Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(11):3007-3021. doi:10.7150/thno.21966 This issue Cite
Research Paper
Flow-dependent epigenetic regulation of IGFBP5 expression by H3K27me3 contributes to endothelial anti-inflammatory effects
Suowen Xu1, Yanni Xu1,2, Meimei Yin1, Shuya Zhang1,3, Peng Liu1,2, Marina Koroleva1, Shuyi Si2, Peter J. Little4,5, Jaroslav Pelisek6, Zheng Gen Jin1, ![]()
1. Aab Cardiovascular Research Institute, Department of Medicine, University of Rochester School of Medicine and Dentistry, Rochester, New York, USA
2. Institute of Medicinal Biotechnology Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing, China
3. Key Laboratory of Fertility Preservation and Maintenance of Ministry of Education, Department of Biochemistry and Molecular Biology, Ningxia Medical University, Yinchuan, China
4. School of Pharmacy, The University of Queensland, Pharmacy Australia Centre of Excellence (PACE), Woolloongabba QLD 4102, Australia
5. Xinhua College of Sun Yat-sen University, Guangzhou, China
6. Department of Vascular and Endovascular Surgery, Klinikum rechts der Isar der Technischen Universitaet Muenchen, Germany
Received 2017-7-17; Accepted 2018-2-17; Published 2018-4-30
Abstract

Rationale: Atherosclerosis is a chronic inflammatory and epigenetic disease that is influenced by different patterns of blood flow. However, the epigenetic mechanism whereby atheroprotective flow controls endothelial gene programming remains elusive. Here, we investigated the possibility that flow alters endothelial gene expression through epigenetic mechanisms.
Methods: En face staining and western blot were used to detect protein expression. Real-time PCR was used to determine relative gene expression. RNA-sequencing of human umbilical vein endothelial cells treated with siRNA of enhancer of zeste homolog 2 (EZH2) or laminar flow was used for transcriptional profiling.
Results: We found that trimethylation of histone 3 lysine 27 (H3K27me3), a repressive epigenetic mark that orchestrates gene repression, was reduced in laminar flow areas of mouse aorta and flow-treated human endothelial cells. The decrease of H3K27me3 paralleled a reduction in the epigenetic “writer”-EZH2, the catalytic subunit of the polycomb repressive complex 2 (PRC2). Moreover, laminar flow decreased expression of EZH2 via mechanosensitive miR101. Genome-wide transcriptome profiling studies in endothelial cells treated with EZH2 siRNA and flow revealed the upregulation of novel mechanosensitive gene IGFBP5 (insulin-like growth factor-binding protein 5), which is epigenetically silenced by H3K27me3. Functionally, inhibition of H3K27me3 by EZH2 siRNA or GSK126 (a specific EZH2 inhibitor) reduced H3K27me3 levels and monocyte adhesion to endothelial cells. Adenoviral overexpression of IGFBP5 also recapitulated the anti-inflammatory effects of H3K27me3 inhibition. More importantly, we observed EZH2 upregulation, and IGFBP5 downregulation, in advanced atherosclerotic plaques from human patients.
Conclusion: Taken together, our findings reveal that atheroprotective flow reduces H3K27me3 as a chromatin-based mechanism to augment the expression of genes that confer an anti-inflammatory response in the endothelium. Our study exemplifies flow-dependent epigenetic regulation of endothelial gene expression, and also suggests that targeting the EZH2/H3K27me3/IGFBP5 pathway may offer novel therapeutics for inflammatory disorders such as atherosclerosis.
Keywords: atherosclerosis, endothelial cells, epigenetic, EZH2, H3K27me3, IGFBP5
Introduction
Atherosclerosis is a devastating global disease in urgent need of novel theranostics [1-7]. Atherosclerotic plaques has a preferential distribution in aortic regions of turbulent/disturbed flow, but regions with steady laminar flow are protected against atherosclerosis development [8-12]. The underlying mechanisms of this geographical distribution of atherosclerotic lesions remain largely unclear, but are likely related to hemodynamic forces-induced signal transduction and gene expression. In particular, laminar flow increases the expression of many anti-atherosclerotic genes, such as kruppel-like factor 2 (KLF2), endothelial nitric oxide synthase (eNOS), and nuclear factor erythroid 2-related factor 2 (Nrf2), which critically regulate endothelial homeostasis, but decreases those “pro-atherogenic” genes, such as monocyte chemoattractant protein-1 (MCP1), and endothelin-1 (ET1) [8-14]. In contrast, disturbed flow skews endothelial cells towards the direction of endothelial dysfunction [8-12]. An understanding of mechanisms that mediate the differential gene expression pattern between laminar flow and disturbed flow regions offers us therapeutic opportunities to intervene in endothelial dysfunction and atherosclerosis.
Atherosclerosis can be regulated by several epigenetic mechanisms, such as DNA methylation/demethylation [15], histone methylation/demethylation and acetylation/deacetylation [16, 17], microRNAs [18] and long non-coding RNAs [19, 20]. Research in the past decade has indicated that these epigenetic mechanisms modulate multiple aspects of endothelial function [21], lineage commitment [22], and atherogenesis [4]. In the context of epigenetic modifications mediated by flow, recent evidence has shown that flow shear stress regulates endothelial gene expression by DNA methylation [23-25], histone acetylation [26], miRNAs [27-29] and long non-coding RNAs [30] . However, it remains elusive whether flow regulates histone methylation and the expression of novel mechanosensitive genes that regulate endothelial function. Among the epigenetic modifiers, EZH2 in the polycomb repressive complex 2 (PRC2) has gained considerable interest for its regulatory role in maintaining pluripotency, cell differentiation, organ development and cancer [31]. PRC2, which directly trimethylates histone 3 lysine 27 (H3K27me3) and causes chromatin compaction and transcriptional silencing, is composed of EED, SUZ12, RbAp46/48, and EZH2 [31]. In contrast, PRC2-mediated H3K27me3 formation can be reversed by two histone demethylases, named UTX (ubiquitously transcribed X-chromosome tetratricopeptide repeat protein, also known as KDM6A) and JMJD3 (jumonji domain containing-3, also known as KDM6B) [31]. Several lines of evidence have implicated EZH2 in the development and progression of atherosclerosis. For example, recent studies observed increased H3K27 dimethylation in endothelial cells from human early and advanced atherosclerotic plaques [16] despite a global decrease in H3K27 methylation level [32, 33]. EZH2 is also responsive to fluid shear stress and involved in laminar flow-mediated cell quiescence [34]. EZH2 regulates transendothelial migration of leukocytes to microvessels [35] as well as tumor angiogenesis [36]. In addition, EZH2 has recently been shown to restrict myocardin-dependent smooth muscle differentiation from the mesothelium [37] and promote the formation of foam cell and atherosclerosis development in hyperlipidemic ApoE-/- mice [38]. Moreover, EZH2 was also upregulated in hyperhomocysteinemic ApoE-/- mice and contributed to foam cell formation [39]. These evidences suggest that EZH2 could play a pro-atherogenic role in atherosclerosis development.
Since EZH2 is a histone methyltransferase that responds to laminar flow [34], it remains unknown whether atheroprotective flow controls EZH2-mediated histone methylation patterns, which in turn alter endothelial gene expression, thus regulating endothelial function. Here, we show that EZH2/H3K27me3 is reduced by laminar flow in endothelial cells both in vivo and in vitro. Through genome-scale RNA sequencing (RNA-seq)-based comparison, chromatin-immunoprecipitation (ChIP) and luciferase assays, we demonstrated that atheroprotective flow upregulates novel mechanosensitive genes, such as IGFBP5 (insulin-like growth factor-binding protein 5) through an H3K27me3-dependent inhibitory mechanism to represses endothelial inflammation. Finally, we observed that EZH2 is upregulated, while IGFBP5 is downregulated in advanced atherosclerotic plaques from patients undergoing carotid endarterectomy, indicating the clinical relevance of EZH2 in regulating atherosclerosis.
Materials and methods
Study population
Human atherosclerotic plaques were obtained from patients undergoing carotid endarterectomy deposited in the Biobank of Department of Vascular and Endovascular Surgery (Klinikum rechts der Isar der Technischen Universität Muenchen, Germany) [16, 33]. Atherosclerotic plaques were histologically characterized according to the American Heart Association (AHA) Stary classification [40, 41] and divided into early (types I-III) or advanced (types V-VII) atherosclerosis stages. In this study, carotid arteries (n=10) with advanced stage of atherosclerosis (types VI) were used. Plaque-free healthy carotid arteries (n=10) were obtained from the Department of Trauma Surgery as previously described [16, 33]. Details of inclusion and exclusion criteria are provided in Supplementary Material. The demographic data of atherosclerotic patients and healthy donors are summarized in Table S1-2. The study was approved by the ethics committee of Technischen Universität Muenchen with patients' written informed consent. The study was performed according to the guidelines of the Declaration of Helsinki.
RNA isolation and quantitative real-time PCR
RNA was extracted from formalin-fixed, paraffin-embedded (FFPE) carotid artery samples using the High Pure RNA Paraffin Kit (#3270289001, Roche, Mannheim, Germany) according to the manufacturer's instructions [16, 33]. Two 10 μm sections of each sample were pooled for RNA isolation. Total RNA from cultured cells and mice tissues were isolated with RNeasy Minikit and TRIzol Reagent, respectively. Total RNA was converted into complementary DNA (cDNA) using the High-Capacity cDNA Reverse Transcription Kit (#4374966, Applied Biosystems, Foster City, CA) following the manufacturer's instructions. Then, quantitative real-time PCR (qRT-PCR) was performed using iQ SYBR Green Supermix (#1708886, Bio-Rad) as previously described in detail [42]. The comparative cycle threshold (Ct) method (2-ΔΔCt) was used to determine the relative gene expression after normalization to housekeeping gene GAPDH. The Ct values of EZH2 in one control sample and one atherosclerotic patient sample were below the detection limit of qRT-PCR and thus not included for analysis. The sequences of primers are listed in Table S3.
En face immunofluorescent staining
En face staining was performed as previously described in detail [43], with minor modifications [42, 44]. Mouse aortas were collected after perfusion with saline and neutrally buffered 4% PFA. Aortic segments were then permeabilized with 0.1% Triton X-100 in PBS for 10 min and blocked with 10% normal goat serum (Invitrogen) in Tris-buffered saline (TBS) containing 2.5% Tween-20 for 30 min at room temperature. Next, aorta segments were incubated with rat anti-VE-Cadherin (1:100; #555289, BD Bioscicence), rabbit anti-EZH2 (#6263, ProSci) or rabbit anti-H3K27me3 (#39155, Active Motif) antibody overnight at 4 °C. After rinsing with washing buffer 3 times, aortic segments were incubated with Alexa Fluor 488-conjugated goat anti-rat and Alexa Fluor 546-conjugated goat anti-rabbit secondary antibodies (1:1,000; Thermo Fisher Scientific) for 1 h in darkness at room temperature. Finally, aortic segments were carefully whole mounted (with lumen side up) in the ProLong Gold-antifade Mounting Media (Invitrogen) for confocal microscopy (IX81, Olympus) with 60x or 40x oil lens. All animal procedures conformed to the Guideline for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of the University of Rochester Medical Center.
Primary isolation of mouse endothelial cells, smooth muscle cells and macrophages
Mouse endothelial cells were isolated from adult C57BL/6J mouse lungs using collagenase I digestion (3 mg/mL in serum-free DMEM) at 37 °C in an incubator for 45 min, followed by CD31- and ICAM2-dynabeads-based double positive selection [45, 46]. The isolated mouse lung endothelial cells were authenticated by endothelial marker VE-cadherin staining. More than 99% of cultured cells were VE-cadherin positive. The isolation of vascular smooth muscle cells (VSMCs) was performed using the explant method as previously described [47], with minor modifications. VSMCs migrated from thoracic aorta explants at around 8 days, then, sprouted VSMCs were digested with 0.05% Trypsin-EDTA and reseeded for confocal microscopy or RNA isolation. More than 85% of cultured cells were smooth muscle alpha-actin (α-SMA) positive. Mouse peritoneal macrophages were isolated from the peritoneum cavity by injecting 5 mL ice-cold PBS/0.1% BSA [48]. Cells were cultured in complete media for 4 h, then, non-adherent cells were removed by washing. Macrophages were stained with CD68, and more than 99% of attached cells were CD68 positive.
Cell culture
Human umbilical vein endothelial cells (HUVECs) were isolated from fresh human umbilical cords and cultured in Medium 200 containing 5% FBS and 1X low serum growth supplement (LSGS) [42, 44]. HUVECs were collected in accordance with the University of Rochester human subjects review board procedures that prescribe to the Declaration of Helsinki. Different donors of human coronary artery endothelial cells (HCAECs) were used in this study, which were from Lifeline Cell Technologies (Frederick, MD, #FC-0032, Lot No. 01181, 01354) and Cell Applications Inc. (#300K-05A, Lot No. 1858, San Diego, CA). HCAECs were cultured in MesoEndo Cell Growth Media supplemented with Growth Supplement (#212K-500, Cell Applications Inc.) plus 5% FBS [42]. For cell authentication, HUVECs and HCAECs were identified by cobblestone cell morphology, staining of endothelial cell-marker VE-Cadherin (BD Bioscicence, #555661) and functional uptake of DiI-oxidized LDL (10 µg/mL, 4 h; Alfa Aesar, #J64164). HCAECs tested negative for mycoplasma, bacteria, yeast, fungi, Hepatitis B and C, as stated on the Vendors' certificates of analysis on individual lots of cells. HUVECs (confluence 80-90%) and HCAECs (confluence 60-70%) were subcultured on the day before experiments. For flow experiments, ECs cultured in fresh complete media were exposed to laminar flow [42] or disturbed flow [49] in a 60 mm or 100 mm cone and plate viscometer. Due to the fact that prolonged culture of ECs renders ECs less responsive to flow [50], HUVECs and HCAECs were passaged on the day before flow treatment. HUVECs and HCAECs at passage 2 to 5 were used for all the experiments. THP-1 cells (gifted by Y. Cai), a pro-monocytic cell line, were cultured as previously described [42]. THP-1 cells were authenticated by the ability to differentiate into macrophages after PMA (100 nM, 48 h) stimulation and form foam cells after loading with oxidized LDL for 24 h. For cell culture studies, all compounds were prepared as stock solution at 10 mM in dimethylsulfoxide (DMSO) and working solutions were freshly prepared before use.
Monocyte adhesion assay
TNFα is a commonly used stimulus in triggering monocyte adhesion/rolling/transmigration [51, 52], by enhancing the production of MCP1 [51, 53] and other pro-inflammatory and pro-migratory cytokines (such as IL6) [54]. Therefore, TNFα was used in the THP-1 monocyte adhesion assay, which was performed as previously described in detail [55]. Quantification of adherent THP-1 monocytes was performed by manual counting of monocytes after washing with pre-warmed HUVEC complete media three times.
siRNA and miRNA transfection
To deplete EZH2, HUVECs at 75-80% confluence were used for transfection. In brief, Lipofectamine 2000 or RNAi Max (6 μL; Thermo Fisher Scientific) [42] was mixed with Opti-MEM (250 μL; Thermo Fisher Scientific), and then two independent siRNA oligos against human EZH2 (20 nM; #SR301494-A, -B, Origene, Rockville, MD) or non-target control siRNA (25 nM; #SR30004, Origene) diluted in 250 μL Opti-MEM was added to the solution, mixed gently, and incubated at room temperature for 20 min. After 3-4 h of transfection, the medium was changed to M200 complete medium and cells were treated 48 h after transfection. Additional Silencer® Select EZH2 siRNA (#s4916, Thermo Fisher Scientific) or Silencer® Select Negative Control #1 siRNA (#S25503, Thermo Fisher Scientific) were used for RNA sequencing. The same method was used to transfect HUVECs with Anti-miRTM negative control (100 nM; #AM17010, Ambion) or Anti-miRTM hsa-miR-101-3p miRNA inhibitor (100 nM; #AM17000, Ambion) using RNAi Max as the transfection reagent.
Adenoviral EZH2 overexpression
EZH2 adenovirus was purchased from Vector Biolab (#1371, Malvern, PA). HUVECs were infected with EZH2 adenovirus as indicated.
Extraction of purified histones from cultured cells
Core histones were purified using a commercial Histone Purification Kit (Active Motif, #40025, Carlsbad, CA, USA) following the manufacturer's instructions, with minor modifications. Briefly, HUVECs were treated with a potent and specific EZH2 inhibitor GSK126 (#A1275, Active Biochem, Hongkong) for the indicated time without changing the media. After washing twice with pre-warmed (37 °C) serum-free M200 media, 0.4 mL ice-cold Extraction Buffer was added to a 60 mm dish. Then, a plastic scraper was used to collect the cell protein extracts. The cells were rotated in Extraction Buffer overnight on a VWR rotating platform at 4 °C to release histones. The following day, cell extracts were transferred to fresh tubes and centrifuged in a microcentrifuge at maximum RCF for 5 min at 4 °C. Crude histones were neutralized with 0.1 mL 5X Neutralization Buffer and purified using columns provided. For Western blots, boiled histone lysates were separated by 12-15% SDS-PAGE. The same membrane was stripped and reprobed with anti-Histone 3 (H3) for equal loading.
Western blot analysis
Cells were harvested in freshly-prepared lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM β-Glycerolphosphate, 50 mM NaF, 1 mM Na3VO4, and 1X protease inhibitor cocktail) by cell lifters. After clarification at 4 °C, 12, 000 g for 10 min, total cell lysate was collected for SDS-PAGE gel analysis as previously described in detail [56]. After transfer, membranes were blocked in PBS-diluted LI-COR blocking buffer at room temperature for 1 h. Then, the blots were incubated with 3% BSA-diluted primary antibodies (listed in Table S4) at 4 °C overnight or room temperature for 1 h, followed by incubation with LI-COR IRDye® 680RD goat anti-mouse IgG (H+L) or IRDye® 800CW goat anti-rabbit IgG (H + L) or IRDye® 680RD donkey anti-goat IgG (H+L) (dilution at 1:10,000) at room temperature for 30 min. Images were visualized using an Odyssey Infrared Imaging System (LI-COR). Densitometric analysis of blots was performed using NIH Image J software (http://imagej.nih.gov/ij/).
Chromatin immunoprecipitation coupled with quantitative real-time PCR (ChIP-qPCR)
ChIP assays were performed using EZ-Magna CHIPTM A/G ChIP kits (#17-10086, EMD Millipore), with slight modifications [57]. Equal amounts of Bioruptor-sheared chromatin were incubated with control rabbit IgG, or rabbit H3K27me3 antibodies at 4 °C overnight. Chromatin-associated proteins were washed and then digested with 1 µL Proteinase K (10 mg/mL) for 2 h at 62 ºC with gentle shaking and incubated at 95 ºC for 10 min for reverse crosslinking and inactivation of Proteinase K. Chromatin DNA were then purified with spin columns provided with the kit. Additional ChIP DNA purification kits were from Active Motif (#58002, Carlsbad, CA). qRT-PCR was performed using ChIP primers encompassing the proximal IGFBP5 gene promoter (Table S3). For each PCR reaction, 1 µL ChIP'ed DNA template was added for each 10 µL reaction. 1% of starting chromatin is used as input. Data are presented as % input using the 2-ΔΔCt method.
Luciferase reporter assay
The predicted IGFBP5 promoter region (-1000 bp to +45 bp relative to transcription start site) was synthesized by GeneScript and subcloned into pGL3-basic plasmid (Promega). Luciferase activity assay was performed as previously described [55]. In brief, 100 ng Luciferase plasmid was transfected into HUVECs for 24 h using Lipofectamine 2000 as described above. COS7 cells were co-transfected with 10 ng pRL-TK plasmid (Promega) for normalization of transfection efficiency. The ratio of luciferase units to pRL-TK units was calculated and normalized. Data are presented as fold over control after normalization.
Genome-wide RNA sequencing study
RNA sequencing (RNA-seq) is a useful approach for probing the pathological mechanisms of vascular diseases [58]. RNA-seq was performed at the University of Rochester Genomics Research Center (URGRC) as previously described [57]. For flow RNA-seq, HUVECs were subjected to atheroprotective laminar flow for 48 h using cells under static conditions as the control (n=2). For RNA-seq in HUVECs depleted of EZH2, cells were transfected with control siRNA (20 nM) or EZH2 siRNA (20 nM) for 48 h before RNA was prepared for RNA-seq (n=3). DESeq2-1.10.1 was used to perform data normalization and differential expression analysis with an adjusted p-value threshold of 0.05. The significance test used in DeSeq2 analysis was the Wald test, and a log2 fold change between groups is presented. Adjusted p-values were derived using the Benjamini and Hochberg multiple test correction.
Accession number
The RNA-seq data reported in this paper were deposited in NCBI Gene Omnibus (http://www.ncbi.nlm.nih.gov/geo/) with the accession number GEO: GSE87534. In Figure S4, published datasets were mined from the GEO database with the accession number of GEO: GDS5083 [59].
Statistics
Data are expressed as mean ± SEM. Statistical analysis was performed using GraphPad Prism software 5.0 (GraphPad, La Jolla, CA). Student's t test and one-way analysis of variance (ANOVA) with post hoc Bonferroni tests were used for comparisons between two groups and multiple comparisons, respectively. A p value less that 0.05 was considered statistically significant.
Results
Laminar flow specifically reduces repressive epigenetic mark H3K27me3 in endothelial cells in vitro and in vivo
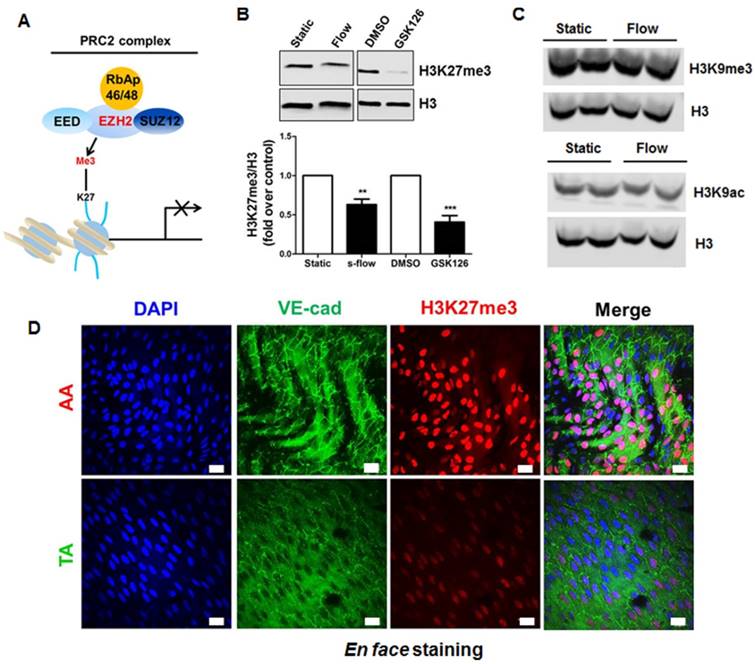
PRC2 is an evolutionarily conserved multi-protein complex that leads to gene silencing through H3K27me3 [31]. To examine the potential involvement of PRC2/H3K27me3 in laminar flow-mediated endothelial gene transcription programming, we first performed a RNA-seq study on human endothelial cells exposed to long-term atheroprotective laminar flow (12 dyne/cm2, 48 h), with an aim to assess the effect of long-term flow on the endothelial transcriptome. We focused on the genes in the PRC2 (Figure 1A), including SUZ12, EED, RbAp46 (RBBP7), RbAp48 (RBBP4), and EZH2. Our data showed that treatment with laminar flow decreased gene expression of EZH2 and RpAp46, without influencing the other three components of PRC2 (Table S5). Since histone methylation is dynamically controlled by the equilibrium of histone methylation and demethylation [60], we also looked at the effect of laminar flow on the expression of the H3K27me3 demethylases including KDM6A (also known as UTX) and KDM6B (also known as JMJD3) [60]. Our RNA-seq data suggest that laminar flow does not significantly alter KDM6A, or KDM6B gene expression (Table S5), implicating that laminar flow could potentially influence net PRC2-dependent H3K27me3 modification. We also observed that, similar to the effect of EZH2 enzymatic inhibitor GSK126 [61], laminar flow treatment significantly downregulated the global level of H3K27me3 in endothelial cells (Figure 1B), indicating that laminar flow influences EZH2-dependent H3K27me3 modification. However, laminar flow does not significantly affect global protein expression of histone 3 lysine 9 trimethylation (H3K9me3) or acetylation (H3K9ac) (Figure 1C), suggesting the specific inhibitory effects of laminar flow on H3K27me3. To address whether our in vitro observations can be translated into in vivo conditions, we compared the differential expression of H3K27me3 in two different regions of mouse aorta: one is the inner curvature of the aortic arch exposed to disturbed flow, and the other is thoracic aorta generally exposed to laminar flow. We observed a marked decrease in H3K27me3 staining in the laminar flow area of mouse aorta (Figure 1D). We also observed that the expression pattern of H3K27me3 in vascular endothelium is heterogeneous.
Laminar flow specifically reduces repressive epigenetic mark H3K27me3 in endothelial cells in vitro and in vivo. (A) Diagram of PRC2. PRC2 is composed of EED, SUZ12, RbAp46/48, and EZH2, which imposes the H3K27me3 epigenetic mark to the target gene promoter, leading to gene silencing. (B) Laminar flow decreases global H3K27me3 levels. HUVECs were exposed to laminar flow for 48 h, and then total histones were purified as described in the methods section. GSK126, a specific inhibitor of EZH2 activity was used as positive control (n=4-5, **P<0.01, ***P<0.001 vs. static control or DMSO). (C) Laminar flow does not affect the level of H3K9me3 or H3K9ac. HUVECs were exposed to laminar flow. Cell alignment in response to flow was observed to ensure successful induction of laminar flow. Then, epigenetic marks H3K9me3 and H3K9ac were determined by Western blot. The same membranes were stripped and incubated with histone 3 (H3) as loading controls (n=3). (D) En face immunofluorescent staining of H3K27me3 in mouse aorta. The aortic arch and thoracic aorta were collected from 3-month-old ApoE-/- mice fed a normal chow diet for en face staining. Red, H3K27me3; green, VE-cadherin; blue, DAPI; scale bar=20 µm (n=5).
Laminar flow decreases the expression of chromatin modifier EZH2 in vitro and in vivo
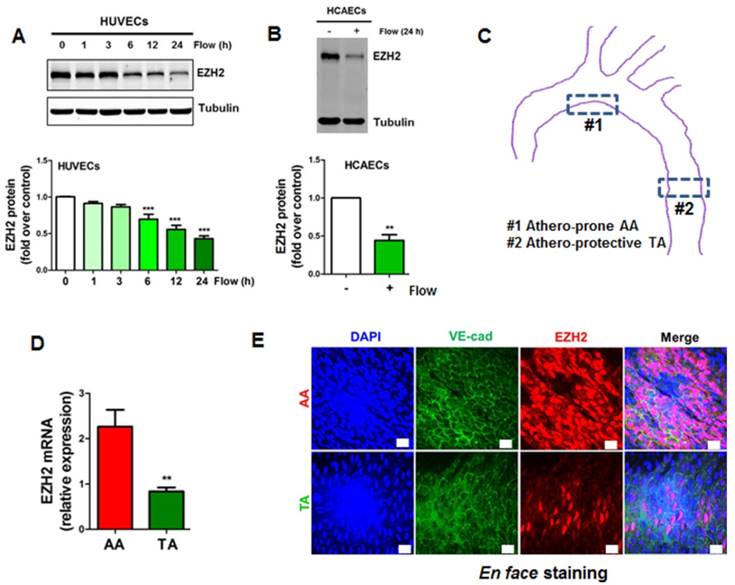
Since EZH2 is the histone methyltransferase that imposes H3K27me3 epigenetic mark, we next asked whether the decrease of H3K27me3 is due to the decrease of EZH2 expression. We first determined the tissue and cell distribution of EZH2. Consistent with the human GTEx database (https://www.gtexportal.org/home/gene/EZH2), we observed that EZH2 was mostly expressed in the spleen, consolidating the key role of EZH2 in immunity [62, 63]. Other organs enriched in EZH2 include lung, brain and aorta, which are endothelial cells-enriched organs (Figure S1A). We next compared EZH2 expression in vascular cells, using primary isolated mouse lung endothelial cells, aortic vascular smooth muscle cells and peritoneal macrophages. We observed that EZH2 was prominently expressed in endothelial cells (Figure S1B). This expression pattern suggests EZH2 may have an important role in regulating immunity and endothelial functions. We then evaluated the time course of laminar flow-induced EZH2 downregulation. We observed that laminar flow significantly decreased EZH2 protein expression starting from 6 h, with maximal reduction at 24 h (Figure 2A). The flow-dependent reduction of EZH2 was recapitulated in HCAECs exposed to laminar flow (Figure 2B). In agreement with the RNA-seq data, we observed that laminar flow does not significantly affect the protein expression of PRC2 components EED and SUZ12 (Figure S2). To examine whether disturbed flow increases EZH2 expression, we compared the protein expression of EZH2 under disturbed flow and laminar flow conditions, using cells under static conditions as a control. We observed that, compared with static conditions, disturbed flow has no significant effect on EZH2 mRNA and protein expression (Figure S3). To corroborate our in vitro study, we analyzed mRNA and protein expression of EZH2 in mouse aorta regions with disturbed flow (inner curvature of aortic arch) and laminar flow (thoracic aorta) (Figure 2C). Interestingly, we observed a significant reduction of EZH2 mRNA and protein expression in arterial regions with laminar flow compared with that of regions with disturbed flow (Figure 2D-E).
Laminar flow decreases the expression of chromatin modifier EZH2 in vitro and in vivo. (A) HUVECs were exposed to laminar flow for the indicated time points, and then EZH2 protein expression was determined by Western blot (*P<0.05, ***P<0.001 vs. static control, n=6). (B) HCAECs were exposed to laminar flow for 24 h, and then EZH2 protein expression was determined by Western blot (**P<0.01, n=3). (C) Illustration of atheroprone (inner curvature of aortic arch, #1) and atheroprotective (thoracic aorta, #2) regions of mouse aorta. The picture was manually drawn by powerpoint. (D) EZH2 mRNA expression in aortic endothelium from atheroprone and atheroprotective regions of mouse aorta. Intima-enriched RNA was isolated from the aortic arch and thoracic aorta of ApoE-/- mice fed a normal chow diet for 3 months. RNA was reverse transcribed and cDNA was used for real-time PCR quantification of EZH2 using GAPDH as internal control (**P<0.01 vs. aortic arch, n=8). (E) En face immunofluorescent staining of EZH2 in mouse aorta. The aortic arch and thoracic aorta were collected from 3-month-old ApoE-/- mice fed a normal chow diet for en face staining. Red, EZH2; green, VE-cadherin; blue, DAPI; scale bar=20 µm (n=5).
miR101 is involved in laminar flow-induced EZH2 downregulation
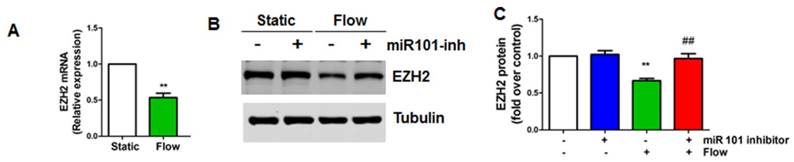
To dissect the mechanism whereby laminar flow reduces EZH2 expression, we first evaluated the effect of laminar flow on EZH2 mRNA expression, and we observed that EZH2 mRNA was significantly decreased in endothelial cells by laminar flow treatment (Figure 3A). Non-coding RNAs, such as microRNAs (miRNAs) are important post-transcriptional mechanisms that induce gene silencing. Mounting studies have shown that several different miRNAs, including miR101, miR26a, miR214, and miR124, can directly target EZH2 expression [64-66]. EZH2 can also be potentially targeted by other miRNAs (Table S6) as predicted by TargetScan [67]. Among these miRNAs, miR101 is the top miRNA that has been reported to be mechanosensitive to date [29, 68]. We then asked whether miRNA 101 might mediate laminar flow-dependent reduction of EZH2 expression. We pre-treated HUVECs with miR101-anti-miR, and found that laminar flow-induced EZH2 downregulation was reversed by miR101-anti-miR treatment (Figure 3B-C), suggesting that miR101 is involved in laminar flow-induced EZH2 downregulation.
miR101 is involved in laminar flow-induced EZH2 downregulation (A) Laminar flow decreases EZH2 mRNA expression. HUVECs were exposed to laminar flow for 24 h, and then RNA was isolated for real-time PCR analysis using GAPDH as loading control (**P<0.01 vs. static control, n=3). (B-C) HUVECs were transfected with miR101 inhibitor (miR101 inh) or control (100 nM) for 48 h and then exposed to laminar flow (L-flow) for 24 h before Western blot was performed to determine EZH2 protein expression (**P<0.01 static+anti-miR control, ##P<0.01 vs. laminar flow+anti-miR control, n=3).
Inhibition of EZH2 expression/activity reduces monocyte adhesion to endothelial cells
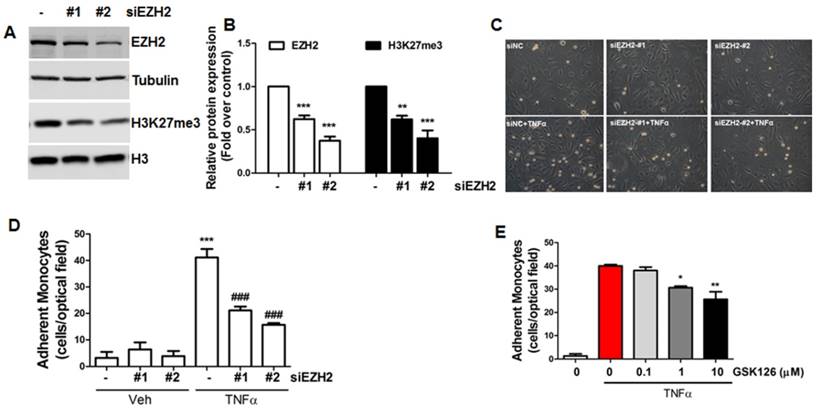
To understand the biological function of EZH2 in endothelial cells, we used two independent siRNAs to deplete EZH2 expression and associated H3K27me3 (Figure 4A-B), then performed monocyte adhesion assays to evaluate the adhesiveness of monocytes to inflamed endothelial cells. Our data showed that EZH2 depletion dramatically inhibited monocyte adhesion to endothelial cells (Figure 4C-D). To confirm this finding, we also incubated cells with the EZH2 inhibitor GSK126 [61]; we observed that GSK126 also inhibited monocyte adhesion to endothelial cells (Figure 4E), suggesting that diminishing H3K27me3 by EZH2 siRNA or pharmacological inhibitors could reduce endothelial inflammation.
siRNA depletion and pharmacological inhibition of EZH2 reduces monocyte adhesion to endothelial cells. (A) HUVECs were transfected with non-targeting control siRNA (siNC, 20 nM) or two independent EZH2 siRNAs (#1, #2, 20 nM). Then, EZH2 and H3K27me3 protein expression was determined by Western blot using tubulin and Histone 3 (H3) as the loading controls, respectively (n=4). (B) Quantification of (A) (**P<0.01, ***P<0.001 vs. siNC, n=4). (C) HUVECs were transfected with non-targeting control siRNA (siNC) or two independent EZH2 siRNAs. Then, a monocyte adhesion assay was performed, original magnification X20. (D) Quantification of (C) (***P<0.001 vs. siNC+vehicle (veh), ###P<0.001 vs. siNC+TNFα, n=4). (E) HUVECs were incubated with different doses of GSK126, then, a monocyte adhesion assay was performed (n=4). Representative data from one experiment is shown in the quantification data (*P<0.05, **P<0.01 vs. TNFα).
Genome-wide transcriptomic profiling identified IGFBP5 as a flow-sensitive and EZH2/H3K27me3-target gene in endothelial cells
To gain mechanistic insights into the anti-inflammatory effects conferred by inhibiting H3K27me3, we first compared the common transcriptome between laminar flow-treated and EZH2-depleted HUVECs (Figure 5A). Since H3K27me3 is a repressive epigenetic mark that leads to gene silencing, we mapped out the upregulated common genes between both treatments (Table S7).
Identification of IGFBP5 as a flow-sensitive and EZH2/H3K27me3-target gene in endothelial cells. (A) Venn diagram analysis of overlapping genes revealed by RNA-seq of HUVECs exposed to laminar flow or EZH2 depletion: a total of 293 genes were identified. A complete list of overlapping genes was presented in Table S7. The RNA-seq data were deposited in NCBI Gene Omnibus (http://www.ncbi.nlm.nih.gov/geo/) with the accession number GEO: GSE87534. (B) Laminar flow upregulates IGFBP5 mRNA expression (n=3). NOS3 gene was used as a positive control for flow treatment (*P<0.05, **P<0.01 vs. static control). (C) Laminar flow upregulates IGFBP5 protein expression in culture supernatant. After exposure to laminar flow for 24 h, culture supernatant was adjusted by cell number, then IGFBP5 protein expression in the media was determined (n=3). (D) Laminar flow reduces H3K27me3 binding to IGFBP5 gene promoter. Chromatin immunoprecipitation (ChIP) coupled with quantitative real-time PCR (ChIP-qPCR) for IGFBP5 was performed in HUVECs exposed to static conditions or laminar flow for 24 h. Representative data from one experiment are presented (as % input; **P<0.01 vs. IgG control, #P<0.05 vs static with H3K27me3 ChIP, n=2). (E) EZH2 overexpression (M.O.I.=2, 48 h) decreases IGFBP5 gene expression, as determined by qRT-PCR (= *P<0.05 vs. Adcon, n=3). (F) EZH2 siRNA depletion (20 nM, 48 h) increases IGFBP5 gene expression, as determined by qRT-PCR (**P<0.01, ***P<0.001 vs. siNC, n=3). (G) Pharmacological inhibition of EZH2 by GSK126 (10 μM, 72 h) increases IGFBP5 gene expression without affecting EZH2 expression, as determined by qRT-PCR (*P<0.05 vs. veh (DMSO), n=4). (H) COS7 cells were transfected with pGL3-IGFBP5-luc (-1000-+45bp). 4 h after transfection, cells were infected with control adenovirus (AdNC) or AdEZH2 (M.O.I=2) for an additional 24 h before luciferase activity assay was performed (*P<0.05 vs. AdNC, n=3).
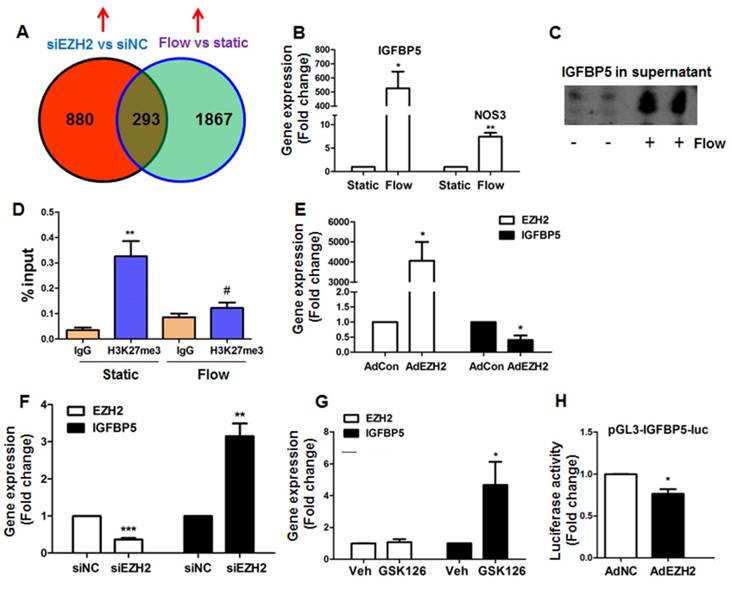
By comparing the common sets of upregulated genes, we identified many anti-atherosclerotic genes, including HMOX1 (heme oxygenase 1), NQO1 (NAD(P)H quinone dehydrogenase 1), GPX3 (glutathione peroxidase 3), KLF4, PLAT (plasminogen activator, tissue type) and IGFBP5 (Table S7). Among genes of interest, we selected a less-characterized new gene IGFBP5 for further studies. Our real-time PCR and western blot analysis indicated that IGFBP5 is a flow-responsive gene that can be upregulated by laminar flow at mRNA and protein levels (Figure 5B-C). To validate whether IGFBP5 is an H3K27me3 target gene, we performed ChIP-qPCR analysis in the promoter region of IGFBP5. We observed enrichment of H3K27me3 at IGFBP5 proximal gene promoter, and binding of H3K27me3 to IGFBP5 gene promoter was reduced by laminar flow treatment (Figure 5D). To confirm whether IGFBP5 is an EZH2/H3K27me3 target gene, we used four approaches: (1) EZH2 gain-of-function by infection with adenoviral EZH2, (2) EZH2 loss-of-function by EZH2 siRNA, (3) pharmacological inhibition of EZH2/H3K27me3 activity by GSK126, and (4) dual-luciferase reporter assay. We observed that EZH2 overexpression decreased, while EZH2 depletion by siRNA or H3K27me3 inhibition by GSK126 increased IGFBP5 expression (Figure 5E-G). Luciferase reporter assay also showed that EZH2 overexpression decreased IGFBP5 promoter activity (Figure 5H), suggesting that IGFBP5 is a bona fide methylation target of H3K27me3. Altogether, our data demonstrate that EZH2 epigenetically silences flow-responsive IGFBP5 expression in endothelial cells.
IGFBP5 inhibits endothelial inflammation
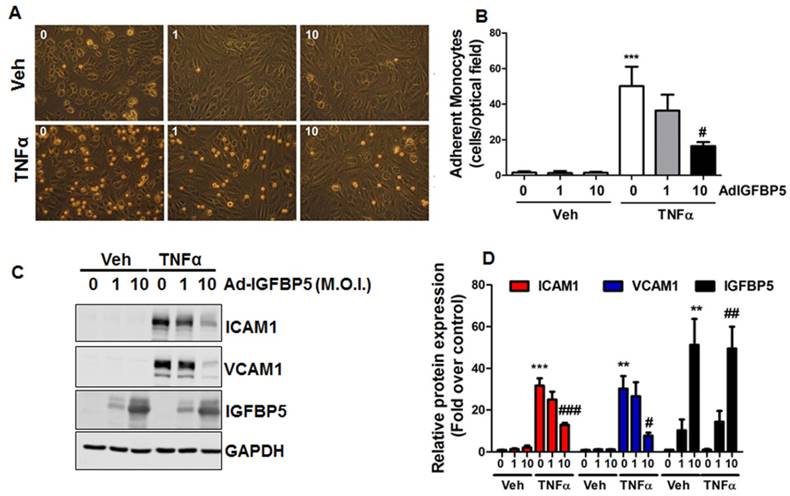
IGFBP5 is an important IGF binding protein that specifically binds to endothelial cell monolayers [69]. Previous studies have shown that IGFBP5 is induced by VEGF and IGF1 treatments [70], and mediated radiation-induced endothelial senescence [71]. However, the effect of IGFBP5 on endothelial inflammation is unknown. Due to the anti-inflammatory profile in laminar flow-treated endothelial cells, we asked whether IGFBP5 could be an anti-inflammatory molecule in endothelial cells. To this end, we performed a monocyte adhesion assay. Our data showed that adenoviral IGFBP5 overexpression attenuated monocyte adhesion to endothelial cells in a dose-dependent manner (Figure 6A-B). IGFBP5 overexpression also decreased the protein expression of pro-adhesive molecules ICAM1 and VCAM1 in TNFα-inflamed endothelial cells (Figure 6C-D).
IGFBP5 inhibits endothelial inflammation. (A-B) HUVECs were infected with increasing doses (M.O.I.=1-10) of IGFBP5 adenovirus for 48 h, then a monocyte adhesion assay was performed in the absence (vehicle) or presence of TNFα. Adherent monocytes were counted and quantified (***P<0.001 vs. veh plus control adenovirus, #P<0.05 vs. TNFα plus control adenovirus, n=4), original magnification X20. (C-D) HUVECs received the same treatments as described in (A), then, whole cell lysates were collected for Western blot analysis using GAPDH as the loading control. Densitometric analysis of (C) is provided in (D) (**P<0.01, ***P<0.001 vs. veh plus control adenovirus, #P<0.05, ##P<0.01, ###P<0.001 vs. TNFα plus control adenovirus, n=3).
EZH2 and IGFBP5 expression in human atherosclerotic plaques
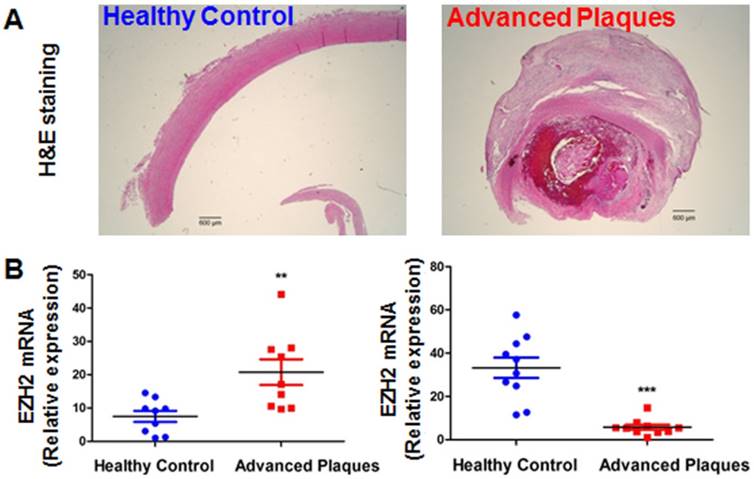
Since our data pointed to the concept that EZH2 depletion and ensuing IGFBP5 upregulation is anti-inflammatory and probably atheroprotective in endothelial cells, we investigated the expression levels of EZH2 and IGFBP5 in atherosclerotic plaques from patients undergoing carotid endarterectomy. We first performed a datamining study on published GEO datasets examining differential gene expression patterns between atherosclerotic plaques and adjacent disease-free regions [59]. We observed a significant upregulation of EZH2 and downregulation of IGFBP5 in plaque areas compared with adjacent plaque-free areas (n=32, Figure S4). To confirm this finding, we also performed real-time PCR analysis using the FFPE samples from healthy controls and atherosclerotic patients. H&E staining showed minimal lesions in control samples; however, extensive plaque hemorrhage was observed in advanced plaques from patients (Figure 7A). Our data also indicated an increase in EZH2 expression in diseased vessels compared with healthy control carotid artery (Figure 7B). Consistent with the proposition that IGFBP5 is epigenetically silenced by EZH2, we observed a significant decrease in IGFBP5 expression in human atherosclerotic plaques (Figure 7B). These observations collectively suggest that the clinical relevance of EZH2 in regulating IGFBP5 expression in human atherosclerotic plaques.
EZH2 and IGFBP5 expression in human atherosclerotic plaques. (A) Hematoxylin and eosin (H&E) staining of healthy control and atherosclerotic carotid arteries (n=3, scale bar=600 µm). (B) mRNA expression levels of EZH2 and IGFBP5 determined in human carotid atherosclerotic plaques and healthy controls using real-time PCR (**P<0.01, ***P<0.001 vs. healthy control, n=9-10).
Discussion
Atherosclerosis is a focal disease that preferentially develops in regions of disturbed flow, but less in regions of atheroprotective laminar flow [8-12]. Several landmark studies [23-25] have recently shown that atheroprone disturbed flow alters endothelial gene expression via DNA methyltransferases (DNMTs)-mediated DNA methylation of key genes involved in endothelial function, such as KLF3, KLF4, and homeobox protein hox-A5 (HoxA5). Disturbed flow also promotes endothelial DNA methylation by repressing the expression of Tet methylcytosine dioxygenase 2 (Tet2) [72]. However, the mechanisms by which atheroprotective laminar flow prevents atherosclerosis at the epigenetic level, especially at the histone methylation level, remain poorly defined. In this study, we uncover that the EZH2-H3K27me3-IGFBP5 pathway contributes to laminar flow-dependent atheroprotective effects.
Through genome-wide RNA-seq, we first identified that laminar flow decreased the expression of two PRC2 components, EZH2 and RpAp46, without affecting the expression of H3K27me3 demethylases-UTX and JMJD3. The net outcome of laminar flow treatment could potentially lead to reduced PRC2 activity, evidenced by decreased global H3K27me3 levels. Recent studies have shown that miR101 is mechanosensitive and EZH2 is a well-defined target of miR101 [29, 68]. By using a miR101 inhibitor treatment of endothelial cells, we observed that miR101 was involved in flow induced EZH2 downregulation, which reproduced a recent study using a different fluid shear system (an ibidi fluidic unit) [34]. In light of the negative feedback loop between miRNA 101 and EZH2 [65], it is plausible that flow sustains low levels of EZH2 through this regulatory loop. We also showed downregulation of EZH2 mRNA and protein expression in laminar flow areas (thoracic aorta) of mouse aorta, compared with disturbed flow areas (inner curvature of aortic arch). The biological function of EZH2 is to impose a repressive epigenetic mark of H3K27me3 onto target gene promoters, thereby leading to gene silencing [31]. Consistent with the proposition that laminar flow exerts strong anti-inflammatory effects; we observed that both EZH2 depletion by siRNA and inhibition by a specific inhibitor GSK126 reduced monocyte adhesion to endothelial cells, suggesting EZH2 and the associated H3K27me3 modifier are pro-inflammatory in endothelial cells. The cellular effects of EZH2 could possibly be mediated by modulating H3K27me3 level and the binding of this repressive epigenetic mark to target gene promoters. To evaluate the effect of atheroprotective flow on the EZH2/H3K27me3-dependent genome-wide transcriptional profile, we performed an RNA-seq transcriptional profiling on laminar flow- and EZH2 siRNA-treated human endothelial cells. A Venn diagram was used to compare the common genes regulated by both laminar flow and EZH2 depletion. We found that atheroprotective flow and EZH2 depletion alter the endothelial gene landscape, which includes upregulating atheroprotective genes (such as IGFBP5) while downregulating pro-atherosclerotic genes. Through studying the overlapping genes between flow and EZH2 depletion, we observed that both treatments upregulate several genes maintaining endothelial homeostasis (HMOX1, NQO1, GPX3, KLF4, and PLAT). Our study extends previous observations showing that laminar flow decreases EZH2 expression in cultured endothelial cells and EZH2 downregulation is involved in flow-induced cell quiescence [34].
Recent studies by Yang et al. [39] and Lv et al. [38] have shown that EZH2 expression is upregulated in mouse models of hyperhomocysteinemia and hyperlipidemia, respectively, unveiling a new functional connection between EZH2 and atherosclerosis. In addition, lentivirus-mediated EZH2 overexpression exacerbates plaque development in ApoE-/- mice fed a high fat diet [38], suggesting that EZH2 may play a pro-atherogenic role in atherosclerosis. However, to date, there are limited EZH2 target genes identified in endothelial cells. Interestingly, our study identifies IGFBP5 as a novel mechanosensitive gene regulated by EZH2-dependent H3K27me3 that may serve as a critical regulator of flow-elicited responses. IGFBPs are important in vascular biology as they regulate gene expression and extracellular matrix accumulation [73]. Our study shows that IGFBP5 is a flow-responsive but EZH2-H3K27me3-repressive gene that confers anti-inflammatory effects. It is possible that laminar flow-induced secretion of IGFBP5 could exert anti-inflammatory effects by modulating IGF-dependent or -independent effects [73]. These data, together with two recent findings showing that atheroprotective drugs, including statins [74] and omega 3-polyunsaturated fatty acids [75], reduce EZH2 expression in cancer cells, suggesting EZH2 is a promising therapeutic target for pharmacological intervention. More importantly, our data-mining study and the analysis of the expression level of EZH2 and IGFBP5 in human atherosclerotic plaques demonstrated an association of EZH2 and IGFBP5 with human atherosclerotic plaques, thus increasing the translational potential of our study.
In the current study, we mainly focused on the role of EZH2/H3K27me3 in endothelial function. To ascertain the anti-inflammatory profile conferred by laminar flow, it will be important to address in future studies whether genetic or pharmacological inhibition of H3K27me3 demethylase activity by JMJD3 and UTX will reverse the effects of laminar flow or EZH2 deficiency on endothelial function. In light of the important role of phosphorylation at Ser21 [76], Thr345 [77] and Thr487 [77] in regulating EZH2 expression/methyltransferase activity, it will be important to evaluate the temporal phosphorylation profile of EZH2 in endothelial cells in response to flow, and the phosphorylation of Ser21 in particular, which can be activated by flow-responsive kinase Akt [78]. Although the precise role of EZH2 in the development of atherosclerosis still requires in-depth investigation using endothelium-specific knockout mice, results presented here indicate that fine-tuned regulation of the levels of EZH2 and H3K27me3 is critical for maintaining endothelial homeostasis. Furthermore, in addition to transcriptional silencing, EZH2 has a non-canonical role in transactivating target genes [31]. This aspect has been well defined in cancer cell biology, showing that EZH2 activates NF-κB target genes [79]. We are currently investigating the role of EZH2 in transactivating endothelial cell-specific genes. We understand that flow-dependent regulation of gene transcriptional programming is a result of the complex interplay between multiple chromatin modifiers (DNA and histone methyltransferases for example [80]). This is particularly intriguing since a recent study has shown that EZH2-dependent PRC2 is associated with multiple long non-coding RNAs [81], thereby participating in gene regulation. We also recognize that laminar shear stress elicits profound changes in endothelial transcriptional programming, which include IGFBP5 and other atherorelevant genes. The study focused on the effects of flow on epigenetic regulation of IGFBP5 expression by H3K27me3 in endothelial cells. All these mechanisms were only confirmed by in vitro studies. It is well established that the cell components are complicated in the advanced plaques, including endothelial cells, macrophages, foam cells and smooth muscle cells. Although EZH2 show higher expression in endothelial cells, compared with other smooth muscle cells and macrophages, it cannot be totally ruled out that EZH2/H3K27me3 could also regulate functions of smooth muscle cells and macrophages, since IGFBP5 is a secreted anti-inflammatory protein, which could possibly impact endothelial cells-smooth muscle cell/macrophage communication. Determining the expression of EZH2/H3K27me3 with different markers of individual vascular cell type in the advanced plaque, will provide further evidence supporting the epigenetic modulation of IGFBP5 in plaque endothelial cells. The elucidation of these novel EZH2-dependent genes that regulate endothelial function will provide us a new direction of cardiovascular research.
In summary, the present study reveals EZH2 as a mechanosensitive and potentially atherogenic chromatin modifier that serves as a molecular switch to determine atheroprotective or atheropromoting outcomes in the endothelium based on distinctive H3K27me3 marks. Defining the link between hemodynamic forces and epigenetic regulation of endothelial gene transcription could deepen our understanding of mechanisms underlying blood flow-dependent gene expression and atherosclerosis, and finally will translate into new remedies for atherosclerosis.
Abbreviations
ChIP: chromatin immunoprecipitation; DNMT: DNA methyltransferase; EZH2: enhancer of zeste homolog 2; FFPE: formalin-fixed, paraffin-embedded; GEO: gene expression omnibus; GPX3: glutathione peroxidase 3; H3K27me3: histone 3 lysine 27; HCAECs: human coronary artery endothelial cells; HMOX1: heme oxygenase 1; HoxA5: homeobox protein hox-A5; HUVECs: human umbilical vein endothelial cells; ICAM1: intracellular adhesion molecule 1; IGFBP5: insulin-like growth factor-binding protein 5; JMJD3: jumonji domain containing-3; KLF: kruppel like factor; NQO1: NAD(P)H quinone dehydrogenase 1; PLAT: plasminogen activator, tissue type; PRC2: polycomb repressive complex 2; RNA-seq: RNA sequencing; Tet2: tet methylcytosine dioxygenase 2; TNFα: tumor necrosis factor-alpha; UTX: ubiquitously transcribed X-chromosome tetratricopeptide repeat protein; VCAM1: vascular adhesion molecule 1.
Supplementary Material
Supplementary methods, figures and tables S1-S4.
Table S5.
Table S6.
Table S7.
Acknowledgements
We are grateful to Jason R. Myers and John Ashton from Genomic Research Center of University of Rochester for RNA-seq data analysis and depositing. Research conducted in these studies was supported by National Institutes of Health RO1 grants (HL109502, R01HL114570, HL128363, HL130167) and American Heart Association Grant-In-Aid Award (GRNT33660671) (to Z.G.J.).
Author contributions
S.X. and Z.G.J. designed the whole study, Y.X., M.Y., P. L, S.Z., M. K. provided important suggestions and input into this project. J.P. provided human control and atherosclerotic samples for the study. S.S. and P.J.L. proofread the manuscript and offered suggestions for the revision. Z.G.J. supervised the whole study.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317-25
2. Xu S, Bai P, Little PJ, Liu P. Poly(ADP-ribose) polymerase 1 (PARP1) in atherosclerosis: from molecular mechanisms to therapeutic implications. Med Res Rev. 2014;34:644-75
3. Wang Y, Chen J, Yang B, Qiao H, Gao L, Su T. et al. In vivo MR and Fluorescence Dual-modality Imaging of Atherosclerosis Characteristics in Mice Using Profilin-1 Targeted Magnetic Nanoparticles. Theranostics. 2016;6:272-86
4. Khyzha N, Alizada A, Wilson MD, Fish JE. Epigenetics of Atherosclerosis: Emerging Mechanisms and Methods. Trends Mol Med. 2017;23:332-47
5. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R. et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;135:e146-e603
6. Fang J, Little PJ, Xu S. Atheroprotective Effects and Molecular Targets of Tanshinones Derived From Herbal Medicine Danshen. Med Res Rev. 2018;38:201-28
7. Son DJ, Jung YY, Seo YS, Park H, Lee DH, Kim S. et al. Interleukin-32alpha Inhibits Endothelial Inflammation, Vascular Smooth Muscle Cell Activation, and Atherosclerosis by Upregulating Timp3 and Reck through suppressing microRNA-205 Biogenesis. Theranostics. 2017;7:2186-203
8. Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91:327-87
9. Rezvan A, Ni CW, Alberts-Grill N, Jo H. Animal, in vitro, and ex vivo models of flow-dependent atherosclerosis: role of oxidative stress. Antioxid Redox Signal. 2011;15:1433-48
10. Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med. 2009;6:16-26
11. Abe J, Berk BC. Novel mechanisms of endothelial mechanotransduction. Arterioscler Thromb Vasc Biol. 2014;34:2378-86
12. Baeyens N, Bandyopadhyay C, Coon BG, Yun S, Schwartz MA. Endothelial fluid shear stress sensing in vascular health and disease. J Clin Invest. 2016;126:821-8
13. Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW. et al. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood. 2006;107:4354-63
14. Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN. et al. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116:49-58
15. Dunn J, Thabet S, Jo H. Flow-Dependent Epigenetic DNA Methylation in Endothelial Gene Expression and Atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35:1562-9
16. Greissel A, Culmes M, Burgkart R, Zimmermann A, Eckstein HH, Zernecke A. et al. Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques. Cardiovasc Pathol. 2016;25:79-86
17. Zhou B, Margariti A, Zeng L, Xu Q. Role of histone deacetylases in vascular cell homeostasis and arteriosclerosis. Cardiovasc Res. 2011;90:413-20
18. Andreou I, Sun X, Stone PH, Edelman ER, Feinberg MW. miRNAs in atherosclerotic plaque initiation, progression, and rupture. Trends Mol Med. 2015;21:307-18
19. Jian L, Jian D, Chen Q, Zhang L. Long Noncoding RNAs in Atherosclerosis. J Atheroscler Thromb. 2016;23:376-84
20. Zhou T, Ding JW, Wang XA, Zheng XX. Long noncoding RNAs and atherosclerosis. Atherosclerosis. 2016;248:51-61
21. Boeckel JN, Guarani V, Koyanagi M, Roexe T, Lengeling A, Schermuly RT. et al. Jumonji domain-containing protein 6 (Jmjd6) is required for angiogenic sprouting and regulates splicing of VEGF-receptor 1. Proc Natl Acad Sci U S A. 2011;108:3276-81
22. Ohtani K, Vlachojannis GJ, Koyanagi M, Boeckel JN, Urbich C, Farcas R. et al. Epigenetic regulation of endothelial lineage committed genes in pro-angiogenic hematopoietic and endothelial progenitor cells. Circ Res. 2011;109:1219-29
23. Dunn J, Qiu H, Kim S, Jjingo D, Hoffman R, Kim CW. et al. Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J Clin Invest. 2014;124:3187-99
24. Zhou J, Li YS, Wang KC, Chien S. Epigenetic Mechanism in Regulation of Endothelial Function by Disturbed Flow: Induction of DNA Hypermethylation by DNMT1. Cell Mol Bioeng. 2014;7:218-24
25. Jiang YZ, Jimenez JM, Ou K, McCormick ME, Zhang LD, Davies PF. Hemodynamic disturbed flow induces differential DNA methylation of endothelial Kruppel-Like Factor 4 promoter in vitro and in vivo. Circ Res. 2014;115:32-43
26. Hastings NE, Simmers MB, McDonald OG, Wamhoff BR, Blackman BR. Atherosclerosis-prone hemodynamics differentially regulates endothelial and smooth muscle cell phenotypes and promotes pro-inflammatory priming. Am J Physiol Cell Physiol. 2007;293:C1824-33
27. Wu W, Xiao H, Laguna-Fernandez A, Villarreal G Jr, Wang KC, Geary GG. et al. Flow-Dependent Regulation of Kruppel-Like Factor 2 Is Mediated by MicroRNA-92a. Circulation. 2011;124:633-41
28. Ni CW, Qiu H, Jo H. MicroRNA-663 upregulated by oscillatory shear stress plays a role in inflammatory response of endothelial cells. Am J Physiol Heart Circ Physiol. 2011;300:H1762-9
29. Qin X, Wang X, Wang Y, Tang Z, Cui Q, Xi J. et al. MicroRNA-19a mediates the suppressive effect of laminar flow on cyclin D1 expression in human umbilical vein endothelial cells. Proc Natl Acad Sci U S A. 2010;107:3240-4
30. Huang TS, Wang KC, Quon S, Nguyen P, Chang TY, Chen Z. et al. LINC00341 exerts an anti-inflammatory effect on endothelial cells by repressing VCAM1. Physiol Genomics. 2017;49:339-45
31. Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128-34
32. Wierda RJ, Rietveld IM, van Eggermond MC, Belien JA, van Zwet EW, Lindeman JH. et al. Global histone H3 lysine 27 triple methylation levels are reduced in vessels with advanced atherosclerotic plaques. Life Sci. 2015;129:3-9
33. Greissel A, Culmes M, Napieralski R, Wagner E, Gebhard H, Schmitt M. et al. Alternation of histone and DNA methylation in human atherosclerotic carotid plaques. Thromb Haemost. 2015;114:390-402
34. Maleszewska M, Vanchin B, Harmsen MC, Krenning G. The decrease in histone methyltransferase EZH2 in response to fluid shear stress alters endothelial gene expression and promotes quiescence. Angiogenesis. 2016;19:9-24
35. Gunawan M, Venkatesan N, Loh JT, Wong JF, Berger H, Neo WH. et al. The methyltransferase Ezh2 controls cell adhesion and migration through direct methylation of the extranuclear regulatory protein talin. Nat Immunol. 2015;16:505-16
36. Lu C, Han HD, Mangala LS, Ali-Fehmi R, Newton CS, Ozbun L. et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell. 2010;18:185-97
37. Snitow M, Lu M, Cheng L, Zhou S, Morrisey EE. Ezh2 restricts the smooth muscle lineage during mouse lung mesothelial development. Development. 2016;143:3733-41
38. Lv YC, Tang YY, Zhang P, Wan W, Yao F, He PP. et al. Histone Methyltransferase Enhancer of Zeste Homolog 2-Mediated ABCA1 Promoter DNA Methylation Contributes to the Progression of Atherosclerosis. PLoS One. 2016;11:e0157265
39. Xiaoling Y, Li Z, ShuQiang L, Shengchao M, Anning Y, Ning D. et al. Hyperhomocysteinemia in ApoE-/- Mice Leads to Overexpression of Enhancer of Zeste Homolog 2 via miR-92a Regulation. PLoS One. 2016;11:e0167744
40. Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr. et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92:1355-74
41. Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W Jr, Rosenfeld ME. et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1994;89:2462-78
42. Xu S, Liu B, Yin M, Koroleva M, Mastrangelo M, Ture S. et al. A novel TRPV4-specific agonist inhibits monocyte adhesion and atherosclerosis. Oncotarget. 2016;7:37622-35
43. Xu S, Ha CH, Wang W, Xu X, Yin M, Jin FQ. et al. PECAM1 regulates flow-mediated Gab1 tyrosine phosphorylation and signaling. Cell Signal. 2016;28:117-24
44. Xu S, Koroleva M, Yin M, Jin ZG. Atheroprotective laminar flow inhibits Hippo pathway effector YAP in endothelial cells. Transl Res. 2016;176:18-28
45. Zhang W, Wang Q, Wu Y, Moriasi C, Liu Z, Dai X. et al. Endothelial cell-specific liver kinase B1 deletion causes endothelial dysfunction and hypertension in mice in vivo. Circulation. 2014;129:1428-39
46. Xu Y, Xu S, Liu P, Koroleva M, Zhang S, Si S. et al. Suberanilohydroxamic Acid as a Pharmacological Kruppel-Like Factor 2 Activator That Represses Vascular Inflammation and Atherosclerosis. J Am Heart Assoc. 2017;6:e007134
47. Xu S, Fu J, Chen J, Xiao P, Lan T, Le K. et al. Development of an optimized protocol for primary culture of smooth muscle cells from rat thoracic aortas. Cytotechnology. 2009;61:65-72
48. Xu S, Huang Y, Xie Y, Lan T, Le K, Chen J. et al. Evaluation of foam cell formation in cultured macrophages: an improved method with Oil Red O staining and DiI-oxLDL uptake. Cytotechnology. 2010;62:473-81
49. Heo KS, Lee H, Nigro P, Thomas T, Le NT, Chang E. et al. PKCzeta mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J Cell Biol. 2011;193:867-84
50. Hamik A, Jain MK. Shear stress: devil's in the details. Blood. 2010;116:2625-6
51. Maus U, Henning S, Wenschuh H, Mayer K, Seeger W, Lohmeyer J. Role of endothelial MCP-1 in monocyte adhesion to inflamed human endothelium under physiological flow. Am J Physiol Heart Circ Physiol. 2002;283:H2584-91
52. Muller WA. How endothelial cells regulate transmigration of leukocytes in the inflammatory response. Am J Pathol. 2014;184:886-96
53. Murao K, Ohyama T, Imachi H, Ishida T, Cao WM, Namihira H. et al. TNF-alpha stimulation of MCP-1 expression is mediated by the Akt/PKB signal transduction pathway in vascular endothelial cells. Biochem Biophys Res Commun. 2000;276:791-6
54. Watson C, Whittaker S, Smith N, Vora AJ, Dumonde DC, Brown KA. IL-6 acts on endothelial cells to preferentially increase their adherence for lymphocytes. Clin Exp Immunol. 1996;105:112-9
55. Xu Y, Liu P, Xu S, Koroleva M, Zhang S, Si S. et al. Tannic acid as a plant-derived polyphenol exerts vasoprotection via enhancing KLF2 expression in endothelial cells. Sci Rep. 2017;7:6686
56. Le K, Li R, Xu S, Wu X, Huang H, Bao Y. et al. PPARalpha activation inhibits endothelin-1-induced cardiomyocyte hypertrophy by prevention of NFATc4 binding to GATA-4. Arch Biochem Biophys. 2012;518:71-8
57. Xu S, Yin M, Koroleva M, Mastrangelo MA, Zhang W, Bai P. et al. SIRT6 protects against endothelial dysfunction and atherosclerosis in mice. Aging (Albany NY). 2016;8:1064-82
58. Xu S. Transcriptome Profiling in Systems Vascular Medicine. Front Pharmacol. 2017;8:563
59. Ayari H, Bricca G. Identification of two genes potentially associated in iron-heme homeostasis in human carotid plaque using microarray analysis. J Biosci. 2013;38:311-5
60. Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, Ge K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci U S A. 2007;104:18439-44
61. McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS. et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108-12
62. Su IH, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT. et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol. 2003;4:124-31
63. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L. et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol. 2016;17:95-103
64. Italiano A. Role of the EZH2 histone methyltransferase as a therapeutic target in cancer. Pharmacol Ther. 2016;165:26-31
65. Vella S, Pomella S, Leoncini PP, Colletti M, Conti B, Marquez VE. et al. MicroRNA-101 is repressed by EZH2 and its restoration inhibits tumorigenic features in embryonal rhabdomyosarcoma. Clin Epigenetics. 2015;7:82
66. Kumar S, Kim CW, Simmons RD, Jo H. Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitive athero-miRs. Arterioscler Thromb Vasc Biol. 2014;34:2206-16
67. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005
68. Chen K, Fan W, Wang X, Ke X, Wu G, Hu C. MicroRNA-101 mediates the suppressive effect of laminar shear stress on mTOR expression in vascular endothelial cells. Biochem Biophys Res Commun. 2012;427:138-42
69. Booth BA, Boes M, Andress DL, Dake BL, Kiefer MC, Maack C. et al. IGFBP-3 and IGFBP-5 association with endothelial cells: role of C-terminal heparin binding domain. Growth Regul. 1995;5:1-17
70. Dahlfors G, Arnqvist HJ. Vascular endothelial growth factor and transforming growth factor-beta1 regulate the expression of insulin-like growth factor-binding protein-3, -4, and -5 in large vessel endothelial cells. Endocrinology. 2000;141:2062-7
71. Rombouts C, Aerts A, Quintens R, Baselet B, El-Saghire H, Harms-Ringdahl M. et al. Transcriptomic profiling suggests a role for IGFBP5 in premature senescence of endothelial cells after chronic low dose rate irradiation. Int J Radiat Biol. 2014;90:560-74
72. Guo S, Long M, Li X, Zhu S, Zhang M, Yang Z. Curcumin activates autophagy and attenuates oxidative damage in EA.hy926 cells via the Akt/mTOR pathway. Mol Med Rep. 2016;13:2187-93
73. Bach LA. Endothelial cells and the IGF system. J Mol Endocrinol. 2015;54:R1-13
74. Ishikawa S, Hayashi H, Kinoshita K, Abe M, Kuroki H, Tokunaga R. et al. Statins inhibit tumor progression via an enhancer of zeste homolog 2-mediated epigenetic alteration in colorectal cancer. Int J Cancer. 2014;135:2528-36
75. Dimri M, Bommi PV, Sahasrabuddhe AA, Khandekar JD, Dimri GP. Dietary omega-3 polyunsaturated fatty acids suppress expression of EZH2 in breast cancer cells. Carcinogenesis. 2010;31:489-95
76. Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT. et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306-10
77. Wu SC, Zhang Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J Biol Chem. 2011;286:28511-9
78. Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601-5
79. Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK. et al. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol Cell. 2011;43:798-810
80. Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C. et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871-4
81. Benetatos L, Voulgaris E, Vartholomatos G, Hatzimichael E. Non-coding RNAs and EZH2 interactions in cancer: long and short tales from the transcriptome. Int J Cancer. 2013;133:267-74
Author contact
![]() Corresponding author: Zheng Gen Jin, Ph.D.; Aab Cardiovascular Research Institute (CVRI), Department of Medicine, University of Rochester School of Medicine and Dentistry; 601 Elmwood Avenue, Box CVRI, Rochester, NY 14642; Phone: 585-276-7691; Fax: 585-276-9829; E-mail Zheng-gen_Jinrochester.edu
Corresponding author: Zheng Gen Jin, Ph.D.; Aab Cardiovascular Research Institute (CVRI), Department of Medicine, University of Rochester School of Medicine and Dentistry; 601 Elmwood Avenue, Box CVRI, Rochester, NY 14642; Phone: 585-276-7691; Fax: 585-276-9829; E-mail Zheng-gen_Jinrochester.edu