Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Overview of EBNA1 functional...
Strategies for blocking EBNA1...
Conclusions
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(19):5307-5319. doi:10.7150/thno.26823 This issue Cite
Review
EBNA1-targeted inhibitors: Novel approaches for the treatment of Epstein-Barr virus-associated cancers
Lijun Jiang1, Chen Xie1, Hong Lok Lung2, Kwok Wai Lo4 ![]() , Ga-Lai Law3
, Ga-Lai Law3 ![]() , Nai-Ki Mak2
, Nai-Ki Mak2 ![]() , Ka-Leung Wong1
, Ka-Leung Wong1 ![]()
1. Department of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong SAR, China.
2. Department of Biology, Hong Kong Baptist University, Kowloon Tong, Hong Kong SAR, China.
3. Department of Applied Biological and Chemical Technology, Hong Kong Polytechnic University, Hung Hom, Hong Kong SAR, China.
4. Department of Anatomical and Cellular Pathology, The Chinese University of Hong Kong, Hong Kong SAR, China.
Received 2018-4-22; Accepted 2018-8-14; Published 2018-10-22
Abstract

Epstein-Barr virus (EBV) infects more than 90% of humans worldwide and establishes lifelong latent infection in the hosts. It is closely associated with endemic forms of a wide range of human cancers and directly contributes to the formation of some. Despite its critical role in cancer development, no EBV- or EBV latent protein-targeted therapy is available. The EBV-encoded latent protein, Epstein-Barr nuclear antigen 1 (EBNA1), is expressed in all EBV-associated tumors and acts as the only latent protein in some of these tumors. This versatile protein functions in the maintenance, replication, and segregation of the EBV genome and can therefore serve as an attractive therapeutic target to treat EBV-associated cancers. In the last decades, efforts have been made for designing specific EBNA1 inhibitors to decrease EBNA1 expression or interfere with EBNA1-dependent functions. In this review, we will briefly introduce the salient features of EBNA1, summarize its functional domains, and focus on the recent developments in the identification and design of EBNA1 inhibitors related to various EBNA1 domains as well as discuss their comparative merits.
Keywords: EBNA1-targeted inhibitor, fluorescent probe, EBV-associated cancers, EBV, EBNA1
Introduction
Epstein-Barr virus (EBV) has been shown to act as a carcinogenic cofactor in the development of several lymphoid and epithelial cancers. Since its discovery in 1964 [1], EBV was found to have a direct association with a wide range of human malignancies and has been conclusively linked to infectious mononucleosis [2]. As a lymphotropic herpesvirus, EBV can establish lifelong latent infection in the host [3]. It preferentially infects B cells and occasionally epithelial cells (Figure 1A) with distinct entry modes [4]. The entry of EBV into B cells is typically receptor mediated [5-8], while the infection of epithelial cells appears to be mediated through cell-to-cell contact [9-14]. A novel quick and efficient “in-cell infection” method is used to infect epithelial cells [15]. It has been reported that non-muscle myosin heavy chain IIA facilitates EBV infection to nasopharyngeal epithelial cells [16]. In some cases, EBV infects CD4+/CD8+ T cells, nature killer cells, smooth muscle cells, as well as the monocytes, but with less infection efficiency [3, 17, 18].
Overview of EBV infection and key diseases developing from EBV infection. (A) EBV lytic and latent infection in EBV tropistic lymphocytes and epithelial cells. (B) Key diseases resulting from different infected cell lines in individuals.
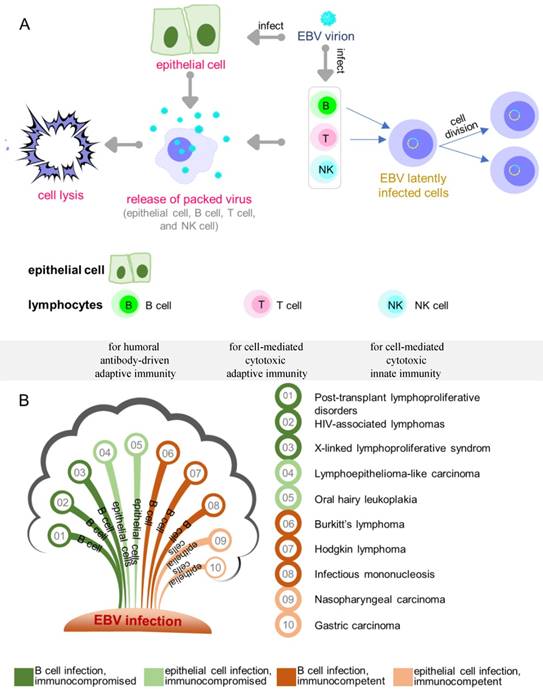
Like all herpesviruses, EBV-infected cells undergo either lytic or latent growth during which only lytic phase produces infectious viruses with concomitant cell lysis (Figure 1A). Depending on the cell type, EBV-infected cells undergo four different latency programs (0, I, II, III). In infected B cells, location and the state of differentiation also determine the form of latency [19]. Naive resting B cells infected by EBV enter latency III (also called growth transcription program) with the expression of all nine known latent proteins, consisting of six nuclear antigens (EBNA 1/2/3A/3B/3C/ EBNA LP) and three membrane proteins (LMP 1/2A/2B). The infected cells exit the resting state to become proliferating lymphoblasts and convert to lymphoblastoid cell lines (LCLs) [20-22]. Latency III is the most-studied latency form as it is expressed when B cells are infected in vitro. While EBV infection in childhood is usually asymptomatic, infection after adolescence frequently causes IM, a type of EBV-associated non-malignancy where latency III is expressed. EBV latency III in immunocompromised or post-transplant individuals is strongly associated with lymphoproliferative disorders. Similar to antigen-activated B blasts [23], EBV-infected lymphoblasts might enter follicles, where some of the cells receive survival signals and undergo differentiation in the germinal-center to become memory B cells. Such signals are believed to drive the cells to enter latency II (also called default transcription program), a typical form of latency observed in nasopharyngeal carcinomas, gastric carcinomas, and Hodgkin's lymphoma [24-26]. This latency is presented with a restricted set of expressed latent proteins: EBNA1, LMP1, LMP2A and LMP2B [27-29]. Notably, LMP1 is not expressed in EBV-associated gastric cancer and the secreted protein BARF1 is expressed in epithelial cells [30-32]. Finally, the cells leave the follicles as resting memory B cells, which express only EBNA1 (latency I, also called EBNA1 only program) or no latent proteins at all (latency 0, also called latency program), and circulate between the peripheral blood and Waldeyer's ring [33]. Although latency I and latency 0 are commonly observed in healthy subjects, latency I is also observed in Burkitt's lymphoma. The major diseases associated with EBV latent infection are summarized in Figure 1B.
As discussed above, EBNA1 is expressed in all latency programs except latency 0, and is also present in all EBV-positive tumors and represents the only latent protein in some of these tumors, such as in latency I Burkitt's lymphoma. EBNA1 is involved in the maintenance of EBV episomes in infected cells [34-36]. The plasmid replication requires the binding of EBNA1 to two distinct regions in origin of replication (oriP) [37], the dyad symmetry (DS) and the family of repeats (FR) [35, 36]. DS contains four known EBNA1 recognition sites and is considered the initiation site of DNA replication [38]. The other functional region, FR, is a cluster of 20 tandem copies with each possessing an 18 bp palindromic EBNA1-binding site. FR primarily functions in mitotic segregation and transcriptional activation [39, 40]. It can also regulate DNA replication through blocking the passage of replication forks. No other latent protein is required for the replication and segregation of the viral genome by EBNA1, which is also known to regulate the transcription of other latent proteins by interacting with specific viral promoters.
The role of EBNA1 in proliferating EBV-infected cells has largely been confirmed. Several reports have shown that specific EBNA1 inhibition, including dominant-negative EBNA1 [41-43], and down-regulating of EBNA1 expression by antisense oligodeoxynucleotide result in growth inhibition [44-46], thus validating EBNA1 as a therapeutic target in EBV-infected cells. A recent study employed computational approaches to systematically investigate the structural details of the “druggable” binding sites on the EBNA1 protein, and suggested the feasibility of EBNA1 as a target for drug discovery [47]. Given the essential and unique roles of EBNA1 in EBV-associated diseases, as well as the computational evidence, it is not surprising that EBNA1 has emerged as one of the intensely studied viral proteins among EBV latent proteins and constitutes an attractive but an elusive target for therapeutic intervention of cancers associated with EBV latent infection.
This review will briefly summarize EBNA1 functional domains together with the recently identified functional moieties and describe various strategies that have been exploited to achieve specific EBNA1 inhibition. The reported compounds will be compared, for their growth-inhibition effect and selectivity as well as the employed cell lines. Inhibitors with in vivo applicability or clearly-identified mechanisms will be highlighted.
Overview of EBNA1 functional domains
EBNA1 is the first identified EBV protein and the only viral protein present in both latent and lytic phases. It is a well-defined DNA-binding protein that contains several functional domains (Figure 2).
Overview of EBNA1 functional domains.
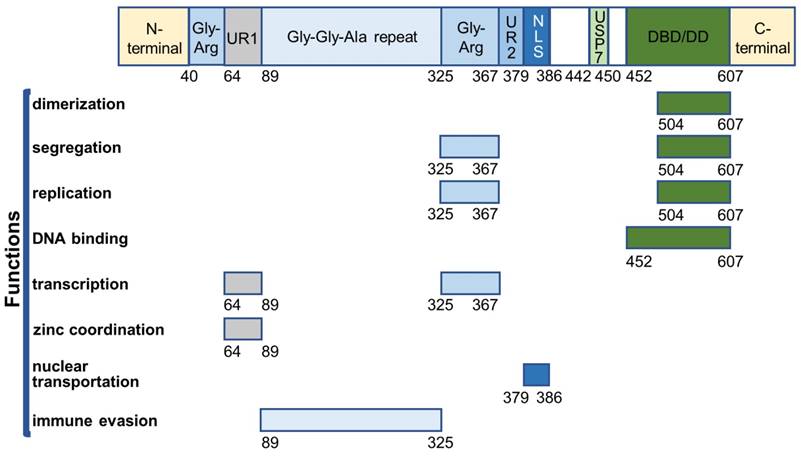
The core DNA-binding and dimerization domain (DBD/DD) of EBNA1 is located at the C-terminus within a.a. 452-607 and is involved in all EBNA1 functions associated with oriP-binding [48-50]. EBNA1 DBD/DD binds oriP at the 18-bp palindromic site as a homodimer [51]. X-ray crystallography of DBD/DD in its apo- and DNA-bound form has been described [52, 53].
EBNA1 can stably interact with ubiquitin-specific protease 7 (USP7/HAUSP), for which the binding domain was mapped to amino acid residues 442-447 [54, 55]. EBNA1-USP7 binding is not required for its functions such as replication, segregation, and transactivation, but it plays an important role in regulating EBV DNA replication [54]. USP7 is known to regulate cell proliferation and apoptosis by interacting with p53 and Mdm2. USP7 can stabilize p53 by deubiquitination, resulting in p53-mediated growth repression and apoptosis [56]. EBNA1 can compete with p53 to bind the N-terminal a.a. 53-328 of USP7 [57] with a 10-fold stronger binding affinity [58]. Thus, EBNA1 can displace USP7 binding with p53 to protect the cells from p53-mediated apoptosis, probably contributing to cell immortalization, proliferation, and survival following infection with EBV [55, 59]. Furthermore, EBNA1 can independently interact with USP7 and casein kinase 2 (CK2) of the host cells to disrupt the formation of promyelocytic leukemia (PML) nuclear bodies (NBs) [60-62]. EBNA1 can enhance the association of CK2 with PML proteins, which, in turn, phosphorylates PML proteins and triggers their degradation. The binding of EBNA1 to USP7 has been shown to mediate the loss of PML. As PML NBs play an important role in p53 activation [63-65], the EBNA1-mediated disruption of PML NBs provides another mechanism for EBNA1 to increase the survival of EBV-infected cells in the development of nasopharyngeal carcinoma and EBV-associated gastric carcinoma [59, 60, 62].
EBNA1 primarily resides in the nucleus of EBV-infected cells. The nuclear localization of EBNA1 is regulated through the interaction between a short EBNA1 sequence (NLS, nucleus localization sequence, a.a. 379-386) and two importins (nuclear import adaptor, α1 and α5) [48, 66-68]. EBNA1 K379 and R380 were found to be necessary for its nuclear translocation [69]. Also, phosphorylation of EBNA1 S385 increased EBNA1 NLS-importin α1/α5 interaction and stimulated EBNA1 nuclear transportation [68, 69]. A recently described S385-phosphorylated EBNA1-importin α1 crystal structure confirmed that its increased nuclear transportation was a result of the enhanced binding between EBNA1 NLS and its minor binding sites on importin α1 [70].
Two linking regions (LR1 a LR2) are located at the N-terminal (a.a. 40-89) and central (a.a. 325-379) regions of EBNA1, each of which contains a Gly-Arg-rich domain (Gly-Arg, a.a. 40-64, a.a. 325-367) and a unique region (UR1, a.a. 64-89; UR2, a.a. 367-379). Gly-Arg domains facilitate DNA looping in vitro [71-73]. The central Gly-Arg domain and UR1 are responsible for transcriptional activation by EBNA1 [74, 75]. Gly-Arg interacts with several nucleosome-associated proteins for transactivation by EBNA1 [54, 76, 77]. The zinc ion is reported to be required for both transcriptional activation and self-association at UR1, which is regarded as a second dimeric interface of EBNA1 [78]. Also, UR1 was shown to associate with the bromodomain-containing protein 4 (Brd4) that facilitates transcriptional activation by EBNA1 [79]. Besides regulating transactivation, another key feature of central Gly-Arg domain is tethering EBNA1 (a.a. 325-376) to chromosomes for segregation of EBV episomes. Several studies have shown that the segregation by EBNA1 is achieved through attaching to EBNA1-binding protein 2 (EBP2) on the mitotic chromosomes [80-82], and both Gly-Arg regions were shown to bind several RNAs in vitro [83].
The Gly-Gly-Ala repeat region (a.a. 89-325) can decrease human leukocyte antigen (HLA) class I presentation of EBNA1 antigens by blocking proteasome-dependent degradation of these antigens, and weakens the T-cell response to the EBV latent infection [84, 85]. This is supported by the evidence that targeting EBNA1 for rapid degradation can enhance CD8+ T cell recognition [86]. Also, the Gly-Gly-Ala repeat domain negatively regulates EBNA1 translational efficiency to maintain low EBNA1 levels for evading the immune system of the host [87, 88]. The reduction in the translational efficiency of EBNA1 could be due to the formation of G-quadruplex in the Gly-Gly-Ala repeat region of the EBNA1 mRNA [89, 90].
Strategies for blocking EBNA1 expression or EBNA1-dependent functions
Small-molecule or peptide inhibitors for proteins or protein-protein interactions have long been investigated with limited success [91-94]. Though the EBV viral protein EBNA1 was identified in 1971, the research was mostly focused on its critical functions (for reviews: [95-97]), and specific inhibition of EBNA1 as a potential therapeutic approach was investigated only in the last decade. Nevertheless, the growing body of studies of EBNA1 inhibition demonstrated that EBNA1 is a potential target for therapeutic intervention (Figure 3).
Recent developments of EBNA1 inhibitors. The inhibitors are shown in their respective EBNA1 domains.
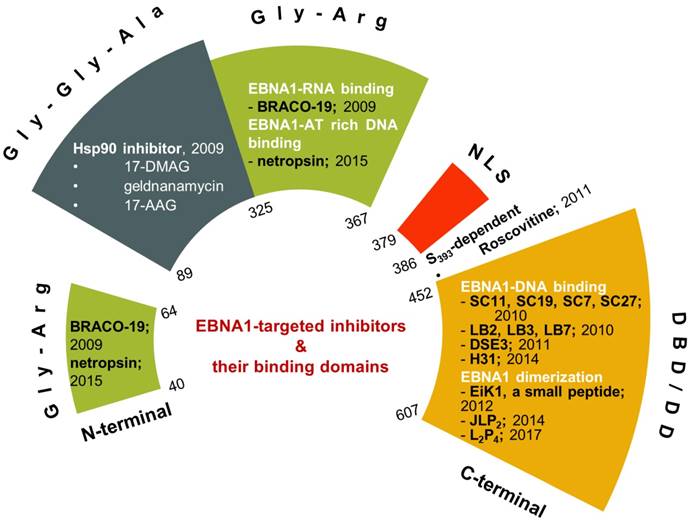
Inhibitors requiring Gly-Gly-Ala repeats
The first series of small-molecule inhibitors for EBNA1 belong to heat shock protein 90 (Hsp90) inhibitors reported by Kenney and colleagues in 2009 (Figure 4) [98, 99]. Hsp90 is known to facilitate folding, stabilization, and some functions of its associated proteins (also called client proteins). The Hsp90 inhibitors decreased EBNA1 expression in Burkitt's lymphoma and NPC cell lines without affecting its stability and half-life. The inhibition of EBNA1 expression was dependent on Gly-Gly-Ala repeats, because no decrease in the expression was observed for mutant EBNA1 lacking Gly-Gly-Ala repeats (EBNA1 ΔGA). Consistent with this observation, EBNA1 translation was decreased by Hsp90 inhibitors for full-length EBNA1, but not EBNA1 ΔGA. Furthermore, Hsp90 inhibitors exhibited significant growth-inhibition in both EBV-immortalized LCLs and EBV-induced lymphoproliferative disease in SCID (severe combined immunodeficient) mice. The inhibitory effect resulted from the decreased expression level of EBNA1. Thus, the Hsp90 inhibitors investigated in this study can be potentially used to treat EBV-associated diseases.
Chemical structures of Hsp90 inhibitors.
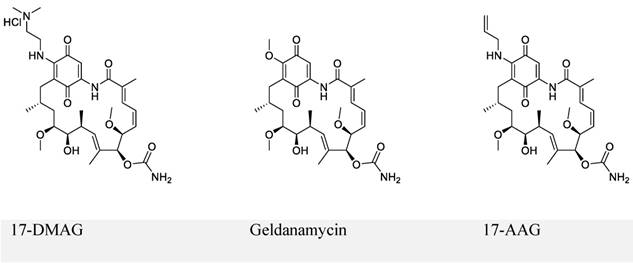
Hsp90 inhibitor (17-DMAG) treatment of LCL-EBNA1 ΔGA lines caused significant growth inhibition, while it did not decrease the expression of EBNA1 ΔGA. Since EBNA1 ΔGA should not affect any of the essential functions of EBNA1, this observation does not support the hypothesis that the decreased EBNA1 expression level contributed to growth-inhibition by Hsp90 inhibitors. This study also failed to describe the detailed mechanism by which Hsp90 inhibitors exhibited significant inhibition. Based on the recent reports that showed Hsp90 inhibitors decreased the expression of some oncogenic Hsp90 clients [98, 100], the authors hypothesized that EBNA1 was a client protein of Hsp90, which was disproved in the immunoprecipitation experiments.
Inhibitors blocking EBNA1-DNA binding
The research related to EBNA1 inhibitors focused on blocking EBNA1-DNA binding. Inhibitors have been reported that either competitively bind to EBNA1 or EBNA1-bound DNA and thus interfere with EBNA1-DNA binding activity. The X-ray crystallographic structure of EBNA1 DBD/DD in the apo- and DNA-bound form [52, 53] also facilitated identification of EBNA1 inhibitors by using screening techniques since EBNA1 has no known ortholog gene in humans.
The first inhibitor series of this kind was identified by Lieberman and colleagues through high-throughput in silico virtual screening [101]. This series identified four small molecules: SC7, SC11, SC19 and SC27 (Figure 5, upper panel), which, except for SC27, inhibited EBNA1-DNA binding (IC50 in the micromolar range). Although all three compounds could almost completely block EBNA1-mediated transcription, selective inhibition was only observed with SC19, as SC7 and SC11 could also non-specifically reduce the unrelated Zta-mediated transcription. Furthermore, SC11 and SC19 could also reduce the EBV genome copy number in the Raji Burkitt lymphoma cell line to 10-25%, while SC7 showed no apparent effect. SC7 and SC19 were recognized as the two top candidates and underwent molecular docking analysis. The simulation results could explain their inhibitory activities (IC50) against EBNA1-DNA binding, which were 23 and 49 μM, respectively. Besides binding to the DNA-binding sites of the EBNA1 protein, SC7 was also predicted to align well with the EBNA1-bound DNA sequence, which was not observed with SC19 and could be due to its bulky phenyl group imparting a different orientation when bound with EBNA1. These docking simulation results of SC7 and SC19 provide valuable information for future development of EBNA1-DNA binding inhibitors to increase the drug potency and selectivity.
Chemical structures of EBNA1-DNA binding inhibitors. Inhibitors in the upper and middle panel are identified via scrrening approaches that could block EBNA1-DNA binding. The inhibitor in the lower panel is a known DNA ligand that competitively binds the EBNA1-bound site on DNA.
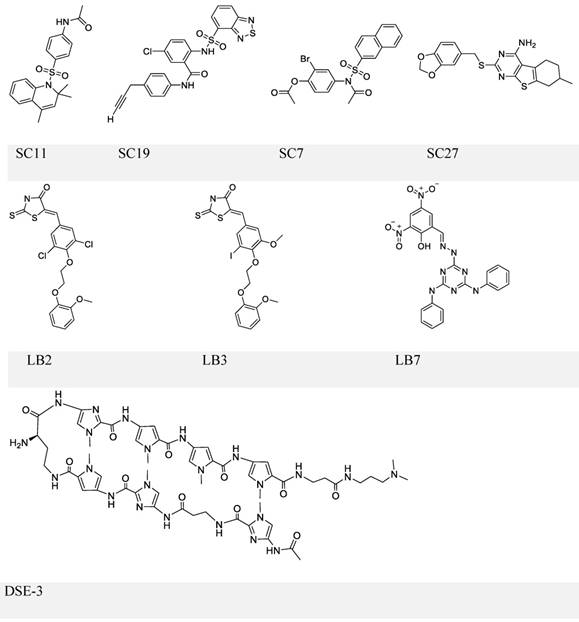
In a separate study, the same group conducted another high-throughput screening and identified three more compounds LB2, LB3, and LB7 (Figure 5, middle panel) [102]. LB7 could selectively inhibit EBNA1-DNA binding, as reflected by its IC50 values (1 μM for EBNA1 binding and no observable inhibition for Zta binding), and its inhibitory activity was more potent than that of SC7 in a parallel comparison (2 μM for EBNA1 and 237 μM for Zta). In addition, LB7 was the only new compound that could reduce the EBV copy number (at 5 μM) but a high dose (100 μM) was required to inhibit EBNA1-induced transcription partially. It should also be noticed that the high concentration used in the transcription repression assay could cause cell death.
Screening approaches have been widely used for the development of small-molecule inhibitors and peptide inhibitors, such as for the identification of inhibitors against EBNA1-DNA binding [103-105]. It is well-documented that EBV viral genome loss causes cell apoptosis [41, 106-108], a phenomenon exhibited by some of the above-mentioned screened compounds. However, the effect of these inhibitors on cell growth was not examined in the previous studies. Furthermore, as pointed out by the investigators, these candidates would not likely be clinically relevant unless significant structural modifications are made to increase their specificity to the protein EBNA1.
An alternative strategy to block EBNA1-DNA binding is to occupy EBNA1 binding sites on viral DNA, typically the 5-'TAGCA-3'. A pyrrole-imidazole series was synthesized and investigated for the ability to target specifically the EBNA1-binding sequence and thereby affect EBNA1-dependent biological functions [109]. Among these DNA ligands, DSE-3 (Figure 5, lower panel) had the best performance in inhibiting EBNA1-DS interaction. DSE-3 showed selective, though not significant, growth inhibition in EBV-positive cells (IC50, ~60 μM in three LCLs and >80 μM in Raji). DSE-3 could also reduce EBV genome copy number, suppress the expression of EBNA1, EBNA2 and LMP2, and prevent EBV-induced transformation of primary B cells. Despite these positive outcomes, DSE-3, and other DNA ligands, could possibly target the genomic DNA of the host cells, and thus affect the expression of some non-viral genes facilitated by EBNA1-host genome interaction [110, 111]. Thus, effects of these DNA ligands on the host genome remain to be further defined. Nevertheless, DSE-3 provided an interesting insight for the blockage of EBNA1-DNA binding.
Inhibitors blocking EBNA1-dependent episome maintenance or transcription
By employing a screening approach, Kang et al. identified two small molecules, roscovitine and H20, against transactivation by EBNA1-oriP interaction (Figure 6, upper panel) [112, 113]. Because roscovitine is a known inhibitor of cyclin-dependent kinases (CDKs), eukaryotic linear motif (ELM) analysis of EBNA1 was performed that suggested serine 393 as a putative CDK site. The S393A mutant of EBNA1 was thereby employed, which confirmed that the transactivation inhibition by roscovitine depends on serine 393. Roscovitine could decrease the nuclear EBNA1 amount and increase cytoplasmic EBNA1, while EBNA1 nuclear/cytoplasmic distribution in the S393 mutant was unaffected by roscovitine treatment. Also, roscovitine could reduce EBV episome DNA and inhibit cell growth of LCLs. This study illustrated the role of S393 in nuclear import of EBNA1. In contrast, a subsequent study showed that mutation of S393 did not affect EBNA1 nuclear localization and its functions related to EBV DNA replication or segregation [114]. Instead, the S393 mutation abrogated PML NBs disruption by EBNA1 by binding to CK2, as mentioned previously [114].
Chemical structures of inhibitors that block EBNA1-oriP transactivation and EBNA1 Gly-Args-dependent functions.
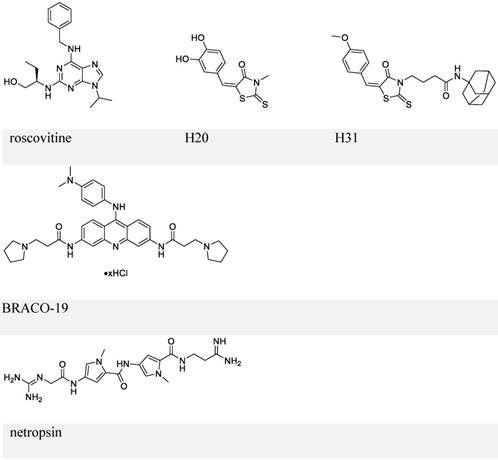
The other small-molecule inhibitor, H20, prevented transactivation by EBNA1 and showed association with EBNA1. However, it failed to inhibit EBNA1-DNA binding directly. This observation was explained by a docking study, which suggested that the docking site for H20 in EBNA1 differs from the oriP FR binding pocket. To improve the ability to block EBNA1-DNA binding, structure-activity relationship analysis of H20 was conducted, which suggested H31 (Figure 6, upper panel) [113]. H31 showed inhibition of DNA binding, transactivation, replication, and EBV episome maintenance by EBNA1. H31 also exhibited selective inhibition of EBV-positive cells (10 μM, decreased by ~70%). Although H31 interfered with EBNA1-DNA binding, it failed to a show direct binding with EBNA1 DBD in a subsequent surface plasmon resonance (SPR) assay.
Inhibitors blocking linking regions (LR1 and LR2)-dependent functions
Since RGG domains are known to bind with RNA [83], a study using electrophoresis mobility shift assay (EMSA) was performed to investigate whether EBNA1 prefers the structured RNA. The analysis confirmed the preference of G-quadruplex RNA to EBNA1 [115]. Since the RGG-like motif in LR1 and LR2 also functions in recruiting origin recognition complex (ORC) to oriP for viral DNA replication, several G-quadruplex-interacting molecules were employed to determine whether interference with EBNA1-RNA binding affects ORC recruitment. EBNA1-ORC2 association was most efficiently disrupted by BRACO-19 (Figure 6, middle panel), suggesting a necessary role of EBNA1-RNA binding for the recruitment of ORC by EBNA1. Thus, BRACO-19 was selected for further investigation. Treatment of Raji Burkitt lymphoma cell line with 10 μM BRACO-19 for three days decreased the EBV genome copy number to ~75%, while a longer treatment of 6 days caused growth inhibition in both Raji and LCLs. It also showed a modest inhibition of mRNA expression of EBNA2 and EBNA3A (~20%), and decreased DNA replication and metaphase attachment by EBNA1.
Since AT-hooks on LR1 and LR2 (two Gly-Args) allow binding between EBNA1 and AT-rich DNA, AT-rich binders may interfere with this binding. Netropsin (Figure 6, lower panel) can bind to the minor groove of the AT-rich sequence of dsDNA [116], and has therefore been studied for its inhibitory activities on EBV genome replication and EBV-positive cell growth. Treatment with 10 μM netropsin could induce ~50% EBV plasmid loss as compared to the control group showing ~30% loss; 50 μM netropsin caused 53% growth inhibition in EBV-positive cells versus 31% inhibition in EBV-negative cells. Therefore, the inhibition by netropsin failed to show selectivity or efficiency. Also, whether netropsin could inhibit EBNA1-AT-rich DNA binding was not studied.
The strategies discussed in this section, as well as the previously described DNA ligands represent indirect approaches to inhibit EBNA1 function. Rather than directly binding to EBNA1, these ligands competitively interact with components EBNA1 associates with, thereby impairing functions exerted by EBNA1. These approaches may have limited applications. Especially, molecules that bind to EBNA1-binding sequences on viral DNA or RNA might also affect normal functions of host cells by binding to host DNA or RNA. From this perspective, small molecules that directly interact with EBNA1 domains are a preferred choice.
Inhibitors based on truncated peptides from EBNA1 dimeric interface
Rather than small-molecule inhibitors, several peptides have been identified by using rational biochemical screening of the EBNA1 DBD/DD domain as EBNA1-DNA-binding inhibitors [117]. Three peptides, P83 (a.a. 552-566), P84 (a.a. 556-570), and P85 (a.a., 560-574), sharing the same a.a. sequence 560-566, showed almost complete blockage of EBNA1-DNA binding. Also, P85 was found to strongly associate with EBNA1 DBD/DD, as indicated by SPR assay, and treatment with P85 caused more than 50% decrease in EBNA1 transcription. Thus, a truncated peptide from EBNA1 DBD/DD was able to interfere with DNA binding and transcription by EBNA1. The study that previously discovered roscovitine also identified another small molecule EiK1 [112]. Like P85, EiK1 weakened DNA binding and transcription by EBNA1; however, it quickly dissociated from the dimeric EBNA1 in the SPR assay. The ability of EiK1 and P85 in interfering EBNA1 dimer formation was also studied via DSS crosslinker-mediated dimerization and yeast two-hybrid assays; the results of these two independent assays suggest that EiK1 seems to disrupt the self-association of the EBNA1 protein. In addition, whether the inhibition of EBNA1 function by P85/EiK1 can cause growth-inhibition in EBV-infected cells remains to be clarified.
The current pool of EBNA1 inhibitors mainly consists of small molecules. The study by Kim and co-workers indicated that EBNA1 DBD/DD is experimentally “druggable” by peptides that can inhibit EBNA1 function [117]. Despite encouraging results, effects of the peptides on cellular growth have not yet been studied. Furthermore, the poor water-solubility of the hydrophobic peptide is still a problem that has to be dealt with.
Inspired by the above study, JLP2 (Figure 7, upper panel) was designed to increase cell permeability and was fluorescently labeled for molecular tracking [118]. The conjugates, containing a peptide inhibitor and a water-soluble fluorophore, ensured selective cellular uptake and growth-inhibition in EBV-positive cells. The EBV-positive nasopharyngeal carcinoma cell line C666-1 treated with 20 μM JLP2 showed a ~50% decrease in cell viability, while no obvious inhibition was observed in the EBV-negative HeLa cells. The higher cytotoxicity of peptide conjugate compared to the unconjugated peptide may be due to the increased cell permeability, as indicated by confocal imaging and cellular uptake results. Molecular docking suggested JLP2 had a stronger EBNA1 binding than either JLP1 or P2. Luminescence titration analysis confirmed the stronger binding by JLP2, where a 1.5-fold emission enhancement was only observed for JLP2 upon addition of EBNA1.
Chemical structures of inhibitors based on peptides from EBNA1 DBD/DD. Adapted with permission from [118, 119], copyright 2014 The Royal Society of Chemistry and 2017 Springer Nature.
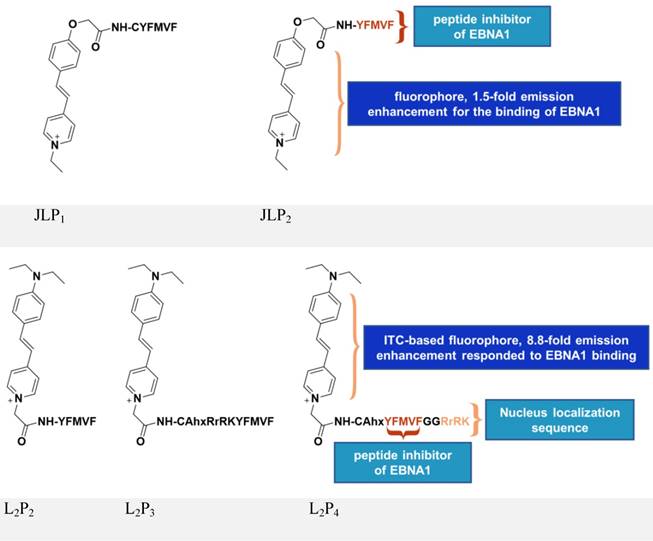
The design of JLP2 represents a step forward, but its profile needs to be optimized including its non-specific cellular localization and its growth-inhibition of Burkitt's lymphoma line. We therefore designed a new series of probes, L2P2/L2P3/L2P4 [119-121], by incorporating an NLS (RrRK) moiety in the probe skeleton, which enabled nuclear localization and targeted nuclear EBNA1 (Figure 7, lower panel) [48, 122]. Molecular dynamics simulations suggested an unexpected role of RrRK, which formed salt bridges among several residues in the aspartate-rich tail of EBNA1 and thus contributed to stabilization of the NLS-containing probe-EBNA1 complex. Nuclear localization was demonstrated by the NLS-containing L2P3 and L2P4, whereas L2P2 only remained in the cytoplasm. The probe, L2P4, responded significantly to binding with EBNA1, exhibiting an 8.8-fold enhancement with a 25 nm blueshift of its emission because of the introduced intramolecular charge transfer-characterized fluorophore. The emission of this kind of fluorophore is highly solvent-dependent, thus protein-binding activity causes a change in its fluorescence, with a more significant change representing stronger binding activity. Among all tested samples, L2P4 exhibited the strongest binding to EBNA1, qualifying it as the best inhibitor in EBV-positive cells including NPC and Burkitt's lymphoma cell lines. In vivo growth inhibition by L2P4 in EBV-positive tumors was also confirmed. Furthermore, cross-linking dimerization assay results indicate that L2P4 likely interferes with the self-association of EBNA1 by direct binding to the dimeric interface. Moreover, the significant emission enhancement of L2P4 could also be potentially used to visualize cellular EBNA1. Thus, by resolving many drawbacks of traditional peptide inhibitors, L2P4 stands out as a novel solution for EBV-infected cells.
The key properties of various EBNA1 inhibitors are summarized and compared in Table 1, including the time required and the employed cell lines. Notably, EBNA1 has a long half-life, which is usually more than 24 h [54, 123], so it requires a relatively long time for treatment by inhibitors in a viability assay.
Comparison of in vitro growth-inhibition by EBNA1 inhibitors
| Inhibitor | Conc. (μM) | Time (days) | EBV-positive cell lines | Cell number (cells/100 μL) | Inhibition (%) | Inhibition towards EBV(-) cells (%) |
|---|---|---|---|---|---|---|
| Hsp90 inhibitor - 17-DMAG | 0.03 | 5 | LCL1 | 1×104 | ~100 | Not found |
| LCL2 | 1×104 | ~100 | ||||
| DNA ligands - DSE3 | 40 | 8 | LCL-1 | Not mentioned | ~70 | Not found |
| LCL-2 | ~70 | |||||
| LCL-3 | ~70 | |||||
| Raji | ~50 | |||||
| roscovitine | 5 | 16 | GM3324 LCL | optimal growth density | ~85 | Not mentioned |
| H31 | 10 | 7 | AKATAEBV(+) | Not mentioned | ~78 | Not found |
| 6 | 1022 LCL | ~66 | ||||
| BRACO-19 | 10 | 6 | Raji | 2.5×104 | ~11 | Not found |
| LCL3456 | ~14 | |||||
| LCL3472 | ~22 | |||||
| netropsin | 50 | 15 | SaII BL | 4-2×104 | ~53 | ~31 |
| JLP2 | 20 | 1 | C666-1 | 1×104 | ~51 | Not found |
| L2P4 | 20 | 1 | C666-1 | 3×103 | ~72 | Not found |
| NPC43 | ~72 | |||||
| Raji | ~53 |
Conclusions
In the past decades, EBV has drawn attention due to its substantial contribution in the development of several lymphoid and epithelial malignancies. The crucial and unique roles of EBNA1 in maintaining EBV infection make it an attractive target for therapeutic intervention of EBV-associated cancers.
Efforts from the past decade in the design or identification of EBNA1 inhibitors have shown some progress on several fronts. For example, small-molecule inhibitors against EBNA1-DNA binding or peptide-based inhibitors from EBNA1 DBD/DD confirmed the “druggability” of EBNA1 for the treatment of EBV-positive cancers. Since inhibitors affecting EBNA1-DNA binding were mainly identified by screening approaches, they are unlikely to be structurally related to EBNA1. Therefore, further structural modifications of these inhibitors are required to increase their specificity to EBNA1. Peptides-based inhibitors possess the advantage of target specificity but suffer other short-comings including poor stability, short half-life and susceptibility to degradation by proteases preventing their effective delivery to the target tumors.
It is important to acknowledge that current EBNA1-targeted inhibitors are far from perfect and not ready for the clinic. Multidisciplinary efforts are required for designing structurally-relevant EBNA1-targeted inhibitors. In this respect, co-immunoprecipitation experiments and X-ray crystallography analysis could provide critical information on the structure of EBNA1 inhibitor complex. Similarly, molecular dynamics simulations as well as docking studies might afford valuable insights. Although L2P4 has been observed in tumor sections by imaging studies, monitoring the whole human body would be more difficult. It would be worthwhile to employ real-time and deep-penetrating imaging modalities, such as positron emission tomography imaging and magnetic resonance imaging, to gain a better understanding of the functions of EBNA1-targeted inhibitors.
Abbreviations
Brd4: bromodomain-containing protein 4; CDKs: cyclin-dependent kinases; CK2: casein kinase 2; DBD/DD: DNA binding and dimerization domain; DS: dyad symmetry; EBNA1: Epstein-Barr nuclear antigen 1; EBP2: EBNA1 binding protein 2; EBV: Epstein-Barr virus; ELM: eukaryotic linear motif; EMSA: electrophoresis mobility shift assay; FR: family of repeats; HLA: human leukocyte antigen; Hsps: heat shock proteins; ITC: intramolecular charge transfer; LCLs: lymphoblastoid cell lines; LR: linking region; NBs: nuclear bodies; NLS: nucleus localization sequence; ORC: origin recognition complex; oriP: origin of replication; PML: promyelocytic leukemia; SCID: severe combined immunodeficient; SPR: surface plasmon resonance; UR: unique region; USP7: ubiquitin specific protease 7.
Acknowledgements
This review was supported by the Hong Kong Research Grants Council (HKBU 22301615 and HKPolyU 153021/18P), The Hong Kong Polytechnic University - University Research Facility in Life Sciences (ULS), and Hong Kong Baptist University (RC-IRMS/1617/1B-CHEM).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1:702-3
2. Henle G, Henle W, Diehl V. Relation of Burkitt's tumor-associated herpes-ytpe virus to infectious mononucleosis. Proc Natl Acad Sci U S A. 1968;59:94-101
3. Fields BN, Knipe DM, Howley PM. Fields virology. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins. 2007
4. Hutt-Fletcher LM. Epstein-Barr virus entry. J Virol. 2007;81:7825-32
5. Fingeroth JD, Weis JJ, Tedder TF, Strominger JL, Biro PA, Fearon DT. Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc Natl Acad Sci U S A. 1984;81:4510-4
6. Nemerow GR, Wolfert R, McNaughton ME, Cooper NR. Identification and characterization of the Epstein-Barr virus receptor on human B lymphocytes and its relationship to the C3d complement receptor (CR2). J Virol. 1985;55:347-51
7. Tanner J, Weis J, Fearon D, Whang Y, Kieff E. Epstein-Barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell. 1987;50:203-13
8. Tanner J, Whang Y, Sample J, Sears A, Kieff E. Soluble gp350/220 and deletion mutant glycoproteins block Epstein-Barr virus adsorption to lymphocytes. J Virol. 1988;62:4452-64
9. Young LS, Dawson CW, Brown KW, Rickinson AB. Identification of a human epithelial cell surface protein sharing an epitope with the C3d/epstein-barr virus receptor molecule of B lymphocytes. Int J Cancer. 1989;43:786-94
10. Sixbey JW, Yao QY. Immunoglobulin A-induced shift of Epstein-Barr virus tissue tropism. Science. 1992;255:1578-80
11. Molesworth SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J Virol. 2000;74:6324-32
12. Oda T, Imai S, Chiba S, Takada K. Epstein-Barr virus lacking glycoprotein gp85 cannot infect B cells and epithelial cells. Virology. 2000;276:52-8
13. Tugizov SM, Berline JW, Palefsky JM. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat Med. 2003;9:307-14
14. Borza CM, Morgan AJ, Turk SM, Hutt-Fletcher LM. Use of gHgL for attachment of Epstein-Barr virus to epithelial cells compromises infection. J Virol. 2004;78:5007-14
15. Ni C, Chen Y, Zeng M, Pei R, Du Y, Tang L. et al. In-cell infection: a novel pathway for Epstein-Barr virus infection mediated by cell-in-cell structures. Cell Res. 2015;25:785-800
16. Xiong D, Du Y, Wang HB, Zhao B, Zhang H, Li Y. et al. Nonmuscle myosin heavy chain IIA mediates Epstein-Barr virus infection of nasopharyngeal epithelial cells. Proc Natl Acad Sci U S A. 2015;112:11036-41
17. Savard M, Belanger C, Tardif M, Gourde P, Flamand L, Gosselin J. Infection of primary human monocytes by Epstein-Barr virus. J Virol. 2000;74:2612-9
18. Guerreiro-Cacais AO, Li L, Donati D, Bejarano MT, Morgan A, Masucci MG. et al. Capacity of Epstein-Barr virus to infect monocytes and inhibit their development into dendritic cells is affected by the cell type supporting virus replication. J Gen Virol. 2004;85:2767-78
19. Babcock GJ, Hochberg D, Thorley-Lawson AD. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity. 2000;13:497-506
20. Aman P, Ehlin-Henriksson B, Klein G. Epstein-Barr virus susceptibility of normal human B lymphocyte populations. J Exp Med. 1984;159:208-20
21. Thorley-Lawson DA, Mann KP. Early events in Epstein-Barr virus infection provide a model for B cell activation. J Exp Med. 1985;162:45-59
22. Pope JH, Horne MK, Scott W. Transformation of foetal human keukocytes in vitro by filtrates of a human leukaemic cell line containing herpes-like virus. Int J Cancer. 1968;3:857-66
23. Liu YJ, Joshua DE, Williams GT, Smith CA, Gordon J, MacLennan IC. Mechanism of antigen-driven selection in germinal centres. Nature. 1989;342:929-31
24. Thorley-Lawson DA, Babcock GJ. A model for persistent infection with Epstein-Barr virus: the stealth virus of human B cells. Life Sci. 1999;65:1433-53
25. Babcock GJ, Thorley-Lawson DA. Tonsillar memory B cells, latently infected with Epstein-Barr virus, express the restricted pattern of latent genes previously found only in Epstein-Barr virus-associated tumors. Proc Natl Acad Sci U S A. 2000;97:12250-5
26. Thorley-Lawson DA. Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol. 2001;1:75-82
27. Fahraeus R, Fu HL, Ernberg I, Finke J, Rowe M, Klein G. et al. Expression of Epstein-Barr virus-encoded proteins in nasopharyngeal carcinoma. Int J Cancer. 1988;42:329-38
28. Brooks L, Yao QY, Rickinson AB, Young LS. Epstein-Barr virus latent gene transcription in nasopharyngeal carcinoma cells: coexpression of EBNA1, LMP1, and LMP2 transcripts. J Virol. 1992;66:2689-97
29. Deacon EM, Pallesen G, Niedobitek G, Crocker J, Brooks L, Rickinson AB. et al. Epstein-Barr virus and Hodgkin's disease: transcriptional analysis of virus latency in the malignant cells. J Exp Med. 1993;177:339-49
30. Imai S, Koizumi S, Sugiura M, Tokunaga M, Uemura Y, Yamamoto N. et al. Gastric carcinoma: monoclonal epithelial malignant cells expressing Epstein-Barr virus latent infection protein. Proc Natl Acad Sci U S A. 1994;91:9131-5
31. Sugiura M, Imai S, Tokunaga M, Koizumi S, Uchizawa M, Okamoto K. et al. Transcriptional analysis of Epstein-Barr virus gene expression in EBV-positive gastric carcinoma: unique viral latency in the tumour cells. Br J Cancer. 1996;74:625-31
32. zur Hausen A, Brink AA, Craanen ME, Middeldorp JM, Meijer CJ, van den Brule AJ. Unique transcription pattern of Epstein-Barr virus (EBV) in EBV-carrying gastric adenocarcinomas: expression of the transforming BARF1 gene. Cancer Res. 2000;60:2745-8
33. Babcock GJ, Decker LL, Freeman RB, Thorley-Lawson DA. Epstein-Barr virus-infected resting memory B cells, not proliferating lymphoblasts, accumulate in the peripheral blood of immunosuppressed patients. J Exp Med. 1999;190:567-76
34. Lupton S, Levine AJ. Mapping genetic elements of Epstein-Barr virus that facilitate extrachromosomal persistence of Epstein-Barr virus-derived plasmids in human cells. Mol Cell Biol. 1985;5:2533-42
35. Reisman D, Yates J, Sugden B. A putative origin of replication of plasmids derived from Epstein-Barr virus is composed of two cis-acting components. Mol Cell Biol. 1985;5:1822-32
36. Rawlins DR, Milman G, Hayward SD, Hayward GS. Sequence-specific DNA binding of the Epstein-Barr virus nuclear antigen (EBNA-1) to clustered sites in the plasmid maintenance region. Cell. 1985;42:859-68
37. Yates J, Warren N, Reisman D, Sugden B. A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc Natl Acad Sci U S A. 1984;81:3806-10
38. Gahn TA, Schildkraut CL. The Epstein-Barr virus origin of plasmid replication, oriP, contains both the initiation and termination sites of DNA replication. Cell. 1989;58:527-35
39. Reisman D, Sugden B. trans activation of an Epstein-Barr viral transcriptional enhancer by the Epstein-Barr viral nuclear antigen 1. Mol Cell Biol. 1986;6:3838-46
40. Krysan PJ, Haase SB, Calos MP. Isolation of human sequences that replicate autonomously in human cells. Mol Cell Biol. 1989;9:1026-33
41. Kennedy G, Komano J, Sugden B. Epstein-Barr virus provides a survival factor to Burkitt's lymphomas. Proc Natl Acad Sci U S A. 2003;100:14269-74
42. Nasimuzzaman M, Kuroda M, Dohno S, Yamamoto T, Iwatsuki K, Matsuzaki S. et al. Eradication of Epstein-Barr virus episome and associated inhibition of infected tumor cell growth by adenovirus vector-mediated transduction of dominant-negative EBNA1. Mol Ther. 2005;11:578-90
43. Imai S, Kuroda M, Yamashita R, Ishiura Y. Therapeutic inhibition of Epstein-Barr virus-associated tumor cell growth by dominant-negative EBNA1. Uirusu. 2005;55:239-49
44. Roth G, Curiel T, Lacy J. Epstein-Barr viral nuclear antigen 1 antisense oligodeoxynucleotide inhibits proliferation of Epstein-Barr virus-immortalized B cells. Blood. 1994;84:582-7
45. Yin Q, Flemington EK. siRNAs against the Epstein Barr virus latency replication factor, EBNA1, inhibit its function and growth of EBV-dependent tumor cells. Virology. 2006;346:385-93
46. Hong M, Murai Y, Kutsuna T, Takahashi H, Nomoto K, Cheng CM. et al. Suppression of Epstein-Barr nuclear antigen 1 (EBNA1) by RNA interference inhibits proliferation of EBV-positive Burkitt's lymphoma cells. J Cancer Res Clin Oncol. 2006;132:1-8
47. Gianti E, Messick TE, Lieberman PM, Zauhar RJ. Computational analysis of EBNA1 "druggability" suggests novel insights for Epstein-Barr virus inhibitor design. J Comput Aided Mol Des. 2016;30:285-303
48. Ambinder RF, Mullen MA, Chang YN, Hayward GS, Hayward SD. Functional domains of Epstein-Barr virus nuclear antigen EBNA-1. J Virol. 1991;65:1466-78
49. Chen MR, Middeldorp JM, Hayward SD. Separation of the complex DNA binding domain of EBNA-1 into DNA recognition and dimerization subdomains of novel structure. J Virol. 1993;67:4875-85
50. Summers H, Barwell JA, Pfuetzner RA, Edwards AM, Frappier L. Cooperative assembly of EBNA1 on the Epstein-Barr virus latent origin of replication. J Virol. 1996;70:1228-31
51. Ambinder RF, Shah WA, Rawlins DR, Hayward GS, Hayward SD. Definition of the sequence requirements for binding of the EBNA-1 protein to its palindromic target sites in Epstein-Barr virus DNA. J Virol. 1990;64:2369-79
52. Bochkarev A, Barwell JA, Pfuetzner RA, Furey W Jr, Edwards AM, Frappier L. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein EBNA 1. Cell. 1995;83:39-46
53. Bochkarev A, Barwell JA, Pfuetzner RA, Bochkareva E, Frappier L, Edwards AM. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein, EBNA1, bound to DNA. Cell. 1996;84:791-800
54. Holowaty MN, Zeghouf M, Wu H, Tellam J, Athanasopoulos V, Greenblatt J. et al. Protein profiling with Epstein-Barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J Biol Chem. 2003;278:29987-94
55. Saridakis V, Sheng Y, Sarkari F, Holowaty MN, Shire K, Nguyen T. et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol Cell. 2005;18:25-36
56. Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J. et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416:648-53
57. Hu M, Li P, Li M, Li W, Yao T, Wu JW. et al. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111:1041-54
58. Holowaty MN, Sheng Y, Nguyen T, Arrowsmith C, Frappier L. Protein interaction domains of the ubiquitin-specific protease, USP7/HAUSP. J Biol Chem. 2003;278:47753-61
59. Frappier L. Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses. 2012;4:1537-47
60. Sivachandran N, Sarkari F, Frappier L. Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of PML nuclear bodies. PLoS Path. 2008;4:e1000170
61. Sivachandran N, Cao JY, Frappier L. Epstein-Barr virus nuclear antigen 1 Hijacks the host kinase CK2 to disrupt PML nuclear bodies. J Virol. 2010;84:11113-23
62. Sivachandran N, Dawson CW, Young LS, Liu FF, Middeldorp J, Frappier L. Contributions of the Epstein-Barr virus EBNA1 protein to gastric carcinoma. J Virol. 2012;86:60-8
63. de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G. et al. PML is a direct p53 target that modulates p53 effector functions. Mol Cell. 2004;13:523-35
64. Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S. et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207-10
65. Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K. et al. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 2000;19:6185-95
66. Fischer N, Kremmer E, Lautscham G, Mueller-Lantzsch N, Grasser FA. Epstein-Barr virus nuclear antigen 1 forms a complex with the nuclear transporter karyopherin alpha2. J Biol Chem. 1997;272:3999-4005
67. Ito S, Ikeda M, Kato N, Matsumoto A, Ishikawa Y, Kumakubo S. et al. Epstein-Barr virus nuclear antigen-1 binds to nuclear transporter karyopherin α1/NPI-1 in addition to karyopherin α2/Rch1. Virology. 2000;266:110-9
68. Nardozzi J, Wenta N, Yasuhara N, Vinkemeier U, Cingolani G. Molecular basis for the recognition of phosphorylated STAT1 by importin alpha5. J Mol Biol. 2010;402:83-100
69. Kitamura R, Sekimoto T, Ito S, Harada S, Yamagata H, Masai H. et al. Nuclear import of Epstein-Barr virus nuclear antigen 1 mediated by NPI-1 (importin α5) is up- and down-regulated by phosphorylation of the nuclear localization signal for which lys379 and arg380 are essential. J Virol. 2006;80:1979-91
70. Nakada R, Hirano H, Matsuura Y. Structural basis for the regulation of nuclear import of Epstein-Barr virus nuclear antigen 1 (EBNA1) by phosphorylation of the nuclear localization signal. Biochem Biophys Res Commun. 2017;484:113-7
71. Su W, Middleton T, Sugden B, Echols H. DNA looping between the origin of replication of Epstein-Barr virus and its enhancer site: stabilization of an origin complex with Epstein-Barr nuclear antigen 1. Proc Natl Acad Sci U S A. 1991;88:10870-4
72. Frappier L, O'Donnell M. Epstein-Barr nuclear antigen 1 mediates a DNA loop within the latent replication origin of Epstein-Barr virus. Proc Natl Acad Sci U S A. 1991;88:10875-9
73. Frappier L, Goldsmith K, Bendell L. Stabilization of the EBNA1 protein on the Epstein-Barr virus latent origin of DNA replication by a DNA looping mechanism. J Biol Chem. 1994;269:1057-62
74. Wu H, Kapoor P, Frappier L. Separation of the DNA replication, segregation, and transcriptional activation functions of Epstein-Barr nuclear antigen 1. J Virol. 2002;76:2480-90
75. Kennedy G, Sugden B. EBNA-1, a bifunctional transcriptional activator. Mol Cell Biol. 2003;23:6901-8
76. Wang S, Frappier L. Nucleosome assembly proteins bind to Epstein-Barr virus nuclear antigen 1 and affect its functions in DNA replication and transcriptional activation. J Virol. 2009;83:11704-14
77. Malik-Soni N, Frappier L. Nucleophosmin contributes to the transcriptional activation function of the Epstein-Barr virus EBNA1 protein. J Virol. 2014;88:2323-6
78. Aras S, Singh G, Johnston K, Foster T, Aiyar A. Zinc coordination is required for and regulates transcription activation by Epstein-Barr nuclear antigen 1. PLoS Pathog. 2009;5:e1000469
79. Lin A, Wang S, Nguyen T, Shire K, Frappier L. The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. J Virol. 2008;82:12009-19
80. Shire K, Ceccarelli DF, Avolio-Hunter TM, Frappier L. EBP2, a human protein that interacts with sequences of the Epstein-Barr virus nuclear antigen 1 important for plasmid maintenance. J Virol. 1999;73:2587-95
81. Wu H, Ceccarelli DF, Frappier L. The DNA segregation mechanism of Epstein-Barr virus nuclear antigen 1. EMBO reports. 2000;1:140-4
82. Kanda T, Otter M, Wahl GM. Coupling of mitotic chromosome tethering and replication competence in Epstein-Barr virus-based plasmids. Mol Cell Biol. 2001;21:3576-88
83. Snudden DK, Hearing J, Smith PR, Grässer FA, Griffin BE. EBNA-1, the major nuclear antigen of Epstein-Barr virus, resembles 'RGG' RNA binding proteins. The EMBO Journal. 1994;13:4840-7
84. Levitskaya J, Coram M, Levitsky V, Imreh S, Steigerwald-Mullen PM, Klein G. et al. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature. 1995;375:685-8
85. Blake N, Lee S, Redchenko I, Thomas W, Steven N, Leese A. et al. Human CD8+ T cell responses to EBV EBNA1: HLA class I presentation of the (Gly-Ala)-containing protein requires exogenous processing. Immunity. 1997;7:791-802
86. Tellam J, Sherritt M, Thomson S, Tellam R, Moss DJ, Burrows SR. et al. Targeting of EBNA1 for rapid intracellular degradation overrides the inhibitory effects of the Gly-Ala repeat domain and restores CD8+ T cell recognition. J Biol Chem. 2001;276:33353-60
87. Yin Y, Manoury B, Fahraeus R. Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus-encoded EBNA1. Science. 2003;301:1371-4
88. Tellam J, Rist M, Connolly G, Webb N, Fazou C, Wang F. et al. Translation efficiency of EBNA1 encoded by lymphocryptoviruses influences endogenous presentation of CD8+ T cell epitopes. Eur J Immunol. 2007;37:328-37
89. Murat P, Zhong J, Lekieffre L, Cowieson NP, Clancy JL, Preiss T. et al. G-quadruplexes regulate Epstein-Barr virus-encoded nuclear antigen 1 mRNA translation. Nat Chem Biol. 2014;10:358-64
90. Tellam JT, Lekieffre L, Zhong J, Lynn DJ, Khanna R. Messenger RNA sequence rather than protein sequence determines the level of self-synthesis and antigen presentation of the EBV-encoded antigen, EBNA1. PLoS Pathog. 2012;8:e1003112
91. Ma Y, Ai G, Zhang C, Zhao M, Dong X, Han Z. et al. Novel Linear Peptides with High Affinity to alphavbeta3 Integrin for Precise Tumor Identification. Theranostics. 2017;7:1511-23
92. Beards F, Jones LE, Charnock J, Forbes K, Harris LK. Placental homing peptide-microRNA inhibitor conjugates for targeted enhancement of intrinsic placental growth signaling. Theranostics. 2017;7:2940-55
93. Debnath B, Xu S, Grande F, Garofalo A, Neamati N. Small molecule inhibitors of CXCR4. Theranostics. 2013;3:47-75
94. Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301-17
95. Frappier L. EBNA1 and host factors in Epstein-Barr virus latent DNA replication. Curr Opin Virol. 2012;2:733-9
96. Westhoff Smith D, Sugden B. Potential cellular functions of Epstein-Barr nuclear antigen 1 (EBNA1) of Epstein-Barr virus. Viruses. 2013;5:226-40
97. Frappier L. Role of EBNA1 in NPC tumourigenesis. Semin Cancer Biol. 2012;22:154-61
98. Sun X, Barlow EA, Ma S, Hagemeier SR, Duellman SJ, Burgess RR. et al. Hsp90 inhibitors block outgrowth of EBV-infected malignant cells in vitro and in vivo through an EBNA1-dependent mechanism. Proc Natl Acad Sci U S A. 2010;107:3146-51
99. Sun X, Kenney SC. Hsp90 inhibitors: a potential treatment for latent EBV infection? Cell Cycle (Georgetown, Tex). 2010;9:1665-6
100. Bonvini P, Gastaldi T, Falini B, Rosolen A. Nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), a novel Hsp90-client tyrosine kinase: down-regulation of NPM-ALK expression and tyrosine phosphorylation in ALK(+) CD30(+) lymphoma cells by the Hsp90 antagonist 17-allylamino,17-demethoxygeldanamycin. Cancer Res. 2002;62:1559-66
101. Li N, Thompson S, Schultz DC, Zhu W, Jiang H, Luo C. et al. Discovery of selective inhibitors against EBNA1 via high throughput in silico virtual screening. PLoS One. 2010;5:e10126
102. Thompson S, Messick T, Schultz DC, Reichman M, Lieberman PM. Development of a high-throughput screen for inhibitors of Epstein-Barr virus EBNA1. J Biomol Screen. 2010;15:1107-15
103. Dietrich U, Durr R, Koch J. Peptides as drugs: from screening to application. Curr Pharm Biotechnol. 2013;14:501-12
104. Geng L, Wang Z, Jia X, Han Q, Xiang Z, Li D. et al. HER2 targeting peptides screening and applications in tumor imaging and drug delivery. Theranostics. 2016;6:1261-73
105. Janzen WP. Screening technologies for small molecule discovery: the state of the art. Chem Biol. 2014;21:1162-70
106. Vereide DT, Sugden B. Lymphomas differ in their dependence on Epstein-Barr virus. Blood. 2011;117:1977-85
107. Vereide DT, Seto E, Chiu YF, Hayes M, Tagawa T, Grundhoff A. et al. Epstein-Barr virus maintains lymphomas via its miRNAs. Oncogene. 2014;33:1258-64
108. Kirchmaier AL, Sugden B. Dominant-negative inhibitors of EBNA-1 of Epstein-Barr virus. J Virol. 1997;71:1766-75
109. Yasuda A, Noguchi K, Minoshima M, Kashiwazaki G, Kanda T, Katayama K. et al. DNA ligand designed to antagonize EBNA1 represses Epstein-Barr virus-induced immortalization. Cancer Sci. 2011;102:2221-30
110. Canaan A, Haviv I, Urban AE, Schulz VP, Hartman S, Zhang Z. et al. EBNA1 regulates cellular gene expression by binding cellular promoters. Proc Natl Acad Sci U S A. 2009;106:22421-6
111. Dresang LR, Vereide DT, Sugden B. Identifying sites bound by Epstein-Barr virus nuclear antigen 1 (EBNA1) in the human genome: defining a position-weighted matrix to predict sites bound by EBNA1 in viral genomes. J Virol. 2009;83:2930-40
112. Kang M-S, Lee EK, Soni V, Lewis TA, Koehler AN, Srinivasan V. et al. Roscovitine inhibits EBNA1 serine 393 phosphorylation, nuclear localization, transcription, and episome maintenance. J Virol. 2011;85:2859-68
113. Lee EK, Kim SY, Noh KW, Joo EH, Zhao B, Kieff E. et al. Small molecule inhibition of Epstein-Barr virus nuclear antigen-1 DNA binding activity interferes with replication and persistence of the viral genome. Antiviral Res. 2014;104:73-83
114. Cao JY, Shire K, Landry C, Gish GD, Pawson T, Frappier L. Identification of a novel protein interaction motif in the regulatory subunit of casein kinase 2. Mol Cell Biol. 2014;34:246-58
115. Norseen J, Johnson FB, Lieberman PM. Role for G-quadruplex RNA binding by Epstein-Barr virus nuclear antigen 1 in DNA replication and metaphase chromosome attachment. J Virol. 2009;83:10336-46
116. Chakravorty A, Sugden B. The AT-hook DNA binding ability of the Epstein Barr virus EBNA1 protein is necessary for the maintenance of viral genomes in latently infected cells. Virology. 2015;484:251-8
117. Kim SY, Song KA, Kieff E, Kang MS. Small molecule and peptide-mediated inhibition of Epstein-Barr virus nuclear antigen 1 dimerization. Biochem Biophys Res Commun. 2012;424:251-6
118. Jiang L, Lui YL, Li H, Chan CF, Lan R, Chan WL. et al. EBNA1-specific luminescent small molecules for the imaging and inhibition of latent EBV-infected tumor cells. Chem Commun. 2014;50:6517-9
119. Jiang L, Lan R, Huang T, Chan C-F, Li H, Lear S. et al. EBNA1-targeted probe for the imaging and growth inhibition of tumours associated with the Epstein-Barr virus. Nat Biomed Eng. 2017;1:0042
120. Wong K-L, Lung H, Mak N-K, Law GL, Wong WT, Cobb SL. New probe for Epstein-Barr virus protein EBNA1. Cell Chem Biol. 2017;24:647-8
121. Kosowicz JG, Lee J, Ambinder RF. Cancer: Seeing the ebb of a tumour virus. Nat Biomed Eng. 2017;1:0059
122. Puckett CA, Barton JK. Targeting a ruthenium complex to the nucleus with short peptides. Biorg Med Chem. 2010;18:3564-9
123. Tellam J, Connolly G, Green KJ, Miles JJ, Moss DJ, Burrows SR. et al. Endogenous presentation of CD8+ T cell epitopes from Epstein-Barr virus-encoded nuclear antigen 1. J Exp Med. 2004;199:1421-31
Author contact
![]() Corresponding authors: Email: klwongedu.hk; nkmakedu.hk; ga-ai.lawedu.hk; kwloedu.hk
Corresponding authors: Email: klwongedu.hk; nkmakedu.hk; ga-ai.lawedu.hk; kwloedu.hk