Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Quantitative data
General aspects of fluorescence...
Detection threshold
Demonstration of value in early...
Summary and conclusion
Abbreviations
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(19):5336-5347. doi:10.7150/thno.27384 This issue Cite
Review
Recommendations for reporting on emerging optical imaging agents to promote clinical approval
Willemieke S. Tummers1,2, Jason M Warram3, Nynke S. van den Berg4, Sarah E. Miller4, Rutger-Jan Swijnenburg2, Alexander L. Vahrmeijer2, Eben L. Rosenthal4 ![]()
1. Department of Radiology, Molecular Imaging Program, Stanford University, Stanford, CA.
2. Department of Surgery, Leiden University Medical Center, Leiden, Netherlands.
3. Department of Otolaryngology, University of Alabama at Birmingham, AL
4. Department of Otolaryngology, Stanford University, Stanford, CA.
Received 2018-5-21; Accepted 2018-8-22; Published 2018-10-22
Abstract

Intraoperative fluorescence imaging is particularly well-suited for surgical applications due to its inherently high sensitivity, resolution, and ability to provide images in real-time. To date, the intraoperative observation of fluorescence has largely been subjective. With the need to show objective evidence in order to demonstrate the benefit of this technique, quantitative data needs to be provided to overseeing regulatory bodies. Standardization of fluorescence imaging protocols would improve reproducibility and minimize inter- and intra-institution variance. This would allow studies to be conducted using the same injection techniques, imaging times, reconstruction methods, and analyses. Here, we provide recommendations for standardized methodologies with the goal of setting a minimum requirement for reporting fluorescence-guided surgery results based on both qualitative and (semi-) quantitative data collection. Clinical trials using fluorescence-guided surgery should present results of three critical elements; 1) intra-operative imaging, 2) specimen mapping and pathology correlation, and 3) target validation. Qualitative analyses should consist of a bright field image, black-and-white fluorescence image, pseudo-colored fluorescence overlay image, and/or heat-map whereby fluorescence signal intensity differences are displayed on a color spectrum. Quantitative analyses should include 1) intraoperative data (consisting of images or video, raw numeric values and ratios); 2) specimen mapping, for correlation of fluorescence with the presence of disease (performed using fresh tissue); and 3) target validation (designed to determine fluorescence intensity relative to receptor density of a specific area). Including the aforementioned methods of both qualitative and quantitative analyses will ensure that trial results are comparable and could be collated in future studies to expedite FDA approval.
Keywords: Optical imaging, fluorescence, clinical trial, standardization, result reporting
Introduction
An incremental improvement in oncologic surgical outcomes can be obtained by successful identification of visually occult disease and tumor-positive margins during surgery, which will have a significant impact on overall cancer survival. Interest in cancer-specific intraoperative molecular imaging has undergone rapid growth. This substantial interest is not surprising considering the robust potential of the technique to achieve subclinical tumor-detection and improved surgical guidance. Fluorescence-guided surgery (FGS) is particularly well-suited for surgical applications due to its inherently high sensitivity, resolution, and ability to provide images in real-time [1]. In recent years, the number of early phase clinical trials using FGS has drastically risen, and shifted towards the use of dyes emitting in the near-infrared rather than in the visible part of the light spectrum. An example of a commonly used near-infrared fluorescent (NIRF) dye is indocyanine green (ICG). ICG achieves its tumor-targeting through “second window imaging,” which is based on the enhanced permeability and retention (EPR) effect [2, 3]. While safe, ICG is a non-specific agent that is rapidly cleared by the liver and excreted in bile. Currently, most strategies use a NIRF dye that is conjugated to a targeting vehicle directed against a tissue of interest, such as a tumor-specific agent, and this combination permits optimal cancer-specific detection [4, 5]. However, human testing using such novel NIRF-guided surgical agents has thus far only been conducted in early phase trials to establish safety and efficacy [6-13]. These trials are unable to show benefit over the current standard-of-care; larger phase II/III trials are needed to demonstrate superiority prior to obtaining regulatory approval and wide-spread adoption into practice. Early stage clinical trials are crucial to providing data to support in vivo application, surgical specimen mapping, and target validation. These three elements should be evaluated using a standardized methodology that can be used to advance these technologies through regulatory pathways into routine clinical use.
Positron emission tomography (PET) imaging represents a clear precedent for the introduction of standardization into FGS. Since its establishment in 1984, numerous articles have been published describing the need for standardization of the technology with regards to clinical application [14-16]. These studies showed that strict standardization of all aspects of imaging and data analysis is required to obtain quantitative, accurate, reliable, precise and reproducible results. Similarly, multiple studies have been performed investigating the concordance of PET results obtained at different institutions [15, 17, 18]. Developing a standardized approach will minimize variability between studies in addition to facilitating the development of multicenter studies, thus allowing for direct comparison of results within and between clinical trials. Moreover, it can potentially allow for future direct translation of results to other centers.
Although often difficult to test in clinical trials, the qualitative representation of fluorescence imaging data is critical, as surgeons make intraoperative decisions based on their own interpretation of images generated and displayed by the imaging device. To be valuable, high-resolution images need to be generated in real-time, without delayed image processing. In many cases, the value and objectivity of qualitative data is questioned, though in the case of FGS its representation is of the utmost importance to the technology's implementation and utility.
Simultaneously, quantitative data plays a key role in the analysis of FGS clinical trials. Currently, quantitative data is difficult to generate as compared to qualitative data, and since FGS is primarily a visual tool for the surgeon, the use of quantitative data has been less emphasized in the first early phase clinical trials. However, with the need to show objective evidence to demonstrate the superiority of FGS techniques, quantitative data should also be provided to the regulatory bodies.
Here, we propose a methodology for reporting results from fluorescence-guided oncologic surgery studies (Figure 1). Our proposed method is based on the minimum requirements for the presented data from all phases of a clinical trial, including intraoperative imaging, ex vivo imaging, and pathologic correlation, which should be represented using both qualitative and quantitative data.
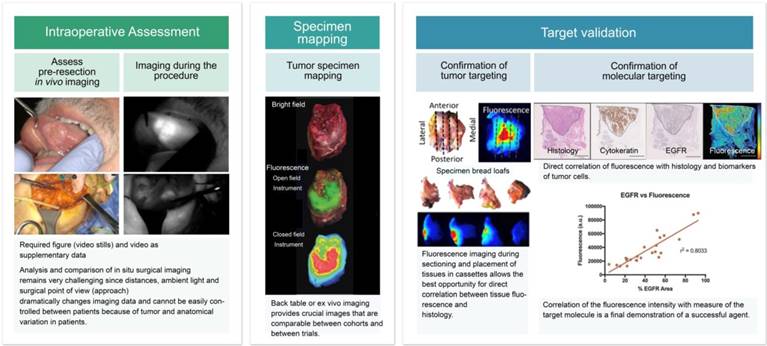
Standardized assessment of reporting results in fluorescence-guided oncologic surgical trials for all aspects of the clinical trial: intraoperative imaging, ex vivo imaging and pathologic assessment. During the intraoperative assessment, focus should remain on imaging at predetermined timepoints intraoperatively. Ex vivo imaging will ensure exact tumor mapping to correlate imaging results to pathology. During pathologic assessment the focus should be on confirmation of fluorophore targeting to tumor tissue [adapted with permission from [48], copyright 2018].
Quantitative data
In vivo fluorescence quantification is challenging, since the measured fluorescence depends not only on the concentration of the imaging agent, but on multiple parameters, such as intrinsic autofluorescence of tissue, the sensitivity of the imaging device, the absorption and scattering properties of the tissue, and photobleaching. All these parameters can influence the accuracy of quantification [19-21]. Standardization of quantification in fluorescence imaging protocols would improve reproducibility and minimize inter- and intra-institution variance, and studies should be conducted using the same injection technique, imaging time, reconstruction method, and analysis [14]. Such strategies are critical for reproducibility in multi-institutional clinical trials but are not easy to achieve. Use of phantoms will be beneficial to determine how well individual cameras and centers perform in the technical aspects of image acquisition, data processing, and analysis [22]. In multicenter studies using PET imaging, the submission of a phantom scan is usually required periodically for scanner calibration and reproducibility [23].
Perhaps the largest research effort to quantify fluorescence in vivo has been focused on the measurement of photosensitizer concentration prior to and during photodynamic therapy (PDT) [20, 21, 24, 25]. Because the effect of PDT depends directly on the concentration of sensitizer used, there is a high need for measuring this quantity quickly and noninvasively [20]. Kim et al. described a noninvasive in vivo method to determine fluorophore concentration in neurosurgery using 5-ALA [19]. Using a fiber-optic probe, the fluorophore concentration is calculated from the quantitative fluorescence spectrum, which is the fluorescence emission spectrum corrected for optical attenuation of the tissue, thus quantifying the observed fluorescence [19]. These handheld fiber-optic probes need to simultaneously collect tissue fluorescence and diffuse reflectance spectra in vivo [26]. The method is performed using specific fiber-optic probes that touch the tissue, which requires a separate device in combination with the fluorescence camera for intra-operative navigation. Another complicating factor of this method is the need to identify the optical properties of tissues and the variation of these properties, which are mostly unknown [26].
Achieving absolute in vivo quantification is unlikely, since the current fluorescence imaging systems are not equipped to generate quantified imaging data, but a semi-quantification of the fluorescence signal should be possible. Similar to PET imaging, where maximum standard uptake values (SUVmax) are calculated based on the administered radioisotope dose and the patient weight, one can also calculate fluorescence signal uptake values by correcting for tissue surface area [27]. In fluorescence imaging, only fluorescence arising from <1 cm depth will contribute to the measured signal. Therefore, signal should be corrected for surface area since corrections either for weight or volume will not necessarily generate a representative value. Consequently, it is recommended for fluorescence imaging to provide standardized reports on surface area-adjusted values where feasibly measured by ex vivo imaging. For example, adjusted from the formula for SUV values, mean fluorescence intensity (MFI) could be calculated as MFIcorrected = fluorescence intensity of tissue of interest / (injected dose/surface area of the tissue) [28].
As described above, quantification of fluorescence signal is still in its infancy. Ultimately, it is expected that this objective measure is going to play a major role in providing evidence of patient benefit. Next to the extensive investments in novel agents and clinical cameras, innovative solutions are being developed for the quantification of fluorescence imaging signals in the ex vivo setting. Several groups are working on the real-time detection of the actual amount and concentration of the fluorescent agent that accumulates in tissues. By scanning part of the tissue or by taking point measurements, the concentration can be determined, leading to exact quantification [19, 29]. Another method to objectify and automate quantification is by machine learning. Here, the software can be trained to identify the fluorescence cut-off value for true-positive fluorescence and true-negative fluorescence. With a learning set, these values can be validated and applied to all patients injected with the same agent and dose [30]. For now, these methods are not commercially available, and further research is required to validate their applicability, but semi-quantification of the fluorescence signal is already possible without these novel devices using methods described below.
Quality control procedures
FGS is intrinsically linked to the sensitivity of the imaging device used. Currently, various imaging systems, ranging from large open-field cameras to minimally-invasive endoscopic cameras, are available for NIRF excitation and emission signal detection during surgery and ex vivo. Consequently, results from different imaging systems are hard to compare. This is one of the main reasons that the FDA primarily reviews combination products, instead of an imaging agent and device separately [31]. This is in contrast to PET agents and scanners, where the implementation of quality control (QC) procedures have led to an established method to verify consistency, linearity, and safety functions of PET scanners. For PET imaging, the National Electrical Manufacturers Association (NEMA) has formulated performance standards, to which manufacturers need to adhere [32]. For fluorescence imaging devices, this is still far from standard practice, with only safety standards currently available. Incorporating documentation on performance testing can help to identify relevant characteristics (e.g., spatial resolution, uniformity, sensitivity, dynamic range), provide guidelines for testing (e.g., phantom material property range/geometry, methods for calculating metrics), and describe viable test methods for the validity of performance characteristics [31].
General aspects of fluorescence data analysis
Ideally, all acquired FGS data should be analyzed for the correlation between imaging results and pathology using a quantitative and qualitative approach.
Detection threshold
Targeted agents are usually present in the tissue of interest at nanomolar concentrations, which requires sensitive cameras with low detection thresholds. Analysis of this threshold for different imaging systems and agents is important when evaluating agents for their intended use. If one aims to detect micrometastases, the sensitivity of the camera and the specificity of the agent should both be extremely high. DSouza et al. performed a study to determine the detection limit of various fluorescent imaging systems using dye concentrations in a phantom and showed that devices with higher bit depths, variable electronic gain settings, and background-light correction during acquisition, had the highest sensitivities [33]. Previously, we have shown that small numbers of cancer cells can be detected in optimized ex vivo settings both preclinically (2.4×104 cells) and clinically (< 5 mm) [34].
Quantification parameters
Significant variations can occur when reporting on fluorescence signal intensities since the value remains dependent on the way the images are analyzed. Below we discuss various methods to quantify fluorescence imaging data.
Mean fluorescence intensity
The mean fluorescence intensity (MFI) is a measurement of the fluorescence signal in a region of interest (ROI) divided by the area within that ROI (either in pixels or cm2). The MFI is typically shown in arbitrary units of fluorescence (AU) or total number of photons detected by the device for each pixel, or per cm2. Arbitrary values are generally used for relative, ratiometric quantification to determine the fold difference or change between various samples that are analyzed in parallel. Devices that use a scaled algorithm for determining fluorescence signal also use AU.
The MFI can also be shown in relative fluorescence units (RFU), which is a relative unit of measurement compared to a reference measurement using the same ROI area. In PET imaging a defined background is determined to compare to the SUVmax value in order to determine the presence of increased uptake. As with PET imaging, it may be recommended to use the maximum fluorescence value, since this is independent from an ROI, less observer-dependent and more reproducible compared to the mean value. The primary disadvantage is that a maximum value is more susceptible to noise. Comparing the maximum fluorescence value to that of a set background (Table 1), may be used to correct for this noise.
Suggestions for tissue to appoint as background for quantitative fluorescence imaging analysis.
| Cancer type and location | Ideal background | Appropriate background |
|---|---|---|
| Breast | Uninvolved breast tissue | Adipose tissue |
| Head and neck | Muscle | Skin or mucosa |
| Colorectal | Uninvolved bowel serosa | Normal bowel mucosa |
| Pancreas | Peritumoral inflammation | Normal pancreatic tissue |
| Lung | Uninvolved lung tissue | Skin |
| Brain | Uninvolved brain tissue | Uninvolved brain tissue |
| Hepatocellular and CRLM | Immature hepatocytes | Uninvolved liver tissue |
| Prostate | Uninvolved prostate tissue | Connective tissue |
| Parathyroid | Thyroid tissue | Muscle |
Tumor-to-background ratio
When addressing the quality of an imaging agent to provide tumor-specific contrast, it has generally been related to a ratio such as tumor-to-background ratio (TBR), signal-to-noise ratio (SNR) or contrast-to-noise ratio (CNR). In the past, studies aimed for a TBR of >2.5, since it was thought that any lower rate was insufficient to identify cancerous tissue intraoperatively [2, 35]. However, an intraoperative TBR of >2.5 is relatively hard to achieve in a clinical setting with currently available agents and imaging systems, although at this TBR the tumor can be clearly distinguished from the surrounding tissue. While a higher signal in the tumor when compared to surrounding tissue is preferred, there are many other variables that contribute to an agent's ability to differentiate normal from diseased tissues. Therefore, it may not be appropriate to identify a specific number as a minimum value before one can determine there is “sufficient contrast”. At a minimum, the difference in signal needs to be high enough for the surgeon to differentiate tumor from normal tissue—in our experience a TBR >3.0 in preclinical studies and >1.5 in clinical studies would be appropriate for further studies.
Another important note is that TBR is highly dependent on the dynamic range of the imaging devices used in addition to the variability in properties of the chosen background tissue. The background will greatly influence the TBR value, and currently no guidelines exist regarding an appropriate background for various cancer types. If unrealistically high values of TBR are set as a standard, it is likely that only surrounding tissues with virtually no fluorescence will be candidates for employing these techniques. However, to ensure clinical applicability of the technique, the background tissue is inherently in close proximity of the tumor even when this tissue displays some intensity of fluorescence. For example, in pancreatic cancer surgery, muscle would not be an appropriate background, whereas for head and neck cancer, muscle would be appropriate based on the surgical field of view (Table 1).
Signal-to-noise ratio (SNR)
The SNR is the quotient of the MFI measured in a ROI and the standard deviation (SD) of the signal intensity in a region outside the boundaries of the object being imaged (i.e., a region from which the tissue's fluorescence signal is zero) [36]. There are two forms of noise in images: random and structured. Random noise (or statistical noise) is directly related to the number of signals coming from the imaging agent. Structured noise refers to non-random variations in counting rates. The latter will adversely affect the interpretation and analysis of the images. Examples of structured noise are organ motion, and imaging instrument non-uniformities [37].
Contrast-to-noise ratio (CNR)
To ensure only contrast through targeted uptake is measured, and contrast through non-specific EPR is excluded, contrast can be quantified by calculating the average signal in the tumor and subtracting the average signal in the selected background tissue. This value can be divided by the standard deviation of the background to estimate the CNR [38]. This technique may be particularly helpful to assess off-target accumulation in non-cancerous tissues that express the biomarker of interest. CNR is especially useful in situations where the background and tumor signals fluctuate widely. In these cases, the TBR value could be the same as compared to a situation with a more constant signal profile. However, the tumor would be more consistently clearly visible in case of a constant pattern. In situations with more fluctuation, CNR would show a difference in value, providing a more representative value that assists in distinguishing true contrast from noise [38].
When detecting lesions in the body, a high SNR alone will not guarantee that sufficient contrast exists to make the lesion detectable. Therefore, SNR should be used to assess one single image. CNR on the other hand can be used to identify fluctuations within one patient over time as it compares a measure of signal fluctuations to noise. In summary, SNR is a good measure to assess data quality of a single image, and CNR provides knowledge regarding how easy or hard it is to detect signal fluctuations over time [36].
Sensitivity and specificity
A topic of constant debate in the field of FGS is the reporting of test characteristics. Certain groups choose not to report sensitivities and specificities in early phase clinical trials, since the number of patients is insufficient for a powered calculation. Some groups use multiple separate specimens from each patient to generate a sufficient sample size for determining test characteristics. We believe that reporting on sensitivity and specificity in early trials is important in order to determine the potential of the fluorescence imaging agent for FGS. In larger clinical trials in later phases, the additional benefit of sensitivity and specificity should focus on improving or complementing the surgeon's assessment.
Demonstration of value in early phase clinical trials: Three critical elements
1. Intraoperative data collection
Real-time fluorescence tumor imaging should demonstrate sufficiently higher contrast enhancement in the tumor relative to surrounding normal structures in order to augment the current standard of care. During minimally-invasive and robotic surgery, this real-time detection of fluorescence is easily integrated as the current technology requires the surgeon to operate using a screen to visualize the surgical field. However, to properly interpret a positive fluorescence signal that will subsequently affect clinical decision-making, consensus should be reached regarding validation of fluorescent markers and imaging devices designed to differentiate tumor from peri-tumoral tissue. To date, an insufficient number of standardized studies have been performed to reach this type of consensus.
Developers of strategies for intraoperative imaging should recognize that the surgical approach varies significantly by tumor type, and that each approach has a unique workflow. The incorporation of intraoperative FGS into the surgical workflow will be individualized to the type of procedure being performed, and the type of cancer being resected. For example, in the case of ovarian cancer, brain cancer or peritoneal metastases during a cytoreduction and hyperthermic intraperitoneal chemotherapy procedure (HIPEC), the primary goal is a more complete debulking [39]. In these cancer types, it is known that extensive debulking leads to prolonged survival for patients [40]. Here, FGS has the potential to be beneficial in identification of subclinical disease, or occult tumor deposits [12]. For other tumor types, complete resection is the key to prolonged survival. In these situations, FGS will play a crucial role in assessing the resection bed for residual tumor after initial resection or to assess the margins of the specimen [6, 9, 13, 41]. Next steps in treatment could be informed by the presence of residual fluorescence, warranting either further resection or, for example, novel techniques such as applying PDT to the resection bed [42].
Intraoperatively, FGS is particularly important for: 1) Identification of tumor; 2) Assessment of resection margins, both before the resection for operative planning and after to assess the wound bed; and 3) Detection of tumor-positive lymph nodes, especially in locations that would suggest metastatic disease. In this case, for example, FGS could inform clinical decision making by aborting a surgery when there is no oncologic benefit.
Margin assessment in the OR
One of the biggest gaps in oncologic surgery remains the high rate of positive margins following surgical resections, which directly correlates with poor survival and locoregional recurrence [43-45]. Both the in vivo assessment of the resection bed and the ex vivo assessment of the specimen are critical to ensure negative margins. The current approach to margin assessment in most tumor types is circumferential analysis of the resected specimen using frozen sections [46]. Therefore, only a fraction of the tumor mass can be analyzed due to the limited sampling that is feasible with frozen sectioning. A tumor-specific fluorescent contrast agent can be leveraged to perform fluorescence mapping of the surgical specimen. Fluorescent 'hot spots' could be used to direct clinicians toward suspicious regions, and thereby reduce sampling error. The detection of a fluorescence signal using back-table benchtop devices could also result in reduction of time needed for specimen analysis, since sampling can be performed based on fluorescence-positive areas, as opposed to sampling all surfaces of a large tumor specimen (i.e., hepatic tumor resections).
Data representation from intraoperative imaging
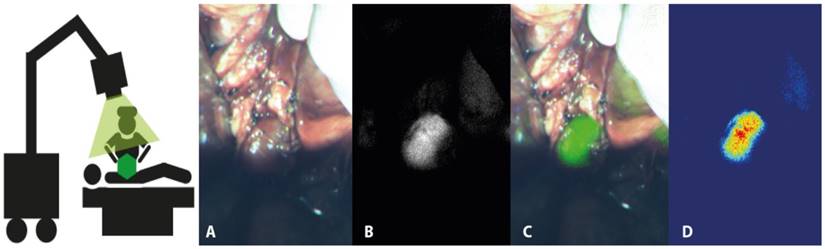
During surgery, fluorescence imaging should be performed at multiple fixed time points as outlined in the clinical trial protocol and standard operation procedure (SOP). For example, one can identify three imaging time points to be used in most trials: First, while scanning the area of interest for the localization of the primary tumor and/or detection of metastases prior to excision. Second, once the tumor has been exposed while still in situ, with surrounding normal tissue in the field of view. And third, after resection of the primary tumor for assessment of the wound bed. During surgery, both endoscopic and open-field imaging systems can be used. In some operative cases, such as an abdominal laparoscopy that is converted to an open procedure, both systems can be used at different timepoints. Since the two devices will generate data that is not necessarily comparable, it is important to clearly state which device's data will be used in a clinical trial and document this in the SOP. Depending on the capabilities of the fluorescence camera, multiple images should be obtained in each setting; a bright field image, a black-and-white fluorescence image, a pseudo-colored fluorescence overlay image, and/or a heat-map whereby fluorescence signal intensity differences are displayed in different colors (Figure 2). Ex vivo imaging of fresh tissue should be presented in a similar manner. Quantitative data of intraoperative images should consist of a numeric value representing fluorescence signal. This can either be reported in MFI values or in a ratio such as TBR.
Suggested method of reporting intra-operative imaging results. This includes a bright field image (A), black-and-white fluorescence image (B), fluorescence overlay image (C), and fluorescence heat-map (D). In this case, an example of a tumor-positive lymph node during abdominal surgery is shown.
The development process of an optimal intraoperative imaging strategy should keep in mind the specific nature of the surgical workflow in order to minimize interruptions and facilitate the adoption of this technology. Early phase clinical trials are designed to demonstrate the safety, appropriate dose and tumor specificity of experimental agents. For these trials, the secondary outcomes primarily focus on the tumor-specific contrast that is generated between tumor tissue and background tissue using the agent-device combination. Regulatory standards ensure the workflow of these early phase trials maintain the standard of care, which makes it difficult to objectively assess the potential clinical value and patient benefit of FGS. For larger clinical trials, clinically relevant endpoints that necessitate experimental assessment will be needed to demonstrate superiority to the current standard of care. Examples of secondary endpoints include improved detection of tumor-positive margins, decreased morbidity, reduced local recurrence, or increased efficiency in the operating room [31]. These endpoints can be directly correlated with known statistics pertaining to survival, thereby demonstrating clinical benefit.
It is important to recognize that the success of these trials will be influenced by the parameters of the clinical imaging devices employed. Not all imaging devices are able to show a TBR in real-time, and the significant differences in sensitivity among the various devices will make it challenging to achieve consistency and compare results regarding the clinical utility of tumor-specific contrast agents. Another limitation that should be recognized is the inability to directly correlate intraoperative images with pathology results in real-time. Therefore, dependence on ex vivo imaging to confirm tumor status will remain a critical component of FGS validation, especially in early phase clinical trials where confirmation of tumor specificity is critical. To circumvent this limitation, ex vivo imaging can be done in the form of back-table fluorescence imaging in the operating room using either a clinical camera or a bench-top device developed for this purpose. These results can later be directly correlated to the pathological assessment [6-9, 12, 13, 27, 41].
2. Specimen mapping
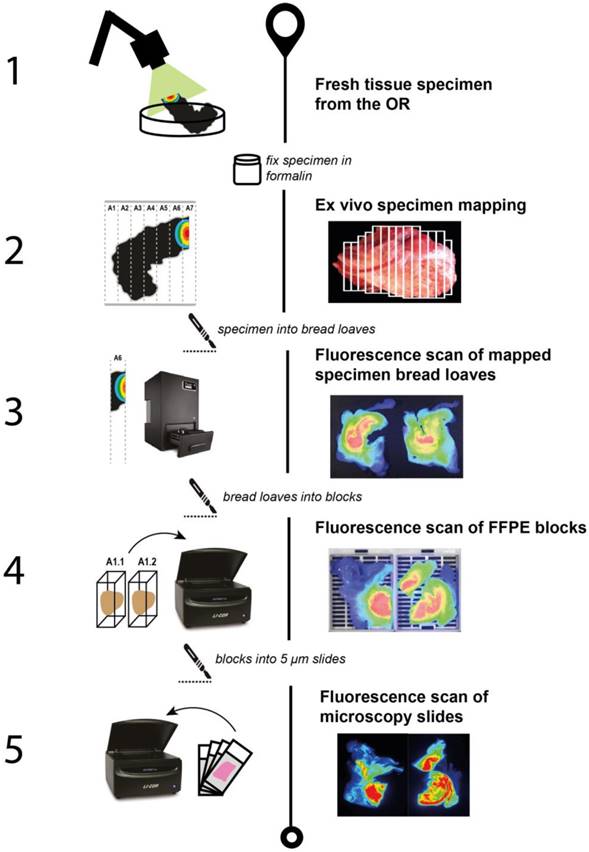
An important stage in the validation of FGS is the correlation of the fluorescence imaging-based results with the pathologic findings, which is used to determine if the fluorescence signal is specifically localized to the tumor. It is recommended that the correlation be performed in a standardized and rigorous method as described in Figure 3. After analysis of the resected fresh tissue, the surgical specimen is processed into bread loaves (either before or after formalin-fixation) and later into blocks to be placed in cassettes (Figure 3). These cassettes are then imaged on a fluorescence scanner both before and after paraffin embedding. Finally, these cassettes are processed into microscopy slides where fluorescence can then be correlated with hematoxylin and eosin (H&E) histopathology. These steps are possible when the fluorescent capabilities of the probe are maintained after formalin fixation. Additional immunohistochemical correlation of the target of interest and H&E should be performed to determine co-localization of the target and fluorescence, which will be discussed later.
Detailed overview using a clinical example of the ex vivo mapping process of a surgical specimen to ensure exact correlation of the surgical specimen with pathologic assessment. During step 1, the fresh tissue specimen is imaged ex vivo to ensure images from all angles. After formalin fixation, the specimen is mapped and sliced into bread loaves in step 2. Fluorescence imaging is subsequently performed of the bread loaves separately. In step 4, cassettes are made that include one bread loaf each and are imaged either on a fluorescence scanner or in a closed-field imaging system. The cassettes are then paraffin-embedded and sectioned to produce microscopy slides. In step 5, these slides are used for pathologic assessment and fluorescence scanning.
Mapping process of the gross surgical specimen
To facilitate the processing of the specimen, a bright-field image of the gross surgical specimen can be acquired and used as a reference map as the specimen is sectioned and placed into cassettes. The origin of the cassette-specific sections can be drawn on a printed or digital image of the primary specimen and saved for later correlation at the time of histopathologic study. This methodology is commonly employed during pathological processing of large surgical specimens. Precise imaging of the gross specimen and close correlation with histopathologic sections increases the efficiency and accuracy of pathologic assessment and enables consistent and accurate interpretation of the technique's specificity. This method is anecdotally used during Mohs surgery and is also described in the standardized pathologic assessment of breast cancer specimens [47]. It is important to maintain the specimen orientation relative to the patient so that the slide-mounted section can be registered back to the primary specimen, and ultimately to the wound bed. This approach will allow the clinician to determine if fluorescence observed on either the resected specimen or in the wound bed is clinically relevant and a source of potential recurrence.
Data representation of ex vivo imaging
After removal from the patient, the fresh specimen is imaged at the back-table using a benchtop device or the surgical camera. Fresh tissue should be imaged when possible and fluorescence intensity should be expressed relative to the surface area of the specimen.
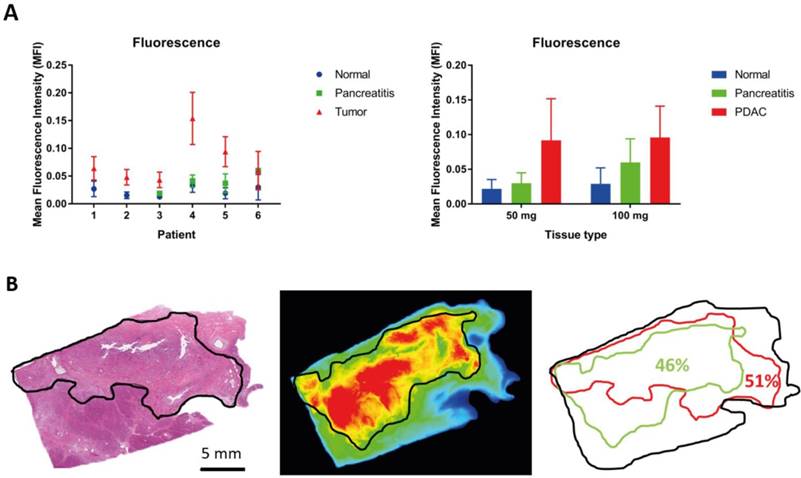
After imaging the fresh surgical specimen, the tissue should be formalin-fixed prior to being processed into bread loaves. Formalin-fixed tissue can still be used for qualitative and quantitative analysis of fluorescence imaging signal, in contrast to paraffin-embedded tissue, which is more suitable for qualitative data analysis. Cassetted, formalin-fixed tissues are ideal for identification of complex tumor-containing tissues since both the orientation and size are matched to the H&E-stained slide of that specific block after paraffin embedding. Quantitative analysis of formalin-fixed tissue can be performed in two ways. First, an absolute quantitative comparison of MFI from the raw data can be performed between tumor tissue and normal surrounding tissue, either per patient or per dose cohort, as demonstrated by an example in pancreatic cancer in Figure 4A. Alternatively, a semi-quantitative scoring method could be used to determine the percentage of fluorescent tissue, compared to the percentage of tumor tissue per tissue block (Figure 4B). The amount of tumor tissue can be assessed on the H&E slide from a particular block, while fluorescence can be measured either with a fluorescence scanner or a closed-field imaging device of a deparaffinized slide.
Quantitative analysis of ex vivo imaging. Quantitative analysis of formalin-fixed tissue can be performed in several ways. Two examples are shown here. (A) A quantitative comparison using the calculated MFI could be performed between tumor tissue and normal surrounding tissue, either per patient or per dose cohort. (B) A semi-quantitative scoring method could be used to determine the percentage of fluorescent tissue, compared to the percentage of tumor tissue per tissue block.
Qualitative representation of ex vivo imaging preferably includes images of the ex vivo specimen, a bread loaf slice and the corresponding cassette to ensure correlation, as also shown in Figure 3. In this context, the bright field image, a black-and-white fluorescence image, and fluorescence overlay image should be shown of each specimen, and ideally also a fluorescence heat-map image.
3. Target validation
After paraffin-embedding of the tissue, the fluorescence signal will be attenuated. This influences the quantification properties of the signal. This, on the other hand, does not influence two other key aspects of data analysis in FGS trials. In the pathologic assessment one should focus on (1) confirmation of tumor targeting using histology, and (2) confirmation of molecular targeting of the biomarkers to the tumor cell. Lastly, correlation of fluorescence intensity with the density of the target molecule can be performed (Figure 1, panel III). Other key factors associated with delivery of the agent can also be assessed in this stage including vascular density, the presence or absence of lymphatic vessels, and the amount of cell proliferation.
Confirmation of tumor cell targeting
As discussed above, this step allows for direct correlation between tissue fluorescence and tumor status on H&E. In early phase trials, this step is critical to ensure the agent is indeed targeting the tumor. Confirmation of tumor status on the H&E slides should be performed in close collaboration with a trained pathologist, who is preferably blinded to the fluorescence imaging results. With the use of a fluorescence scanner, the location of fluorescence can be detected with a µm resolution. Because the tumor is often composed of stromal elements, it will be necessary to distinguish tumor/cancer cell targeting and targeting of other structures within the tumor mass.
If the mapping process (as described above) is performed in a rigorous manner, the correlation generated at this step can be traced back to the images of the intact gross surgical specimen acquired during back-table imaging. It is even possible to apply this correlation back to the intraoperative findings; however, this is complicated by the necessary preservation of the specimen's orientation at each step in addition to the influence of ambient light on intraoperative fluorescence.
Confirmation of molecular targeting
In the case of a targeted agent, the confirmation of precise tumor targeting should be a required step of the data analysis. Here, co-localization of the target of interest and fluorescence signal shows that the agent truly targets the biomarker, as compared to fluorescence signal seen in the tumor as a result of the EPR effect. A (semi-)quantitative analysis can be performed to analyze the fluorescence intensity relative to the receptor density of a specific tissue section (Figure 1). Co-localization can be performed using serial tissue sections, where one slide is used for immunohistochemistry (IHC) to confirm the target biomarker and one is used for fluorescence scanning. Additional IHC may be performed to gain a better understanding of the behavior of the factors influencing the tissue target; for example, for markers that influence delivery of the agent such as vascular density, the presence of lymphatic vessels, and the amount of cell turnover. Therefore, this staining will uniquely be performed during early phase trials when this information is relevant to advancing the agent and further characterizing its potential as a fluorescent biomarker. In later phase trials, one could choose to perform additional staining if the imaging results of a patient are out of the range of what is expected but would not necessarily be performed routinely. Fluorescence microscopy may be introduced into future analyses in FGS clinical trials in order to determine the position of the imaging agent in the tumor, e.g., membrane-bound, internalized, or in stroma.
Summary and conclusion
To ensure wide-spread use and FDA approval of FGS, the quality and standardization of reporting is paramount. Therefore, we present here recommendations for standardized reporting with the goal of establishing a set of minimum requirements for reporting FGS clinical trial results. In this approach, both qualitative and quantitative data play a major role. Three critical elements of early phase clinical trials can be identified and should be reported:
1) Intra-operative data collection: FGS has the potential to play a unique role in the identification of tumor tissue, assessment of resection margins, and detection of tumor-positive lymph nodes, especially in locations that would suggest metastatic disease. In early phase trials, qualitative data should consist of multiple images obtained in each setting, which include a bright field image, a black-and-white fluorescence image, a pseudo-colored fluorescence overlay image, and/or a heat-map. Quantitative data should consist of numerical values reporting fluorescence, preferably both raw MFIs and a ratio such as TBR.
2) Specimen mapping: This is critical to the validation of FGS to correlate fluorescence imaging-based results with the pathologic findings, preferably using fresh tissue. After imaging the fresh surgical specimen, the specimen should be formalin fixed. Formalin-fixed tissue can be used for qualitative and quantitative analysis of fluorescence signal, in contrast to paraffin-embedded tissue, which is more suitable for qualitative data analysis.
3) Target validation: Co-localization of the target of interest and fluorescence signal needs to demonstrate that the agent truly targets the biomarker. A semi-quantitative analysis can be performed to analyze the fluorescence intensity relative to the receptor density of a specific area.
In summary, if these guidelines are widely accepted, trial results could be comparable and collated to expedite future FDA approval.
Abbreviations
AU: arbitrary units; CNR: contrast-to-noise ratio; EPR: enhanced permeability and retention; FDA: Food and Drug Administration; FGS: Fluorescence-guided surgery; H&E: hematoxylin and eosin; ICG: indocyanine green; IHC: immunohistochemistry; SUVmax: maximum standard uptake values; MFI: mean fluorescence intensity; NEMA: National Electrical Manufacturers Association; NIRF: near-infrared fluorescent; PDT: photodynamic therapy; PET: Positron emission tomography; QC: quality control; ROI: region of interest; RFU: relative fluorescence units; SNR: signal-to-noise ratio; SD: standard deviation; SOP: standard operation procedure; TBR: tumor-to-background ratio.
Conflict of interest
No conflicts to state.
References
1. Vahrmeijer AL, Hutteman M, van der Vorst JR, van de Velde CJ, Frangioni JV. Image-guided cancer surgery using near-infrared fluorescence. Nat Rev Clin Oncol. 2013;10:507-18
2. Jiang JX, Keating JJ, Jesus EM, Judy RP, Madajewski B, Venegas O. et al. Optimization of the enhanced permeability and retention effect for near-infrared imaging of solid tumors with indocyanine green. Am J Nucl Med Mol Imaging. 2015;5:390-400
3. Madajewski B, Judy BF, Mouchli A, Kapoor V, Holt D, Wang MD. et al. Intraoperative near-infrared imaging of surgical wounds after tumor resections can detect residual disease. Clin Cancer Res. 2012;18:5741-51
4. Zhang RR, Schroeder AB, Grudzinski JJ, Rosenthal EL, Warram JM, Pinchuk AN. et al. Beyond the margins: Real-time detection of cancer using targeted fluorophores. Nat Rev Clin Oncol. 2017;14:347-64
5. Rosenthal EL, Warram JM, Bland KI, Zinn KR. The status of contemporary image-guided modalities in oncologic surgery. Ann Surg. 2015;261:46-55
6. Rosenthal EL, Warram JM, de Boer E, Chung TK, Korb ML, Brandwein-Gensler M. et al. Safety and tumor specificity of cetuximab-irdye800 for surgical navigation in head and neck cancer. Clin Cancer Res. 2015;21:3658-66
7. Burggraaf J, Kamerling IM, Gordon PB, Schrier L, de Kam ML, Kales AJ. et al. Detection of colorectal polyps in humans using an intravenously administered fluorescent peptide targeted against c-met. Nat Med. 2015;21:955-61
8. Hoogstins CE, Tummers QR, Gaarenstroom KN, de Kroon CD, Trimbos JB, Bosse T. et al. A novel tumor-specific agent for intraoperative near-infrared fluorescence imaging: A translational study in healthy volunteers and patients with ovarian cancer. Clin Cancer Res. 2016;22:2929-38
9. Boogerd LSF, Hoogstins CES, Schaap DP, Kusters M, Handgraaf HJM, van der Valk MJM. et al. Safety and effectiveness of sgm-101, a fluorescent antibody targeting carcinoembryonic antigen, for intraoperative detection of colorectal cancer: A dose-escalation pilot study. Lancet Gastroenterol Hepatol. 2018;3:181-91
10. Miller SE, Tummers WS, Teraphongphom N, van den Berg NS, Hasan A, Ertsey RD. et al. First-in-human intraoperative near-infrared fluorescence imaging of glioblastoma using cetuximab-irdye800. J Neurooncol. 2018;139:135-43
11. Tummers WS, Miller SE, Teraphongphom NT, Gomez A, Steinberg I, Huland DM. et al. Intraoperative pancreatic cancer detection using tumor-specific multimodality molecular imaging. Ann Surg Oncol. 2018;25:1880-8
12. van Dam GM, Themelis G, Crane LM, Harlaar NJ, Pleijhuis RG, Kelder W. et al. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-alpha targeting: First in-human results. Nat Med. 2011;17:1315-9
13. Lamberts LE, Koch M, de Jong JS, Adams ALL, Glatz J, Kranendonk MEG. et al. Tumor-specific uptake of fluorescent bevacizumab-irdye800cw microdosing in patients with primary breast cancer: A phase i feasibility study. Clin Cancer Res. 2017;23:2730-41
14. Shankar LK, Hoffman JM, Bacharach S, Graham MM, Karp J, Lammertsma AA. et al. Consensus recommendations for the use of 18f-fdg pet as an indicator of therapeutic response in patients in national cancer institute trials. J Nucl Med. 2006;47:1059-66
15. Barrington SF, Qian W, Somer EJ, Franceschetto A, Bagni B, Brun E. et al. Concordance between four european centres of pet reporting criteria designed for use in multicentre trials in hodgkin lymphoma. Eur J Nucl Med Mol Imaging. 2010;37:1824-33
16. Boellaard R, Krak NC, Hoekstra OS, Lammertsma AA. Effects of noise, image resolution, and roi definition on the accuracy of standard uptake values: A simulation study. J Nucl Med. 2004;45:1519-27
17. Antunovic L, Rodari M, Rossi P, Chiti A. Standardization and quantification in pet/ct imaging: Tracers beyond fdg. PET Clin. 2014;9:259-66
18. Kinahan PE, Fletcher JW. Positron emission tomography-computed tomography standardized uptake values in clinical practice and assessing response to therapy. Semin Ultrasound CT MR. 2010;31:496-505
19. Kim A, Khurana M, Moriyama Y, Wilson BC. Quantification of in vivo fluorescence decoupled from the effects of tissue optical properties using fiber-optic spectroscopy measurements. J Biomed Opt. 2010;15:067006
20. Weersink R, Patterson MS, Diamond K, Silver S, Padgett N. Noninvasive measurement of fluorophore concentration in turbid media with a simple fluorescence /reflectance ratio technique. Appl Opt. 2001;40:6389-95
21. Pogue BW, Burke G. Fiber-optic bundle design for quantitative fluorescence measurement from tissue. Appl Opt. 1998;37:7429-36
22. Westerterp M, Pruim J, Oyen W, Hoekstra O, Paans A, Visser E. et al. Quantification of fdg pet studies using standardised uptake values in multi-centre trials: Effects of image reconstruction, resolution and roi definition parameters. Eur J Nucl Med Mol Imaging. 2007;34:392-404
23. Scheuermann JS, Saffer JR, Karp JS, Levering AM, Siegel BA. Qualification of pet scanners for use in multicenter cancer clinical trials: The american college of radiology imaging network experience. J Nucl Med. 2009;50:1187-93
24. Wu J, Feld MS, Rava RP. Analytical model for extracting intrinsic fluorescence in turbid media. Appl Opt. 1993;32:3585-95
25. Patterson MS, Pogue BW. Mathematical model for time-resolved and frequency-domain fluorescence spectroscopy in biological tissues. Appl Opt. 1994;33:1963-74
26. Huang Z, Shi S, Qiu H, Li D, Zou J, Hu S. Fluorescence-guided resection of brain tumor: Review of the significance of intraoperative quantification of protoporphyrin ix fluorescence. Neurophotonics. 2017;4:011011
27. Miller SE, Tummers WS, Teraphongphom N, van den Berg NS, Hasan A, Ertsey RD. et al. First-in-human intraoperative near-infrared fluorescence imaging of glioblastoma using cetuximab-irdye800. J Neurooncol. 2018
28. Tahari AK, Chien D, Azadi JR, Wahl RL. Optimum lean body formulation for correction of standardized uptake value in pet imaging. J Nucl Med. 2014;55:1481-4
29. Valdes PA, Angelo JP, Choi HS, Gioux S. Qf-ssop: Real-time optical property corrected fluorescence imaging. Biomed Opt Express. 2017;8:3597-605
30. Choi H. Deep learning in nuclear medicine and molecular imaging: Current perspectives and future directions. Nucl Med Mol Imaging. 2018;52:109-18
31. Tummers WS, Warram JM, Tipirneni KE, Fengler J, Jacobs P, Shankar L. et al. Regulatory aspects of optical methods and exogenous targets for cancer detection. Cancer Res. 2017;77:2197-206
32. Daube-Witherspoon ME, Karp JS, Casey ME, DiFilippo FP, Hines H, Muehllehner G. et al. Pet performance measurements using the nema nu 2-2001 standard. J Nucl Med. 2002;43:1398-409
33. Souza AV, Lin H, Henderson ER, Samkoe KS, Pogue BW. Review of fluorescence guided surgery systems: Identification of key performance capabilities beyond indocyanine green imaging. J Biomed Opt. 2016;21:80901
34. Prince AC, Jani A, Korb M, Tipirneni KE, Kasten BB, Rosenthal EL. et al. Characterizing the detection threshold for optical imaging in surgical oncology. J Surg Oncol. 2017;116:898-906
35. Zhang C, Liu T, Su Y, Luo S, Zhu Y, Tan X. et al. A near-infrared fluorescent heptamethine indocyanine dye with preferential tumor accumulation for in vivo imaging. Biomaterials. 2010;31:6612-7
36. Welvaert M, Rosseel Y. On the definition of signal-to-noise ratio and contrast-to-noise ratio for fmri data. PLoS One. 2013;8:e77089
37. Weissleder R, Ross B, Rehemtulla A, Gambhir SS. Molecular imaging: Principles and practice. 1st ed. Raleigh, USA: People's Medical Publishing House. 2010
38. Tichauer KM, Holt RW, Samkoe KS, El-Ghussein F, Gunn JR, Jermyn M. et al. Computed tomography-guided time-domain diffuse fluorescence tomography in small animals for localization of cancer biomarkers. J Vis Exp. 2012;65(e):4050
39. Tipirneni KE, Warram JM, Moore LS, Prince AC, de Boer E, Jani AH. et al. Oncologic procedures amenable to fluorescence-guided surgery. Ann Surg. 2017;266:36-47
40. Stummer W, Novotny A, Stepp H, Goetz C, Bise K, Reulen HJ. Fluorescence-guided resection of glioblastoma multiforme by using 5-aminolevulinic acid-induced porphyrins: A prospective study in 52 consecutive patients. J Neurosurg. 2000;93:1003-13
41. Tummers WS, Miller SE, Teraphongphom NT, Gomez A, Steinberg I, Huland DM. et al. Intraoperative pancreatic cancer detection using tumor-specific multimodality molecular imaging. Ann Surg Oncol. 2018
42. Dolmans DE, Fukumura D, Jain RK. Photodynamic therapy for cancer. Nat Rev Cancer. 2003;3:380-7
43. Sadot E, Groot Koerkamp B, Leal JN, Shia J, Gonen M, Allen PJ. et al. Resection margin and survival in 2368 patients undergoing hepatic resection for metastatic colorectal cancer: Surgical technique or biologic surrogate? Ann Surg. 2015;262:476-85 discussion 83-5
44. Pawlik TM, Scoggins CR, Zorzi D, Abdalla EK, Andres A, Eng C. et al. Effect of surgical margin status on survival and site of recurrence after hepatic resection for colorectal metastases. Ann Surg. 2005;241:715-22 discussion 22-4
45. O'Kelly Priddy CM, Forte VA, Lang JE. The importance of surgical margins in breast cancer. J Surg Oncol. 2016;113:256-63
46. Black C, Marotti J, Zarovnaya E, Paydarfar J. Critical evaluation of frozen section margins in head and neck cancer resections. Cancer. 2006;107:2792-800
47. Provenzano E, Bossuyt V, Viale G, Cameron D, Badve S, Denkert C. et al. Standardization of pathologic evaluation and reporting of postneoadjuvant specimens in clinical trials of breast cancer: Recommendations from an international working group. Mod Pathol. 2015;28:1185-201
48. Gao RW, Teraphongphom NT, van den Berg NS, Martin BA, Oberhelman NJ, Divi V. et al. Determination of tumor margins with surgical specimen mapping using near-infrared fluorescence. Cancer Res. 2018
Author contact
![]() Corresponding author: E.L. Rosenthal, MD. Professor of Otolaryngology and Radiology, Ann & John Doerr Medical Director, Stanford Cancer Center, Stanford, CA. Tel: 650 723 4250; Fax: 650 723 2225; E-mail:elredu
Corresponding author: E.L. Rosenthal, MD. Professor of Otolaryngology and Radiology, Ann & John Doerr Medical Director, Stanford Cancer Center, Stanford, CA. Tel: 650 723 4250; Fax: 650 723 2225; E-mail:elredu