Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods and Materials
Results
Discussion
Conclusion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(19):5434-5451. doi:10.7150/thno.27882 This issue Cite
Research Paper
Microglial TLR4-dependent autophagy induces ischemic white matter damage via STAT1/6 pathway
Chuan Qin1*, Qian Liu1*, Zi-Wei Hu1, Luo-Qi Zhou1, Ke Shang1, Dale B. Bosco2, Long-Jun Wu2, Dai-Shi Tian1 ![]() , Wei Wang1
, Wei Wang1 ![]()
1. Department of Neurology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China
2. Department of Neurology, Mayo Clinic, Rochester, MN 55905
* These two authors contributed equally to this work.
Received 2018-6-14; Accepted 2018-10-3; Published 2018-10-29
Abstract

Rationale: Ischemic white matter damage frequently results in myelin loss, accompanied with microglial activation. We previously found that directing microglia towards an anti-inflammatory phenotype provided a beneficial microenvironment and helped maintain white matter integrity during chronic cerebral hypoperfusion. However, the molecular mechanisms underlying microglial polarization remain elusive.
Methods: Hypoperfusion induced white matter damage mice model and lipopolysaccharide (LPS) induced primary cultured microglia were established. Autophagy activation in microglia was detected both in vivo and in vitro by immunofluorescence, Western blot and electron microscopy. Autophagy inhibitors/agonist were administrated to investigate the role of autophagic process in modulating microglial phenotypes. Quantitative real time-polymerase chain reaction and Western blot were carried out to investigate the possible pathway.
Results: We identified rapid accumulation of autophagosomes in primary cultured microglia exposed to LPS and within activated microglia during white matter ischemic damage. Autophagy inhibitors switched microglial function from pro-inflammatory to anti-inflammatory phenotype. Furthermore, we found TLR4, one of the major receptors binding LPS, was most highly expressed on microglia in corpus callosum during white matter ischemic damage, and TLR4 deficiency could mimic the phenomenon in microglial functional transformation, and exhibit a protective activity in chronic cerebral hypoperfusion. Whereas, the anti-inflammatory phenotype of microglia in TLR4 deficiency group was largely abolished by the activation of autophagic process. Finally, our transcriptional analysis confirmed that the up-regulation of STAT1 and down-regulation of STAT6 in microglia exposure to LPS could be reversed by autophagy inhibition.
Conclusion: These results indicated that TLR4-dependent autophagy regulates microglial polarization and induces ischemic white matter damage via STAT1/6 pathway.
Keywords: TLR4, autophagy, white matter ischemic injury, microglia, STAT
Introduction
Patients with carotid artery stenosis often present with white matter (WM) lesions and related cognitive deficits [1, 2]. Chronic cerebral hypoperfusion-induced WM damage has recently arisen as a key pathology for various diseases including ischemic stroke and vascular dementia [3, 4]. Characterized by loss of axon-glial integrity and demyelination, WM injury is an important contributor to the cognitive deficits that follow cerebral hypoperfusion [5]. This pathology can also result in microglial activation and increase neuroinflammation [6].
Microglia are the resident immune cells of the central nervous system (CNS), which become activated in brain pathologies such as ischemic stroke and intracerebral hemorrhage. Activation of microglia features changes in morphology and receptor expression, cytokine release, migration, and phagocytosis, and it ranges from pro-inflammatory (classical activation) and potentially neurotoxic to anti-inflammatory (alternative activation) and neuroprotective phenotypes [7-9]. The balance between classically and alternatively activated microglial phenotypes can dictate disease progression in the CNS. Microglial phenotype is often affected by sensing of pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), or modified endogenous ligands via innate pattern recognition receptors, such as Toll-like receptors (TLRs) [9, 10]. Of interest is TLR4, which is mainly expressed in microglia, and acts with cluster of differentiation 14 (CD14), myeloid differentiation protein 2 (MD-2), and lipopolysaccharide (LPS)-binding protein to recognize LPS [9, 11]. TLR4 has attracted particular attention in several inflammatory CNS diseases, including stroke and multiple sclerosis [12, 13]. Both pharmacological inhibition and genetic deficiency of TLR4 exerts a neuroprotective effect within experimental stroke conditions [12, 14, 15]. TLR4 is also thought to be critical for recurrent immune-driven microglia activation and inflammatory response within multiple sclerosis [13, 16]. However, despite extensive research, it is still largely unknown how TLR4 influences microglial phenotype and its importance to WM stroke.
Autophagy is an evolutionarily conserved degradation pathway, which primarily functions as a cell survival adaptive mechanism during stress conditions [17]. Recently, TLR4 dependent autophagy has been reported to be critical for macrophage associated inflammatory response [18-20]. These studies indicate that pathogen-sensing by TLR4 triggers multiple signaling events that converge on pathogen and organelle-specific autophagy in several types of cells including mononuclear macrophage system [21]. Emerging work suggests that autophagy may also contribute to glial cell function [22, 23]. However, interactions and mechanisms between microglial autophagy with neuro-inflammation and myelin integrity have been little explored.
In this study, we investigated the molecular mechanisms of autophagy on TLR4 signal pathway in microglial polarization. To mimic clinical conditions with chronic cerebral hypoperfusion, a murine model of bilateral carotid artery stenosis (BCAS) was used to determine the role of TLR4 in WM injury and repair. The effect of TLR4-mediated autophagy upon microglia phenotype was also explored in vitro with primary microglia. Additionally, we used transcriptional profiling to determine that modulation of microglial phenotype by autophagy was mediated by the STAT1/6 signaling pathway. Taken together, our results indicate that TLR4-dependent autophagy might have a role in regulation of microglial polarization and could induce ischemic white matter damage via STAT1/6 pathway.
Methods and Materials
Animals
All animal studies were approved by the Institute of Animal Care Committee of Tongji Medical College, Huazhong University of Science and Technology, China. Adult male mice were used to reduce sex and age influences on white matter ischemic injury. C57BL/6J (wild-type, WT) mice (20-25g; 10-12 weeks old) were obtained from Hunan SJA Laboratory Animal Co. Ltd., Hunan, China. The murine strain CB57/10Scnj (TLR4 knockout, TLR4 KO, Jackson Laboratory, Bar Harbor, ME, USA), which does not express TLR4, was used for deletion studies [24]. Mice were housed in groups of 2-4 mice and kept in a 12-hour light/12-hour dark cycle at the standard conditions of 22 °C temperature with ad libitum access to water and food.
Bilateral Carotid Artery Stenosis (BCAS) procedure
Chronic cerebral hypoperfusion was induced by BCAS procedure as described [25]. Briefly, a midline cervical incision was made and bilateral common carotid arteries were exposed under isoflurane anesthesia (induced at 5%, and maintained at 1.2-1.6%). Then, microcoils (0.18 mm internal diameter, Sawane Spring Co, Shizuoka, Japan) were twined around both common carotid arteries under an operating microscope. Mice were remaining in a hypoperfusion state after the surgery for the remainder of the study. For the sham-operated animals, bilateral common carotid arteries were exposed without coils placed around the arteries.
Experimental groups were randomized and analyzed by independent investigators blinded to treatment condition. Because BCAS leads to consistent WM lesions, as previously reported [6, 25] and based on our preliminary studies, we detected an obvious changing between sham and BCAS groups. Sample size was calculated with the Graphpad random number generator for α value of 0.05 and power value of 0.80. A total of 82 wild-type and 84 TLR4 knockout male mice weighing 20 to 25 g were used for the experiment.
Primary Cell Culture
Primary microglia were isolated from the brain of neonatal wild-type and TLR4 knockout mice at P1-P2 as described previously [6, 26] with minor modifications. Briefly, mixed glial cells were cultured in high glucose DMEM medium supplemented with 20% fetal bovine serum (FBS) at 37 °C in a 95% O2 and 5% CO2 incubator for 8-10 days, with the medium refreshed every 3 days. Microglia were dislodged by shaking at 200 rpm at 37°C for 2 h. The obtained microglia were seeded into 6-well plates in high glucose DMEM medium supplemented with 20% FBS at a density of 1×106/well for 24 h before further treatment. The purity of cultured microglia was identified to be more than 98% by immunofluorescent staining for anti-Iba1 (Wako, 1:500).
LPS (100ng/ml) or IL-4 (20 ng/mL) was added to microglial cultures for 24 hours to induce different phenotypes. To determine the role of autophagy in microglial activation, cultured microglia stimulated by LPS were also incubated with the autophagy inhibitors bafilomycin (10nm, HY-100558, MedChem Express, MCE)[27, 28] or wortmannin (100nm, HY-10197, MedChem Express, MCE)[18], or the autophagy agonist rapamycin (10nm, HY-10219, MedChem Express, MCE) [28].
Eight-arm radial maze test
Working memory was assessed by an eight-arm maze as described [29] one month after BCAS. During pre-training, the mice were deprived of food 12 hours per day, and then placed in the central starting platform and allowed to consume for a 5-min period (one session per mouse) food pellets scattered over the whole maze extending from the center to the distal end of each arm. In the spatial working memory task of the eight-arm radial maze, food pellets were placed in the distal end of each arm. Each mouse was placed in the central starting platform to consume food in each arm, until all eight pellets were consumed or 15min had elapsed. A working memory error was scored when a mouse reentered an arm. The animals completed one trial per day (7 trials total). For the reference memory task, four of the eight arms were constantly baited by one pellet in a food well and a trial was terminated immediately after four pellets were consumed. The animals completed one trial per day (9 trials total). The whole experiments were performed in a separate testing room that was quiet and dark, and recorded by an observer who was blinded to different treatment groups.
Tissue preparation
At one month post-surgery, mice were euthanized with 5% isoflurane and transcardially perfused with 20 ml of 0.9% heparinized PBS. For Western blot, brains were removed and rapidly frozen in nitrogen-cooled isopentane. For immunohistochemistry and immunofluorescence, mice were then perfused with 20 ml of 4% paraformaldehyde (PFA) in 0.1% phosphate buffer (PBS). The fixed brains were removed and post-fixed in 4% PFA for 6h (4°C) and gradually dehydrated using 15%, 20%, and 30% sucrose in PBS. Coronal slices of 12 μm was performed at -20°C using a constant temperature freezing microtome (CM 1900, Leica, Germany). They were then gently transferred to the microslides and stored in -80°C until processing.
Luxol fast blue staining
Luxol fast blue (LFB) staining was conducted to observe histological changes in the white matter [25]. Briefly, brain slices were incubated in a preheated 0.1% LFB (G1030, Goodbio technology CO. LTD, Wuhan, China) at 60oC for 8-10 hours. After washing, brain sections were differentiated in lithium carbonate solution, and then dehydrated by 75%-90%-100% ethanol. Images were taken by light microscope (DP 50, Olympus, Japan). The white matter lesions were evaluated in 6 regions: the medial part of corpus callosum (CCm), fiber bundles of the caudoputamen (CPu), fimbria of hippocampus (F), anterior commissure (AC), internal capsule (IC), and optic tract (OT). Data was shown as median ± interquartile. Mann-Whitney U test was used for the comparison between the groups.
Immunofluorescent staining
For immunofluorescent staining, brain slices and cell cultures were fixed for 15 min in ice-cold 4% PFA, washed three times with 0.1M PBS, and then permeablized by 0.25 % Triton-X100 in PBS. After blocking with 10% bovine serum albumin (Sigma-Aldrich) for 1h, slides were incubated with primary antibodies overnight at 4oC followed by corresponding antibody incubation for 1 h without light at room temperature. Sections were counterstained with 4,6-diamidino-2-phenylindole (DAPI) (10 µg/mL, Sigma-Aldrich).
Primary antibodies used for immunohistochemistry included those against: myelin associated glycoprotein (MAG, 1:200, L20, sc-9543; Santa Cruz Biotechnology), myelin basic protein (MBP,1:100, MAB386; Millipore), Neurofilament-L, M and H (NF-L, 2835, NF-M, 2838, and NF-H, 2836, 1:1000, Cell Signaling), panNeurofascin (PanNfasc, 1:1000, kindly given by Prof. Peter Brophy), type Ⅵ sodium channel (Nav1.6, 1:200, AB5580, Millipore), contactin-associated protein (Caspr, 1:100, clone K65/35, UC Davies NIH NeuroMab), ionized calcium binding adapter molecule1 (Iba1,1:500,Wako Pure Chemical Industries), Iba1 (1:200, 234004, Synaptic Systems), adenomatous polyposis coli (APC, 1:200, OP80, Millipore), GFAP (1:200, CST#3670, CST), TLR4 (1:100, spc-200, StressMarq Biosciences Inc), Fc RII/III receptor (CD16/32, 1:50, 553142, BD Pharmingen), macrophage mannose receptor (CD206, 1:200, AF2535, R&D system). Secondary antibodies labeled with FITC, Cy3, AlexaFluor488, or AlexaFluor647 were purchased from Jackson ImmunoResearch Laboratories. Images were acquired by confocal microscopy (FV1200, Olympus, Japan).
In the sections immunostained for LC3, Iba-1, CD16/32, and CD206, the number of immunopositive cells were counted by an investigator who was blinded to different treatment groups in six fields (cells/mm2) in the median part of the corpus callosum at 0 to 0.5 mm anterior from the bregma.
The images of nodes of Ranvier were taken with a 100× oil immersion objective at 1024 × 1024 pixel resolution. Length measurements and co-localization assessment were performed using Imaris (Bitplane AG, Switzerland) and ImageJ (NIH, USA) software as described [30]. The sodium channel cluster (Nav1.6) length measurements were performed using ImageJ. For Caspr and panNfasc, common epitope labeling co-localization analysis was measured using Imaris and Pearson coefficients. To optimize the co-localization of Caspr and panNfasc in 3-dimention graphs, images were rendered in blend mode with grey background, and the parameters including image contrast, brightness and blend opacity were adjusted using Imaris. The investigator performed the measurements with blinded to different treatment groups.
Monodansylcadaverine staining
Monodansylcadaverine (MDC) (D4008, Sigma-Aldrich) was a special autophagolysosome marker to analyze the process of autophagy [31]. Cell plates were stained with MDC at a finial concentration of 50 μM for 10 min at 37oC. After incubation, cells were washed three times with PBS, then fixed in 4% PFA for 30 min, and immediately observed under the light microscope (DP 50, Olympus, Japan).
In MDC staining, the percentage of cells with characteristic punctate MDC staining indicative of autophagy was assessed by an investigator who was blinded to different treatment groups in six random fields.
Electron Microscopy
For brain tissue, mice were anesthetized and subjected to intracardial perfusion with 2.5% glutaraldehyde in 4% paraformaldehyde. Brain were removed and cut into 1mm thick coronal slice in ice, then the corpus callosum was cut into 1mm section and post-fixed with 2.5% glutaraldehyde and stored at 4 oC. In cell, microglia in a 10cm dish were fixed with 2.5% glutaraldehyde for 2 h and scraped down to centrifuged in 2500 rpm for 5 min, then stored at 4 oC. The samples were processed for routine EM observation and examined with an electron microscope (Hitachi, HT7700) at 120 kV. At 1000x magnification, the ratio of myelinated axon thickness to axon diameter (g-ratio) was measured for 200 fibers (40 axons per mouse, 5 mice per group) in electronmicro-graphs using the Image J (National Institute of Health, Bethesda, MD, USA). The g-ratio was plotted against the diameter of axons to directly compare the extent of myelination around an axon size. At 5000x magnification, the autophagic vacuole in microglia was identified as double membrane bound vesicle within residual.
RNA Extraction and Quantitative Real-Time PCR
Total RNA was extracted from the primary microglia cultures from different treatment groups using Trizol (Invitrogen, Carlsbad, CA, USA). 1ug RNA was synthesized using ReverTra Ace qPCR RT Kit (TOYOBO, Japan) according to the manufacturer's instructions. Quantitative PCR was performed using synthetic primers and SYBR gene PCR Master Mix (TOYOBO, Japan). After incubation at 95oC for 30s, samples were subjected to 40 cycles of 95 oC for 3s and 72oC for 30s. Related primers were listed in Table 1. β-actin was used as an internal reference. The expression of target genes were analyzed according to the 2-∆∆CT method [32].
Related primers of target genes in quantitative PCR.
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| β-Actin | TGGAATCCTGTGGCATCCATGA | AATGCCTGGGTACATGGTGGTA |
| CD86 | GACCGTTGTGTGTGTTCTGG | GATGAGCAGCATCACAAGGA |
| iNOS | GCTTGTCTCTGGGTCCTCTG | CTCACTGGGACAGCACAGAA |
| TNF-α | ACGGCATGGATCTCAAAGAC | AGATAGCAAATCGGCTGACG |
| IL-1β | TGTCTTGGCCGAGGACTAAGG | TGGGCTGGACTGTTTCTAATGC |
| TGF-β | TGCGCTTGCAGAGATTAAAA | CGTCAAAAGACAGCCACTCA |
| YM | ACCCCTGCCTGTGTACTCACCT | CACTGAACGGGGCAGGTCCAAA |
| Arg1 | TTAGGCCAAGGTGCTTGCTGCC | TACCATGGCCCTGAGGAGGTTC |
| IL-10 | GGCAGAGAACCATGGCCCAGAA | AATCGATGACAGCGCCTCAGCC |
| JAK1 | AGACTGAGGTGAAGCAGGTGG | CAGGGCGAAGAGGTTGTGA |
| JAK2 | GCTCCTCTGCTTGATGACTTTG | ACAGCCAGTGGAGTCTGGTCT |
| JAK3 | CCTGCGGTTGGTGATGGA | GCAGTAGGCCGTCGGTCGGTGT |
| STAT1 | TGCCTATGATGTCTCGTTTGC | ATCTGTACGGGATCTTCTTGGA |
| STAT3 | GTACAAAGGCACTCCATCAGC | CCCCATATCGTCTGAAACTCC |
| STAT5 | AATGAGAACACCCGCAATGAGT | TGCCAACGCTGAACTGAGACT |
| STAT6 | GCTGGACAGACCTACAGACCC | GGCGTTGTGGAAAGTCAACATA |
| IRF3 | ACGGCAGGACGACACGAT | TCAGCAGCTAACCGCAACAC |
| IRF4 | AGGTGACTCTGTGCTTTGGTGA | GGGAGCGGTGGTAATCTGG |
| IRF5 | GGGGACAACACCATCTTCAAG | CGTTGGAGCAGACCTCGTAGA |
| IRF8 | CGAACAGATCGACAGCAGCA | AGCACAGCGTAACCTCGTCTT |
| NF-кB | TGAACTTGTGGGGAAGGACTG | GGAACGTGAAAGGGGTTATTGT |
| MSX3 | GCCGCTGATGCTCCTGGTA | TGGGCAAGGTTTTAGGAAGGT |
| PPARG | CCACAGTTGATTTCTCCAGCAT | TCCCCACAGACTCGGCAC |
| NLRP3 | TCTTCTCAAGTCTAAGCACCAAC | ACAGCAATCTGATTCCAAAGTC |
| TRAF6 | GATCCAGGGCTACGATGTGG | CTTGTGCCCTGCATCCCTTA |
Western blot analysis
The protein of white matter tissue and primary microglia was obtained for Western blot analysis as described [30, 33]. White matter tissue has been dissected to homogenize in ice with RIPA buffer solution (Beyotime, China) for protein extraction. Cells were lysed in buffer (Beyotime, China) supplemented with phosphatase inhibitors (Beyotime, China). The concentration of protein was measured by using bovine serum albumin (BSA, Beyotime, China) as a standard.
Equivalent amounts of protein (40ug) of each sample were resolved on 10-15% sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel electrophoresis. After electrophoresis, the proteins were transferred to PVDF membranes. The membranes incubated overnight at 4oC with the primary antibodies. Primary antibodies used for Western blot included those against: MAG (1:1000, L20, sc-9543; Santa Cruz Biotechnology), MBP (1:500, MAB386; Millipore), NF-L, NF-M and NF-H (#2835, #2838, and #2836, 1:1000, Cell Signaling), inducible nitric oxide synthetase (iNOS, 1:10 ab15326, Abcam), CD16 (1:2000, ab109223, Abcam), Arginase-1 (Arg-1, 1:200, sc-18351, Santa Cruz Biotechnology), CD206 (1:1000, AF2535, R&D systems), LC3B (1:1000, L7543, Sigma-Aldrich), P62/SQSTM1 (1:1000, p0067, Sigma-Aldrich), Beclin-1 (1:1000, B6061, Sigma-Aldrich), and β-actin (1:8000, Boster, China). Then, membranes were incubated with DelightTM 800 Conjugate anti-mouse (H+L), anti-goat (H+L), anti-rabbit (H+L) or anti-rat (H+L) secondary antibody (1:10000, CST, Germany), and images were captured using Odyssey CLx Imager (LI-COR Biosciences). Finally, the intensity of blots was semi-quantified by ImageJ. β-actin was used as the internal control.
Statistical analysis
All numerical values and error bars in the quantification graphs are expressed as mean ± SEM except for LFB staining. Statistical analyses were performed using SPSS 19.0. Significance was determined by two-way analysis of variance (ANOVA) with repeated analysis, Mann-Whitney U test, or two-way ANOVA with Dunnett's post-hoc test to compare differences between groups. All tests were considered statistically significant at P<0.05.
Results
Cerebral hypoperfusion-induced white matter injury and cognitive dysfunction was attenuated by TLR4 deficiency
Cognitive impairment was detected in mice with chronic cerebral hypoperfusion [6, 34]. TLR4 deficiency was shown to delay neuronal death and neurological impairment in mouse models of brain injuries [35, 36]. As such, we hypothesized that knockout of TLR4 would reduce BCAS-induced cognitive impairment. Working memory was assessed using an eight-arm maze test. Sham control mice quickly improved their performance over the training period, while mice at 1 month post-BCAS made significantly more revisiting errors and less different arm choices in the first 8 entries, with no change in revisiting errors (Figure 1A). There was no difference between TLR4 knockout and wild-type mice within the sham condition. However, after BCAS, TLR4 deficiency resulted in fewer revisiting errors (Figure 1A), and a greater number of different arm choices in the first eight entries compared to the wild type controls (Figure 1A). No obvious differences in spatial reference memory were detected between the groups (Figure 1A).
TLR4 deficiency attenuated cognitive impairment and white matter lesions induced by BCAS. (A) The working memory and reference memory of mice were assessed by the 8-arm maze test at 1-month post injury. WT mice suffered from BCAS made much more revisiting errors (P<0.001) and less different arm choices (P<0.001) comparing to the sham-operated mice. TLR4 knockout mice with BCAS made much less revisiting errors (P<0.001) and more different arm choices (P=0.007) comparing to the WT group. No impairment in spatial reference memory was revealed between different groups (P>0.05). Two-way analysis of variance (ANOVA) with repeated analysis, n=9 in each group. (B) White matter lesions were detected by LFB staining in different groups. Scale bar, 200 µm. Dot-plot with median and interquartile range of the severity of white matter lesions in in CCm, CPu, AC, IC, F and OT in histogram. Two-way ANOVA with Dunnett's post-hoc test, **P<0.01 versus WT Sham, #P<0.05, ##P<0.01 versus WT BCAS. n=6 per group. (C) Representative images depicting immunofluorescent labeling of myelin-associated glycoprotein (MAG), myelin basic protein (MBP), and nerve fiber (NF) in the corpus callosum of coronal slices from different groups. Scale bar, 100µm. The expression of MAG, MBP and NF (three major NF subunits based upon their molecular mass: the lowest (NF-L), the middle (NF-M) and the highest (NF-H)) was determined by Western blot in mice from different groups. Quantitative analysis of Western blot results was performed. Two-way ANOVA with Dunnett's post-hoc test, **P˂0.01 versus WT Sham, ##P<0.01 versus WT BCAS. n=8 per group.
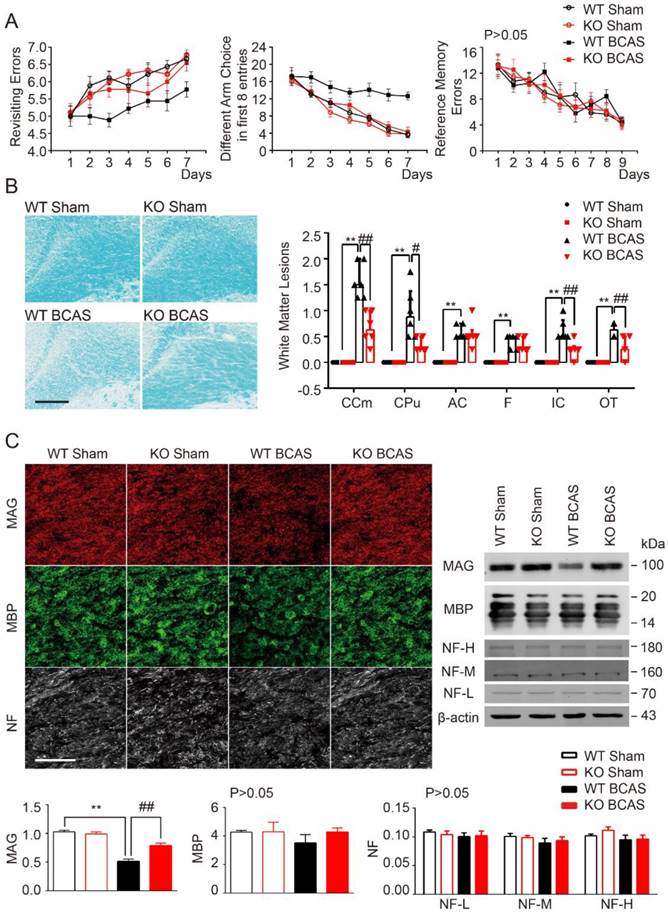
Our previous studies noted that working memory deficits in BCAS was due to the injury of frontal-subcortical circuits [6]. Luxol fast blue (LFB) staining was used as a highly sensitive marker of myelin loss. The disruption of WM integrity was detected in the medial part of corpus callosum (CCm), caudoputamen (CPu), anterior commissure (AC), the internal capsule (IC), fimbria of hippocampus (F), and optic tract (OT) 1 month after hypoperfusion (Figure 1B). Interestingly, myelin integrity was preserved in TLR4 knockout mice, which was in agreement with the function of TLR4 in demyelination and cognitive impairment (Figure 1B).
To further explore the function of TLR4 in myelin integrity after BCAS, we investigated the expression of myelin-associated glycoprotein (MAG), a marker for myelin-axon integrity, as well as myelin basic protein (MBP) and neurofilament (NF), general markers for myelin and axons respectively. We quantified their expression at 1 month post-injury in the BCAS group, using immunochemistry and Western blot (Figure 1C). MAG was sharply compromised 1 month after BCAS, whereas MBP tended to be lower in BCAS mice, but no statistical difference between the groups detected, and expression of pan-axon marker NF appeared consistent (Figure 1C).
Together, these results showed that BCAS impaired working memory and disrupted myelin integrity. These impairments could be mitigated in TLR4 knockout mice, suggesting a role for TLR4 in mediating BCAS-triggered deficits (Figure 1).
TLR4 deletion protected white matter against ischemic disorganization of Ranvier's Nodes and ultrastructural damage of myelin
The nodes of Ranvier, which are flanked by myelinated axon segments, are important sites for action potential propagation based on the clustering of voltage-gated sodium channels (Nav1.6) [30]. Previous studies have shown that nodes of Ranvier structure is vulnerable to ischemia and is a sensitive indicator of WM ischemic injury [5, 30]. Indeed, we found a significant reduction in the co-localization of Caspr and panNfasc within the paranodes of wild type mice 1-month post BCSA, suggesting a breakdown in paranodal structure (Figure 2A). In addition, a significant increase to the length of Nav1.6 clusters in fibers within the corpus callosum was observed (Figure 2B). TLR4 knockout improved the length of Nav1.6 channels and selective delocalization of neurofascin and Caspr in response to hypoperfusion injury (Figure 2A and 2B). No difference was detected between the TLR4 knockout mice and their wild type controls under sham conditions.
TLR4 deficiency alleviated the disruption of myelin sheath and white matter integrity due to chronic cerebral hypoperfusion. (A) Representative 3 dimensional confocal images labeled with panNfasc/Caspr. Scale bar, 10µm. Quantitative analysis of panNfasc/Caspr colocalization was shown in the histogram. Two-way ANOVA with Dunnett's post-hoc test, **P˂0.01 versus WT Sham, ##P˂0.01 versus WT BCAS. n=5 per group. (B) Representative confocal images labeled with Nav1.6/Caspr. Scale bar, 10µm. Summary of the length of Nav1.6 domain was shown in curve. Two-way ANOVA with Dunnett's post-hoc test, P˂0.0001 WT BCAS versus WT Sham, KO BCAS versus WT BCAS. n=5 mice per group and 100 Nav1.6 domains for each mouse. (C) Representative electron microscopy images in the corpus callosum from different groups. Scale bar, 2µm. (D) Quantitative analysis of the g ratio was performed. Two-way ANOVA with Dunnett's post-hoc test, **P˂0.01 versus WT Sham, #P˂0.05 versus WT BCAS, n=200 myelinated axons (40 axons per mouse, 5 mice per group) for each group.
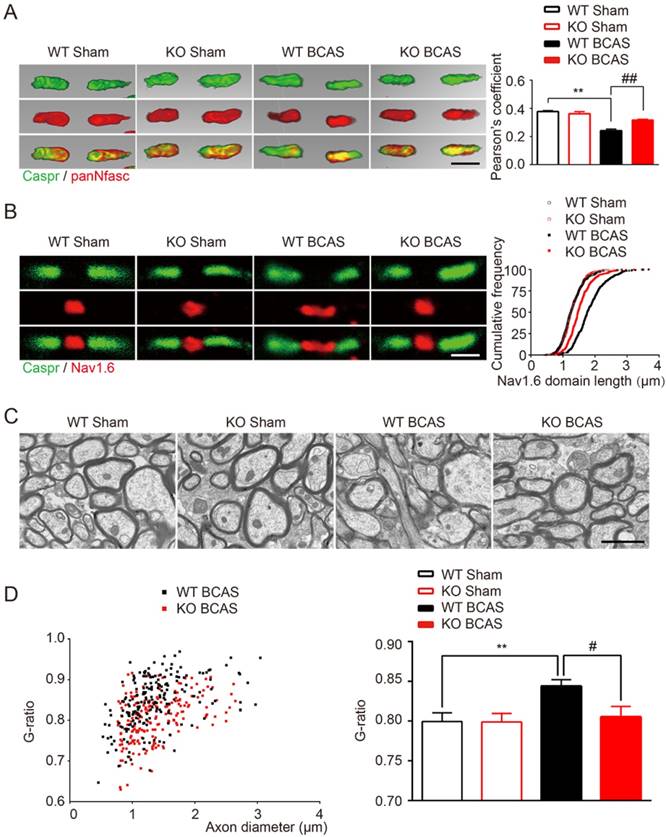
Ultrastructural damage of myelin in corpus callosum was also detected in the hypoperfused mice, as previously reported [6]. Disruption of myelinated structures, manifested as vacuoles and delamination, was observed in wild-type BCAS mice, compared with intact lamellar myelin sheath in sham control group (Figure 2C). Quantitative analysis of g-ratios confirmed the ultrastructural disruption of myelin 1-month post BCAS (Figure 2D). We also found that the g-ratio was significantly decreased in TLR4 knockout mice that underwent BCAS, suggesting that the disruption of the myelin sheath was relieved by deletion of TLR4 (Figure 2D).
Together, these results indicated that chronic cerebral hypoperfusion induces the disorganization of Ranvier's Nodes and ultrastructural damage of myelin. More importantly, we found that hypoperfusion induced damage could be partially ameliorated by TLR4 deficiency.
TLR4 deletion facilitated microglial polarization towards an anti-inflammatory profile
TLR4 is probably the most versatile and wildly expressed member of the TLR family [37]. Ionized calcium binding adapter molecule 1 (Iba1), glial fibrillary acidic protein (GFAP), and adenomatous polyposis coli (APC), the classical markers for microglia, astrocyte, and oligodendrocyte, respectively, were used to explore the cellular location of TLR4 in WM. TLR4 was expressed on all glia types, but was most highly expressed on microglia in corpus callosum (Figure 3A).
TLR4 deletion facilitated microglial polarization towards an anti-inflammatory profile. (A) Representative images showing cellular localization of TLR4 in glial cells in the corpus callosum. Iba-1, a marker for microglia; GFAP for astrocytes; APC for oligodendrocytes. Scale bar, 50 µm. (B) Representative confocal images of coronal sections labeled with Iba-1, CD16/32 and CD206 at 1 month post BCAS. Scale bar, 50µm. Quantitative analysis of Iba-1, CD16/32 double positive cells and Iba-1, CD206 double positive cells was shown in the histogram. Two-way ANOVA with Dunnett's post-hoc test, **P˂0.01 versus WT Sham, ##P˂0.01 versus WT BCAS. n=6 per group. (C) The protein expression of pro-inflammatory markers (iNOS, CD16/32) and anti-inflammatory markers (Arg-1, CD206) in primary microglia was detected by Western blot. Quantitative analysis was performed. Two-way ANOVA with Dunnett's post-hoc test, **P˂0.01 versus WT Control, #P˂0.05 ##P˂0.01 versus WT LPS. n=6 per group.
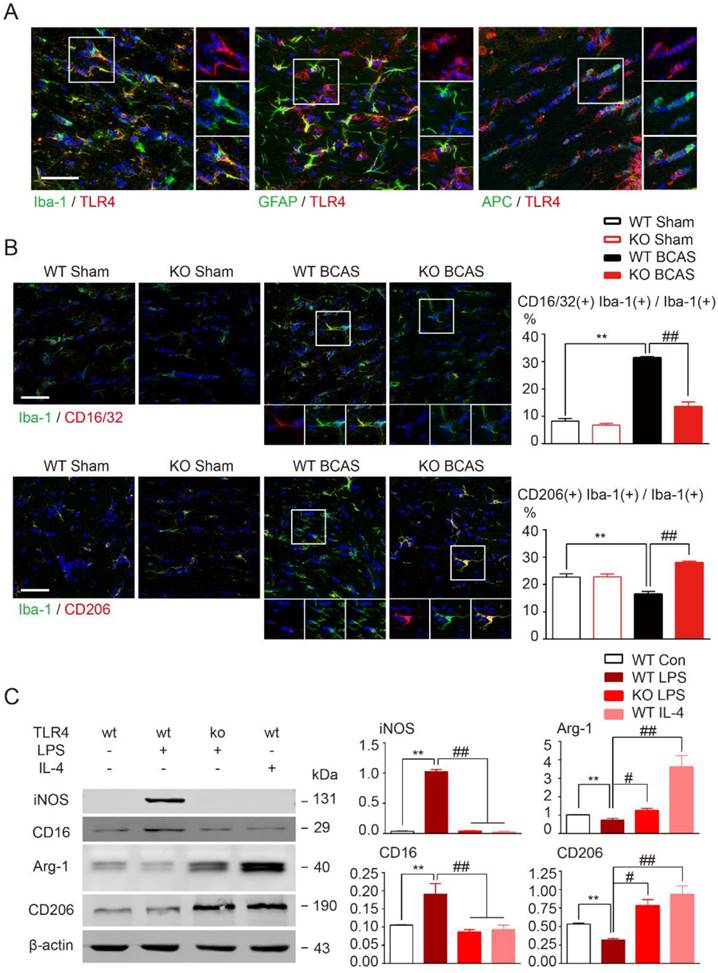
TLR4-mediated pathway activation is involved in the production of pro-inflammatory mediators [36, 38]. Therefore, we investigated whether TLR4 was required for microglial polarization towards a pro-inflammatory phenotype in our BCAS model. Pro-inflammatory microglia are characterized by upregulation of CD16 Fc receptors and inducible nitric oxide synthase (iNOS), whereas microglia undergoing anti-inflammatory activation upregulate arginase (Arg)-1 and mannose receptor (CD206) [6, 39]. For wild-type mice prolonged hypoperfusion resulted in the percentage of CD16/32, Iba-1 double positive microglia to substantially increase while CD206, Iba-1 double positive microglia number significantly decreased when compared to sham controls (Figure 3B). However, induction of pro-inflammatory microglia was largely inhibited by TLR4 knockout and more anti-inflammatory microglia were observed (Figure 3B). Loss of TLR4 in sham controls did not change the functional state of microglia. These results indicate that deletion of TLR4 switched microglia polarization from pro-inflammatory to anti-inflammatory type in WM ischemic injury.
While the previous results indicate a pathological role for TLR4, it is not expressed exclusively by microglia following CNS injury [40]. To investigate TLR4 in microglia alone, we used LPS stimulation in primary cultured microglia to mimic the neuroinflammation observed in WM injuries. Western blot results revealed that pro-inflammatory markers (CD16 and iNOS) were significantly increased by LPS stimulation and anti-inflammatory markers (arginase1 and CD206) were greatly suppressed (Figure 3C). Consistent with in vivo data, these alterations in microglial markers were significantly attenuated by TLR4 deletion (Figure 3C), indicating that TLR4 facilitated microglial polarization towards a pro-inflammatory profile both in vivo and in vitro.
Inflammation induced microglial autophagy could be blocked by TLR4 deletion
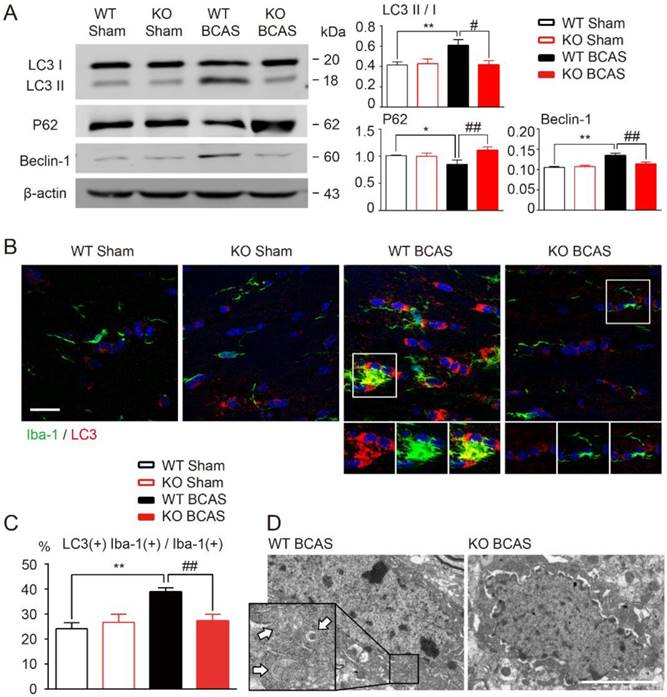
It has been reported that TLR4 might participate in the process of autophagy activation in several different tissues [19, 41], including mononuclear macrophage system [18, 20]. We hypothesized that TLR4 is also involved in the development of microglial autophagy during WM hypoperfusion. To test this hypothesis, we compared the expression profiles of autophagy related proteins in the WM of wild-type and TLR4 knockout mice after BCAS. Immunofluorescent staining and Western blot analysis showed that chronic hypoperfusion increased the expression of LC3 II and beclin-1, and down regulated p62 expression in white matter (Figure 4A). LC3 and Iba-1 double positive microglia elevated significantly at one month after BCAS (Figure 4B and 4C). TLR4 knockout suppressed ischemia induced conversion of LC3 I to LC3 II, inhibited beclin-1 up-regulation and increased p62 protein expression in white matter (Figure 4A). The increase of LC3 and Iba-1 double positive microglia after BCAS was reversed in TLR4 knockout mice (Figure 4B and 4C). In addition, transmission electron microscopy revealed an increase in the formation of autophagic vesicles in microglia after BCAS, which was also reduced in TLR4 knockout mice (Figure 4D).
TLR4 deficiency suppressed microglial autophagy after BCAS. (A) The expression of autophagy related proteins (LC3, P62 and Beclin-1) in white matter at 1 month post BCAS was detected by Western blot. Quantitative analysis was performed. Two-way ANOVA with Dunnett's post-hoc test, *P˂0.05 **P˂0.01 versus WT Control, #P˂0.05 ##P˂0.01 versus WT LPS. n=8 per group. (B) Representative images of microglia labeled with Iba-1 and LC3 in the corpus callosum at 1 month post BCAS. Scale bar, 20 μm. (C) Quantitative analysis was performed as percentage of Iba-1, LC3 double positive cells in Iba-1 positive microglia. Two-way ANOVA with Dunnett's post-hoc test, **P˂0.01 versus WT Sham, ##P˂0.01 versus WT BCAS. n=6 per group. (D) Representative electron microscopy images showing accumulation of autophagosomes in microglia in the corpus callosum at 1 month post BCAS. Arrows indicate classic autophagosomes. Scale bar, 4µm.
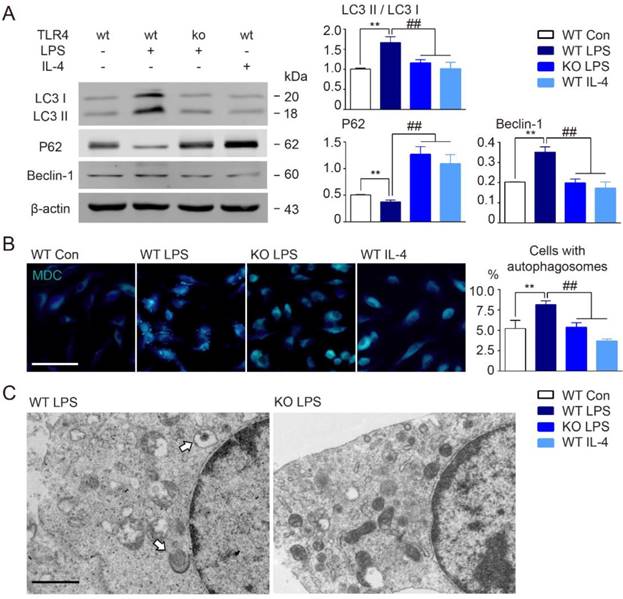
Our data clearly suggested that TLR4 might be involved in microglial autophagy activation following BCAS. To further examine this possibility, we performed Western blot analysis of autophagy related proteins in primary cultured microglia using LPS to activate TLR4. In line with in vivo results, we found that cultured microglia stimulated by LPS mimicked those in our BCAS model, including up-regulation in autophagy signaling elements LC3 II and beclin-1, and decreased p62 (Figure 5A). We also used monodansylcadaverine (MDC) dye, which specifically accumulates in autophagic vacuoles and cells with autophagosomes [42]. Consistent with our other results, LPS stimulation enhanced MDC staining, as well as, the accumulation of autophagosomes as observed by electron microscopy (Figure 5B and 5C). However, TLR4 deletion in microglia suppressed LPS-induced conversion of LC3 I to LC3 II, inhibited beclin-1 up-regulation, prevented decreases to p62 expression, and suppressed autophagosome accumulation (Figure 5A-C). Additionally, considered that LPS is a ligand of TLR4 and has a direct interaction with TLR4, we also chose interferon gamma (IFN-γ) as another stimulator to investigate whether IFN-γ could exhibit similar tendency of autophagy activity in wild-type derived or TLR4-deficient microglia. We found that 100ng/ml IFN-γ could also induce the activation of autophagy in microglia, presented by up-regulation of autophagy signaling elements LC3 II and beclin-1, and down-regulation of p62 expression (Figure S1). Both of pro-inflammatory activators, LPS and IFN-γ, were confirmed to activate the state of microglial inflammation and autophagy. Whereas, TLR4 deficiency in microglia could also suppress IFN-γ-induced conversion of LC3 I to LC3 II, inhibit beclin-1 up-regulation, and prevent decreases to p62 expression (Figure S1). Thus, the data indicated that chronic cerebral hypoperfusion could induce the activation of microglial autophagy, and that this process was dependent on TLR4 signaling.
TLR4 deficiency suppressed microglial autophagy in culture. (A) The expression of autophagy related proteins (LC3, P62 and Beclin-1) in cultured microglia were detected by Western blot. Quantitative analysis was performed. Two-way ANOVA with Dunnett's post-hoc test, **P˂0.01 versus WT Control, ##P˂0.01 versus WT LPS. n=8 per group. (B) Representative images of MDC staining for autophagosomes in cultured microglia. Scale bar, 50 μm. Quantitative analysis of cells with autophagosomes was performed. Two-way ANOVA with Dunnett's post-hoc test, **P˂0.01 versus WT Control, ##P˂0.01 versus WT LPS. n=6 per group. (C) Representative electron microscopy images showing accumulation of autophagosomes in cultured microglia under LPS stimulation. Arrows indicate classic autophagosomes. Scale bar, 1µm.
TLR4-mediated microglia pro-inflammatory polarization via autophagic pathway
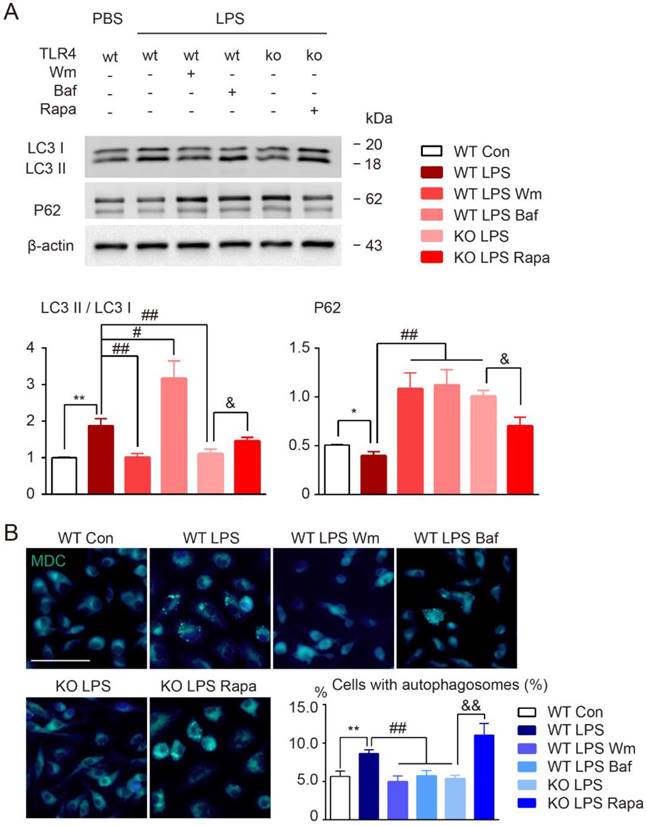
To determine the role of autophagy in modulation of microglia polarization, microglia were first incubated with autophagy inhibitors wortmannin (Wm) or bafilomycin A1 (Baf) during LPS stimulation. Wm inhibits starvation induced autophagy by inhibiting class III PI3K [43] and Baf inhibits maturation of autophagosomes by targeting the H+-ATPase of the vacuolar system [27]. Our results showed that both Wm (100 nM) and Baf (100 nM) were able to block LPS-induced autophagy efficiently as detected by Western blot analysis of autophagy related proteins LC3 and p62, and MDC staining (Figure 6). To address whether the autophagy process was involved in modulation of microglia polarization, we then tested different functional markers of microglia in the presence of autophagy inhibition. We found that LPS induced up-regulation of pro-inflammatory transcripts (CD86, iNOS, TNF-α, IL-β) and proteins (iNOS, CD16) were blocked in response to Wm and Baf (Figure 7). However, the expression levels of anti-inflammatory markers in microglia at transcriptional level (TGF-α, YM, Arg-1, interleukin-10 (IL-10)) and protein level (Arg-1, CD206) were unaltered by the autophagy inhibitors (Figure 7). These results indicated that autophagy was involved in the pro-inflammatory activation of microglia.
Autophagy modulation of cultured microglia under LPS stimulation. (A) The expression of autophagy related proteins (LC3 and P62) in cultured microglia were detected by Western blot. Quantitative analysis was performed. Two-way ANOVA with Dunnett's post-hoc test, *P˂0.05 **P˂0.01 versus WT Control, #P˂0.05 ##P˂0.01 versus WT LPS, &P˂0.05 versus KO LPS. n=8 per group. (B) Representative images showing MDC staining in microglia. Scale bar, 10 µm, 50 µm. Quantitative analysis of cells with autophagosomes were shown in the histogram. Two-way ANOVA with Dunnett's post-hoc test, **P˂0.01 versus WT Control, ##P˂0.01 versus WT LPS, &&P˂0.01 versus KO LPS. n=8 per group.
TLR4 dependent autophagy mediated pro-inflammatory phenotype in cultured microglia. (A) The protein expression of pro-inflammatory markers (iNOS, CD16/32), anti-inflammtory markers (Arg-1, CD206) in primary cultured microglia was detected by Western blot. (B) Quantitative analysis of Western blot was performed. Two-way ANOVA with Dunnett's post-hoc test, **P˂0.01 versus WT Control, #P˂0.05 ##P˂0.01 versus WT LPS, &P˂0.05 &&P˂0.01 versus KO LPS. n=8 per group. (C) The mRNA expression of pro-inflammatory markers (CD86, TNF-α, iNOS and IL-1β) and anti-inflammatory markers (TGF-β, YM, Arg-1 and IL-10) in primary cultured microglia in response to different stimulation and treatment was detected by quantitative real-time PCR. n.s. no significant changes between different groups. Two-way ANOVA with Dunnett's post-hoc test, #P˂0.05 ##P˂0.01 versus WT LPS, &P˂0.05 &&P˂0.01 versus KO LPS, ФP˂0.05 ФФP˂0.01 versus KO IL-4, $P˂0.05 $$P˂0.01 versus KO IL-4 Rapa. n=6 per group.
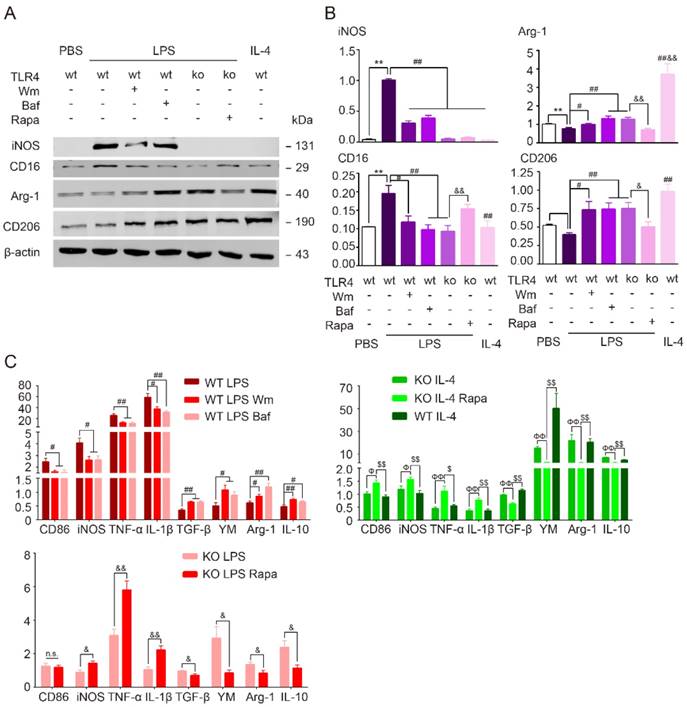
Our results have shown that TLR4 deficiency switched microglia polarization from pro-inflammatory to anti-inflammatory phenotype in the presence of LPS. To examine whether TLR4 modulates microglia polarization via autophagic pathway, we incubated TLR4 knockout microglia with the autophagy agonist rapamycin (Rapa) during LPS or IL-4 stimulation. Deficiency in TLR4 blocked the LPS-induced upregulation of LC3, preserved p62, and inhibited MDC-labeled autophagosome formation (Figure 6). Rapa reversed the suppressive effects of TLR4 deficiency on autophagy activation (Figure 6). Additionally, the TLR4 knockout microglia exposure to both LPS and Rapa, exhibited markedly pro-inflammatory phenotype similar to wild-type cells exposed to just LPS (Figure 7). In line with this, the autophagy agonist Rapa reversed the anti-inflammatory activity of TLR4 knockout microglia exposure to IL-4, shown by the up-regulation of pro-inflammatory profiles and down-regulation of anti-inflammatory expression (Figure 7C). The data indicated that TLR4 was the up-stream of autophagy induced pro-inflammatory activation.
In addition, the widely used autophagy inhibitors, wortmannin and bafilomycin, were also chose to investigate the effects of modulation of autophagy activation on inflammatory polarization and cognitive impairment in BCAS model. We found that after BCAS, administration of autophagy inhibitors, Wm and Baf, resulted in fewer revisiting errors and a greater number of different arm choices in the first eight entries compared to the vehicle group (Figure S2A). No obvious differences in spatial reference memory were detected between the groups (Figure S2A). LFB staining showed that the disruption of WM integrity was reversed after Wm and Baf administration, which was in agreement with the cognitive function improvement (Figure S2B). Meanwhile, prolonged hypoperfusion induced pro-inflammatory microglia (CD16/32 positive) was largely inhibited by autophagy inhibitors and more anti-inflammatory microglia (CD206 positive) were observed (Figure S2C). Together, the data suggested that modifying autophagy in BCAS model might change the inflammatory polarization, decrease demyelination, and alleviate cognitive impairment.
TLR4-dependent autophagy regulated microglial polarization via the STAT1/6 pathway
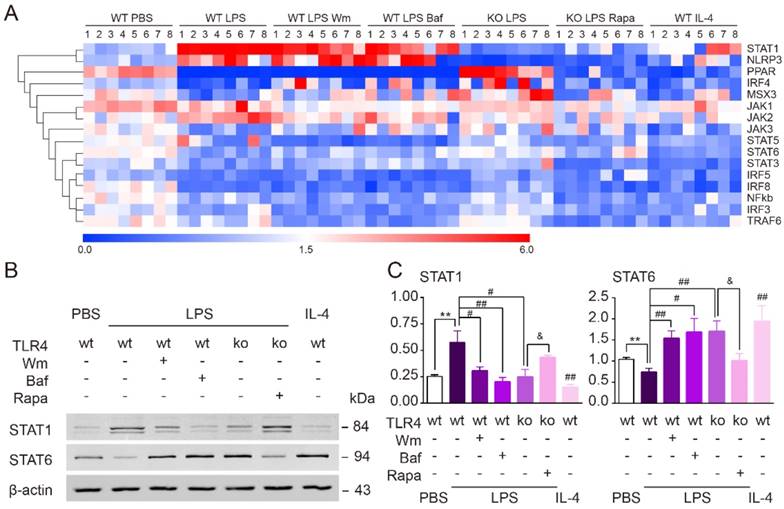
To explore the signaling pathways by which TLR4-dependent autophagy induces microglial polarization towards a pro-inflammatory phenotype, we performed quantitative real-time PCR to screen various pathway targets (IRF3, IRF4, IRF5, IRF8, MSX3, PPAR, STAT1, STAT3, STAT5, STAT6, NF-кB, NLRP3, TRAF6, JAK1, JAK2 and JAK3) that have been previously reported to be involved in autophagy [44-46] (Figure 8A). We found that the up-regulation of STAT1 and down-regulation of STAT6 in cultured microglia exposure to LPS were both reversed by the two autophagy inhibitors, Wm and Baf (Figure 8A). In line with our other results, TLR4 deficiency suppressed the effects of LPS upon these two factors. Administration of the autophagy agonist Rapa, on the other hand, could rescue the effects of LPS within TLR4 deficient microglia (Figure 8A). Western blot further confirmed that the protein expression level of STAT1/6 was changed consistently with PCR result (Figure 8B-C). Therefore, these results indicated that TLR4-dependent autophagy was likely to shift microglia from anti-inflammation to pro-inflammation state via STAT1/6 signaling pathway.
TLR4-dependent autophagy regulated microglial polarization via STAT1/6 pathway. (A) Microglial polarization related gene profiles (IRF3, IRF4, IRF5, IRF8, MSX3, PPAR, STAT1, STAT3, STAT5, STAT6, NF-кb, NLRP3, TRAF6, JAK1, JAK2 and JAK3) were detected by quantitative real-time PCR. (B) The protein expression of STAT1 and STAT6 in primary cultured microglia was detected by Western blot. (C) Quantitative analysis of Western blot was performed. Two-way ANOVA with Dunnett's post-hoc test, **P˂0.01 versus WT Control, #P˂0.05 ##P˂0.01 versus WT LPS, &P˂0.05 versus KO LPS. n=8 per group.
Discussion
The importance of the TLR4 pathway in microglial activation is compellingly illustrated by the frequency of pathway disruption in the pathology of stroke and other CNS disorders [9-14]. However, the underlying basis of TLR4 mediated functional change in microglial phenotype remains unknown. The present study reveals the consequences of TLR4 deficiency in chronic cerebral hypoperfusion-induced WM damage, and identifies unique molecular and cellular outcomes that modulate microglial polarization. Our key findings included: (a) TLR4 deletion facilitated microglial polarization towards an anti-inflammatory profile and exhibited a protective activity against myelin damage and cognitive deficits after WM injury; (b) TLR4 pathway could modulate microglia to pro-inflammation via autophagic pathway under neuroinflammation; (c) TLR4-dependent autophagy may regulate microglial polarization via STAT1/6 pathway. Taken together, our results uncover the TLR4-dependent microglial function in WM ischemic damage induced by chronic cerebral hypoperfusion, and provide the first insight into the putative mechanism of TLR4-induced autophagy of activated microglia in demyelination/remyelination after CNS disorders.
The balance between microglial inflammatory states influences the progression of a multitude of CNS disorders [29, 47, 48]. It has been demonstrated that microglial production of pro-inflammatory cytokines could contribute to ischemic brain damage and neurological dysfunction after acute ischemic stroke [49, 50]. Microglia mediated inflammatory response could also disrupt the integrity of WM following chronic cerebral hypoperfusion [6]. Anti-inflammatory microglia are an essential part of the regenerative response in CNS demyelinating diseases, promoting oligodendrocyte differentiation [47]. In our recent report, we showed that prolonged cerebral hypoperfusion significantly induced microglia activation in preferentially pro-inflammatory polarization state [6]. A switch to anti-inflammatory polarization was stably detected after treatment with an immunomodulator fingolimod[6]. Pathogen recognition receptors, such as TLRs, serve as the sensor of pathogen- or danger-associated molecular patterns (PAMPs or DAMPs) from microbes or damaged/dying cells [51], and play a fundamental role in regulating innate immunity and inflammatory response [9, 10, 52]. TLR4 is mainly expressed in microglia in CNS that is the central part of the inflammatory responses accumulating pathological damage [9, 10]. Our current study demonstrated that TLR4 deficiency could modulate microglial polarization towards anti-inflammatory state, alleviate white matter demyelination, and eventually ameliorate cognitive dysfunction in chronic cerebral hypoperfusion.
Recently, the mechanisms of crosstalk between autophagy and inflammatory signaling cascades have been investigated in several studies, but still remain unclear [21, 23, 53-55]. Generally, autophagy is considered to inhibit inflammasome activation by sequestering damaged mitochondria, which can avoid the release of factors such as reactive oxygen species (ROS) and mitochondrial DNA that activate the NLRP3 inflammasome. Besides, autophagy isolates the NLRP3 binding protein ASC to prevent caspase 1 mediated cleavage of pro-IL-1β and pro-IL-18 into the active forms [21]. What's more, the autophagy machinery engulfs and eradicates pro-IL-1β proteins, representing another level of IL-1β regulation [23]. Though these results demonstrate that autophagy can inhibit inflammation, opposite opinions were also mentioned in other researches [56-59]. Autophagy was found to function as a double-edged sword in diabetic retinopathy [57]. Under mild stress, autophagic activity can lead to cell survival. However, during severe stress, dysregulated autophagy results in massive cell death and may have a role in initiation and exacerbation of diabetic retinopathy [57]. In addition, autophagy suppresses inflammation was also reported in airway disease [58]. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyper-production in airway epithelium. And genetic blockage of autophagy markedly reduced particle material-induced expression of inflammatory cytokines, e.g. IL-8 and IL-6, and MUC5AC in HBE cells [58]. In another study, phagocytic uptake of cells dying through autophagy by macrophages leads to a pro-inflammatory response characterized by the induction and secretion of IL-6, TNF-α, IL-8 and IL-10 [59]. Besides, it is reported that basal autophagy inhibits IL-1β secretion; however, enhancement of autophagy augments IL-1β secretion. These results demonstrated that inflammasome and the autophagy apparatus synergize during IL-1β secretion in cells stimulated to undergo autophagy [56]. Autophagy induction could also cooperate with GRASP and Rab8a (a GTPase controlling post-Golgi polarized sorting and exocytosis) in driving IL-1β secretion, which is a type of unconventional way [56]. Overall, the interactions between autophagy and inflammation are much more complex than we thought. In fact, autophagy has been linked to macrophages polarization and balances the beneficial and detrimental effects of immunity and inflammation. In the present study, we also investigated the relation between autophagy and regulation of microglial inflammatory phenotype, which is known to contribute to ischemic white matter damage. Interestingly, we revealed that enhancement of autophagy induces microglial pro-inflammatory activation, whereas decreased autophagy promote microglia toward anti-inflammatory state. Our results were consistent with several other studies [60, 61]. It has been reported that autophagy induced by AGEs or rapamycin stimulated macrophage polarization to M1 with gene expressions of IL-1β, iNOS and TNF-α; in contrast, autophagy inhibitor reduced M1 polarized macrophages [60]. Autophagic agonist rapamycin could also stimulate human derived monocytes to differentiate into M1 state and release more IL-12 and less IL-10, whereas activated mTOR by TSC2 knockdown caused M2 polarization [61].
Autophagy and inflammatory signaling pathways balance host defense and homeostasis [21]. Autophagy is an important component of the innate immune response, and TLR4 serves as a previously unrecognized environmental sensor for autophagy [18-20, 62]. However, whether LPS stimulation results in consistent activation of TLR4-mediated autophagy remains controversial. For example, it was reported that TLR4 stimulated by LPS could activate the VPS34-dependent formation of cytoplasmic LC3 aggregates, possibly indicative of autophagosomes, and enhance the autophagic elimination of phagocytosed mycobacteria in macrophages [18]. Some studies supported the concept as LPS stimulation could consistently activate autophagy in primary macrophages culture [63]. However, other reports found high concentrations of LPS had a differential LC3 response in microglia [64], which was inconsistent with TLR4's role in autophagy. Regardless, our data support the idea that microglial autophagy appears to be consistently activated via TLR4 signaling pathway under neuroinflammation after chronic cerebral hypoperfusion.
Information on precise signaling mechanisms of TLR4-mediated autophagy remains unclear. Although TLR-activated autophagy is regulated by the interaction of a Toll-interleukin-1 receptor domain-containing adaptor-inducing interferon-b (TRIF) or myeloid differentiation factor 88 (MyD88) with beclin-1 [52], previous studies demonstrated that TRIF, but not MyD88, is required for the formation of LC3 aggregates. The interaction of TLR adaptor proteins with beclin-1 seems to be mediated by TNF receptor associated factor 6 (TRAF6) [65]. Upon LPS exposure, TLR4 promotes the assembly of the phosphoinositide-3-kinase class 3 (PI3KC3) complex through TRAF6-mediated K63 ubiquitination of beclin-1 to free it from an inhibitory interaction with B-cell lymphoma-2 (BCL-2) [65]. Here we showed that TLR4 activation was a necessary step in recruiting autophagy proteins, and TLR4 deficiency could efficiently suppress autophagic related process both in the primary microglia and BCAS model in vivo. These results indicated that trigger of microglial autophagy machinery was dependent on TLR4 signaling pathway after ischemic white matter damage.
Autophagy and related processes regulate intracellular trafficking of pathogens, production of inflammatory mediators, and viability of cells that coordinate immunity[21]. Through these functions, the autophagy machinery controls complex multicellular immune inflammation responses at the whole organism level. The distinct phenotypic features of pro-inflammatory and anti-inflammatory microglia are controlled by a network of molecular pathways that relay environmental signals via signaling cascades to impact gene expression and cellular metabolism. However, whether and how TLR4 dependent autophagy regulates microglial polarization remain to be elucidated. In the present study, we screened various related gene profiles involved in the transition of microglial polarization. The results indicated that TLR4-dependent autophagy was likely to modulate inflammatory state of microglia via STAT1/6 signaling pathway.
Signal transducers and activators of transcription (STATs) are latent cytoplasmic transcription factors that are phosphorylated at a single tyrosine residue via members of the Jak kinase family following ligand-receptor interaction, assemble in dimeric form, translocate to the nucleus, and bind to specific DNA sequence motifs [66, 67]. STAT1 and STAT6 are reported to be associated with pro-inflammatory and anti-inflammatory microglial phenotype, respectively [68]. Although STAT1 and STAT6 were reported to regulate reciprocally in T cells [69], STAT6 activation was strictly IL-4-dependent and could regulate STAT1 activation in macrophages [70]. Therefore, it is plausible that the up-regulation of STAT1 and down-regulation of STAT6 are coordinated to modulate microglia polarization and LPS neuroinflammation and possible under chronic cerebral hypoperfusion.
Conclusion
Our results demonstrate that up-regulation of TLR4 induced by chronic cerebral hypoperfusion triggers microglial autophagy machinery events, subsequently results in microglia polarization towards pro-inflammatory phenotype via STAT1/6 pathway, and consequently contributes to WM ischemic damage and cognitive deficit.
Abbreviations
Lipopolysaccharide (LPS), white matter (WM), Toll-like receptors (TLRs), lipopolysaccharide (LPS), bilateral carotid artery stenosis (BCAS), the medial part of corpus callosum (CCm), fiber bundles of the caudoputamen (CPu), fimbria of hippocampus (F), anterior commissure (AC), internal capsule (IC), and optic tract (OT), Monodansylcadaverine (MDC), myelin associated glycoprotein (MAG), myelin basic protein (MBP), neurofilament (NF) , voltage-gated sodium channels (Nav1.6), Ionized calcium binding adapter molecule 1 (Iba1), glial fibrillary acidic protein (GFAP), and adenomatous polyposis coli (APC), cluster of differentiation 16 (CD16), inducible nitric oxide synthase (iNOS), arginase (Arg)-1, mannose receptor (CD206), wortmannin (Wm), bafilomycin A1 (Baf), rapamycin (Rapa), Signal transducers and activators of transcription (STATs).
Acknowledgements
This work was supported by National Natural Science Foundation of China (61327902-6, 91332108 to W. Wang, 81571132, 81873743 to D.S. Tian, 81801223 to C. Qin), Fundamental Research Funds for the Central Universities (2017KFYXJJ107 to D.S. Tian, 2017KFYXJJ124 to C. Qin), and National Institutes of Health (R01NS088627 to L.J.Wu).
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Johnston SC, O'Meara ES, Manolio TA, Lefkowitz D, O'Leary DH, Goldstein S. et al. Cognitive impairment and decline are associated with carotid artery disease in patients without clinically evident cerebrovascular disease. Ann Intern Med. 2004;140:237-47
2. Buratti L, Balucani C, Viticchi G, Falsetti L, Altamura C, Avitabile E. et al. Cognitive deterioration in bilateral asymptomatic severe carotid stenosis. Stroke. 2014;45:2072-7
3. Verden D, Macklin WB. Neuroprotection by central nervous system remyelination: Molecular, cellular, and functional considerations. J Neurosci Res. 2016;94:1411-20
4. Miyamoto N, Maki T, Pham LD, Hayakawa K, Seo JH, Mandeville ET. et al. Oxidative stress interferes with white matter renewal after prolonged cerebral hypoperfusion in mice. Stroke. 2013;44:3516-21
5. Choi BR, Kim DH, Back DB, Kang CH, Moon WJ, Han JS. et al. Characterization of White Matter Injury in a Rat Model of Chronic Cerebral Hypoperfusion. Stroke. 2016;47:542-7
6. Qin C, Fan W-H, Liu Q, Shang K, Murugan M, Wu L-J. et al. Fingolimod Protects Against Ischemic White Matter Damage by Modulating Microglia Toward M2 Polarization via STAT3 Pathway. Stroke. 2017;48:3336-46
7. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387-94
8. Biber K, Owens T, Boddeke E. What is microglia neurotoxicity (Not)? Glia. 2014;62:841-54
9. Papageorgiou IE, Lewen A, Galow LV, Cesetti T, Scheffel J, Regen T. et al. TLR4-activated microglia require IFN-gamma to induce severe neuronal dysfunction and death in situ. Proc Natl Acad Sci U S A. 2016;113:212-7
10. Hakimizadeh E, Kazemi Arababadi M, Shamsizadeh A, Roohbakhsh A, Allahtavakoli M. The Possible Role of Toll-Like Receptor 4 in the Pathology of Stroke. Neuroimmunomodulation. 2016;23:131-6
11. Regen T, van Rossum D, Scheffel J, Kastriti ME, Revelo NH, Prinz M. et al. CD14 and TRIF govern distinct responsiveness and responses in mouse microglial TLR4 challenges by structural variants of LPS. Brain Behav Immun. 2011;25:957-70
12. Burguillos MA, Svensson M, Schulte T, Boza-Serrano A, Garcia-Quintanilla A, Kavanagh E. et al. Microglia-Secreted Galectin-3 Acts as a Toll-like Receptor 4 Ligand and Contributes to Microglial Activation. Cell Rep. 2015;10:1626-38
13. Okada Y, Ochi H, Fujii C, Hashi Y, Hamatani M, Ashida S. et al. Signaling via toll-like receptor 4 and CD40 in B cells plays a regulatory role in the pathogenesis of multiple sclerosis through interleukin-10 production. J Autoimmun. 2017;88:103-13
14. Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599-608
15. Hyakkoku K, Hamanaka J, Tsuruma K, Shimazawa M, Tanaka H, Uematsu S. et al. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience. 2010;171:258-67
16. Lobo-Silva D, Carriche GM, Castro AG, Roque S, Saraiva M. Interferon-beta regulates the production of IL-10 by toll-like receptor-activated microglia. Glia. 2017;65:1439-51
17. Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, Vandenabeele P. Autophagy: for better or for worse. Cell Res. 2012;22:43-61
18. Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135-44
19. Zhan Z, Xie X, Cao H, Zhou X, Zhang XD, Fan H. et al. Autophagy facilitates TLR4- and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy. 2014;10:257-68
20. Chen S, Yuan J, Yao S, Jin Y, Chen G, Tian W. et al. Lipopolysaccharides may aggravate apoptosis through accumulation of autophagosomes in alveolar macrophages of human silicosis. Autophagy. 2015;11:2346-57
21. Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol. 2016;16:661-75
22. Dello Russo C, Lisi L, Feinstein DL, Navarra P. mTOR kinase, a key player in the regulation of glial functions: relevance for the therapy of multiple sclerosis. Glia. 2013;61:301-11
23. Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy. 2016;12:245-60
24. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X. et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085-8
25. Shibata M, Ohtani R, Ihara M, Tomimoto H. White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke. 2004;35:2598-603
26. Serdar M, Herz J, Kempe K, Lumpe K, Reinboth BS, Sizonenko SV. et al. Fingolimod protects against neonatal white matter damage and long-term cognitive deficits caused by hyperoxia. Brain Behav Immun. 2016;52:106-19
27. Qin AP, Liu CF, Qin YY, Hong LZ, Xu M, Yang L. et al. Autophagy was activated in injured astrocytes and mildly decreased cell survival following glucose and oxygen deprivation and focal cerebral ischemia. Autophagy. 2010;6:738-53
28. Yang Z, Zhao TZ, Zou YJ, Zhang JH, Feng H. Hypoxia Induces autophagic cell death through hypoxia-inducible factor 1alpha in microglia. PloS one. 2014;9:e96509
29. Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S. et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063-70
30. Reimer MM, McQueen J, Searcy L, Scullion G, Zonta B, Desmazieres A. et al. Rapid disruption of axon-glial integrity in response to mild cerebral hypoperfusion. J Neurosci. 2011;31:18185-94
31. Munafo DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114:3619-29
32. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 2001;25:402-8
33. Qu WS, Tian DS, Guo ZB, Fang J, Zhang Q, Yu ZY. et al. Inhibition of EGFR/MAPK signaling reduces microglial inflammatory response and the associated secondary damage in rats after spinal cord injury. J Neuroinflamm. 2012:9
34. Shibata M, Yamasaki N, Miyakawa T, Kalaria RN, Fujita Y, Ohtani R. et al. Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke. 2007;38:2826-32
35. McDonough A, Lee RV, Noor S, Lee C, Le T, Iorga M. et al. Ischemia/Reperfusion Induces Interferon-Stimulated Gene Expression in Microglia. J Neurosci. 2017;37:8292-308
36. García-Culebras A, Palma-Tortosa S, Moraga A, García-Yébenes I, Durán-Laforet V, Cuartero MI. et al. Toll-Like Receptor 4 Mediates Hemorrhagic Transformation After Delayed Tissue Plasminogen Activator Administration in In Situ Thromboembolic Stroke. Stroke. 2017;48:1695-9
37. Döring C, Regen T, Gertig U, van Rossum D, Winkler A, Saiepour N. et al. A presumed antagonistic LPS identifies distinct functional organization of TLR4 in mouse microglia. Glia. 2017;65:1176-85
38. Woodward NC, Levine MC, Haghani A, Shirmohammadi F, Saffari A, Sioutas C. et al. Toll-like receptor 4 in glial inflammatory responses to air pollution in vitro and in vivo. J Neuroinflamm. 2017:14
39. Kimura T, Nada S, Takegahara N, Okuno T, Nojima S, Kang S. et al. Polarization of M2 macrophages requires Lamtor1 that integrates cytokine and amino-acid signals. Nat Commun. 2016;7:13130
40. Okun E, Griffioen KJ, Mattson MP. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011;34:269-81
41. Yu T, Guo F, Yu Y, Sun T, Ma D, Han J. et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell. 2017;170:548-63.e16
42. Song W, Ma Z, Zhang Y, Yang C. Autophagy plays a dual role during intracellular siRNA delivery by lipoplex and polyplex nanoparticles. Acta Biomater. 2017;58:196-204
43. Xu Y, Jagannath C, Liu X-D, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like Receptor 4 Is a Sensor for Autophagy Associated with Innate Immunity. Immunity. 2007;27:135-44
44. Cao L, He C. Polarization of macrophages and microglia in inflammatory demyelination. Neurosci Bull. 2013;29:189-98
45. Holtman IR, Skola D, Glass CK. Transcriptional control of microglia phenotypes in health and disease. J Clin Invest. 2017;127:3220-9
46. Yu Z, Sun D, Feng J, Tan W, Fang X, Zhao M. et al. MSX3 Switches Microglia Polarization and Protects from Inflammation-Induced Demyelination. J Neurosci. 2015;35:6350-65
47. Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL. et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211-8
48. Tian DS, Li CY, Qin C, Murugan M, Wu LJ, Liu JL. Deficiency in the voltage-gated proton channel Hv1 increases M2 polarization of microglia and attenuates brain damage from photothrombotic ischemic stroke. J Neurochem. 2016;139:96-105
49. Wu LJ, Wu G, Akhavan Sharif MR, Baker A, Jia Y, Fahey FH. et al. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat Neurosci. 2012;15:565-73
50. Fu Y, Liu Q, Anrather J, Shi FD. Immune interventions in stroke. Nat Rev Neurol. 2015;11:524-35
51. Amici SA, Dong J, Guerau-de-Arellano M. Molecular Mechanisms Modulating the Phenotype of Macrophages and Microglia. Front Immunol. 2017;8:1520
52. Shi CS, Kehrl JH. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem. 2008;283:33175-82
53. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722-37
54. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323-35
55. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell. 2016;166:288-98
56. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011;30:4701-11
57. Dehdashtian E, Mehrzadi S, Yousefi B, Hosseinzadeh A, Reiter RJ, Safa M. et al. Diabetic retinopathy pathogenesis and the ameliorating effects of melatonin; involvement of autophagy, inflammation and oxidative stress. Life Sci. 2018;193:20-33
58. Chen ZH, Wu YF, Wang PL, Wu YP, Li ZY, Zhao Y. et al. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyperproduction in airway epithelium. Autophagy. 2016;12:297-311
59. Petrovski G, Zahuczky G, Majai G, Fesus L. Phagocytosis of cells dying through autophagy evokes a pro-inflammatory response in macrophages. Autophagy. 2007;3:509-11
60. Guo Y, Lin C, Xu P, Wu S, Fu X, Xia W. et al. AGEs Induced Autophagy Impairs Cutaneous Wound Healing via Stimulating Macrophage Polarization to M1 in Diabetes. Sci Rep. 2016;6:36416
61. Chen W, Ma T, Shen XN, Xia XF, Xu GD, Bai XL. et al. Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res. 2012;72:1363-72
62. Xu Y, Liu XD, Gong X, Eissa NT. Signaling pathway of autophagy associated with innate immunity. Autophagy. 2008;4:110-2
63. Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27:1110-21
64. He Y, She H, Zhang T, Xu H, Cheng L, Yepes M. et al. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J Cell Biol. 2018;217:315-28
65. Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3:ra42
66. Ihle JN, Thierfelder W, Teglund S, Stravopodis D, Wang D, Feng J. et al. Signaling by the cytokine receptor superfamily. Ann N Y Acad Sci. 1998;865:1-9
67. Ohmori Y, Hamilton TA. Interleukin-4/STAT6 represses STAT1 and NF-kappa B-dependent transcription through distinct mechanisms. J Biol Chem. 2000;275:38095-103
68. Jha MK, Lee WH, Suk K. Functional polarization of neuroglia: Implications in neuroinflammation and neurological disorders. Biochem Pharmacol. 2016;103:1-16
69. Yu CR, Mahdi RM, Ebong S, Vistica BP, Chen J, Guo Y. et al. Cell proliferation and STAT6 pathways are negatively regulated in T cells by STAT1 and suppressors of cytokine signaling. J Immunol. 2004;173:737-46
70. Miyamoto H, Katsuyama E, Miyauchi Y, Hoshi H, Miyamoto K, Sato Y. et al. An essential role for STAT6-STAT1 protein signaling in promoting macrophage cell-cell fusion. J Biol Chem. 2012;287:32479-84
Author contact
![]() Corresponding authors: Dai-Shi Tian MD, PhD E-mail: tiandstjmu.edu.cn Or Wei Wang MD, PhD E-mail: wwangtjmu.edu.cn Department of Neurology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, P.R. China. Phone: +86-27-83663337
Corresponding authors: Dai-Shi Tian MD, PhD E-mail: tiandstjmu.edu.cn Or Wei Wang MD, PhD E-mail: wwangtjmu.edu.cn Department of Neurology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, P.R. China. Phone: +86-27-83663337