Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(11):2896-2908. doi:10.7150/thno.24378 This issue Cite
Research Paper
Site-specific stabilization of minigastrin analogs against enzymatic degradation for enhanced cholecystokinin-2 receptor targeting
Maximilian Klingler1, Clemens Decristoforo1, Christine Rangger1, Dominik Summer1, Julie Foster2, Jane K Sosabowski2, Elisabeth von Guggenberg1 ![]()
1. Department of Nuclear Medicine, Medical University of Innsbruck, Innsbruck, Austria
2. Centre for Molecular Oncology, Barts Cancer Institute, Queen Mary University of London, London, United Kingdom
Received 2017-12-15; Accepted 2018-3-1; Published 2018-4-16
Abstract

Minigastrin (MG) analogs show high affinity to the cholecystokinin-2 receptor (CCK2R) and have therefore been intensively studied to find a suitable analog for imaging and treatment of CCK2R-expressing tumors. The clinical translation of the radioligands developed thus far has been hampered by high kidney uptake or low enzymatic stability. In this study, we aimed to develop new MG analogs with improved targeting properties stabilized against degradation through site-specific amino acid modifications.
Method: Based on the lead structure of a truncated MG analog, four new MG derivatives with substitutions in the C-terminal part of the peptide (Trp-Met-Asp-Phe-NH2) were synthesized and derivatized with DOTA at the N-terminus for radiolabeling with trivalent radiometals. The in vitro properties of the new analogs were characterized by analyzing the lipophilicity, the protein binding, and the stability of the Indium-111 (111In)-labeled analogs in different media. Two different cell lines, AR42J cells physiologically expressing the rat CCK2R and A431 cells transfected with human CCK2R (A431-CCK2R), were used to study the receptor affinity and cell uptake. For the two most promising MG analogs, metabolic studies in normal BALB/c mice were carried out as well as biodistribution and imaging studies in tumor xenografted athymic BALB/c nude mice.
Results: Two out of four synthesized peptide analogs (DOTA-MGS1 and DOTA-MGS4) showed retained receptor affinity and cell uptake when radiolabeled with 111In. These two peptide analogs, however, showed a different stability against enzymatic degradation in vitro and in vivo. When injected to normal BALB/c mice, for 111In-DOTA-MGS1 at 10 min post injection (p.i.) no intact radiopeptide was found in the blood, whereas for 111In-DOTA-MGS4 more than 75% was still intact. 111In-DOTA-MGS4 showed a clear increase in injected activity per gram tissue (IA/g) for A431-CCK2R xenografts (10.40±2.21% IA/g 4 h p.i.) when compared to 111In-DOTA-MGS1 (1.23±0.15% IA/g 4 h p.i.). The tumor uptake of 111In-DOTA-MGS4 was also combined with a low uptake in stomach and kidney leading to high-contrast NanoSPECT/CT images.
Conclusion: Of the four new MG analogs developed, the best results in terms of enzymatic stability and increased tumor targeting were obtained with 111In-DOTA-MGS4 showing two substitutions with N-methylated amino acids. 111In-DOTA-MGS4 was also superior to other MG analogs reported thus far and seems therefore an extremely promising targeting molecule for theranostic use with alternative radiometals.
Keywords: minigastrin, cholecystokinin-2 receptor, metabolic stabilization, molecular imaging, radiometals
Introduction
G-protein-coupled receptors, which are found on the cell membrane of different tumor cells, are an ideal target for the detection and treatment of cancer [1]. Radiolabeled peptide receptor agonists and antagonists have been successfully used to target somatostatin receptors in neuroendocrine tumors and the gastrin-releasing peptide receptor in prostate and breast cancer lesions [2-5]. Another attractive target is the cholecystokinin-2 receptor (CCK2R) overexpressed in different neoplasms, including medullary thyroid carcinoma (MTC) and small cell lung cancer [6]. However, CCK2R targeting with radiolabeled peptide analogs, based on the natural ligands minigastrin (MG) and cholecystokinin (CCK), has not yet resulted in a widely accepted diagnostic and therapeutic application in nuclear medicine [7-9].
Many attempts have been made to find CCK2R specific ligands suitable for single photon emission tomography (SPECT), positron emission tomography (PET), and therapeutic applications [9-17]. Using [DGlu1]minigastrin (MG0) conjugated to the linear chelator diethylenetriaminepentaacetic acid (DTPA) a high tumor uptake was seen in scintigraphic imaging with indium-111 (111In), but the uptake in the kidneys was also very high leading to nephrotoxic side effects in therapeutic applications with yttrium-90 [14,18].
The N-terminal penta-Glu sequence in the peptide chain of MG0 was related to the high kidney uptake [19]. Using [DGlu1,desGlu2-6]MG (MG11) conjugated to the macrocyclic chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) a large reduction in kidney uptake could be achieved; however, low enzymatic stability seemed to impair the tumor uptake [8,20-22].
To address the problems related to high kidney uptake and low bioavailability, a number of working groups combined under a concerted European COST Action (BM0607 Targeted Radionuclide Therapy) evaluated 12 new CCK2R targeting peptide analogs. Besides high CCK2R receptor affinity and high cell uptake, metabolic studies in vivo revealed a rapid enzymatic degradation of the different radiolabeled peptide derivatives [23,24]. In comparative biodistribution studies, 111In-labeled CP04 (formerly PP-F11 or PP11-D), a MG analog with the penta-Glu sequence substituted by a penta-DGlu sequence, showed the most advantageous properties in terms of tumor uptake and tumor-to-kidney ratio [25,26]. First clinical studies with 111In-CP04 and 177Lu-DOTA-PP-F11N, a MG analog derived from CP04 by substitution of Met with Nle, have demonstrated the feasibility of CCK2R targeting and identified stomach and kidneys as possible dose-limiting organs [27,28].
It has been shown that radiolabeled MG analogs can be stabilized in vivo by co-injection of enzyme inhibitors such as the neutral endopeptidase inhibitor phosphoramidon [29]. Especially for 111In-DOTA-MG11, the stabilization against degradation was correlated with a significant increase in tumor uptake [30]. Clinical evidence that this highly promising new approach also leads to increased bioavailability and tumor uptake in patients is still missing. So far, the different chemical modifications introduced in CCK2R-targeting peptide analogs mainly focused on the N-terminal part of the peptide and were not successful in stabilizing the peptide against degradation [23]. No modifications other than substitution of oxidation-prone Met were applied in the C-terminal sequence Trp-Met-Asp-Phe-NH2, which is essential for receptor binding [14]. The aim of this study was to investigate substitutions in this particular region of the peptide. For this purpose, N-methylated amino acids or amino acids with bulky aromatic side chains were introduced in positions 6 and 8 of the peptide sequence of truncated DOTA-MG11. Based on data available in the literature, the applied substitutions retained CCK2R affinity and metabolically stabilized CCK analogs [31,32], and we have translated these modifications to MG analogs. We report here on the development of four new radiolabeled MG analogs for possible theranostic use with different radiometals. The initial characterization in vitro and in vivo has been carried out with the 111In-labeled peptide derivatives and includes receptor affinity, cell uptake, and stability studies, as well as biodistribution and imaging studies in a mouse tumor xenograft model.
Materials and Methods
Materials
All commercially obtained chemicals were of analytical grade and used without further purification unless otherwise stated. 111InCl3 was purchased from Mallinckrodt Medical (Petten, The Netherlands). DOTA-MG11 used for comparative studies was purchased from piCHEM (Raaba-Grambach, Austria).
Peptide synthesis
Using MG11 (DGlu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2) as lead structure, four different DOTA-MG analogs with specific amino acid substitutions were synthesised by standard solid phase peptide synthesis using 9-fluorenylmethoxycarbonyl (Fmoc)-protected amino acids, as previously described [33]. The peptides were assembled on a rink amide MBHA resin with capacity of 0.5 mmol/g resin (Novabiochem, Hohenbrunn, Germany). Coupling of the Fmoc amino acids following (N-Me)Nle or (N-Me)Phe was repeated twice.
Purification was performed by reversed phase high performance liquid chromatography (RP-HPLC) on a Dionex P680 chromatography system (Dionex, Gemering, Germany) with a Dionex UVD170U multi-wavelength UV detector, equipped with a Nucleosil 300-5-C18 column (8 × 250 mm), using a gradient system starting from water containing 0.1% trifluoroacetic acid (TFA) (solvent A) and increasing the concentration of acetonitrile (ACN) containing 0.1% TFA (solvent B), with flow rate of 3 mL/min: 0-3 min 0% B, 3-5 min 0-25% B, 5-27 min 25-47% B, 27-32 min 47-60% B, 32-35 min 60-0% B, 35-40 min 0% B (HPLC method 1). The peptide conjugates were analyzed on a Dionex UltiMate 3000 chromatography system, consisting of a HPLC pump, a variable UV-detector (UV-VIS at λ = 280 nm), an autosampler, a radiodetector (GabiStar, Raytest, Straubenhardt, Germany), a Nucleosil 120-5-C18 column (Macherey-Nagel, Düren, Germany), and a gradient system with a flow rate of 1 mL/min: 0-3 min 0% B, 3-5 min 0-25% B, 5-20 min 25-40% B, 20-25 min 40-60% B, 25-28 min 60-0% B, 28-33 min 0% B (HPLC method 2). The synthesized peptide conjugates with confirmed purity were characterized by MALDI-TOF MS and lyophilized.
Radiolabeling and radiochemical analysis
For labeling with 111In, the DOTA-peptides were incubated with 111InCl3 solution at a specific activity of 2-10 GBq/µmol. The reaction solution was adjusted to pH 5 using a 1.2-fold volume of 0.4 M sodium acetate/0.24 M gentisic acid (final peptide concentration >40 µM). For lower peptide concentration of 10 µM and higher specific activity >30 GBq/µmol, 0.4 M 2-(N-morpholino)ethanesulfonic acid (MES) was used for pH adjustment. The reaction solution was incubated at elevated temperature in a low protein binding microcentrifuge tube (Eppendorf, Hamburg, D). Radiochemical analysis of the radiolabeled peptide conjugates was performed using HPLC method 2. For in vitro and in vivo assays, the radiolabeled peptides were purified by solid phase extraction (SPE), as previously described [35]. For NanoSPECT/CT imaging studies, the reaction solution was quenched with 0.1 M EDTA (1/20th of the reaction volume) and the radioligands were used without SPE purification.
Characterization of the radiolabeled peptides in vitro
For determination of the distribution coefficient (log D), the radiolabeled DOTA-peptides diluted in PBS were mixed with an equal volume of octanol in a low protein binding microcentrifuge tube (~20 pmol/ml). The mixture was vigorously vortexed at room temperature (RT) over a period of 15 min using a small shaker (MS3 Basic, IKA, Staufen, Germany) with a speed of 1,500 rpm. After a waiting time of 10 min, sufficient for the separation of the two phases, 200 µL aliquots of both layers were measured in a 2480 Wizard2 automatic gamma-counter (PerkinElmer Life Sciences and Analytical Sciences, Turku, Finland) and the log D value (n=6) was calculated.
To get first insights into the metabolic stability, the radiolabeled DOTA-peptides were incubated in human serum (~2,000 pmol/mL) as well as rat liver and kidney homogenates (~ 500 pmol/mL), according to previously established protocols [10]. At different time points after incubation at 37 °C, the degradation of the radioligands was assessed by radio-HPLC (HPLC method 2). The percentage of intact radiopeptide during incubation was calculated in relation to the radiochemical purity (RCP) after SPE purification. Additionally, the radiolabeled DOTA-peptides were incubated in PBS (~2,000 pmol peptide/mL) at RT and the stability assessed by radio-HPLC.
Protein binding was determined from serum samples using Sephadex G-50 size-exclusion chromatography (GE Healthcare Illustra, Little Chalfont, UK) [10].
Receptor binding und cell uptake studies
AR42J rat pancreatic tumor cells (ECACC, Salisbury, UK), physiologically expressing the rat CCK2R, and A431 human epidermoid carcinoma cells, stably transfected with the plasmid pCR3.1 containing the full coding sequence for the human CCK2R (A431-CCK2R) and with the empty vector alone (A431-mock) [34], were used for receptor binding and cell uptake studies. AR42J cells were cultured in RPMI 1640 medium and A431 cells in DMEM, each supplemented with 10% (v/v) fetal bovine serum and 5 mL of a 100× penicillin-streptomycin-glutamine mixture, at 37 °C in a humidified 95% air/5% CO2 atmosphere. Media and supplements were purchased from Invitrogen Corporation (Lofer, AUT).
Internalization experiments with the radiolabeled DOTA-peptides were carried out as previously described [35], using 1.5×106 AR42J cells and 1.0×106 A431-CCK2R and A431-mock cells per well incubated with ~50,000 cpm of radioligand (final peptide concentration of ~0.4 nM). Non-specific binding was determined by blocking with 1 µM pentagastrin in AR42J cells or by using A431-mock cells without receptor expression. The binding specificity of A431-CCK2R cells was confirmed by additional blocking studies. For different time points up to 2 h incubation, the specific cell uptake was calculated by subtracting the non-specifically internalized radioligand fraction found in blocked AR42J cells or A431-mock cells from the internalized radioligand fraction in AR42J or A431-CCK2R cells and expressed in relation to the total activity added (% of specific cell uptake). For externalization experiments, after 2 h incubation the supernatant was replaced with 3.0 mL medium (unblocked) or medium containing 1 µM pentagastrin (blocked). A 300 µL sample of supernatant was taken at different time points. After 4 h the remaining medium and the cells were collected and all fractions measured in the gamma counter. The percentage of externalized radioactivity over time was calculated in relation to the internalized radioligand fraction determined before medium replacement.
The binding affinity of the DOTA-peptides for the CCK2R was tested in a competition assay against [125I]Tyr12-gastrin I (Perkin Elmer Life Science, Boston, MA). The cells were prepared as described for internalization experiments and incubated in triplicates with the radioligand (~30,000 cpm in 100 µL PBS/0.5%BSA) and increasing concentrations (0.001-1,000 nM in 150 µL PBS/0.5%BSA) of the DOTA-peptides or DOTA-MG11 for comparison. Incubation was performed at RT under which conditions a limited extent of internalization may affect the binding equilibrium. We therefore describe the binding affinity measurements obtained from this assay as apparent half maximal inhibitory concentration (IC50) [35,36]. The apparent IC50 value was calculated following non-linear regression with Origin software (Origin 6.1, OriginLab, Northampton, MA).
Metabolic studies and tumor targeting in vivo
All animal experiments were conducted in compliance with the Austrian animal protection laws and with the approval of the Austrian Ministry of Science (BMWF-66.011/0083-II/3b/2012). As different reports on the metabolic stability and biodistribution of 111In-DOTA-MG11 are already available in the literature, these studies were carried out only with 111In-DOTA-MGS1 and 111In-DOTA-MGS4.
Metabolic studies evaluating the stability of the radioligands in vivo were carried out in 5-6-week-old female BALB/c mice (Charles River, Sulzfeld, Germany; n = 2). To allow monitoring of the metabolites by radio-HPLC, mice were injected intravenously with a higher amount of radioactivity (10-15 MBq, corresponding to 3-4 nmol total peptide) through a lateral tail vein. Mice were euthanized by cervical dislocation 10 min post injection (p.i.). A sample of urine and blood was collected, and liver and kidneys were dissected. All samples were processed according to previously established protocols [35] and analyzed by radio-HPLC (HPLC method 2).
Biodistribution studies were performed in 6-week-old female athymic BALB/c nude mice (Charles River, Sulzfeld, Germany). For the induction of tumor xenografts, A431-CCK2R and A431-mock cells (2×106 in 200 μL DMEM) were injected subcutaneously in the left and right flank, respectively. After 10-12 days, when tumors had reached a size of ~0.3 mL (with medium tumor weights of 0.27±0.17 g for A431-CCK2R tumors and 0.35±0.25 g for A431-mock tumors), mice were randomly divided into groups of three and injected intravenously via a lateral tail vein with the 111In-labeled DOTA-peptides. For comparison with previous studies, an injected activity of 0.3 MBq corresponding to 0.03 nmol peptide was used [25]. The groups of animals were sacrificed by cervical dislocation 1 or 4 h p.i. and the tumors and other tissues (blood, lung, heart, muscle, spleen, intestine, liver, kidney, stomach, and pancreas) were removed. The samples were weighed and measured in the gamma counter to calculate the percentage of injected activity per gram tissue (%IA/g) as well as tumor-to-organ activity ratios. Statistical analysis was performed using independent two population t-test (significance level 0.05) and Origin software.
Whole body imaging studies were carried out using a NanoSPECT/CT four-headed camera (Bioscan, Washington, DC). The xenografted male athymic BALB/c nude mice (Charles River, Harlow, UK) with medium tumor weights of 0.38±0.15 g for A431-CCK2R tumors and 0.27±0.10 g for A431-mock tumors were injected intravenously with 10 MBq (0.3 nmol) of either 111In-DOTA-MGS1 (n = 2) or 111In-DOTA-MGS4 (n = 3). Whole body SPECT/CT images were obtained at 1 and 4 h p.i. under inhaled anesthesia (1 L/min 1.5% isoflurane in O2) in prone position on a heated bed to maintain a body temperature (scanning time 45 min). SPECT images were obtained in 24 projections with 1.4 mm pinhole collimators in helical scanning mode and a voxel size of 100×100 µm per voxel. CTs were imaged with a 45 kVp X-ray source in 180 projections over 6 min. Images were reconstructed (CT recon: InVivoScope inviCRO LLC, Boston, USA. SPECT recon: HiSPECT, Scivis GmbH, Göttingen, Germany), then merged, and region of interest analysis in tumors and kidneys was performed using VivoQuant software (inviCRO LLC, Boston, USA). After scanning (approximately 5 h p.i.) the mice were sacrificed by cervical dislocation. Tumors and other tissues were removed, weighed, and their radioactivity measured in a gamma counter (1282 Compugamma CS, LKB Wallac).
Results
Peptide synthesis
The peptide derivatives were synthesized with 20-35% yield, starting from 50 µmol resin, using 250 µmol of each Fmoc-protected amino acid and 150 µmol of Boc-protected DOTA. The peptide conjugates were obtained at a chemical purity ≥95% as confirmed by RP-HPLC and characterized by MALDI-TOF MS. The peptide structure, amino acid sequence, and analytical data for the four DOTA-peptides are presented in Figure 1 and Table 1.
Structure of (A) DOTA-MG11, (B) DOTA-MGS1, (C) DOTA-MGS2, (D) DOTA-MGS3, (E) DOTA-MGS4; R = DOTA-DGlu-Ala-Tyr-Gly
Analytical data for the different DOTA-MG derivatives.
| Peptide conjugate | Peptide sequence | Yield (%) | Purity (%) | tR (min) | MW calculated, m/z [M+H]+ | MW found, m/z [M+H]+ |
|---|---|---|---|---|---|---|
| DOTA-MGS1 | DOTA-DGlu-Ala-Tyr-Gly-Trp-Met-Asp-1-NaI-NH2 | 35 | ≥ 97 | 19.0 | 1453.6 | 1453.5 |
| DOTA-MGS2 | DOTA-DGlu-Ala-Tyr-Gly-Trp-Met-Asp-(N-Me)Phe-NH2 | 35 | ≥ 98 | 15.8 | 1417.6 | 1417.6 |
| DOTA-MGS3 | DOTA-DGlu-Ala-Tyr-Gly-Trp-Phg-Asp-1-Nal-NH2 | 30 | ≥ 98 | 20.0 | 1455.6 | 1455.5 |
| DOTA-MGS4 | DOTA-DGlu-Ala-Tyr-Gly-Trp-(N-Me)Nle-Asp-(N-Me)Phe-NH2 | 20 | ≥ 98 | 19.8 | 1413.7 | 1413.6 |
Radiolabeling
Radiolabeling with 111In resulted in a RCP of 90-99%. For the Met-containing peptide conjugates DOTA-MGS1, DOTA-MGS2 and DOTA-MG11 using milder reaction conditions of 80 °C and a shorter incubation time of 15 min, the oxidative side products in the reaction could be reduced to 5-10%, reaching a RCP ≥90%. In the radio-HPLC profiles, retention times (tR) in the order of 111In-DOTA-MG11 (15.5 min) < 111In-DOTA-MGS2 (15.8 min) < 111In-DOTA-MGS1 (19.0 min) < 111In-DOTA-MGS4 (19.8 min) < 111In-DOTA-MGS3 (20.0 min) were found.
Characterization in vitro and stability studies
A summary of the in vitro characteristics of the different 111In-labeled MG derivatives in comparison with 111In-DOTA-MG11 is shown in Table 2.
In vitro characteristics of the 111In-labeled DOTA-peptides.
| Radiolabeled peptide | log D | Protein binding 4 h after incubation (%) | Stability in PBS 24 h after incubation (% intact radiopeptide) | Stability in serum 24 h after incubation (% intact radiopeptide) |
|---|---|---|---|---|
| 111In-DOTA-MG11 | -3.55±0.23 | 10.1±2.7 | 94.3 | 16.1±0.5 |
| 111In-DOTA-MGS1 | -3.00±0.17 | 20.0±1.8 | 90.8 | 58.7±2.0 |
| 111In-DOTA-MGS2 | -3.46±0.19 | 11.3±7.3 | 93.2 | 87.4±0.3 |
| 111In-DOTA-MGS3 | -2.58±0.13 | 26.2±9.5 | 95.4 | 94.0±1.2 |
| 111In-DOTA-MGS4 | -2.49±0.06 | 8.9±4.5 | 96.2 | 96.7±0.2 |
According to log D values, the radioligands showed a hydrophilicity profile in the order of 111In-DOTA-MG11 > 111In-DOTA-MGS2 > 111In-DOTA-MGS1 > 111In-DOTA-MGS3 and 111In-DOTA-MGS4. A similar order was observed in the HPLC analysis. Protein binding analyzed at 2, 4 and 24 h did not vary considerably over time. After 4 h incubation, values of 11.3±7.3% and 8.9±4.5% were found for 111In-DOTA-MGS2 and 111In-DOTA-MGS4, respectively, which compare well to 111In-DOTA-MG11 (10.1±2.7%). 111In-DOTA-MGS1 (20.0±1.8%) and 111In-DOTA-MGS3 (26.2±9.5%) showed a slightly increased protein binding.
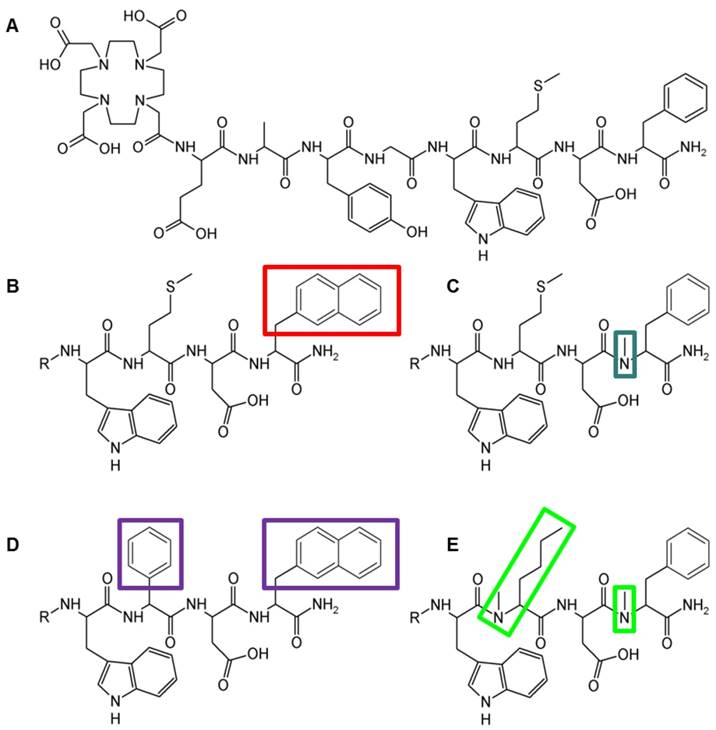
A high chemical stability of all radiolabeled complexes was found in PBS solution, including 111In-DOTA-MG11. For all Met-containing compounds the levels of oxidative side products remained <10% up to 24 h after incubation. The enzymatic stability in human serum as well as in rat liver and kidney homogenates is summarized in Figure 2. In human serum, a low stability was found for 111In-DOTA-MG11 and 111In-DOTA-MGS1. After 24 h incubation only ~16% of intact 111In-DOTA-MG11 and ~60% of intact 111In-DOTA-MGS1 was detected. 111In-DOTA-MGS2 showed a much lower degradation of 13%; for 111In-DOTA-MGS3 and 111In-DOTA-MGS4 only a limited amount (6% and 3%, respectively) was degraded after 24 h incubation. In rat liver and kidney homogenates, a higher degree of degradation was observed for all radioligands. When comparing the incubation in rat liver homogenate of the new designed MG analogs to 111In-DOTA-MG11, all four compounds showed a higher stability. However, only for 111In-DOTA-MGS3 and 111In-DOTA-MGS4, still some intact radiopeptide could be detected at 2 h after incubation. The values of intact radioligand after 2 h incubation in liver homogenate were in the order of 111In-DOTA-MGS3 (54.5±3.7%) > 111In-DOTA-MGS4 (19.4±2.2%) > 111In-DOTA-MGS2 (1.3±0.3%) > 111In-DOTA-MGS1 and 111In-DOTA-MG11 (both <1%). In rat kidney homogenate a stability increase was observed only for 111In-DOTA-MGS3 and 111In-DOTA-MGS4, whereas 111In-DOTA-MGS1 and 111In-DOTA-MGS2 showed a very rapid and complete degradation comparable to 111In-DOTA-MG11. The values of intact radioligand after 2 h incubation in kidney homogenate were in the order of 111In-DOTA-MGS3 (34.3±0.8%) > 111In-DOTA-MGS4 (11.0±1.1%) > 111In-DOTA-MGS2, 111In-DOTA-MGS1 and 111In-DOTA-MG11 (all three <1%).
Intact radiopeptide after incubation at 37 °C in (A) human serum as well as rat tissue homogenates of (B) liver and (C) kidney for 111In-DOTA-MG11 (open squares), 111In-DOTA-MGS1 (red), 111In-DOTA-MGS2 (dark cyan), 111In-DOTA-MGS3 (purple) and 111In-DOTA-MGS4 (green).
CCK2R affinity and specific cell internalization
The apparent IC50 values determined from competitive binding studies are summarized in Table 3. In AR42J cells, DOTA-MGS1 showed a similar binding affinity to DOTA-MG11, while the binding affinity of DOTA-MGS4 was lower. In A431-CCK2R cells the three peptide derivatives showed comparable affinities. DOTA-MGS2 and DOTA-MGS3 showed an impaired binding affinity in both cell lines.
Apparent IC50 values of the new MG analogs and DOTA-MG11 in AR42J and A431-CCK2R cells.
| Peptide conjugate | AR42J IC50 (nM) | A431-CCK2R IC50 (nM) |
|---|---|---|
| DOTA-MG11 | 2.24 | 4.96 |
| DOTA-MGS1 | 1.92 | 3.51 |
| DOTA-MGS2 | 22.6 | 40.4 |
| DOTA-MGS3 | 56.8 | 184 |
| DOTA-MGS4 | 4.53 | 4.98 |
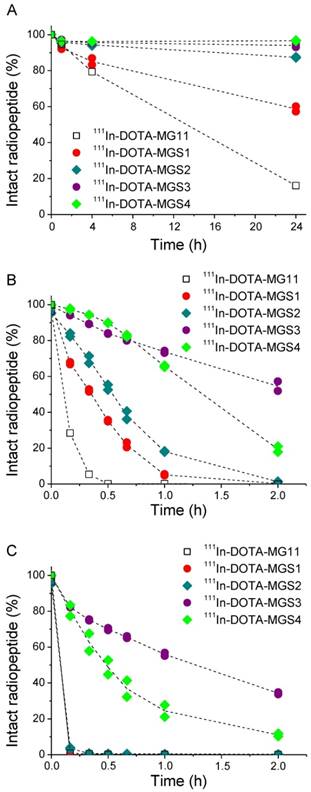
A similar trend was found in the internalization assays. In AR42J cells, 111In-DOTA-MG11 (18.2±0.9%) and 111In-DOTA-MGS1 (29.1±0.3%) showed high specific cell uptake after 2 h, while the uptake of 111In-DOTA-MGS4 (9.4±0.8%) was lower. In A431-CCK2R cells all radioligands showed similar results, with specific uptake values of 26.2±3.4% for 111In-DOTA-MGS1, 24.9±3.7% for 111In-DOTA-MGS4, and 29.4±1.5% for 111In-DOTA-MG11 at 2 h after incubation. A much lower specific cell uptake was observed for 111In-DOTA-MGS2 (AR42J: 4.6±0.5%; A431-CCK2R: 3.9±0.5%) and 111In-DOTA-MGS3 (AR42J: 0.8±0.1%; A431-CCK2R: 1.5±0.7%). In the blocking studies performed for all radioligands with both cell lines, at 2 h after incubation a very low unspecific cell uptake was observed (AR42J: <0.4%; A431-CCK2R: <0.6%) and also the uptake in A431-mock cells was negligible (<0.2%), confirming a highly receptor-specific cell uptake. The specific cell uptake determined over time is displayed in Figure 3.
Cell uptake in (A) AR42J cells and (B) A431-CCK2R cells (internalization expressed as % of specific cell uptake) for 111In-DOTA-MG11 (open squares), 111In-DOTA-MGS1 (red), 111In-DOTA-MGS2 (dark cyan), 111In-DOTA-MGS3 (purple) and 111In-DOTA-MGS4 (green).
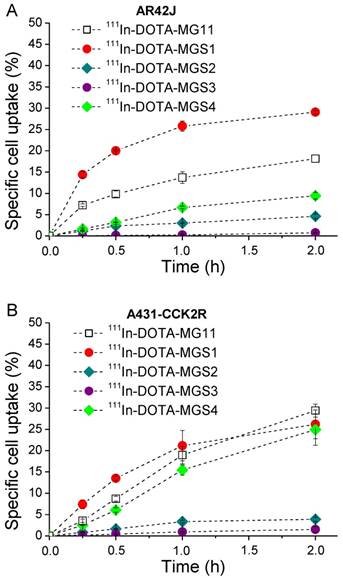
Externalization studies, performed under blocked and unblocked assay conditions, with 111In-DOTA-MGS1, 111In-DOTA-MGS4, and 111In-DOTA-MG11, showed varying results in the two cell lines (Figure 4). In AR42J cells a low and comparable externalization of radioactivity over time was observed under blocked assay conditions, with values of 11.7±1.7% for 111In-DOTA-MGS1, 16.3±2.3% for 111In-DOTA-MGS4 and 15.8±0.4% for 111In-DOTA-MG11 after 4 h. Also under unblocked assay conditions, very similar values were found (111In-DOTA-MGS1: 9.6±0.4%; 111In-DOTA-MGS4: 11.1±4.4%; 111In-DOTA-MG11: 14.3±1.5%). In A431-CCK2R a similar externalization was observed under unblocked assay conditions (111In-DOTA-MGS1: 21.4±0.7%; 111In-DOTA-MGS4: 14.1±2.6%; 111In-DOTA-MG11: 22.3 ±0.2%), while a much higher degree of externalization was observed under blocked assay conditions with values of 33.0±0.6% for 111In-DOTA-MGS1, 53.8±5.6% for 111In-DOTA-MGS4 and 33.7±0.9% for 111In-DOTA-MG11.
Externalized radioactivity from AR42J cells under unblocked (A) and blocked (B) assay conditions, as well as A431-CCK2R cells under unblocked (C) and blocked (D) assay conditions for 111In-DOTA-MG11 (open squares), 111In-DOTA-MGS1 (red) and 111In-DOTA-MGS4 (green).
Metabolic studies and tumor targeting in vivo
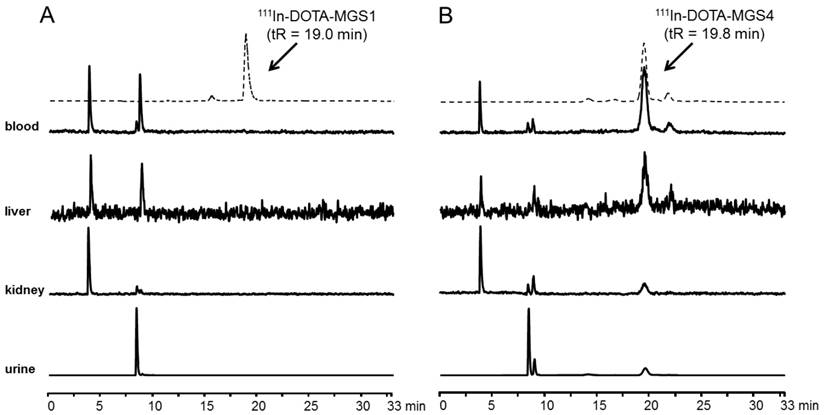
In Figure 5 representative radiochromatograms are displayed showing the metabolites found in blood, liver, kidney and urine of BALB/c mice injected with 111In-DOTA-MGS1 and 111In-DOTA-MGS4. 111In-DOTA-MGS1 showed a very fast enzymatic degradation in vivo. Already at 10 min p.i., no intact radiopeptide (tR = 19.0 min) was detectable in all the examined samples. The metabolites with tR ~4 min and ~9 min indicate that the radiopeptide was rapidly degraded to short peptide fragments. For 111In-DOTA-MGS4 a much higher stability was observed. In blood and liver of mice sacrificed 10 min p.i., still >75% of intact radiopeptide (tR = 19.8 min) and similar metabolites to 111In-DOTA-MGS1 could be detected. A higher degree of degradation was observed in kidney and urine, where only 25-40% of intact 111In-DOTA-MGS4 was found.
Metabolites as analyzed by radio-HPLC from blood, liver, kidney and urine of BALB/c mice injected with (A) 111In-DOTA-MGS1 and (B) 111In-DOTA-MGS4 (10 min p.i.); dashed line showing the radiochromatogram after labeling.
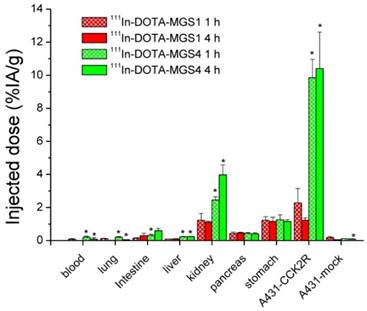
The results of the biodistribution studies are summarized in Table 4 and Figure 6. 111In-DOTA-MGS1 and 111In-DOTA-MGS4 were both rapidly cleared from the body mainly through the kidneys and showed a low unspecific uptake in most tissues and organs. At 4 h p.i., significantly increased uptake values of 111In-DOTA-MGS4 compared to 111In-DOTA-MGS1 could be observed in kidney, liver, spleen, lung, and heart. 111In-DOTA-MGS4 showed a clearly increased uptake in A431-CCK2R xenografts (9.85±1.11 and 10.40±2.21% IA/g at 1 and 4 h p.i.). This uptake was increased by a factor of 4 to 8 when compared with the rather low tumor uptake of 111In-DOTA-MGS1 (2.27±0.88 and 1.23±0.15% IA/g at 1 and 4 h p.i.). For both radioligands, the uptake in A431-mock xenografts with values below 0.30 % IA/g was negligible, confirming that the uptake in A431-CCK2R xenografts was CCK2R-specific. Interestingly, the difference in CCK2R-specific tumor uptake between 111In-DOTA-MGS1 and 111In-DOTA-MGS4 did not translate into a similar effect in stomach and pancreas, two organs with physiological CCK2R expression. Both radioligands showed a rather low kidney uptake. The kidney uptake of 111In-DOTA-MGS4 (2.45±0.20 and 3.98±0.59% IA/g at 1 and 4 h p.i.) was increased by a factor of 2 to 4 in comparison with 111In-DOTA-MGS1 (1.23±0.41 and 1.11±0.07% IA/g at 1 and 4 h p.i.). The uptake values determined from mice sacrificed after SPECT/CT imaging, injected with a higher peptide dose, show a similar picture. For 111In-DOTA-MGS1 a comparable uptake in A431-CCK2R xenografts (1.97±0.40% IA/g) and kidney (1.05±0.09% IA/g) was found, whereas for 111In-DOTA-MGS4 the tumor uptake (7.11±1.01% IA/g) and kidney uptake (2.46±0.48% IA/g) was reduced by >30%. For both radioligands, the uptake in stomach and pancreas was also decreased. Tumor-to-organ activity ratios for blood, kidney, and stomach, as displayed in Table 5, were favorable for both radioligands, but due to the higher tumor uptake clearly superior for 111In-DOTA-MGS4.
Biodistribution of 111In-DOTA-MGS1 and 111In-DOTA-MGS4 in A431-CCK2R and A431-mock tumor-xenografted nude mice at 1 h and 4 h p.i. (0.3 MBq, 0.03 nmol) including also animals from imaging studies dissected after 5 h p.i. (10 MBq, 0.3 nmol). Values are expressed as % IA/g (mean±SD, n = 3; for 111In-DOTA-MGS1 after imaging n = 2).
| 111In-DOTA-MGS1 | 111In-DOTA-MGS1 | 111In-DOTA-MGS1 | 111In-DOTA-MGS4 | 111In-DOTA-MGS4 | 111In-DOTA-MGS4 | |
|---|---|---|---|---|---|---|
| Time p.i. | 1 h | 4 h | after imaging (5 h) | 1 h | 4 h | after imaging (5 h) |
| blood | 0.08±0.04 | 0.01±0.003 | 0.03±0.01 | 0.20±0.07 | 0.08±0.09 | 0.01±0.001* |
| lung | 0.12±0.03 | 0.02±0.002 | 0.04±0.01 | 0.20±0.04* | 0.05±0.003* | 0.03±0.002 |
| heart | 0.05±0.01 | 0.02±0.001 | 0.02±0.004 | 0.10±0.03* | 0.04±0.004* | 0.02±0.001 |
| muscle | 0.04±0.04 | 0.02±0.01 | 0.15±0.19 | 0.05±0.01 | 0.04±0.02 | 0.33±0.16 |
| spleen | 0.07±0.02 | 0.06±0.01 | 0.09±0.04 | 0.12±0.01* | 0.10±0.01* | 0.06±0.02 |
| intestine | 0.15±0.02 | 0.29±0.16 | 0.20±0.08 | 0.31±0.07* | 0.60±0.14 | 0.34±0.21 |
| liver | 0.09±0.01 | 0.10±0.02 | 0.12±0.001 | 0.23±0.02* | 0.24±0.02* | 0.14±0.003* |
| kidney | 1.23±0.41 | 1.11±0.07 | 1.05±0.09 | 2.45±0.20* | 3.98±0.59* | 2.46±0.48* |
| pancreas | 0.43±0.08 | 0.46±0.04 | 0.08±0.01 | 0.43±0.06 | 0.41±0.06 | 0.04±0.004* |
| stomach | 1.23±0.22 | 1.16±0.25 | 0.64±0.12 | 1.26±0.30 | 1.16±0.11 | 0.41±0.02* |
| tumor xenograft | ||||||
| A431-CCK2R | 2.27±0.88 | 1.23±0.15 | 1.96±0.40 | 9.85±1.11* | 10.40±2.21* | 7.11±1.01* |
| A431-mock | 0.18±0.06 | 0.05±0.002 | 0.07±0.02 | 0.11±0.01 | 0.09±0.02* | 0.08±0.01 |
* statistically significant when comparing 111In-DOTA-MGS4 to111In-DOTA-MGS1 (p<0.05)
Selected tumor-to-organ ratios for A431-CCK2R tumor-xenografts at different time points p.i. of 111In-DOTA-MGS1 and 111In-DOTA-MGS4, including also animals dissected after imaging studies
| Time p.i. | 111In-DOTA-MGS1 1 h | 111In-DOTA-MGS1 4 h | 111In-DOTA-MGS1 5 h (after imaging) | 111In-DOTA-MGS4 1 h | 111In-DOTA-MGS4 4 h | 111In-DOTA-MGS4 5 h (after imaging) |
|---|---|---|---|---|---|---|
| Tumor/blood | 30.8±13.2 | 139±31 | 65.3±0.7 | 50.8±11.0 | 241±177 | 480±24 |
| Tumor/kidney | 1.85±0.29 | 1.12±0.17 | 1.86±0.21 | 4.04±0.71 | 2.68±0.86 | 2.92±0.32 |
| Tumor/stomach | 1.81±0.43 | 1.12±0.39 | 3.07±0.03 | 8.04±1.46 | 8.97±1.53 | 17.2±2.3 |
Biodistribution in A431-CCK2R and A431-mock tumor-xenografted nude mice of 111In-DOTA-MGS1 and 111In-DOTA-MGS4 at 1 h and 4 h p.i. Values are expressed as % IA/g (mean±SD, n = 3) and are given in Table 4 (*statistically significant when comparing 111In-DOTA-MGS4 to 111In-DOTA-MGS1; p<0.05).
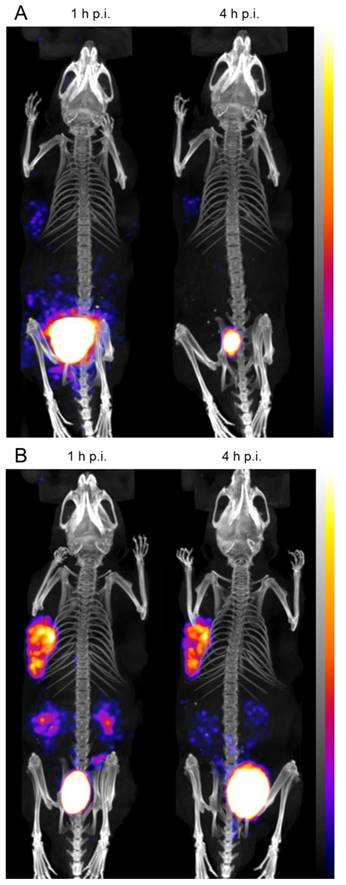
In the whole-body SPECT/CT images, a high signal-to-noise ratio was confirmed for both radioligands (Figure 7). The A431-CCK2R tumors and the bladder were the primarily visible structures, whereas the A431-mock tumors could be detected only on the CT images. From the region-of-interest analysis performed in two mice for each radiopeptide, a tumor-to-kidney ratio of 1.23±0.27 and 1.33±0.23 was calculated for 111In-DOTA-MGS1, whereas 111In-DOTA-MGS4 showed higher ratios of 2.56±0.30 and 2.78±0.50, at 1 h and 4 h p.i., respectively. When interpreting the maximum intensity projection (MIP) at a defined SPECT image intensity scale from 0.1 Bq to 0.9 Bq, tumor visualization of 111In-DOTA-MGS4 was clearly superior to that of 111In-DOTA-MGS1.
Representative SPECT/CT images at 1 h p.i. and 4 h p.i. of BALB/c nude mice xenografted with A431-CCK2R (left) and A431-mock cells (right) injected with (A) 111In-DOTA-MGS1 and (B) 111In-DOTA-MGS4; SPECT in color scale from 0.1 Bq to 0.9 Bq; CT in grey scale (high to low signals: top to bottom).
Discussion
The concept of CCK2R targeting for diagnostic and therapeutic application in MTC and other tumor entities has been proven many years ago using radiolabeled peptide analogs derived from natural ligands [14,18]. However, drawbacks related to high kidney uptake or low enzymatic stability of the radioligands developed so far have limited a wider clinical application [8,18]. In the aim to improve the stability against degradation in vivo and the targeting properties, different modifications of the peptide sequence have been investigated. These include modifications in the N-terminal peptide sequence, such as introduction of D-Glu, D-Gln, and His chains of different length, as well as cyclisation and dimerization [24,25,37]. No stabilization of the C-terminal receptor-specific sequence has been investigated so far, which was shown to be a major site of metabolization in vivo [24]. Therefore, in this study we have investigated site-specific chemical modifications at the C-terminal receptor-binding part of the peptide.
It has already been shown that substitution of Met against non-canonical amino acids such as Nle [11,14], homopropargyl glycine [23], and methoxinine [38] to avoid oxidation is feasible without losing receptor affinity. Based on a literature screening beside Met we also identified C-terminal Phe as a possible site for modification. Using DOTA-MG11 as lead structure we introduced unnatural N-methylated or aromatic amino acids in these two positions of the peptide sequence. Some of the applied substitutions have already been described for CCK-4 analogs leading to potent CCK2R ligands with improved stability in crude rat brain membranes [31]. The four new MG analogs developed either show a single substitution with 1-Nal or (N-Me)Phe at position 8 or an additional substitution with Phg or (N-Me)Nle at position 6. Substitution with aromatic amino acids had a higher effect on the lipophilicity and protein binding of the 111In-labeled DOTA-peptides than substitution with N-methylated amino acids. All radiopeptides showed a high stability of the metal complex and resistance to Met-oxidation when incubated in PBS.
The binding assays and cell uptake studies performed in two different cell lines confirmed a high CCK2R affinity only for DOTA-MGS1 and DOTA-MGS4, whereas the binding of DOTA-MGS2 and DOTA-MGS3 was strongly impaired. The observed differences in binding affinity also led to varying cell internalization of the 111In-labeled DOTA-peptides. A similar difference has been found also for a cyclic MG analog [39,40] and could be related to interspecies or post-translational differences between AR42J cells, physiologically expressing rat CCK2R, and A431-CCK2R cells, transfected with human CCK2R [23]. Also, a higher efflux of radioactivity over time was found for A431-CCK2R cells, especially under blocked assay conditions. Although DOTA-MGS1, DOTA-MGS4 and DOTA-MG11 showed nearly the same affinity and cell uptake when radiolabeled with 111In, a much higher degree of externalized radioactivity was found for 111In-DOTA-MGS4 in this cell line. A possible explanation of this observation could be a higher stability against degradation leading to the efflux of intact radiopeptide, which can rebind to the receptor under unblocked conditions.
We performed extensive stability studies evaluating the stability of the radioligands in human serum and rat tissue homogenates as well as in metabolic studies in vivo. As reported previously, incubation in tissue homogenates allowed for a more conclusive assessment of the enzymatic degradation than studies in serum [41]. In human serum, a stabilizing effect was found for all four radioligands. In rat tissue homogenates, 111In-DOTA-MGS1 and 111In-DOTA-MGS2, showing only one substitution in position 8, in analogy to 111In-DOTA-MG11, were rapidly digested. 111In-DOTA-MGS3, containing two bulky aromatic amino acids, showed the highest stability and also the stability of 111In-DOTA-MGS4, with two N-methylated peptide bonds, was remarkably increased. Therefore, a combination of two substitutions in the C-terminal sequence seems mandatory to reach stabilization against enzymatic degradation. In metabolic studies in vivo carried out with 111In-DOTA-MGS1 and 111In-DOTA-MGS4, no intact radiopeptide was detectable for 111In-DOTA-MGS1 in all analyzed samples (blood, urine, liver homogenate, kidney homogenate). For 111In-DOTA-MGS4, in contrast, we were able to detect high levels of intact radioligand in blood and liver (>75%), and also in kidney and urine up to 30% intact radiopeptide was found, confirming a partial excretion of the intact radioligand. When comparing our results to other data available from the literature, we found varying results on the enzymatic stability of MG analogs. Ocak et al. reported a very fast degradation with no intact radiopeptide already 10 min p.i. for different 177Lu-labeled peptide analogs, including also 177Lu-CP04 [24]. Maina et al. evaluated the same peptide analog labeled with 111In and reported a high stability with up to 70% intact radiopeptide in blood 5 min p.i. [42]. Thus, the results of these studies need to be interpreted with caution as they depend on different factors such as the peptide amount injected to the animals, the time point of taking the samples as well as processing of samples for analysis. In this study we found a good agreement between the results obtained with rat tissue homogenates and metabolic studies performed in mice.
Biodistribution studies evaluating the pharmacokinetics and tumor targeting properties were carried out in BALB/c nude mice xenografted with A431-CCK2R and A431-mock cells. This tumor model has been used for the evaluation of a wide variety of CCK2R-targeting peptide analogs and allows the comparison of our results with data generated by other groups. We have confirmed the specificity of CCK2R targeting for A431-CCK2R cells in vitro and no additional blocking studies were performed in vivo. Based on the similar cell uptake of 111In-DOTA-MGS1 and 111In-DOTA-MGS4 in this cell line, we could directly compare the influence of stabilization on the targeting properties for these two radioligands. For both investigated radiopeptides, a very low unspecific uptake below 1% IA/g was observed in most tissues except kidney, the main excretory organ, and stomach, an organ with physiological CCK2R expression [43]. Only a very limited excretion may additionally occur through feces [20,43]. As expected, 111In-DOTA-MGS1, based on its low enzymatic resistance, showed a low tumor uptake of ~3% IA/g at 1 h p.i. A similar tumor uptake has been reported for 111In-DOTA-MG11 [25,30]. The improved bioavailability of 111In-DOTA-MGS4 led to a considerable increase in tumor uptake (~10% IA/g), whereas kidney retention was only two times higher. Thus, a favorable tumor-to-kidney ratio of ~4 was reached with this radioligand. Also, when injecting a 10-fold higher peptide amount for imaging studies, a favorable biodistribution with reduced kidney uptake as well as low uptake in receptor-expressing organs, stomach and pancreas, was found, leading to high-contrast imaging. Some saturation effects were observed, reducing the uptake of 111In-DOTA-MGS4 in A431-CCK2R xenografts by ~30%, and also the uptake in stomach was considerably reduced. Similar findings have been reported also for 111In-CP04 [44]. The tumor uptake of 111In-DOTA-MGS4 is comparable with other 111In-labeled MG analogs previously studied. Under the same conditions (tumor model, peptide amount), 111In-DOTA-MG0 and 111In-sargastrin, a DOTA-conjugated analog of human gastrin-I, also showed a tumor uptake of ~10% IA/g, combined with a very high kidney retention (>50% IA/g) [25]. 111In-DOTA-MGS4, in contrast, showed a strongly reduced kidney uptake leading to a high tumor-to-kidney ratio. 111In-DOTA-MGS4 further showed a stable tumor uptake at 4 h p.i. and, therefore, a higher tumor retention when compared to 111In-CP04 (~6 %IA/g) [25] and 177Lu-DOTA-PP-F11N (~7 %IA/g) [45]. Our approach also withstands the comparison with co-injection of enzyme inhibitors. Co-injection of the enzyme inhibitor phosphoramidon (300 µg) led to a highly improved tumor uptake of ~15% IA/g at 4 h p.i. for 111In-DOTA-MG11 and 111In-CP04, though combined with a higher uptake in stomach (~5% IA/g for 111In-DOTA-MG11) or kidney (~8% IA/g for 111In-DOTA-CP04) [30]. When using enzyme inhibitors suitable for clinical use, such as racecadotril and its metabolite tiorphan, a much lower increase in tumor uptake was found [46].
Altogether, the stabilizing amino acid modifications introduced in the C-terminal region of 111In-DOTA-MGS4 led to an overall improved biodistribution profile. In addition to a highly increased tumor-uptake, 111In-DOTA-MGS4 also shows very favorable tumor-to-organ ratios for both kidney and stomach and therefore seems superior to other MG analogs previously studied [37,47].
Conclusion
In this study we have shown that MG analogs can be stabilized against enzymatic degradation in vivo through site-specific modifications of amino acids in the C-terminal binding sequence without losing CCK2R affinity. This new strategy not only leads to MG analogs with improved bioavailability, but more importantly leads to significantly increased tumor uptake. Of the radioligands studied, 111In-DOTA-MGS4, with two N-methylated amino acids in position 6 and 8, showed the most promising properties in terms of enzymatic stability, tumor targeting, and tumor-to-organ ratios, and therefore seems most suitable for CCK2R targeting and possible theranostic use with alternative radiometals.
Abbreviations
ACN: acetonitrile; Boc: tert-butyl-oxycarbonyl; CCK: cholecystokinin; CCK2R: cholecystokinin-2 receptor; DTPA: diethylenetriaminepentaacetic acid; DOTA: 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid; Fmoc: 9-fluorenylmethoxycarbonyl; 111In: Indium-111; MG: minigastrin; MG0: [DGlu1]minigastrin; MG11: [DGlu1,desGlu2-6]MG; MIP: maximum intensity projection; MTC: medullary thyroid carcinoma; NEP: neutral endopeptidase; PBS: phosphate-buffered saline; PET: positron emission tomography; RCP: radiochemical purity; RP-HPLC: reversed phase high performance liquid chromatography; RT: room temperature; SPE: solid phase extraction; SPECT: single photon emission tomography; TFA: trifluoroacetic acid.
Acknowledgements
This study was financially supported by the Austrian Science Fund (FWF): project number P 27844. Technical assistance of Chuangyan Zhai in peptide synthesis, Amal Shaaban in stability assays, and Martina Wick in receptor affinity and cell uptake studies is greatly acknowledged. Heinz Zoller kindly provided access to MALDI-TOF mass spectrometry at the Hepatology Laboratory of the Medical University of Innsbruck.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fani M, Maecke HR. Radiopharmaceutical development of radiolabelled peptides. Eur J Nucl Med Mol Imaging. 2012;39:11-30
2. Bison SM, Konijnenberg MW, Melis M, Pool SE, Bernsen MR, Teunissen JJM. et al. Peptide receptor radionuclide therapy using radiolabeled somatostatin analogs: focus on future developments. Clin Transl Imaging. 2014;2:55-66
3. Wild D, Fani M, Fischer R, Del Pozzo L, Kaul F, Krebs S. et al. Comparison of somatostatin receptor agonist and antagonist for peptide receptor radionuclide therapy: a pilot study. J Nucl Med. 2014;55:1248-52
4. Sancho V, Di Florio A, Moody TW, Jensen RT. Bombesin receptor-mediated imaging and cytotoxicity: review and current status. Curr Drug Deliv. 2011;8:79-134
5. Nock BA, Kaloudi A, Lymperis E, Giarika A, Kulkarni HR, Klette I. et al. Theranostic perspectives in prostate cancer with the gastrin-releasing peptide receptor antagonist NeoBOMB1: preclinical and first clinical results. J Nucl Med. 2017;58:75-80
6. Reubi JC, Schaer JC, Waser B. Cholecystokinin(CCK)-A and CCK-B/gastrin receptors in human tumors. Cancer Res. 1997;57:1377-86
7. Gotthardt M, Béhé MP, Beuter D, Battmann A, Bauhofer A, Schurrat T. et al. Improved tumour detection by gastrin receptor scintigraphy in patients with metastasised medullary thyroid carcinoma. Eur J Nucl Med Mol Imaging. 2006;33:1273-9
8. Fröberg AC, De Jong M, Nock BA, Breeman WAP, Erion JL, Maina T. et al. Comparison of three radiolabelled peptide analogues for CCK-2 receptor scintigraphy in medullary thyroid carcinoma. Eur J Nucl Med Mol Imaging. 2009;36:1265-72
9. Roosenburg S, Laverman P, van Delft FL, Boerman OC. Radiolabeled CCK/gastrin peptides for imaging and therapy of CCK2 receptor-expressing tumors. Amino Acids. 2011;41:1049-58
10. von Guggenberg E, Behe M, Behr TM, Saurer M, Seppi T, Decristoforo C. 99mTc-labeling, in vitro and in vivo evaluation of HYNIC and (Nalpha-His)-acetic acid modified [D-Glu1]-minigastrin. Bioconjug Chem. 2004;15:864-871
11. von Guggenberg E, Dietrich H, Skvortsova I, Gabriel M, Virgolini IJ, Decristoforo C. 99mTc-labelled HYNIC-minigastrin with reduced kidney uptake for targeting of CCK-2 receptor-positive tumours. Eur J Nucl Med Mol Imaging. 2007;34:1209-18
12. Roosenburg S, Laverman P, Joosten L, Cooper MS, Kolenc-Peitl P, Foster JM. et al. PET and SPECT imaging of a radiolabeled minigastrin analogue conjugated with DOTA, NOTA, and NODAGA and labeled with 64Cu, 68Ga, and 111In. Mol Pharm. 2014;11:3930-7
13. Mather SJ, Mckenzie AJ, Sosabowski JK, Morris TM, Watson SA. Selection of radiolabeled gastrin analogs for peptide receptor - targeted radionuclide therapy. J Nucl Med. 2007;48:615-22
14. Behr TM, Jenner N, Behe M, Angerstein C, Gratz S, Raue F. et al. Radiolabeled peptides for targeting cholecystokinin-B/gastrin receptor-expressing tumors. J Nucl Med. 1999;40:1029-44
15. Wayua C, Low PS. Evaluation of a nonpeptidic ligand for imaging of cholecystokinin 2 receptor-expressing cancers. J Nucl Med. 2015;56:113-9
16. Kolenc-Peitl P, Mansi R, Tamma M, Gmeiner-Stopar T, Sollner-Dolenc M, Waser B. et al. Highly improved metabolic stability and pharmacokinetics of indium-111-DOTA-gastrin conjugates for targeting of the gastrin receptor. J Med Chem. 2011;54:2602-9
17. Helbok A, Decristoforo C, Behe M, Rangger C, Guggenberg E. Preclinical Evaluation of In-111 and Ga-68 labelled minigastrin analogues for CCK-2 receptor imaging. Curr Radiopharm. 2009;2:304-10
18. Béhé M, Behr TM. Cholecystokinin-B (CCK-B)/gastrin receptor targeting peptides for staging and therapy of medullary thyroid cancer and other CCK-B receptor expressing malignancies. Biopolymers. 2002;66:399-418
19. Good S, Walter MA, Waser B, Wang X, Müller-Brand J, Béhé M. et al. Macrocyclic chelator-coupled gastrin-based radiopharmaceuticals for targeting of gastrin receptor-expressing tumours. Eur J Nucl Med Mol Imaging. 2008;35:1868-77
20. Trejtnar F, Laznicek M, Laznickova A, Kopecky M, Petrik M, Béhé M. et al. Biodistribution and elimination characteristics of two 111In-labeled CCK-2/gastrin receptor-specific peptides in rats. Anticancer Res. 2007;27:907-12
21. Tornesello AL, Aurilio M, Accardo A, Tarallo L, Barbieri A, Arra C. et al. Gastrin and cholecystokinin peptide-based radiopharmaceuticals: an in vivo and in vitro comparison. J Pept Sci. 2011;17:405-12
22. Breeman WAP, Fröberg AC, de Blois E, van Gameren A, Melis M, de Jong M. et al. Optimised labeling, preclinical and initial clinical aspects of CCK-2 receptor-targeting with 3 radiolabeled peptides. Nucl Med Biol. 2008;35:839-49
23. Aloj L, Aurilio M, Rinaldi V, D'ambrosio L, Tesauro D, Peitl PK. et al. Comparison of the binding and internalization properties of 12 DOTA-coupled and 111In-labelled CCK2/gastrin receptor binding peptides: a collaborative project under COST action BM0607. Eur J Nucl Med Mol Imaging. 2011;38:1417-25
24. Ocak M, Helbok A, Rangger C, Peitl PK, Nock BA, Morelli G. et al. Comparison of biological stability and metabolism of CCK2 receptor targeting peptides, a collaborative project under COST BM0607. Eur J Nucl Med Mol Imaging. 2011;38:1426-35
25. Laverman P, Joosten L, Eek A, Roosenburg S, Peitl PK, Maina T. et al. Comparative biodistribution of 12 111In-labelled gastrin/CCK2 receptor-targeting peptides. Eur J Nucl Med Mol Imaging. 2011;38:1410-6
26. Kolenc Peitl P, Tamma M, Kroselj M, Braun F, Waser B, Reubi JC. et al. Stereochemistry of amino acid spacers determines the pharmacokinetics of 111In-DOTA-minigastrin analogues for targeting the CCK2/gastrin receptor. Bioconjug Chem. 2015;26:1113-9
27. Konijnenberg M, Erba PA, Mikolajczak R, Decristoforo C, Maecke H, Maina-Nock T. et al. First biosafety, biodistribution and dosimetry study of the gastrin analogue 111In-CP04 in medullary thyroid cancer. Phase I clinical trial, GRANT-T-MTC. Eur J Nucl Med Mol Imaging. 2017;44(Suppl 2):S258
28. Rottenberger C, Nicolas G, McDougall L, Kaul F, Christ E, Schibli R. et al. First-in-human administration of the CCK-2 receptor agonist 177Lu-PP-F11N in patients with metastasized medullary thyroid carcinoma - preliminary results of the “Lumed” trial. Eur J Nucl Med Mol Imaging. 2017;44(Suppl 2):S260
29. Nock BA1, Maina T, Krenning EP, de Jong M. To serve and protect: enzyme inhibitors as radiopeptide escorts promote tumor targeting. J Nucl Med. 2014;55:121-7
30. Kaloudi A, Nock BA, Lymperis E, Krenning EP, de Jong M, Maina T. Improving the in vivo profile of minigastrin radiotracers: a comparative study involving the neutral endopeptidase inhibitor phosphoramidon. Cancer Biother Radiopharm. 2016;31:20-8
31. Corringer PJ, Weng JH, Ducos B, Durieux C, Boudeau P, Bohme A. et al. CCK-B agonist or antagonist activities of structurally hindered and peptidase-resistant Boc-CCK4 derivatives. J Med Chem. 1993;36:166-72
32. Agnes RS, Lee YS, Davis P, Ma S-W, Badghisi H, Porreca F. et al. Structure-activity relationships of bifunctional peptides based on overlapping pharmacophores at opioid and cholecystokinin receptors. J Med Chem. 2006;49:2868-75
33. Pfister J, Summer D, Rangger C, Petrik M, von Guggenberg E, Minazzi P. et al. Influence of a novel, versatile bifunctional chelator on theranostic properties of a minigastrin analogue. EJNMMI Research. 2015;5:74
34. Aloj L, Caracò C, Panico M, Zannetti A, Del Vecchio S, Tesauro D. et al. In vitro and in vivo evaluation of 111In-DTPAGlu-G-CCK8 for cholecystokinin-B receptor imaging. J Nucl Med. 2004;45:485-94
35. von Guggenberg E, Sallegger W, Helbok A, Ocak M, King R, Mather SJ. et al. Cyclic minigastrin analogues for gastrin receptor scintigraphy with technetium-99m: preclinical evaluation. J Med Chem. 2009;52:4786-93
36. Brom M, Joosten L, Laverman P, Oyen WJ, Béhé M, Gotthardt M. et al. Preclinical evaluation of 68Ga-DOTA-minigastrin for the detection of cholecystokinin-2/gastrin receptor-positive tumors. Mol Imaging. 2011;10:144-52
37. Laverman P, Sosabowski JK, Boerman OC, Oyen WJ. Radiolabelled peptides for oncological diagnosis. Eur J Nucl Med Mol Imaging. 2012;39(Suppl 1):78-92
38. Grob NM, Behe M, von Guggenberg E, Schibli R, Mindt TL. Methoxinine - an alternative stable amino acid substitute for oxidation-sensitive methionine in radiolabelled peptide conjugates. J Pept Sci. 2017;23:38-44
39. von Guggenberg E, Rangger C, Sosabowski JK, Laverman P, Reubi JC, Virgolini IJ. et al. Preclinical evaluation of radiolabeled DOTA-derivatized cyclic minigastrin analogs for targeting cholecystokinin receptor expressing malignancies. Mol Imaging Biol. 2012;14:366-75
40. Rangger C, Klingler M, Balogh L, Pöstényi Z, Polyak A, Pawlak D. et al. 177Lu labeled cyclic minigastrin analogues with therapeutic activity in CCK2R expressing tumors: preclinical evaluation of a kit formulation. Mol Pharm. 2017;14:3045-58
41. Ocak M, Helbok A, von Guggenberg E, Ozsoy Y, Kabasakal L, Kremser L. et al. Influence of biological assay conditions on stability assessment of radiometal-labelled peptides exemplified using a 177Lu-DOTA-minigastrin derivative. Nucl Med Biol. 2011;38:171-9
42. Maina T, Konijnenberg MW, KolencPeitl P, Garnuszek P, Nock BA, Kaloudi A. et al. Preclinical pharmacokinetics, biodistribution, radiation dosimetry and toxicity studies required for regulatory approval of a phase I clinical trial with 111In-CP04 in medullary thyroid carcinoma patients. Eur J Pharm Sci. 2016;91:236-42
43. Melicharova L1, Laznickova A, Laznicek M. Preclinical evaluation of gastrin derivatives labelled with 111In: radiolabelling, affinity profile and pharmacokinetics in rats. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:544-51
44. Konijnenberg MW, Breeman WAP, de Blois E, Chan HS, Boerman OC, Laverman P. et al. Therapeutic application of CCK2R-targeting PP-F11: influence of particle range, activity and peptide amount. EJNMMI Res. 2014;4:47
45. Sauter AW, Mansi R, Valverde I, Del Pozzo L, Béhé M, Wild D. et al. Does the use of protease inhibitors outperform the chemical stabilization approach on targeting the cholecystokinin 2 receptor in vivo with radiolabelled gastrin analogues? Eur J Nucl Med Mol Imaging. 2016;43(Suppl 1):S238-S239
46. Kaloudi A, Nock BA, Lymperis E, Valkema R, Krenning EP, de Jong M. et al. Impact of clinically tested NEP/ACE inhibitors on tumor uptake of [111In-DOTA]MG11-first estimates for clinical translation. EJNMMI Research. 2016;6:15
47. Fani M, Peitl PK, Velikyan I. Current status of radiopharmaceuticals for the theranostics of neuroendocrine neoplasms. Pharmaceuticals (Basel). 2017;10:30
Author contact
![]() Corresponding author: E-mail: elisabeth.von-guggenbergac.at; Tel: +4351250480960; Fax: +435125046780960
Corresponding author: E-mail: elisabeth.von-guggenbergac.at; Tel: +4351250480960; Fax: +435125046780960