Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(19):5246-5258. doi:10.7150/thno.28676 This issue Cite
Research Paper
Small Heterodimer Partner Regulates Circadian Cytochromes p450 and Drug-Induced Hepatotoxicity
Tianpeng Zhang1,2,3, Fangjun Yu1, Lianxia Guo1, Min Chen1, Xue Yuan1, Baojian Wu1,3 ![]()
1. Research Center for Biopharmaceutics and Pharmacokinetics, College of Pharmacy, Jinan University, 601 Huangpu Avenue West, Guangzhou, China
2. Integrated Chinese and Western Medicine Postdoctoral research station, Jinan University, 601 Huangpu Avenue West, Guangzhou, China
3. Guangdong Province Key Laboratory of Pharmacodynamic Constituents of TCM and New Drugs Research, Jinan University, 601 Huangpu Avenue West, Guangzhou, China.
Received 2018-7-22; Accepted 2018-9-24; Published 2018-10-22
Abstract

The role of small heterodimer partner (SHP) in regulation of xenobiotic detoxification remains elusive. Here, we uncover a critical role for SHP in circadian regulation of cytochromes P450 (CYPs) and drug-induced hepatotoxicity.
Methods: The mRNA and protein levels of CYPs in the livers of wild-type and SHP-/- mice were measured by quantitative real-time polymerase chain reaction and Western blotting, respectively. Regulation of CYP by SHP was investigated using luciferase reporter, mobility shift, chromatin immunoprecipitation, and/or co-immunoprecipitation assays.
Results: The circadian rhythmicities of xenobiotic-detoxifying CYP mRNAs and proteins were disrupted in SHP-deficient mice. Of note, SHP ablation up-regulated Cyp2c38 and Cyp2c39, whereas it down-regulated all other CYP genes. Moreover, SHP regulated the expression of CYP genes through different mechanisms. SHP repressed Lrh-1/Hnf4α to down-regulate Cyp2c38, E4bp4 to up-regulate Cyp2a5, Dec2/HNF1α axis to up-regulate Cyp1a2, Cyp2e1 and Cyp3a11, and Rev-erbα to up-regulate Cyp2b10, Cyp4a10 and Cyp4a14. Furthermore, SHP ablation sensitized mice to theophylline (or mitoxantrone)-induced toxicity. Higher level of toxicity was correlated with down-regulated metabolism and clearance of theophylline (or mitoxantrone). In contrast, SHP ablation blunted the circadian rhythmicity of acetaminophen-induced hepatotoxicity and alleviated the toxicity by down-regulating Cyp2e1-mediated metabolism and reducing formation of the toxic metabolite. Toxicity alleviation by SHP ablation was also observed for aflatoxin B1 due to reduced formation of the toxic epoxide metabolite.
Conclusion: SHP participates in circadian regulation of CYP enzymes, thereby impacting xenobiotic metabolism and drug-induced hepatotoxicity.
Keywords: SHP, E4BP4, REV-ERB, CYP, hepatotoxicity
Introduction
Mammalian physiology (e.g., blood pressure, body temperature and heart beat) and behaviors (e.g., wake-sleep and feeding) are subject to circadian rhythms. Circadian rhythms are generated and regulated by the molecular clock machineries consisting of central clock (located in the suprachiasmatic nucleus (SCN)) and peripheral clocks (in peripheral organs) [1]. BMAL1 (brain and muscle ARNT-like 1) and CLOCK (circadian locomotor output cycles kaput) or NPAS2 (neuronal PAS domain protein 2) are two core components of mammalian clock system [2]. They form a heterodimer that activates the transcription of clock-controlled genes including cryptochrome (CRY), period (PER), and REV-ERBs [2]. In turn, CRY and PER proteins inhibit the transcriptional activity of BMAL1/CLOCK, and REV-ERBs repress the expression of BMAL1 [2]. By using this transcription-translation feedback mechanism, the clock system generates the circadian oscillations in gene expressions.
Metabolism (biotransformation) is the main mechanism for the body's defense against xenobiotic threats [3]. Xenobiotic metabolism is generally divided into three phases, namely, phase I modification, phase II conjugation and phase III excretion [4]. Cytochromes P450 (CYPs) belong to phase I enzymes and are responsible for phase I metabolism of up to 75% of clinically used drugs [5]. CYP-mediated metabolism is generally a detoxification pathway for drugs (e.g., theophylline and mitoxantrone, two hepatotoxic drugs) as the metabolites are usually biologically inactive or less active [6,7]. However, in some cases CYP metabolism elicits hepatotoxicity because of generation of toxic metabolites (e.g., APAP and aflatoxin B1) [8,9]. There is accumulating evidence that xenobiotic metabolism and tolerability is under the control of circadian clock [10]. PARbZip transcription factors DBP (albumin site D-binding protein), TEF (thyrotroph embryonic factor) and HLF (hepatocyte leukemia factor) control circadian expression of Cyp2b10 through regulation of constitutive androstane receptor (CAR) [11]. Circadian sensitivity to cyclophosphamide (a chemotherapeutic drug)-induced toxicity is determined by the functional status of CLOCK/ BMAL1 complex [12]. CLOCK controls circadian rhythm of intestinal Mdr1a and digoxin uptake by regulating HLF and E4BP4 (adenovirus E4 promoter-binding protein) [13].
The small heterodimer partner (SHP/NR0B2) is an atypical nuclear receptor (NR) that lacks a DNA-binding domain [14]. SHP, expressed abundantly in the liver, regulates gene expression by interacting with other NRs (e.g., CAR, PXR, ER, LRH-1, and HNF4α) and negatively modulating their transcriptional activities [15,16,17,18,19]. Three distinct mechanisms have been proposed for SHP repression of NR activities: 1) competition for coactivator binding to NRs; 2) active repression via recruitment of corepressors, and 3) inhibition of NR binding to DNA [15]. Inhibition of a particular NR by SHP may involve two or more mechanisms [11]. SHP has been implicated in regulation of diverse physiological pathways including bile acid metabolism, lipogenesis, gluconeogenesis and steroidogenesis [15,20,21]. Conforming to its role in lipogenesis, SHP is identified as an important regulator of development of fatty liver diseases in a recent study [22].
SHP is a circadian gene whose expression is under the control of BMAL1 and CLOCK/NPAS2 [23,24]. Liver receptor homologue-1 (LRH-1) may also be involved in circadian regulation of SHP [25]. Due to its crosstalk with BMAL1, NPAS, RORα/γ and REV-ERBα, SHP is regarded as an essential component of the liver circadian clock machinery [19,26]. In fact, SHP is a potential mediator connecting nutrient signaling with the circadian clock [21]. In the present study, we investigated the role of SHP in circadian regulation of CYPs and determined the regulatory mechanisms. We further clarified the impact of SHP on xenobiotic metabolism and drug-induced hepatotoxicity.
Results
Disrupted rhythmicity of CYP expression in SHP-deficient mice
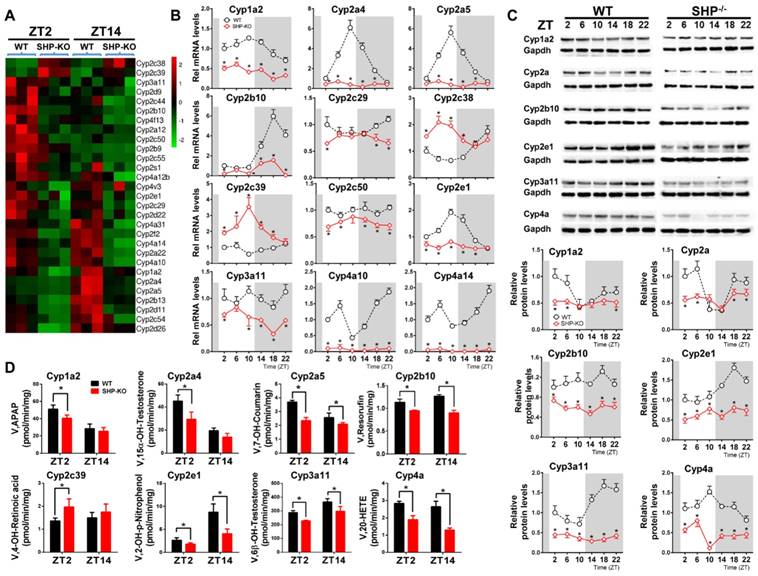
We first confirmed that SHP was absent in SHP-knockout (SHP-KO) mice (Figure S1). As expected, SHP deficiency leads to up-regulation of its target genes including Cyp7a1 and Cyp8b1 [15] (Figure S1). qPCR analyses indicated that hepatic expression of many xenobiotic-detoxifying CYP genes (e.g., those from Cyp1a, 2a, 2b, 2c, 2d, 2e and 3a subfamilies) were under the control of SHP [27] (Figure 1A). Cyp2c38 and 2c39 were significantly up-regulated in SHP-deficient mice, whereas all other CYP genes were down-regulated (Figure 1A). Further, circadian rhythms of CYP mRNAs (Cyp1a2, 2a4, 2a5, 2b10, 2c29, 2c38, 2c39, 2c50, 2e1, 4a10 and 4a14) were blunted (Figure 1B). The circadian rhythmicities of CYP proteins (Cyp1a2, 2a, 2b10, 2e1, 3a11 and 4a) were also disrupted in the liver (Figure 1C). In addition, SHP-KO mice showed altered enzymatic activities that were in a good agreement with the protein changes of CYPs (Figure 1D). Taken together, these data revealed a critical role of SHP in circadian regulation of CYP enzymes.
SHP deficiency alters the rhythmicities of CYP enzymes in mouse liver. (A) Heatmap comparing hepatic mRNA expression of CYP enzymes in WT vs. SHP-/- (or SHP-KO) mice at ZT2 and ZT14. The heatmap was generated using Cluster3 and Java Treeview. (B) qPCR analysis of rhythmic expression of CYP enzymes in livers from WT and SHP-/- mice. (C) Western blotting analysis of rhythmic expression of CYP enzymes in livers from WT and SHP-/- mice. (D) Activity analysis of CYP enzymes in livers from WT and SHP-/- mice at ZT2 and ZT14. The specific substrates for probing CYP activities were as follows: phenacetin (Cyp1a2), testosterone (Cyp2a4), coumarin (Cyp2a5), pentoxyresorufin (Cyp2b10), retinoic acid (Cyp2c39), testosterone (Cyp3a11), 4-nitrophenol (Cyp2e1) and arachidonic acid (Cyp4a). APAP: acetaminophen; 20-HETE: 20-hydroxy arachidonic acid. Data are mean ± SD from n = 5 mice for each time point, *p < 0.05 versus WT group (comparisons made at individual circadian times for (B-C)).
SHP induces expression of Cyp1a2, 2e1 and 3a11 through repression of Dec2/HNF1α axis
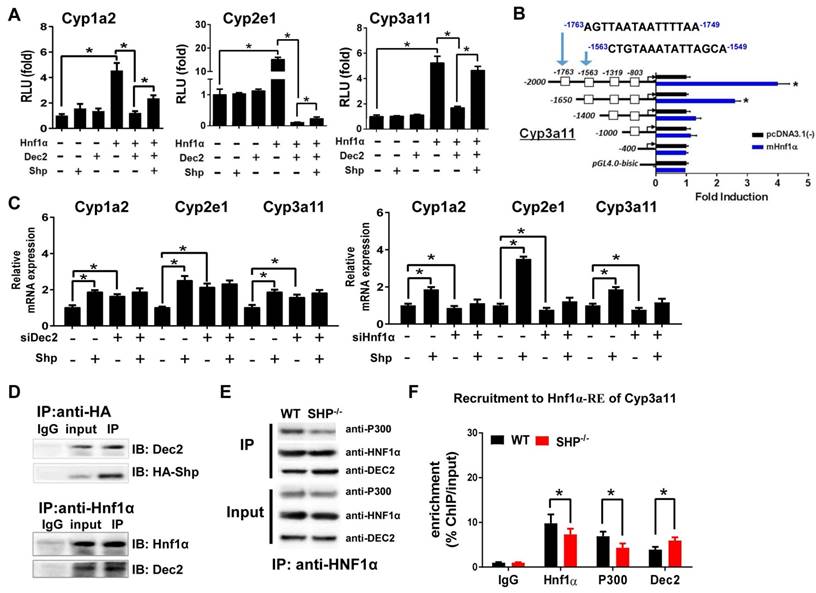
Due to the lack of a DNA-binding domain, direct regulation of gene transcription by SHP is unlikely [10]. Consistent with this notion, SHP cannot directly act on Cyp1a2, 2e1 and 3a11 (Figure 2A). HNF1α is a known activator of Cyp1a2 [28,29] and Cyp2e1 [30,31], and a potential activator of Cyp3a11 [24]. We confirmed that HNF1α induces the transcription of Cyp1a2, 2e1 and 3a11 in luciferase reporter assays (Figure 2A). Also, we identified the regions (-1763/-1749 bp and -1563/-1549 bp) within Cyp3a11 promoter responsible for specific binding of HNF1α (Figure 2B). Interestingly, Dec2, a clock-controlled protein previously shown to repress C/EBPα-induced transactivation of CYP2D6 gene [32], inhibited HNF1α transactivation of Cyp1a2, Cyp2e1 and Cyp3a11 (Figure 2A). SHP antagonized the repressive action of Dec2 on HNF1α, thereby enhancing the transcription of CYPs (Figure 2A). Moreover, the activation effects of SHP on Cyp1a2, Cyp2e1 and Cyp3a11 were lost when Dec2 or HNF1α was knocked down (Figure 2C). Furthermore, Co-IP experiments indicated protein-protein interactions between SHP and Dec2, and between Dec2 and HNF1α (Figure 2D). SHP ablation attenuated the interaction of p300 (a coactivator) with HNF1α (Figure 2E). This was accompanied by an enhanced interaction between Dec2 and HNF1α (Figure 2E). In addition, ChIP assays showed significant recruitments of Dec2, HNF1α and p300 to the HNF1α binding site of Cyp2e1, and SHP ablation increased the amount of Dec2 bound to the HNF1α binding site, but decreased the amount of bound p300 (Figure 2F and Figure S2A). Taken together, SHP up-regulates expressions of Cyp1a2, 2e1 and 3a11 through repression of the Dec2/HNF1α axis.
SHP induces Cyp1a2, Cyp2e1 and Cyp3a11 expression through repression of the Dec2/Hnf1α axis. (A) Luciferase reporter assays in HEK293T cells transfected with Cyp1a2, Cyp2e1 and Cyp3a11-Luc reporter (100 ng) and expression plasmids (100 ng) as indicated. (B) Luciferase reporter assays with truncated versions of Cyp3a11 promoter (100 ng). (C) Dec2 or Hnf1α knockdown abrogates the activation effects of SHP on Cyp1a2, Cyp2e1 and Cyp3a11. Hepa1c1c7 cells were transfected with expression plasmids and cultured for 36 h. (D) Co-IP assays showing protein-protein interactions of SHP with Dec2, and of Dec2 with Hnf1α. HEK293T cells were transfected with expression plasmids and cultured for 48 h. (E) Co-IP assays showing the effect of SHP on the protein-protein interactions of Hnf1α with Dec2 and P300 in mouse liver. The mouse liver nuclear extracts were derived from WT and SHP-/- mice. (F) ChIP assay showing the relative enrichment of Dec2, Hnf1α and P300 to the promoter region of Cyp3a11 in livers of WT and SHP-/- mice. Data are presented as mean ± SD (n = 5). *p < 0.05 versus control (t-test).
SHP regulates Cyp2a5 through repression of E4bp4
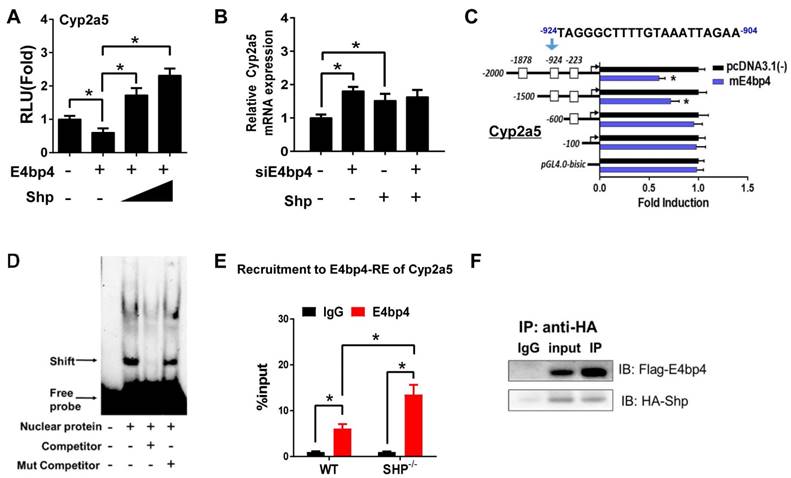
E4bp4/E4BP4 is a clock output gene that plays a role in circadian regulation of Cyp/CYP enzymes such as Cyp7a1 [33] and CYP3A4 [34]. We found that E4bp4 repressed the transcription of Cyp2a5, and SHP dose-dependently antagonized the repressive action of E4bp4 (Figure 3A). The activation effect of SHP on Cyp2a5 was lost when E4bp4 was knocked down (Figure 3B). Promoter analysis showed that E4bp4 repressed the transcription of Cyp2a5 by binding to the DNA sequence of -924/-904 bp (a D-box) upstream of the transcriptional start site (TSS) (Figure 3C). EMSA indicated direct DNA-protein interactions of the D-box of Cyp2a5 with E4bp4 (Figure 3D). ChIP experiments confirmed in vivo recruitment of E4bp4 protein to the D-box of Cyp2a5, and SHP ablation increased the amount of E4bp4 bound to the D-box (Figure 3E and Figure S2B). We also observed direct protein-protein interaction between SHP and E4bp4 (Figure 3F). The data overall suggest that SHP up-regulates Cyp2a5 through suppression of E4bp4, a repressor of the enzyme.
SHP induces Cyp2a5 expression through repression of E4bp4. (A) Luciferase reporter assays in HEK293T cells transfected with Cyp2a5-Luc reporter (100 ng) and expression plasmids (100 or 200 ng) as indicated. (B) E4bp4 knockdown abrogates the activation effects of SHP on Cyp2a5. Hepa1c1c7 cells were transfected with expression plasmids and cultured for 36 h. (C) Luciferase reporter assays with truncated versions of Cyp2a5 promoter (100 ng). (D) EMSA assay showing binding of E4bp4 to its response element of Cyp2a5. The nuclear extracts were obtained from HEK293T cells transfected with E4bp4. (E) ChIP assay showing the relative enrichment of E4bp4 to the promoter region of Cyp2a5 in liver of WT and SHP-/- mice. (F) Co-IP assay showing interaction of SHP with E4bp4. HEK293T cells were transfected with HA-tagged SHP and E4bp4 and cultured for 48 h. Data are presented as mean ± SD (n = 5). *p < 0.05 versus control (t-test).
SHP represses Cyp2c38 through repression of Lrh-1 and Hnf4α
Up-regulation of Cyp2c38 in SHP-KO mice suggested a negative control of SHP on Cyp2c38 expression (Figure 1). In luciferase reporter assays, Lrh-1 and Hnf4α significantly activated the promoter activity of Cyp2c38 (Figure 4A). Activation of Cyp2c38 (and 2c39) by Lrh-1 was confirmed using genetic mice lacking hepatic Lrh-1 (Figure 4B). Promoter analyses identified a specific Lrh-1-binding region (-1040/-1026 bp, Cyp2c38-LrhRE) and a specific Hnf4α-binding region (-120/-101 bp, Cyp2c38-Hnf4RE) within Cyp2c38 promoter (Figure 4C). EMSA assays supported direct binding of Cyp2c38-LrhRE sequence to Lrh-1 and binding of Cyp2c38-HnfRE to Hnf4α (Figure 4D). In vivo interactions of the two activators with Cyp2c38 were confirmed using ChIP assays (Figure 4E). There were significant recruitments of Lrh-1 and Hnf4α to Cyp2c38 promoter (Figure 4E). The data indicated that Lrh-1 and Hnf4α trans-activated Cyp2c38 through direct binding to their respective response elements. However, Lrh-1/HNF4α transactivation of Cyp2c38 was inhibited by SHP, a known repressor of Lrh-1 and HNF4α [35,36] (Figure 4A). Consistently, the effect of SHP on Cyp2c38 was attenuated when Lrh-1 or Hnf4α was knocked down (Figure 4F). ChIP assays showed significant increased protein amounts of Lrh-1/Hnf4a bound to Cyp2c38 promoter in SHP-KO mice (Figure 4G). Taken together, SHP repressed the transcription of Cyp2c38 through its suppressive actions on Lrh-1 and Hnf4α. Mouse Cyp2c38 and Cyp2c39 are highly homologous and have a 91.8% identity [37]. Sequence analysis revealed an LrhRE (identical to that of Cyp2c38) and an Hnf4RE (highly similar to that of Cyp2c38) in Cyp2c39 promoter (Figure 4H), suggesting that SHP regulates Cyp2c39 using the same mechanism as it does for Cyp2c38.
SHP represses Cyp2c38 through repression of Lrh-1 and Hnf4α. (A) Luciferase reporter assays in HEK293T cells transfected with Cyp2c38-Luc reporter (100 ng) and expression plasmids (100 ng) as indicated. (B) qPCR analysis of expression of Cyp2c38 and Cyp2c39 in the livers from WT and LRH-1hep-/- mice. (C) Luciferase reporter assays with truncated versions of Cyp2c38 promoter (100 ng). (D) EMSA assays for Lrh-1 and Hnf4α DNA-binding activity on nuclear extracts from HEK293T cells transfected with Lrh-1 or Hnf4α. (E) ChIP assay showing the relative enrichment of Lrh-1 and Hnf4α to the promoter region of Cyp2c38 in the liver of WT mice. (F) Lrh-1 or Hnf4α knockdown attenuates the repressive actions of SHP on Cyp2c38. Hepa1c1c7 cells were transfected with expression plasmids and cultured for 36 h. (G) ChIP assay showing the relative enrichment of Lrh-1 and Hnf4α to the promoter region of Cyp2c38 in the livers of WT and SHP-/- mice. (H) Sequence comparisons of HNF4α and LRH-1 binding sites between Cyp2c38 and Cyp2c39. Data are presented as mean ± SD (n = 5). *p < 0.05 versus control (t-test).

SHP represses Rev-erbα to up-regulate Cyp2b10, Cyp4a10 and Cyp4a14
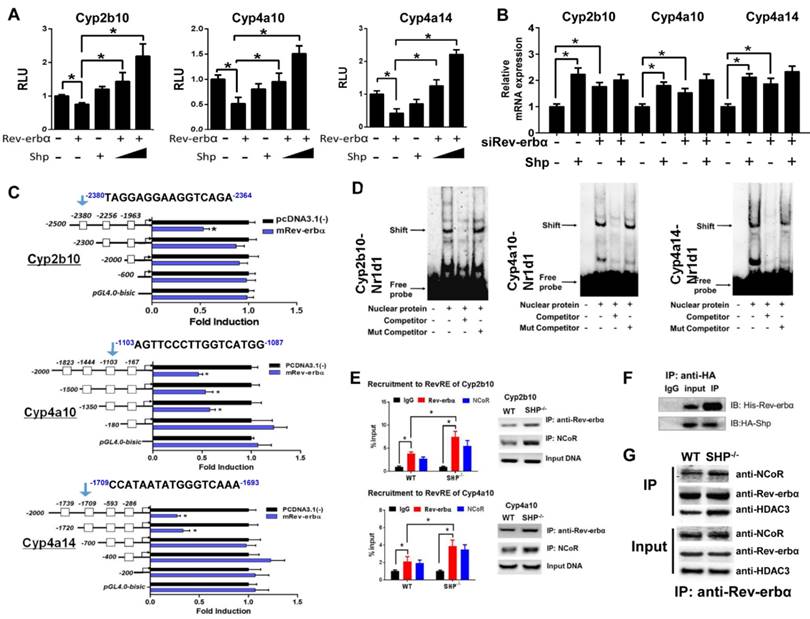
Luciferase reporter assays showed that Rev-erbα repressed the transcription of Cyp2b10, Cyp4a10 and Cyp4a14, suggesting a suppressive role of Rev-erbα in CYP expression (Figure 5A). This agrees well with the observations that the expressions of Cyp2b10, Cyp4a10 and Cyp4a14 are markedly up-regulated in Rev-erbα-deficient mice (Figure S3A), and that Rev-erbα can be recruited onto the RevRE (Rev-erbα response element) sites of Cyp2b10, Cyp4a10 and Cyp4a14 (Figure S3B). However, SHP co-transfection blocked the action of Rev-erbα and eliminated its repressor activity (Figure 5A). Moreover, the activation effects of SHP on Cyp2b10, Cyp4a10 and Cyp4a14 were lost when Rev-erbα was knocked down (Figure 5B). We further identified the Rev-erbα binding sites in Cyp2b10, Cyp4a10 and Cyp4a14 promoters (-2380/-2364 bp for Cyp2b10, -1103/-1087 bp for Cyp4a10, and -1709/-1693 bp for Cyp4a14) using the serial deletion method (Figure 5C). EMSA experiments with biotinylated oligonucleotides showed that Rev-erbα bound directly to each of Rev-erbα binding sites, forming a distinct DNA-protein complex (Figure 5D). The complex bands became faint in the presence of unlabeled competitor but unaffected by the addition of mutated competitor (Figure 5D). ChIP assays with mouse liver extracts showed significant recruitments of both Rev-erbα and NcoR (a corepressor) to the RevREs of Cyp2b10 and Cyp4a10, and the extents of recruitment were higher in SHP-KO than in wild-type mice (Figure 5E and Figure S4). Co-IP experiments confirmed a protein-protein interaction between SHP and Rev-erbα (Figure 5F) that is consistent with a previous study [19]. Further, SHP ablation led to enhanced interactions between Rev-erbα and the corepressors (NcoR and HDAC3) (Figure 5G). Taken together, SHP up-regulates Cyp2b10, Cyp4a10 and Cyp4a14 through inhibition of Rev-erbα, a transcriptional repressor of the three enzymes.
SHP represses Rev-erbα to up-regulate Cyp2b10, Cyp4a10 and Cyp4a14. (A) Luciferase reporter assays in HEK293T cells transfected with Cyp2b10, Cyp4a10 and Cyp4a14-Luc reporter (100 ng) and expression plasmids (100 ng or 200 ng) as indicated. (B) Rev-erbα knockdown abrogates the activation effects of SHP on Cyp2b10, Cyp4a10 and Cyp4a14. Hepa1c1c7 cells were transfected with expression plasmids and cultured for 36 h. (C) Luciferase reporter assays with truncated versions of Cyp2b10, Cyp4a10 and Cyp4a14 promoters (100 ng). (D) EMSA assays for Rev-erbα DNA-binding activity on nuclear extracts from HEK293T cells transfected with Rev-erbα. (E) ChIP assay showing the relative enrichment of Rev-erbα and NCoR to the promoter region of Cyp4a10 in livers of WT and SHP-/- mice. (F) Co-IP assay showing an interaction of SHP with Rev-erbα. HEK293T cells were transfected with HA-tagged SHP and His-tagged Rev-erbα. (G) Co-IP assays showing the effect of SHP on the protein-protein interactions of Rev-erbα with NCoR and HDAC3 in mouse liver. The mouse liver nuclear extracts were derived from WT and SHP-/- mice. Data are presented as mean ± SD (n = 5). *p < 0.05 versus control (t-test).
SHP ablation sensitizes mice to theophylline toxicity by down-regulating metabolism
Theophylline is a bronchodilating agent commonly used in the treatment of respiratory diseases. CYP-mediated metabolism is the primary pathway for clearance and detoxification of theophylline in humans and rodents with 1,3-dimethyluric acid (1,3-DMU) and 1-methylxanthine (1-MU) as major metabolites [38] (Figure 6A). In mice, 1,3-DMU is the dominant metabolite generated by Cyp1a2 and Cyp2e1 [6,33]. Administration of theophylline (150 mg/kg, i.p.) to wild-type mice induced hepatic and cardiac toxicities that were independent of the time of dosing (Figure 6B). We then investigated the impact of SHP ablation on the toxicity of theophylline dosed at single circadian time point (ZT2). Compared with wild-type, SHP-KO mice showed a higher mortality in response to theophylline treatment (200 mg/kg, i.p.) (Figure 6C). Also, the plasma levels of ALT, AST, CK, and LDH were much higher in SHP-KO than in wild-type mice (Figure 6C). More severe steatosis to the livers of SHP-KO mice was confirmed by histopathological examination (Figure 6D). Pharmacokinetic analyses showed decreased levels of plasma theophylline but increased levels of plasma 1,3-DMU in SHP-KO mice consistent with reduced microsomal metabolism of theophylline (Figure 6E). Reduced metabolism of theophylline in SHP-KO mice was ascribed to down-regulated expressions of Cyp1a2 and Cyp2e1, two principal enzymes responsible for theophylline metabolism (Figure 1). This was further supported by the fact that SHP overexpression in primary hepatocytes isolated from SHP-KO mice up-regulated Cyp1a2 and Cyp2e1, and enhanced theophylline metabolism (Figure 6F). Taken together, SHP ablation sensitized mice to theophylline toxicity by down-regulating the detoxification pathway.
SHP ablation sensitizes mice to theophylline toxicity by down-regulating metabolism. (A) Major metabolic pathways for theophylline. (B) Serum ALT and AST levels in WT and SHP-/- mice 12 h after vehicle or theophylline administration (150 mg/kg, i.p.) at ZT2 and ZT14. Data presented as mean ± SD (n = 5); *p < 0.05 between treatment and vehicle control. (C) The survival rates of WT and SHP-/- mice after theophylline administration (200 mg/kg, i.p., n = 10) (left). Serum ALT and AST levels in WT and SHP-/- mice 2 h after theophylline administration (n = 10) (right). (D) Representative H&E and Oil-red O staining of histological sections of livers from WT and SHP-/- mice 12 h after theophylline administration (200 mg/kg, i.p.) at ZT2 (top). Histopathological scores and lipid contents of livers from WT and SHP-/- mice 12 h after theophylline administration (200 mg/kg, i.p.) at ZT2 (bottom). (E) Plasma concentrations of theophylline and 1,3-DMU in WT and SHP-/- mice at 0.17, 0.5, 2, and 4 h after theophylline treatment (150 mg/kg, i.p., n = 3) (left and middle). Liver microsomal metabolism of theophylline in WT and SHP-/- mice at ZT2 and ZT14 (n = 5) (right). *p < 0.05 versus WT group (comparisons made at individual time points for pharmacokinetic curve). (F) qPCR analyses of Cyp1a2 and Cyp2e1 mRNAs in primary mouse hepatocytes (isolated from SHP-/- mice) transiently overexpressed with SHP or blank plasmid (left). Metabolism of theophylline by primary mouse hepatocytes (right). *p < 0.05 versus control. (G) Survival rates of WT and SHP-/- mice after mitoxantrone administration (15 mg/kg, i.p., n = 10) (left). Serum CK and LDH levels in WT and SHP-/- mice 4 days after mitoxantrone administration (right). (H) Plasma concentrations of mitoxantrone in WT and SHP-/- mice at 0.5, 2, 4, and 6 h after mitoxantrone treatment (5 mg/kg, i.p., n = 3) at ZT14 (left). Liver microsomal metabolism of mitoxantrone in WT and SHP-/- mice at ZT2 and ZT14 (n = 5). All data presented as mean ± SD, *p < 0.05 versus WT group (comparisons made at individual time points for pharmacokinetic curves).
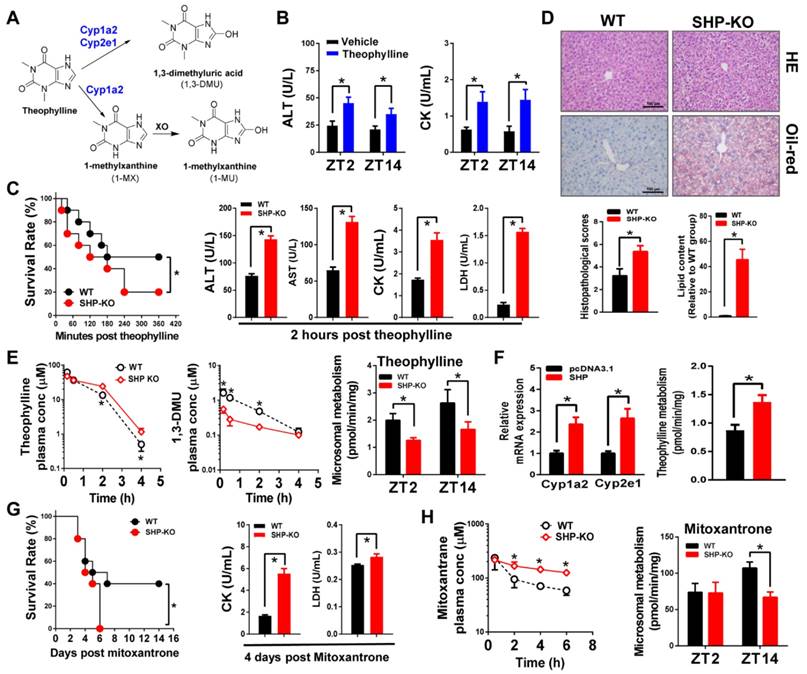
In addition to theophylline, we also clarified the impact of SHP ablation on toxicity and metabolism of the chemotherapeutic agent mitoxantrone. A previous study has established an inverse relationship between the severity of toxicity and the extent of metabolism for mitoxantrone [7]. Multiple CYP enzymes are involved in metabolism of mitoxantrone including Cyp2e1 [39] and Cyp3a11 in mice (Figure S5A). As observed for theophylline, SHP ablation sensitized mice to mitoxantrone toxicity (Figure 6G). Exacerbated toxicity was probably accounted for by the elevated levels of mitoxantrone due to down-regulated enzymes and reduced metabolism (Figure 1 and Figure 6H).
SHP ablation alleviates APAP hepatotoxicity in mice by down-regulating metabolism
Acetaminophen (APAP) is primarily metabolized and inactivated through conjugative reactions (glucuronidation and sulfation). However, a small portion of APAP is converted to the toxic metabolite N-acetyl-p-benzoquinone imine (NAPQI) by CYP2E1 [8]. NAPQI is unstable and rapidly conjugated with glutathione (GSH), and further transformed to APAP cysteine (APAP-Cys) and APAP acetylcysteine (APAP-NAC) (Figure 7A). Consistent with the literature [40], APAP toxicity exhibited circadian rhythmicity in wild-type mice (Figure 7B-C). APAP injection (500 mg/kg, i.p.) at ZT14 induced higher levels of toxicity compared with ZT2 (Figure 7B-C). We observed extensive multifocal centrilobular necrosis, hepatocellular dropout and neutrophil inflammation in mouse livers with APAP injection at ZT14 (Figure 7C). Higher levels of toxicity at ZT14 were correlated with a higher amount of GSH depletion (Figure S6A). Interestingly, SHP ablation blunted circadian rhythmicity of APAP toxicity (Figure 7B-C). This was because APAP toxicity of ZT14 was greatly alleviated in SHP-KO mice (Figure 7B-C). Additionally, a high APAP dose (800 mg/kg, i.p.) was injected to mice at ZT14 to assess survival rates. SHP ablation markedly reduced mortality (Figure 7D). Consistently, the serum levels of AST and ALT were much lower in SHP-KO than in wild-type mice (Figure 7D). Also, SHP-KO mice showed less GSH depletion in the liver (Figure S6B). Pharmacokinetic analysis showed that SHP ablation decreased the plasma levels of APAP-Cys and APAP-NAC (two stable products of NAPQI), suggesting reduced formation of NAPQI (Figure 7E). Formation of APAP glucuronide and sulfate was unaffected by SHP ablation (Figure S7). Moreover, overexpression of SHP led to a significant increase in Cyp2e1 expression as well as increases in the formation of APAP-Cys and APAP-NAC in primary hepatocytes isolated from SHP-KO mice (Figure 7F). These data indicated that SHP ablation alleviates APAP hepatotoxicity in mice by down-regulating Cyp2e1-mediated metabolism.
SHP ablation alleviates APAP hepatotoxicity in mice by down-regulating metabolism. (A) Major metabolic pathways for APAP. (B) Serum ALT and AST levels in WT and SHP-/- mice 12 h after vehicle or APAP administration (500 mg/kg, i.p., n = 5) at ZT2 and ZT14. Data presented as mean ± SD; *p < 0.05 between treatment and vehicle control. (C) Representative H&E staining of histological sections of livers from WT and SHP-/- mice 12 h after vehicle or APAP administration (500 mg/kg, i.p., n = 5) at ZT2 and ZT14. (D) Survival rates of WT and SHP-/- mice after APAP administration at ZT14 (left). Serum ALT and AST levels in WT and SHP-/- mice 12 h after APAP administration (800 mg/kg, i.p., n = 10) at ZT14 (right). Data presented as mean ± SD, *p < 0.05 versus WT group. (E) Plasma concentrations of APAP-Cys and APAP-NAC in WT and SHP-/- mice at 0.5, 1, 2, and 4 h after APAP treatment (500 mg/kg, i.p., n = 3). Data presented as mean ± SD, *p < 0.05 versus WT group (comparisons made at individual time points for pharmacokinetic curve). (F) qPCR analysis of Cyp2e1 mRNA in primary mouse hepatocytes (isolated from SHP-/- mice) transiently overexpressed with SHP or blank plasmid (left). Metabolism of APAP by primary mouse hepatocytes (middle and right). *p < 0.05 versus control. (G) Survival rates of WT and SHP-/- mice after AFB1 administration (15 mg/kg, i.p., n = 10) at ZT2 (left). Plasma concentrations of AFB1 in WT and SHP-/- mice at 5, 15, 30, and 60 min after AFB1 treatment (5 mg/kg, i.p., n = 3) (right). Liver microsomal metabolism of AFB1 in WT and SHP-/- mice at ZT2 and ZT14 (n = 5) (right). All data presented as mean ± SD, *p < 0.05; versus WT group (comparisons made at individual time points for pharmacokinetic curves).
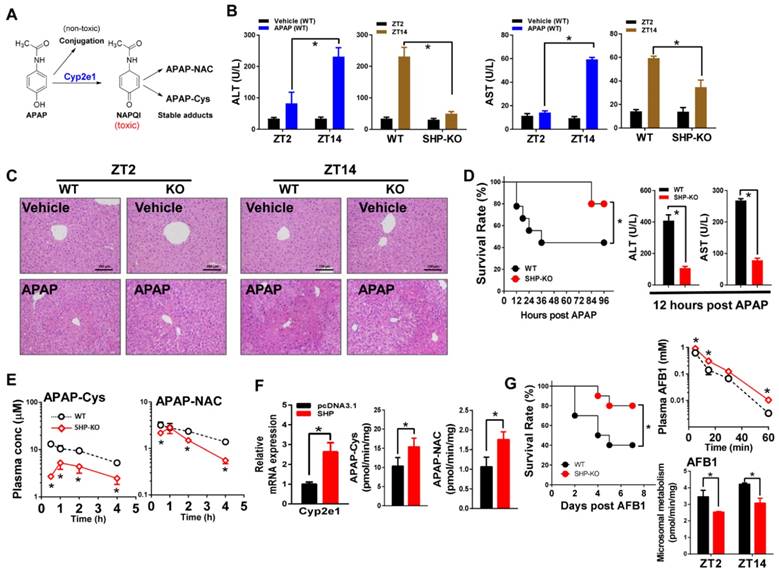
We also investigated a potential role of SHP in aflatoxin B1 (AFB1, a hepatocarcinogenic mycotoxin) metabolism and toxicity. AFB1 is metabolized to a toxic epoxide (AFB1-8,9-epoxide) with significant contribution from Cyp2a5 [9,41,42] (Figure S5B). SHP ablation led to a reduction in AFB1-induced mortality (Figure 7G). Pharmacokinetic study showed elevated levels of plasma AFB1 in SHP-KO mice because of reduced metabolism (Figure 7G). The data overall suggested that SHP ablation down-regulates AFB1 metabolism to reduce formation of toxic metabolite and alleviate AFB1 toxicity.
Discussion
Contrasting with the notion that SHP should be a negative regulator of xenobiotic metabolism because of its repressive actions on the xenobiotic response receptors such as CAR and PXR [6], we demonstrated in this study that SHP is a positive transcriptional regulator of many xenobiotic-detoxifying CYP enzymes including Cyp1a2, Cyp2a5, Cyp2b10, and Cyp3a11 (Figure 1). As SHP is a transcriptional repressor, positive regulation of CYPs is attained through inhibition of other CYP repressors such as Rev-erbα, E4bp4 and Dec2 (Figures 2-5). Consistent with down-regulated CYP expressions, CYP-mediated metabolism of drugs (e.g., theophylline and APAP) was reduced in SHP-KO mice (Figures 6-7). Decreased metabolic activities can be solely attributed to the changes in CYP expression because aminolevulinic acid synthase (ALAS1, the rate-limiting enzyme in the synthesis of heme) was unaffected and P450 oxidoreductase (Por, CYP redox partner) was slightly up-regulated (Figure S8A). It was noteworthy that in addition to CYP, several phase II enzymes (Ugts and Sults) were probably regulated by SHP (Figure S8B).
Our data lend a strong support to the proposal that SHP is an essential component of liver circadian clock machinery [17,21]. SHP is not only a clock-controlled gene (cyclically expressed in the liver, Figure S9) [17,18], it also interacts with other transcription factors (e.g., Rorα/γ, Rev-erbα and Lrh-1) to regulate clock genes (e.g., Npas2 and Bmal1), thereby controlling lipid metabolism and nutrient signaling [19,21]. The current study showed that SHP ablation blunted circadian oscillations of CYP enzymes. SHP regulates CYP expression through its crosstalk with the core clock genes Rev-erbα, E4bp4 and Dec2 [43], and the circadian genes Lrh-1/Hnf4α (Figure S9). Therefore, we argued that SHP is an integral component of the liver clock system, and controls circadian expression of CYP through a two-pronged mechanism, acting through its own rhythmic expression, and by modulating the functions of core clock genes (and circadian transcriptional factors).
CYP-mediated metabolism is a detoxification pathway for theophylline and mitoxantrone. However, CYP metabolism of APAP and aflatoxin B1 elicits toxicity because of generation of toxic metabolites. Therefore, it was not surprising that SHP ablation led to differential alterations in toxicity sensitivity to theophylline/mitoxantrone and APAP/aflatoxin B1 despite a metabolism change consensus (Figures 6-7). Although the enzymes Cyp1a2 and Cyp2e1 showed expression variations between ZT2 and ZT14, theophylline toxicity was independent of the time of dosing (Figure 6B). This was probably because the additive metabolism of Cyp1a2 (with a higher protein level at ZT2 and a lower level at ZT14) and Cyp2e1 (with a higher protein level at ZT14 and a lower level at ZT2) in the liver would not show a circadian difference, as evidenced by the in vitro microsomal metabolism assays (Figure 1C and Figure 6E). On the other hand, APAP toxicity was circadian time dependent with a higher level of toxicity at ZT14 (Figure 7B). This agrees well with the circadian pattern of Cyp2e1 (the enzyme responsible for production of the toxic metabolite NAPQI) with a higher expression in dark phase than in light phase (Figure 1C). We observed a slight change at 4 h alone in the pharmacokinetic curve of parent APAP because Cyp2e1 metabolism is a minor clearance pathway and the impact of this pathway on systemic exposure of parent drug is rather limited (Figure 6E and Figure S10).
SHP ablation caused a slight up-regulation of Lrh-1 consistent with the literature [21], whereas its effects on the expressions of other CYP-regulatory nuclear receptors/transcriptional factors including Car were minimal (Figure S11). Cyp2b10 is a known target gene of Car [7,11]. Since SHP represses Car-mediated transactivation of Cyp2b10 [11], one may expect to see up-regulated Cyp2b10 in SHP-KO mice. This seems to contradict the actual down-regulation of Cyp2b10 (Figure 1). It is uncertain whether Car mediates regulation of Cyp2b10 by SHP in vivo. If it does, the extent of Rev-erbα-mediated up-regulation of Cyp2b10 in SHP-KO mice would be more evident than actually observed because of the counteracting effect from Car.
Our study identified Dec2, E4bp4, and Rev-erbα as transcriptional repressors of xenobiotic-metabolizing CYP enzymes. Regulation of CYP family enzymes by these three factors was also noted previously in the literature. DEC2 interacts with C/EBPα to repress CYP2D6 in HepG2 cells [27]. E4BP4 suppresses the transcription of CYP3A4 through direct binding to the D-box within the promoter region [29]. Rev-erbα down-regulates Lrh-1 to repress Cyp7a1 (the rate limiting enzyme for bile acid biosynthesis) expression [44]. Collectively, circadian regulation of CYP family enzymes are rather complex, and multiple and differential mechanisms are usually involved [28].
In summary, SHP actively participates in circadian regulation of CYP enzymes via crosstalk with multiple circadian genes (Dec2, E4bp4, Rev-erbα, and Lrh-1/Hnf4α), thereby impacting xenobiotic metabolism and drug-induced hepatotoxicity.
Methods
Materials
The materials, primers and plasmids are provided in Supplementary Material.
Animal studies
SHP-/- mice (or SHP-KO) (C57BL/6 background) were kind gifts from the laboratory of Dr. David M. Moore (Baylor College of Medicine, Houston, TX) [45]. All mice were bred and housed in the Institute of Laboratory Animal Science (Jinan University, Guangzhou, China). All mice were maintained on a 12 h light: 12 h dark cycle (light on 7:00 AM to 7:00 PM), with free access to food and water. For the analyses of mRNA, proteins and liver microsomal CYP enzyme activity, wild-type (C57BL/6) or SHP-/- male mice (8-12 weeks of age, n = 5 per group) were sacrificed at ZT2, ZT6, ZT10, ZT14, ZT18, ZT22, and livers were harvested.
In toxicological studies, wild-type or SHP-/- male mice (8-12 weeks of age, n = 5 per group) received a single intraperitoneal injection of theophylline (150 mg/kg) and acetaminophen (500 mg/kg) at ZT2 or ZT14 and killed by CO2 inhalation at specific time points. Plasma ALT (alanine aminotransferase), AST (aspartate aminotransferase), CK (creatine kinase) and LDH (lactate dehydrogenase) levels were measured using enzymatic assay kits (Jiancheng Bioengineering Institute, Nanjing, China). Liver tissues were fixed in 4% paraformaldehyde. Hematoxylin-eosin staining (H&E) was performed as previously described [46]. Additionally, 6 μm-thick frozen liver sections were stained with Oil Red O to illustrate hepatic lipid accumulation. The lipid content in liver was measured as previously described [47]. For survival experiments, wild-type or SHP-/- male mice (8-12 weeks of age, n = 10 per group) received a single intraperitoneal injection of theophylline (200 mg/kg), mitoxantrone (15 mg/kg), acetaminophen (800 mg/kg) or Aflatoxin B1 (12 mg/kg).
For pharmacokinetic studies, theophylline (150 mg/kg), mitoxantrone (5 mg/kg), acetaminophen (500 mg/kg) or Aflatoxin B1 (5 mg/kg) was administered to wild-type or SHP-/- male mice (22-24 g, n = 3 per time point) by intraperitoneal injection. At each time point, 3 mice were rendered unconscious with isoflurane for blood and liver sampling. The blood was collected by cardiac puncture. All animal experimental procedures were approved by the Jinan University Institutional Animal Care and Use Committee and were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Isolation and culture of primary mouse hepatocytes
Primary hepatocytes were isolated from SHP-/- mice as described previously [48]. In brief, the liver was perfused with HBSS and digested with collagenase IV by perfusion through the inferior vena cava. After washing with HBSS, hepatocytes were collected and cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). After 2 h, the medium was changed to serum-free DMEM. The cells were maintained for additional 24 h prior to transient transfection and cell lysate preparation.
Cell culture and transfection
Hepa-1c1c7 cells were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). The cells were cultured in Minimum Essential Medium Alpha Basic supplemented with 10% FBS. Hepa-1c1c7 cells were seeded onto 12-well plates and transfected with indicated plasmids and/or siRNA (sequences summarized in Table S1) using JetPrime (Polyplus Transfection, Ill kirch, France) according to the manufacturer's protocol.
qPCR
Total RNA was isolated from liver samples and cells using RNAiso Plus (Takara), and quantitative reverse transcriptase PCR were performed as previously described [43]. Gene expression levels were normalized to cyclophilin b. All primer sequences are summarized in Table S2.
Western blotting
Total liver protein extracts were prepared, and the lysates were subjected to Western blotting as described previously [49]. Blots were probed with anti-Cyp1a2 (Abcam), anti-Cyp2a (Abcam), anti-Cyp2b10 (OriGene), anti-Cyp2e1 (Abcam), anti-Cyp3a11 (Abcam), anti-Cyp4a (Abcam), anti-Dec2 (Proteintech), anti-Rev-erbα (Sigma), anti-E4bp4 (Santa), anti-Shp (OriGene), anti-Hnf1α (Proteintech), anti-His tag (Santa) and anti-Gapdh (Abcam) antibodies.
Liver microsomal CYP enzyme activity
Mouse liver microsomes were prepared as previously described [43]. The microsomal Cyp1a2, Cyp2a4, Cyp2a5, Cyp2b10, Cyp2c39, Cyp2e1, Cyp3a11 and Cyp4a activities were determined using published procedures [50]. Enzymatic activity was determined by LC-MS analysis of the generated metabolites of the respective substrate. Incubation and analytical conditions are listed in Table S3 and Table S4.
Pharmacokinetic analysis
The plasma samples were prepared as previously described [43].The concentrations of drugs and their metabolites were determined by UPLC-QTOF/MS (Waters, Milford, MA) [43]. The analytical conditions, retention time and pseudomolecuar ions are listed in Table S4. Representative chromatograms for chemical quantification are shown in Figure S12.
Luciferase reporter, Co-IP, EMSA and ChIP assays
All these assays were performed as previously described [43,45]. In brief, in luciferase reporter assays, HEK293T cells were transfected with indicated plasmids. After 24 h treatment, cells were lysed and assayed for luciferase activities. In Co-IP, HEK293T cells were transfected with the expression plasmids (Shp, E4bp4, Rev-erbα, Dec2 and/or Hnf1α). HEK293T cells or mouse liver were lysed in IP lysis buffer (Beyotime, Shanghai, China). Immunoprecipitation was performed as previously described [51]. Lystate was incubated with 2 μg anti-Hnf1α (Proteintech, Wuhan, China), anti-HA-tag (Abcam, Cambridge, MA), anti-Rev-erbα (CST, Beverly, MA) or anti-normal rabbit IgG (CST, Beverly, MA) antibody. For EMSA, HEK293T cells were transfected with E4bp4, Lrh-1, Hnf4α, or Rev-erbα plasmids. After 48 h treatment, the nuclear extracts were incubated with the probes (Table S5). For ChIP, mouse liver was fixed in 1% formaldehyde and digested with micrococcal nuclease. The sheared chromatin was immunoprecipitated with anti-E4bp4 (Santa Cruz, CA), anti-Rev-erbα (CST, Beverly, MA), Lrh-1 (Abcam, Cambridge, MA), anti-Hnf4α (Abcam, Cambridge, MA), anti-NCoR (Abcam, Cambridge, MA), anti-p300 (Santa Cruz, CA), anti-Dec2 (Proteintech Group, Wuhan, China), anti-Hnf1α (Proteintech Group, Wuhan, China) or normal rabbit IgG (control) at 4 ℃ overnight. The purified DNAs were analyzed by qPCR with the primers (Table S6). qPCR products were run on an agarose gel (2%) and were analyzed using an Omega-Lum G imaging system.
Statistical analysis
Data are presented as mean ± SD (standard deviation). Statistical analyses for survival were performed with the logrank test. Statistical analyses on all other data were performed using a Student's t-test comparing levels of measured parameters of wild-type vs. SHP-KO mice. The level of significance was set at p < 0.05.
Abbreviations
ALAS1: aminolevulinic acid synthase; APAP: acetaminophen; AFB1: aflatoxin B1; C/EBPα: CCAAT/enhancer-binding protein α; ChIP: chromatin immunoprecipitation; CYP: Cytochrome P450 family; DEC2: differentiated embryonic chondrocyte gene 2; EMSA: electrophoretic mobility shift assay; HBSS: Hanks' balanced salt solution; HDAC3: histone deacetylase 3; HNF1α: hepatocyte nuclear factor 1 alpha; HNF4α: hepatocyte nuclear factor 4 alpha; HnfRE: HNF4α response element; LRH-1: liver receptor homolog 1; LrhRE: Lrh-1 response element; NCoR: nuclear receptor co-repressor; NR: nuclear receptor; p300: histone acetyltransferase p300; Por: P450 oxidoreductase; RevRE: Rev-erbα response element; SHP: small heterodimer partner; 1,3-DMU: 1,3-dimethyluric acid; ZT: Zeitgeber time.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grants 81573488 and 81503210).
Contributions
Participated in research design: Zhang and Wu. Conducted experiments: Zhang, Yu, Guo, Yuan, Chen. Performed data analysis: Zhang, Yu, and Wu. Wrote or contributed to the writing of the manuscript: Zhang and Wu.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mohawk JA, Green CB, Takahashi JS. Central and peripheral circadian clocks in mammals. Annu Rev Neurosci. 2012;35:445-462
2. Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014;24:90-99
3. Benet LZ. The role of BCS (biopharmaceutics classification system) and BDDCS (biopharmaceutics drug disposition classification system) in drug development. J Pharm Sci. 2013;102:34-42
4. Xu C, Li CY, Kong AN. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res. 2005;28:249-268
5. Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science. 1999;286:487-491
6. Minton NA, Henry JA. Acute and chronic human toxicity of theophylline. Hum Exp Toxicol. 1996;15:471-481
7. Lévi F, Tampellini M, Metzger G. et al. Circadian changes in mitoxantrone toxicity in mice: relationship with plasma pharmacokinetics. Int J Cancer. 1994;59:543-547
8. Chen C, Krausz KW, Idle JR. et al. Identification of novel toxicity-associated metabolites by metabolomics and mass isotopomer analysis of acetaminophen metabolism in wild-type and Cyp2e1-null mice. J Biol Chem. 2008;283:4543-4559
9. Dohnal V, Wu Q, Kuča K. Metabolism of aflatoxins: key enzymes and interindividual as well as interspecies differences. Arch Toxicol. 2014;88:1635-1644
10. Claudel T, Cretenet G, Saumet A. et al. Crosstalk between xenobiotics metabolism and circadian clock. FEBS Lett. 2007;581:3626-3633
11. Gachon F, Olela FF, Schaad O. et al. The circadian PAR-domain basic leucine zipper transcription factors DBP, TEF, and HLF modulate basal and inducible xenobiotic detoxification. Cell Metab. 2006;4:25-36
12. Gorbacheva VY, Kondratov RV, Zhang R. et al. Circadian sensitivity to the chemotherapeutic agent cyclophosphamide depends on the functional status of the CLOCK/BMAL1 transactivation complex. Proc Natl Acad Sci U S A. 2005;102:3407-3412
13. Murakami Y, Higashi Y, Matsunaga N. et al. Circadian clock-controlled intestinal expression of the multidrug-resistance gene mdr1a in mice. Gastroenterology. 2008;135:1636-1644
14. Seol W, Choi HS, Moore DD. An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science. 1996;272:1336-1339
15. Bae Y, Kemper JK, Kemper B. Repression of CAR-mediated transactivation of CYP2B genes by the orphan nuclear receptor, short heterodimer partner (SHP). DNA Cell Biol. 2004;23:81-91
16. Ourlin JC, Lasserre F, Pineau T. et al. The small heterodimer partner interacts with the pregnane X receptor and represses its transcriptional activity. Mol Endocrinol. 2003;17:1693-1703
17. Goodwin B, Jones SA, Price RR. et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517-526
18. Johansson L, Thomsen JS, Damdimopoulos AE. et al. The orphan nuclear receptor SHP inhibits agonist-dependent transcriptional activity of estrogen receptors ERalpha and ERbeta. J Biol Chem. 1999;274:345-353
19. Båvner A, Sanyal S, Gustafsson JA. et al. Transcriptional corepression by SHP: molecular mechanisms and physiological consequences. Trends Endocrinol Metab. 2005;16:478-488
20. Huang J, Iqbal J, Saha PK. et al. Molecular characterization of the role of orphan receptor small heterodimer partner in development of fatty liver. Hepatology. 2007;46:147-157
21. Wang L, Han Y, Kim CS. et al. Resistance of SHP-null mice to bile acid-induced liver damage. J Biol Chem. 2003;278:44475-44481
22. Yang Z, Tsuchiya H, Zhang Y. et al. REV-ERBα Activates C/EBP Homologous Protein to Control Small Heterodimer Partner-Mediated Oscillation of Alcoholic Fatty Liver. Am J Pathol. 2016;186:2909-2920
23. Pan X, Zhang Y, Wang L. et al. Diurnal regulation of MTP and plasma triglyceride by CLOCK is mediated by SHP. Cell Metab. 2010;12:174-186
24. Lee SM, Zhang Y, Tsuchiya H. et al. Small heterodimer partner/neuronal PAS domain protein 2 axis regulates the oscillation of liver lipid metabolism. Hepatology. 2015;61:497-505
25. Oiwa A, Kakizawa T, Miyamoto T. et al. Synergistic regulation of the mouse orphan nuclear receptor SHP gene promoter by CLOCK-BMAL1 and LRH-1. Biochem Biophys Res Commun. 2007;353:895-901
26. Wu N, Kim KH, Zhou Y. et al. Small Heterodimer Partner (NR0B2) Coordinates Nutrient Signaling and the Circadian Clock in Mice. Mol Endocrinol. 2016;30:988-995
27. Hrycay EG, Bandiera SM. Expression, function and regulation of mouse cytochrome P450 enzymes: comparison with human P450 enzymes. Curr Drug Metab. 2009;10:1151-1183
28. Chung I, Bresnick E. Identification of positive and negative regulatory elements of the human cytochrome P4501A2 (CYP1A2) gene. Arch Biochem Biophys. 1997;338:220-226
29. Cheung C, Akiyama TE, Kudo G. et al. Hepatic expression of cytochrome P450s in hepatocyte nuclear factor 1-alpha (HNF1alpha)-deficient mice. Biochem Pharmacol. 2003;66:2011-2020
30. Liu SY, Gonzalez FJ. Role of the liver-enriched transcription factor HNF-1 alpha in e xpression of the CYP2E1 gene. DNA Cell Biol. 1995;14:285-293
31. Matsunaga N, Ikeda M, Takiguchi T. et al. The molecular mechanism regulating 24-hour rhythm of CYP2E1 expression in the mouse liver. Hepatology. 2008;48:240-251
32. Matsunaga N, Inoue M, Kusunose N. et al. Time-dependent interaction between differentiated embryo chondrocyte-2 and CCAAT/enhancer-binding protein α underlies the circadian expression of CYP2D6 in serum-shocked HepG2 cells. Mol Pharmacol. 2012;81:739-747
33. Noshiro M, Usui E, Kawamoto T. et al. Multiple mechanisms regulate circadian expression of the gene for cholesterol 7alpha-hydroxylase (Cyp7a), a key enzyme in hepatic bile acid biosynthesis. J Biol Rhythms. 2007;22:299-311
34. Takiguchi Tomita M, Matsunaga N et al. Molecular basis for rhythmic expression of CYP3A4 in serum-shocked HepG2 cells. Pharmacogenet Genomics. 2007;17:1047-1056
35. Chanda D, Xie YB, Choi HS. Transcriptional corepressor SHP recruits SIRT1 histone deacetylase to inhibit LRH-1 transactivation. Nucleic Acids Res. 2010;38:4607-4619
36. Lee YK, Dell H, Dowhan DH. et al. The orphan nuclear receptor SHP inhibits hepatocyte nuclear factor 4 and retinoid X receptor transactivation: two mechanisms for repression. Mol Cell Biol. 2000;20:187-195
37. Meng XY, Zheng QC, Zhang HX. A comparative analysis of binding sites between mouse CYP2C38 and CYP2C39 based on homology modeling, molecular dynamics simulation and docking studies. Biochim Biophys Acta. 2009;1794:1066-1072
38. Derkenne S, Curran CP, Shertzer HG. et al. Theophylline pharmacokinetics: comparison of Cyp1a1(-/-) and Cyp1a2(-/-) knockout mice, humanized hCYP1A1_1A2 knock-in mice lacking either the mouse Cyp1a1 or Cyp1a2 gene, and Cyp1(+/+) wild-type mice. Pharmacogenet Genomics. 2005;15:503-511
39. Rossato LG, Costa VM, de Pinho PG. et al. The metabolic profile of mitoxantrone and its relation with mitoxantrone-induced cardiotoxicity. Arch Toxicol. 2013;87:1809-1820
40. DeBruyne JP, Weaver DR, Dallmann R. The hepatic circadian clock modulates xenobiotic metabolism in mice. J Biol Rhythms. 2014;29:277-287
41. Pelkonen P, Kirby GM, Wild CP. et al. Metabolism of nitrosamines and aflatoxin B1 by hamster liver CYP2A enzymes. Chem Biol Interact. 1994;93:41-50
42. Kirby GM, Chemin I, Montesano R. et al. Induction of specific cytochrome P450s involved in aflatoxin B1 metabolism in hepatitis B virus transgenic mice. Mol Carcinog. 1994;11:74-80
43. Honma S, Kawamoto T, Takagi Y. et al. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature. 2002;419:841-844
44. Zhang T, Zhao M, Lu D. et al. REV-ERBa regulates CYP7A1 through repression of liver receptor homolog-1. Drug Metab Dispos. 2018;46:248-258
45. Wang L, Lee YK, Bundman D. et al. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell. 2002;2:721-731
46. Johnson BP, Walisser JA, Liu Y. et al. Hepatocyte circadian clock controls acetaminophen bioactivation through NADPH cytochrome P450 oxidoreductase. Proc Natl Acad Sci U S A. 2014;111:18757-18762
47. Green H, Kehinde O. An established preadipose cell line and its differentiation in culture. II. Factors affecting the adipose conversion. Cell. 1975;5:19-27
48. Zhang W, Sargis RM, Volden PA. et al. PCB 126 and other dioxin-like PCBs specifically suppress hepatic PEPCK expression via the aryl hydrocarbon receptor. PLoS One. 2012;7:e37103
49. Lu D, Wang S, Xie Q. et al. Transcriptional regulation of human UDP-glucuronosyltransferase 2B10 by farnesoid X receptor in human hepatoma HepG2 cells. Mol Pharmaceutics. 2017;14:2899-2907
50. Lu D, Dong D, Liu Z. et al. Metabolism elucidation of BJ-B11 (a heat shock protein 90 inhibitor) by human liver microsomes: identification of main contributing enzymes. Expert Opin Drug Metab Toxicol. 2015;11:1029-1040
51. Wang S, Yuan X, Lu D. et al. Farnesoid X receptor regulates SULT1E1 expression through inhibition of PGC1α binding to HNF4α. Biochem Pharmacol. 2017;145:202-209
Author contact
![]() Corresponding author: Baojian Wu, Ph.D. College of Pharmacy, Jinan University. E-mail: bj.wucom or tbaojianwuedu.cn
Corresponding author: Baojian Wu, Ph.D. College of Pharmacy, Jinan University. E-mail: bj.wucom or tbaojianwuedu.cn