Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(15):4170-4180. doi:10.7150/thno.25798 This issue Cite
Research Paper
Enhanced autophagy contributes to protective effects of IL-22 against acetaminophen-induced liver injury
Ruidong Mo1,2#, Rongtao Lai1,2#, Jie Lu1,2#, Yan Zhuang1, Tianhui Zhou1,2, Shaowen Jiang1,2, Peipei Ren1,2, Ziqiang Li1,2, Zhujun Cao1,2, Yuhan Liu1,2, Lichang Chen1,2, Lifu Xiong1,2, Peng Wang1,2, Hui Wang1, Wei Cai1, Xiaogang Xiang1,2 ![]() , Shisan Bao3
, Shisan Bao3 ![]() , Qing Xie1,2
, Qing Xie1,2 ![]()
1. Department of Infectious Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
2. Translational Lab of Liver Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
3. Discipline of Pathology, School of Medical Sciences and Bosch Institute, Charles Perkins Centre, The University of Sydney, New South Wales 2006, Australia
#These authors contributed equally to this work.
Received 2018-2-28; Accepted 2018-7-4; Published 2018-7-30
Abstract

Acute or acute-on-chronic liver failure is a leading cause of death in liver diseases without effective treatment. Interleukin-22 (IL-22) is currently in clinical trials for the treatment of severe alcoholic hepatitis, but the underlying mechanisms remain to be explored. Autophagy plays a critical role in alleviating liver injury. The aim of the current study is to explore the role of autophagy in IL-22-mediated hepato-protective effect against acetaminophen (APAP)-induced liver injury.
Methods: A model of acute liver injury induced by APAP was used in vivo. IL-22 was administrated to the APAP-treated mice. Hepatocytes were pre-incubated with IL-22, followed by exposure to APAP for in vitro analyses.
Results: IL-22 administration significantly reduced serum ALT and AST, hepatic reactive oxygen species, and liver necrosis in APAP-challenged mice. APAP treatment increased hepatic autophagosomes, which was further intensified by IL-22 co-treatment. Hepatic LC3-II was moderately upregulated after APAP administration without obvious alteration of phosphorylation of AMP-activated kinase (p-AMPK). IL-22 pretreatment significantly upregulated hepatic LC3-II and p-AMPK in APAP-treated mice. IL-22 also alleviated APAP-induced cytotoxicity and upregulated LC3-II and p-AMPK expression in cultured hepatocytes treated with APAP in vitro. When p-AMPK was blocked with compound C (an AMPK inhibitor), IL-22-mediated LC3-II conversion and protection against APAP-induced cytotoxicity was weakened.
Conclusions: Enhanced AMPK-dependent autophagy contributes to protective effects of IL-22 against APAP-induced liver injury.
Keywords: acetaminophen, IL-22, autophagy, p62/SQSTM1, liver injury.
Introduction
Acetaminophen (APAP) is probably the most common analgesic and antipyretic over-the-counter drug used worldwide. APAP overdose is the leading cause of drug-induced acute liver failure [1]. APAP toxicity is due to the conversion of APAP to a reactive metabolite, N-acetyl-p-amino-benzoquinone imine (NAPQI) by CYP450 enzymes [2]. Though NAPQI could be detoxified by glutathione, excessive NAPQI depletes cellular glutathione and subsequently forms APAP protein adduct. APAP adduct interrupts mitochondrial respiratory chain complex, contributing to hepatocyte necrosis via the induction of mitochondrial oxidative stress [3].
Autophagy is involved in protein recycling, disposing of damaged organelles and denatured proteins through encapsulation in a double-membraned autophagosome, which subsequently fuses with lysosomes for proteolytic degradation [4]. When autophagy was enhanced, microtubule-associated protein 1 light chain 3 (LC3) was promoted to conjugate with phosphatidylethanolamine (PE). The PE-conjugated form of LC3 (LC3-II) translocates to the autophagosomal membrane, contributing to the formation of a double-membrane autophagosome [5]. The activity of autophagy is enhanced in low nutrient or inflammatory environments [6, 7] for maintaining cell homeostasis by selectively removing damaged or unwanted organelles and proteins. Autophagy protects the liver from acute injury in several acute liver injury animal models including ischemia-reperfusion liver injury [8] and APAP-induced liver injury [9, 10]. It is reported that inhibition of autophagy markedly exacerbated APAP-induced liver injury [11]. The protective function of autophagy in APAP-induced liver injury is mediated through removal of the APAP adduct [9, 10]. In addition, mitochondrial ROS generation, mitochondrial dysfunction, and inflammation play important roles in the pathogenesis of APAP-induced hepatocytes necrosis [3, 12]. Autophagy also plays a critical role in removing the damaged mitochondria, thereby alleviating APAP-induced cell injury [13].
Recently it has been reported that IL-10 promotes mitophagy, which eliminates dysfunctional mitochondria likely through a mammalian target of rapamycin inhibitor [14]. IL-22 is mainly secreted by Th17, Th22, γδ T cells, NK cells, neutrophils or group 3 innate lymphoid cells (ILC3) [15], as well as macrophages [16]. IL-22 protects against liver injury in a variety of mouse models including adenovirus-induced or T cell-mediated hepatitis [15, 17]. Furthermore, we have demonstrated previously that IL-22 protects the host from drug-induced hepatocellular injury [18] and may play a hepato-protective role in liver inflammation/fibrosis during chronic hepatitis B [19]. Because IL-22 has many beneficial effects in protecting against liver injury and promoting liver regeneration, and has minimal side effects due to restricted IL-22 receptor expression on epithelial cells [20], recombinant IL-22 protein is currently in clinical trials for the treatment of severe alcoholic hepatitis [21]. Recently, the phase I clinical trial of IL-22 (recombinant human IL-22 IgG2-Fc, F-652) has been performed. In the trial, F-652 is well-tolerated and demonstrated favorable pharmacokinetics and pharmacodynamics properties when intravenously administrated to healthy male volunteers [22].
Though the hepato-protective function of IL-22 is well-documented, the underlying mechanisms are not fully understood. It is reported that IL-22 alleviated oxidative and endoplasmic reticulum (ER) stress induced by palmitate through activation of autophagy in INS-1 cells [23]. However, it is unclear whether IL-22 exerts the hepato-protective effect through autophagy activation during APAP-induced liver injury. The present study investigated the hepato-protective mechanism of IL-22 in APAP overdose-induced liver injury models. Interestingly, autophagic activities were induced in APAP-treated mice and primary mouse hepatocytes/human non-tumor hepatic L02 cells. IL-22 pretreatment further enhanced autophagic activities to attenuate liver injury induced by APAP. These findings suggest a novel mechanism by which IL-22 protects against liver injury, which may provide useful information for both basic research and clinical practice in management of liver failure.
Methods
Animal model and experimental protocol
Male C57BL/6J mice (7 weeks old, weighing 18-22 g) were obtained from the Shanghai SLAC Laboratory Animal Company (Shanghai, China). The mice were kept under 12 h light/dark cycles specific pathogen-free conditions at 20-25 °C, with free access to water and standard chow. The animal experiments were approved by The Animal Ethics Committee of Ruijin Hospital, Shanghai Jiao Tong University School of Medicine and were carried out according to the institutional guidelines.
Mice were randomly assigned to three groups: 1) Vehicle (normal saline, NS), 2) APAP (400 mg/kg in normal saline), 3) IL-22-Fc (1 mg/kg in PBS) plus APAP (400 mg/kg in normal saline) [24]. A APAP-induced liver injury was developed as described previously [24]. Briefly, these mice were injected intraperitoneally (i.p.) with 400 mg/kg APAP or vehicle (NS) following overnight fasting. The mice in group 3 were pretreated with 1 mg/kg recombinant IL-22-Fc i.p. (Generon Corporation, Shanghai, China) 2 h prior to APAP challenge. The group 3 represented the procedure of IL-22 plus APAP treatment, unless otherwise indicated. The sham-treated mice were i.p. injected with an equivalent volume of PBS. To investigate IL-22 therapeutic effect for APAP intoxication, at 2 h post-APAP treatment, mice were administrated 1 mg/kg IL-22 or an equal volume of PBS. Serum and liver were collected at various time points.
Additional protocols and procedures are described in Supplementary Material.
Results
IL-22 attenuates APAP-induced hepatotoxicity in mice
Serum ALT and AST levels and liver histology were determined to evaluate the severity of liver injury. ALT and AST increased gradually and peaked in APAP-challenged mice at 24 h post APAP injection (Figure S1A-B) compared to the vehicle treatment. Elevation of serum ALT and AST levels were ameliorated in IL-22 plus APAP-treated mice compared to APAP-challenged mice, especially at 24 h post-APAP administration (Figure 1A-B).
IL-22 pretreatment protects mice from APAP-induced liver injury. (A-B) Serum ALT and AST from normal control (normal saline, NS), APAP-challenged and IL-22 plus APAP-treated mice at 6 h and 24 h. (C-F) Representative liver sections (H&E staining) from three groups after different treatments as indicated (D-F). Necrotic areas in liver sections were quantified (C). Data are expressed as mean ± SEM; p value was calculated by un-paired Student's t test in (A-C), *p < 0.05 and ***p < 0.001.
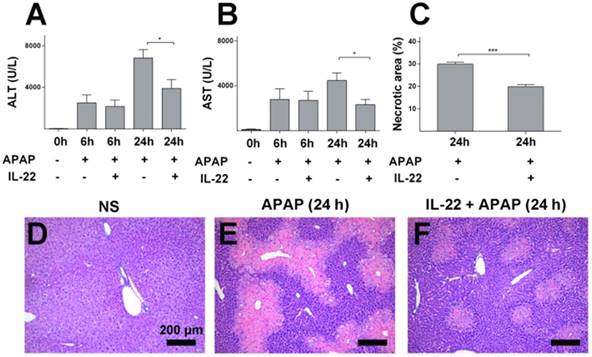
Histopathology demonstrated obvious centrilobular necrosis in livers from the APAP-challenged mice in a time-dependent manner, consistent with the serum ALT/AST alteration (Figure S1C-E). The areas of centrilobular necrosis were significantly reduced in IL-22 pretreatment plus APAP-treated mice (Figure 1C-F). The potential therapeutic effect of IL-22 was determined by adding the post-treatment with IL-22 approach. Though IL-22 post-treatment (2 h after APAP) mediated a moderate reduction in serum ALT and AST compared to APAP-challenged mice, no significant difference was observed between the two groups (Figure S2).
IL-22 alleviates APAP-induced hepatic oxidative stress and reduces mRNA expression of hepatic inflammatory mediators without alteration of hepatic APAP metabolism
Intracellular ROS was increased significantly in the APAP-challenged mice compared to vehicle treatment. However, intracellular ROS was dramatically decreased in IL-22 plus APAP-treated mice compared to APAP-challenged at 24 h (Figure 2A).
IL-22 attenuates APAP-induced hepatic oxidative stress and inflammation. (A) Oxidative stress is indicated in liver sections from three experimental groups at 24 h post-APAP (original magnification 100×) by histological ROS staining. Yellow arrows denote the ROS-positive areas in mice livers. (B) Representative images of TUNEL-stained liver sections from the three experimental groups indicated in (A). (C) Hepatic IL-6, IL-1β and TNF mRNA expressions were detected at 0, 6 and 24 h in mice from the three experimental groups, as indicated. (D) Serum levels of IL-6, TNF and IL-22 were measured by CBA (IL-6, TNF) and ELISA (IL-22). Data are presented as means ± SEM; p value was calculated by Kruskal-Wallis test (C) and one-way ANOVA (D), *p < 0.05, **p < 0.01 and ***p < 0.001.
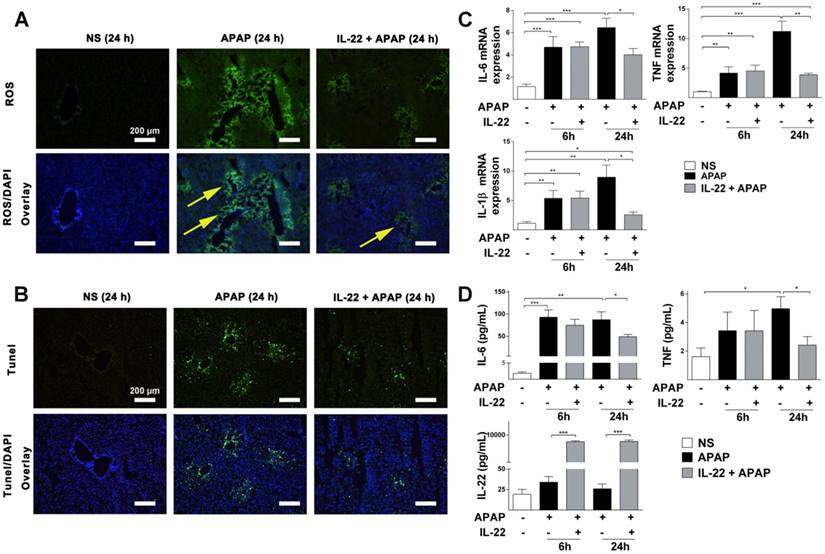
To determine whether the protective role of IL-22 against APAP-induced hepatotoxicity was due to altered APAP metabolism, hepatic CYP2E1 was determined in the different treatment groups. Comparable hepatic CYP2E1 expression was detected in the APAP group and IL-22 plus APAP group (Figure S3A). Hepatic glutathione (GSH) levels were depleted significantly at early stage (2 h), and recovered gradually at later stages (12 h and 24 h) (Figure S3B). Our data are in line with previous studies, showing similar patterns of GSH changes following APAP challenge [25-27]. There was no significant difference in the recovery of hepatic GSH levels between APAP-challenged and IL-22 plus APAP-treated mice (Figure S3C), suggesting that IL-22 pretreatment did not alter hepatic APAP metabolism.
Furthermore, hepatocyte proliferation was also determined in mice with different treatments. Hepatic Ki-67+ and PCNA+ cells were increased in both APAP-challenged and IL-22 plus APAP-treated mice compared to the control mice. Interestingly, hepatic Ki-67+ and PCNA+ cells were not increased significantly in the IL-22 plus APAP treatment group compared to the APAP-challenged group (Figure S4). It was speculated that in the IL-22 plus APAP treatment group, less hepatic necrosis appeared due to protection by IL-22, which led to less hepatic proliferation, reducing the difference of hepatic proliferation between the IL-22 plus APAP treatment group and the APAP-challenged group.
TUNEL staining was significantly increased in APAP-challenged mice compared to vehicle-treated mice at 24 h (Figure S5 and Figure 2B). However, TUNEL staining was decreased in IL-22 plus APAP-treated mice compared to APAP-challenged mice at 24 h (Figure 2B).
The mRNA expressions of inflammatory cytokines (IL-6, IL-1β and TNF) were upregulated in APAP-challenged mice at 6 and 24 h. However, in IL-22 plus APAP-treated mice, hepatic mRNA expressions of IL-6, IL-1β and TNF were significantly decreased compared to those of APAP-challenged mice at 24 h (Figure 2C).
Serum protein levels of IL-6 and TNF were significantly increased in APAP-challenged mice compared to vehicle-treated at 24 h (Figure 2D). Serum levels of IL-6 and TNF in IL-22 plus APAP-treated mice were down-regulated compared to those of the APAP-challenged group (Figure 2D). Serum IL-22 was upregulated significantly in IL-22 plus APAP-treated mice compared to APAP or vehicle treatment at both 6 and 24 h (Figure 2D). In addition, hepatic endogenous IL-22 mRNA was detected. Hepatic IL-22 mRNA expression was significantly increased in mice challenged with APAP compared to control group, consistent with Kleinschmidt et al.'s report that hepatic IL-22 mRNA expression was increased significantly 3 h and 6 h after APAP challenge [28]. More interestingly, the expression of IL-22 mRNA in liver was decreased with IL-22 plus APAP treatment compared to that of the APAP-challenged group at 24 h post-APAP (Figure S6).
Divergent expression of hepatic transcriptomes in vehicle-, APAP- and IL-22 plus APAP-treated mice
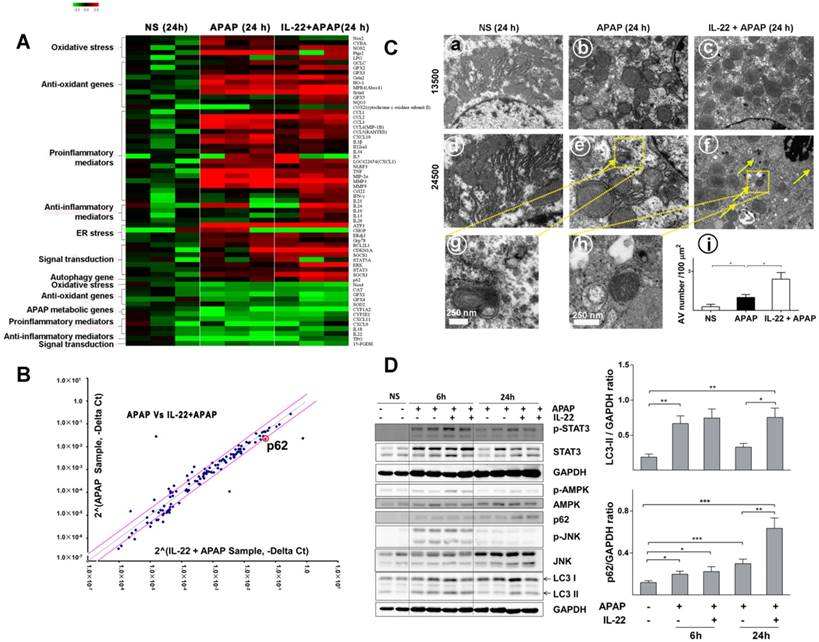
To determine the alteration of hepatic transcriptomes following IL-22 pretreatment, mRNA expression profile was detected in vehicle-treated, APAP-challenged and IL-22 plus APAP-treated mice groups at 24 h using PCR array. Hepatic mRNA levels of oxidative stress and inflammatory-related genes were upregulated in APAP-challenged compared to vehicle-treated groups (Figure 3A). The mRNA levels of oxidative stress and inflammatory mediators in IL-22 plus APAP-treated mice were down-regulated compared to those of the APAP-challenge alone group. In addition, the expression levels of anti-oxidant-related genes in IL-22 plus APAP-treated mice were upregulated compared to those of the APAP-challenged alone group (Figure 3A). Furthermore, the mRNA level of autophagy-associated gene p62 (SQSTM1) was increased about 2-fold in IL-22 plus APAP-treated mice compared to APAP-challenged mice (Figure 3B).
Divergent transcriptome expression patterns of hepatic oxidative stress, inflammation and autophagy genes in vehicle-, APAP- or IL-22 plus APAP-treated mice. (A) Heatmap displaying the relative expression of oxidative stress-, inflammation- and autophagy-related genes in normal control, APAP-treated and IL-22 plus APAP-treated groups (n = 3 per group) at 24 h. (B) Relative gene expression profiles between APAP-treated vs. IL-22 plus APAP-treated mice (n = 3). The red lines indicate positive or negative fold changes (upregulated 2-fold or down-regulated 0.5-fold) for selected genes in APAP- vs. IL-22 plus APAP-treated mice. (C) Mice liver samples were analyzed by TEM. (a, d) Mice treated with normal saline (NS); (b, e, g) mice treated with APAP; (c, f, h) mice treated with IL-22 plus APAP; (i) the number of autophagosomes in hepatocytes per 100 μm2 were compared among three groups. Yellow arrows indicate autophagosomes. (D) The protein expressions of p-STAT3, STAT3, p-AMPK, AMPK, p62, p-JNK, JNK and LC3-I/II in livers from normal control (NS), APAP (400 mg/kg)-treated, and IL-22 plus APAP-treated mice were detected at 6 and 24 h post-APAP by western blot. GAPDH served as a loading control. Quantification of the LC3-II and p62 to GAPDH expression ratios. Data are presented as means ± SEM; p value was calculated by Kruskal-Wallis test (C-D), *p < 0.05, **p < 0.01 and ***p < 0.001.
Enhanced hepatic autophagy is observed in IL-22-pretreated mice
Quantitative RT-PCR confirmed that p62 mRNA expression was significantly upregulated in IL-22-pretreated mice compared to APAP-challenged mice (Figure S7). Electron microscopy revealed that hepatic autophagosomes were increased following APAP challenge (Figure 3C). In addition, more hepatic autophagosomes were formed in IL-22 plus APAP-treated mice compared to APAP-challenged mice (Figure 3C).
IL-22 ameliorates APAP-induced liver injury and activates AMPK-dependent autophagy
To further understand the mechanism by which IL-22 activates autophagy, we examined AMPK, a representative upstream effector of autophagy. Hepatic AMPK phosphorylation (p-AMPK) was upregulated at both 6 and 24 h in IL-22 plus APAP-treated mice compared to APAP-challenged mice (Figure 3D). STAT3, which is widely accepted as a major downstream effector of the IL-22 signaling pathway [17, 29], was notably phosphorylated at 6 and 24 h in IL-22 plus APAP-treated mice (Figure 3D). Hepatic microtubule-associated protein light chain 3-II (LC3-II) was increased at 6 h following APAP challenge (Figure 3D). In addition, the expressions of hepatic endogenous LC3-II and p62 were enhanced in IL-22 plus APAP-treated mice compared to APAP-challenged mice, especially at 24 h (Figure 3D). Moreover, p-JNK expression was moderately decreased in IL-22 plus APAP-treated mice compared to APAP-challenged mice at 24 h (Figure 3D).
Based on the above results, the extent of hepatic autophagosomes formation was further determined by immunohistochemistry. Cleaved LC3 was tested in the livers from IL-22-, APAP-, or IL-22 plus APAP-treated mice. Hepatic cleaved LC3 expression was increased with IL-22 plus APAP treatment compared with APAP challenge at 6 h (Figure S8).
IL-22 attenuates APAP-induced cytotoxicity and enhances autophagy in L02 cells treated with APAP in vitro
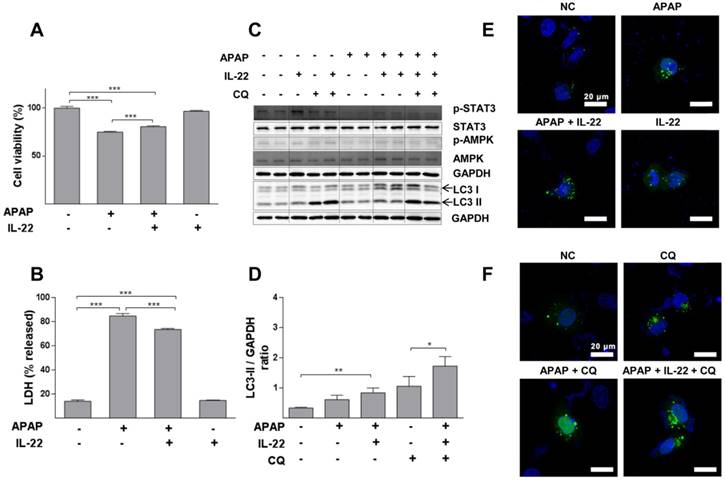
Cell viability was decreased in a dose-dependent way in L02 cells with APAP exposure (Figure S9). APAP significantly decreased cell viability, which was partially restored with IL-22 pretreatment (Figure 4A). Moreover, LDH (a necrotic marker) release was significantly increased in culture medium of L02 cells exposed to APAP, which was inhibited with IL-22 pretreatment (Figure 4B). The cell viability and the LDH levels in culture medium from L02 cells under IL-22 treatment alone were similar to those of the control, suggesting that IL-22 did not exert a toxic effect on L02 cells (Figure 4A-B). LC3-II expression was moderately increased in L02 cells exposed to APAP. LC3-II expression was further increased in IL-22 plus APAP-treated L02 cells compared to APAP-treated cells. p-AMPK protein was upregulated with IL-22 plus APAP treatment compared to APAP alone in L02 cells. IL-22 plus APAP-mediated LC3-II induction was further increased in the presence of chloroquine (Figure 4C-D). Moreover, GFP-LC3 puncta formation was increased following APAP exposure in GFP-LC3-transfected L02 cells. Increased GFP-LC3 puncta formation was observed with IL-22 plus APAP treatment compared to APAP alone in L02 cells (Figure 4E). After treating with chloroquine, GFP-LC3 puncta formation in L02 cells was further augmented with IL-22 plus APAP treatment when compared to APAP treatment (Figure 4F). IL-22 treatment alone upregulated the expression of p62 in L02 cells (Figure S10).
IL-22 alleviates APAP-induced cytotoxicity and enhances autophagy in L02 cells treated with APAP. (A) L02 cells were treated with IL-22 (400 ng/mL) for 1 h, followed by treatment with APAP (5 mM). The cell viability was measured at 24 h after APAP treatment by MTT. (B) LDH release in the culture medium was measured when L02 cells were treated as indicated in (A). (C) L02 cells were incubated with IL-22 (400 ng/mL) for 1 h, followed by treatment with APAP (5 mM) for 6 h. Immunoblot determined p-STAT3, STAT3, p-AMPK, AMPK and LC3-I/II expressions in L02 cells. GAPDH served as a loading control. (D) Quantification of the LC3-II to GAPDH expression ratio. (E) L02 cells were transfected with GFP-LC3, exposed to IL-22 (400 ng/mL) or not for 1 h, followed by treatment with APAP (5 mM) or not for 6 h. (F) L02 cells were treated with IL-22 (400 ng/mL) for 1 h, followed by treatment with APAP (5 mM) in the presence of chloroquine (CQ) (20 μM) or not for 6 h. Distribution of GFP-LC3 puncta in L02 cells was evaluated using a Zeiss fluorescence microscope. Data are presented as means ± SEM; p value was calculated by Kruskal-Wallis test (A-B, D), *p < 0.05, **p < 0.01 and ***p < 0.001.
IL-22 promotes autophagy by an AMPK-dependent pathway in primary mouse hepatocytes
The expression of p-AMPK was increased in primary mouse hepatocytes after treating with IL-22. p-AMPK was moderately decreased for 6 h after APAP challenge, which was partly reversed by IL-22 pretreatment (Figure 5A-B). LC3-II expression was also increased with IL-22 plus APAP treatment compared to APAP treatment alone in primary mouse hepatocytes. In addition, IL-22 plus APAP-induced LC3-II expression was further enhanced in the presence of chloroquine (Figure 5A-B).
AMPK pathway is involved in IL-22-regulated autophagy in primary mouse hepatocytes treated with APAP. (A) Cultured primary mouse hepatocytes were treated with IL-22 (400 ng/mL) for 0.5, 1, 2 and 6 h in lane 2, 3 4 and 5 respectively. The other cultured primary mouse hepatocytes were treated with IL-22 (400 ng/mL) or not for 1 h, followed by addition of APAP (5 mM) for 6 h in the presence or absence of chloroquine (CQ) as indicated. Western blot analysis showed cellular protein expressions of p-STAT3, STAT3, p-AMPK, AMPK and LC3-I/II. (B) Quantification of the LC3-II to GAPDH expression ratio. (C) Primary mouse hepatocytes were pretreated with compound C (4 μM) for 6 h, incubated with IL-22 (400 ng/mL) for another 1 h, then APAP (5 mM) for 6 h. Western blot analysis showing cellular protein expressions of p-STAT3, STAT3, p-AMPK, AMPK and LC3-I/II. GAPDH served as a loading control. (D) Quantification of the p-AMPK to GAPDH expression ratio. (E) Quantification of the LC3 II to GAPDH expression ratio. Data are presented as means ± SEM; p value was calculated by one-way ANOVA (B, D-E). *p < 0.05, **p < 0.01 and ***p < 0.001.
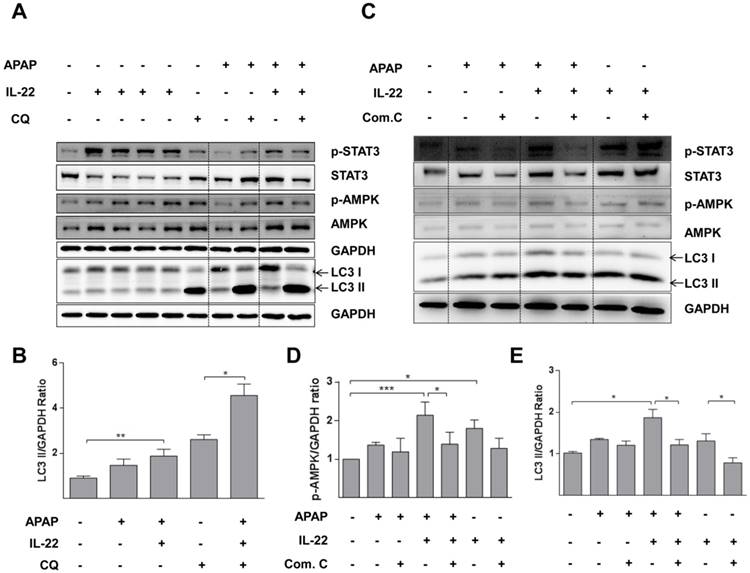
To confirm the importance of p-AMPK in IL-22-promoting autophagy, hepatocytes were pretreated with compound C, an AMPK inhibitor in vitro. In hepatocytes treated with IL-22 plus APAP, the enhancement of LC3-II formation was abolished by pretreatment with compound C (Figure 5C-E).
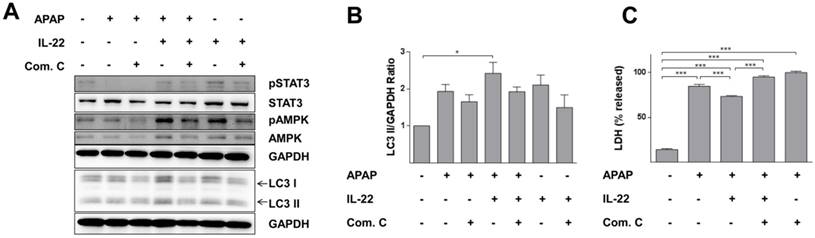
Protective effect of IL-22 against APAP-induced cell injury is associated with AMPK pathway-dependent autophagy in L02 cells
LC3-II expression was further increased with IL-22 plus APAP treatment compared to APAP exposure alone in L02 cells. Phosphorylation of AMPK was enhanced with IL-22 plus APAP treatment compared to APAP alone. However, pretreatment with AMPK inhibitor (compound C) decreased p-AMPK up-regulation, as well as the formation of LC3-II induced by IL-22 plus APAP (Figure 6A-B). Importantly, the protective effect of IL-22 against APAP-induced cell damage was also abolished by compound C (Figure 6C).
AMPK-dependent pathway activation is necessary for IL-22-mediated autophagy and cytoprotection in L02 cells treated with APAP. (A) L02 cells were pretreated with compound C (4 μM) for 6 h, incubated with IL-22 (400 ng/mL) for another 1 h, followed by treatment with APAP (5 mM) for 6 h. Western blot analysis showed cellular protein expressions of p-STAT3, STAT3, p-AMPK, AMPK, and LC3-I/II. GAPDH served as a loading control. (B) Quantification of the LC3-II to GAPDH expression ratio. (C) L02 cells were pretreated with compound C (4 μM) for 6 h, incubated with IL-22 (400 ng/mL) for another 1 h, followed by treatment with APAP (5 mM) for 24 h. Then, LDH levels released in culture medium from L02 cells were measured. Data are presented as means ± SEM; p value was calculated by one-way ANOVA (B-C), *p < 0.05, ***p < 0.001.
Discussion
APAP overdose-induced liver injury is probably one of the most common causes of drug-induced acute liver failure [30] without effective medication. N-acetylcysteine (NAC) currently is the only drug approved for treatment of APAP overdose-induced liver injury via suppressing ROS at a relatively early stage [31]. IL-22 has been demonstrated to alleviate APAP-induced liver injury; however, the underlying mechanisms remain incompletely understood. The present study aimed to investigate whether IL-22 plays a hepato-protective effect on APAP-induced liver injury by regulation of hepatic autophagy. We hypothesized that upregulated AMPK expression and promoted autophagy by IL-22 might be a potential therapeutic strategy for APAP-induced liver injury. In this study, it was demonstrated that the AMPK pathway-dependent autophagy induced by IL-22 protected against APAP-induced hepatotoxicity.
IL-22 also promotes tissue repair by directly activating anti-apoptotic, proliferative programs and antioxidants in various types of cells, including hepatocytes [17, 32]. Previous studies have indicated that IL-22 plays a protective role in preventing hepatocellular damage induced by alcohol [33], concanavalin A (Con A)[17] , or carbon tetrachloride (CCl4) [34]. In addition, though recombinant IL-22 has been reported to have a beneficial role in acute liver injury induced by APAP [35], whether autophagy was involved in the hepato-protective effect of IL-22 against APAP-induced hepatic injury has not been assessed. In addition to the above-mentioned alternative protective mechanisms—including pro-survival, anti-inflammation and metabolic regulation [32, 35]—the present study suggests that activation of autophagy contributes to the protective effect of IL-22 against APAP-induced hepatotoxicity.
Mitochondrial dysfunction and excessive ROS initiate APAP-induced liver injury [25], subsequently triggering cell necrosis. The present study demonstrated that IL-22 pretreatment attenuated APAP-induced liver injury, showing less hepatocyte necrosis and apoptosis, consistent with previous studies [24, 35]. As expected, APAP-induced serum/hepatic pro-inflammatory cytokines and excessive ROS were ameliorated following IL-22 pretreatment. Consistent with our studies, Ju and colleagues have reported that LiposIA (liposome-based nanoparticle)-delivered IL-22 improves the protective efficacy of IL-22 against APAP-induced liver injury, accompanied with inhibited generation of ROS and TNF, and prevented mitochondrial dysfunction [36]. Hasnain et al.'s study has shown that IL-22 plays a role in relieving cytokines- or glucolipotoxicity-mediated oxidative stress in pancreatic islets [32]. However, Scheierman et al. reported [35] that inflammatory cytokines are not affected by IL-22 in endotoxemic mice. The discrepancy between Scheierman et al.'s study and ours might due to the different animal models, i.e., local liver injury in our model vs. a more systemic injury in endotoxemic mice. In addition, different species and/or experimental environment may also contribute to such difference.
Hepatic IL-22 mRNA expression was decreased in the IL-22 pretreatment group. We speculated the following reasons may account for the decrease of IL-22 mRNA: Firstly, hepatic IL-22 mRNA expression may be negatively regulated by the high amount of circulating IL-22. Secondly, since IL-22 treatment has a protective effect on APAP-induced liver injury, the alleviated liver injury may affect the expression pattern of hepatic IL-22 mRNA in mice exposed to APAP.
Autophagy, as an adaptive process, is involved in protein recycling and disposal of damaged organelles and proteins for maintaining liver homeostasis [8]. Recent studies have suggested that autophagy, serving as an adaptive mechanism, is activated in response to APAP treatment to relieve liver injury induced by APAP [9]. Autophagy plays a protective role by removing damaged mitochondria and APAP-adducted proteins [10, 11]. Such findings support our study, showing that autophagy enhancement (autophagosomes formation and increased expression of LC3-II [37]) has been detected following APAP challenge.
Autophagy has been considered a likely therapy target by different chemical compounds to protect against APAP-induced liver injury [10, 38]. It is reported that autophagy is activated by IL-22 to alleviate palmitate-induced oxidative and ER stress in INS-1 cells [23]. In addition, mycobacterium tuberculosis intracellular growth is suppressed by IL-22 via enhancing phagolysosomal fusion through down-regulating Rab14 and enhancing Rab7 production (Rab14, Rab7, member RAS oncogene family) [39]. Importantly, Rab7 is required for completing autophagic flux in mouse embryonic fibroblasts [40]. Thus, it is speculated that autophagy is involved in the biological effect of IL-22.
Previous studies have shown that autophagy plays a role in relieving oxidative stress in severe acute pancreatitis [41] and hepatic ER stress in mice [42]. P62, an autophagy-related gene involved in selective autophagy [43], interacts with LC3 and acts as a cargo adapter to deliver the damaged mitochondria into autophagosomes. P62 knock-down exacerbates APAP-induced hepatocytes necrosis [9]. In the present study, PCR array and RT-qPCR results suggested that p62 was upregulated in mice pretreated with IL-22. Therefore, we investigated the protective role of IL-22-mediated autophagy in APAP-induced hepatotoxicity. IL-22 pretreatment further enhanced LC3-II protein expression and formation of autophagosomes in vivo, which indicated that hepatic autophagy was enhanced in the IL-22 pre-treated group. The up-regulation of LC3-II by IL-22 administration in APAP-treated L02 cells failed to reach statistical significance, suggesting there is the possible involvement of other cells in IL-22-induced up-regulation of autophagy in the hepatocytes of APAP-treated mice. Autophagy attenuates cellular damage via selectively removing damaged mitochondria [44], and dysregulation of autophagy leads to increase of oxidative stress in APAP-induced liver injury [10]. Such finding is in line with our current study, showing APAP-induced ROS was attenuated by IL-22 pretreatment. Chloroquine, a potent inhibitor of autophagosome-lysosome fusion (by increasing lysosomal pH), is often used to evaluate the function of autophagy or autophagic flux. Following treatment with chloroquine, LC3-II accumulation is due to blockage of degradation of LC3-II. Thus, the increase of LC3-II following treatment of chloroquine compared with the mock treatment would reflect the amount of LC3-II delivered to lysosomes for degradation (autophagic flux). LC3-II was increased further in IL-22 plus APAP-treated cells in the presence of chloroquine compared to chloroquine treatment alone (Figure 4C), suggesting that IL-22 promotes autophagic flux of APAP-treated hepatocytes. Similarly, suppression of autophagosome-lysosome fusion by chloroquine remarkably increased GFP-LC3 puncta induced by IL-22 pretreatment, indicating that IL-22 enhanced autophagic flux rather than impaired autophagosomes clearance. It was intriguing that IL-22 alone plus chloroquine did not further increase LC3-II levels in L02 cells obviously. It was speculated that IL-22 would not obviously promote autophagy of hepatocytes in the absence of APAP, which might be due to the function of APAP as an autophagy initiator. These results suggest that enhanced autophagy of APAP-treated hepatocytes by IL-22 may require the existence of APAP.
Furthermore, our current data demonstrated that IL-22 pretreatment upregulated autophagy, but activation of autophagy was suppressed by AMPK inhibitor compound C. IL-22 was not able to attenuate APAP-induced cytotoxicity when hepatocytes were pretreated with compound C (accompanied with suppressed autophagy). These results suggest that inhibition of autophagy abolishes the protective effects of IL-22 against APAP-induced cytotoxicity. Thus, our data implied that enhanced autophagy was involved in the IL-22-protective effect against APAP-induced liver injury.
AMPK, an important energy sensor, can regulate cellular metabolism to maintain energy homeostasis [45]. Importantly, AMPK also plays a cytoprotective role in hepatocytes by promoting cell survival, alleviating mitochondrial injury and activating autophagy [46]. In the present study, IL-22-induced autophagy activation and protective effect could be abolished by compound C, as shown by the LDH release experiment. Taken together, our data implied that AMPK pathway-dependent autophagy was involved in the IL-22-mediated protective effect on APAP-induced liver injury. In addition to the enhanced AMPK-dependent autophagy involved in IL-22-mediated hepatic protection, other effects upon AMPK activation may also contribute to the hepatic protection. Activation of AMPK-mediated protective mechanisms may be involved in a variety of growth, metabolism, and cell-polarity, as well as mitochondrial homeostasis processes [46-49].
In the present study, it is also worth mentioning that the expression of p62 increased with IL-22 pretreatment. It is generally accepted that p62 accumulation suggests autophagy impairment, and the decrease of p62 indicates enhanced autophagic process. On the other hand, p62 protein level is not only regulated by degradation but also affected by regulation of p62 mRNA expression [50]. P62 expression would be upregulated in circumstances where there is an increase of autophagic flux [51, 52]. This is supported in cardiac proteinopathy, where p62 protects cardiomyocytes against proteotoxic stress by promoting aggresome formation and autophagy activation [52]. We acknowledge that there are some limitations to our study. Post-treatment with IL-22 tended to alleviate the liver injury induced by APAP, but didn't reach statistical significance. This result was also consistent with Scheiermann et al.'s study [35]. The precise role of IL-22 as a therapeutic agent is still being determined.
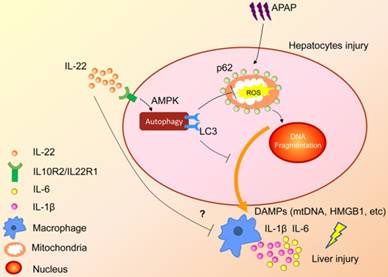
In summary, the present study demonstrated that upregulated hepatocellular autophagy induced by IL-22 contributed to the alleviation of APAP-induced liver injury. The hepato-protective role of IL-22 against APAP-induced liver injury is related to AMPK pathway-dependent autophagy activation, which inhibits the expression of oxidative and inflammatory mediators (Figure 7). Our data may provide some constructive insights into specific targeting of APAP-induced liver injury.
A proposed model for enhanced autophagy involving the protective role of IL-22 against APAP-induced hepatotoxicity. During APAP-induced liver injury, excess mitochondrial ROS production is caused when NAPQI binds to mitochondrial proteins, subsequently resulting in loss of the membrane potential and release of intermembrane proteins. The intermembrane proteins translocate to the nucleus and may participate in APAP-induced nuclear fragmentation. IL-22 activates p-AMPK pathway and may promote autophagic removal of damaged mitochondrion and protects against APAP-induced liver injury. The dotted lines indicate the underlying alternative mechanisms that were not investigated in the current study. DAMPs: damage associated molecular patterns; HMGB1: high mobility group box 1 protein; mtDNA: mitochondrial DNA; ROS: reactive oxygen species.
Abbreviations
APAP: acetaminophen; APAP-AD: acetaminophen protein adduct; AMPK: AMP-activated kinase; CYP2E1: cytochrome P450 2E1; JNK: c-Jun N-terminal kinase; LC3-II: microtubule-associated protein light chain 3-II; LDH: lactate dehydrogenase; NAPQI: n-acetyl-p-amino-benzoquinone imine; ROS: reactive oxygen species; TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling.
Supplementary Material
Supplementary materials and methods, table and figures.
Acknowledgements
The authors thank Dr Jia-ying Fan (Laboratory of Morphology, Shanghai Jiao Tong University School of Medicine) for assistance with frozen sectioning. This study was supported by the National Natural Science Foundation of China (No. 81570535, No. 81770587, No. 81600463), The National Key Programs on Infectious Diseases of China (2017ZX10202202-005-004, 2017ZX10203201-008, 2018ZX09206005-004), Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support (20172008), The Shanghai Three-Year Plan of the Clinical Skills and Innovations (16CR1002A), National Clinical Key Specialty Construction Project of China (Infectious Diseases) and The Three-year Action Plan of Public Health in Shanghai (15GW2K0102).
Author Contributions
R. Mo, R. Lai, J. Lu, X. Xiang, S. Bao and Q. Xie designed the study and drafted the manuscript. R. Mo, R. Lai, J. Lu, Y. Zhuang, T. Zhou, S. Jiang, Z. Cao, L. Chen, L. Xiong performed animal experiments. Isolation of primary mouse hepatocytes and in vitro cytotoxic assays were performed by R. Mo, Y. Liu, P. Ren, Z. Li, P Wang. All other experiments were performed by R. Mo, R. Lai, J. Lu, Y. Zhuang, T. Zhou and S. Jiang. R. Mo, R. Lai, J. Lu, X. Xiang, S. Bao, Q. Xie, H. Wang, W. Cai interpreted the data.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet. 2010;376:190-201
2. Nelson SD. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin Liver Dis. 1990;10:267-78
3. Han D, Dara L, Win S, Than TA, Yuan L, Abbasi SQ. et al. Regulation of drug-induced liver injury by signal transduction pathways: Critical role of mitochondria. Trends Pharmacol Sci. 2013;34:243-53
4. Tanida I. Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal. 2011;14:2201-14
5. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T. et al. Lc3, a mammalian homologue of yeast apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720-8
6. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W. et al. Phosphorylation of ulk1 (hatg1) by amp-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456-61
7. Whelan KA, Merves JF, Giroux V, Tanaka K, Guo A, Chandramouleeswaran PM. et al. Autophagy mediates epithelial cytoprotection in eosinophilic oesophagitis. Gut. 2017;66:1197-207
8. Rautou PE, Mansouri A, Lebrec D, Durand F, Valla D, Moreau R. Autophagy in liver diseases. J Hepatol. 2010;53:1123-34
9. Ni HM, McGill MR, Chao X, Du K, Williams JA, Xie Y. et al. Removal of acetaminophen protein adducts by autophagy protects against acetaminophen-induced liver injury in mice. J Hepatol. 2016;65:354-62
10. Lin Z, Wu F, Lin S, Pan X, Jin L, Lu T. et al. Adiponectin protects against acetaminophen-induced mitochondrial dysfunction and acute liver injury by promoting autophagy in mice. J Hepatol. 2014;61:825-31
11. Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55:222-32
12. Win S, Than TA, Min RW, Aghajan M, Kaplowitz N. C-jun n-terminal kinase mediates mouse liver injury through a novel sab (sh3bp5)-dependent pathway leading to inactivation of intramitochondrial src. Hepatology. 2016;63:1987-2003
13. Williams JA, Ni HM, Haynes A, Manley S, Li Y, Jaeschke H. et al. Chronic deletion and acute knockdown of parkin have differential responses to acetaminophen-induced mitophagy and liver injury in mice. J Biol Chem. 2015;290:10934-46
14. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of il-10 mediated by metabolic reprogramming of macrophages. Science. 2017;356:513-9
15. Jie Z, Liang Y, Yi P, Tang H, Soong L, Cong Y. et al. Retinoic acid regulates immune responses by promoting il-22 and modulating s100 proteins in viral hepatitis. J Immunol. 2017;198:3448-60
16. Liu Y, Verma VK, Malhi H, Gores GJ, Kamath PS, Sanyal A. et al. Lipopolysaccharide downregulates macrophage-derived il-22 to modulate alcohol-induced hepatocyte cell death. Am J Physiol Cell Physiol. 2017;313:C305-C13
17. Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (il-22) plays a protective role in t cell-mediated murine hepatitis: Il-22 is a survival factor for hepatocytes via stat3 activation. Hepatology. 2004;39:1332-42
18. Lai R, Xiang X, Mo R, Bao R, Wang P, Guo S. et al. Protective effect of th22 cells and intrahepatic il-22 in drug induced hepatocellular injury. J Hepatol. 2015;63:148-55
19. Xiang X, Gui H, King NJ, Cole L, Wang H, Xie Q. et al. Il-22 and non-elr-cxc chemokine expression in chronic hepatitis b virus-infected liver. Immunol Cell Biol. 2012;90:611-9
20. Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the il-22-il-22r1 system. Nature reviews Drug discovery. 2014;13:21-38
21. Gao B, Shah VH. Combination therapy: New hope for alcoholic hepatitis? Clin Res Hepatol Gastroenterol. 2015;39(Suppl 1):S7-S11
22. Tang KY, Lickliter J, Huang ZH, Xian ZS, Chen HY, Huang C. et al. Safety, pharmacokinetics, and biomarkers of f-652, a recombinant human interleukin-22 dimer, in healthy subjects. Cell Mol Immunol. 2018
23. Hu M, Yang S, Yang L, Cheng Y, Zhang H. Interleukin-22 alleviated palmitate-induced endoplasmic reticulum stress in ins-1 cells through activation of autophagy. PloS one. 2016;11:e0146818
24. Feng D, Wang Y, Wang H, Weng H, Kong X, Martin-Murphy BV. et al. Acute and chronic effects of il-22 on acetaminophen-induced liver injury. J Immunol. 2014;193:2512-8
25. Furuta K, Yoshida Y, Ogura S, Kurahashi T, Kizu T, Maeda S. et al. Gab1 adaptor protein acts as a gatekeeper to balance hepatocyte death and proliferation during acetaminophen-induced liver injury in mice. Hepatology. 2016;63:1340-55
26. Kurahashi T, Lee J, Nabeshima A, Homma T, Kang ES, Saito Y. et al. Ascorbic acid prevents acetaminophen-induced hepatotoxicity in mice by ameliorating glutathione recovery and autophagy. Arch Biochem Biophys. 2016;604:36-46
27. Lundback P, Lea JD, Sowinska A, Ottosson L, Furst CM, Steen J. et al. A novel high mobility group box 1 neutralizing chimeric antibody attenuates drug-induced liver injury and postinjury inflammation in mice. Hepatology. 2016;64:1699-710
28. Kleinschmidt D, Giannou AD, McGee HM, Kempski J, Steglich B, Huber FJ. et al. A protective function of il-22bp in ischemia reperfusion and acetaminophen-induced liver injury. J Immunol. 2017;199:4078-90
29. Feng D, Kong X, Weng H, Park O, Wang H, Dooley S. et al. Interleukin-22 promotes proliferation of liver stem/progenitor cells in mice and patients with chronic hepatitis b virus infection. Gastroenterology. 2012;143:188-98 e7
30. Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS. et al. Acetaminophen-induced acute liver failure: Results of a united states multicenter, prospective study. Hepatology. 2005;42:1364-72
31. Paridaens A, Raevens S, Colle I, Bogaerts E, Vandewynckel YP, Verhelst X. et al. Combination of tauroursodeoxycholic acid and n-acetylcysteine exceeds standard treatment for acetaminophen intoxication. Liver international: official journal of the International Association for the Study of the Liver. 2017;37:748-56
32. Hasnain SZ, Borg DJ, Harcourt BE, Tong H, Sheng YH, Ng CP. et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nat Med. 2014;20:1417-26
33. Ki SH, Park O, Zheng M, Morales-Ibanez O, Kolls JK, Bataller R. et al. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: Role of signal transducer and activator of transcription 3. Hepatology. 2010;52:1291-300
34. Pan H, Hong F, Radaeva S, Gao B. Hydrodynamic gene delivery of interleukin-22 protects the mouse liver from concanavalin a-, carbon tetrachloride-, and fas ligand-induced injury via activation of stat3. Cell Mol Immunol. 2004;1:43-9
35. Scheiermann P, Bachmann M, Goren I, Zwissler B, Pfeilschifter J, Muhl H. Application of interleukin-22 mediates protection in experimental acetaminophen-induced acute liver injury. Am J Pathol. 2013;182:1107-13
36. Chen W, Zhang X, Fan J, Zai W, Luan J, Li Y. et al. Tethering interleukin-22 to apolipoprotein a-i ameliorates mice from acetaminophen-induced liver injury. Theranostics. 2017;7:4135-48
37. Schneider JL, Cuervo AM. Liver autophagy: Much more than just taking out the trash. Nat Rev Gastroenterol Hepatol. 2014;11:187-200
38. Sun QZ, Lin GF, Li LL, Jin XT, Huang LY, Zhang G. et al. Discovery of potent and selective inhibitors of cdc2-like kinase 1 (clk1) as a new class of autophagy inducers. J Med Chem. 2017;60:6337-52
39. Dhiman R, Venkatasubramanian S, Paidipally P, Barnes PF, Tvinnereim A, Vankayalapati R. Interleukin 22 inhibits intracellular growth of mycobacterium tuberculosis by enhancing calgranulin a expression. J Infect Dis. 2014;209:578-87
40. Ganley IG, Wong PM, Gammoh N, Jiang X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell. 2011;42:731-43
41. Huang L, Jiang Y, Sun Z, Gao Z, Wang J, Zhang D. Autophagy strengthens intestinal mucosal barrier by attenuating oxidative stress in severe acute pancreatitis. Dig Dis Sci. 2018;63:910-9
42. Yang H, Ni HM, Guo F, Ding Y, Shi YH, Lahiri P. et al. Sequestosome 1/p62 protein is associated with autophagic removal of excess hepatic endoplasmic reticulum in mice. J Biol Chem. 2016;291:18663-74
43. Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ. et al. Pink1/parkin-mediated mitophagy is dependent on vdac1 and p62/sqstm1. Nat Cell Biol. 2010;12:119-31
44. Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem J. 2012;441:523-40
45. Yang YM, Han CY, Kim YJ, Kim SG. Ampk-associated signaling to bridge the gap between fuel metabolism and hepatocyte viability. World journal of gastroenterology: WJG. 2010;16:3731-42
46. Saberi B, Ybanez MD, Johnson HS, Gaarde WA, Han D, Kaplowitz N. Protein kinase c (pkc) participates in acetaminophen hepatotoxicity through c-jun-n-terminal kinase (jnk)-dependent and -independent signaling pathways. Hepatology. 2014;59:1543-54
47. Mihaylova MM, Shaw RJ. The ampk signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016-23
48. Kim J, Kundu M, Viollet B, Guan KL. Ampk and mtor regulate autophagy through direct phosphorylation of ulk1. Nat Cell Biol. 2011;13:132-41
49. Kang SW, Haydar G, Taniane C, Farrell G, Arias IM, Lippincott-Schwartz J. et al. Ampk activation prevents and reverses drug-induced mitochondrial and hepatocyte injury by promoting mitochondrial fusion and function. PloS one. 2016;11:e0165638
50. Mizushima N, Yoshimori T. How to interpret lc3 immunoblotting. Autophagy. 2007;3:542-5
51. Wang GY, Bi YG, Liu XD, Han JF, Wei M, Zhang QY. Upregulation of connexin 43 and apoptosisassociated protein expression by high glucose in h9c2 cells was improved by resveratrol via the autophagy signaling pathway. Mol Med Rep. 2017;16:3262-8
52. Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and p62 in cardiac proteinopathy. Circ Res. 2011;109:296-308
Author contact
![]() Corresponding authors: Qing Xie MD, PhD, Department of Infectious Diseases, Translational Lab of Liver Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Building 36, 197 Ruijin Er Road, Huangpu District, Shanghai, 200025, China. Tel: +86 21 64370045 x 680403; Fax: +86 21 6445 4930; E-mail: xieqingrjhcom, Shisan Bao MD, PhD, Discipline of Pathology, School of Medical Sciences and Bosch Institute, Charles Perkins Centre, The University of Sydney, NSW 2006, Australia. Tel: +61 2 9351 6156; Fax: +61 2 9351 3429; E-mail: bob.baoedu.au and Xiaogang Xiang MD, PhD, Department of Infectious Diseases, Translational Lab of Liver Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Building 36, 197 Ruijin Er Road, Huangpu District, Shanghai, 200025, China. Tel: +86 21 64370045 x 681021; Fax: +86 21 6445 4930; E-mail: shine-xxgcom
Corresponding authors: Qing Xie MD, PhD, Department of Infectious Diseases, Translational Lab of Liver Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Building 36, 197 Ruijin Er Road, Huangpu District, Shanghai, 200025, China. Tel: +86 21 64370045 x 680403; Fax: +86 21 6445 4930; E-mail: xieqingrjhcom, Shisan Bao MD, PhD, Discipline of Pathology, School of Medical Sciences and Bosch Institute, Charles Perkins Centre, The University of Sydney, NSW 2006, Australia. Tel: +61 2 9351 6156; Fax: +61 2 9351 3429; E-mail: bob.baoedu.au and Xiaogang Xiang MD, PhD, Department of Infectious Diseases, Translational Lab of Liver Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Building 36, 197 Ruijin Er Road, Huangpu District, Shanghai, 200025, China. Tel: +86 21 64370045 x 681021; Fax: +86 21 6445 4930; E-mail: shine-xxgcom