Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(19):5276-5288. doi:10.7150/thno.27847 This issue Cite
Research Paper
Increasing lean muscle mass in mice via nanoparticle-mediated hepatic delivery of follistatin mRNA
Canan Schumann1, Duc X. Nguyen1, Mason Norgard2, Yulia Bortnyak1, Tetiana Korzun1, Stephanie Chan1, Anna St. Lorenz1, Abraham S. Moses1, Hassan A. Albarqi1, Leon Wong1, Katherine Michaelis2, Xinxia Zhu2, Adam W. G. Alani1, Olena R. Taratula1, Stephanie Krasnow1, Daniel L. Marks2 ![]() , Oleh Taratula1
, Oleh Taratula1 ![]()
1. Department of Pharmaceutical Sciences, College of Pharmacy, Oregon State University, Portland, OR 97201, USA
2. Department of Pediatrics, Papé Family Pediatric Research Institute, Oregon Health & Science University, Portland, Oregon 97239, USA
Received 2018-6-12; Accepted 2018-10-2; Published 2018-10-22
Abstract

Muscle atrophy occurs during chronic diseases, resulting in diminished quality of life and compromised treatment outcomes. There is a high demand for therapeutics that increase muscle mass while abrogating the need for special dietary and exercise requirements. Therefore, we developed an efficient nanomedicine approach capable of increasing muscle mass.
Methods: The therapy is based on nanoparticle-mediated delivery of follistatin messenger RNA (mRNA) to the liver after subcutaneous administration. The delivered mRNA directs hepatic cellular machinery to produce follistatin, a glycoprotein that increases lean mass through inhibition of negative regulators of muscle mass (myostatin and activin A). These factors are elevated in numerous disease states, thereby providing a target for therapeutic intervention.
Results: Animal studies validated that mRNA-loaded nanoparticles enter systemic circulation following subcutaneous injection, accumulate and internalize in the liver, where the mRNA is translated into follistatin. Follistatin serum levels were elevated for 72 h post injection and efficiently reduced activin A and myostatin serum concentrations. After eight weeks of repeated injections, the lean mass of mice in the treatment group was ~10% higher when compared to that of the controls.
Conclusion: Based on the obtained results demonstrating an increased muscle mass as well as restricted fat accumulation, this nanoplatform might be a milestone in the development of mRNA technologies and the treatment of muscle wasting disorders.
Keywords: mRNA, polymeric nanoparticles, muscle atrophy, follistatin, activin A, myostatin
Introduction
Muscle atrophy is a detrimental condition with multifactorial etiology that is found in myriad disease states including cancer, AIDS, cardiac failure, and muscular dystrophies [1]. Beyond its tremendous burden on quality of life, the extent of muscle atrophy is a prognostic indicator of poor treatment outcomes and often is a determining factor in disease mortality [1]. In many patient populations, muscle wasting cannot be simply reversed by nutritional support or physical exercise [2] and, therefore, efficient therapies that preserve or increase skeletal muscle mass independent of exercise and special diets are highly warranted.
In the past decades, research has been done to identify the mechanisms that underlie muscle growth as well as muscle tissue regeneration [3-10]. Consequently, various muscle atrophy-associated signaling pathways, and their key regulators are actively considered as therapeutic targets for preventing muscle wasting and stimulating muscle growth [1]. Among these, significant attention is paid to transforming growth factor-β (TGF-β) superfamily proteins that play a critical role in negatively regulating muscle mass in health and disease [11, 12]. TGF-β superfamily proteins are primarily expressed and secreted by skeletal muscle, [13] and they negatively regulate muscle growth by binding to the activin type IIB receptor (ActRIIB) on muscle cells [14]. ActRIIB signaling increases muscle proteolysis, reduces protein synthesis, and inhibits myoblast differentiation and proliferation [15]. Recently, Latres et al. concluded that two TGF-β superfamily proteins, myostatin and activin A, are key negative regulators of muscle growth in primates and mice, and activin A plays a more significant role than myostatin in regulating muscle mass in primates [16]. Previous reports also demonstrated that circulating or intramuscular activin A and myostatin levels are elevated in many pathological wasting states [9, 12, 17-20]. For example, it was reported that circulating activin A levels are higher in humans with cancer cachexia and are directly proportional to the degree of muscle wasting [9, 12, 20]. Recent preclinical studies suggested that production of activin A by tumors contributes to cachexia [9, 21] and its blockade by a soluble form of the ActRIIB receptor prevents muscle atrophy in mice [21]. The therapeutic potential of ActRIIB decoy receptor was evaluated in clinical trials in patients with cancer or degenerative muscle disorders, but these studies yielded mixed results with respect to safety [22-24]. For example, bleeding complications related to the administration of decoy receptors in clinical trials with muscular dystrophy patients resulted in early termination of these studies [25]. Preclinical studies further confirmed that administration of ActRIIB decoy receptors is associated with increased pancreas weight, elevated blood glucose levels, and impaired glucose tolerance. The observed side effects were not related to the inhibition of activin A or myostatin [16].
Follistatin is a natural circulating glycoprotein that functions as an endogenous antagonist for several members of the TGF-β superfamily proteins (e.g., activin A, myostatin, growth differentiation factor 11) by binding to them in serum and preventing their interaction with the ActRIIB receptor [12, 14, 26-28]. It has two isoforms generated by alternative splicing: FS-317 and FS-344 [29, 30]. Posttranslational modification of each isoform gives rise to FS-288 and FS-315 isoforms, respectively [30]. FS-315 is the predominant isoform that is found primarily in the blood, [31] and therefore, it is perfect for inhibiting circulating ligands of TGF-β superfamily [32, 33]. To date, two small open-label studies demonstrated that follistatin gene therapy based on delivery of FS-315 encoding cDNA via an adenoviral vector to a patient is efficient in increasing muscle mass in humans with inclusion body myositis and Becker muscular dystrophy [30, 34]. The viral delivery of the cDNA, however, encompasses an array of technical and safety issues including [35-37] (1) adenoviral vector-associated immunogenicity, inflammation, and toxicity; (2) potential for permanent integration of cDNA into the host genome; and, (3) numerous challenges in production and scaling-up of clinical-grade viral vectors.
To overcome these limitations, we have developed and evaluated a novel follistatin gene therapy approach based on the delivery of FS-344 mRNA. In general, mRNA-based strategies intended to supplement deficient proteins in patients have significant potential for the treatment of various diseases, including muscle atrophy [38-40]. After delivery of the required mRNA (e.g., FS-344 mRNA) to the targeted cells, the desired protein can be endogenously produced and post-translationally modified by the machinery of the transfected cells during an extended period of time [40]. However, application of mRNA in the development of novel therapeutic modalities, including follistatin-based therapy, is still limited due to the numerous challenges associated with mRNA delivery to the targeted cells after systemic administration [41]. The major limitations include the degradation of naked mRNA in blood, immunogenicity, poor accumulation in the targeted tissue, low internalization into the cytoplasm, and inflammatory reactions after repeated treatment [39-41]. Therefore, we have developed polymeric nanoparticles that efficiently protect and deliver FS-344 mRNA to the liver after subcutaneous administration with no adverse side effects. Our decision to target liver cells is based on the fact that in humans the liver is a major contributor to circulating follistatin, implying that these cells are equipped to produce and secrete the fully mature and glycosylated follistatin protein [42]. We demonstrated that the fully functional follistatin glycoprotein is synthesized from the mRNA templates delivered to the targeted cells. Subsequent secretion of the expressed follistatin into the bloodstream leads to inhibition of both myostatin and activin A, thereby causing an increase in lean muscle mass.
Methods
Materials
mRNA coding for a green fluorescent protein (GFP mRNA) and FS-315 follistatin protein (FS-344 mRNA) were obtained from TriLink BioTechnologies (San Diego, CA). The obtained mRNA molecules were modified with an anti-reverse cap analog (ARCA cap) and 5-methylcytidine (5meC) that increased ribosomal translation efficiency and nuclease stability, respectively. N-hydroxysuccinimide (NHS)-functionalized polyethylene glycol (PEG, 2 kDa) was obtained from NOF Corporation (White Plains, NY). Dulbecco's phosphate buffered saline (DPBS) and Roswell Park Memorial Institute (RPMI) medium were purchased from Mediatech (Manassas, VA). All other chemicals and supplies were obtained from VWR International (Radnor, PA).
Preparation and characterization of mRNA-loaded nanoparticles
The employed PEG[Glu(DET)]2 polymer was synthesized and characterized according to the procedure described in Supplementary Material (Figure S1-S4) [43]. To prepare the mRNA-loaded nanoparticles, mRNA molecules were complexed with the polymer molecules via electrostatic interactions at the following amine/phosphate (N/P) ratios: 0.5, 1, 2, 4, 6 and 8. The N/P ratio was calculated by relating the number of positively charged primary amine groups on the PEG[Glu(DET)]2 polymer (90 groups per molecule) with the number of negatively charged phosphate groups of mRNA (1231 groups per molecule). mRNA at a constant concentration of 0.24 μM (0.1 mg/mL) was mixed in MilliQ water with the polymer at various concentrations such as 1.65 μM (0.033 mg/mL), 3.3 μM (0.066 mg/mL), 6.6 μM (0.132 mg/mL), 13.2 μM (0.264 mg/mL), 19.8 μM (0.395 mg/mL) and 26.4 μM (0.527 mg/mL) to prepare nanoparticles at the N/P ratios of 0.5, 1, 2, 4, 6 and 8, respectively. The reaction mixtures were vortexed at 1500 rpm for 30 min at room temperature. The efficiency of the polymer to complex mRNA into nanoparticles at various N/P ratios was evaluated by a gel retardation assay (Figure S5). Of note, nanoparticles are only formed when the polymer molecules interact with the mRNA molecules. To modify the surface of the nanoparticles with PEG, the 2 kDa NHS-activated PEG was added to each reaction mixture (final concentration 5 mg/mL) and then vortexed at 1500 rpm for an additional 30 min at room temperature (final mRNA concentration 0.1 mg/mL). Cryogenic transmission electron microscopy (cryo-TEM), dynamic light scattering (DLS), and Fourier transform infrared spectroscopy (FTIR) were used to characterize the synthesized polymer, and the final nanoparticles. The detailed procedures are provided in Supplementary Material.
Cell lines
The HEK293 human embryonic kidney cell line was obtained from ATCC (Manassas, VA) and the Huh7 hepatocyte-derived cellular carcinoma cell line was kindly provided by OHSU RIPPS (Portland, OR). Cells were cultured using RPMI 1640 medium supplemented with fetal bovine serum (10%) and penicillin-streptomycin (1.2%). Since the developed nanoparticles are aimed to deliver mRNA to liver cells, the Huh7 hepatocytes were employed to evaluate their potential toxicity and efficiency. In addition, kidneys are the major organs for nanoparticle excretion, and therefore, HEK293 kidney cells were also used to assess the toxicity of the developed nanoparticles on these organs and evaluate the ability of kidney cells to produce follistatin protein coded by the delivered mRNA.
Cellular cytotoxicity assay
To evaluate the cytotoxicity of the developed nanoparticles, Huh7 cells were seeded in 96-well plates at a density of 1×104 cells/well and incubated for 24 h at 37 °C in a humidified atmosphere of 5% CO2 (v/v). Subsequently, the cells were incubated for 48 h with 200 μL of RPMI 1640 medium containing either non- or PEG-modified nanoparticles prepared at the following N/P ratios: 0.5, 1, 2, 4, 6 and 8. The final FS-344 mRNA concentration in each well was 500 ng/mL. Cells incubated in fresh medium were employed as controls, and cell viability was determined using a modified Calcein AM assay according to a previously published procedure [44].
Cellular internalization and protein production assays
To evaluate cellular internalization and protein production, Huh7 cells were seeded in 6-well plates at a density of 2×105 cells/well and incubated for 48 h with the PEG-modified nanoparticles (N/P = 2) containing Cy5-labeled GFP mRNA. The final mRNA concentration was 500 ng/mL in each well. Cells were subsequently rinsed in DPBS, and fluorescence images were acquired using the EVOS FL System (Grand Island, NY, Life Technologies).
Follistatin ELISA assay
To verify that the delivered FS-344 mRNA is capable of secreted follistatin protein production, Huh7 and HEK293 cells seeded at 1×104 cells/well density in 96-well plates were incubated for 48 h with two different concentrations of FS-344 mRNA (250 and 500 ng/mL) loaded in the PEG-modified nanoparticles (N/P = 2). Then, cell culture medium was collected from each well and analyzed for follistatin concentration using an ELISA assay according to the manufacturer's protocol (RayBiotech, Norcross, GA).
Animals
Male C57BL/6J and Swiss Webster mice were obtained from Charles River Laboratories (Wilmington, MA, USA) and animal studies were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Oregon Health & Science University.
Evaluation of nanoplatform acute toxicity
Fifteen Swiss Webster mice aged 6-7 weeks were subcutaneously injected into the right flank with 0.1 mL of the nanoparticles containing FS-344 mRNA at a dose of 0.5 mg/kg. In the control group, five mice were injected with saline. Blood samples were collected by cardiac puncture at 8, 48 and 72 h post injection (5 mice per time point) and submitted for blood analysis to IDEXX laboratory (Veterinary Diagnostic, Portland, OR). To evaluate the effect of mRNA-loaded nanoparticles on acute liver, renal and muscle toxicity, we measured the concentrations of surrogate biomarkers in the blood for liver function (alanine aminotransferase (ALT), alkaline phosphatase (ALP), aspartate aminotransferase (AST), and gamma-glutamyl transferase (GGT)), muscle and muscle function (creatine kinase (CK)), and kidney function (blood urea nitrogen (BUN)). In addition, the serum levels of proteins and blood electrolytes were evaluated as an indicator of major organ toxicity [45].
Evaluation of the biodistribution of GFP-mRNA nanoparticles in vivo
Thirty Swiss Webster mice were injected subcutaneously into the right flank with 0.1 mL of the nanoparticles loaded with GFP mRNA at a dose of 0.5 mg/kg. Three mice were euthanized at 8, 12, 24, 48, 72, 96, 120, 144 and 168 h following injection and major organs were collected. At each time point, the GFP production and biodistribution were evaluated by measuring green fluorescence intensity in the resected organs with the IVIS imaging system (PerkinElmer, Waltham, MA, USA). Regions of interest were drawn over the livers and kidneys in the fluorescence images, and the average fluorescence signal for each organ was measured using IVIS Living Image™ software. Finally, the livers and kidneys were fixed with formalin, embedded in paraffin, sectioned, and mounted on slides according to previously reported protocols [46]. After staining with DAPI for nuclei visualization, fluorescence images were recorded with an EVOS FL Cell Imaging System.
Evaluation of follistatin production efficiency of the FS-344 mRNA-loaded nanoparticles
Twenty seven Swiss Webster mice aged 6-7 weeks were subcutaneously injected into the right flank with 0.1 mL of the nanoparticles loaded with FS-344 mRNA at a dose of 0.5 mg/kg. Blood samples were collected by cardiac puncture at 8, 12, 24, 48, 72, 96, 120, 144 and 168 h post injection (3 mice per time point) and follistatin serum concentrations were analyzed with an ELISA kit according to the manufacturer's protocol (RayBiotech, Norcross, GA). In the control group, mice were injected with saline.
In a separate experiment, C57BL/6J mice aged 5-6 weeks were injected subcutaneously with nanoparticles loaded with FS-344 mRNA (0.5 mg/kg) every 72 h for 8 weeks, and follistatin serum concentrations were measured 24 h after the last injection as described above.
Evaluation of nanoparticle efficiency to increase lean muscle mass
17-week-old C57BL/6L mice were used to assess the efficiency of the developed nanoparticles to increase lean muscle mass. After baseline muscle and fat mass were measured by magnetic resonance relaxometry (EchoMRI, Echo Medical Systems, Houston, TX), mice in the control and treatment groups (five mice per group) were subcutaneously injected every 72 h for eight weeks (total 16 injections) with saline or with nanoparticles containing FS-344 mRNA (0.5 mg/kg), respectively. Animals were then monitored, and body composition (lean and fat mass) was measured weekly. Upon completion of the treatment, mice were euthanized, and the major organs and gastrocnemius and triceps muscles were resected, and wet weight was measured. Finally, blood samples were collected and myostatin and activin A serum concentrations were analyzed with ELISA kits according to the manufacturers' protocol (RayBiotech, Norcross, GA).
Quantitative qRT-PCR
RNA was extracted and purified with RNeasy Mini kits (Qiagen Inc.), then reverse transcribed into cDNA with a High Capacity cDNA Reverse Transcription Kit (Life Technologies). qRT-PCR was performed using TaqMan reagents from Life Technologies (assay numbers: Tnfα - Mm00443260_g1, Il-6 - Mm00446190_m1, Il-1b - Mm01336189_m1, Crp - Mm00432680_g1, Apcs - Mm00488099_g1). Inflammatory response genes in liver (tumor necrosis factor α, Tnfα; interleukin 6, Il-6; interleukin 1 beta, Il-1b; C-reactive protein, Crp; and amyloid P component, serum, Apcs) were normalized to 18S and compared between FS-344 mRNA nanoparticle-treated mice and untreated controls.
Statistical analysis
The data were analyzed using descriptive statistics and presented as mean values ± standard deviation (SD) from 3-5 separate measurements. Groups were compared using independent sample Student's t-tests. Differences between groups were considered statistically significant at p < 0.05.
Results
Preparation and in vitro characterization of mRNA-loaded nanoparticles
mRNA-loaded polymeric nanoparticles were prepared by complexation of mRNA and N-substituted polyethylene glycol-diblock-polyglutamide (PEG[Glu(DET)]2) containing 1,2-diaminoethane (DET) as side chains (Figure 1A).
Preparation of mRNA-loaded polymeric nanoparticles. (A) Complexation of negatively charged FS-344 mRNA by positively charged PEG[pGlu(DET)]2 polymers into nanoparticles. (B) Modification of the mRNA-loaded nanoparticles by conjugation of PEG to polymer amino groups on the nanoparticle surface via amide bonds.
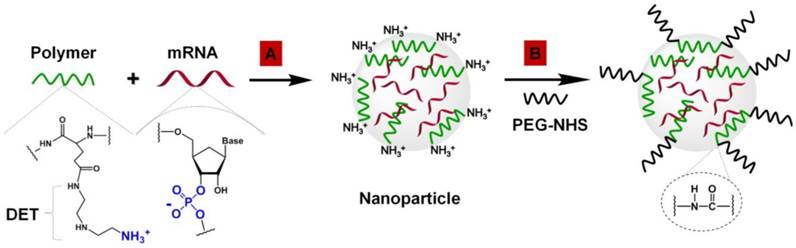
A gel retardation assay validated the high efficiency of the PEG[Glu(DET)]2 polymer to complex mRNA at various N/P (positive polymer amine/negative mRNA phosphate) ratios via spontaneous electrostatic interactions between the negatively charged phosphate groups of the mRNA and positively charged primary amines of the DET (Figure S5).
The protonated amines in the polymer structure (Figure 1A), which are important for mRNA complexation, introduced a positive charge onto the surface of the nanoparticles. For example, the nanoparticles prepared at an N/P ratio of 2 have a zeta potential of +19.2 ± 3.7 mV. In vitro studies suggest that the positively charged nanoparticles prepared at the different N/P ratios substantially decreased the viability of Huh7 hepatocytes (Figure 2A). To diminish the toxicity associated with high positive charge and to improve the biocompatibility of the prepared nanoparticles, their surfaces were modified with a 2 kDa PEG by coupling NHS groups located on the terminal end of PEG to DET amines on the nanoparticle surface via amide bonds (Figure 1B). As a result, the zeta potential of the PEG-modified nanoparticles (N/P = 2) decreased from +19.2 ± 3.7 mV to +4.3 ± 1.5 mV, and these nanoparticles did not compromise the viability of Huh7 and HEK293 cells (Figure 2A and Figure S6). The final nanoparticles (N/P = 2) have a slightly positive surface charge (+4.3 ± 1.5 mV), spherical shape (Figure 2B), a hydrodynamic diameter of 80.0 ± 1.5 nm (Figure 2C), and a relatively narrow size distribution (polydispersity index (PDI) = 0.23 ± 0.02).
(A) Viability of Huh7 human liver cells treated for 48 h with non- and PEG-modified mRNA nanoparticles prepared at the following N/P ratios: 0.5, 1, 2, 4, 6 and 8. Representative cryo-TEM image (B) and dynamic light scattering profile (C) of the PEG-modified, mRNA-loaded nanoparticles (N/P = 2). (D-E) Representative fluorescence microscopy images of Huh7 cells incubated for 48 h with nanoparticles loaded with the Cy5-labeled mRNA encoding for GFP. Images represent red fluorescence signal from Cy5-labeled mRNA (D) and green fluorescence signal generated by the expressed GFP (E). (F) Follistatin concentrations in the cell culture media quantified using ELISA after incubation of Huh7 and HEK293 human kidney cells for 48 h with and without nanoparticles containing FS-344 mRNA (250 ng/mL and 500 ng/mL). Results are not normalized for nanoparticle cell uptake differences. *p < 0.05 when compared with follistatin concentration produced by non-treated cells at the same time point.
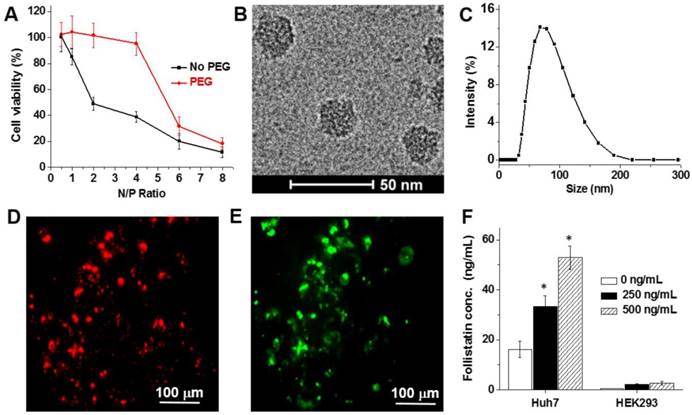
In vitro studies demonstrated that the prepared nanoparticles effectively delivered encapsulated mRNA into the hepatic cells, and the released mRNA molecules were translated into the corresponding protein. After a 48 h incubation period of Huh7 cells with nanoparticles containing Cy5-labeled mRNA coding for a green fluorescent protein (GFP mRNA), strong red (Cy5) and green (GFP) fluorescence signals were detected in the cells (Figure 2D-E). Finally, the in vitro studies validated that the FS-344 mRNA delivered by the developed nanoparticles is capable of secreted follistatin production and control of protein concentration in a dose-dependent manner (Figure 2F). When compared to non-treated cells, 2.1- and 3.3-fold increases in follistatin protein secretion were detected after incubation of Huh7 cells with 250 ng/mL and 500 ng/mL mRNA concentrations loaded into the nanoparticles, respectively.
Evaluation of biodistribution, protein production efficiency, and acute toxicity of mRNA-loaded nanoparticles
The green fluorescent signal generated by GFP was detected in the liver and kidneys (Figure 3 and Figures S7-S8) following a single subcutaneous injection of GFP mRNA-loaded nanoparticles, and the intensity of GFP fluorescence was significantly higher and prolonged in the liver when compared to the kidneys. Fluorescence signal was observed in the liver as early as 4 h following administration, reached its maximum intensity at 12 h, and was still detectable at 168 h post-injection. In contrast, negligible fluorescence was detected in the kidneys 4 h after administration, reaching its maximum intensity at 24 h, and disappearing at 144 h (Figure 3 and Figure S7). Fluorescence imaging of tissue sections confirmed the results obtained with the IVIS Imaging System (Figure S9).
GFP production in the liver and kidneys at various time points following subcutaneous injection of the nanoparticles loaded with GFP mRNA at a dose of 0.5 mg/kg.
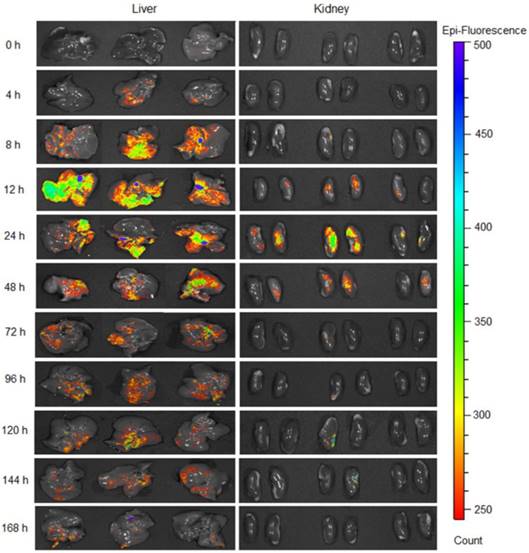
The observed nanoparticle biodistribution is related to the fact that a predominant portion of any systemically injected nanoparticle faces either sequestration by the liver or filtration by the kidney [47, 48]. The detected difference in GFP production between the kidney and the liver can be explained by both the physical and physiological characteristics of the different organs. The liver has a larger surface area than the kidneys, and is the main metabolic detoxification organ of the body. Therefore, it is reasonable that a higher amount of nanoparticles rapidly accumulated in the liver and therefore the GFP signal was detected 4 h after nanoparticle administration. Finally, the in vitro data suggested that the liver cells outperformed the kidney cells by providing a 20-fold increase in protein production (Figure 2F).
We also demonstrated that the FS-344 mRNA-loaded nanoplatform is effective at increasing serum follistatin levels in mice following subcutaneous administration. It was observed that 8 h after injection, a 2.4-fold increase in follistatin serum level was achieved (Figure 4A) and the elevated follistatin level persisted up to 72 h. A minimal decrease (3.9 ng/mL) in follistatin serum level was observed from the 8 h time point (19.9 ng/mL) until the 72 h time point (16.0 ng/mL). Of note, follistatin levels returned to baseline at ~96 h. These data justified an every 3-day (72 h) subcutaneous injection dosing schedule to keep follistatin levels elevated in the serum (Figure 4A).
(A) Follistatin serum levels at various time points after a single subcutaneous injection of nanoparticles containing FS-344 mRNA (0.5 mg/kg). (B) Follistatin serum levels 24 h after the 16th injection. Mice were injected every 72 h for eight weeks with the nanoparticles (treatment) or saline (control). Serum concentrations of follistatin were analyzed using ELISA. *p < 0.05 when compared with the protein level in serum of non-treated animals.
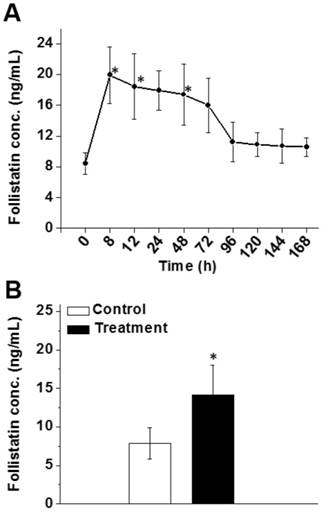
We also validated the efficiency of PEG-modified nanoparticles to elevate serum follistatin levels after chronic treatment (16 injections over 8 weeks). The increase in follistatin serum level was 1.8-fold following the last injection (Figure 4B).
Despite the accumulation of the developed nanoplatform in the liver and kidneys and efficient production of secreted follistatin protein, mice displayed no sign of toxicity (e.g., change in behavior, appearance, etc.) after injection of the nanoparticles loaded with FS-344 mRNA (0.5 mg/kg). The measured serum levels of blood electrolytes, proteins and surrogate biomarkers for liver (ALP, ALT, AST, and GGT), muscle (CK), and kidney (BUN) functions in the mice treated with FS-344 mRNA-loaded nanoparticles at various time points post-injection were not different than those of non-treated mice, suggesting that the prepared nanoplatform does not exhibit any acute toxicity (Figure 5A-B and Figure S10).
The blood levels of alkaline phosphatase (ALP), alanine aminotransferase (ALT), aspartate aminotransferase (AST), and gamma-glutamyl transferase (GGT) illustrating liver function (A), creatine kinase (CK) illustrating heart and skeletal muscle function (A), and blood urea nitrogen (BUN) illustrating kidney function (B) in non-treated mice (0 h) and mice subcutaneously injected with FS-344 mRNA-loaded nanoparticles (0.5 mg/kg), assessed at different time points. (C) Expressions of inflammatory genes in the livers of non-treated mice (control) and mice injected every 72 h for 8 weeks with the FS-344 mRNA-loaded nanoparticles (0.5 mg/kg).
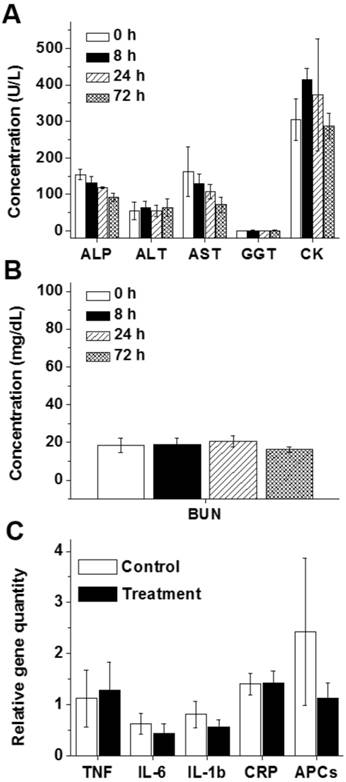
Finally, to evaluate whether the developed therapy can cause liver inflammation, expression of a set of inflammatory genes was measured in the livers of mice after 16 repeated injections of FS-344 mRNA-loaded nanoparticles. We did not detect any significant increase in the expressions of the tested inflammatory genes in the livers of the treated mice when compared to those of the control group (Figure 5C).
Evaluation of nanoparticles efficiency to increase lean muscle mass
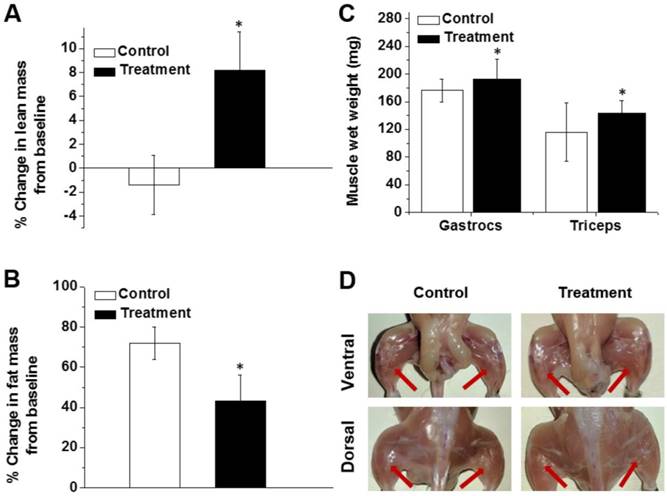
After 8 weeks of treatment, the lean mass content of the mice in the treatment group was 8.2% higher than their baseline values (Figure 6A). In contrast, the relative lean muscle mass content of the animals in the control group decreased by 1.4% during the same period of time (Figure 6A). Body composition analysis further demonstrated that body fat was 28.6% lower in the nanoparticle-treated mice than the non-treated controls, indicating that follistatin is also involved, directly or indirectly, in the regulation of fat metabolism (Figure 6B). The gastrocnemius and triceps wet weights were 9.6% and 23.9% greater in treated mice than the control group (Figure 6C), supporting the hypothesis that administration of mRNA-loaded nanoparticles stimulates muscle mass gain. Gross observation of nanoparticles-treated animals also demonstrated an increase in the size of muscles compared with control animals (Figure 6D).
Percent change in lean muscle mass (A) and fat mass (B) from baseline in non-treated mice (control) and mice treated with FS-344 mRNA-loaded nanoparticles (0.5 mg/kg). Wild-type 17-week-old C57BL/6L mice were subcutaneously injected with either saline (control) or the nanoparticles every three days for 8 weeks. The lean and fat mass values obtained prior to treatment were used as a baseline for subsequent analyses. The average baseline values were not significantly different among the treatment and control groups. *p < 0.05 when compared with non-treated animals. (C) Average weights of gastrocnemius and triceps muscles resected from non-treated and treated mice. *p < 0.05 when compared with tissue weights resected from non-treated mice. (D) Photographs of hind leg muscles of non-treated and treated mice. The red arrows denote the gastrocnemius muscles.
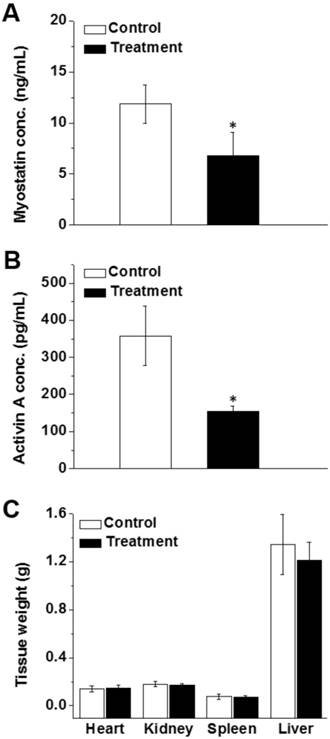
Analysis of blood samples at the end of treatment revealed that the serum levels of activin A and myostatin in mice treated with mRNA-loaded nanoparticles were 1.8 times lower than those of the control animals (Figure 7A-B). These data suggest that the produced follistatin leads to inhibition of circulating activin A and myostatin, thereby causing an increase in lean muscle mass. Finally, the obtained results demonstrate no significant difference in the weights of heart, liver, spleen, and kidneys (Figure 7C).
Serum concentrations of myostatin (A) and activin A (B) after eight weeks of treatment with saline (control) and FS-344 mRNA-loaded nanoparticles (treatment). Serum concentrations of myostatin and activin A were analyzed using ELISA assay. *p < 0.05 when compared with protein levels in the serum of non-treated animals. (C) Average weights of hearts, kidneys, spleens, and livers resected from non-treated and treated mice. *p < 0.05 when compared with non-treated mice.
Discussion
Many preclinical and clinical studies demonstrate that a therapeutic modality based on inhibition of TGF-β superfamily proteins/ ActRIIB signaling by follistatin (FS-315) is a promising strategy for treating muscle wasting [43, 49]. Several pharmacological approaches are presently being investigated to increase FS-315 level in the body including the administration of (i) recombinant follistatin [27, 50] and (ii) adenoviral vectors carrying an expression cassette for FS-315 [30, 49, 51, 52]. These approaches, however, have various technical and safety limitations and therefore, we developed a novel strategy for elevating FS-315 serum levels based on mRNA-loaded nanoparticles that can overcome most of the existing barriers. Since mRNA-based follistatin therapy requires repeated injections, an important reason for developing the subcutaneously administered nanoparticles is the potential for self-administration by patients and a prolonged therapeutic window as a result of the depot effect. To the best of our knowledge, this is the first demonstration that mRNA-loaded nanoparticles are able to enter into systemic circulation following a subcutaneous injection, accumulate in the liver, and internalize into the hepatic cells where the released mRNA is translated into the encoded protein. The intravenous route is predominantly used for administration of various lipid nanoparticles that are currently considered the most efficient non-viral vectors for mRNA delivery to the liver [39, 53, 54]. Two separate studies revealed that subcutaneous administration of mRNA-loaded lipid nanoparticles resulted in protein production only at the site of injection [55, 56]. In contrast, our in vivo results clearly demonstrated that within 4 h after subcutaneous injection, the developed polymeric nanoparticles provide delivery of the GFP mRNA to the liver where the encoded protein is robustly expressed (Figure 3 and Figure S8). We also validated that the subcutaneously administered nanoparticles offer sustained GFP production in the liver over seven days (168 h) following a single injection (Figure 3). Our results are in good agreement with previous reports demonstrating that subcutaneously injected nanoparticles loaded with therapeutic agents different than mRNA (e.g., insulin and siRNA) can enter systemic circulation and accumulate in the liver [57, 58].
Our in vivo results further confirmed that repeated subcutaneous injections of the nanoparticles loaded with a low dose of FS-344 mRNA (0.5 mg/kg) provide sustained and robust production of secreted FS-315 (Figure 4A-B) that is capable of decreasing serum levels of both myostatin and activin A (Figure 7A-B), thereby increasing muscle mass in 17-week-old C57BL/6L mice (Figure 6A). The rationale behind utilizing post-adolescence mice in our study was to monitor changes in lean and fat body mass in adults with a static body composition rather than treating younger mice whose growth could be attributed to anabolic growth typically found in adolescent mice [59].
We also validated that the mRNA-loaded polymeric nanoparticles are safe and do not cause obvious liver toxicity or inflammation upon repeated administration (Figure 5). Finally, our results indicate that the follistatin gene therapy is selective to skeletal muscle tissue and does not increase the weights of the rest of the organs including the heart (Figure 7C). This is beneficial due to the fact that cardiac hypertrophy leads to decreases in cardiac output and function leading to eventual death.
The observed high efficiency and low toxicity of the developed nanoparticles are related to the structure of the employed PEG[pGlu(DET)]2 polymer (Figure 1A and Figure S1). Previous studies demonstrated that siRNA-loaded nanoparticles assembled from the polymers containing DET as side chains have remarkable gene transfection efficiency without toxicity within various cell types [60-63]. This efficiency is attributed to the unique DET structure (Figure 1A), where the mono-protonated form of DET at physiological pH is converted to a di-protonated form upon exposure to the acidic pH of the late endosome and lysosome [64]. The DET-containing polymer exhibits pH-selective membrane destabilization in the endosomes/lysosomes and efficiently exits into the cytoplasm with no compromise to the membrane integrity of cytoplasmic organelles. In addition, modification of the prepared PEG[pGlu(DET)]2-mRNA nanoparticles with PEG further improves their safety by diminishing positive surface charges (Figure 1B and Figure 2A).
The developed mRNA therapy designed to express endogenous follistatin is a more efficacious therapeutic route than systemic administration of recombinant protein for several reasons. Datta-Mannan et al. reported that after a single intravenous administration to mice, FST-315 protein is cleared rapidly from circulation (half-life ~2 h) [65]. In contrast, by injecting nanoparticles loaded with a follistatin mRNA, we demonstrated that a protein reservoir is created in the liver that produces follistatin over a 72 h period (Figure 4A). Therefore, multiple daily injections of recombinant follistatin would be required to achieve similarly elevated serum levels. The cost of protein production and purification is high, and previous studies revealed that the intrinsic pharmacokinetic properties of native FST-315 are poorly suited for acting as a parenteral biotherapeutic agent [65]. Therefore, the cost savings of delivering native follistatin via nanoparticle mRNA delivery are likely to be substantial. Finally, the possibility of an immunogenic reaction to an injected recombinant follistatin dramatically increases when compared to an endogenously produced protein.
To date, follistatin gene therapy based on delivery of cDNA via an adenoviral vector to patients demonstrated great potential for increasing muscle mass in humans with degenerative muscle disorders [30, 49, 51, 52]. Viral delivery of the cDNA, however, has many technical and safety issues, including the necessity for multiple direct site muscular injections that are reported to be extremely painful. In the clinical trial, the viral vector was injected into three muscle groups of the quadriceps, and four injections were delivered per muscle [30]. Furthermore, immunosuppressive drugs including prednisone must be administered to avoid harmful immune and inflammatory responses associated with the viral vector [30]. It is also apparent that due to differences in immune responses within each patient, viral clearance is variable and therefore follistatin treatment efficacy is inconsistent. Finally, there are concerns regarding the potential for permanent genomic insertion of the viral gene therapy cargo, and the production and scaling-up of clinical grade viral vectors for gene therapy is known to be technically challenging and expensive [66, 67]. The developed mRNA-based approach circumvents the majority of the problems associated with viral follistatin DNA delivery. Our data demonstrate that after a simple subcutaneous injection there is no evidence of immune activation, which eliminates the need for immunosuppressive therapy. We also demonstrated the response to dose titration (Figure 2F), although it is likely that the relationship between mRNA delivery and protein production will be complex and specific to each protein product. Obviously, there are no concerns about permanent genomic insertion with this method, and the reagent is technically straightforward to prepare, greatly facilitating clinical translation. Finally, an important advantage of our approach compared to viral delivery is the ability to cease a therapy at any time point. Most data suggest that viral delivery will result in the production of the protein of interest for months to years after the last injection [68]. If an adverse effect is caused by the produced protein, the reaction would likely persist for an extended time with the chance of causing life-threatening complications. The therapy reported herein allows follistatin levels to return to baseline 4 days after a single injection, greatly decreasing the dangers of a persistent therapy (Figure 4A).
Finally, our studies demonstrated that subcutaneous administration of FS-344 mRNA-loaded nanoparticles reduced fat accumulation in the body (Figure 6B). These results are in agreement with previous reports indicating that overexpression of the FS-344 transgene in pigs and mice enhanced muscle mass while reducing fat accumulation [69-71]. Studies have suggested that inhibition of myostatin in muscle is responsible for the decreased fat mass. For example, Nakatani et al. demonstrated that transgenic expression of a follistatin-derived peptide that retains myostatin inhibitory activity while not affecting the bioactivity of activins resulted in decreased fat accumulation in mice [72].
Conclusions
We designed and evaluated a novel nanoplatform for efficient delivery of FS-344 mRNA to the liver following subcutaneous administration. The obtained results validated that the delivered mRNA directs the hepatic cellular machinery to produce follistatin, a secreted glycoprotein that inhibits TGF-β superfamily proteins. The developed nanoplatform is safe and provides a therapeutic pathway to increase muscle mass and to restrict fat accumulation, which is significant for the treatment of a variety of muscle wasting disorders including muscular dystrophies. The obtained results advocate for further evaluation of the developed nanoparticle-based gene therapy in various animal models of muscle atrophy.
Abbreviations
ActRIIB: activin type IIB receptor; ALT: alanine transaminase; ALP: alkaline phosphatase; AST: aspartate aminotransferase; BUN: blood urea nitrogen; CK: creatine kinase; cryoTEM: cryogenic transmission electron microscopy; DLS: dynamic light scattering; FS: follistatin; GGT: gamma-glutamyl transferase; IACUC: the Institutional Animal Care and Use Committee; mRNA: messenger RNA; PEG: polyethylene glycol; TGF-β: transforming growth factor-β.
Acknowledgements
This research was supported by the College of Pharmacy at Oregon State University (OSU), the National Pancreas Foundation, NIH/NBIB (1R15EB020351-01A1), and OHSU Knight Cancer Institute and Friends of Doernbecher. The funding sources had no involvement in the collection, analysis and interpretation of the data or in the decision to submit the article for publication. Electron microscopy was performed using the Multiscale Microscopy Core (MMC) at Oregon Health & Science University with technical support from the (OHSU)-FEI Living Lab and the Center for Spatial Systems Biomedicine (OCSSB).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov. 2015;14:58-74
2. Gallagher IJ, Stephens NA, MacDonald AJ, Skipworth RJ, Husi H, Greig CA. et al. Suppression of skeletal muscle turnover in cancer cachexia: evidence from the transcriptome in sequential human muscle biopsies. Clin Cancer Res. 2012;18:2817-27
3. Du Y, Ge J, Li Y, Ma PX, Lei B. Biomimetic elastomeric, conductive and biodegradable polycitrate-based nanocomposites for guiding myogenic differentiation and skeletal muscle regeneration. Biomaterials. 2018;157:40-50
4. Ge J, Liu K, Niu W, Chen M, Wang M, Xue Y. et al. Gold and gold-silver alloy nanoparticles enhance the myogenic differentiation of myoblasts through p38 MAPK signaling pathway and promote in vivo skeletal muscle regeneration. Biomaterials. 2018;175:19-29
5. Kwee BJ, Mooney DJ. Biomaterials for skeletal muscle tissue engineering. Curr Opin Biotechnol. 2017;47:16-22
6. Liu J, Saul D, Boker KO, Ernst J, Lehman W, Schilling AF. Current methods for skeletal muscle tissue repair and regeneration. Biomed Res Int. 2018;2018:1984879
7. Mao AS, Mooney DJ. Regenerative medicine: Current therapies and future directions. Proc Natl Acad Sci USA. 2015;112:14452-9
8. Marcinczyk M, Elmashhady H, Talovic M, Dunn A, Bugis F, Garg K. Laminin-111 enriched fibrin hydrogels for skeletal muscle regeneration. Biomaterials. 2017;141:233-42
9. Schiaffino S, Pereira MG, Ciciliot S, Rovere-Querini P. Regulatory T cells and skeletal muscle regeneration. FEBS J. 2017;284:517-24
10. Tan SJ, Fang JY, Wu Y, Yang Z, Liang G, Han B. Muscle tissue engineering and regeneration through epigenetic reprogramming and scaffold manipulation. Sci Rep. 2015;5:16333
11. Han HQ, Mitch WE. Targeting the myostatin signaling pathway to treat muscle wasting diseases. Curr Opin Support Palliat Care. 2011;5:334-41
12. Loumaye A, de Barsy M, Nachit M, Lause P, Frateur L, van Maanen A. et al. Role of Activin A and myostatin in human cancer cachexia. J Clin Endocrinol Metab. 2015;100:2030-8
13. McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83-90
14. Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA. 2001;98:9306-11
15. Elkina Y, von Haehling S, Anker SD, Springer J. The role of myostatin in muscle wasting: an overview. J Cachexia Sarcopenia Muscle. 2011;2:143-51
16. Latres E, Mastaitis J, Fury W, Miloscio L, Trejos J, Pangilinan J. et al. Activin A more prominently regulates muscle mass in primates than does GDF8. Nat Commun. 2017;8:15153
17. Costelli P, Muscaritoli M, Bonetto A, Penna F, Reffo P, Bossola M. et al. Muscle myostatin signalling is enhanced in experimental cancer cachexia. Eur J Clin Invest. 2008;38:531-8
18. Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S. et al. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci USA. 1998;95:14938-43
19. Lenk K, Schur R, Linke A, Erbs S, Matsumoto Y, Adams V. et al. Impact of exercise training on myostatin expression in the myocardium and skeletal muscle in a chronic heart failure model. Eur J Heart Fail. 2009;11:342-8
20. Togashi Y, Kogita A, Sakamoto H, Hayashi H, Terashima M, de Velasco MA. et al. Activin signal promotes cancer progression and is involved in cachexia in a subset of pancreatic cancer. Cancer Lett. 2015;356:819-27
21. Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q. et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142:531-43
22. Amato AA, Sivakumar K, Goyal N, David WS, Salajegheh M, Praestgaard J. et al. Treatment of sporadic inclusion body myositis with bimagrumab. Neurology. 2014;83:2239-46
23. Smith RC, Lin BK. Myostatin inhibitors as therapies for muscle wasting associated with cancer and other disorders. Curr Opin Support Palliat Care. 2013;7:352-60
24. Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM. et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63:561-71
25. Aversa Z, Costelli P, Muscaritoli M. Cancer-induced muscle wasting: latest findings in prevention and treatment. Ther Adv Med Oncol. 2017;9:369-82
26. Gilson H, Schakman O, Kalista S, Lause P, Tsuchida K, Thissen JP. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am J Physiol Endocrinol Metab. 2009;297:E157-64
27. Yaden BC, Croy JE, Wang Y, Wilson JM, Datta-Mannan A, Shetler P. et al. Follistatin: a novel therapeutic for the improvement of muscle regeneration. J Pharmacol Exp Ther. 2014;349:355-71
28. Xia Y, Schneyer AL. The biology of activin: recent advances in structure, regulation and function. J Endocrinol. 2009;202:1-12
29. Inouye S, Guo Y, DePaolo L, Shimonaka M, Ling N, Shimasaki S. Recombinant expression of human follistatin with 315 and 288 amino acids: chemical and biological comparison with native porcine follistatin. Endocrinology. 1991;129:815-22
30. Mendell JR, Sahenk Z, Malik V, Gomez AM, Flanigan KM, Lowes LP. et al. A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol Ther. 2015;23:192-201
31. Keutmann HT, Schneyer AL, Sidis Y. The role of follistatin domains in follistatin biological action. Mol Endocrinol. 2004;18:228-40
32. Amthor H, Nicholas G, McKinnell I, Kemp CF, Sharma M, Kambadur R. et al. Follistatin complexes Myostatin and antagonises Myostatin-mediated inhibition of myogenesis. Dev Biol. 2004;270:19-30
33. Lerch TF, Shimasaki S, Woodruff TK, Jardetzky TS. Structural and biophysical coupling of heparin and activin binding to follistatin isoform functions. J Biol Chem. 2007;282:15930-9
34. Mendell JR, Sahenk Z, Al-Zaidy S, Rodino-Klapac LR, Lowes LP, Alfano LN. et al. Follistatin gene therapy for sporadic inclusion body myositis improves functional outcomes. Mol Ther. 2017;25:870-9
35. David RM, Doherty AT. Viral Vectors: The road to reducing genotoxicity. Toxicol Sci. 2017;155:315-25
36. van der Loo JC, Wright JF. Progress and challenges in viral vector manufacturing. Hum Mol Genet. 2016;25:R42-52
37. Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15:541-55
38. Sahin U, Kariko K, Tureci O. mRNA-based therapeutics-developing a new class of drugs. Nat Rev Drug Discov. 2014;13:759-80
39. Ramaswamy S, Tonnu N, Tachikawa K, Limphong P, Vega JB, Karmali PP. et al. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc Natl Acad Sci USA. 2017;114:E1941-E50
40. Kaczmarek JC, Kowalski PS, Anderson DG. Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med. 2017;9:60
41. Islam MA, Reesor EK, Xu Y, Zope HR, Zetter BR, Shi J. Biomaterials for mRNA delivery. Biomater Sci. 2015;3:1519-33
42. Hansen JS, Rutti S, Arous C, Clemmesen JO, Secher NH, Drescher A. et al. Circulating Follistatin is liver-derived and regulated by the glucagon-to-insulin ratio. J Clin Endocrinol Metab. 2016;101:550-60
43. Itaka K, Ishii T, Hasegawa Y, Kataoka K. Biodegradable polyamino acid-based polycations as safe and effective gene carrier minimizing cumulative toxicity. Biomaterials. 2010;31:3707-14
44. Taratula O, Kuzmov A, Shah M, Garbuzenko OB, Minko T. Nanostructured lipid carriers as multifunctional nanomedicine platform for pulmonary co-delivery of anticancer drugs and siRNA. J Control Release. 2013;171:349-57
45. Shah VM, Nguyen DX, Alfatease A, Bracha S, Alani AW. Characterization of pegylated and non-pegylated liposomal formulation for the delivery of hypoxia activated vinblastine-N-oxide for the treatment of solid tumors. J Control Release. 2017;253:37-45
46. Schumann C, Taratula O, Khalimonchuk O, Palmer AL, Cronk LM, Jones CV. et al. ROS-induced nanotherapeutic approach for ovarian cancer treatment based on the combinatorial effect of photodynamic therapy and DJ-1 gene suppression. Nanomedicine. 2015:11 1961-70
47. Tsoi KM, MacParland SA, Ma XZ, Spetzler VN, Echeverri J, Ouyang B. et al. Mechanism of hard-nanomaterial clearance by the liver. Nat Mater. 2016;15:1212-21
48. Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-liver interactions: Cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332-48
49. Rodino-Klapac LR, Haidet AM, Kota J, Handy C, Kaspar BK, Mendell JR. Inhibition of myostatin with emphasis on follistatin as a therapy for muscle disease. Muscle Nerve. 2009;39:283-96
50. D'Orlando C, Marzetti E, Francois S, Lorenzi M, Conti V, di Stasio E. et al. Gastric cancer does not affect the expression of atrophy-related genes in human skeletal muscle. Muscle Nerve. 2014;49:528-33
51. Kota J, Handy CR, Haidet AM, Montgomery CL, Eagle A, Rodino-Klapac LR. et al. Follistatin gene delivery enhances muscle growth and strength in nonhuman primates. Sci Transl Med. 2009;1:6ra15
52. Mendell JR, Rodino-Klapac L, Sahenk Z, Malik V, Kaspar BK, Walker CM. et al. Gene therapy for muscular dystrophy: lessons learned and path forward. Neurosci Lett. 2012;527:90-9
53. DeRosa F, Guild B, Karve S, Smith L, Love K, Dorkin JR. et al. Therapeutic efficacy in a hemophilia B model using a biosynthetic mRNA liver depot system. Gene Ther. 2016;23:699-707
54. An D, Schneller JL, Frassetto A, Liang S, Zhu X, Park JS. et al. Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep. 2017;21:3548-58
55. Phua KK, Leong KW, Nair SK. Transfection efficiency and transgene expression kinetics of mRNA delivered in naked and nanoparticle format. J Control Release. 2013;166:227-33
56. Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK. et al. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release. 2015;217:345-51
57. Chen S, Tam YY, Lin PJ, Leung AK, Tam YK, Cullis PR. Development of lipid nanoparticle formulations of siRNA for hepatocyte gene silencing following subcutaneous administration. J Controlled Release. 2014;196:106-12
58. Zhang Z, Cai H, Liu Z, Yao P. Effective enhancement of hypoglycemic effect of insulin by liver-targeted nanoparticles containing cholic acid-modified chitosan derivative. Mol Pharm. 2016;13:2433-42
59. Body weight information for C57BL/6J by the Jackson Laboratory. https://www.jax.org/jax-mice-and-services/strain-data-sheet-pages/body-weight-chart-000664
60. Kanayama N, Fukushima S, Nishiyama N, Itaka K, Jang WD, Miyata K. et al. A PEG-based biocompatible block catiomer with high buffering capacity for the construction of polyplex micelles showing efficient gene transfer toward primary cells. ChemMedChem. 2006;1:439-44
61. Masago K, Itaka K, Nishiyama N, Chung UI, Kataoka K. Gene delivery with biocompatible cationic polymer: pharmacogenomic analysis on cell bioactivity. Biomaterials. 2007;28:5169-75
62. Kim HJ, Ishii A, Miyata K, Lee Y, Wu S, Oba M. et al. Introduction of stearoyl moieties into a biocompatible cationic polyaspartamide derivative, PAsp(DET), with endosomal escaping function for enhanced siRNA-mediated gene knockdown. J Control Release. 2010;145:141-8
63. Kagaya H, Oba M, Miura Y, Koyama H, Ishii T, Shimada T. et al. Impact of polyplex micelles installed with cyclic RGD peptide as ligand on gene delivery to vascular lesions. Gene Ther. 2012;19:61-9
64. Miyata K, Oba M, Nakanishi M, Fukushima S, Yamasaki Y, Koyama H. et al. Polyplexes from poly(aspartamide) bearing 1,2-diaminoethane side chains induce pH-selective, endosomal membrane destabilization with amplified transfection and negligible cytotoxicity. J Am Chem Soc. 2008;130:16287-94
65. Datta-Mannan A, Yaden B, Krishnan V, Jones BE, Croy JE. An engineered human follistatin variant: insights into the pharmacokinetic and pharmocodynamic relationships of a novel molecule with broad therapeutic potential. J Pharmacol Exp Ther. 2013;344:616-23
66. Alba R, Baker AH, Nicklin SA. Vector systems for prenatal gene therapy: principles of adenovirus design and production. Methods Mol Biol. 2012;891:55-84
67. Nadeau I, Kamen A. Production of adenovirus vector for gene therapy. Biotechnol Adv. 2003;20:475-89
68. Haidet AM, Rizo L, Handy C, Umapathi P, Eagle A, Shilling C. et al. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci USA. 2008;105:4318-22
69. Chang F, Fang R, Wang M, Zhao X, Chang W, Zhang Z. et al. The transgenic expression of human follistatin-344 increases skeletal muscle mass in pigs. Transgenic Res. 2017;26:25-36
70. Singh R, Braga M, Reddy ST, Lee SJ, Parveen M, Grijalva V. et al. Follistatin targets distinct pathways to promote brown adipocyte characteristics in brown and white adipose tissues. Endocrinology. 2017;158:1217-30
71. McPherron AC, Lee SJ. Suppression of body fat accumulation in myostatin-deficient mice. J Clin Invest. 2002;109:595-601
72. Nakatani M, Kokubo M, Ohsawa Y, Sunada Y, Tsuchida K. Follistatin-derived peptide expression in muscle decreases adipose tissue mass and prevents hepatic steatosis. Am J Physiol Endocrinol Metabol. 2011;300:E543-53
Author contact
![]() Corresponding author: Oleh Taratula, E-mail address: Oleh.Taratulaedu Tel: 503-346-4704 Collaborative Life Science Building 2730 SW Moody Ave., Mail Code: CL5CP Portland, OR 97201-5042 Or Daniel L. Marks, E-mail address: marksdedu Tel.: 503-494-8307 3181 SW Sam Jackson Park Rd, Mail Code L481 Portland, OR, 97239
Corresponding author: Oleh Taratula, E-mail address: Oleh.Taratulaedu Tel: 503-346-4704 Collaborative Life Science Building 2730 SW Moody Ave., Mail Code: CL5CP Portland, OR 97201-5042 Or Daniel L. Marks, E-mail address: marksdedu Tel.: 503-494-8307 3181 SW Sam Jackson Park Rd, Mail Code L481 Portland, OR, 97239