Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(15):4050-4061. doi:10.7150/thno.21524 This issue Cite
Research Paper
Wild-type IDH2 promotes the Warburg effect and tumor growth through HIF1α in lung cancer
Jiangjiang Li1,2,3,4, Ya He1,2,3, Zheqiong Tan1,2,3, Jingchen Lu1,2,3, Liling Li1,2,3, Xin Song5, Feng Shi1,2,3, Longlong Xie1,2,3, Shuo You6, Xiangjian Luo1,2,3, Namei Li1,2,3, Yueshuo Li1,2,3, Xiaolan Liu1,2,3, Min Tang1,2,3, Xinxian Weng1,2,3, Wei Yi1,2,3, Jia Fan7, Jian Zhou7, Gao Qiang7, ShuangJian Qiu7, Weizhong Wu7, Ann M. Bode8, Ya Cao1,2,3,4,9 ![]()
1. Key Laboratory of Carcinogenesis and Invasion, Chinese Ministry of Education, Xiangya Hospital, Central South University, Changsha 410078, China
2. Cancer Research Institute, Xiangya School of Medicine, Central South University, Changsha 410078, China
3. Key Laboratory of Carcinogenesis, Chinese Ministry of Health, Changsha 410078, China
4. Research Center for Technologies of Nucleic Acid-Based Diagnostics and Therapeutics Hunan Province, Changsha 410078, China
5. Cancer Biotherapy Center, The Third Affiliated Hospital of Kunming Medical University (Tumor Hospital of Yunnan Province), Kunming 650118, China
6. Second Affiliated Hospital of Xiangya, Central South University, Changsha 410000, China
7. Key Laboratory for Carcinogenesis and Cancer Invasion, Chinese Ministry of Education, Zhongshan Hospital, Shanghai Medical School, Fudan University, Shanghai 200000, China
8. The Hormel Institute, University of Minnesota, Austin, MN 55912, USA
9. National Joint Engineering Research Center for Genetic Diagnostics of Infectious Diseases and Cancer, Changsha 410078, China
Received 2017-6-19; Accepted 2018-6-11; Published 2018-7-16
Abstract

Hotspot mutations of isocitrate dehydrogenase 1 and 2 (IDH1/2) have been studied in several cancers. However, the function of wild-type IDH2 in lung cancer and the mechanism of its contribution to growth of cancer cells remain unknown. Here, we explored the role and mechanism of wild-type IDH2 in promoting growth of lung cancer.
Methods: Information regarding genomic and clinical application focusing on IDH2 in cancer was examined in several databases of more than 1,000 tumor samples. IDH2 expression was assessed by immunohistochemistry in tissues from lung cancer patients. The biological functions of IDH2 were evaluated by using cell-based assays and in vivo xenograft mouse models.
Results: Here we reported that wild-type IDH2 is up-regulated and is an indicator of poor survival in lung cancer and several other cancers. Targeting IDH2 with shRNA resulted in decreased HIF1α expression, leading to the attenuation of lung cancer cell proliferation and tumor growth. Treatment of lung cancer cells with AGI-6780 (a small molecule inhibitor of IDH2), PX-478 (an inhibitor of HIF1α) or incubation with octyl-α-KG inhibited lung cancer cell proliferation.
Conclusion: IDH2 promotes the Warburg effect and lung cancer cell growth, which is mediated through HIF1α activation followed by decreased α-KG. Therefore, IDH2 could possibly serve as a novel therapeutic target for lung cancer.
Keywords: isocitrate dehydrogenase 2, lung cancer, Warburg effect, tumor growth, HIF1-α
Introduction
Lung cancer is the leading cause of cancer-related death worldwide [1]. Non-small cell lung cancer (NSCLC) accounts for 80% to 85% of all lung cancer cases, including lung adenocarcinoma, squamous cell lung carcinoma and large cell lung carcinoma [2]. The four main genetic mutations of NSCLC include K-RAS (24%), EGFR (13%), ALK (5%) and TP53 (5%) [3]. EGFR inhibitors have been used as first-line treatment drugs against lung cancer [4]. Unfortunately, the clinical outcome of NSCLC patients remains poor, with a 5-year overall survival (OS) rate of 18% in the USA and 10-15% in China [5; 6].
Reprogramming energy metabolism is proving to be a common survival mechanism in cancer cells and is recognized as a hallmark of cancer [7]. Cancer cells take up glucose and glutamine at high rates as the two most important nutrients to support their survival and growth [8]. Glucose and glutamine are utilized for biosynthesis, proliferation and NADPH production in cancer cells and both can be controlled by oncogenes such as K-RAS and C-MYC [9-14].
Interestingly, gain-of-function mutations of the isocitrate dehydrogenase 1 and 2 (IDH1/2) enzymes have been identified recently in several cancers [15]. IDH1/2 catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG) or the reverse reaction. Gain-of-function mutations in IDH1/2 have been shown to activate oncogenes and regulate the expression level of multiple genes [16]. These mutations produce the “onco-metabolite” 2-hydroxyglutarate (2-HG) which is from α-KG [17; 18]. 2-HG is a competitive inhibitor of α-KG-dependent dioxygenases [17; 18]. Thus, IDH1/2 mutations impair the function of α-KG-dependent dioxygenases by consuming α-KG and competitively substituting α-KG with 2-HG. α-KG was shown to have antitumor effects through inhibition of angiogenesis in a murine tumor model [19]. The α-KG-dependent dioxygenases use α-KG as a substrate. The dioxygenases include KDM, TET2, PHD2 and PLOD1-3, which control histone demethylation and hypoxia-inducible factor-1α (HIF1α)-dependent cellular signaling and collagen formation [15]. HIF1α is broadly expressed and correlates with poor prognosis in human cancers by regulating genes involved in glycolysis, angiogenesis, cell cycle progression and other cellular pathways [20]. HIF1α was shown to be a positive factor in solid tumor growth and was shown to be required for tumor growth and metastasis of NSCLC [21-23].
In addition to the well-known function of the mutant IDH2, the wild-type IDH2 protein (IDH2wt) was found to participate in glutamine metabolism and promoted cell survival. IDH2 carboxylates α-KG from glutamine to citrate in hypoxia to promote glioblastoma cell growth and increased viability [24]. IDH1/2 participate in reductive carboxylation of glutamine to support redox homeostasis during anchorage-independent tumor spheroid formation [25]. The protein level of wild-type IDH2 was found to be markedly up-regulated in esophageal squamous cell carcinoma (ESCC) tissues and is associated with worse overall survival and reduced progression-free survival of ESCC patients [26]. IDH2wt suppresses melanoma cell growth, increases tumor-free survival in animal models and contribute to 5-hmC loss [27]. High expression of 5-hydroxymethylcytosine and IDH2wt was associated with favorable prognosis after curative resection of hepatocellular carcinoma [28].
Here we reported that wild-type IDH2 is highly expressed in multiple cancers, especially in lung cancer, and this high level of IDH2 correlates with poor survival. We found that IDH2 enhances the Warburg effect and increases cell growth by promoting a lower intracellular level of α-KG. Targeting IDH2 with shRNA resulted in decreased HIF1α levels, leading to attenuated cell viability, proliferation and tumor growth. Treatment of cells with AGI-6780 (a small molecule inhibitor of IDH2), PX-478 (an inhibitor of HIF1α) or incubation with octyl-α-KG inhibited cell proliferation. Overall, these results suggest that high expression of wild-type IDH2 has an oncogenic function and might be a new therapeutic target against lung cancer.
Methods
Reagents and antibodies
CoCl2, dihydroethidium and propidium iodide were from Sigma/Aldrich. Octyl-α-ketoglutarate was from Cayman and PX-478 and AGI-6780 were from Selleck. Antibodies to detect IDH2, HIF1α, LDHA, ALDOA, PDK1, and total ubiquitination and the NADP/NADPH kit were from Abcam and the β-action antibody was from Sigma.
Immunohistochemistry analysis
Lung cancer tissues and paired normal tissues were from Xiangya Hospital and the Third Affiliated Hospital of Kunming Medical University and were validated by a pathologist from the Xiangya Hospital. The study protocol was approved by the hospital ethical review committees. Immunohistochemistry was performed as previously described [14; 29; 30].
Western blot analysis
Cells were harvested and washed with PBS twice, disrupted in IP buffer (Thermo) and centrifuged at 12,000 × g for 20 min. Protein (50 μg) from the supernatant fraction (quantified by the BCA Protein Assay Kit, Thermo) was subjected to SDS-PAGE, and transferred to a PVDF membrane (Millipore). Membranes were blocked with 5% non-fat milk for 1 h at room temperature and then incubated with the specific primary antibody, followed by the corresponding HRP-conjugated anti-mouse or anti-rabbit secondary antibody. Protein bands were visualized by the Western lightening plus-ECL kit (Pierce).
Generation of IDH2-overexpressing and IDH2-knockdown cell pools
For lentivirus production, an IDH2-overexpressing plasmid LV105-IDH2, its empty vector LV105 (Genecopoeia), shRNA plasmids, GV-248-sh-IDH2#1 and GV-248-sh-IDH2#2, and control vector GV-248 (Genechem,) were separately co-transfected with packaging (psPAX2) and envelope (pMD2.G) vectors into HEK293T cells. Lentivirus was harvested at 48 h post transfection from the supernatant fraction, and mixed with 5 mg/mL polybrene (Selleck) to increase the infection efficiency. H460 and A549 lung cancer cells were infected with the lentivirus and selected in 1 mg/mL puromycin (Selleck) for 1 week. For rescue experiments, an IDH2-overexpressing plasmid, LV105-IDH2-res (Genecopoeia), was transfected into lung cancer cells using Lipofectamine 2000 reagent (Invitrogen).
Cell culture
A549 lung cancer cells were originally purchased from American Type Culture Collection (kind gifts from Dr. Xingming Deng, Emory University Atlanta, Georgia [31]). The H1299, H460, H520, H522, PC9 and MRC5 cells were obtained from Cancer Research Institute of Central South University. The H460 and A549 stable IDH2-overexpressing and IDH2-knockdown cells were maintained in RPMI 1640 with 10% fetal calf serum and penicillin/streptomycin/gentamicin antibiotics. Cells were grown in a 37 °C incubator with 5% CO2.
Cell viability and cell growth assays
Cells (5×104) were seeded in a 6-well plate and cell numbers were counted by Trypan blue exclusion using a blood counting chamber. Cells (2×103) were seeded in a 96-well plate and cell viability was estimated by MTS assay (Promega). Cells were treated with different concentrations of octyl-α-KG, PX-478, or AGI-6780 and then cell counting and cell viability estimation were performed as indicated above.
Cell death assay
Cell death was measured using propidium iodide (100 ng/mL) and a BD FACSCantoTM II flow cytometer (BD Biosciences).
Intracellular glucose uptake, lactate production, ATP assays
Cells (5×105) were seeded in a 6-well plate and culture medium was replaced after 8 h. The culture medium was collected after another 16 h. Glucose and lactate were measured using the Beckman AU680 Automatic Biochemistry Analyzer normalized by cell number. Cells (5×105) were seeded in a 6-well plate and treated with octyl-α-KG (1 mM) and PX-478 (10 uM) for 24 h and then glucose and lactate were measured as indicated above. Intracellular ATP concentration was measured by using an ATP bioluminescent somatic cell assay kit (Perkin Elmer). Cells (2×104 cells/well) were plated in a 96-well plate and harvested after 12 h. Luminescent intensity as the ATP value from each well was measured using an EnSpireTM Multilabel reader (Perkin Elmer).
ROS, α-KG, 2-HG and oxygen consumption rate assays
Intracellular levels of reactive oxygen species (ROS) were measured by flow cytometry. Cells were collected and suspended at 1×106 cells/mL in PBS and incubated with 1 μM dihydroethidium (Sigma) for 30 min before analysis by a BD FACSCantoTM II flow cytometer (BD Biosciences). The mean values were set as the ROS level. α-KG and 2-HG was measured using an alpha-ketoglutarate colorimetric fluorometric assay kit and D-2-hydroxyglutarate colorimetric assay kit (Biovision). The oxygen consumption rate (OCR) was measured using the MitoXpress® Xtra Oxygen Consumption Assay kit (Luxcel) and the slope of the fluorescence curve was set as the OCR. The OCR was also analyzed by Seahorse XF24 at baseline and after adding oligomycin, FCCP, and rotenone.
Database search
Clinical data were from publically available databases, including cBioprotal (TCGA) and Oncomine, which were used to explore the genomic changes and mRNA expression of IDH2 in cancers [32-34]. Kmplot was used to compare survival profiles of cancer patients who were divided by high and low IDH2 expression [35]. Survexpress was utilized to analyze the mRNA of IDH2 in differential pathological types and clinical stage of lung cancer tissues [36]. The Cancer Cell Line Encyclopedia (CCLE) was employed to analysis the correlation of copy number and mRNA level.
Ubiquitination analysis
Cells incubated with CoCl2 (500 μM) for 24 h were harvested and the HIF1α antibody was used to immunoprecipitate total HIF1α. Ubiquitination of HIF1α was examined using a ubiquitination primary antibody and its corresponding secondary antibody.
Carbon-labeled isotopologues analysis
Cells were seeded and the culture medium was replaced with carbon-labeled [U-13C] glucose or [U-13C] glutamine for 24 h and then GC-MS was used for analysis.
Xenograft studies
Animal experiments were conducted according to the protocols approved by the Central South University Animal Care Committee. Nude mice (BCLB/c-nu, male, 4-6 weeks old) were subcutaneously injected with H460 stable IDH2 overexpressing, IDH2 knockdown cells or paired control cells. Tumor growth was recorded by measuring length and width of the tumors. Tumor size was calculated using the formula length×width2/2 and the tumors were harvested and weighed at the experimental endpoint. Tumor cell proliferation was determined by Ki-67 IHC staining.
RT-qPCR
Total mRNA was isolated utilizing the Trizol reagent (Invitrogen) and used for reverse transcription (Thermo Fisher Scientific). Then qRT-PCR was performed using the ABI7500 (Applied Biosystems) by mixing cDNA, primers and SYBR®Green Real-Time PCR Master Mixes (Invitrogen). The primer sequences are included in Supplementary Material [37].
Statistical analysis
Differences between lung cancer tissues and paired normal lung tissues were assessed by a paired student's t-test. Two-tailed unpaired t-tests and correlation analysis were conducted using GraphPad Prism 5. Statistical significance is reported with a p value of < 0.05.
Results
IDH2 is up-regulated in lung cancer tissues and is a negative prognostic factor for lung cancer patients
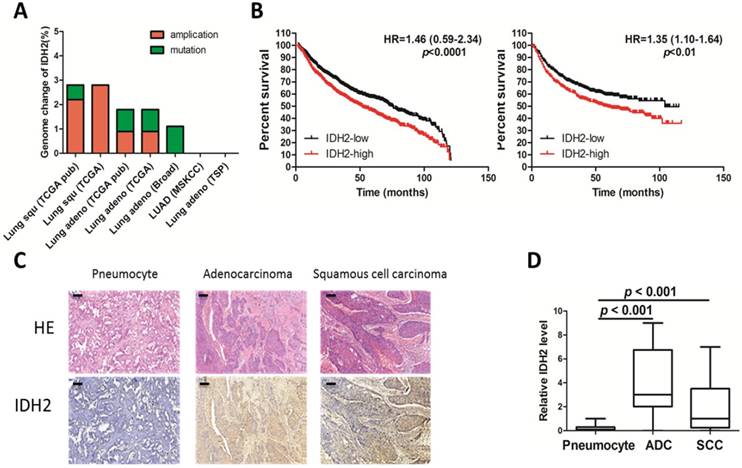
To obtain genomic and clinical outcome data regarding IDH2 expression in cancers, a search for the IDH2 gene was conducted in several databases containing more than 1,000 tumor samples. Amplification was the main genomic change occurring in the IDH2 gene and was found in 1.6% of the total samples (Figure 1A) [32; 33]. IDH2 expression was found to be higher in lung cancer than in normal lung tissues (Table 1 and Table S1) and is the most altered of all the tricarboxylic acid cycle (TCA) genes (Table 2) [34]. High levels of IDH2 correlated with poor overall survival and shorter progression-free survival in lung cancer (Figure 1B) [35]. Analysis of the correlation of IDH2 expression with clinical characteristics revealed that IDH2 is not associated with clinical stage but instead with differentiation and pathological type (Figure S1A). The correlation of IDH2 expression and overall survival of lung cancer patients was more obvious in adenocarcinomas in non-smoker patients (Figure S1B).
IDH2 is up-regulated in lung cancer. (A) Genomic amplification and mutation of the IDH2 gene in lung cancer (data from the TCGA Research Network). (B) Kaplan-Meier survival curves of censored Cox analysis lung cancer stratified by IDH2-expression risk group. The total number of samples analyzed was 1928 for overall survival (left) and 965 for progression-free survival (right); the median value was used as the cut-off (Kmplot). (C-D) Immunohistochemistry analysis (200×) was used to determine IDH2 protein levels in adenocarcinoma (ADC, n = 16), squamous cell carcinoma (SCC, n = 17) and normal pneumocyte tissues (n = 33), Bar scale, 100 μm.
IDH2 expression profiles across multiple cancer types compared to normal tissue.
| Analysis by Cancer type | Cancer vs. Normal | |
|---|---|---|
| Increase | Decrease | |
| Breast | 21 | 9 |
| Lung | 12 | 0 |
| Ovarian | 11 | 0 |
| Gastric | 0 | 9 |
| Kidney | 1 | 9 |
Numbers are datasets containing IDH2 mRNA expression (Oncomine).
TCA cycle gene expression profiles in lung cancer compared to normal tissue.
| TCA cycle genes | Cancer vs. Normal | |
|---|---|---|
| Increase | Decrease | |
| IDH2 | 12 | 9 |
| FH | 7 | 0 |
| MDH2 | 6 | 1 |
| IDH1 | 5 | 1 |
| OGDH | 0 | 0 |
Numbers are from datasets containing TCA cycle gene mRNA expression (Oncomine).
Additionally, we performed immunohistochemistry analysis of IDH2 in lung cancer and in paired noncancerous tissues derived from lung cancer patients (n = 33). IDH2 was mainly distributed in the cytoplasm of NSCLC cells (Figure 1C) and was significantly (p < 0.001) higher in adenocarcinoma and squamous cell carcinoma compared to the paired noncancerous pneumocyte tissues (Figure 1D).
From the TCGA and other databases, we found that the IDH2 mRNA level is positively correlated with its copy number in lung cancer cells (Figure S1C) and, in addition, breast cancer and pancreatic cancer carried the IDH2 amplification (Figure S1D). IDH2 is also an indicator of poor survival in these two cancers (Figure S1E). These data suggest an important role of IDH2 in multiple cancers.
Wild-type IDH2 controls intracellular α-KG concentration in lung cancer
To confirm that IDH2 is not mutated in lung cancer cells, we sequenced the genome of the NCI-H460 and A549 lung cancer cell lines and no mutations were found, which indicated that IDH2 is wild-type in these cells (Figure S2A-B). To determine the influence of wild-type IDH2 on intracellular α-KG concentration, H460 and A549 lung cancer cells stably overexpressing IDH2 (H460-IDH2 and A549-IDH2) or knockdown IDH2 (H460-shIDH2#1, #2 and A549-shIDH2 #1, #2) were generated by the lentivirus method and IDH2 expression was confirmed by Western blotting (Figure 2A).
Wild-type IDH2 controls intracellular concentrations of α-KG, 2-HG and redox status. (A) Western blot showing IDH2 levels in H460 and A549 lung cancer cells overexpressing IDH or expressing knockdown IDH2. β-Actin was used as a loading control. (B-C) Intracellular α-KG levels were determined in H460 and A549 lung cancer cells overexpressing IDH or expressing knockdown IDH2 (B), and in xenograft tissues from mice implanted with H460 cells overexpressing IDH2 or expressing knockdown IDH2 (C). Data are shown as mean values ± S.D. from 3 replicates of each sample, and p values were determined by a two-tailed Student's t test (* p < 0.05; **p < 0.01). (D-F) Intracellular 2-HG levels (D), total ROS levels (E) and the NADP+/NADPH ratio (F) were determined in H460 and A549 lung cancer cells overexpressing IDH2 or expressing knockdown IDH2 (* p < 0.05; **p < 0.01). (G) Carbon-labeled isotopologue TCA cycle metabolites from [U-13C] glucose and [U-13C] glutamine (* p < 0.05; **p < 0.01).
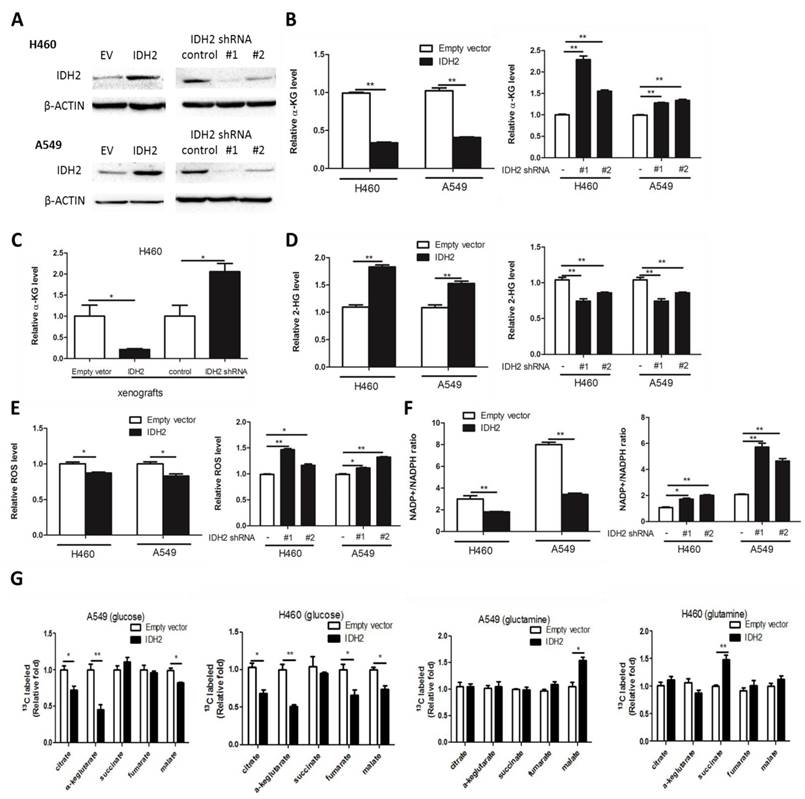
The intracellular α-KG concentration was then analyzed. We found that the α-KG concentration was markedly decreased in H460-IDH2 and A549-IDH2 cells, but was increased in H460 and A549 IDH2 knockdown cells (Figure 2B). Additionally, we performed a xenograft experiment in which nude mice were subcutaneously injected with H460-IDH2 or H460-shIDH2 #1 cells and found that the α-KG concentration was also lower in H460-IDH2 xenograft tumor tissues, but higher in H460-shIDH2 #1 xenograft tumor tissues (Figure 2C). This suggests that IDH2 might consume α-KG in lung cancer cells.
In breast cancer cells, IDH2 can carboxylate α-KG from glutamine to citrate in hypoxic conditions and increase the 2-HG level [38]. Hypoxia could induce production of 2-HG by LDHA and MDH1/2 [39]. We found that 2-HG was elevated in H460-IDH2 and A549-IDH2 cells and decreased in H460 and A549 IDH2 knockdown lung cancer cells under normal oxygen conditions (Figure 2D). Thus, wild-type IDH2 can decrease α-KG concentration and enhance the production of 2-HG. Reductive carboxylation of glutaminolysis can generate NADPH, which could be used to eliminate cellular ROS [25]. We measured the total ROS level and the NADP/NADPH ratio by flow cytometry in IDH2-overexpressing cells. IDH2 overexpression reduced cellular ROS levels (Figure 2E) along with the NADP/NADPH ratio (Figure 2F), whereas knocking down IDH2 enhanced the total cellular ROS levels (Figure 2E) and increased the NADP/NADPH ratio (Figure 2F). These results suggest that the reverse reaction of wild-type IDH2 might consume intracellular α-KG, produce 2-HG and lower the level of cellular ROS under normal oxygen conditions in lung cancer cells.
To identify the metabolic flux that contributes to changes in α-KG, carbon-labeled glucose or glutamine was added in the culture medium and TCA cycle metabolites were measured. IDH2 overexpression decreased all carbon-labeled isotopologue TCA cycle metabolites generated from [U-13C] glucose but increased slightly those from [U-13C] glutamine (Figure 2G). The ratio of carbon-labeled α-KG from [U-13C] glucose was the most altered metabolite of all the tricarboxylic acid cycle metabolites (Figure 2G).
Wild-type IDH2 promotes the Warburg effect
Expression of IDH2 correlates with time to first progression and overall survival time, which indicates that IDH2 might influence key hallmarks of cancer, such as aerobic glycolysis, cell proliferation and death. Wild-type IDH2 regulates the concentration of α-KG, which is a substrate of PHD2 that degrades HIF1α. HIF1α plays a key role in the Warburg effect by transcriptionally regulating multiple genes, and specifically glycolytic genes, including aldolase A (ALDOA), lactate dehydrogenase A (LDHA) and pyruvate dehydrogenase kinase 1 (PDK1) [22; 37]. We first analyzed the mRNA levels of IDH2 and HIF1α-targeted genes in lung cancer tissues taken from the database [36]. IDH2 mRNA was positively correlated with 44 of a total of 74 HIF1α target genes analyzed in lung cancer tissues (Table S2).
By examining glucose uptake and lactate production of NSCLC cells, we next determined whether IDH2 could influence the Warburg effect. We found that IDH2 overexpression promoted cellular glucose uptake, lactate production and lowered cellular ATP content (Figure 3A-C). In contrast, knockdown of IDH2 decreased cellular glucose uptake, lactate production and increased cellular ATP content (Figure 3A-C).
Wild-type IDH2 promotes the Warburg effect in lung cancer cells. (A-B) Glucose uptake (A) and lactate production (B) were determined in H460 and A549 lung cancer cells cells overexpressing IDH or expressing knockdown IDH2. Data are shown as mean values ± S.D. from 3 replicates of each sample, and p values were determined by a two-tailed Student's t test (**p < 0.01). (C-E) ATP content (C) and oxygen consumption rate (OCR, D, E) were determined in H460 and A549 lung cancer cells overexpressing IDH or expressing knockdown IDH2 (* p < 0.05; **p < 0.01). (F) RT-qPCR analysis of HIF1α-targeted glycolysis genes in H460 and A549 lung cancer cells overexpressing IDH or expressing knockdown IDH2. Data are shown as mean values ± S.D. (n = 3) for each group. (**p < 0.01) was compared with empty vector control group by Student's t-test).
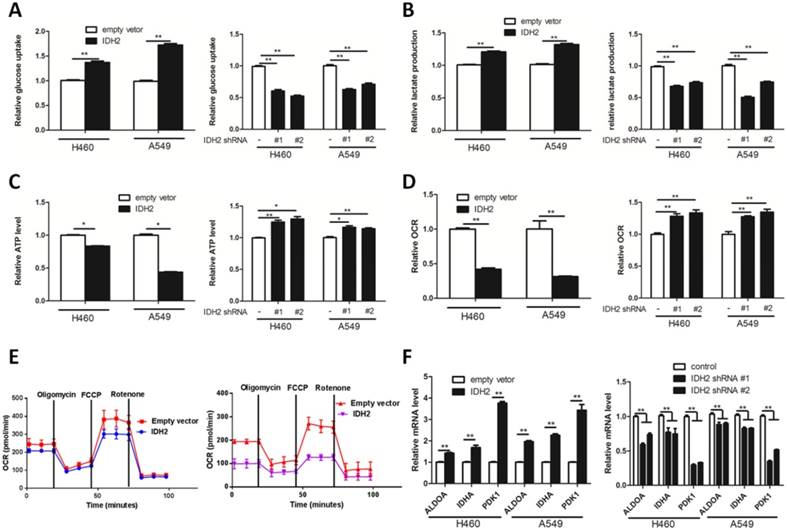
Enhancing cellular glycolysis and glutaminolysis might lower the amount of α-KG available for oxidative phosphorylation, thus decreasing the cellular ATP concentration and oxygen consumption rate (OCR). We found that IDH2 overexpression reduced the OCR, whereas knockdown IDH2 increased the OCR of lung cancer cells (Figure 3D-E).
We then determined the effect of IDH2 expression on HIF1α-targeted glycolytic gene expression. Lung cancer cells overexpressing IDH2 potentiated the mRNA expression of the HIF1α-targeted glycolytic genes ALDOA, LDHA and PDK1 (Figure 3E), whereas knockdown IDH2 had lower ALDOA, LDHA and PDK1 mRNA expression (Figure 3E). The protein level of ALDOA, LDHA and PDK1 was further analyzed in xenograft tissues. The expression of these three proteins was higher in H460-IDH2 xenograft tumor tissues and lower in H460-shIDH2 #1 cell xenograft tumor tissues (Figure S5C-D).
Wild-type IDH2 promotes lung cancer cell proliferation and tumor growth
We next examined the role of IDH2wt in cell proliferation and tumor growth. IDH2 overexpression enhanced cell viability and promoted proliferation. Knocking down IDH2 significantly decreased cell viability and inhibited proliferation (Figure 4A-B). IDH2 also diminished lung cancer cell death (Figure 4C and Figure S3A). We also found that IDH2 overexpression enhanced colony formation (Figure 4D and Figure S3B) and reduced apoptosis and TSZ-induced necroptosis in these cancer cells (Figure S4A-B).
Wild-type IDH2 promotes lung cancer cell growth in vitro and in vivo. (A-B) Cell viability (A) and cell number (B) were determined in H460 and A549 lung cancer cells overexpressing IDH or expressing knockdown IDH2. Data are presented as mean values ± S.D. (n = 3) for each group. (* p < 0.05, ** p < 0.01). (C) Cell death was determined in H460 and A549 lung cancer cells overexpressing IDH or expressing knockdown IDH2 (** p < 0.01). (D) Colony number was determined in H460 and A549 lung cancer cells overexpressing IDH2 and compared with cells expressing an empty vector (** p < 0.01). (E) Tumor volume and tumor weight were determined in xenografts from H460 lung cancer cells overexpressing IDH2. Data are presented as mean values ± S.D. (n = 5) for each group. (* p < 0.05). (F) Tumor volume and tumor weight were determined in xenografts from H460 lung cancer cells expressing knockdown IDH2. Data are presented as mean values ± S.D. (n = 3) for each group. (* p < 0.05). (G) Rescue experiments: cell number and western blot analysis of IDH2 in H460 and A549 lung cancer cells stably expressing IDH2 knockdown and overexpression plasmid (* p < 0.05, ** p < 0.01).
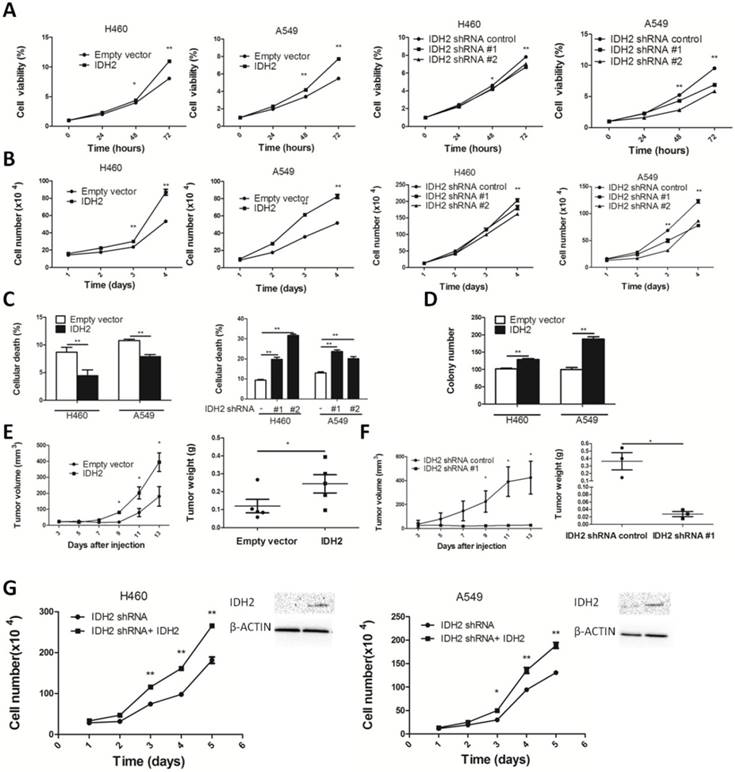
Additionally, we performed a xenograft experiment in which nude mice were subcutaneously injected with H460-IDH2 cells or H460-shIDH2 #1 cells. Results showed that volume and weight of tumors from mice implanted with H460-IDH2 cells were higher compared with tumors from mice injected with empty vector cells (Figure 4E and Figure S5A). In contrast, volume and tumor weight of tumors from mice implanted with H460-shIDH2 #1 cell were lower compared with tumors from mice injected with control cells (Figure 4F and Figure S5B). These results overall demonstrated that IDH2 could promote tumor growth.
To confirm the specificity of shRNA manipulations, rescue experiments were conducted. H460 and A549 lung cancer cells stably expressing sh-IDH2 RNA were transiently transfected with an IDH2 wild type plasmid. Cell proliferation rates were significantly higher in IDH2 wild-type plasmid-transfected cells compared to controls (Figure 4G).
Wild-type IDH2 promotes the Warburg effect and cell growth mediated through HIF1α
We have shown that IDH2wt could promote the Warburg effect and cell growth, and α-KG might be a primary contributing molecular metabolite. One important α-KG-dependent dioxygenase is hypoxia-inducible factor prolyl hydroxylase 2 (PHD2), which can affect cellular metabolism by degrading HIF1α. HIF1α-targeted glycolytic gene expression was found positively correlated with IDH2 expression. We thus determined whether IDH2 could promote the Warburg effect and cell growth by modulating HIF1α. First, IDH2 and HIF1α expression was analyzed and found to be positively correlated in a panel of lung cancer cells and in normal MRC5 lung fibroblasts (Figure S6A). H460 and A549 IDH2-overexpressing or -knockdown cells were treated with CoCl2 and HIF1α expression was assessed. HIF1α protein levels were higher in H460 and A549 IDH2-overexpressing cell lines and decreased in IDH2-knockdown cell lines (Figure 5A). The total HIF1α ubiquitination levels were decreased in IDH2-overexpressing cell lines, which indicated that IDH2 increased HIF1α levels by lowering HIF1 α ubiquitination (Figure 5A).
Wild-type IDH2 promotes the Warburg effect and cell growth through HIF1α. (A) Ubiquitinated and total HIF1α protein levels were determined by Western blotting in H460 and A549 lung cancer cells overexpressing IDH or expressing knockdown IDH2. β-Actin served as a loading control. (B) HIF1α protein levels were determined by Western blotting in H460 lung cancer cells overexpressing IDH2 and treated with DMSO or 1 mM octyl-α-KG. β-Actin served as a loading control. (C) Intracellular α-KG levels were determined in H460 lung cancer cells overexpressing IDH2 and treated with DMSO or 1 mM octyl-α-KG (** p < 0.01). (D) Glucose uptake and lactate production were determined in H460 and A549 lung cancer cells in the presence of the HIF1α inhibitor, PX-478 (10 μM) (** p < 0.01).(E) Cell number was determined by Trypan blue staining in H460 and A549 lung cancer cells overexpressing IDH2 in the presence of PX-478 (10 μM) (** p < 0.01).
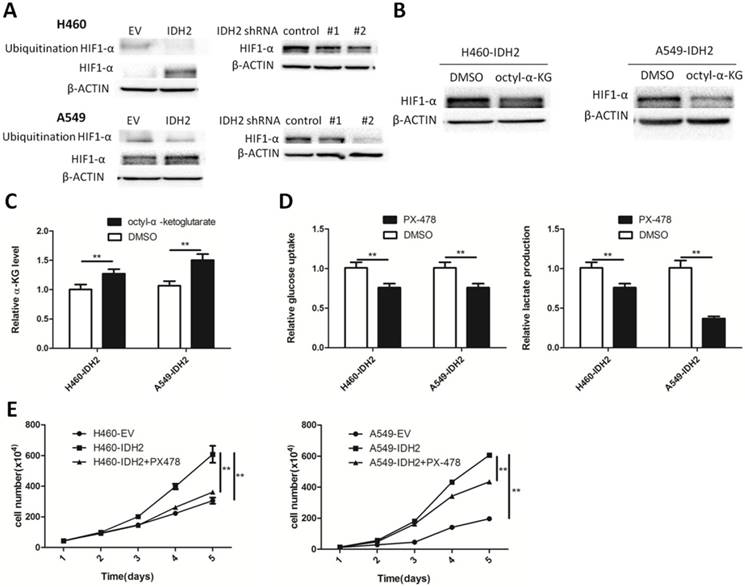
Next, we determined whether changing the concentration of α-KG could affect the expression of the HIF1α protein. Cell-permeable octyl-α-KG and CoCl2 were added to the culture medium and at 24 h, the intracellular concentration of α-KG and the HIF1α protein level were analyzed. Increased amounts of intracellular α-KG were associated with lower cellular HIF1α protein levels (Figure 5B-C).
We further examined whether inhibiting HIF1α would influence the Warburg effect. Treatment of H460-IDH2 and A549-IDH2 cells with the HIF1α inhibitor, PX-478, reduced glucose uptake and lactate production rate (Figure 5D). This suggested that IDH2 promotes the Warburg effect by modulating the cellular HIF1α protein.
The effect of IDH2 on cell growth might be due to its effect on α-KG, 2-HG or NADPH. Hypoxia can induce production of 2-HG by LDHA, MDH1/2, but not wild type IDH1/2 [39]. The 2-HG concentration should be low in those cells with wild type IDH1/2 and not be a primary contributor to IDH2 function. Treatment with the ROS scavenger, N-acetyl-cysteine (NAC), had little effect on cell viability or number (Figure S6B) and could not restore the effect of the IDH2 inhibitor on cell viability (Figure S6C). We next examined the effect of the HIF1α inhibitor, PX-478, on cell proliferation. Incubation with PX-478 obviously diminished cell number at various time points (Figure 5E). These results overall suggest that the oncogenic function of wild-type IDH2 in lung cancer is mainly due to its effect on α-KG and IDH2 promotes cell growth by mediating the cellular HIF1α protein.
Targeting wild-type IDH2 with AGI-6780 and replenishing cellular α-KG inhibits NSCLC cell viability and growth
IDH2 was up-regulated in human cancers and attenuation of IDH2 inhibited lung cancer cell proliferation and tumor growth, which implicated IDH2 as a potential anticancer target. AGI-6780 is a commercially available IDH2 inhibitor that can target both the IDH2 mutant and wild type enzymes with different IC50s [40]. We found AGI-6780 selectively inhibited proliferation of lung cancer cells, but did not affect normal MRC5 lung fibroblasts (Figure 6A). H460 and A549 cells treated with 20 μM AGI-6780 for 24 h had a 30-40% reduction in IDH activity (Figure 6B). AGI-6780 decreased lung cancer cell viability (Figure 6C) and proliferation (Figure 6D) in a time- and dose-dependent manner.
Targeting wild-type IDH2 and replenishing cellular α-KG decreases cell viability and growth. (A) The growth inhibition rates of different human lung cancer cells and normal MRC5 lung fibroblasts in the presence of AGI-6780. (B) IDH activity was analyzed in H460 and A549 lung cancer cells treated with DMSO or AGI-6780 (20 μM) for 24 h (** p < 0.01). (C) Cell viability and cell number of H460 and A549 lung cancer cells treated with different concentrations of AGI-6780 (0-50 μM). (D) Cell number was determined by Trypan blue staining in H460 and A549 lung cancer cells in the presence of 5 μM AGI-6780 (** p < 0.01). (E-F) Glucose uptake, lactate production and cell number were determined in H460 and A549 lung cancer cells treated with 1 mM octyl-α-KG (** p < 0.01). (G) Cell number was determined by Trypan blue staining in H460 and A549 cells transfected with siRNA targeting HIF1α (* p < 0.05, ** p < 0.01).
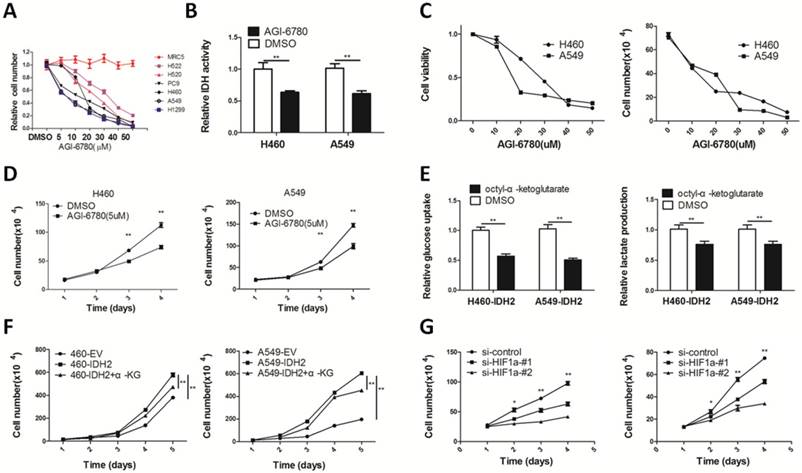
We found that incubation with the cell-permeable octyl-α-ketoglutarate diminished cellular glucose uptake and lactate production (Figure 6E). Additionally, this compound inhibited viability and proliferation of lung cancer cells (Figure 6F).
Discussion
IDH1 and IDH2 mutations have greatly altered our understanding of cancer biology by the discovery that they are new, unconventional oncogenes. Metabolic enzymes, including IDHs, have now gained a rightful place on the 'oncogene hit list' along with kinases and transcription factors [41]. IDHs are highly mutated in glioma, cartilaginous tumors, acute leukemia and thyroid carcinomas. In a previous study, we showed that IDH2 rs11540478 is a risk factor for lung cancer [42]. Here, we found that IDH2wt is up-regulated in lung cancer; and, wild-type IDH2 also performs a function similar to that of mutant IDHs. Wild-type IDH2 decreased the level of intracellular α-KG to promote the Warburg effect and cell growth, which is mediated by increasing HIF1α protein expression. HIF1α inhibited PDH by activating PDKs to decrease the flux of pyruvate to the TCA cycle. At the same time, LDHA also transcriptionally activated HIF1α and more lactate was produced from pyruvate, which contributes to the Warburg effect and cell growth.
Hypoxia can induce production of 2-HG by LDHA and MDH1/2 [39]. Because mutation-induced 2-HG is more than 100-fold higher than that found in normal cells, while in IDH2-overexpressed lung cancer cells is up to 2-fold than its control cells; the 2-HG may be produced by LDHA or MDH1/2. From these results, the function of IDH2wt seems to rely more on the effect of α-KG rather than 2-HG or ROS. The reduction of α-KG by IDH2wt might have an oncogenic function in lung tumorigenesis.
Interestingly, the AMPK/α-ketoglutarate axis dynamically regulates differentiation of progenitor cells [43]. We noted that IDH2 expression is the highest in poorly differentiated lung cancer tissues (Figure S1A). We also found that IDH2 expression is the highest in large cell carcinoma (LCC). LCC is defined as undifferentiated NSCLC that lacks differentiation, which also provides clues for the relationship of IDH2 and cellular differentiation [44]. Mechanistically, wild-type IDH2 might inhibit lung cancer cell differentiation through α-KG-dependent dioxygenases.
More importantly, we found that treatment of cells with AGI-6780 reduced IDH2 activity and decreased cellular viability and proliferation. Replenishing cellular α-KG also decreased cellular viability and growth in vivo. The inhibitors of IDHs have been used in clinical trials and α-ketoglutarate salts have been used as clinical nutrition and in metabolic care [45-49], which provides new clues for lung cancer treatment. However, survival data generated using public datasets have limitations, such as lack of confounding variables (such as gender and smoking status). In lung cancer, IDH1 and IDH2 are both overexpressed (Table 2) and play the same role. IDH1 was also shown to promote tumor growth [50]. IDH2 expression levels in gastric and kidney cancer are opposite to those of other cancers. The role and mechanism of IDH2 may be reversed in different types of cancer, which needs further study.
In summary, we reported a growth promotion mechanism of wild-type IDH2 in lung cancer and identified the role of α-KG and HIF1α in IDH2wt's oncogenic function. These results suggested that treatment of lung cancer cells with AGI-6780 (a small molecule inhibitor of IDH2), PX-478 (an inhibitor of HIF1α), and replenishing cellular α-KG might be a new strategy for the treatment of lung cancer in the future.
Abbreviations
2-HG: 2-hydroxyglutarate; α-KG: alpha ketoglutarate; HIF1α: hypoxia inducible factor-1 alpha; IDH2: isocitrate dehydrogenase 2; NSCLC: non-small cell lung cancer; ROS: reactive oxygen species.
Supplementary Material
Supplementary figures, tables and experimental procedures.
Acknowledgements
The results published here are partly based upon data generated by The Cancer Genome Atlas pilot project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions that constitute the TCGA research network can be found at http://cancergenome.nih.gov.
Author contributions
Ya Cao supervised the study. Jiangjiang Li, Ya He, Zheqiong Tan, Jingchen Lu, Liling Li Namei Li, Yueshuo Li, performed experiments and Xiangjian Luo analyzed data. Xin Song, Jia Fan, Jian Zhou, Gao Qiang, ShuangJian Qiu and Weizhong Wu provided clinical samples and clinical information. Xiaolan Liu, Min Tang, Xinxian Weng, Wei Yi, Shuo You provided technical and material support. Jiangjiang Li wrote the manuscript. Ann M. Bode, Feng Shi and Longlong Xie revised the manuscript.
Financial support
This work was supported by The National Basic Research Program of China (No. 2011CB504305) and the Natural Science Foundation of Hunan Province (No. 14JJ3039).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Bray F, Center MM. et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
2. Giladi M, Weinberg U, Schneiderman RS. et al. Alternating electric fields (tumor-treating fields therapy) can improve chemotherapy treatment efficacy in non-small cell lung cancer both in vitro and in vivo. Semin Oncol. 2014;41(Suppl 6):S35-41
3. Sequist LV, Heist RS, Shaw AT. et al. Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann Oncol. 2011;22:2616-24
4. Reck M, Heigener DF, Mok T. et al. Management of non-small-cell lung cancer: recent developments. Lancet. 2013;382:709-19
5. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30
6. Chen W, Zheng R, Baade PD. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
7. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297-308
8. Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012;2:881-98
9. DeBerardinis RJ, Mancuso A, Daikhin E. et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345-50
10. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27-47
11. Xiao L, Hu ZY, Dong X. et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene. 2014;33:4568-78
12. Cao Y, DePinho RA, Ernst M. et al. Cancer research: past, present and future. Nat Rev Cancer. 2011;11:749-54
13. Tan Z, Luo X, Xiao L. et al. The Role of PGC1alpha in Cancer Metabolism and its Therapeutic Implications. Mol Cancer Ther. 2016;15:774-82
14. Yu X, Li W, Deng Q. et al. Neoalbaconol inhibits angiogenesis and tumor growth by suppressing EGFR-mediated VEGF production. Mol Carcinog. 2017;56:1414-26
15. Clark O, Yen K, Mellinghoff IK. Molecular Pathways: Isocitrate Dehydrogenase Mutations in Cancer. Clin Cancer Res. 2016
16. Flavahan WA, Drier Y, Liau BB. et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110-4
17. Ward PS, Patel J, Wise DR. et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225-34
18. Xu W, Yang H, Liu Y. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17-30
19. Matsumoto K, Obara N, Ema M. et al. Antitumor effects of 2-oxoglutarate through inhibition of angiogenesis in a murine tumor model. Cancer Sci. 2009;100:1639-47
20. Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12:9-22
21. Ryan HE, Poloni M, McNulty W. et al. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res. 2000;60:4010-5
22. Martinengo C, Poggio T, Menotti M. et al. ALK-dependent control of hypoxia-inducible factors mediates tumor growth and metastasis. Cancer Res. 2014;74:6094-106
23. Lin YJ, Shyu WC, Chang CW. et al. Tumor Hypoxia Regulates Forkhead Box C1 to Promote Lung Cancer Progression. Theranostics. 2017;7:1177-91
24. Wise DR, Ward PS, Shay JE. et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108:19611-6
25. Jiang L, Shestov AA, Swain P. et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature. 2016;532:255-8
26. Chen X, Xu W, Wang C. et al. The clinical significance of isocitrate dehydrogenase 2 in esophageal squamous cell carcinoma. Am J Cancer Res. 2017;7:700-14
27. Lian CG, Xu Y, Ceol C. et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150:1135-46
28. Liu WR, Tian MX, Jin L. et al. High expression of 5-hydroxymethylcytosine and isocitrate dehydrogenase 2 is associated with favorable prognosis after curative resection of hepatocellular carcinoma. J Exp Clin Cancer Res. 2014;33:32
29. Hu J, Deng X, Bian X. et al. The expression of functional chemokine receptor CXCR4 is associated with the metastatic potential of human nasopharyngeal carcinoma. Clin Cancer Res. 2005;11:4658-65
30. Liu S, Li H, Chen L. et al. (-)-Epigallocatechin-3-gallate inhibition of Epstein-Barr virus spontaneous lytic infection involves ERK1/2 and PI3-K/Akt signaling in EBV-positive cells. Carcinogenesis. 2013;34:627-37
31. You S, Li R, Park D. et al. Disruption of STAT3 by niclosamide reverses radioresistance of human lung cancer. Mol Cancer Ther. 2014;13:606-16
32. Gao J, Aksoy BA, Dogrusoz U. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
33. Cerami E, Gao J, Dogrusoz U. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
34. Rhodes DR, Yu J, Shanker K. et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1-6
35. Gyorffy B, Surowiak P, Budczies J. et al. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013;8:e82241
36. Aguirre-Gamboa R, Gomez-Rueda H, Martinez-Ledesma E. et al. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS One. 2013;8:e74250
37. Faubert B, Boily G, Izreig S. et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113-24
38. Terunuma A, Putluri N, Mishra P. et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J Clin Invest. 2014;124:398-412
39. Intlekofer AM, Dematteo RG, Venneti S. et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015;22:304-11
40. Wang F, Travins J, DeLaBarre B. et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622-6
41. Prensner JR, Chinnaiyan AM. Metabolism unhinged: IDH mutations in cancer. Nat Med. 2011;17:291-3
42. Li J, Lu J, He Y. et al. A new functional IDH2 genetic variant is associated with the risk of lung cancer. Mol Carcinog. 2017;56:1082-7
43. Yang Q, Liang X, Sun X. et al. AMPK/alpha-Ketoglutarate Axis Dynamically Mediates DNA Demethylation in the Prdm16 Promoter and Brown Adipogenesis. Cell Metab. 2016;24:542-54
44. Rossi G, Mengoli MC, Cavazza A. et al. Large cell carcinoma of the lung: clinically oriented classification integrating immunohistochemistry and molecular biology. Virchows Arch. 2014;464:61-8
45. Burris H MI, Maher E, Wen P, Beeram M, Touat M, Faris J, Azad N, Cloughesy T, Gore L, Trent J, Hoff Dv, Goldwasser M, Fan B, Liu H, Agresta S. The first reported results of AG-120, a first-in-class, potent inhibitor of the IDH1 mutant protein, in a Phase I study of patients with advanced IDH1-mutant solid tumors, including gliomas. Mol Cancer Ther. 2015:14 (12, Suppl 2):Abstract PL04-05
46. Dinardo C DS, Pollyea D, Stein E, Fathi A, Roboz G, Collins R, Swords R, Flinn I, Altman J, Tallman M, Kantarjian H, Derti A, Goldwasser M, Prahl M, Wu B, Yen K, Agresta S, Stone R. Molecular profiling and relationship with clinical response in patients with idh1 mutation-positive hematologic malignancies receiving AG-120, a first-in-class potent inhibitor of mutant IDH1, in addition to data from the completed dose escalation portion of the phase 1 study. Blood. 2015;126(23S):Abstract 1306
47. Fujii T, Khawaja MR, DiNardo CD. et al. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016;21:373-80
48. Mondesir J, Willekens C, Touat M. et al. IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives. J Blood Med. 2016;7:171-80
49. Cynober LA. The use of alpha-ketoglutarate salts in clinical nutrition and metabolic care. Curr Opin Clin Nutr Metab Care. 1999;2:33-7
50. Tan F, Jiang Y, Sun N. et al. Identification of isocitrate dehydrogenase 1 as a potential diagnostic and prognostic biomarker for non-small cell lung cancer by proteomic analysis. Mol Cell Proteomics. 2012;11:M111 008821
Author contact
![]() Corresponding author: Prof. Ya Cao, Key Laboratory of Carcinogenesis and Invasion, Chinese Ministry of Education, Xiangya Hospital, Central South University, Changsha 410078, China E-mail: ycao98sina.com. Telephone: +86-731-84805448
Corresponding author: Prof. Ya Cao, Key Laboratory of Carcinogenesis and Invasion, Chinese Ministry of Education, Xiangya Hospital, Central South University, Changsha 410078, China E-mail: ycao98sina.com. Telephone: +86-731-84805448