Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results and Discussion
Methods
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(19):5469-5481. doi:10.7150/thno.28295 This issue Cite
Research Paper
“Cell-addictive” dual-target traceable nanodrug for Parkinson's disease treatment via flotillins pathway
YanHui Li1,2, ZiXuan Chen1, ZhiGuo Lu2, QingHu Yang1, LinYing Liu2, ZhaoTan Jiang1, LiQun Zhang3, Xin Zhang2 ![]() , Hong Qing1
, Hong Qing1 ![]()
1. School of Life Science, Beijing Institute of Technology, Beijing 100081, PR China
2. State Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, PR China
3. Beijing Chest Hospital, Capital Medical University, Beijing 101149, PR China
Received 2018-7-4; Accepted 2018-9-30; Published 2018-10-29
Abstract

α-synclein (αS) aggregation is a representative molecular feature of the pathogenesis of Parkinson's disease (PD). Epigallocatechin gallate (EGCG) can prevent αS aggregation in vitro. However, the in vivo effects of PD treatment are poor due to the obstacles of EGCG accumulation in dopaminergic neurons, such as the blood brain barrier and high binding affinities between EGCG and membrane proteins. Therefore, the key to PD treatment lies in visual examination of EGCG accumulation in dopaminergic neurons.
Methods: DSPE-PEG-B6, DSPE-PEG-MA, DSPE-PEG-phenylboronic acid, and superparamagnetic iron oxide nanocubes were self-assembled into tracing nanoparticles (NPs). EGCG was then conjugated on the surface of the NPs through the formation of boronate ester bonds to form a “cell-addictive” dual-target traceable nanodrug (B6ME-NPs). B6ME-NPs were then used for PD treatment via intravenous injection.
Results: After treatment with B6ME-NPs, the PD-like characteristics was alleviated significantly. First, the amount of EGCG accumulation in PD lesions was markedly enhanced and traced via magnetic resonance imaging. Further, αS aggregation was greatly inhibited. Finally, the dopaminergic neurons were considerably increased.
Conclusion: Due to their low price, simple preparation, safety, and excellent therapeutic effect on PD, B6ME-NPs are expected to have potential application in PD treatment.
Keywords: Parkinson's disease, dopaminergic neurons, EGCG, nanoparticles, α-synclein aggregation
Introduction
Parkinson's disease (PD) is arguably one of the most recognizable brain disorders and is a growing concern in the modern world [1,2]. The majority of the currently available treatments for PD are dopamine replacement therapies that are not suitable for long-term use [3]. Cytoplasmic α-synclein (αS) aggregation in dopaminergic neurons is one of the molecular mechanisms of the pathogenesis of PD. The toxicity due to protein aggregation leads to the loss of dopaminergic neurons in the substantia nigra (SN), ultimately resulting in early PD symptoms. Fortunately, research on this mechanism has provided new insights into PD therapeutic strategies. Accordingly, preventing αS aggregation has become an attractive, alternative method for PD therapy [4].
Epigallocatechin gallate (EGCG), a safe and cheap compound, can prevent αS aggregation in vitro [5-7]. Additionally, EGCG reduces the loss of dopaminergic neurons in the SN and prevents tyrosine hydroxylase (TH) protein depletion in vivo [8]. Regrettably, there are many challenges in the targeting of EGCG to its acting site. It is difficult to accumulate EGCG in the brain due to the blood-brain barrier (BBB). Importantly, EGCG cannot be internalized into dopaminergic neurons as a result of its high binding affinities with some membrane proteins [9-11]. These high binding affinities can hijack EGCG and hinder endocytosis. Moreover, the accumulation of EGCG cannot be evaluated during treatments. Therefore, the key to successful PD treatment lies in visualizing EGCG accumulation in dopaminergic neurons.
Nanoparticles (NPs) have the potential to overcome these shortcomings. One target molecule B6, a representative peptide with high affinity for transferrin receptor (TfR) on the BBB, can be conjugated to the surface of NPs to facilitate their entry into the brain [12,13]. Dopamine transporter (DAT), expressed on dopaminergic neurons specifically, can be chosen as the NPs' target. Mazindol (MA) is a potent inhibitor of DA uptake, as it promotes DAT internalization. It has the same binding site as cocaine, but the binding affinity of MA to DAT is 11-fold higher than that of cocaine. It is considered an attractive molecular target for the development of novel drug abuse treatments [14-16]. Hence, the “MA-DAT” model can be used for intracellular delivery, similar to the “cocaine-DAT” mechanism of addiction. Therefore, MA was conjugated on the surface of the NPs and innovative cell-addictive NPs were developed. The reactive oxygen species (ROS)-labile boronate ester was used as a linker between DSPE-PEG2000 and EGCG, because the dopaminergic neurons maintain high ROS status in PD patients. Moreover, superparamagnetic iron oxide nanocubes (SPIONs) with high r2 relativity were loaded in the NPs for magnetic resonance imaging (MRI), to trace the accumulation of NPs in the brain. This kind of NP is different from conventional NPs. Conventional NPs undergo passive uptake by cells; they are spontaneously entrapped by cells [17]. However, the innovative cell-addictive NPs increase the affinity of the cells to NPs, enabling easier uptake by cells and facilitating the accumulation of EGCG in dopaminergic neurons. The DAT-induced internalization of nanoparticle will be promoted by the surface-modification of MA on nanoparticles. This process will re-accelerate the internalization of EGCG, which would be hindered by the high-affinity of EGFR occurred on the surface of cells. The two different processed mentioned above would differ the retention time of nanoparticles on cell surface, and further the signal pathway through cell membrane.
Herein, “cell-addictive”, traceable, ROS-responsive NPs with dual targets were prepared to enhance the accumulation of EGCG in dopaminergic neurons for the treatment of PD. As shown in Scheme 1A, 1,2-dioleoylsn-glycero -3-phosphoethanolamine-n-[poly(ethylene glycol)]2000-B6 (DSPE-PEG2000-B6), DSPE-PEG2000-mazindol (DSPE-PEG2000-MA), DSPE-PEG2000-phenylboronic acid, and SPIONs were self-assembled into tracing NPs. The hydrophilic drug EGCG was then conjugated on the surface of the NPs through the formation of boronate ester bonds and these EGCG-loaded NPs were named B6ME-NPs.
(A) The structural composition and preparation of B6ME-NPs. DSPE-PEG-B6, DSPE-PEG-MA and DSPE-PEG-phenylboronic acid were used to improve the biocompatibility of magnetic nanoparticles through a micelle formation procedure. EGCG was then grafted onto the surface of the nanoparticles through the formation of a boronate ester bond. (B) The schematic diagram of B6ME-NPs in vivo: (1) B6ME-NPs target TfR on BBB and cross the BBB. (2) The NPs can target DAT and accumulate in the lesions area. (3) The NPs are internalized by dopaminergic neurons via DAT. (4) EGCG is released from the NPs due to high ROS response. (5) Free EGCG inhibits αS aggregation. (6) The high concentration of EGCG induces flotillins, the formation of membrane curvature and vesicle budding.
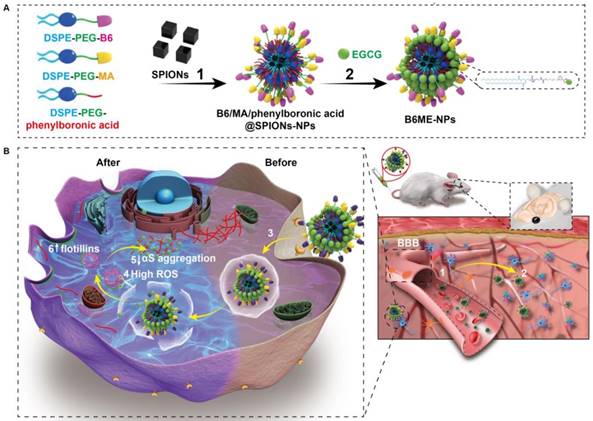
The treatment mechanisms of B6ME-NPs are shown in Scheme 1B. (1) B6ME-NPs could cross the BBB by receptor-mediated endocytosis via B6- and TfR-mediated endocytosis [12,18]. (2) B6ME-NPs are targeted to dopaminergic neurons via the interaction between DAT and MA. (3) The B6ME-NPs are internalized into dopaminergic neurons via MA and DAT receptor-mediated endocytosis. (4) EGCG is released from B6ME-NPs, which is induced by the ROS condition in dopaminergic neurons. (5) The released EGCG inhibits αS aggregation and reduces the toxicity of dopaminergic neurons. (6) Free EGCG also induces flotillins and leads to membrane curvature and vesicle budding. This could contribute to synaptic-vesicle recycling and promote neurotransmitter circulation. Further, the NPs accumulation can be traced via SPIONs monitoring. In addition, B6ME-NPs showed obvious advantages and improved therapeutic efficacy for PD compared to free EGCG.
Results and Discussion
Synthesis and characterization of B6ME-NPs
To prepare B6ME-NPs, DSPE-PEG2000-B6, DSPE-PEG2000-MA, and DSPE-PEG2000-phenylboronic acid were synthesized. DSPE-PEG2000-Mal was chemically modified by B6 via an addition reaction (Figure S1). The obtained lipid was then assessed by matrix-assisted laser desorption ionization time of flight mass spectrometry. Successful synthesis of DSPE-PEG2000-B6 was confirmed through the peak of the DSPE-PEG2000, which shifted to around 3970.4097 Da (Figure S2). DSPE-PEG2000-NHS was chemically modified with MA via an esterification reaction (Figure S3). The 1H nuclear magnetic resonance (NMR) spectra in Figure S4 confirmed the successful synthesis of DSPE-PEG2000-MA. DSPE-PEG2000-Mal was chemically modified with 4-mercaptophenylboronic acid via an addition reaction (Figure S5). The 1H NMR spectra in Figure S3 confirmed that the peroxide responsive linker was successfully linked to DSPE-PEG-Mal. The hydrophobic SPIONs were synthesized via high temperature thermal decomposition. The morphology and size of SPIONs were measure by transmission electron microscopy (TEM). As shown in Figure S6, the SPIONs were cuboid with diameters of approximately 20 nm.
After synthesis of the abovementioned ingredients, B6ME-NPs were constructed via hydrophilic and hydrophobic interactions. As displayed in Table S1, NPs without B6(MA+EGCG+SPIONs) NPs (ME-NPs), non-MA(B6+EGCG+SPIONs) NPs (B6E-NPs), NPs without EGCG(B6+MA+SPIONs) (B6M-NPs) and whole traceable NPs(B6+MA+EGCG+SPIONs) (B6ME-NPs) were prepared for comprehensive study. As shown in Figure 1A-B, the average diameters of all samples were approximately 80 nm and the zeta potential ranged from -0.20 to -0.70 mV. B6ME-NPs presented a mono-disperse structure according to the TEM image (Figure 1C). Additionally, the morphology of the SPIONs was unchanged after encapsulation.
Synthesis and characterization of B6ME-NPs. (A) Average sizes of different nanoparticles. The polydispersity indices of B6ME-NPs, B6ME-NPs, B6ME-NPs, and B6ME-NPs were 0.195, 0.224, 0.106, and 0.249, respectively. (B) Zeta potentials of different nanoparticle dispersions in 0.01 × 10-3 M PBS at 25 ºC. (C) TEM image of B6ME-NPs. Scale bar: 50 nm. Inset: TEM image with negative staining. Scale bar: 50 nm. (D) r2 as a function of Fe concentration from B6ME-NPs. The slope of Fe concentration-R2 regression curve is r2 relaxivity. (E) Serum stability of B6ME-NPs after incubation in culture medium supplemented with 10% FBS. (F) EGCG released from B6ME-NPs triggered by different concentrations of H2O2. Error bars indicate mean ± SD.
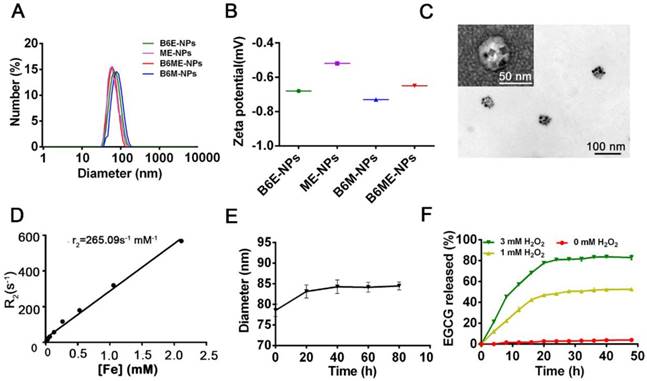
The SPIONs with hydrophobic ligand presented a low MR relaxivity due to the limited access of water protons. It was observed that B6ME-NPs' dispersity in water was significantly enhanced by the surface hydrophilic modifications (Figure S7). To assess the magnetic properties of the traceable NPs for MRI applications, relaxation rate (r2) values were measured and calculated by measuring the change in the spin-spin r2 per unit iron concentration. As shown in Figure 1D, the r2 value of the B6ME-NPs was up to 265.09 mM-1s-1, which was high enough for in vivo MRI application in brain imaging. As displayed in Figure 1E, due to the PEG modification, B6ME-NPs exhibited good serum stability in 10% fetal bovine serum, which further indicated the feasibility of intravenous injection of B6ME-NPs (Figure S8). Release experiments were performed in PBS (0.01 M, pH 7.4) with different concentrations of H2O2. The release of EGCG depended on the degradation of boronate ester bonds, which was induced by H2O2. Figure 1F shows that the amount of released EGCG reached about 85% and 43% after 24 h of incubation in 3 and 1 mM H2O2 solutions, but only 2% of the EGCG was released without H2O2. These results indicated that the ROS could induce the release of EGCG.
Transportation of NPs across an in vitro BBB model via B6 modification
As shown in Figure S9, we conducted subsequent experiments after determining that the nanoparticles were non-toxic. Transport of NPs across the BBB is restricted because of microvessel endothelial cells, which are linked together by tight junctions. TfR is considered a highly specific target for mediating NPs delivery because of its high expression in the cerebral microvascular endothelial cells [19,20]. Therefore, B6 was modified to targeted TfR. To measure the permeability of B6, a cell-based in vitro BBB model was constructed [19,21] (Figure S10). This kind of in vitro model is commonly used to assess the mechanisms of cellular uptake, the internalization profiles of nanocarriers, and their permeability across cell monolayers. In our study, a two-chamber trans-well system was prepared. In vitro permeability assays were carried out in 24-well plates, where the two compartments simulated the blood (apical) and brain (basolateral) sides of the BBB. These two sides are separated by a microporous filter. bEND.3 cells, a type of cerebral microvascular endothelial cells, were seeded on this filter and cultured to form a monolayer (Figure 2A) and trans-endothelial electrical resistance was used to evaluate the integrity of bEND.3 monolayer [19-21]. Subsequently, B6ME-NPs and ME-NPs labeled with Cy5 were added in the upper chamber when the resistance value reached 200 Ω cm2. Flow cytometry was used to measure cellular uptake of the NPs.
Transport of EGCG-loaded NPs across an in vitro BBB model. (A) The structure of the trans-well chambers. SH-SY5Y and bEND.3 cells were co-cultured in trans-well chambers to imitate the BBB. Cellular uptake of different NPs in (B) bEND.3 and (C) SH-SY5Y cells was observed by flow cytometry. (D) B6ME-NPs and ME-NPs in SH-SY5Y cells was detected via CLSM. Nuclei: DAPI, blue; NPs: Cy5, red; lysosome: LysoTracker, green. Scale bar: 100 μm. (E) Quantitation of (D). Error bars indicate mean ± SD (n= 3). *P < 0.05, **P < 0.01, ***P < 0.001.
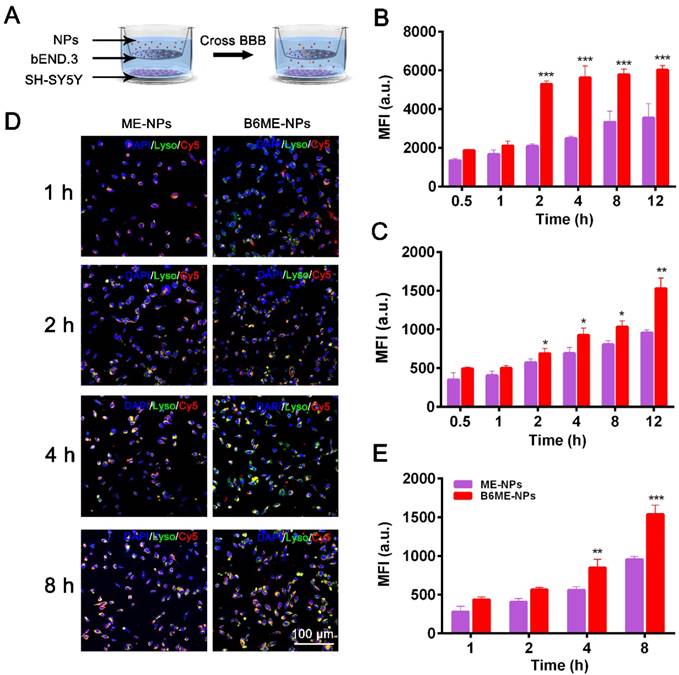
As shown in Figure 2B-C, the mean fluorescence intensities of B6ME-NPs-treated bEND.3 and SH-SY5Y cells were much higher than that of ME-NPs-treated cells at all times. This result indicates that modification of the NPs with B6 significantly enhanced the permeability of the BBB model to the NPs. In addition, to visually evaluate the permeability of the BBB model to the NPs, confocal laser scanning microscopy (CLSM) was performed to detect uptake of the NPs by SH-SY5Y cells. As shown in Figure 2D, red fluorescence was observed in B6ME-NPs-treated cells after 1 h, while fluorescence was observed in the ME-NPs-treated cells after 2 h. This result indicates that modification with B6 accelerated permeation of the NPs across the BBB model. Furthermore, after 8 h, the red fluorescence intensity in the B6ME-NPs-treated cells was significantly higher than that in the ME-NPs-treated cells (Figure 2E). These results indicate that modification of the NPs with B6 could enhance their permeation across the BBB.
Effect of B6ME-NPs on dopaminergic neurons and comparison with B6E-NPs
DAT is a specific transport protein on the membrane of dopaminergic neurons. The physiological role of DAT is the reuptake of released dopamine (DA) into presynaptic DA-terminals [16]. Dopaminergic neurons are important PD therapeutic targets. MA has been explored as a possible agent in cocaine addiction pharmacotherapy, as it has the same binding site as cocaine and a stronger binding ability to DAT [14,15,22]. Thus, NPs were modified with MA to target dopaminergic neurons. SH-SY5Y cells were used to check the targeting ability of MA-modified NPs. Before treatment, the NPs were labeled with Cy5. As shown in Figure 3A-B, after 2 h and 8 h, the fluorescence intensity of Cy5 in B6ME-NPs samples was much higher than that in B6E-NPs (NPs without MA) samples, which indicates that MA could facilitate endocytosis of NPs into dopaminergic neurons. The intracellular concentration of EGCG in SH-SY5Y cells after treatment with different samples (bulk EGCG, B6E-NPs, or B6ME-NPs) was evaluated via LC-MS. These results also verify the abovementioned conclusions (Figure S11).
Dopaminergic neurons-specific drug delivery analysis. (A) Cell uptake of B6ME-NPs and B6E-NPs in SH-SY5Y cells observed by CLSM. Nuclei: DAPI, blue; NPs: Cy5, red; lysosome: LysoTracker, green. Scale bar: 50 μm. (B) Quantitation of (A). (C) Cellular uptake of B6ME-NPs, B6E-NPs and MA in SH-SY5Y cells (2 h incubation) observed via flow cytometry. (D) Ultrathin sections of SH-SY5Y cells incubated with B6ME-NPs and B6E-NPs for 8 h were observed via TEM. Scale bar: 2 μm. (E) Quantitation of (D). Error bars indicate mean ± SD (n= 3). *P < 0.05, **P < 0.01, ***P < 0.001; (Tg + PBS), (Tg + B6ME-NPs) vs. WT. #P < 0.05, ##P < 0.01, ###P < 0.001; (Tg + EGCG) vs. (Tg + B6ME-NPs).
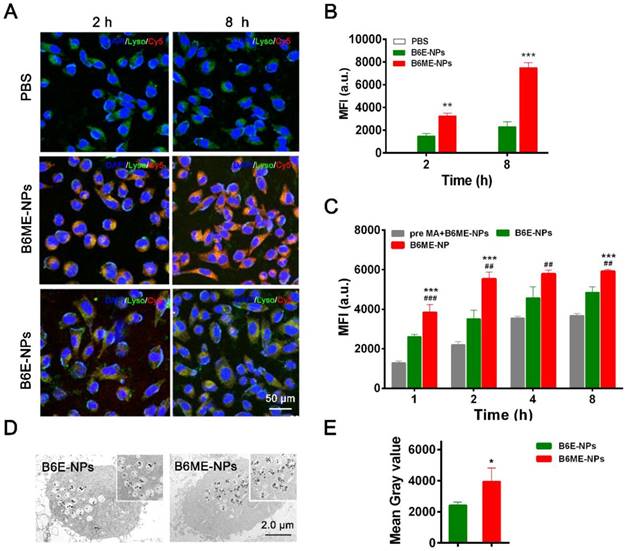
In order to confirm whether MA promoted cellular uptake of the NPs, the cells were pretreated with free MA for 2 h. As shown in Figure 3C, the fluorescence intensity of Cy5 was significantly decreased after pretreatment with free MA. These results indicate that the targeting ability of the NPs is attributable to MA. The sensitivity of MRI depends on the accumulation of SPIONs. Therefore, the amount of SPIONs in the SH-SY5Y cells was analyzed. As shown in Figure 3D-E, ultrathin sections of SH-SY5Y cells were imaged using TEM and the amount of SPIONs in the cells was quantified. After incubation for 8 h, the amount of SPIONs was significantly increased in the B6ME-NPs-treated cells compared with the amount in the B6E-NPs-treated cells. Therefore, MA-modified NPs may possess better therapeutic and tracing activities.
Comparison of EGCG with B6ME-NPs in vitro
In order to compare the ability of free EGCG and B6ME-NPs to inhibit aggregation in vitro, the aggregation of αS in αS-overexpressing SH-SY5Y cells (αS-SH) was studied. Intracellular αS aggregation was detected using CLSM after co-culture with different samples for 48 h. Cells treated with B6ME-NPs showed reduced αS aggregation as compared to the cells treated with only EGCG (Figure 4A and C). Interestingly, Kovtun and his colleagues revealed that some DAT internalization arises from lipid rafts [23]. As the NPs were designed to target DAT, it was presumed that B6ME-NPs might affect the cell membrane. Therefore, the morphology of SH-SY5Y cell membrane was observed via TEM analysis. As shown in Figure 4B and the quantitative result in Figure 4D, B6ME-NPs changed the cell curvature and number of vesicles as compared to B6M-NPs after 24 h treatment. These results indicate that EGCG-modified NPs promoted the transport of vesicles. In order to validate whether these NPs impact lipid raft-mediated endocytosis after treatment, the levels of some lipid raft proteins, including EGFR, contactin 1, flotillin 1, flotillin 2, and neurotrimin, were tested via western blotting. As shown in Figure 4E-J, there were significant differences in the expressions of EGFR, contactin 1, flotillin 1, and flotillin 2, but not neurotrimin. In particular, the effects of EGCG on flotillin 1 and flotillin 2 were much more obvious than those on other proteins. Furthermore, reverse transcription-PCR results confirmed the differences in gene expression between all groups. As is shown in Figure S12, EGFR, contactin 1, flotillin 1, and flotillin 2 showed highest mRNA expression in the cells treated with B6ME-NPs. Manfred and his co-workers found that co-assembly of flotillin 1 and flotillin 2 induced membrane curvature and vesicle budding. Therefore, EGCG-loaded NPs could first increase the expression of flotillin 1 and flotillin 2 and then their co-assembly. The co-assembly could then induce the formation and transport of membrane vesicles. Finally, the transport of vesicles could facilitate the endocytosis of EGCG-loaded NPs.
The role of B6ME-NPs in vitro. (A) αS-overexpression in SH-SY5Y cells as a PD cell model was detected via CLSM. Nuclei: DAPI, blue; control cells: mCherry, red; αS-SH: mCherry-SNAC, red. Scale bar: 20 μm. (B) The TEM images showing the differences of of vesicles and cell membrane curvature induced by B6M-NPs or B6MA-NPs in SH-SY5Y cells (48 h). Red triangles mark curvature, blue triangles mark vesicles. Scale bar: 2 μm. (C) Quantitation of (A). (D) Quantitation of vesicular NPs in (B). (E) Expressions of lipid raft-related proteins (EGFR, Contactin1, flotillin1, flotillin2 and neurotrimin) detected by Western Blot. (F-J) Quantitation of each protein in (E). Error bars represent mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001; (Tg + PBS), (Tg + B6ME-NPs) vs. WT. #P < 0.05, ##P < 0.01, ###P < 0.001; (Tg + EGCG) vs. (Tg + B6ME-NPs).
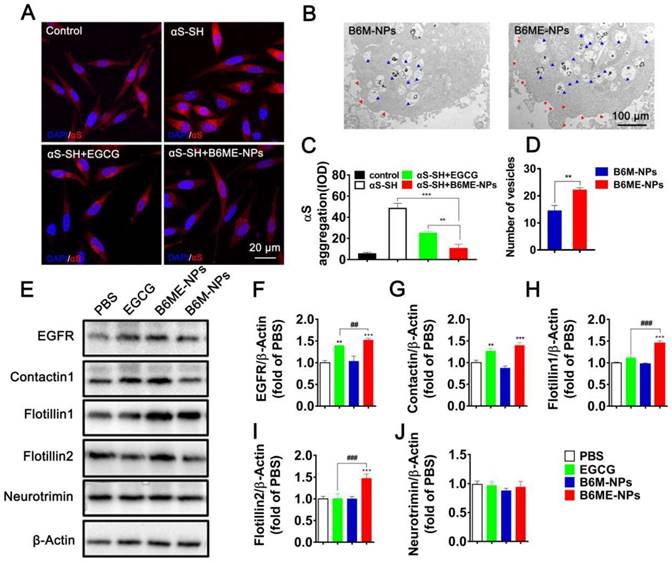
Increased αS levels disrupt neurotransmitter release via decreased synaptic-vesicle recycling-pool size and mobility [24]. Therefore, increased membrane curvature and vesicle budding after B6ME-NPs treatment may play a positive role in PD therapy. For the first time, we found that EGCG-loaded NPs induced the expression of certain genes and proteins (such as EGFR and contactin) in vitro. B6ME-NPs affected the expression of lipid raft proteins. There is enormous interest in finding a new mechanism of achieving high intracellular EGCG concentration using B6ME-NPs. B6ME-NPs individually affected other intriguing lipid raft proteins; both the genes and proteins of flotillins (flotillin1, flotillin2) were induced by these NPs. These results suggested that B6ME-NPs could efficiently treat PD via a specific flotillins pathway as shown in Figure S13.
Biodistribution and pharmacokinetics in vivo
In order to determine whether NPs were delivered into the brain, the biodistribution of NPs was investigated in all organs using the Kodak in vivo imaging system and NPs labeled with Cy7. As shown in Figure 5A and Figure S14, most of the ME-NPs accumulated in the kidneys. This result suggests that ME-NPs could not stay in the body for a long time. Moreover, there was no fluorescence of Cy7-labeled ME-NPs in the brain. This result shows that ME-NPs could not be transported into the brain. In contrast, a majority of the Cy7-labeled B6ME-NPs were delivered to the brain. These results illustrate that B6 could enable the NPs to permeate into the brain in vivo. The precise biodistribution in the brain was measured using MRI. The cuboid SPIONs possess high sensitivity for MRI due to their high values of r2. As shown in Figure 5B, many dark spots were observed in the brain after the mice were treated with B6ME-NPs for 24 h via intravenous injection. In contrast, there were no dark spots in the brain of ME-NPs-treated mice. These results confirmed the permeability of the BBB to B6-modified B6ME-NPs. In addition, the SN concentrations were measured to evaluate the SN targeting ability of MA. As shown in Figure 5C, the EGCG concentrations in the SN of the B6ME-NPs group were significantly higher than the EGCG concentrations in the B6E-NP group after treatment for 24 h. These results showed that the modification with MA could enhance targeting to the SN.
Pharmacokinetics and accumulation of the traceable NPs in the SN region. (A) Representative fluorescence images of mice 24 h after i.v. treatment with different nanoparticles (PBS, Cy7-labeled ME-NPs and Cy7-labeled B6ME-NPs). (B) T2* MR images of mice brains 24 h after i.v. treatment with different nanoparticles (PBS, SPIONs-labeled ME-NPs and SPIONs-labeled B6ME-NPs). (C) SN concentration of EGCG from different nanoparticles at different time points post i.v. injection detected with liquid MS. (D) Plasma concentration of EGCG from different nanoparticles at different time points post i.v. injection detected with HPLC. Error bars represent mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.005.
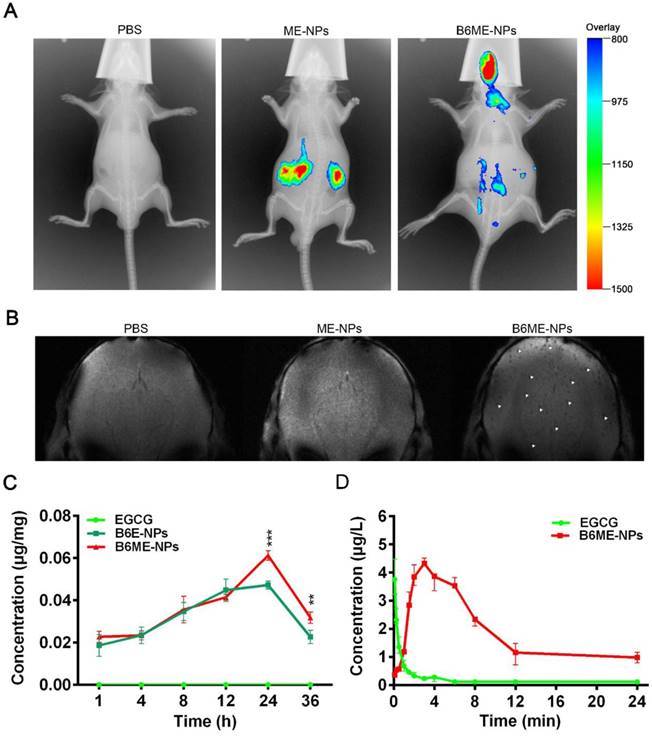
In addition, the pharmacokinetics of EGCG were evaluated. As shown in Figure 5D and Table S3, the EGCG concentration in the plasma was close to 0 after the mice were treated with free EGCG for 8 h. However, the EGCG concentration in the plasma was up to 2.3 μg/L in B6ME-NPs-treated mice. The area under the curve (AUC) values were calculated and are shown in Figure 5D. As shown in Table S3, the AUC value in the free EGCG group was significantly higher than that in the B6ME-NPs group. These results suggest that B6ME-NPs could significantly prolong the retention time of EGCG in the body.
In vivo therapeutic studies
As shown in Figure S15, behavioral tests and biochemical analyses demonstrated that there were significant differences in αS aggregation and behavioral performances between 3- and 5-month-old αS-overexpressing transgenic mice; in particular, the 5-month-old mice showed symptoms of PD. Therefore, we choose 3.5 months as the intervention timepoint for the treatment of PD and investigated the function of EGCG alone and B6ME-NPs on PD progression. The typical symptom of PD is dyskinesia and the open field test, rota-rod test, and pole test are the most commonly used methods for behavioral testing in PD mice. These methods are used to analyze whether dyskinesia is relieved with respect to aspects of exercise ability (open field test) and motor coordination ability (rota-rod test, pole test, and coat hanger test) after drug treatment in PD mice.
Open field test, rota-rod test, and pole test were performed to evaluate the therapeutic ability of B6ME-NPs in vivo. Additionally, immunofluorescence staining of dopaminergic neurons from the SN region and immunohistochemistry of the brain tissues were performed to analyze the protective effect of B6ME-NPs on dopaminergic neurons. The results of the rota-rod test and pole test (Figure 6A and B) and open field tests were conducted to observe walking trails (Figure 6A) and the final distance of every group was quantified (Figure 6B)indicate the better therapeutic efficacy of B6ME-NPs in PD. These results showed that there was not much difference between the normal wild type (WT) and B6ME-NPs-treated mice. Further, the results revealed that B6ME-NPs positively affected the PD mouse and were advantageous in the alleviation of PD symptoms. On the other hand, PBS-treated and EGCG-treated mice did not show significant differences, which proves that EGCG alone does not accumulate in the body. Figure 6C shows aggregation of αS in dopaminergic neurons from mice after treatment with different samples. We found that dopaminergic neurons of mice treated with B6ME-NPs showed reduced αS aggregation; the levels were comparable with those in the dopaminergic neurons of WT mice. On the other hand, the dopaminergic neurons of the PD model showed high aggregation of αS, while EGCG-treated mice showed moderate aggregation of αS.
In vivo PD therapy studies. The WT mice as control. The representative after different samples (PBS, bulk EGCG and B6ME-NPs) treating for 1.5 months. αS overexpression transgene mice as PD model. Both EGCG and the NPs via intravenous injection. (A) Rota-rod test is for the coordinated motion detection. (B) Pole test is used to assess motor function. (C) Immunofluorescence staining of the SN region of treated mouse brain. Nuclei: DAPI, blue; DAergic neuronal: TH, red; aggregation: ThT, green. Scale bar: 20 μm. (D) Quantitation of (C). (E) TH immunohistochemical slices of the SN region of treated mouse brain. Scale bar: 100 μm. (F) Quantitation of (E). Error bars represent mean ± SD (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001; (Tg + PBS), (Tg + B6ME-NPs) vs. WT. #P < 0.05, ##P < 0.01, ###P < 0.001; (Tg + EGCG) vs. (Tg + B6ME-NPs).
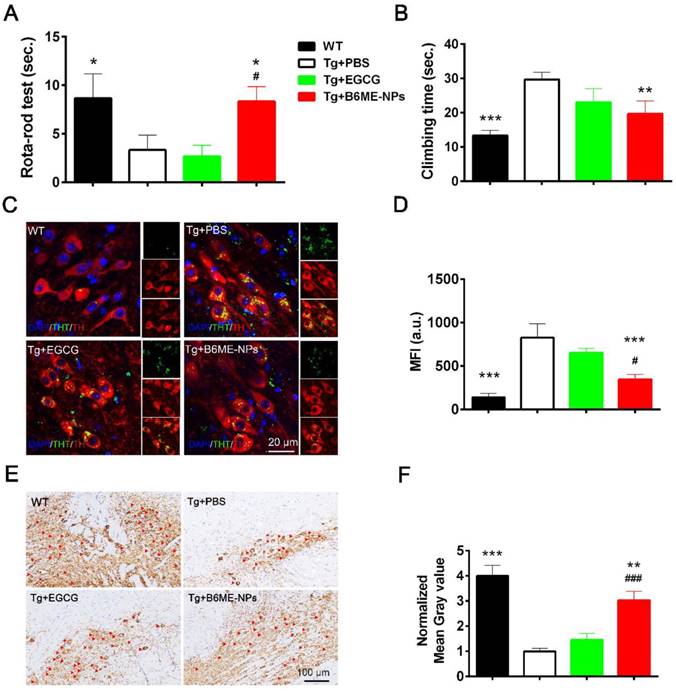
The loss of pigmented dopaminergic neurons in the SN is known to be a major pathological hallmark in PD. Here, we tested the biomarker of dopaminergic neurons by immunohistochemistry and western blotting. Immunohistochemistry of the SN of mice from different groups showed that B6ME-NPs could protect dopaminergic neurons (Figure 6E). We found a significant difference between the brain tissues of B6ME-NPs-treated mice and the brain tissues of the PD model and the EGCG-treated mice. The SN of B6ME-NPs-treated mice was found to be much healthier than those of the PD model and EGCG-treated mice. This further validated the previous results (Figure 6E-F are the quantitation of Figure 6C-D). Western blot analysis of DAT and TH (Figure S17) further validated these findings. We found the highest expression of both proteins in the samples from 6ME-NPs-treated PD mice. Richardson and coworkers suggested that cell-surface DAT levels are sensitive to membrane potential changes, which can rapidly drive DAT internalization from and insertion into the cell membrane, thus affecting the capacity of DAT to regulate extracellular DA levels [25]. Taken together, our results suggest that our system may act as a neuroprotective agent through the membrane. These therapeutic effects on PD model mice further confirm that B6ME-NPs could be promising agents for early treatment of PD and could slow down disease progression. While these results are robust and promising, additional in vitro and in vivo studies will be needed to further validate these findings and better understand the anti-PD effect of B6ME-NPs in other model systems.
Methods
Materials
1,2-dioleoylsn-glycero-3-phosphoethanolamine-n-[poly(ethylene glycol)]2000 (DSPE-PEG2000) ,1,2-dioleylsn-glycero-3-phosphoethanolamine-n-[poly(ethylenenglycol)]2000-maleimide(DSPE-PEG2000-Mal) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-n-[poly(ethyleneglycol)]-hydroxy succinamide (DSPE-PEG-NHS,PEG Mw = 2,000 Da) were purchased from Shanghai Advance Vehicle Technology Pharmaceutical L.T.D (Shanghai, China). Cell-penetrating peptide B6 was from GL Biochem Ltd. Co (Shanghai, China). (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) were purchased from Sigma-Aldrich. Epigallocatechin-3-gallate and Mazindol were purchased from Sigma. 4'-Hydroxy phenylboronic acid was purchased from Solarbio Science and Technology co. L.T.D (Beijing, China). Cell culture products were purchased from Gibco-BRL-Life Technologies (UK). Western Blot products were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Synthesis and Purification of each component and superparamagnetic iron oxide nanocubes (SPIONs)
DSPE-PEG2000-B6, DSPE-PEG2000-phenylboronic acid, and SPIONs were synthesized and purified as previously reported [26,27]. Briefly, DSPE-PEG2000-B6 was tested by MALDI-TOFMS. 1H NMR (Bruker 600 MHz, DMSO-d6, δ ppm) was carried out to characterize the obtained DSPE-PEG2000-mazindol and DSPE-PEG2000-phenylboronic acid. The core size and morphology of SPIONs were studied using a transmission electron microscope (TEM) (JEM-2100 electron microscope) operated at an acceleration voltage of 200 kV.
Synthesis and purification of DSPE-PEG2000-Mazindol
DSPE-PEG2000-NHS (500 mg) and Mazindol were dissolved in DMF. The reaction mixture was stirred gently at room temperature for 24 h. The unreacted Mazindol was removed by dialyzing in a Cellu SepH1-membrane (MWCO 2,000 Da) against anhydrous ethanol for 4 h then against deionized water for 12 h, and lyophilized to obtain the final product DSPE-PEG2000-Mazindol. 1H NMR (Bruker 600 MHz, DMSO-d6, δ ppm) was carried out to characterize the obtained product.
Preparation of traceable nanoparticles (NPs)
SPIONs (10.0 mg), DSPE-PEG-B6 (4.0 mg), DSPE-PEG-Mazindol (4.0 mg) and DSPE-PEG-phenylboronic acid (10.0 mg) were mixed in 5 mL chloroform, followed by slow evaporation of the solvent. 5 mL phosphate buffered saline (PBS 0.01 M, pH 7.4) was added to the solid under sonication until the solution became clear. 10 mg of EGCG was added into the solution, stirring at room temperature for 24 h. The mixture was dialyzed extensively (dialysis bag MWCO 3,500 Da) against distilled water for 24 h to remove all impurities. The concentration of EGCG was measured via HPLC assay. For the HPLC analysis, we used a mobile phase with water: formic acid (99.7%/0.3% v/v) / methanol: formic acid (99.7%/0.3% v/v). Water phase: organic phase (75%/25% v/v) at a flow rate of 1.0 mL/min. The concentration of EGCG was determined based on the peak area at the retention time of 12 min at 272 nm. The concentration of Fe was measured via ICP.
Characterization of the NPs
Dynamic light scattering (DLS; Zetasizer Nano ZS, Malvern Instruments) was used to measure the sizes and zeta potentials of the NPs. The morphology of the nanoparticles was measured by transmission electron microscopy (JEM-2100). The transverse relaxation property of the nanoparticles was measured with a 1.5 T magnetic resonance analyzer (minispec mq60, Bruker).
ROS-responsive EGCG release from NPs
The in vitro release of EGCG was measured in PBS (20% vitamin C) with 0, 1, and 3 mM H2O2. Briefly, drug-loaded NPs were suspended in PBS and dialyzed at 37 °C under horizontal shaking (150 rpm). The concentration of EGCG in PBS was detected using HPLC assay (Agilent, Japan).
Cell culture
SH-SY5Y neuroblastoma cells were originally obtained from American Tissue Type Cell Collection. The cells overexpressing αS-SH were gifted by Prof. Zhang. bEND.3 cells were obtained from Shanghai Enzyme Biological Technology Co., Ltd (Shanghai, China). All of these cells were cultured in DMEM (HyClone, Logan, UT) supplemented with 5% horse serum, 10% FBS, and 1% antibiotics (penicillin/streptomycin) at 37 °C in a humidified 95% air/5% CO2 incubator.
Cell toxicity assays
96-well polystyrene plates were used to plate SH-SY5Y neuroblastoma cells at ~10,000 cells per 100 µL of medium per well. Cells were incubated at 37 °C, for 24 h to allow the cells to attach, then treated with different NPs (B6ME-NPs, B6M-NPs, B6E-NPs, ME-NPs,), bulk EGCG and PBS as control for 48 h. Then, the cells were treated with MTT for 4 h at 37 °C. The absorbance at 490 nm was measured using a Tecan Safire microplate reader. Averages from six replicate wells were used for each sample.
Transport across an in vitro blood-brain barrier (BBB) model
An in vitro model of BBB was established as previously reported [19]. Briefly, bEND.3 cells were seeded in the upper chambers. SH-SY5Y cells were seeded on the lower chamber with a cover slip. The in vitro BBB models were exposed to different drug formulations and PBS as control. After 0.5, 1, 2, 4, 6, 8 and 12 h of incubation, the cover slips were moved to new 24-well plates. The bEND.3 and SH-SY5Y cells were then digested with trypsin to be measured by flow cytometry (BD Co., USA) or washed and fixed with 4% paraformaldehyde, stained with DAPI and analyzed by CLSM (Zeiss Co., Germany).
MA impact on cellular uptake
To analyze the effects of MA of NPs uptake, SH-SY5Y cells were incubated with Cy5 dye-loaded NPs (Cy5-B6ME-NPs, Cy5-B6E-NPs) and PBS as control. After 2 and 8 h of incubation, cells were washed and stained with Lyso Tracker green for 1 h at 37 °C. The cells were then fixed with 4% paraformaldehyde and stained with DAPI. The samples were analyzed with CLSM (Zeiss Co., Germany).
In vivo MR brain imaging
To investigate whether the B6-modified NPs can cross the BBB in vivo, T2* MR images of 3.5-month-old human αS-overexpressing mice were collected 24 h after 25 mg EGCG/kg i.v. administration of different drug formulations. During in vivo imaging, the mice were anesthetized with 2.5% isoflurane in 75% NO2 plus 22% O2 and the isoflurane was reduced to 1% for maintenance of anesthesia. MRI studies of this line of transgenic mice were carried out on a 7 T MRI system. The imaging parameters were: 100 µm isotropic spatial resolution; TR = 3000 ms; TE = 50.0 ms; FA = 90°.
Blood pharmacokinetics
To measure pharmacokinetics, PD model mice were randomly divided into two groups and injected intravenously through the tail vein with the traceable NPs and free drugs at an equivalent dose of 100 mg EGCG/kg. Blood samples (100 μL) were collected in a heparinized tube from the orbital venous plexus at 5 min, 15 min, 0.5, 1, 2, 4, 6, 8 and 24 h post injection. To extract and protect EGCG, methanol and 20% vitamin C were added to the blood. The samples were vortexed and the solution was immediately centrifuged at 1,800 ×g for 10 min. The concentration of EGCG in the supernatant was measured by HPLC.
In vivo biodistribution
To measure the in vivo biodistribution, PD model mice were intravenously administrated Cy7-labelled drug formulations and PBS as control at an equivalent dose of 100 mg EGCG/kg. 24 h after treatment, Cy7 fluorescence was detected via a Kodak in vivo imaging system. Then, mice were sacrificed and their organs (brain, heart, liver, spleen, lung and kidney) were rapidly excised, washed three times with PBS, and imaged.
SN EGCG concentration
To determine the SN drug concentration, PD model mice were randomly divided into three groups and injected intravenously through the tail vein B6ME-NPs, B6E-NPs and free drugs, at an equivalent dose of 100 mg EGCG/kg. Then, mice were sacrificed and the SN was rapidly excised. To extract and protect EGCG, methanol and 20% vitamin C were added to the tissue. The samples were grinded, vortexed and the solution was immediately centrifuged at 1,800 ×g for 10 min. The concentration of EGCG in the supernatant was measured by LC-MS.
Behavior tests
Open field test, Pole Test and Rotarod were performed as previously reported [28,29]. The total distance, time period and mice remained were used as measures of motor function.
Double co-immunofluorescence analysis of the SN
Double immunofluorescence analysis was carried out on the SN to examine the effects of CME-NPs on α-syn aggregation in dopaminergic neurons. 2 N HCl was used to denature the free-floating sections/NSC cultures, and 3% NGS, 0.1% Triton X-100, and 0.5% BSA solution were used to block the cultures for 2 h. The sections were incubated with mouse anti-tyrosine hydroxylase (1:200) for 24 h at 4 ℃. Anti-mouse Alexa Fluor 594 was used as the secondary antibody (1:200). Later, the sections were incubated with ThT (10 mM) for 5 min. Sections were then mounted with DAPI-containing hard set antifade mounting medium (Vectashield, Vector Laboratories, CA, USA). Sections were analyzed by CLSM (Zeiss Co., Germany).
Gene expression analysis by qRT-PCR
Primers were procured from Sangon Biotech (Shanghai) Co., Ltd. qRT-PCR analysis was carried out to measure the expressions of genes in the lipid raft. Total RNA was isolated from the cultures of control and treated cells using TriZol reagent. Detection and quantification of the targets was done in real time using the ABI Prism 7900 sequence detector system (PE Applied Biosystems; Foster City CA, USA) and SYBR green chemistry (Applied Biosystems, USA). ΔΔCt method was used to calculate relative expression.
Statistical analysis
All data are expressed as mean ± SD unless otherwise indicated. Statistical significance was analyzed using one-way ANOVA. Statistical differences in behavioral data were determined using two-way repeated measures ANOVA.
Conclusion
In this study, “cell-addictive” traceable NPs were prepared for PD treatment. The NPs were named B6ME-NPs. They were electrically neutral with homogeneous size and morphology; therefore, their serum stability was outstanding. Furthermore, the MRI sensitivity of B6ME-NPs was excellent due to their high r2 value. PD mice exhibited good treatment results after they were treated with B6ME-NPs via intravenous injection and PD-like characteristics were significantly alleviated. The amount of EGCG accumulated in PD lesions was notably enhanced and traced via magnetic resonance imaging. In addition, αS aggregation was considerably inhibited. Furthermore, dopaminergic neurons were markedly increased. Due to its low price, simple preparation, safety, and excellent therapeutic effect on PD, B6ME-NPs are expected to have potential application in PD treatment.
Abbreviations
αS: α-synclein aggregation; PD: Parkinson's disease; EGCG: Epigallocatechin gallate; NPs: nanoparticles; SN: substantia nigra; TH: tyrosine hydroxylase; BBB: blood-brain barrier; TfR: transferrin receptor; DAT: Dopamine transporter; MA: Mazindol; ROS: reactive oxygen species; SPIONs: superparamagnetic iron oxide nanocubes; MRI: magnetic resonance imaging; TEM: transmission electron microscopy; r2: relaxation rate; CLSM: confocal laser scanning microscopy; AUC: area under the curve; WT: wild type.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was financially supported by the Beijing Municipal Science & Technology Commission (No. Z161100002616015), National Natural Science Foundation of China (No. 81671268), the Brain cognition and brain medicine of Beijing Municipal Science & Technology Commission (Z161100002616020), the National natural science foundation of China (81701260), the National High Technology Research and Development Program (2016YFA0200303) and the Beijing Natural Science Foundation (L172046) .
Competing Interests
The authors have declared that no competing interest exists.
References
1. Reeve A, Simcox E, Turnbull D. Ageing and Parkinson's disease: why is advancing age the biggest risk factor? Ageing Res Rev. 2014;14:19-30
2. Saraiva C, Praca C, Ferreira R, Santos T, Ferreira L, Bernardino L. Nanoparticle-mediated brain drug delivery: Overcoming blood-brain barrier to treat neurodegenerative diseases. J Control Release. 2016;235:34-47
3. Kalfon L, Youdim MBH, Mandel SA. Green tea polyphenol (-)-epigallocatechin-3-gallate promotes the rapid protein kinase C- and proteasome-mediated degradation of Bad: Implications for neuroprotection. J Neurochem. 2007;100:992-1002
4. Wong YC, Krainc D. Alpha-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23:1-13
5. Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K. et al. EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc Natl Acad Sci. 2010;107:7710-5
6. Yoshida W, Kobayashi N, Sasaki Y, Ikebukuro K, Sode K. Partial peptide of alpha-synuclein modified with small-molecule inhibitors specifically inhibits amyloid fibrillation of alpha-synuclein. Int J Mol Sci. 2013;14:2590-600
7. Xu Y, Zhang Y, Quan Z, Wong W, Guo J, Zhang R. et al. Epigallocatechin Gallate (EGCG) Inhibits Alpha-Synuclein Aggregation: A Potential Agent for Parkinson's Disease. Neurochem Res. 2016;41:2788-96
8. Levites Y, Weinreb O, Maor G, Youdim MB, Mandel S. Green tea polyphenol (-)-epigallocatechin-3-gallate prevents N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic neurodegeneration. J Neurochem. 2001;78:1073-82
9. Gundimeda U, McNeill TH, Fan TK, Deng R, Rayudu D, Chen Z. et al. Green tea catechins potentiate the neuritogenic action of brain-derived neurotrophic factor: role of 67-kDa laminin receptor and hydrogen peroxide. Biochem Biophys Res Commun. 2014;445:218-24
10. Li Y-F, Wang H, Fan Y, Shi H-J, Wang Q-M, Chen B-R. et al. Epigallocatechin-3-Gallate Inhibits Matrix Metalloproteinase-9 and Monocyte Chemotactic Protein-1 Expression Through the 67-kappaDa Laminin Receptor and the TLR4/MAPK/NF-kappaB Signalling Pathway in Lipopolysaccharide-Induced Macrophages. Cell Physiol Biochem. 2017;43:926-36
11. Huang Y, Sumida M, Kumazoe M, Sugihara K, Suemasu Y, Yamada S. et al. Oligomer formation of a tea polyphenol, EGCG, on its sensing molecule 67 kDa laminin receptor. Chem Commun (Camb). 2017;53:1941-4
12. Yin T, Yang L, Liu Y, Zhou X, Sun J, Liu J. Sialic acid (SA)-modified selenium nanoparticles coated with a high blood-brain barrier permeability peptide-B6 peptide for potential use in Alzheimer's disease. Acta Biomater. 2015;25:172-83
13. Liu Z, Gao X, Kang T, Jiang M, Miao D, Gu G. et al. B6 peptide-modified PEG-PLA nanoparticles for enhanced brain delivery of neuroprotective peptide. Bioconjug Chem. 2013;24:997-1007
14. Dersch CM, Akunne HC, Partilla JS, Char GU, de Costa BR, Rice KC. et al. Studies of the biogenic amine transporters. 1. Dopamine reuptake blockers inhibit [3H]mazindol binding to the dopamine transporter by a competitive mechanism: preliminary evidence for different binding domains. Neurochem Res. 1994;19:201-8
15. Severinsen K, Koldso H, Thorup KAV, Schjoth-Eskesen C, Moller PT, Wiborg O. et al. Binding of mazindol and analogs to the human serotonin and dopamine transporters. Mol Pharmacol. 2014;85:208-17
16. Houlihan WJ, Boja JW, Parrino VA, Kopajtic TA, Kuhar MJ. Halogenated mazindol analogs as potential inhibitors of the cocaine binding site at the dopamine transporter. J Med Chem. 1996;39:4935-41
17. Torchilin VP. Passive and active drug targeting: drug delivery to tumors as an example. Handb Exp Pharmacol. 2010;197:3-53
18. Zhang J, Zhou X, Yu Q, Yang L, Sun D, Zhou Y. et al. Epigallocatechin-3-gallate (EGCG)-stabilized selenium nanoparticles coated with Tet-1 peptide to reduce amyloid-?? aggregation and cytotoxicity. ACS Appl Mater Interfaces. 2014;6:8475-87
19. Zhang N, Yan F, Liang X, Wu M, Shen Y, Chen M. et al. Localized delivery of curcumin into brain with polysorbate 80-modified cerasomes by ultrasound-targeted microbubble destruction for improved Parkinson's disease therapy. Theranostics. 2018;8:2264-77
20. Omidi Y, Campbell L, Barar J, Connell D, Akhtar S, Gumbleton M. Evaluation of the immortalised mouse brain capillary endothelial cell line, b.End3, as an in vitro blood-brain barrier model for drug uptake and transport studies. Brain Res. 2003;990:95-112
21. Qiao R, Jia Q, Huwel S, Xia R, Liu T, Gao F. et al. Receptor-mediated delivery of magnetic nanoparticles across the blood-brain barrier. ACS Nano. 2012;6:3304-10
22. Houlihan WJ, Kelly L, Pankuch J, Koletar J, Brand L, Janowsky A. et al. Mazindol analogues as potential inhibitors of the cocaine binding site at the dopamine transporter. J Med Chem. 2002;45:4097-109
23. Kovtun O, Sakrikar D, Tomlinson ID, Chang JC, Arzeta-Ferrer X, Blakely RD. et al. Single-quantum-dot tracking reveals altered membrane dynamics of an attention-deficit/hyperactivity-disorder-derived dopamine transporter coding variant. ACS Chem Neurosci. 2015;6:526-34
24. Scott D, Roy S. alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci. 2012;32:10129-35
25. Richardson BD, Saha K, Krout D, Cabrera E, Felts B, Henry LK. et al. Membrane potential shapes regulation of dopamine transporter trafficking at the plasma membrane. Nat Commun. England. 2016;7:10423
26. Hu B, Dai F, Fan Z, Ma G, Tang Q, Zhang X. Nanotheranostics: Congo Red/Rutin-MNPs with Enhanced Magnetic Resonance Imaging and H2O2-Responsive Therapy of Alzheimer's Disease in APPswe/PS1dE9 Transgenic Mice. Adv Mater. 2015;27:5499-505
27. Lu Z, Zheng Y, Zhang T, Shen J, Xiao Z, Hu J. et al. Effects of Photo-Activated Nanomicelles Loaded with Fragrance on Central Nervous System. J Biomed Nanotechnol. 2018;14:1675-87
28. Sedelis M, Schwarting RK, Huston JP. Behavioral phenotyping of the MPTP mouse model of Parkinson's disease. Behav Brain Res. 2001;125:109-25
29. Rabl R, Breitschaedel C, Flunkert S, Duller S, Amschl D, Neddens J. et al. Early start of progressive motor deficits in Line 61 alpha-synuclein transgenic mice. BMC Neurosci. 2017;18:22
Author contact
![]() Corresponding authors: E-mail: xzhangac.cn; hqingedu.cn
Corresponding authors: E-mail: xzhangac.cn; hqingedu.cn