong to the family of secreted or membrane-bound zinc- and calcium-dependent endopeptidases [102]. Most of the MMPs are secreted in their inactive (zymogen) form. After secretion into the extracellular space they become rapidly activated through proteolytic processing of the cysteine switch by plasmin, trypsin, kallikrein, mast cell tryptase and other MMPs, as well as ROS and hypochlorous acid from inflammatory processes [102-105]. MMPs have been implicated in a variety of physiological events such as neoangiogenesis, neural plasticity, wound healing, tissue remodeling, embryogenesis and apoptosis [104, 106]. Besides the physiological functions of MMPs, different pathologies have been strongly linked to MMP function, i.e., rheumatoid arthritis, osteoarthritis, cardiovascular disease, fibrosis, tumorigenesis, tumor neovascularization, invasion, metastasis, blood brain barrier damage, atherosclerosis, multiple sclerosis, meningitis, Alzheimer's disease, stroke and many more [102, 104, 107-111]. MMPs contribute to the acute (neuro-)inflammatory reaction by modulating the expression, activity, and integrity of chemokines, inflammatory cytokines, growth factors, and matrix components and by affecting receptor turnover and leukocyte transmigration (diapedesis) [104, 106, 112, 113].
In ischemic stroke, MMPs are known to play a key role during disease progression. They are associated with BBB damage, hemorrhagic transformation, and vasogenic edema formation [110, 114-116]. It should be pointed out that over-simplification of MMPs as purely extracellular matrix (ECM)-degrading enzymes should be avoided since in vivo evidence that MMPs degrade ECM is lacking [117]. Several MMPs have been described to modulate the inflammatory response in cerebral ischemia via a network of complex functions and sources, including glial cells and neutrophils as important cellular sources. In particular, the gelatinases MMP-2 and MMP-9 are elevated after cerebral ischemia and have been correlated with BBB opening and hemorrhagic transformation [118, 119]. Interestingly, early inhibition of MMP-9 activity in experimental stroke, as well as genetic knockout, has beneficial effects on stroke outcome [120-124]. In contrast to acute MMP inhibition, it was shown that late inhibition can have detrimental effects, leading to increased brain damage characterized by reductions in numbers of neurons and newly formed blood vessels, supporting the hypothesis of dynamic multi-phasic effects of MMPs [119, 125-127].
Moreover, it has been reported that MMP inhibition leads to reduced migration of doublecortin (microtubule-associated protein expressed by neuronal precursor cells)-positive cells from the subventricular zone to infarcted areas after stroke [128]. Problems with the study design, definition of endpoints, the design of MMP inhibitors and heterogeneous nature of MMPs might so far explain the lack of efficacy and adverse effects of MMP inhibitors in clinical studies (for review: [129]).The multitude of available data on beneficial effects of MMP modulation in different disease settings makes MMPs attractive as biomarkers and therapeutic targets. Thus, there is a strong need for MMP inhibitors with increased specificity and selectivity for MMP subtypes, as well as sophisticated methods to detect MMPs in vivo in real time.
Most of the currently available preclinical imaging probes for tracking MMP activity in stroke rely on fluorescently labeled broad spectrum inhibitors or activatable (smart) probes. Smart probes are designed to remain in a quenched state until the quencher is degraded by activated MMPs under pathophysiological conditions, resulting in emission of near-infrared fluorescence (NIRF) [130].
Klohs and colleagues were the first to describe the use of an activatable, near-infrared fluorescent probe to image general MMP activity after stroke 24 hours after injection of the tracer. Treatment with the MMP inhibitor GM6001 resulted in significantly lower tracer signal and lesion volume, suggesting successful imaging of in vivo MMP activity [131]. Another study made use of MMPsense 750 FAST with improved characteristics including a shorter optical imaging time for early (within hours) visualization of MMP activity. Increased changes in fluorescence intensities were found in early ischemic reperfusion and at 24 hours post ischemia, but not in sham controls. The results correlated with MMP-9 protein levels, 5 and 24 hours after ischemia reperfusion. Overall MMP activity could be attenuated by hypothermia [132]. In contrast to these planar imaging approaches, Martín and colleagues further applied tomographic optical imaging describing an increase of MMP activity as early as 3 to 6 hours correlating with the volume of the ischemic damage [133]. Next to smart probes, a recent study suggested gelatinase-activatable cell-penetrating peptides (ACPP) with a cleavable l-amino acid linker linked to Cy5 or gadolinium for imaging MMP activity and therapy response in ischemic tissue in the femtomolar range [134]. These studies emphasize the potential of in vivo assessment of MMP activity after stroke and investigation of therapy response. The drawback of both probes is, however, the relatively long circulation required before imaging and/or the relatively low signal intensities due to physical limitations due to tissue scattering and limited penetration depth of light, requiring invasive measurements [131, 132]. To address this, the probe MMP-P12 with improved characteristics including optimized stability, blood half-life and enzyme susceptibility was developed. The probe allowed observation of significant signal enhancement from as early as 15 minutes to 24 hours after injection, with a peak 1 hour after injection. The spatiotemporal signal was followed for up to 14 days after stroke. Treatment with the MMP inhibitor GM6001 reduced the MMP-P12 fluorescence signal, suggesting MMP substrate specificity, though no complete blocking of the signal was observed. The feasibility of using this probe for drug development purposes was further tested by treating mice with RWJ67657, an inhibitor of the p38 mitogen-activated protein kinase (MAPK) signaling pathway, which resulted in modulation of MMP activity after stroke accompanied by improved functional outcome. Moreover, p38 MAPK inhibition rescued MMP-P12 NIRF intensity [130]. A direct comparison of NIRF probes is difficult due to the differences in stroke models being used and the different imaging time points. Despite the various advantages of NIRF imaging for real-time visualization of MMP activity in preclinical drug discovery including high sensitivity, low cost, non-ionizing radiation and vast applicability [135], there is a lack of clinical translatability due to the physical limitations of light only allowing for intra-operative applications.
Another important step towards clinical translation is the use of radioactively labeled compounds for nuclear imaging applications. The first radiofluorinated hydroxamate-based MMP inhibitor, the I-2-(N-benzyl-4-(2-[18F]fluoroethoxy)phenylsulphonamido)-N-hydroxy-3-methylbutanamide compound CGS 27023A 1 ([18F]BR-351), was developed by Wagner and colleagues. Due to the underlying radiochemistry, [18F]BR-351 can only bind the activated forms of MMP-2 (IC50 = 4 nM), -8 (IC50 = 2 nM), -9 (IC50 = 50 nM), and -13 (IC50 = 11 nM) [136].
In a multi-tracer study [18F]BR-351 was used in combination with the TSPO ligand [18F]DPA-714 for spatio-temporal assessment of MMP activity in a murine model of ischemic stroke (Figure 2) [60]. Interestingly, MMP expression, as measured by [18F]BR-351, appeared already 24 hours after tMCAo, peaked at 7 days post stroke, and was only seen in a subset of mice at later time points (14-21 days post stroke) [60]. Although this study did not focus on targeted MMP therapy, the results raise the intriguing possibility that [18F]BR-351 might be useful in selecting individuals who may benefit from MMP inhibitory therapy. Future studies should focus on how radioligands like [18F]BR-351 can be used to select and assess MMP-based therapies in stroke. Encouraging results come from a recent clinical study in patients with multiple sclerosis where [18F]BR-351 was successfully applied to monitor the early signs of multiple sclerosis and for monitoring anti-inflammatory treatment, thus opening new avenues for personalized therapies [137].
The combination of longitudinal MMP imaging in conjunction with improved inhibitors, which selectively modulate MMPs, is needed to rejuvenate the idea of using MMP modulation as a personalized therapy approach after stroke.
Cell-based therapies and in vivo cell tracking
Molecular imaging has been applied to track different cell populations after stroke. This is important to improve our understanding of the spatiotemporal immune cell infiltration and activation after cerebral ischemia. One example is the early clinical study by Price et al. [138], in which migration of [111In]troponin-labeled neutrophils in stroke patients was successfully tracked by gamma scintigraphy. Additionally, molecular imaging provides a powerful tool to evaluate the success of (stem) cell-based therapies, aiming to stimulate tissue repair after stroke. Investigated cell types showing promising therapeutic success in preclinical studies include, but are not limited to, neural stem cells (NSCs) [139], mesenchymal stem cells (MSCs) [140] and bone marrow mononuclear cells (BMMNCs) [141]. Interestingly, the reported success of those therapies is attributed to paracrine effects rather than to the replacement of destroyed neurological tissue. Observed effects include increased neurological plasticity [142] and stimulation of angiogenesis and, therefore, revascularization of the ischemic tissue [143-147]. Another important mode of action is immunomodulation induced by the transplanted cells. Brenneman et al. showed a downregulation of inflammatory cytokines in the brains of rats treated with BMMNCs as well as upregulation of Il-10 in acute stroke [148]. Yoo et al. reported immune suppression via TGF-β produced by transplanted MSCs themselves [149]. Interestingly, the immunomodulatory effects were systemic rather than restricted to the brain, possibly explaining why intravenous application of stem cells has therapeutic effects, even though only very few cells reach the brain [140, 150]. Next to MSCs and BMMNCs, specialized regulatory B- and T-lymphocytes may also importantly contribute to therapeutic capacity in ischemic stroke and should be pursued as imaging and therapeutic targets (for review: [151]).
Non-invasive imaging is ideally suited to investigate the migration and engraftment of transplanted cells. For all, direct or indirect labeling of the transplanted cells is necessary. Long term tracking and engraftment of transplanted MSCs or BMMNCs has been successfully conducted in animal models and first human stroke patients using the SPECT tracers [99mTc]HMPAO and [111In]Oxine [152, 153] as well as small iron oxide-based agents such as SPIO-based particles for MRI [154, 155]. The severe limitation of direct labeling approaches for both nuclear imaging as well as MRI is the low retention rate and accompanied tracer leakage from the transplanted cells [156, 157]. In the case of SPIOs, leaked particles can be taken up by phagocytic cells in the ischemic area and might result in positive signals in the absence of transplanted cells [156]. Alternatively, cells can be labeled indirectly, e.g., by using reporter genes that are only expressed by transplanted cells [158]. Bioluminescence imaging (BLI) has been applied to visualize and monitor endogenous stem cell vitality and response post stroke [159] and to image the fate of fluc-transduced MSCs [157, 160], NSCs [159] and monocytes/macrophages [161]. BLI is limited by the low penetration depth of light and, while visualization of cells through the skull is possible, imaging remains restricted to surface structures. Genetic reporters have also been investigated for tracking of cells using nuclear imaging. The best characterized reporter is based on the herpes simplex virus type 1 thymidine kinase (HSV-1-tk) that can phosphorylate isotope-labeled pyrimidine analogs like [124I]FIAU or [18F]FHBG and thereby trap them in transplanted cells [162]. Severe disadvantages of the HSV-1-tk-system are possible immune reactions against the viral protein and the relatively low blood brain barrier penetration of the applied imaging probes [163]. A promising alternative is the human sodium iodide symporter (hNIS) that can transport all radioactive forms of I-. While hNIS so far has not been applied after stroke, it has been successfully used to track MSCs, for example, in animal models of breast cancer [164], glioma [165] and myocardial infarction [166]. Similarly, indirect labeling of cells, e.g., via reporters based on CEST, has been investigated for tracking cells by MRI [167]. However, to date this approach has not been applied to track cells after cerebral ischemia.
Novel imaging approaches will focus not only on tracking the fate of transplanted cells, but at the same time monitoring therapy responses in the form of immunomodulation, changes in the microenvironment and activation of endogenous neural stem cells. In a promising approach Daadi et al. applied longitudinal multimodal imaging to track NSCs labeled both directly (SPIOs for tracking with MRI) and indirectly (HSV-1-tk reporter for PET imaging with [18F]FHBG) [168]. They elegantly combined tracking of grafted cells with analysis of infarct size by MRI and neurological metabolic activity by [18F]FDG-PET over the course of several months. In the future, multimodal- and multitracer imaging assessing microglia and tissue remodeling, in combination with cell tracking will help to monitor immunomodulation and changes in brain microenvironment [60]. In addition, monitoring of endogenous neural stem cells, for example by 3'-deoxy-3'-[18F]fluoro-L-thymidin ([18F]FLT) -PET, could elucidate reparative processes induced by stem cell therapies. Rueger et al. applied [18F]FLT to monitor endogenous NSC proliferation in the subventricular zone following minocycline treatment in rats with cerebral ischemia [169]. In this multimodal- and multitracer imaging study, [18F]FLT imaging was combined with [11C]PK11195-PET to assess neuroinflammation and MRI to monitor infarct size.
Future perspectives
Imaging the brain-gut axis
Investigation of the role of intestinal microbiota in neurological diseases and recovery has drawn a lot of attention for microbiota as possible disease-modulating entities in stroke (for review: [170, 171]). Immune cells, hormones and neurotransmitters originating from the gut may reach the brain via a recently discovered lymphatic subsystem, which allows for direct or indirect transport of inflammatory mediators from the gut to the brain (Figure 1). This specialized lymphatic system drains from the cerebrospinal fluid in the adjacent subarachnoid space and the interstitial fluid to the deep cervical lymph nodes and may be visualized by molecular imaging [172-174].
Another aspect of intestinal microbiota is their role in regulating the permeability of the BBB. Germ-free animals have been shown to exhibit reduced expression levels of tight junction proteins, which results in increased permeability of the BBB for molecules [174-176]. In line with these observations, complete depletion of gut microbiota with a broad-spectrum antibiotic cocktail led to increased mortality rates and development of severe colitis in a mouse model of stroke [177].
In response to stroke stages, various reports exemplify significant changes in the composition of gut microbiota in preclinical and clinical stroke that partially correlate with disease severity [178-181]. This raises the intriguing possibility of selective modulation of the gut microbiome to treat stroke. In line with this notion, beneficial effects on stroke severity were observed by therapeutic transplantation of fecal microbiota partially restoring bacterial species diversity and improving stroke outcome likely via a lymphocyte-dependent mechanism [179]. Further evidence for a link between microbiota and the lymphocytic response after stroke was deduced by studies of Benakis and colleagues. They induced microbiota dysbiosis by antibiotic treatment, which led to an increase in regulatory T cells, suppression of IL-17-positive γδ T cells and thus reduced trafficking of effector T cells from the gut to the leptomeninges after stroke, resulting in smaller infarcts and improved behavioral outcome [182]. Little is known about the in vivo dynamics of specialized gut microbiota and trafficking cells (i.e, lymphocytes) after stroke. Therefore, the in vivo follow-up by molecular imaging of bacterial dysbiosis and trafficking cells may have strong indications in patient stratification and selection for therapy. Novel imaging agents are needed to study gut microbiota non-invasively over time.
Imaging astrocytes
Astrocytes are the key cells within the neurovascular unit mediating exchange of signals between the vascular system and neurons and are an important mediator of damage and repair after stroke [183-185]. As for many other immune cells, astrocytes also have detrimental functions through hyperreactivity and glial scar formation, as well as beneficial functions through neuroprotective, angiogenic, immunomodulatory, neurogenic, and antioxidant functions (for review: [186]). Therefore, astrocytes also represent an important therapeutic and thus imaging target. Despite encouraging data on monoamine oxidase B (MAO-B) binding PET ligands like [11C]deuterium-L-deprenyl (DED) in Alzheimer's disease [187, 188], in vivo imaging data on astrocyte distribution and activity after stroke still represents an underexplored aspect of the disease. Besides DED, TSPO and α4β2 nicotinic acetylcholine receptor-targeted PET ligands are able to visualize astrocytic activation, depending on the imaging time point after stroke [54, 77, 189]. Nevertheless, there is still a need for more specific astrocytic imaging biomarkers with longer half-life isotopes for monitoring disease progression and therapy responses.
Discussion
Multi-modality and multi-tracer imaging have the potential to support drug development processes by answering key questions about location, extent, temporal profile, and therapy effect on an imaging biomarker after stroke, thereby facilitating our understanding of disease progression. By simultaneously providing profiles of different markers after stroke, multi-modality and multi-tracer imaging can assist in development, selection, application and follow-up of targeted therapies for stroke patients at specific time-points (personalized medicine). These types of technologies are important, especially in diseases such as stroke with its highly dynamic disease process and with heterogenous and variable molecular signatures within different parts of the diseased brain.
Current imaging paradigms aim at identifying the location and extent of the stroke lesion, estimation of the penumbra and patient selection for recanalization therapy. CT and several MRI techniques are used to obtain physiological data and to exclude hemorrhagic transformation [190, 191]. However, next to physiological and anatomical information, molecular parameters are important for targeted and personalized therapies. This review was not dedicated to pathophysiological and molecular markers such as cerebral blood flow, oxygen and glucose consumption, but on imaging biomarkers that may allow the assessment of further consequences after stroke such as neuroinflammatory and tissue repair processes. Current tracers fall short in discriminating between different inflammatory cell types and/or polarization states. Thus, further imaging biomarkers and novel techniques, e.g., optogenetics, specifically targeting these differences in cell activation and function need to be developed and are currently under evaluation [72, 77, 100]. Ideally, these novel markers should be longitudinally studied (intra-individual imaging) and combined with, e.g., other imaging and molecular techniques including “-omics” and liquid biopsies to obtain a patient-specific detailed characterization of disease. This will allow stratification of patients for treatment and drug development paradigms. As target availability and resulting patient stratification is a key determinant of sample sizes in clinical trials, molecular imaging will provide important information to determine sample sizes and to define inclusion and exclusion information earlier than standard clinical assessments and at the same time increase the sensitivity and predictivity of clinical studies [192].
In parallel with defining new imaging targets, innovative disease-resolving and neurorestorative therapies need to be developed. These may include immunomodulatory therapies, dietary interventions and related alterations of the microbiota-gut-brain axis [82, 193].
Despite the multitude of advances in imaging-guided therapies, there is still a strong need to further improve the quality of research to obtain predictive and clinically translatable data. To this end, research activities should involve collaborative community efforts, including multi-center studies as suggested [10, 194] and applied recently [54, 195-198]. The concept of reverse-translating quality standards from clinical studies into preclinical work has shown promise and overall acceptance to further enhance translatability of research [199].
Next to general quality measures in stroke research, imaging research must overcome additional issues specific to the field. Obtaining meaningful, quantifiable data remains an important challenge in preclinical and clinical research. Nuclear imaging techniques provide the essential pharmacodynamic data of radiolabeled compounds, but they also need to be harmonized [200-202]. In particular, quantification in small animals remains challenging, since pharmacokinetic/pharmacodynamic modeling usually requires determination of an arterial input function by repeated blood sampling. The choice of the right mathematical model, either model- or data-based, applied for determination of transfer rate constants, volumes of distribution or binding potentials may also vary between different ligands and targets [201, 202]. This problem can be exemplified by means of TSPO tracer studies, which suffer from a lack of a suitable reference region in the brain. Multiple approaches have been undertaken to overcome these limitations to create parametric images including population-based approaches, Logan analysis and spectral analysis [203-205].
In summary, over the last couple of years, the molecular imaging field has rapidly evolved and many novel approaches to dissect molecular processes over time and during the course of a disease process in various tissue compartments in vivo are under way. Molecular imaging will therefore represent an important asset to the drug development process by providing essential information about patient-specific disease progression and therapy response, while allowing for patient selection, stratification and personalized therapy.
Acknowledgements
This review was partly funded by the EU 7th Framework Programme (FP7/2007-2013) under grant agreement n° 278850 (INMiND) and Horizon2020 Programme under grant agreement n° 675417 (PET3D) as well as the 'Cells-in-Motion' Cluster of Excellence (DFG EXC1003 - CiM), the Interdisciplinary Center for Clinical Research (IZKF core unit PIX), Münster, Germany.
The authors thank Nina Knubel for the design and production of the artwork.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Feigin VL, Forouzanfar MH, Krishnamurthi R, Mensah GA, Connor M, Bennett DA. et al. Global and regional burden of stroke during 1990-2010: findings from the Global Burden of Disease Study 2010. Lancet. 2014;383:245-55 doi:10.1016/S0140-6736(13)61953-4
2. Feigin VL, Mensah GA, Norrving B, Murray CJL, Roth GA, GBD 2013 Stroke Panel Experts Group. Atlas of the Global Burden of Stroke (1990-2013): The GBD 2013 Study. Neuroepidemiology. 2015;45:230-6 doi:10.1159/000441106
3. O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59:467-77 doi:10.1002/ana.20741
4. Wardlaw JM, Murray V, Berge E, Del Zoppo GJ. Thrombolysis for acute ischaemic stroke. Cochrane database Syst Rev. 2009:CD000213. doi:10.1002/14651858.CD000213.pub2
5. Yarbrough CK, Ong CJ, Beyer AB, Lipsey K, Derdeyn CP. Endovascular Thrombectomy for Anterior Circulation Stroke: Systematic Review and Meta-Analysis. Stroke. 2015;46:3177-83 doi:10.1161/STROKEAHA.115.009847
6. Grønberg NV, Johansen FF, Kristiansen U, Hasseldam H. Leukocyte infiltration in experimental stroke. J Neuroinflammation. 2013;10:115. doi:10.1186/1742-2094-10-115
7. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe C-U, Siler DA. et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849-57 doi:10.1161/STROKEAHA.108.534503
8. DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: New estimates of R&D costs. J Health Econ. 2016;47:20-33 doi:10.1016/j.jhealeco.2016.01.012
9. Dirnagl U, Endres M. Found in Translation: Preclinical Stroke Research Predicts Human Pathophysiology, Clinical Phenotypes, and Therapeutic Outcomes. Stroke. 2014;45:1510-8 doi:10.1161/STROKEAHA.113.004075
10. Dirnagl U. Thomas Willis Lecture. Stroke. 2016;47:2148-53 doi:10.1161/STROKEAHA.116.013244
11. Peruzzotti-Jametti L, Donegá M, Giusto E, Mallucci G, Marchetti B, Pluchino S. The role of the immune system in central nervous system plasticity after acute injury. Neuroscience. 2014;283:210-21 doi:10.1016/j.neuroscience.2014.04.036
12. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796-808 doi:10.1038/nm.2399
13. Willmann JK, van Bruggen N, Dinkelborg LM, Gambhir SS. Molecular imaging in drug development. Nat Rev Drug Discov. 2008;7:591-607 doi:10.1038/nrd2290
14. Rahmim A, Zaidi H. PET versus SPECT: strengths, limitations and challenges. Nucl Med Commun. 2008;29:193-207 doi:10.1097/MNM.0b013e3282f3a515
15. Lammertsma AA. PET/SPECT: functional imaging beyond flow. Vision Res. 2001;41:1277-81
16. Gauberti M, Montagne A, Quenault A, Vivien D. Molecular magnetic resonance imaging of brain-immune interactions. Front Cell Neurosci. 2014;8:389. doi:10.3389/fncel.2014.00389
17. Lee C-M, Farde L. Using positron emission tomography to facilitate CNS drug development. Trends Pharmacol Sci. 2006;27:310-6 doi:10.1016/j.tips.2006.04.004
18. Smith JJ, Sorensen AG, Thrall JH. Biomarkers in Imaging: Realizing Radiology's Future. Radiology. 2003;227:633-8 doi:10.1148/radiol.2273020518
19. Gunn RN, Rabiner EA. Imaging in Central Nervous System Drug Discovery. 2016.
20. Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci. 2017;20:136-44 doi:10.1038/nn.4475
21. Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391-7 http://www.ncbi.nlm.nih.gov/pubmed/10441299. Accessed 2 Nov 2016
22. Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S. et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063-70 doi:10.1161/STROKEAHA.112.659656STROKEAHA.112.659656 [pii]
23. Fu Y, Liu Q, Anrather J, Shi F-D. Immune interventions in stroke. Nat Rev Neurol. 2015;11:524-35 doi:10.1038/nrneurol.2015.144
24. Perez-de-Puig I, Miró-Mur F, Ferrer-Ferrer M, Gelpi E, Pedragosa J, Justicia C. et al. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. 2015;129:239-57 doi:10.1007/s00401-014-1381-0
25. Yilmaz G, Granger DN. Cell adhesion molecules and ischemic stroke. Neurol Res. 2008;30:783-93 doi:10.1179/174313208X341085
26. Jacobs AH, Tavitian B, consortium Inm. Noninvasive molecular imaging of neuroinflammation. J Cereb Blood Flow Metab. 2012;32:1393-415 doi:10.1038/jcbfm.2012.53
27. Pulli B, Chen JW. Imaging Neuroinflammation - from Bench to Bedside. J Clin Cell Immunol. 2014:5 doi:10.4172/2155-9899.1000226
28. de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol. 2016;16:378-91 doi:10.1038/nri.2016.49
29. Gauberti M, De Lizarrondo SM, Vivien D. The “inflammatory penumbra” in ischemic stroke: From clinical data to experimental evidence. Eur Stroke J. 2016;1:20-7 doi:10.1177/2396987316630249
30. Kelly KA, Allport JR, Tsourkas A, Shinde-Patil VR, Josephson L, Weissleder R. Detection of Vascular Adhesion Molecule-1 Expression Using a Novel Multimodal Nanoparticle. Circ Res. 2005;96:327-36 doi:10.1161/01.RES.0000155722.17881.dd
31. McAteer MA, Sibson NR, von zur Muhlen C, Schneider JE, Lowe AS, Warrick N. et al. In vivo magnetic resonance imaging of acute brain inflammation using microparticles of iron oxide. Nat Med. 2007;13:1253-8 doi:10.1038/nm1631
32. Tsourkas A, Shinde-Patil VR, Kelly KA, Patel P, Wolley A, Allport JR. et al. In Vivo Imaging of Activated Endothelium Using an Anti-VCAM-1 Magnetooptical Probe. Bioconjug Chem. 2005;16:576-81 doi:10.1021/bc050002e
33. Hoyte LC, Brooks KJ, Nagel S, Akhtar A, Chen R, Mardiguian S. et al. Molecular magnetic resonance imaging of acute vascular cell adhesion molecule-1 expression in a mouse model of cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:1178-87 doi:10.1038/jcbfm.2009.287
34. Gauberti M, Montagne A, Marcos-Contreras OA, Le Béhot A, Maubert E, Vivien D. Ultra-sensitive molecular MRI of vascular cell adhesion molecule-1 reveals a dynamic inflammatory penumbra after strokes. Stroke. 2013;44:1988-96 doi:10.1161/STROKEAHA.111.000544
35. Montagne A, Gauberti M, Macrez R, Jullienne A, Briens A, Raynaud J-S. et al. Ultra-sensitive molecular MRI of cerebrovascular cell activation enables early detection of chronic central nervous system disorders. Neuroimage. 2012;63:760-70 doi:10.1016/j.neuroimage.2012.07.018
36. Liu H, Jablonska A, Li Y, Cao S, Liu D, Chen H. et al. Label-free CEST MRI Detection of Citicoline-Liposome Drug Delivery in Ischemic Stroke. Theranostics. 2016;6:1588-600 doi:10.7150/thno.15492
37. Pagel MM. The Pursuit of Theranostics with CEST MRI. Theranostics. 2016;6:1601-2 doi:10.7150/thno.16337
38. van Kasteren SI, Campbell SJ, Serres S, Anthony DC, Sibson NR, Davis BG. Glyconanoparticles allow pre-symptomatic in vivo imaging of brain disease. Proc Natl Acad Sci U S A. 2009;106:18-23 doi:10.1073/pnas.0806787106
39. Jin AY, Tuor UI, Rushforth D, Kaur J, Muller RN, Petterson JL. et al. Reduced blood brain barrier breakdown in P-selectin deficient mice following transient ischemic stroke: a future therapeutic target for treatment of stroke. BMC Neurosci. 2010;11:12. doi:10.1186/1471-2202-11-12
40. Quenault A, Martinez de Lizarrondo S, Etard O, Gauberti M, Orset C, Haelewyn B. et al. Molecular magnetic resonance imaging discloses endothelial activation after transient ischaemic attack. Brain. 2016:aww260. doi:10.1093/brain/aww260
41. Deddens LH, Van Tilborg GAF, Mulder WJM, De Vries HE, Dijkhuizen RM. Imaging neuroinflammation after stroke: current status of cellular and molecular MRI strategies. Cerebrovasc Dis. 2012;33:392-402 doi:10.1159/000336116
42. Breckwoldt MO, Chen JW, Stangenberg L, Aikawa E, Rodriguez E, Qiu S. et al. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc Natl Acad Sci U S A. 2008;105:18584-9 doi:10.1073/pnas.0803945105
43. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314-8 doi:10.1126/science.1110647
44. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19:987-91 doi:10.1038/nn.4338
45. Gerhard A, Schwarz J, Myers R, Wise R, Banati RB. Evolution of microglial activation in patients after ischemic stroke: a [11C](R)-PK11195 PET study. Neuroimage. 2005;24:591-5 doi:10.1016/j.neuroimage.2004.09.034
46. Thiel A, Cechetto DF, Heiss W-D, Hachinski V, Whitehead SN. Amyloid Burden, Neuroinflammation, and Links to Cognitive Decline After Ischemic Stroke. Stroke. 2014;45:2825-9 doi:10.1161/STROKEAHA.114.004285
47. Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapère JJ, Lindemann P. et al. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27:402-9 doi:10.1016/j.tips.2006.06.005
48. Chen MK, Guilarte TR. Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther. 2008;118:1-17 doi:10.1016/j.pharmthera.2007.12.004
49. Scarf AM, Kassiou M. The translocator protein. J Nucl Med. 2011;52:677-80 doi:10.2967/jnumed.110.086629jnumed.110.086629 [pii]
50. Chauveau F, Van Camp N, Dollé F, Kuhnast B, Hinnen F, Damont A. et al. Comparative evaluation of the translocator protein radioligands 11C-DPA-713, 18F-DPA-714, and 11C-PK11195 in a rat model of acute neuroinflammation. J Nucl Med. 2009;50:468-76 doi:10.2967/jnumed.108.058669
51. Winkeler A, Boisgard R, Awde AR, Dubois A, Thézé B, Zheng J. et al. The translocator protein ligand [18F]DPA-714 images glioma and activated microglia in vivo. Eur J Nucl Med Mol Imaging. 2012;39:811-23 doi:10.1007/s00259-011-2041-4
52. Rojas S, Martin A, Arranz MJ, Pareto D, Purroy J, Verdaguer E. et al. Imaging brain inflammation with [(11)C]PK11195 by PET and induction of the peripheral-type benzodiazepine receptor after transient focal ischemia in rats. J Cereb Blood Flow Metab. 2007;27:1975-86 doi:9600500 [pii]10.1038/sj.jcbfm.9600500
53. Martín A, Boisgard R, Thézé B, Van Camp N, Kuhnast B, Damont A. et al. Evaluation of the PBR/TSPO radioligand [(18)F]DPA-714 in a rat model of focal cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:230-41 doi:10.1038/jcbfm.2009.205
54. Wiesmann M, Zinnhardt B, Reinhardt D, Eligehausen S, Wachsmuth L, Hermann S. et al. A specific dietary intervention to restore brain structure and function after ischemic stroke. Theranostics. 2017;7:493-512 doi:10.7150/thno.17559
55. Zinnhardt B, Pigeon H, Thézé B, Viel T, Wachsmuth L, Fricke IB. et al. Combined PET Imaging of the Inflammatory Tumor Microenvironment Identifies Margins of Unique Radiotracer Uptake. Cancer Res. 2017 doi:10.1158/0008-5472.CAN-16-2628
56. Stephenson DT, Schober DA, Smalstig EB, Mincy RE, Gehlert DR, Clemens JA. Peripheral benzodiazepine receptors are colocalized with activated microglia following transient global forebrain ischemia in the rat. J Neurosci. 1995:15 7 Pt 2:5263-74. http://www.ncbi.nlm.nih.gov/pubmed/7623150
57. Gerhard A, Neumaier B, Elitok E, Glatting G, Ries V, Tomczak R. et al. In vivo imaging of activated microglia using [11C]PK11195 and positron emission tomography in patients after ischemic stroke. Neuroreport. 2000;11:2957-60 http://www.ncbi.nlm.nih.gov/pubmed/11006973. Accessed 20 Aug 2014
58. Pappata S, Levasseur M, Gunn RN, Myers R, Crouzel C, Syrota A. et al. Thalamic microglial activation in ischemic stroke detected in vivo by PET and [11C]PK1195. Neurology. 2000;55:1052-4 http://www.ncbi.nlm.nih.gov/pubmed/11061271. Accessed 15 Aug 2016
59. Cosenza-Nashat M, Zhao M-LL, Suh H-SS, Morgan J, Natividad R, Morgello S. et al. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol. 2009;35:306-28 doi:10.1111/j.1365-2990.2008.01006.x
60. Zinnhardt B, Viel T, Wachsmuth L, Vrachimis A, Wagner S, Breyholz H-J. et al. Multimodal imaging reveals temporal and spatial microglia and matrix metalloproteinase activity after experimental stroke. J Cereb Blood Flow Metab. 2015;35:1711-21 doi:10.1038/jcbfm.2015.149
61. Imaizumi M, Kim H-J, Zoghbi SS, Briard E, Hong J, Musachio JL. et al. PET imaging with [ 11 C]PBR28 can localize and quantify upregulated peripheral benzodiazepine receptors associated with cerebral ischemia in rat. Neurosci Lett. 2007;411:200-5
62. Lartey FM, Ahn GO, Shen B, Cord KT, Smith T, Chua JY. et al. PET Imaging of Stroke-Induced Neuroinflammation in Mice Using [F]PBR06. Mol Imaging Biol. 2013 doi:10.1007/s11307-013-0664-5
63. Boutin H, Prenant C, Maroy R, Galea J, Greenhalgh AD, Smigova A. et al. [18F]DPA-714: direct comparison with [11C]PK11195 in a model of cerebral ischemia in rats. PLoS One. 2013;8:e56441. doi:10.1371/journal.pone.0056441PONE-D-11-25095 [pii]
64. Ribeiro M-J, Vercouillie J, Debiais S, Cottier J-P, Bonnaud I, Camus V. et al. Could (18) F-DPA-714 PET imaging be interesting to use in the early post-stroke period? EJNMMI Res. 2014;4:28. doi:10.1186/s13550-014-0028-4
65. Feng L, Svarer C, Thomsen G, de Nijs R, Larsen VA, Jensen P. et al. In Vivo Quantification of Cerebral Translocator Protein Binding in Humans Using 6-Chloro-2-(4'-123I-Iodophenyl)-3-(N,N-Diethyl)-Imidazo[1,2-a]Pyridine-3-Acetamide SPECT. J Nucl Med. 2014;55:1966-72 doi:10.2967/jnumed.114.143727
66. Dupont A-C, Largeau B, Santiago Ribeiro M, Guilloteau D, Tronel C, Arlicot N. Translocator Protein-18 kDa (TSPO) Positron Emission Tomography (PET) Imaging and Its Clinical Impact in Neurodegenerative Diseases. Int J Mol Sci. 2017;18:785. doi:10.3390/ijms18040785
67. Ikawa M, Lohith TG, Shrestha S, Telu S, Zoghbi SS, Castellano S. et al. 11 C-ER176, a Radioligand for 18-kDa Translocator Protein, Has Adequate Sensitivity to Robustly Image All Three Affinity Genotypes in Human Brain. J Nucl Med. 2017;58:320-5 doi:10.2967/jnumed.116.178996
68. Feeney C, Scott G, Raffel J, Roberts S, Coello C, Jolly A. et al. Kinetic analysis of the translocator protein positron emission tomography ligand [(18)F]GE-180 in the human brain. Eur J Nucl Med Mol Imaging. 2016;43:2201-10 doi:10.1007/s00259-016-3444-z
69. Lartey FM, Ahn G-O, Ali R, Rosenblum S, Miao Z, Arksey N. et al. The Relationship Between Serial [(18) F]PBR06 PET Imaging of Microglial Activation and Motor Function Following Stroke in Mice. Mol Imaging Biol. 2014;16:821-9 doi:10.1007/s11307-014-0745-0
70. Wang Y, Yue X, Kiesewetter DO, Wang Z, Lu J, Niu G. et al. [(18)F]DPA-714 PET imaging of AMD3100 treatment in a mouse model of stroke. Mol Pharm. 2014;11:3463-70 doi:10.1021/mp500234d
71. Tóth M, Little P, Arnberg F, Häggkvist J, Mulder J, Halldin C. et al. Acute neuroinflammation in a clinically relevant focal cortical ischemic stroke model in rat: longitudinal positron emission tomography and immunofluorescent tracking. Brain Struct Funct. 2016;221:1279-90 doi:10.1007/s00429-014-0970-y
72. Domercq M, Szczupak B, Gejo J, Gómez-Vallejo V, Padro D, Gona KB. et al. PET Imaging with [ 18 F]FSPG Evidences the Role of System xc - on Brain Inflammation Following Cerebral Ischemia in Rats. Theranostics. 2016;6:1753-67 doi:10.7150/thno.15616
73. Walberer M, Jantzen SU, Backes H, Rueger MA, Keuters MH, Neumaier B. et al. In-vivo detection of inflammation and neurodegeneration in the chronic phase after permanent embolic stroke in rats. Brain Res. 2014;1581:80-8 doi:10.1016/j.brainres.2014.05.030
74. Thiel A, Heiss W-D. Imaging of Microglia Activation in Stroke. Stroke. 2011:42
75. Murata Y, Rosell A, Scannevin RH, Rhodes KJ, Wang X, Lo EH. Extension of the Thrombolytic Time Window With Minocycline in Experimental Stroke. Stroke. 2008:39
76. Martin A, Boisgard R, Kassiou M, Dolle F, Tavitian B. Reduced PBR/TSPO expression after minocycline treatment in a rat model of focal cerebral ischemia: a PET study using [(18)F]DPA-714. Mol Imaging Biol. 2011;13:10-5 doi:10.1007/s11307-010-0324-y
77. Martín A, Szczupak B, Gómez-Vallejo V, Domercq M, Cano A, Padro D. et al. In vivo PET imaging of the α4β2 nicotinic acetylcholine receptor as a marker for brain inflammation after cerebral ischemia. J Neurosci. 2015;35:5998-6009 doi:10.1523/JNEUROSCI.3670-14.2015
78. Wiesmann M, Zerbi V, Jansen D, Haast R, Lütjohann D, Broersen LM. et al. A Dietary Treatment Improves Cerebral Blood Flow and Brain Connectivity in Aging apoE4 Mice. Neural Plast. 2016;2016:6846721. doi:10.1155/2016/6846721
79. Tremoleda JL, Thau-Zuchman O, Davies M, Foster J, Khan I, Vadivelu KC. et al. In vivo PET imaging of the neuroinflammatory response in rat spinal cord injury using the TSPO tracer [(18)F]GE-180 and effect of docosahexaenoic acid. Eur J Nucl Med Mol Imaging. 2016;43:1710-22 doi:10.1007/s00259-016-3391-8
80. Pelgrim CE, Franx BAA, Snabel J, Kleemann R, Arnoldussen IAC, Kiliaan AJ. Butyrate Reduces HFD-Induced Adipocyte Hypertrophy and Metabolic Risk Factors in Obese LDLr-/-.Leiden Mice. Nutrients. 2017;9:714. doi:10.3390/nu9070714
81. Arnoldussen IAC, Wiesmann M, Pelgrim CE, Wielemaker EM, van Duyvenvoorde W, Amaral-Santos PL. et al. Butyrate restores HFD-induced adaptations in brain function and metabolism in mid-adult obese mice. Int J Obes (Lond). 2017;41:935-44 doi:10.1038/ijo.2017.52
82. Vauzour D, Camprubi-Robles M, Miquel-Kergoat S, Andres-Lacueva C, Bánáti D, Barberger-Gateau P. et al. Nutrition for the ageing brain: Towards evidence for an optimal diet. Ageing Res Rev. 2017;35:222-40 doi:10.1016/j.arr.2016.09.010
83. Janssen CIF, Jansen D, Mutsaers MPC, Dederen PJWC, Geenen B, Mulder MT. et al. The Effect of a High-Fat Diet on Brain Plasticity, Inflammation and Cognition in Female ApoE4-Knockin and ApoE-Knockout Mice. PLoS One. 2016;11:e0155307. doi:10.1371/journal.pone.0155307
84. Scheltens P, Twisk JWR, Blesa R, Scarpini E, von Arnim CAF, Bongers A. et al. Efficacy of Souvenaid in mild Alzheimer's disease: results from a randomized, controlled trial. J Alzheimers Dis. 2012;31:225-36 doi:10.3233/JAD-2012-121189
85. Olde Rikkert MGM, Verhey FR, Blesa R, von Arnim CAF, Bongers A, Harrison J. et al. Tolerability and safety of Souvenaid in patients with mild Alzheimer's disease: results of multi-center, 24-week, open-label extension study. J Alzheimers Dis. 2015;44:471-80 doi:10.3233/JAD-141305
86. Hjorth E, Freund-Levi Y. Immunomodulation of microglia by docosahexaenoic acid and eicosapentaenoic acid. Curr Opin Clin Nutr Metab Care. 2012;15:1. doi:10.1097/MCO.0b013e32835017cc
87. Heras-Sandoval D, Pedraza-Chaverri J, Pérez-Rojas JM. Role of docosahexaenoic acid in the modulation of glial cells in Alzheimer's disease. J Neuroinflammation. 2016;13:61. doi:10.1186/s12974-016-0525-7
88. Price CJS, Wang D, Menon DK, Guadagno J V, Cleij M, Fryer T. et al. Intrinsic activated microglia map to the peri-infarct zone in the subacute phase of ischemic stroke. Stroke. 2006;37:1749-53 doi:10.1161/01.STR.0000226980.95389.0b
89. Radlinska BA, Ghinani SA, Lyon P, Jolly D, Soucy J-P, Minuk J. et al. Multimodal microglia imaging of fiber tracts in acute subcortical stroke. Ann Neurol. 2009;66:825-32 doi:10.1002/ana.21796
90. Gulyás B, Tóth M, Schain M, Airaksinen A, Vas Á, Kostulas K. et al. Evolution of microglial activation in ischaemic core and peri-infarct regions after stroke: A PET study with the TSPO molecular imaging biomarker [11C]vinpocetine. J Neurol Sci. 2012;320:110-7
91. Gulyas B, Toth M, Vas A, Shchukin E, Kostulas K, Hillert J. et al. Visualising neuroinflammation in post-stroke patients: a comparative PET study with the TSPO molecular imaging biomarkers [11C]PK11195 and [11C]vinpocetine. Curr Radiopharm. 2012;5:19-28 http://www.ncbi.nlm.nih.gov/pubmed/22074478. Accessed 15 Aug 2016
92. Kreisl WC, Mbeo G, Fujita M, Zoghbi SS, Pike VW, Innis RB. et al. Stroke incidentally identified using improved positron emission tomography for microglial activation. Arch Neurol. 2009;66:1288-9 doi:10.1001/archneurol.2009.208
93. Kreisl WC, Jenko KJ, Hines CS, Lyoo CH, Corona W, Morse CL. et al. A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J Cereb Blood Flow Metab. 2013;33:53-8 doi:10.1038/jcbfm.2012.131
94. Owen DRJ, Gunn RN, Rabiner EA, Bennacef I, Fujita M, Kreisl WC. et al. Mixed-Affinity Binding in Humans with 18-kDa Translocator Protein Ligands. J Nucl Med. 2011;52:24-32 doi:10.2967/jnumed.110.079459
95. Owen DR, Howell OW, Tang S-P, Wells LA, Bennacef I, Bergstrom M. et al. Two binding sites for [3H]PBR28 in human brain: implications for TSPO PET imaging of neuroinflammation. J Cereb Blood Flow Metab. 2010;30:1608-18 doi:10.1038/jcbfm.2010.63
96. Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C. et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626-30 doi:10.1126/science.1236062
97. Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A. et al. An 18-kDa Translocator Protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2012;32:1-5 doi:10.1038/jcbfm.2011.147
98. Owen DR, Guo Q, Rabiner EA, Gunn RN. The impact of the rs6971 polymorphism in TSPO for quantification and study design. Clin Transl Imaging. 2015;3:417-22 doi:10.1007/s40336-015-0141-z
99. Felsky D, De Jager PL, Schneider JA, Arfanakis K, Fleischman DA, Arvanitakis Z. et al. Cerebrovascular and microglial states are not altered by functional neuroinflammatory gene variant. J Cereb Blood Flow Metab. 2016;36:819-30 doi:10.1177/0271678X15626719
100. Janssen B, Vugts DJ, Funke U, Molenaar GT, Kruijer PS, van Berckel BNM. et al. Imaging of neuroinflammation in Alzheimer's disease, multiple sclerosis and stroke: Recent developments in positron emission tomography. Biochim Biophys Acta - Mol Basis Dis. 2016;1862:425-41 doi:10.1016/j.bbadis.2015.11.011
101. Sokias R, Werry E, Chau SW, Reekie T, Munoz L, Wong E. et al. Determination and reduction of translocator protein (TSPO) ligand rs6971 discrimination. Med Chem Commun. 2016 doi:10.1039/C6MD00523C
102. Gomis-Rüth FX. Structural aspects of the metzincin clan of metalloendopeptidases. Mol Biotechnol. 2003;24:157-202 doi:10.1385/MB:24:2:157
103. Klein T, Bischoff R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids. 2011;41:271-90 doi:10.1007/s00726-010-0689-x
104. Nissinen L, Kähäri V-M. Matrix metalloproteinases in inflammation. Biochim Biophys Acta. 2014;1840:2571-80 doi:10.1016/j.bbagen.2014.03.007
105. Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid generated by myeloperoxidase modifies adjacent tryptophan and glycine residues in the catalytic domain of matrix metalloproteinase-7 (matrilysin): an oxidative mechanism for restraining proteolytic activity during inflammation. J Biol Chem. 2003;278:28403-9 doi:10.1074/jbc.M304739200
106. Rempe RG, Hartz AM, Bauer B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J Cereb Blood Flow Metab. 2016;36:1481-507 doi:10.1177/0271678X16655551
107. Overall CM, López-Otín C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2:657-72 doi:10.1038/nrc884
108. Manicone AM, McGuire JK. Matrix metalloproteinases as modulators of inflammation. Semin Cell Dev Biol. 2008;19:34-41 doi:10.1016/j.semcdb.2007.07.003
109. John A, Tuszynski G. The role of matrix metalloproteinases in tumor angiogenesis and tumor metastasis. Pathol Oncol Res. 2001;7:14-23 http://www.ncbi.nlm.nih.gov/pubmed/11349215. Accessed 21 Jul 2014
110. del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Berg GI. et al. Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke. 2007:38 2 Suppl:646-51. doi:10.1161/01.STR.0000254477.34231.cb
111. Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci. 2006;11:529-43 http://www.ncbi.nlm.nih.gov/pubmed/16146751. Accessed 31 Jul 2014
112. McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark-Lewis I, Overall CM. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. 2002;100:1160-7 http://www.ncbi.nlm.nih.gov/pubmed/12149192
113. Rodriguez D, Morrison CJ, Overall CM, Rodríguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803:39-54 doi:10.1016/j.bbamcr.2009.09.015 S0167-4889(09)00240-7 [pii]
114. Montaner J, Alvarez-Sabin J, Molina CA, Angles A, Abilleira S, Arenillas J. et al. Matrix metalloproteinase expression is related to hemorrhagic transformation after cardioembolic stroke. Stroke. 2001;32:2762-7 http://www.ncbi.nlm.nih.gov/pubmed/11739970
115. del Zoppo GJ, Frankowski H, Gu YH, Osada T, Kanazawa M, Milner R. et al. Microglial cell activation is a source of metalloproteinase generation during hemorrhagic transformation. J Cereb Blood Flow Metab. 2012;32:919-32 doi:10.1038/jcbfm.2012.11
116. Kwon I, Kim EH, del Zoppo GJ, Heo JH. Ultrastructural and temporal changes of the microvascular basement membrane and astrocyte interface following focal cerebral ischemia. J Neurosci Res. 2009;87:668-76 doi:10.1002/jnr.21877
117. Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol. 2010;10:712-23 doi:10.1038/nri2852nri2852 [pii]
118. Lakhan SE, Kirchgessner A, Tepper D, Leonard A. Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front Neurol. 2013;4:32. doi:10.3389/fneur.2013.00032
119. Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ. et al. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12:441-5 doi:10.1038/nm1387
120. Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:1020-30 http://www.ncbi.nlm.nih.gov/pubmed/9596253
121. Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY. et al. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005;25:6401-8 doi:10.1523/JNEUROSCI.1563-05.2005
122. Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20:1681-9 doi:10.1097/00004647-200012000-00007
123. Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA. et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724-32 http://www.ncbi.nlm.nih.gov/pubmed/11567062. Accessed 14 Aug 2014
124. Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz MA. et al. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J Neurosci. 2000;20:7037-42 http://www.ncbi.nlm.nih.gov/pubmed/10995849. Accessed 14 Aug 2014
125. Zlokovic B V. Remodeling after stroke. Nat Med. 2006;12:390-1 doi:10.1038/nm0406-390
126. Rosell A, Lo EH. Multiphasic roles for matrix metalloproteinases after stroke. Curr Opin Pharmacol. 2008;8:82-9 doi:10.1016/j.coph.2007.12.001
127. Yang Y, Rosenberg GA. Matrix metalloproteinases as therapeutic targets for stroke. Brain Res. 2015;1623:30-8 doi:10.1016/j.brainres.2015.04.024
128. Lee S-R, Kim H-Y, Rogowska J, Zhao B-Q, Bhide P, Parent JM. et al. Involvement of matrix metalloproteinase in neuroblast cell migration from the subventricular zone after stroke. J Neurosci. 2006;26:3491-5 doi:10.1523/JNEUROSCI.4085-05.2006
129. Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov. 2014;13:904-27 doi:10.1038/nrd4390
130. Chang D, Wang Y-C, Bai Y-Y, Lu C-Q, Xu T-T, Zhu L. et al. Role of P38 MAPK on MMP Activity in Photothrombotic Stroke Mice as Measured using an Ultrafast MMP Activatable Probe. Sci Rep. 2015;5:16951. doi:10.1038/srep16951
131. Klohs J, Baeva N, Steinbrink J, Bourayou R, Boettcher C, Royl G. et al. In vivo near-infrared fluorescence imaging of matrix metalloproteinase activity after cerebral ischemia. J Cereb Blood Flow Metab. 2009;29:1284-92 doi:10.1038/jcbfm.2009.51
132. Barber PA, Rushforth D, Agrawal S, Tuor UI. Infrared optical imaging of matrix metalloproteinases (MMPs) up regulation following ischemia reperfusion is ameliorated by hypothermia. BMC Neurosci. 2012;13:76. doi:10.1186/1471-2202-13-761471-2202-13-76 [pii]
133. Martín A, Garofalakis A, Tavitian B. In vivo evidence that the increase in matrix metalloproteinase activity occurs early after cerebral ischemia. Mol Imaging. 2012;11:22-6 http://www.ncbi.nlm.nih.gov/pubmed/22418024. Accessed 4 Jan 2018
134. Chen S, Cui J, Jiang T, Olson ES, Cai Q-Y, Yang M. et al. Gelatinase activity imaged by activatable cell-penetrating peptides in cell-based and in vivo models of stroke. J Cereb Blood Flow Metab. 2017;37:188-200 doi:10.1177/0271678X15621573
135. Luo S, Zhang E, Su Y, Cheng T, Shi C. A review of NIR dyes in cancer targeting and imaging. Biomaterials. 2011;32:7127-38 doi:10.1016/j.biomaterials.2011.06.024
136. Wagner S, Breyholz HJ, Höltke C, Faust A, Schober O, Schäfers M. et al. A new 18F-labelled derivative of the MMP inhibitor CGS 27023A for PET: radiosynthesis and initial small-animal PET studies. Appl Radiat Isot. 2009;67:606-10 doi:10.1016/j.apradiso.2008.12.009
137. Gerwien H, Hermann S, Zhang X, Korpos E, Song J, Kopka K. et al. Imaging matrix metalloproteinase activity in multiple sclerosis as a specific marker of leukocyte penetration of the blood-brain barrier. Sci Transl Med. 2016:8
138. Price CJS, Menon DK, Peters AM, Ballinger JR, Barber RW, Balan KK. et al. Cerebral neutrophil recruitment, histology, and outcome in acute ischemic stroke: An imaging-based study. Stroke. 2004;35:1659-64 doi:10.1161/01.STR.0000130592.71028.92
139. Chen L, Zhang G, Gu Y, Guo X. Meta-Analysis and Systematic Review of Neural Stem Cells therapy for experimental ischemia stroke in preclinical studies. 2016;6:32291. http://dx.doi.org/10.1038/srep32291.
140. Vu Q, Xie K, Eckert M, Zhao W, Cramer SC. Meta-analysis of preclinical studies of mesenchymal stromal cells for ischemic stroke. Neurology. 2014;82:1277-86
141. Vahidy FS, Rahbar MH, Zhu H, Rowan PJ, Bambhroliya AB, Savitz SI. Systematic Review and Meta-Analysis of Bone Marrow-Derived Mononuclear Cells in Animal Models of Ischemic Stroke. Stroke. 2016;47:1632-9 doi:10.1161/STROKEAHA.116.012701
142. Bacigaluppi M, Pluchino S, Peruzzotti-Jametti L, Jametti LP, Kilic E, Kilic U. et al. Delayed post-ischaemic neuroprotection following systemic neural stem cell transplantation involves multiple mechanisms. Brain. 2009:132 Pt 8:2239-51. doi:10.1093/brain/awp174
143. Shyu W-C, Lin S-Z, Chiang M-F, Su C-Y, Li H. Intracerebral peripheral blood stem cell (CD34+) implantation induces neuroplasticity by enhancing beta1 integrin-mediated angiogenesis in chronic stroke rats. J Neurosci. 2006;26:3444-53 doi:10.1523/JNEUROSCI.5165-05.2006
144. Nakano-Doi A, Nakagomi T, Fujikawa M, Nakagomi N, Kubo S, Lu S. et al. Bone marrow mononuclear cells promote proliferation of endogenous neural stem cells through vascular niches after cerebral infarction. Stem Cells. 2010;28:1292-302
145. Wang J, Yu L, Jiang C, Chen M, Ou C, Wang J. Bone marrow mononuclear cells exert long-term neuroprotection in a rat model of ischemic stroke by promoting arteriogenesis and angiogenesis. Brain Behav Immun. 2013;34:56-66 doi:10.1016/j.bbi.2013.07.010
146. Kubis N, Tomita Y, Tran-Dinh A, Planat-Benard V, André M, Karaszewski B. et al. Vascular fate of adipose tissue-derived adult stromal cells in the ischemic murine brain: A combined imaging-histological study. Neuroimage. 2007;34:1-11 doi:10.1016/j.neuroimage.2006.09.014
147. Liao W, Xie J, Zhong J, Liu Y, Du L, Zhou B. et al. Therapeutic effect of human umbilical cord multipotent mesenchymal stromal cells in a rat model of stroke. Transplantation. 2009;87:350-9 doi:10.1097/TP.0b013e318195742e
148. Brenneman M, Sharma S, Harting M, Strong R, Cox CS, Aronowski J. et al. Autologous bone marrow mononuclear cells enhance recovery after acute ischemic stroke in young and middle-aged rats. J Cereb Blood Flow Metab. 2010;30:140-9 doi:10.1038/jcbfm.2009.198
149. Yoo SW, Chang DY, Lee HS, Kim GH, Park JS, Ryu BY. et al. Immune following suppression mesenchymal stem cell transplantation in the ischemic brain is mediated by TGF-?? Neurobiol Dis. 2013;58:249-57 doi:10.1016/j.nbd.2013.06.001
150. Yang B, Hamilton JA, Valenzuela KS, Bogaerts A, Xi X, Aronowski J. et al. Multipotent Adult Progenitor Cells Enhance Recovery After Stroke by Modulating the Immune Response from the Spleen. Stem Cells. 2017;35:1290-302 doi:10.1002/stem.2600
151. Liesz A, Hu X, Kleinschnitz C, Offner H. Functional role of regulatory lymphocytes in stroke: facts and controversies. Stroke. 2015;46:1422-30 doi:10.1161/STROKEAHA.114.008608
152. Mitkari B, Kerkelä E, Nystedt J, Korhonen M, Mikkonen V, Huhtala T. et al. Intra-arterial infusion of human bone marrow-derived mesenchymal stem cells results in transient localization in the brain after cerebral ischemia in rats. Exp Neurol. 2013;239:158-62 doi:10.1016/j.expneurol.2012.09.018
153. Correa PL, Mesquita CT, Felix RM, Azevedo JC, Barbirato GB, Falcão CH. et al. Assessment of intra-arterial injected autologous bone marrow mononuclear cell distribution by radioactive labeling in acute ischemic stroke. Clin Nucl Med. 2007;32:839-41 doi:10.1097/RLU.0b013e318156b980
154. Hoehn M, Kustermann E, Blunk J, Wiedermann D, Trapp T, Wecker S. et al. Monitoring of implanted stem cell migration in vivo magnetic resonance imaging investigation of rat. Proc Natl Acad Sci U S A. 2002:99
155. Grudzenski S, Baier S, Ebert A, Pullens P, Lemke A, Bieback K. et al. The effect of adipose tissue-derived stem cells in a middle cerebral artery occlusion stroke model depends on their engraftment rate. Stem Cell Res Ther. 2017;8:96. doi:10.1186/s13287-017-0545-y
156. Bulte JWM. In Vivo MRI Cell Tracking: Clinical Studies. 2009; August:314-25.
157. Wolfs E, Holvoet B, Gijsbers R, Casteels C, Roberts SJ, Struys T. et al. Optimization of multimodal imaging of mesenchymal stem cells using the human sodium iodide symporter for pet and cerenkov luminescence imaging. PLoS One. 2014;9:1-12
158. Waerzeggers Y, Klein M, Miletic H, Himmelreich U, Li H, Monfared P. et al. Multimodal imaging of neural progenitor cell fate in rodents. Mol Imaging. 7:77-91. http://www.ncbi.nlm.nih.gov/pubmed/18706290. Accessed 8 Aug. 2017
159. Vandeputte C, Reumers V, Aelvoet S, Thiry I, Swaef S De, Haute C Van Den. et al. Bioluminescence imaging of stroke-induced endogenous neural stem cell response. Neurobiol Dis. 2014;69:144-55 doi:10.1016/j.nbd.2014.05.014
160. Kim DE, Schellingerhout D, Ishii K, Shah K, Weissleder R. Imaging of Stem Cell Recruitment to Ischemic Infarcts in a Murine Model. Stroke. 2004;35:952-7
161. Selt M, Tennstaedt A, Beyrau A, Nelles M, Schneider G, Löwik C. et al. In vivo non-invasive tracking of macrophage recruitment to experimental stroke. PLoS One. 2016;11:1-21
162. Tjuvajev JG, Avril N, Oku T, Sasajima T, Miyagawa T, Joshi R. et al. Imaging herpes virus thymidine kinase gene transfer and expression by positron emission tomography. Cancer Res. 1998;58:4333-41 http://www.ncbi.nlm.nih.gov/pubmed/9766661
163. Jacobs A, Voges J, Reszka R, Lercher M, Gossmann A, Kracht L. et al. Positron-emission tomography of vector-mediated gene expression in gene therapy for gliomas. Lancet (London, England). 2001;358:727-9 http://www.ncbi.nlm.nih.gov/pubmed/11551583. Accessed 8 Aug 2017
164. Dwyer RM, Ryan J, Havelin RJ, Morris JC, Miller BW, Liu Z. et al. Mesenchymal stem cell-mediated delivery of the sodium iodide symporter supports radionuclide imaging and treatment of breast cancer. Stem Cells. 2011;29:1149-57
165. Chien YC, Chen JCH, Lin WC, Ding HJ, Wang HE, Kao CHK. et al. Using [18F]FBAU for imaging brain tumor progression in an F98/tk-luc glioma-bearing rat model. Oncol Rep. 2014;32:691-9
166. Perin EC, Tian M, Marini FC, Silva G V, Zheng Y, Baimbridge F. et al. Imaging long-term fate of intramyocardially implanted mesenchymal stem cells in a porcine myocardial infarction model. PLoS One. 2011;6:e22949. doi:10.1371/journal.pone.0022949
167. Vandsburger MH, Radoul M, Cohen B, Neeman M. MRI Reporter Genes: Applications For Imaging Of Cell Survival, Proliferation, Migration and Differentiation. NMR Biomed. 2013;26:872-84
168. Daadi MM, Hu S, Klausner J, Li Z, Sofilos M, Sun G. et al. Imaging Neural Stem Cell Graft-Induced Structural Repair in Stroke. Cell Transplant. 2013;22:881-92 doi:10.3727/096368912X656144
169. Rueger MA, Muesken S, Walberer M, Jantzen SU, Schnakenburg K, Backes H. et al. Effects of minocycline on endogenous neural stem cells after experimental stroke. Neuroscience. 2012;215:174-83 doi:10.1016/j.neuroscience.2012.04.036
170. Winek K, Dirnagl U, Meisel A. The Gut Microbiome as Therapeutic Target in Central Nervous System Diseases: Implications for Stroke. Neurotherapeutics. 2016;13:762-74 doi:10.1007/s13311-016-0475-x
171. Winek K, Meisel A, Dirnagl U. Gut microbiota impact on stroke outcome: Fad or fact? J Cereb Blood Flow Metab. 2016;36:891-8 doi:10.1177/0271678X16636890
172. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M. et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015:212
173. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD. et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337-41 doi:10.1038/nature14432
174. Sharon G, Sampson TR, Geschwind DH, Mazmanian SK. The Central Nervous System and the Gut Microbiome. Cell. 2016;167:915-32 doi:10.1016/j.cell.2016.10.027
175. Schroeder BO, Bäckhed F. Signals from the gut microbiota to distant organs in physiology and disease. Nat Med. 2016;22:1079-89 doi:10.1038/nm.4185
176. Luczynski P, McVey Neufeld K-A, Oriach CS, Clarke G, Dinan TG, Cryan JF. Growing up in a Bubble: Using Germ-Free Animals to Assess the Influence of the Gut Microbiota on Brain and Behavior. Int J Neuropsychopharmacol. 2016;19:pyw020. doi:10.1093/ijnp/pyw020
177. Winek K, Engel O, Koduah P, Heimesaat MM, Fischer A, Bereswill S. et al. Depletion of Cultivatable Gut Microbiota by Broad-Spectrum Antibiotic Pretreatment Worsens Outcome After Murine Stroke. Stroke. 2016;47:1354-63 doi:10.1161/STROKEAHA.115.011800
178. Houlden A, Goldrick M, Brough D, Vizi ES, Lénárt N, Martinecz B. et al. Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav Immun. 2016;57:10-20 doi:10.1016/j.bbi.2016.04.003
179. Singh V, Roth S, Llovera G, Sadler R, Garzetti D, Stecher B. et al. Microbiota Dysbiosis Controls the Neuroinflammatory Response after Stroke. J Neurosci. 2016:36
180. Yin J, Liao S, He Y, Wang S, Xia G, Liu F. et al. Dysbiosis of Gut Microbiota With Reduced Trimethylamine-N-Oxide Level in Patients With Large-Artery Atherosclerotic Stroke or Transient Ischemic Attack. J Am Heart Assoc. 2015;4:e002699. doi:10.1161/JAHA.115.002699
181. Swidsinski A, Loening-Baucke V, Krüger M, Kirsch S, Grau A, Urbanek C. et al. Central Nervous System and the Colonic Bioreactor: Analysis of Colonic Microbiota in Patients with Stroke Unravels Unknown Mechanisms of the Host Defense after Brain Injury. Intest Res. 2012;10:332. doi:10.5217/ir.2012.10.4.332
182. Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M. et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat Med. 2016;22:516-23 doi:10.1038/nm.4068
183. Kisler K, Nelson AR, Montagne A, Zlokovic B V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci. 2017;18:419-34 doi:10.1038/nrn.2017.48
184. Becerra-Calixto A, Cardona-Gómez GP. The Role of Astrocytes in Neuroprotection after Brain Stroke: Potential in Cell Therapy. Front Mol Neurosci. 2017;10:88. doi:10.3389/fnmol.2017.00088
185. Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232-43 doi:10.1038/nature09613
186. Becerra-Calixto A, Cardona-Gómez GP. The Role of Astrocytes in Neuroprotection after Brain Stroke: Potential in Cell Therapy. Front Mol Neurosci. 2017;10:88. doi:10.3389/fnmol.2017.00088
187. Rodriguez-Vieitez E, Saint-Aubert L, Carter SF, Almkvist O, Farid K, Schöll M. et al. Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer's disease. Brain. 2016;139:922-36 doi:10.1093/brain/awv404
188. Schöll M, Carter SF, Westman E, Rodriguez-Vieitez E, Almkvist O, Thordardottir S. et al. Early astrocytosis in autosomal dominant Alzheimer's disease measured in vivo by multi-tracer positron emission tomography. Sci Rep. 2015;5:16404. doi:10.1038/srep16404
189. Lavisse S, Guillermier M, Herard AS, Petit F, Delahaye M, Van Camp N. et al. Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci. 2012;32:10809-18 doi:10.1523/JNEUROSCI.1487-12.201232/32/10809 [pii]
190. Wintermark M, Sanelli PC, Albers GW, Bello JA, Derdeyn CP, Hetts SW. et al. Imaging recommendations for acute stroke and transient ischemic attack patients: a joint statement by the American Society of Neuroradiology, the American College of Radiology and the Society of NeuroInterventional Surgery. J Am Coll Radiol. 2013;10:828-32 doi:10.1016/j.jacr.2013.06.019
191. Gonzalez RG. Current State of Acute Stroke Imaging. Stroke. 2013;44:3260-4 doi:10.1161/STROKEAHA.113.003229
192. Cook D, Brown D, Alexander R, March R, Morgan P, Satterthwaite G. et al. Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 2014;13:419-31 doi:10.1038/nrd4309
193. Anrather J, Iadecola C. Inflammation and Stroke: An Overview. Neurotherapeutics. 2016;13:661-70 doi:10.1007/s13311-016-0483-x
194. Dirnagl U, Hakim A, Macleod M, Fisher M, Howells D, Alan SM. et al. A concerted appeal for international cooperation in preclinical stroke research. Stroke. 2013;44:1754-60 doi:10.1161/STROKEAHA.113.000734
195. Milidonis X, Lennen RJ, Jansen MA, Mueller S, Boehm-Sturm P, Holmes WM. et al. Multicenter Evaluation of Geometric Accuracy of MRI Protocols Used in Experimental Stroke. PLoS One. 2016;11:e0162545. doi:10.1371/journal.pone.0162545
196. Llovera G, Hofmann K, Roth S, Salas-Pérdomo A, Ferrer-Ferrer M, Perego C. et al. Results of a preclinical randomized controlled multicenter trial (pRCT): Anti-CD49d treatment for acute brain ischemia. Sci Transl Med. 2015:7
197. Maysami S, Wong R, Pradillo JM, Denes A, Dhungana H, Malm T. et al. A cross-laboratory preclinical study on the effectiveness of interleukin-1 receptor antagonist in stroke. J Cereb Blood Flow Metab. 2016;36:596-605 doi:10.1177/0271678X15606714
198. Kleikers PWM, Hooijmans C, Göb E, Langhauser F, Rewell SSJ, Radermacher K. et al. A combined pre-clinical meta-analysis and randomized confirmatory trial approach to improve data validity for therapeutic target validation. Sci Rep. 2015;5:13428. doi:10.1038/srep13428
199. Boltze J, Wagner D-C, Henninger N, Plesnila N, Ayata C. Phase III Preclinical Trials in Translational Stroke Research: Community Response on Framework and Guidelines. Transl Stroke Res. 2016;7:241-7 doi:10.1007/s12975-016-0474-6
200. Takano A, Varrone A, Gulyás B, Salvadori P, Gee A, Windhorst A. et al. Guidelines to PET measurements of the target occupancy in the brain for drug development. Eur J Nucl Med Mol Imaging. 2016;43:2255-62 doi:10.1007/s00259-016-3476-4
201. Gunn RN, Gunn SR, Cunningham VJ. Positron emission tomography compartmental models. J Cereb Blood Flow Metab. 2001;21:635-52 doi:10.1097/00004647-200106000-00002
202. Kuntner C, Stout D. Quantitative preclinical PET imaging: opportunities and challenges. Front Phys. 2014;2:12. doi:10.3389/fphy.2014.00012
203. Lavisse S, García-Lorenzo D, Peyronneau M-A, Bodini B, Thiriez C, Kuhnast B. et al. Optimized Quantification of Translocator Protein Radioligand 18F-DPA-714 Uptake in the Brain of Genotyped Healthy Volunteers. J Nucl Med. 2015;56:1048-54 doi:10.2967/jnumed.115.156083
204. Golla SS V, Boellaard R, Oikonen V, Hoffmann A, van Berckel BNM, Windhorst AD. et al. Parametric Binding Images of the TSPO Ligand 18F-DPA-714. J Nucl Med. 2016;57:1543-7 doi:10.2967/jnumed.116.173013
205. Golla SS V, Boellaard R, Oikonen V, Hoffmann A, van Berckel BNM, Windhorst AD. et al. Quantification of [18F]DPA-714 binding in the human brain: initial studies in healthy controls and Alzheimer's disease patients. J Cereb Blood Flow Metab. 2015;35:766-72 doi:10.1038/jcbfm.2014.261
Author contact
![]() Corresponding author: Andreas H Jacobs and Bastian Zinnhardt, European Institute for Molecular Imaging (EIMI), Westfälische Wilhelms University Münster, Waldeyerstraße 15, 48149 Münster, Germany. Phone: +492518349300; Fax: +492518349312; Email: ahjacobsde; zinnhardtde
Corresponding author: Andreas H Jacobs and Bastian Zinnhardt, European Institute for Molecular Imaging (EIMI), Westfälische Wilhelms University Münster, Waldeyerstraße 15, 48149 Münster, Germany. Phone: +492518349300; Fax: +492518349312; Email: ahjacobsde; zinnhardtde