Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(1):210-231. doi:10.7150/thno.28434 This issue Cite
Research Paper
An IgM antibody targeting the receptor binding site of influenza B blocks viral infection with great breadth and potency
Chenguang Shen1,2*, Minwei Zhang1,3*, Yuanzhi Chen1*, Limin Zhang1, Guosong Wang1, Junyu Chen1, Siyuan Chen1, Zizhen Li1, Feixue Wei1, Jing Chen3, Kunyu Yang4, Shuxin Guo1, Yujing Wang1, Qingbing Zheng1, Hai Yu1, Wenxin Luo1, Jun Zhang1, Honglin Chen1,5, Yixin Chen1 ![]() , Ningshao Xia1
, Ningshao Xia1
1. State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics, National Institute of Diagnostics and Vaccine Development in Infectious Diseases, School of Life Sciences, School of Public Health, Xiamen University, Xiamen 361102, Fujian, China;
2. Shenzhen Key Laboratory of Pathogen and Immunity, State Key Discipline of Infectious Disease, Shenzhen Third People's Hospital, Shenzhen, China;
3. Department of Emergency and Critical Care Medicine, First Affiliated Hospital of Xiamen University, Xiamen 361002, Fujian, China;
4. Xiamen International Travel Healthcare Centre, Xiamen 361012, Fujian, China;
5. Department of Microbiology, State Key Laboratory of Emerging Infectious Diseases, University of Hong Kong, Hong Kong, China.
* These authors contributed equally to this work.
Received 2018-7-10; Accepted 2018-11-30; Published 2019-1-1
Abstract

Broadly neutralizing antibodies (bnAbs) targeting the receptor binding site (RBS) of hemagglutinin (HA) have potential for developing into powerful anti-influenza agents. Several previously reported influenza B bnAbs are nevertheless unable to neutralize a portion of influenza B virus variants. HA-specific bnAbs with hemagglutination inhibition (HI) activity may possess the ability to block virus entry directly. Polymeric IgM antibodies are expected to more effectively inhibit virus attachment and entry into target cells due to their higher avidity and/or steric hindrance. We therefore hypothesized that certain RBS-targeted IgM antibodies with strong cross-lineage HI activity might display broader and more potent antiviral activity against rapidly evolving influenza B viruses.
Methods: In this study, we generated IgM and IgG bnAbs targeting the RBS of influenza B virus using the murine hybridoma technique. IgM and IgG versions of the same antibodies were then developed by isotype switching and characterized in subsequent in vitro and in vivo experiments.
Results: Two IgM and two IgG bnAbs against influenza B virus HA were identified. Of these, one IgM subtype antibody, C7G6-IgM, showed strong HI and neutralization activities against all 20 representative influenza B strains tested, with higher potency and broader breadth of anti-influenza activity in vitro than the IgG subtype variant of itself, or other previously-reported influenza B bnAbs. Furthermore, C7G6-IgM conferred excellent cross-protection against distinct lineages of influenza B viruses in mice and ferrets, performing better than the anti-influenza drug oseltamivir, and showed an additive antiviral effect when administered in combination with oseltamivir. Mechanistically, C7G6-IgM potently inhibits infection with influenza B virus strains from different lineages by blocking viral entry.
Conclusion: In summary, our study highlights the potential of IgM subtype antibodies in combatting pathogenic microbes. Moreover, C7G6-IgM is a promising candidate for the development of prophylactics or therapeutics against influenza B infection.
Keywords: influenza B virus, hemagglutinin, receptor binding site, broadly neutralizing antibodies, IgM
Introduction
Influenza B viruses (IBVs) cause a considerable public health burden each season, contributing to significant morbidity and mortality globally [1-3], particularly in children and infants [4, 5]. Two genetically and antigenically distinct lineages of IBVs, namely Victoria and Yamagata, are currently co-circulating worldwide, infecting people of all ages [6]. Seasonal influenza (Influenza A and B) epidemics result in approximately 3 to 5 million cases of severe illness yearly, with 250,000-500,000 deaths [7, 8]. IBVs account for 20%-25% of annual influenza infections; however, infection rates can rise above 60% in some seasons [9, 10]. The clinical presentation, complications and death rate of seasonal influenza A and B virus infections are clinically indistinguishable; infections with these two types of influenza virus pose equal risks [11]. Influenza virus vaccines are currently the most effective countermeasure against influenza B virus infections, but these vaccines have limited efficacy because they induce only strain-matched humoral immune responses, while the globular head domain of the viral hemagglutinin (HA) continues to evolve rapidly [12]. Few anti-influenza drugs are available and the treatment window of these drugs is very narrow [13, 14]. In addition, one of these antiviral medications, oseltamivir, is reported to be less effective against IBVs than against influenza A viruses [15, 16]. In view of this, 60% of patients admitted to intensive care units with influenza B do not receive specific influenza antiviral therapy, resulting in poor outcomes [9]. This highlights the urgent need to develop more effective medical approaches to treat influenza B virus infections.
Passive immune protection using broadly neutralizing antibodies (bnAbs) is a promising approach for treating viral infections [17]. In recent years, we and other groups have developed several bnAbs against the HA protein of influenza B virus to confer protection against multiple influenza B virus strains from distinct lineages in vivo [18-21]. These antibodies constitute a novel and promising approach for the treatment and prevention of influenza B infections and strengthen the achievability of the development of universal influenza B vaccines. These antibodies bind to distinct epitopes on the HA and inhibit virus infection through different mechanisms. Two of these antibodies, CR8033 and C12G6, target the receptor binding site (RBS) domain on the HA to directly prevent viral entry, showing strong cross-lineage hemagglutination inhibition (HI) activity against influenza B viruses. However both CR8033 and C12G6 fail to potently inhibit the viral entry of some influenza B virus strains, as determined by measuring the level of HI activity [18, 21]. Considering that prevention of virus binding to the cell receptor is the first key step in inhibiting viral entry and infection, development of more effective influenza B bnAbs which can directly block viral entry is important to facilitate the formulation of antiviral antibody cocktails against rapidly evolving influenza B viruses.
Immunoglobulin G (IgG), IgM and IgA antibodies are the main types of antibody produced in mice and humans; they all offer protection from pathogenic infections [22, 23]. IgM antibodies appear early in the course of an infection, whereas IgG antibodies are generated following subsequent class switching and antibody maturation [24]. IgG is secreted as a monomer that is small in size, while the predominant form of IgM is pentameric and the largest antibody in the body [23]. Due to the high avidity and/or steric hindrance of IgM antibodies [23], we hypothesized that certain IgM subtype antibodies which target specific epitopes on the RBS domain of influenza B virus might be more likely to block virus binding to cell receptors and effectively prevent different variant strains from causing infection, thus displaying broader and more potent antiviral activity than IgG subtype antibodies of the same specificity.
In this study, we found that antisera from mice intranasally immunized with one influenza B live virus strain contained large proportions of IgM antibodies that were able to neutralize both lineages of influenza B viruses and also possessed cross-lineage HI activity. Through HI and MN (microneutralization) screening of antibodies in mice sequentially immunized with representative Yamagata and Victoria strains, we discovered two murine IgM bnAbs (7G6-IgM and 3G10-IgM) and two murine lgG bnAbs (11B10-IgG and 10H10-IgG) which bind to the receptor binding domain of influenza B virus. Chimeric versions of these antibodies containing a human IgG1 or IgM Fc fragment were also generated. Noteworthily, an IgM subtype chimeric antibody, C7G6-IgM, exhibits significantly higher potency and greater breadth of antiviral activity in vitro than the other tested antibodies, including bnAbs reported on previously, and confers strong cross-protection against distinct lineages of IBVs in mice and ferrets. These findings provide innovative insights relevant to the development of high-efficacy antiviral agents and universal vaccines against influenza viruses and other pathogens.
Results
Generation of high-efficiency HA head-specific bnAbs against influenza B
To generate antibody responses against influenza B viruses, six-week-old female BALB/c mice were infected intranasally with the Yamagata lineage virus strain, B/Florida/4/2006 (FL/2006), and the antisera from these mice collected 7 and 14 days after immunization for reactivity analysis (Figure 1A). Vaccination with B/Florida/4/2006 alone can elicit cross-lineage IgG and IgM antibody-based immune responses in mice, as evidenced using an enzyme-linked immunosorbent assay (ELISA) against the HA proteins of influenza B virus strains representing both lineages, namely B/Florida/4/2006 and the Victoria virus strain B/Brisbane/60/2008 (BR/2008). For the 7-day sera, higher levels of IgM antibodies specific to FL/2006 or BR/2008 were induced compared to the titers of antigen-specific IgG antibodies (Figure 1B, left panel). For the 14-day sera, antigen-specific IgG titers were significantly increased, whereas relatively lower levels of IgM antibodies were detected (Figure 1B, right panel). Interestingly, the 7-day sera, which contains large proportions of influenza B virus-specific IgM, possessed detectable cross-lineage HI and MN activities against both lineages of influenza B viruses (Figure 1B, left panel), whereas the 14-day sera, which contains more IgG antibodies, exhibited HI and MN activities against only the immunogen, Yamagata lineage strain FL/2006 (Figure 1B, right panel). Furthermore, the antisera from mice infected with the Victoria lineage virus strain (B/Brisbane/60/2008) displayed a similarly distinct pattern of differing antiviral reactivity against the immunizing strain and the Yamagata lineage strain, FL/2006 (Figure S1). These results suggest that some antigen-specific IgM isotype antibodies induced in the early phase of the immune response possess stronger and broader cross-lineage HI and MN activities against influenza B viruses than the antigen-specific IgG isotype antibodies induced subsequently.
Schematic showing the generation of bnAbs. (A) Serum collection from mice (n=6) immunized intranasally with the Yamagata lineage virus strain FL/2006 (B/Florida/4/2006, blue cartoon particle) at 1×104 TCID50 per mouse. The mice were immunized at day 0 and sera were collected 7 and 14 days after each immunization. (B) Characterization of 7-day and 14-day anti-influenza sera following intranasal immunization with the Yamagata lineage strain of influenza B virus. Shown are data for serum total IgG titers, serum total IgM titers, serum HI titers and serum MN titers of FL/2006-immunized sera against two representative influenza B viruses, FL/2006 and BR/2008 (B/Brisbane/60/2008, Victoria lineage), analyzed in parallel. Recombinant HA proteins of FL/2006 and BR/2008 were used as ELISA plate-coating antigens. IgG and IgM titers were determined with quantitative ELISA and are expressed in ng/mL. Bars represent averages and standard errors. (C) A schematic depicting the generation of cross-reactive antibodies. Mice (n=8) were sequentially infected intranasally with FL/2006 and BR/2008 at 1×104 TCID50 per mouse. 21 or 28 days after the first immunization, spleen cells were collected from infected mice and fused with mouse myeloma Sp2/0 cells. (D) Hybridomas from the 21-day fusion groups were screened for production of cross-lineage mAbs specific to influenza B virus using HI and MN assays against FL/2006 and BR/2008 viruses. (E) Hybridomas from the 28-day fusion groups were screened for production of cross-lineage mAbs specific to influenza B virus using HI and MN assays against FL/2006 and BR/2008 viruses. +++, strong reactivity; ++, moderate reactivity; +, weak reactivity; -, no reactivity. PC: positive control, antibody CR8033-like. NC: negative control, antibody CR9114-like.
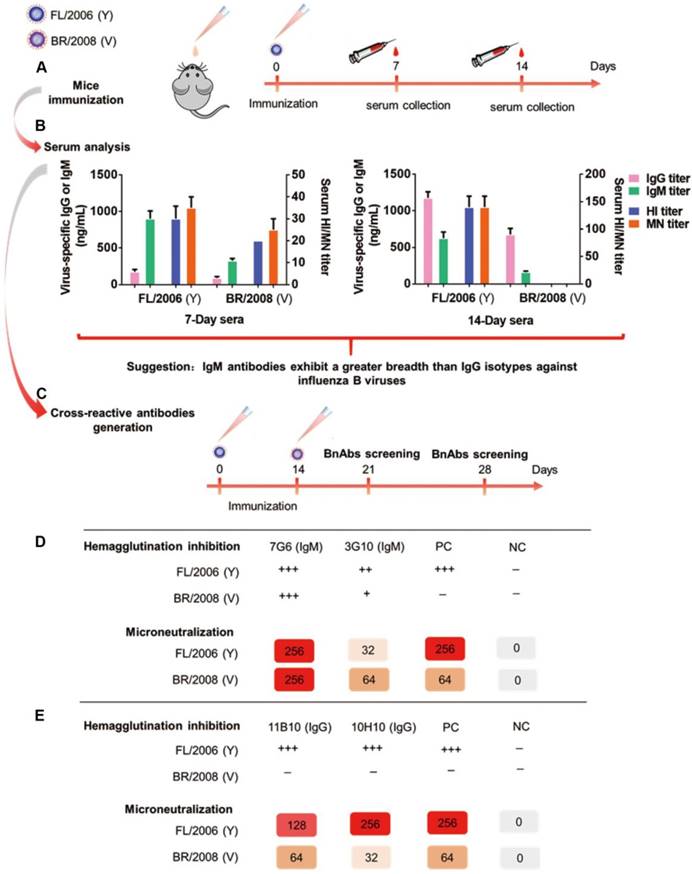
To verify our hypothesis and generate high-efficiency bnAbs against influenza B, mice were sequentially immunized intranasally with live B/Florida/4/2006 and B/Brisbane/60/2008 viruses at 14-day intervals (Figure 1C). On days 21 and 28 after the first immunization, spleen cells collected from immunized mice were fused with mouse myeloma Sp2/0 cells to generate hybridomas, as described previously [25]. The hybridomas were screened for the secretion of influenza B virus-specific mAbs in HI and MN assays against both B/Florida/4/2006 (Yamagata) and B/Brisbane/60/2008 (Victoria) in parallel, and hybridomas which showed positive MN activity against both viruses were selected. From the day 21 fusions two broadly neutralizing IgM isotype antibodies, 7G6-IgM and 3G10-IgM, were generated and both showed HI activity against both lineages of influenza B viruses (Figure 1D). Although we also generated two broadly neutralizing IgG isotype antibodies, 11B10-IgG and 10H10-IgG, from the fusions performed at day 28, these antibodies only showed HI activity against the Yamagata virus strain (Figure 1E).
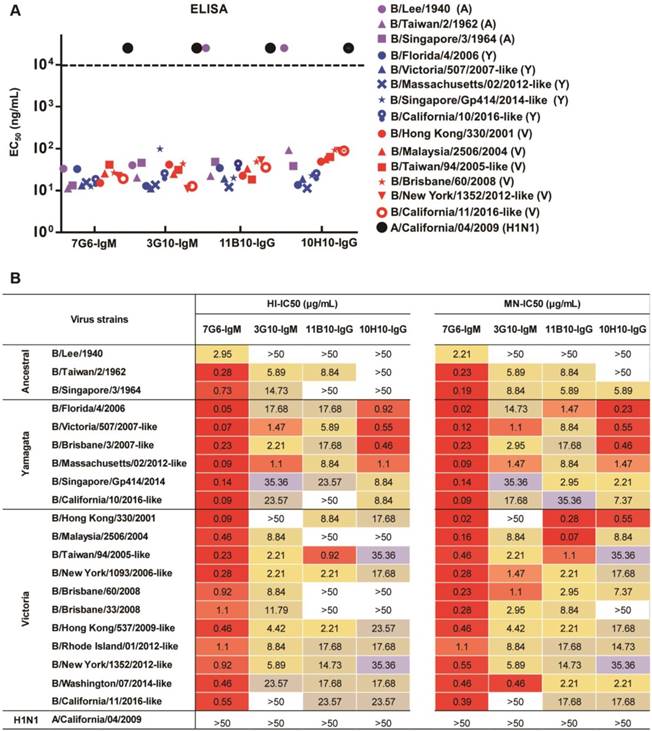
The four antibodies (7G6-IgM, 3G10-IgM, 11B10-IgG and 10H10-IgG) were purified from mouse ascites, and their reactivities against distinct lineages of influenza B virus strains were determined in ELISA, HI and MN assays. In the binding test, the 7G6-IgM, 3G10-IgM, 11B10-IgG and 10H10-IgG antibodies reacted specifically with all fourteen tested viruses from three diverse lineages of influenza B viruses, except for 11B10-IgG and 10H10-IgG, which did not react with B/Lee/1940 (Figure 2A). In the HI and MN assays, the four antibodies all demonstrated cross-lineage activities against the three lineages of influenza B viruses, except that 10H10-IgG lacked HI activity against the Ancestral lineage viruses, and of these, the IgM subtype 7G6-IgM antibody showed the strongest and broadest HI and MN activities against the 20 tested influenza B viruses (Figure 2B). To further evaluate the potential clinical use of these four bnAbs and confirm whether the IgM subtype HA head-specific bnAbs were superior to the IgG subtype bnAbs, variants of the 7G6, 3G10, 10H10 and CR9114-like [18] (a non-neutralizing antibody targeting the HA stem region of influenza B, included for comparison) antibodies with human IgM or IgG1 Fc regions were constructed and designated C7G6-IgM, C7G6-IgG, C3G10-IgM, C3G10-IgG, C10H10-IgM, C10H10-IgG, CR9114-like-IgM [18] and CR9114-like-IgG [18]. The IgM and IgG subtype antibodies were analyzed using HPLC, together with purified human IgM and protein standards (440 kDa and 158 kDa) as controls; the molecular weights of these eight variants were in the expected range (Figure S2). To further compare the functional activities of the antibodies generated in this study with those previously described, chimeric human IgG1 Fc region-versions of an additional six cross-lineage neutralizing influenza B HA-specific bnAbs were also constructed and prepared, being designated C11B10, C12G6 [21], CR8033-like [18], CR8071-like [18], 5A7-like [19] and 46B8-like [20]. The chimeric antibodies constructed and expressed in this study are not completely identical to the 'original' versions of the CR9114, CR8033, CR8071, 5A7 and 46B8 antibodies described previously, and have thus been designated CR9114-like, CR8033-like, CR8071-like, 5A7-like and 46B8-like, respectively.
In vitro binding and HI and MN activities of mouse antibodies 7G6-IgM, 3G10-IgM, 11B10-IgG and 10H10-IgG. (A) Binding (EC50 ELISA values) of 7G6-IgM, 3G10-IgM, 11B10-IgG and 10H10-IgG antibodies to purified viruses representing the three influenza B lineages. EC50 values above 104 ng/mL (dashed line) were scored as negative. Yamagata lineage strains are indicated in blue, Victoria lineage strains are indicated in red, the Ancestral lineage strains are indicated in purple and influenza A virus is indicated in black. (B) Fifty percent inhibitory concentrations (IC50) of the indicated antibodies against representative strains from the three influenza B lineages were determined by performing HI and MN assays. The values are the averages of three independent experiments and are reported in μg/mL. Values below 50 µg/mL are color-filled: red-orange shades, strong reactivity; yellow shades, moderate reactivity; beige-light blue shades, weak reactivity; >50, no reactivity.
C7G6-IgM antibody demonstrates unprecedentedly strong and broad cross-lineage HI and MN activities against influenza B viruses
In the binding test, C7G6-IgM, C7G6-IgG, C3G10-IgM and C3G10-IgG antibodies reacted specifically with all nine tested viruses from the three influenza B lineages. The C10H10-IgM, C10H10-IgG and C11B10 antibodies reacted with eight of the tested viruses, but not the ancestral strain B/Lee/1940 (Figure 3A). The results also demonstrated that there were no significant differences in binding activity against influenza B viruses between the IgM and IgG variants of each antibody (Figure 3A). Next, we directly compared the in vitro HI and MN activities of the 14 bnAbs constructed above, using a panel of 20 representative influenza B virus strains from three distinct lineages. C7G6-IgM was the only antibody tested that possessed HI activity against all 20 influenza B viruses tested, with a median fifty percent inhibitory concentration (IC50) of 0.95 µg/mL. In contrast, C7G6-IgG did not show HI activity against three influenza B virus strains: B/Lee/1940, B/Singapore/3/1964 and B/California/11/2016-like. C3G10-IgM, C11B10 and C12G6 displayed HI activity against most of the tested viruses from three lineages. C3G10-IgG, C10H10-IgM and C10H10-IgG exhibited HI activity against some strains from the Yamagata and Victoria lineages, but not the ancestral lineage. CR8033-like displayed HI activity against Yamagata and Ancestral lineage strains, but not Victoria viruses. The other antibodies did not exhibit HI activity against any influenza B strain (Figure 3B). Median IC50 values were 0.95, 19.89, 19.15, 20.79, 4.72, 13.75, 18.59, 12.87 and 2.83 µg/mL for C7G6-IgM, C7G6-IgG, C3G10-IgM, C3G10-IgG, C10H10-IgM, C10H10-IgG, C11B10, C12G6 and CR8033-like antibodies, respectively, when non-neutralized viruses were excluded from the evaluation.
In vitro binding and HI and MN activities of chimeric bnAbs against influenza B. (A) Binding (EC50 ELISA values) of the indicated antibodies to representative purified viruses from the three influenza B lineages. EC50 values greater than 104 ng/mL (dashed line) were scored as negative. (B-C) Fifty percent inhibitory concentrations (IC50) of the indicated antibodies against 20 representative strains from the three influenza B lineages were determined from three independent HI (B) or neutralization assays (C), each plotted as a single symbol. Bars represent averages and standard errors. The viruses were selected to cover the major phylogenetic branches of influenza B viruses. Yamagata lineage strains are indicated in blue, Victoria lineage strains are indicated in red, Ancestral lineage strains are indicated in purple, and influenza A virus is indicated in black.
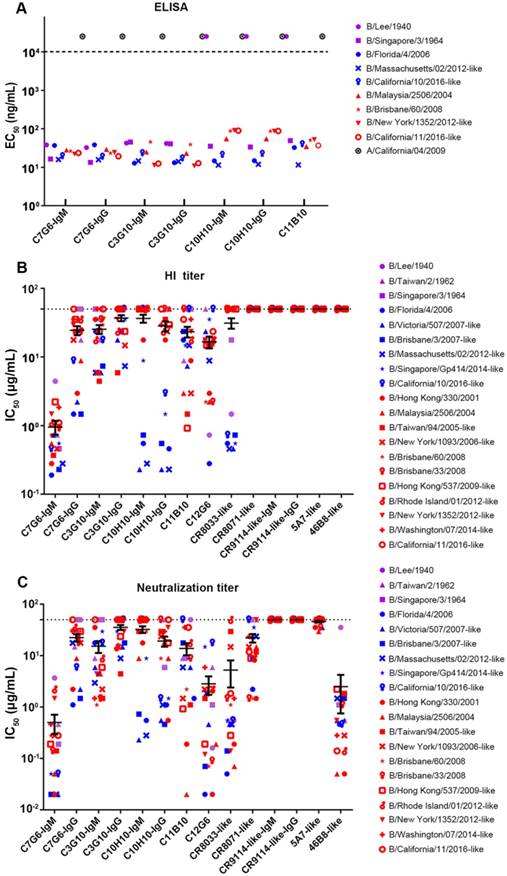
In the MN assay, all the antibodies except for CR9114-like-IgG and CR9114-like-IgM (a non-neutralizing antibody against influenza B) neutralized virus strains from the Yamagata and Victoria lineages, with differences in both potency and breadth of reactivity. C7G6-IgM, C12G6 and 46B8-like neutralized all 20 representative viruses, with a median IC50 of 0.50, 2.83 and 2.49 µg/mL, respectively. CR8033-like failed to neutralize the Victoria virus strain B/Rhode Island/01/2012-like, C11B10 failed to neutralize the Ancestral virus strain B/Lee/1940 and the Yamagata virus strain B/California/10/2016-like, C7G6-IgG and C3G10-IgM failed to neutralize two Ancestral virus strains and the Victoria virus isolated in 2016, while C10H10-IgG was unable to neutralize two Ancestral virus strains and two Victoria virus strains. The remaining antibodies, C3G10-IgG, C10H10-IgM, CR8071-like and 5A7-like were able to neutralize some virus strains from the Yamagata and Victoria lineages, but not Ancestral viruses (Figure 3C). Median IC50 values were 0.50, 17.40, 9.03, 17.52, 10.68, 11.47, 9.74, 2.83, 2.87, 12.82, 34.18 and 2.49 µg/mL for C7G6-IgM, C7G6-IgG, C3G10-IgM, C3G10-IgG, C10H10-IgM, C10H10-IgG, C11B10, C12G6, CR8033-like, CR8071-like, 5A7-like and 46B8-like antibodies, respectively, when non-neutralized viruses were excluded from the evaluation. Therefore, C7G6-IgM exhibits greater potency and broader breadth of HI and MN activity against influenza B viruses than its IgG variant C7G6-IgG and all the other influenza B bnAbs tested.
To explore the reason why C7G6-IgM possesses superior antiviral activity against influenza B viruses to its IgG variant, we directly compared the affinity of C7G6-IgM and C7G6-IgG for B/Florida/4/2006 HA. The affinity of C7G6-IgG to B/Florida/4/2006 HA is 24.67 nM, whereas the corresponding affinity of C7G6-IgM is 0.58 nM, which is approximately 42 times greater than that of C7G6-IgG (Figure S3). To further determine whether the polymeric form of C7G6 is important for its potency and breadth of viral neutralization, we constructed a monomeric form of C7G6-IgM and directly compared the in vitro neutralization activities and breadth of reactivity of the pentameric IgM, monomeric IgM and monomeric IgG forms of the C7G6 antibody. The result revealed that C7G6 pentameric IgM exhibits higher potency and breadth of neutralization against influenza B viruses than either of the two monomeric antibodies (Figure S4). These data demonstrated that the pentameric IgM form is responsible for the excellent antiviral activity of the C7G6 antibody.
C7G6-IgM demonstrates high-efficacy prophylactic and therapeutic activity in mice and ferrets
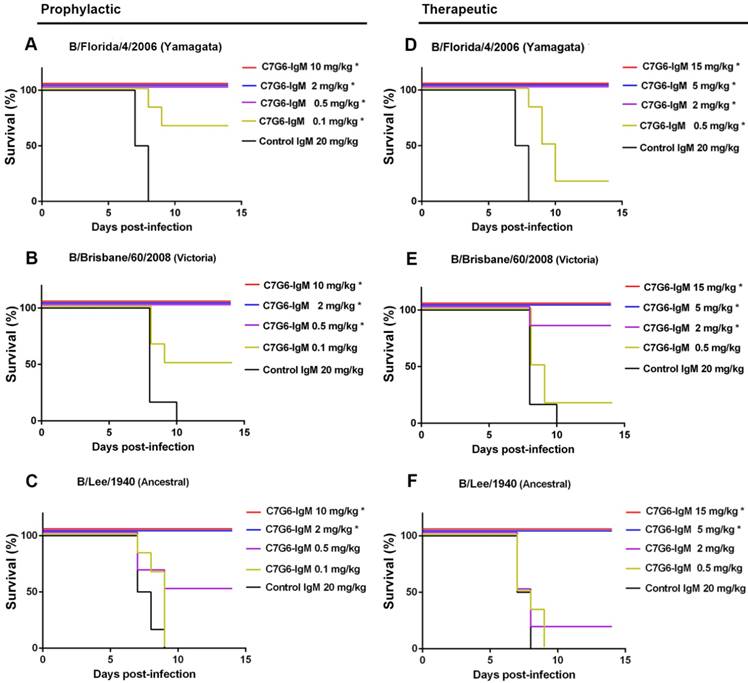
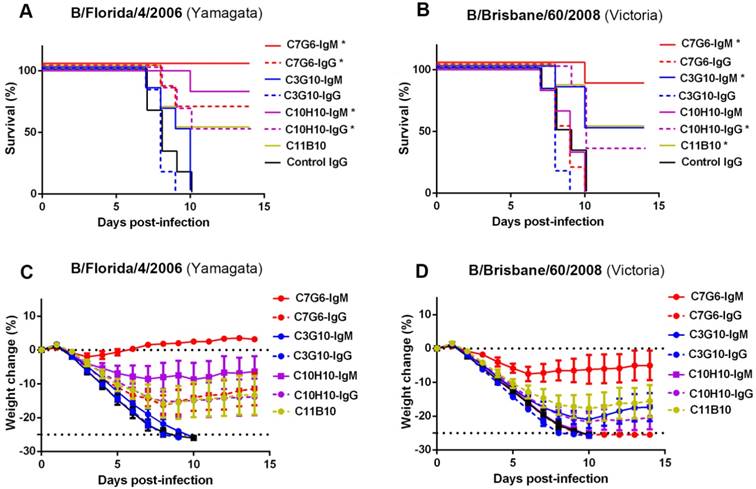
Following in vitro characterization of the HI and MN activities of the antibodies generated in this study against influenza B, a comprehensive evaluation of their capacities to cross-protect against virus strains representing the three distinct influenza B lineages in vivo was required. It has been reported that the half-lives of IgM molecules are far shorter than those of IgGs in vivo [26], so we first compared the half-lives of C7G6-IgM and C7G6-IgG in mice. The half-life of C7G6-IgG in serum is 4.500 days, and in contrast, the corresponding half-life of C7G6-IgM is 1.479 days, which is approximately three times shorter than that of C7G6-IgG (Figure S5). We next determined whether C7G6-IgM could provide efficient protection against challenge with distinct influenza B lineages in mice and ferrets, despite having such a short in vivo half-life. Three mouse-adapted (MA) influenza B viruses, MA-B/Lee/1940 (Ancestral lineage), MA-B/Florida/4/2006 (Yamagata lineage) and MA-B/Brisbane/60/2008 (Victoria lineage) were used to characterize these antibodies in vivo [21]. First, we evaluated the prophylactic and therapeutic antiviral activities of these antibodies against infection with influenza B viruses in the mouse model. For the prophylactic groups, 10 or 2 mg/kg of C7G6-IgM completely protected mice against infection with viruses from all three lineages (Figure 4A-C), with increased body weight being observed at the end of the study (Figure S6A-C). A 0.5 mg/kg dose of C7G6-IgM completely protected mice from lethal infection with Yamagata and Victoria lineage viruses and provided partial protection (50% survival) against the Ancestral virus, with moderate weight loss. Even 0.1 mg/kg of C7G6-IgM still partially protected mice against the Yamagata and Victoria viruses. In the therapeutic setting, C7G6-IgM doses greater than or equal to 5 mg/kg ensured survival of all mice infected with any of the three viruses (Figure 4D-F), with increased weight or only slight body weight loss at the end of the study (Figure S6D-F). The 2 and 0.5 mg/kg doses of C7G6-IgM also had a partial therapeutic effect against the three influenza B viruses in mice, with increased survival rates and reduced body weight loss compared to the control antibody groups. Consistent with the survival and body weight data, lung viral titers were considerably reduced in mice given C7G6-IgM, for all three virus infections, and both prophylactic and therapeutic protocols, when compared with the IgM control group (Figure S7). We also evaluated the therapeutic efficacy of C7G6-IgG, C3G10-IgM, C11H10-IgG and C11B10 in infections with the mouse-adapted Yamagata and Victoria viruses. Mice treated therapeutically with a single 10 mg/kg dose of any of these antibodies one day after infection all survived lethal challenge with either of the viruses, lost little, if any, body weight and exhibited lower lung viral titers compared to those of the control animals (Figure S8).
In vivo prophylactic and therapeutic efficacy of C7G6-IgM in mice. (A-C) Prophylactic efficacy of C7G6-IgM against lethal challenge with 25 MLD50 (50% mouse lethal dose) of MA-B/Florida/4/2006 (A), MA-B/Brisbane/60/2008 (B), or MA-B/Lee/1940 virus (C). The survival curves of BALB/c mice (n = 6 per group) treated with C7G6-IgM (10, 2, 0.5, or 0.1 mg/kg) or control IgM (20 mg/kg) 1 day before lethal challenge are shown. (D-F) For the therapeutic groups, survival curves for BALB/c mice (n = 6 per group) that received C7G6-IgM (15, 5, 2 or 0.5 mg/kg) or control IgM (20 mg/kg) 1 day after lethal challenge with 25 MLD50 of MA-B/Florida/4/2006 (D), MA-B/Brisbane/60/2008 (E), or MA-B/Lee/1940 (F) virus are shown. This experiment was repeated three times; one representative dataset is shown. The log-rank test was used to assess the significance (*P < 0.05) of survival outcome. The control IgM is C5G6-IgM (a mAb against the 2009 pandemic H1N1 influenza A virus).
To extend the evaluation, we directly compared the in vivo therapeutic efficacy of C7G6-IgM in mice with that of its IgG variant C7G6-IgG and other bnAbs we have generated. All mice receiving C7G6-IgM (1 mg/kg) survived lethal challenge with the Yamagata lineage strain. In contrast, 66.7, 0, 0, 83.3, 50, 50 or 0% of mice survived infection with the Yamagata virus strain MA-B/Florida/4/2006 1 day before receiving a 1 mg/kg dose of C7G6-IgG, C3G10-IgM, C3G10-IgG, C10H10-IgM, C10H10-IgG, C11B10 or control IgG antibody, respectively (Figure 5A). For the Victoria strain MA-B/Brisbane/60/2008, the same doses of C7G6-IgM, C7G6-IgG, C3G10-IgM, C3G10-IgG, C10H10-IgM, C10H10-IgG, C11B10 or C5G6 antibody protected 83.3, 0, 50, 0, 0, 33.3, 50 or 0% of mice, respectively (Figure 5B). Reduced weight loss in C7G6-IgM-treated mice also reflected the better protective potency of C7G6-IgM when compared to C7G6-IgG and the other influenza B-specific bnAbs (Figure 5C-D).
Comparison of therapeutic efficacies of C7G6-IgM and other bnAbs in mice. Survival curves (A-B) and body weight change (C-D) for BALB/c mice (n = 6 per group) treated intraperitoneally with the indicated antibodies (1 mg/kg) 24 h after lethal challenge with 25 MLD50 of MA-B/Florida/4/2006 or B/Brisbane/60/2008. This experiment was repeated three times; one representative dataset is shown. The body weight curves represent mean ± 95% confidence intervals of the mean. For (A-B), statistical analysis was performed by log-rank test (*P < 0.05, compared to the control IgG-treated group). The control IgG is C5G6.
In view of the potent and broad antiviral effects of C7G6-IgM in vitro and in vivo, C7G6-IgM was further investigated to determine its therapeutic potential for future clinical applications. Oseltamivir is a widely used antiviral drug recommended by the United States' Centers for Disease Control and Prevention (CDC) for the prevention and treatment of influenza infection [27]. Despite the efficacy of oseltamivir being limited, especially when treatment is delayed, severely ill influenza patients are generally given oseltamivir upon hospital admission. We thus determined the efficacy of C7G6-IgM in comparison to and in combination with the anti-influenza drug oseltamivir in mice. For the comparison experiment, mice were treated with either a single dose of 15 mg/kg C7G6-IgM or a four-day course of twice-daily doses of 25 mg/kg oseltamivir, with dosing regimens beginning at day 1, 3 or 5 after infection with mouse-adapted B/Florida/4/2006 or B/Brisbane/60/2008 virus. When administered 1 day after infection, C7G6-IgM treatment provided complete protection against both virus lineage strains, with mice gaining body weight, whereas oseltamivir only partially protected animals, with significant weight loss and survival rates of 83 and 67% for B/Florida/4/2006 and B/Brisbane/60/2008 infections, respectively (Figure 6A-B and Figure S9A-B). Even when administration of C7G6-IgM was delayed until 3 days after lethal challenge with either lineage influenza B virus, survival rates of 83% were achieved. Inspiringly, beginning treatment with C7G6-IgM at 5 days after infection still permitted more than 30% of mice to survive lethal challenge with either of the influenza B lineage strains. In contrast, all mice starting treatment with oseltamivir at 3 or 5 days post-infection died within 10 days of infection (Figure 6A-B and Figure S9A-B).
Efficacy of C7G6-IgM compared to and in combination with oseltamivir in mice. (A-B) Kaplan-Meier survival curves of mice that received a single dose of C7G6-IgM (15 mg/kg) (closed symbols), a four-day course of oseltamivir (25 mg/kg, two doses a day) (open symbols), or control IgM (25 mg/kg) (no symbols) on the indicated day after intranasal infection with 25 MLD50 of MA-B/Florida/4/2006 (A) or MA-B/Brisbane/60/2008 (B). d.p.i: days post-infection. (C-F) Survival curves (C-D) and lung viral titers (E-F) of BALB/c mice (n = 6 per group) that received a single treatment with C7G6-IgM or a control IgM intraperitoneally at 5 mg/kg, oseltamivir orally at 25 mg/kg twice a day for 4 days, or a combined treatment of C7G6-IgM and oseltamivir, starting from 2 days after intranasal infection with 25 MLD50 of MA-B/Florida/4/2006 or MA-B/Brisbane/60/2008. The virus titers in lungs were determined 4 days after infection. The results are representative of three independent experiments; one representative dataset is shown. The black bars indicate mean values and error bars represent SE. Statistically significant differences in survival outcome were estimated with the log-rank test. The t-test was used to assess the significance of lung viral titers. *P < 0.05 and ***P < 0.001, compared to the control IgM-treated group. The control IgM is C5G6-IgM.
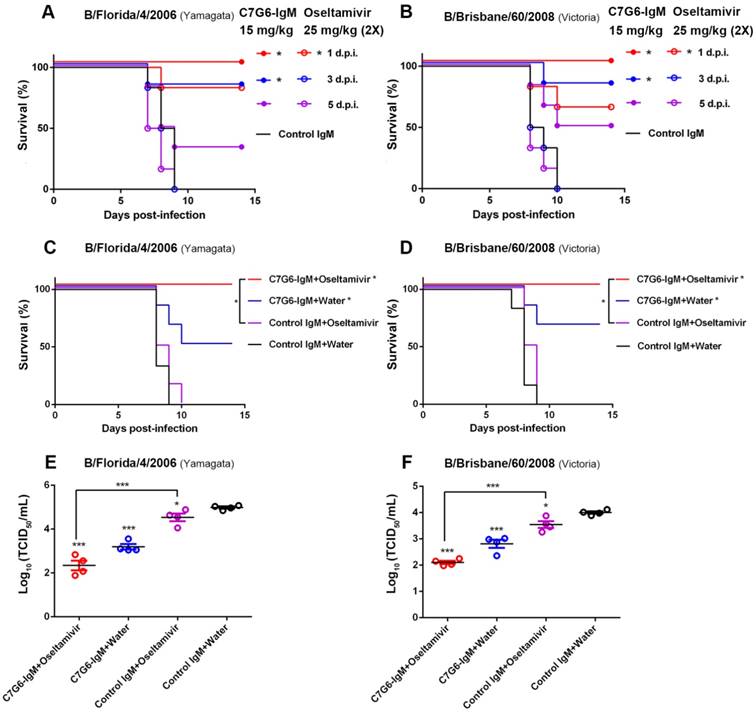
For the antibody-drug combination study, mice were administered C7G6-IgM and oseltamivir, either jointly or alone. Mice infected with a lethal dose of MA-B/Florida/4/2006 or MA-B/Brisbane/60/2008 were treated, starting at 2 days after infection, with a single 5 mg/kg dose of C7G6-IgM, 25 mg/kg oseltamivir twice a day for 4 days, or a combination of these two therapies. Co-administration of C7G6-IgM with oseltamivir completely protected mice lethally challenged with either virus strain, with reduced weight loss, compared to treatment with either active agent alone (Figure 6C-D and Figure S9C-D). In contrast, mice receiving oseltamivir plus control antibody exhibited 100% mortality within 10 days of lethal challenge for both lineages of influenza B virus, similar to the control group. Administration of C7G6-IgM plus water (to mimic the oseltamivir dosing regimen) only partially protected animals, with survival rates of 50 and 67% for the Yamagata and Victoria strains, respectively (Figure 6C-D). At the same time, consistent with the survival and body weight data, mice receiving the combined treatment demonstrated lower lung viral titers at day 4 after infection, compared to those treated with C7G6-IgM or oseltamivir alone (Figure 6E-F).
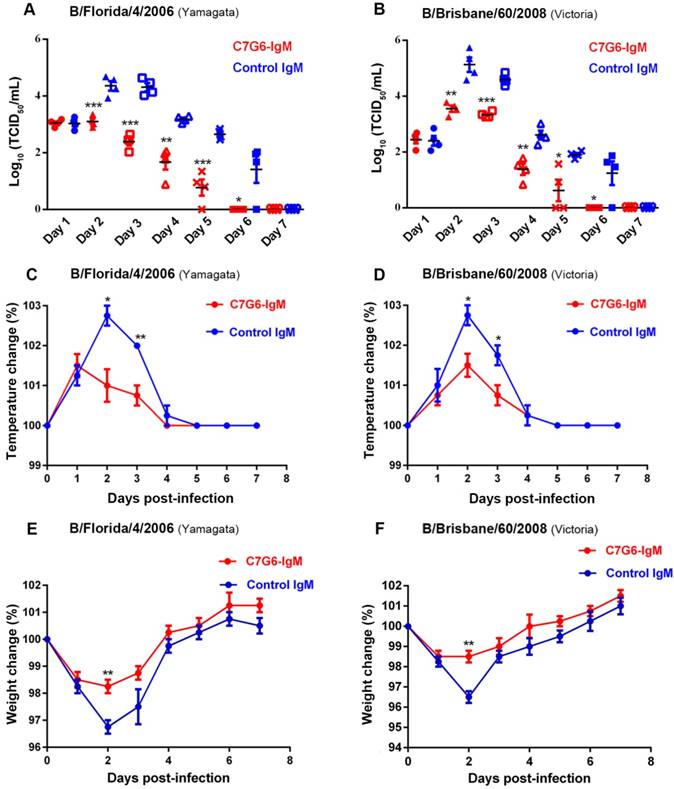
Finally, we determined the therapeutic efficacy of C7G6-IgM for treating ferrets infected with MA-B/Florida/4/2006 or MA-B/Brisbane/60/2008. As expected, C7G6-IgM exhibited considerable antiviral efficacy against challenge with both lineages of influenza B viruses in ferrets (Figure 7). The nasal wash viral titers of ferrets treated with C7G6-IgM were significantly lower than those of ferrets treated with control antibody for both the virus infections (Figure 7A-B). Similar to the nasal wash data, C7G6-IgM treatment resulted in fever reduction after infection with either of the two viruses, compared with control antibody-treated animals (Figure 7C-D). Moreover, infected animals administered C7G6-IgM experienced only slight body weight loss during both types of infection (Figure 7E-F). In contrast, significant body weight loss was observed among control antibody-treated animals (Figure 7E-F).
Therapeutic efficacy of C7G6-IgM in ferrets. (A-B) Virus titers in nasal washes from ferrets (n = 4 per group) treated with 20 mg/kg C7G6-IgM or control IgM (C5G6-IgM) 1 day after intranasal infection with 1×107 TCID50 of MA-B/Florida/4/2006 (A) or MA-B/Brisbane/60/2008 (B). Nasal washes were collected on the indicated days and titrated by TCID50 assay. (C-D) Changes in body temperatures of ferrets treated with 20 mg/kg C7G6-IgM or control IgM (C5G6-IgM) 1 day after intranasal infection with 1×107 TCID50 of MA-B/Florida/4/2006 (C) or MA-B/Brisbane/60/2008 (D). (E-F) Body weight change curves of ferrets treated with 20 mg/kg C7G6-IgM or control IgM (C5G6-IgM) 1 day after intranasal infection with 1×107 TCID50 of MA-B/Florida/4/2006 (E) or MA-B/Brisbane/60/2008 (F). This experiment was repeated three times; one representative dataset is shown. Body temperatures and body weight change are expressed as the percentage of baseline values. The black bars indicate mean values and error bars represent SE. Statistical analysis was performed by t-test. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to the control IgM-treated group.
C7G6-IgM, C3G10-IgM, C11B10 and C10H10-IgG recognize epitopes that overlap with the RBS domain of influenza B HA
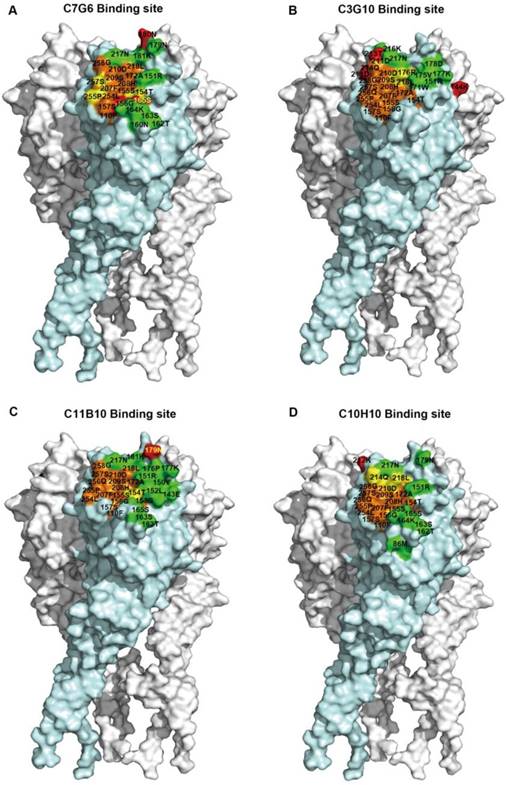
Given the cross-lineage HI and MN activities of the antibodies generated in this study, these four bnAbs may recognize highly conserved epitopes near the RBS domain of the influenza B virus HA head. To determine the specific epitopes recognized by C7G6-IgM, C3G10-IgM, C11B10 and C10H10-IgG, we attempted to generate escape mutants of eight influenza B viruses: B/Taiwan/2/1962, B/Singapore/3/1964, B/Florida/4/2006, B/Taiwan/94/2005-like, B/Victoria/507/2007-like, B/Hong Kong/537/2009-like, B/Massachusetts/02/2012-like and B/Brisbane/60/2008. Although these antibodies most likely bind to the more variable HA head domain, only a small number of escape variants were generated from three independent escape mutant induction experiments, all with mutations in the HA. For C7G6-IgM, three escape mutants of the B/Taiwan/2/1962 virus (G156R, K165E, N180T) were generated; for C3G10-IgM, three escape mutants of the B/Taiwan/94/2005-like virus (N144D, N211S, T213I) were generated; for C11B10, one escape mutant of B/Hong Kong/537/2009-like (N179K) was generated; and for C10H10-IgG, one escape mutant of B/Singapore/3/1964 (E212T) was generated (Table 1). G156 is a highly conserved residue (99.4%) among the 2,000 full-length influenza B HA sequences from the National Center for Biotechnology Information (NCBI) database (Table 2). The E165 variant occurs in only 0.1% of influenza B virus strains. All of the other strains (99.9%) have K165, I165, N165 or S165 and can be neutralized by C7G6-IgM (Figure 3C). The residue at position 180 in the HA of all influenza B viruses is either N or Y; such virus strains are sensitive to neutralization by C7G6-IgM (Figure 3C). For C3G10-IgM, K144 exists in 58.2% of influenza B virus strains. In addition, 37.2% of strains have N144 and are also sensitive to this antibody. N211 and T213 are relatively conserved (94.9% and 90.1%, respectively) among the 2,000 HA sequences examined. The N179K and E212T mutations have not been observed in any naturally arising influenza B isolates. All of the substituted residues are located in or near the HA receptor-binding pocket of influenza B [28]. We also evaluated the changes in binding activities of C7G6-IgM, C3G10-IgM, C11B10 and C10H10-IgG to their corresponding escape mutants using an ELISA assay. Interestingly, C7G6-IgM bound the three escape mutants of the B/Taiwan/2/1962 virus (G156R, K165E and N180T) similarly to the wild type (WT) virus (Figure S10A), while C3G10-IgM, C11B10 and C10H10-IgG showed reduced binding activities to their corresponding escape mutants when compared to the WT virus (Figure S10B-D).
Amino acid substitutions found in the HA of C7G6-IgM, C3G10-IgM, C11B10 and C10H10-IgG-induced escape mutants.
| Antibody | Influenza B virus | Residue mutations in the HA induced with the antibody |
|---|---|---|
| C7G6-IgM | B/Taiwan/2/1962 (Ancestral) | G156R,K165E,N180T |
| C3G10-IgM | B/Taiwan/94/2005-like (Victoria) | N144D,N211S,T213I |
| C11B10 | B/Hong Kong/537/2009-like (Victoria) | N179K |
| C10H10-IgG | B/Singapore/3/1964 (Ancestral) | E212T |
Frequency of potential C7G6-IgM, C3G10-IgM, C11B10 or C10H10-IgG interacting residues.
| Antibody | Residue | Frequency (%) | Total (%) |
|---|---|---|---|
| C7G6-IgM | G156 | 99.4 | 99.4 |
| N165 | 52.3 | 99.9 | |
| S165 | 30.2 | ||
| I165 | 17.1 | ||
| K165 | 0.3 | ||
| N180 | 81.5 | 100 | |
| Y180 | 18.5 | ||
| C3G10-IgM | K144 | 58.2 | 95.4 |
| N144 | 37.2 | ||
| N211 | 94.9 | 96.7 | |
| D211 | 1.8 | ||
| T213 | 90.1 | 90.1 | |
| C11B10 | N179 | 81.5 | 100 |
| Y179 | 18.5 | ||
| C10H10-IgG | E212 | 52.0 | 100 |
| K212 | 48.0 |
A multiple sequence alignment of 2000 full-length influenza B HA sequences from the NCBI database was used to assess the genetic diversity and to calculate the frequencies of potential C7G6-IgM, C3G10-IgM, C11B10 and C10H10-IgG interacting residues. All of the residues listed were found in the HA proteins of influenza B viruses, all of which can be neutralized by the indicated antibodies.
We next identified the epitopes using a molecular docking method. The candidate epitope residues were determined based on three-dimensional HA trimer models. We first compared the predicted CR8033 epitope obtained using this molecular docking method with the published CR8033 epitope to evaluate the accuracy of this method; the result showed that most of the predicted contact residues were accordant with the corresponding published contact residues (Figure S11). For the epitopes of the antibodies generated in this study, the results indicated that the epitopes of the four antibodies were at the top of the HA head and overlapped with the RBS region, with the four epitopes being distinct, but overlapping each other to a certain degree (Figure 8A-D). We also analyzed the antigenic conservation of these epitopes; at least half of the amino acids within the epitopes of C7G6, C3G10 and C11B10 are more than 97% conserved (Figure S12A-C). Although some amino acids forming the epitopes of these four antibodies are less than 90% conserved, C7G6-IgM, C3G10-IgM, C11B10 and C10H10-IgG can neutralize the vast majority of virus strains with these distinct mutations (Figure S13). Finally, we conducted a competition ELISA assay to further confirm the epitope domain recognized by each antibody; the results showed that each antibody overlapped with the RBS-targeting CR8033-like antibody epitope, competing for binding to HA with the CR8033-like antibody but not competing for binding with the CR9114-like antibody (Figure S14).
Epitope mapping of C7G6, C3G10, C11B10 and C10H10. (A-D) Surface representation illustrations of the neutralizing epitopes recognized by C7G6 (A), C3G10 (B), C11B10 (C) and C10H10 (D) on the HA trimer model of B/Florida/4/2006, generated using DS Visualizer 1.7 (version 1.7, Accelrys, Inc.). The structure was constructed via homology modeling, as described previously [18]. The epitope was determined using a molecular docking strategy. Red, amino acid substitutions; green, contact residues unique to the antibody epitope; orange, contact residues unique to RBS; yellow, common contact residues of the antibody epitope and the RBS. Residue numbers are shown; contact residues of the epitope with amino acid substitutions are in yellow and others are in black.
Neutralization mechanisms of C7G6-IgM, C3G10-IgM, C11B10 and C10H10-IgG
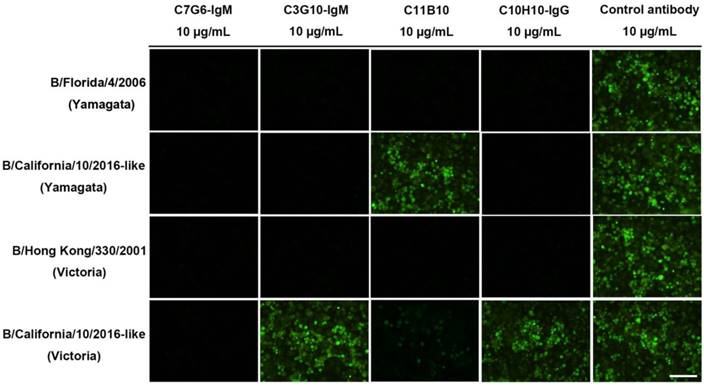
Broadly neutralizing influenza antibodies such as CR8033, which binds to the RBS domain in the HA of influenza virus, exhibit HI activity against Yamagata lineage strains and neutralize virus strains of this lineage through viral entry inhibition [18]. The C7G6-IgM, C3G10-IgM, C11B10 and C10H10-IgG antibodies exhibit broad HI activity against distinct lineages of influenza B viruses, suggesting that these antibodies could directly prevent viruses from infecting cells. A comparative study of the neutralizing mechanisms of these four antibodies was performed, using four influenza B virus strains: B/Florida/4/2006 (Yamagata), B/California/10/2016-like (Yamagata), B/Hong Kong/330/2001 (Victoria) and B/California/11/2016-like (Victoria). Consistent with the HI activities exerted by these antibodies against both influenza B lineages, C7G6-IgM strongly inhibited infection by all four viruses, C3G10-IgM and C10H10-IgG effectively prevented infections with three of the viruses (B/Florida/4/2006, B/California/10/2016-like and B/Hong Kong/330/2001), while C11B10 blocked all B/Florida/4/2006 and B/Hong Kong/330/2001 viruses and a portion of B/California/11/2016-like virus from attaching to cells (Figure 9). In contrast, the control antibody did not prevent cells from being infected by any of the viruses (Figure 9). It has been reported that some anti-HA head antibodies are able to neutralize influenza virus by inhibiting membrane fusion [19]. We performed a red blood cell (RBC) fusion assay to determine whether these antibodies inhibit membrane fusion in both lineages of influenza B viruses. Fusion was determined by measuring the release of NADPH through RBC lysis. We observed that C7G6-IgM, C3G10-IgM, C11B10 and C10H10-IgG antibodies inhibited lysis of Victoria lineage virus-infected RBCs to varying degrees, indicating that a portion of viral particles were prevented from fusing with RBC membranes, but that none of these antibodies inhibited lysis of RBCs infected with Yamagata lineage virus, whereas the positive control antibody C12G6 was able to inhibit the lysis of cells infected with either virus lineage (Figure S15).
Neutralization mechanisms of C7G6-IgM, C3G10-IgM, C11B10 and C10H10-IgG. MDCK cells were inoculated with B/Florida/4/2006 (Yamagata), B/California/10/2016-like (Yamagata), B/Hong Kong/330/2001 (Victoria) or B/California/11/2016-like (Victoria) virus preincubated with the indicated antibody. The expression of influenza B NP protein in MDCK cell monolayers 16-18 h after inoculation was detected by immunofluorescence using a monoclonal anti-NP primary antibody. Green, infected cells positive for NP protein; black, infected cells negative for NP protein. Scale bars = 100 μm. This experiment was repeated three times; one representative dataset is shown.
Discussion
BnAbs have been evaluated as promising agents in the fight against influenza and other infectious diseases. Four classes of influenza B bnAbs with high-efficacy antiviral activities have been developed in recent years; classified according to epitope distribution, these include the RBS-reactive antibodies, vestigial esterase domain-reactive antibodies, HA stem-reactive antibodies and NA head-reactive antibodies [18, 29-31]. Although all these antibodies can efficiently neutralize different lineages of influenza B viruses both in vitro and in vivo, only the RBS-reactive antibodies are able to directly inhibit viruses to prevent cell infection and have consequently been confirmed to confer stronger cross-protection against influenza B viruses than the other three classes of bnAbs, which exert antiviral effects only after infection has been initiated [18, 21]. Additionally, influenza B viruses evolve more slowly than influenza A viruses, and the receptor binding site domain is conserved among the three lineages of influenza B; indeed, representative RBS-reactive antibodies are able to neutralize various virus strains isolated since influenza B was first identified in 1940 [21]. Therefore, RBS-reactive bnAbs appear to be the most promising candidate out of the four classes of influenza B bnAbs for the development of novel high-efficacy therapeutics against influenza B infection. On the other hand, it will also be meaningful to develop the other three classes of bnAbs, as the HA vestigial esterase, HA stem and NA head domains are more conserved than the HA globular head domain; the development of various classes of bnAbs recognizing distinct epitopes will facilitate the formulation of broader-spectrum and more potent therapeutic antibody cocktails against influenza B virus infection. We and other groups have reported several RBS-targeted HA bnAbs that broadly cross-protect against influenza B viruses with great potency [18, 21]. However, these bnAbs still fail to neutralize some influenza B virus variants. Thus, it is highly desirable to find a new approach to identify and/or engineer bnAbs with even greater potency and breadth of recognition to counteract the rapidly evolving threat from influenza and other virus infections.
In this study, we generated and identified two IgM bnAbs (7G6-IgM and 3G10-IgM) and two IgG bnAbs (10H10-IgG and 11B10-IgG) using the murine hybridoma technique. We further switched the classes of these bnAbs to IgG and IgM, respectively, to understand the biological properties of IgG and IgM versions of the same antibody. We report here seven bnAbs exhibiting cross-lineage HI and MN activities against the RBS domain of influenza B viruses, designated C7G6-IgM, C7G6-IgG, C3G10-IgM, C3G10-IgG, C10H10-IgM, C10H10-IgG and C11B10. These antibodies effectively inhibit both Yamagata and Victoria lineage influenza B viruses in in vitro and in vivo experiments by blocking the attachment of these viruses to cells. One of these antibodies, C7G6-IgM, shows superior HI and MN activities to the other bnAbs, including its IgG and monomeric IgM variants, and is able to neutralize a panel of influenza B viruses representing strains isolated since 1940. C7G6-IgM blocks virus infection by preventing viral entry and confers stronger cross-protection against both of the currently circulating genetic lineages of influenza B viruses in mice and ferrets than its IgG variant and other antibodies in this study. Importantly, C7G6-IgM partially protected mice from lethal challenges of influenza B virus strains from both lineages, even when given at 5 days post infection, and a single treatment of C7G6-IgM confers stronger cross-protection against influenza B virus than oseltamivir. Interestingly, coadministration of C7G6-IgM and oseltamivir provided better protective efficacy than either treatment alone, most likely because they target distinct viral proteins and different steps of the viral life cycle. Because of its high therapeutic efficacy, C7G6-IgM is a promising candidate for the development of high-efficacy immunotherapy against influenza B.
We demonstrated that isotype switching from IgM to IgG had an impact on the ability to inhibit viral infection, and that the polymeric form of C7G6 is critical to its strong potency and broad breadth of viral neutralization. This was verified by data revealing that the pentameric form of C7G6-IgM has stronger affinity for influenza B HA than the monomeric form of C7G6-IgG. Thus, due to steric hindrance and high avidity, the large and bulky pentamers of IgM antibodies are likely to inhibit viral attachment and entry even more effectively and potently block virus infection. These results are consistent with the data generated during investigations into the use of the IgM antibody PAT-SM6 in various cancer treatments; the pentameric structure of this IgM antibody demonstrated high avidity and bound to targets much more strongly than monomeric IgGs [32]. The advantages of IgM antibodies in blocking viral infection may also inform the development of broad-spectrum prophylactics or therapeutics to utilize against other pathogenic microbes.
At present, most antibody-based therapeutics are IgGs. However, extensive research in the past years has led to the realization that IgM antibodies have potential as therapeutics; several monoclonal IgM antibodies have been developed as therapeutic agents and administered in clinical trials to collect evidence regarding efficacy and safety [33-36]. An IgM-based therapeutic product, Pentaglobin, is now available from Biotest [37]. Pentaglobin is unique in its ability to neutralize infectious pathogens, being far superior to normal IgG preparations; a result consistent with our data on C7G6-IgM. Although most IgM-based agents are still either in laboratory or clinical trials, the above studies clearly support the role of monoclonal IgMs as therapeutic agents. Despite the growing clinical promise, IgM antibodies have so far failed to be widely used as therapeutics because these antibodies are considered difficult to produce and purify. Due to the large size of these proteins, the production of sufficient quantities of functionally active recombinant IgMs is still a challenge. However, the availability of human therapeutic IgM is now becoming more feasible due to the implementation of recombinant expression systems like Per.C6 [38] and successful increases in fermentation scale [39], making IgM generation economically competitive with that of IgG antibodies. The Per.C6 cell derived IgM antibody PAT-SM6 has proved to be safe in humans, with no reported serious adverse events during a phase I study [40]. The half-lives of IgMs are short, ranging from approximately 1.76 h to 5 days in clinical studies [26], however, we found that C7G6-IgM confers potent cross-protection against both lineages of influenza B viruses in mice and ferrets even though it has a half-life of only 1.479 days. In conclusion, despite C7G6-IgM being of the IgM isotype, it will not be difficult to produce and purify this type of antibody, and the superior cross-lineage protection of C7G6-IgM ensures that it is a promising candidate for the development of novel anti-influenza B virus agents.
It is relatively difficult to develop bnAbs recognizing the RBS domain of influenza virus, due to the high variability in the HA head region. We have previously demonstrated that influenza B bnAbs can be generated when mice are sequentially immunized intranasally with live influenza B viruses. In this study, we developed four bnAbs from mice using the same immunization strategy, reconfirming that influenza B bnAbs can be generated from mice sequentially immunized with live B/Florida/4/2006 (Yamagata) and B/Brisbane/60/2008 (Victoria) viruses via the intranasal route. Another study, utilizing intramuscular immunization, reported that priming with Victoria and boosting with Yamagata induced more responses to both lineages than priming with Yamagata and boosting with Victoria [41]. It has been shown that the route of administration strongly influences both the quantity and quality of vaccine-induced immunity [42, 43]. Thus, a rational design for sequential and mucosal immunization strategies should be considered in generating bnAbs. For the first time, we demonstrated that antiviral polyclonal sera containing a large amount of IgM antibodies has cross-lineage HI and MN activities against IBVs. Hence, a more efficient way to screen bnAbs may be to select IgM, rather than IgG, subtype antibodies. The subsequent generation of two IgM subtype bnAbs, 7G6-IgM and 3G10-IgM, further confirmed our hypothesis that IgM bnAbs can have strong neutralizing ability. The superior antiviral effects of these two antibodies can probably be attributed to the properties inherent to the pentameric form of the IgM antibody, as the chimeric IgM forms of the two antibodies retain potent antiviral activity, whereas an obvious decrease in activity was observed for the chimeric IgG and monomeric IgM versions. These results demonstrate that specific IgM antibodies with multivalent fragments are effective in neutralizing pathogens. However, not all of the IgM antibodies exhibited stronger antiviral activity than their IgG variants, for example, the IgG variant of 10H10 antibody is superior to its IgM variant in terms of both HI and MN activity. Therefore, the ability of IgM antibodies to broadly and potently inhibit viruses might depend on which epitope they target. The strong broad-spectrum antiviral effects exerted by C7G6-IgM in this study suggest that there are specific epitopes near the RBS domain of IBVs which are suitable targets for IgM subtype bnAbs.
There are certain limitations associated with this study. Further confirmation of the protective potential of humanized versions of the antibodies generated in this study is necessary. Identification of the epitopes is important for understanding the distinct antiviral activities of the IgM and IgG variants of these antibodies, and the epitopes bound by the IgM and IgG subtype antibodies still need to be characterized precisely. The exact structure of the epitopes targeted by these antibodies will also provide guidance for the design of universal vaccines against influenza B.
Methods
Viruses and recombinant HA
The influenza virus strains B/Lee/1940, B/Taiwan/2/1962, B/Singapore/3/1964, B/Florida/4/2006, B/Hong Kong/330/2001, B/Malaysia/2506/2004, B/Brisbane/60/2008, B/Brisbane/33/2008 and A/California/04/2009 were kindly provided by BEI Resources. The virus strains B/Victoria/507/2007-like, B/Brisbane/3/2007-like, B/Massachusetts/02/2012-like, B/Singapore/Gp414/2014-like, B/California/10/2016-like, B/Taiwan/94/2005-like, B/New York/1093/2006-like, B/Hong Kong/537/2009-like, B/Rhode Island/01/2012-like, B/New York/1352/2012-like, B/Washington/07/2014-like and B/California/11/2016-like were from the collection at the First Affiliated Hospital of Xiamen University and the Xiamen International Travel Healthcare Center. The mouse-adapted strains MA-B/Florida/4/2006 (Yamagata), MA-B/Brisbane/60/2008 (Victoria) and MA-B/Lee/1940 (Ancestral) were generated in our laboratory through successive passaging in the lungs of mice; the MLD50 (50% mouse lethal dose) of MA-B/Florida/4/2006 is 3.60×103 TCID50, the MLD50 of B/Brisbane/60/2008 is 4.74×104 TCID50, the MLD50 of MA-B/Lee/1940 is 2.57×104 TCID50. All viruses were grown in Madin-Darby Canine Kidney (MDCK) cells using standard viral culturing techniques. Viruses used in ELISA assays were concentrated and purified via high-speed centrifugation of infected cell supernatants through a 30% sucrose cushion, as described previously [44]. The purified viruses were resuspended in phosphate-buffered saline (PBS) and stored at -80 °C until use. MDCK cells were maintained in Dulbecco Modified Eagle's Medium (DMEM) supplemented with 10% fetal calf serum (FCS). Recombinant HA proteins of B/Florida/4/2006 and B/Brisbane/60/2008 with no foldon domain were prepared using a baculovirus vector in insect cells, as described previously [45].
Production of mouse immune sera and monoclonal antibodies
For the serum collection and characterization, six-week-old female BALB/c mice were immunized intranasally on day 0 with live Yamagata lineage virus strain B/Florida/4/2006 at 1×104 TCID50 per mouse [46]. Sera were collected at 7 and 14 days after immunization. Sera were treated with Receptor Destroying Enzyme II (RDE II, Denka Seiken, Japan) before ELISA, HI and MN assays were performed, according to the manufacturer's instructions. For the generation of cross-reactive antibodies, six-week-old female BALB/c mice were sequentially immunized intranasally with B/Florida/4/2006 and B/Brisbane/60/2008 at days 0 and 14, respectively, at 1×104 TCID50 per mouse. 7 or 14 days after the last immunization, spleen cells were collected from immunized mice, fused with mouse myeloma Sp2/0 cells and maintained according to a standard procedure [25]. The hybridomas were screened for the secretion of influenza B virus-specific mAbs in HI and MN assays against both B/Florida/4/2006 (Yamagata) and B/Brisbane/60/2008 (Victoria) in parallel. Two IgM subtype cross-reactive mAbs (7G6-IgM and 3G10-IgM) from the first fusion detection and two IgG subtype cross-reactive mAbs (11B10-IgG and 10H10-IgG) from the second fusion detection were identified. The hybridomas producing the four antibodies were cloned three times via limiting dilution, then the IgG antibodies and IgM antibodies were purified from mouse ascites using protein A agarose columns (Bio-Rad, Hercules, USA) and protein L affinity chromatography (GE Healthcare, Marlborough, USA), respectively, performed according to the producer's instructions.
Cloning and sequence analysis
Total RNA from 107 hybridoma cells was extracted using a MiniBEST Universal RNA Extraction Kit (TaKaRa, Dalian, China). The extracted RNA was then subjected to a reverse transcription reaction with the following primers: 5'-CCGTTTGKATYTCCAGCTTGGTSCC-3' for reverse transcription of the light chain variable region gene and 5'-CGGTGACCGWGGTBCCTTGRCCCCA-3' for reverse transcription of the heavy chain variable region gene. The coding regions of the H- and L-chains of the antibodies were amplified by PCR using the following primers: 5'-ATGGACTCCAGGCTCAATTTAGTTTTCCT-3' (H-chain-forward) and 5'-CGGTGACCGWGGTBCCTTGRCCCCA-3' (H-chain-reverse); and 5'-ATGAAGTTGCCTGTTAGGCTGTTGGTGCT-3' (L-chain-forward) and 5'-CCGTTTGKATYTCCAGCTTGGTSCC-3' (L-chain-reverse). The PCR products were cloned into a pMD18-T vector and sent to the Shanghai Boya Company for sequencing. The sequences of the antibody variable regions were confirmed using BLAST alignment, and the corresponding amino acid sequences were determined. The RNA of influenza virus strains used in this study was extracted from samples using a QIAamp Viral RNA Mini Kit (Qiagen, Dusseldorf, Germany), according to manufacturer's instructions. The extracted viral RNA was subjected to one-step RT-PCR (QIAGEN One-Step RT-PCR Kit) with influenza B HA primer sets, as described previously [47]. The PCR products were gel purified using a Universal DNA Purification Kit (TianGen, Beijing, China) and were sent to the Shanghai Boya Company for sequencing.
Construction of chimeric antibodies
The construction of IgG chimeric antibodies containing human Fc fragments was performed as described previously [48]. Briefly, the variable gene regions of each antibody were inserted into a pTT5 vector containing the constant region of human IgG1 gamma-heavy or kappa-light chain. The recombinant antibodies were expressed in Chinese hamster ovary (CHO) cells by transient transfection and purified from culture media via MabSelect XtraTM affinity chromatography (Amersham GE Health, Boston, USA).
For the pentameric IgM chimeric antibodies, the pTT5-based human IgM expression vectors contain a murine immunoglobulin signal peptide, as described [49], and variable-gene cloning sites upstream of the human IgM heavy chain constant region or human kappa-light constant region were constructed. The IgM kappa-light chain and human immunoglobulin J chain cDNA (a gift from Dr. Jiahuai Han) were inserted into one pTT5 vector via a P2A (GSGATNFSLLKQAGDVEENPGP)-based bicistronic expression strategy; the light chain encoding vector was constructed without J chain cDNA to produce monomeric IgM. The IgM heavy chain constant region was amplified from the human IgM-secreting lymphoblastoid cell line, Daudi (ATCC® CCL-213™), with RT-PCR primers 5'-ATGGGGAGTGCATCCGCCCCAAC-3' (upper) and 5'-TCAGTAGCAGGTGCCAGCTG-3' (lower) and inserted into another pTT5 vector. The variable genes of each antibody were cloned into these vectors in frame with the signal peptide and constant region genes. The same heavy chain encoding plasmid was shared for expression of both pentameric and monomeric IgM antibodies. These vectors were then co-transfected into CHO cells for expression at a heavy to light chain ratio of 1:1.5. Both pentameric and monomeric IgM antibodies in supernatants were subsequently purified via protein L affinity chromatography. Detailed information on the monoclonal antibodies used in this study is listed in Table S1.
High performance size-exclusion chromatography (HPSEC)
The experiment was performed as described previously [50]. Purified human IgM (Luoyang Bai Aotong Experimental Materials Center, Luoyang, China), protein standards (440 kDa and 158 kDa from GE Healthcare), and purified IgM subtype and IgG subtype antibodies were subjected to HPLC (Agilent Technologies 1200 series, Beijing, China) through a TSK Gel G3000 PWXL 7.8×300 mm column (TOSOH, Shanghai, China) equilibrated in phosphate buffer (pH 6.5) with 0.5 M NaCl. The column flow rate was maintained at 0.5 mL/min, and proteins in the eluates were detected at 280 nm.
ELISA
For the determination of ELISA EC50 (50% effective concentration) values of antibodies against influenza B virus strains, microtiter plates (Wantai BioPharm, Beijing, China) were coated overnight at 4 °C with 5 μg/mL purified viruses or 2 μg/mL recombinant HA proteins (50 μL per well). The plates were washed thrice with PBS containing 0.1% v/v Tween-20 (PBST) and blocked with blocking solution (phosphate-buffered saline with 2% sucrose, 0.2% casein-Na, and 2% gelatin) for 2 h at 37 °C. The plates were then washed with PBST. Serial 2-fold dilutions of sera or purified antibody were added to the wells and incubated at 37 °C for 30 min. The antibodies tested in this assay were the mouse antibodies 7G6-IgM, 3G10-IgM, 11B10-IgG and 10H10-IgG, and the chimeric antibodies C7G6-IgM, C7G6-IgG, C3G10-IgM, C3G10-IgG, C10H10-IgM, C10H10-IgG and C11B10. After three washes, 100 μL of horseradish peroxidase (HRP)-conjugated goat anti-mouse (or anti-human) antibody solution was added to each well and incubated at 37 °C for 30 min. After five washes, 100 μL of tetramethylbenzidine (TMB) substrate (Wantai BioPharm) was added at room temperature in the dark. After 15 min, the reaction was stopped with a 2 M H2SO4 solution. The absorbance was measured at 450 nm. All samples were run in triplicate. The relative affinity of antibody binding to purified viruses was determined by measuring the concentration of antibody required to achieve the EC50.
For competition ELISA, an additional preincubation with a 10-fold molar excess of unlabeled competitor antibodies was performed before adding the mAbs to the plate. Competition levels were calculated as the percentage inhibition of the half-maximal binding concentration of test antibody relative to the absorbance without added competitor [51]. For the determination of serum antibody responses, influenza virus-specific total IgG and IgM concentrations were determined as described previously [52]. Purified mouse IgG and IgM were used as standards to determine the relative antibody concentrations in immune sera from optical spectrophotometer readings at 450 nm.
Affinity measurement by bio-layer interferometry (BLI)
B/Florida/04/2006 HA protein was biotinylated using an EZ-Link™ Sulfo-NHS-LC-Biotin kit (Thermo Fisher Scientific, Waltham, MA, USA, Cat No: 21335). Octet system K2 (Part No: 30-5050), Streptavidin (SA) Biosensor (Part No: 18-5019) and kinetics buffer (Part No: 18-1105) were purchased from Pall Life Sciences (Menlo Park, CA, USA). Biotinylated FL/2006 HA proteins (30 µg/mL) were loaded onto SA biosensors for 180 s, following by soaking the biosensors in kinetics buffer for 10 s. Next, the loaded biosensors were exposed to a series of different dilutions of C7G6-IgG and C7G6-IgM antibodies (1.23-33.3 nM) for 90 s, followed by dissociation for 30 s. Background subtraction was used to correct for sensor drifting. All experiments were performed with shaking at 1,000 rpm. Background wavelength shifts were measured from reference biosensors that were loaded only with kinetics buffer. ForteBio's data analysis 9.0 software was used to fit the data to a 1:1 binding model to extract an association rate and dissociation rate. The KD was calculated using the ratio kdis(Koff)/kon [53].
HI assay
The HI assay was carried out as described previously [54], with slight modifications. Briefly, virus was diluted to 8 HA units and combined with an equal volume of serially diluted antibody or sera and incubated for 1 h at room temperature. An equal volume of 0.5% turkey red blood cells (TRBCs) was added to the wells, followed by another 1-h incubation at room temperature. The lowest concentration of mAb or sera that completely inhibited hemagglutination was designated the HI titer.
Micro-neutralization assay
Micro-neutralization (MN) assays were carried out as described previously [18]. Briefly, MDCK cells were maintained in DMEM supplemented with 10% FCS at 37 °C, 5% CO2. On the day of the experiment, MDCK cells in a 96-well format plate were washed twice with PBS and incubated in DMEM supplemented with 3 μg/mL TPCK-treated trypsin (Sigma-Aldrich, St Louis, MO, USA, Cat No: T1426). Serial 2-fold dilutions of mAb or sera were mixed with an equal volume of virus and incubated for 2 h at 37 °C. After incubation, 35 µL of the mixture, containing 100 TCID50 (50% tissue culture infectious dose) of virus, was then added to MDCK cells and incubated for 1 h. The viral supernatant was removed and replaced with DMEM plus antibiotics. The cells were cultured for 72 h at 37 °C in the presence of 5% CO2, and the neutralizing titer was determined using the HA test. For the HA test, 50 µL of 0.5% TRBCs was added to 50 µL of cell culture supernatant, and the mixture was incubated at room temperature for 1 h. The neutralization titer was the highest mAb or sera dilution that was negative for hemagglutination. The assay was performed in quadruplicate. IC50 values were determined using the Spearman-Kärber formula.
Evaluation of antibody half-lives in mice
Antibody half-lives were measured as described previously [21]. Briefly, 6- to 8-week-old female BALB/c mice were injected intravenously (i.v.) with purified antibody (C7G6-IgM or C7G6-IgG) at a concentration of 10 mg/kg. The mice (n=5/group) were bled on days 1, 4, 7, 11 and 14, and human IgM and IgG concentrations measured by ELISA. The t1/2 of the elimination phase was determined with a one-phase exponential decay model using data points between days 1 and 14 post-injection. GraphPad Prism software was used for this analysis.
Prophylactic and therapeutic efficacy studies in mice
All in vivo studies were performed in accordance with Institutional Animal Care and Use Committee guidelines and were approved by the Ethics Committee of Xiamen University Laboratory Animal Center. In a prophylactic setting, groups of 6 female BALB/c mice aged 6 to 8 weeks were injected intraperitoneally (i.p.) with 200 μL of antibody. One day later, the mice were deeply anaesthetized and challenged intranasally (i.n.) with 25 MLD50 of virus. In a therapeutic setting, the mice received the antibody 1 day after infection, or were administered with the antibody or oseltamivir at the indicated doses starting from 1, 3, or 5 days after infection. For oseltamivir comparison studies, mice received oseltamivir (25 mg/kg) orally twice daily for 4 days. For the co-administration study, the mice were administered with a single treatment of C7G6-IgM intraperitoneally at 5 mg/kg (with water administered orally to mimic the oseltamivir treatment), oseltamivir orally at 25 mg/kg twice a day for 4 days (accompanied by a single dose of control antibody at 5 mg/kg), or a combined treatment of C7G6-IgM and oseltamivir, starting at 2 days after infection. The animals were observed daily for mortality and morbidity, and body weight was measured for up to 14 days after infection. Animals that lost more than 25% of their initial body weight were euthanized in accordance with our animal study protocol. The lungs of an additional 4 mice per experimental condition were collected at 4 days after infection for virus titration.
Therapeutic efficacy studies in ferrets
The ferret experiments were carried out as described previously [21]. Fourteen-week-old male ferrets, certified by the supplier to be free of any evidence of infectious, contagious or communicable disease, were kept in the BSL2 laboratories for at least 14 days prior to the experiment for acclimatization. All ferrets were subcutaneously implanted with a microchip (implantable, programmable temperature transponder IPTT-300, Bio Medic Data Systems, Seaford, USA) between the shoulder blades, with subcutaneous body temperature measured by the microchip, and data reported by the transponder chip twice a day. Baseline body weight and temperature levels were measured for at least 7 days prior to the assay. Ferrets were anesthetized with isoflurane and received 500 μL of the virus per naris (1 mL in total) containing 1×107 TCID50. Ferrets were held upright with their head tilted slightly back for about 1 minute to avoid the likelihood of inoculum dripping from the nares. The ferrets were then returned to their home cage and observed for righting reflex. A day later, the ferrets were anesthetized with isoflurane and oxygen and injected i.p. with the antibody. Ferrets were observed twice daily, with nasal discharge, sneezing, activity level and weight of animals being measured on day 0, prior to infection, and each day thereafter. Nasal washes were collected daily for the first 7 days following infection for virus titration.
Selection of escape mutants
1×107 PFU of influenza B virus was incubated with an excess of antibody (100 μg/mL) for an hour at room temperature and the mixture was injected into 10-day-old embryonated chicken eggs. After 48-72 h of culture, the harvested allantoic fluid was plaqued in the presence of the antibody (10 μg/mL): viruses that grew in these conditions were collected and the complete HA sequence of these viruses determined.
Sequence analysis of antibody epitope conservation
A total of 2,000 full-length, non-redundant influenza B HA sequences were downloaded from the NCBI FLU database [55]. All 2,000 sequences were aligned and analyzed using BIOEDIT software. In this analysis, a residue was considered conserved only if it was the same between two sequences. The values reported for percent conservation are the number of sequences with an identical residue at a position divided by the total number of sequences.
Homology modeling and molecular docking
The B/Florida/4/2006 HA structure was constructed using Discovery Studio software, based on the crystal structure of B/Hong Kong/8/1973 (PDB ID: 3BT6). The 3D antibody model was determined by individually selecting the optimal framework and complementarity-determining region (CDR) template fragments from the crystal structures of known antibodies, based on sequence alignment. Through energy minimization, the CDR loops were grafted to a matched framework. The antibody and the HA were analyzed using ZDOCK in the ZDOCKpro module of the Insight II software package [56]. The antibody was used as the ligand, and the HA was used as the receptor. To save computing time, the HA2 regions and the constant regions of the antibody Fab were blocked. From the ZDOCK output file, the first 30 poses were selected for further RDOCK analysis [57]. Some escape mutants were used to narrow the range of choices. Based on the RDOCK scores and subjective judgment, a likely antibody-antigen complex structure was deduced.
Cell-based entry inhibition assay
The assay was performed as described previously [21]. Briefly, viruses were incubated with 10 μg/mL purified antibody for 1 h. After incubation, the mixture was added to MDCK cell monolayers cultured in 96-well format in infection medium. The inoculum was subsequently removed and replaced with medium supplemented with 10 μg/mL antibody, then cells cultured for 16-18 h at 37 °C in the presence of 5% CO2. The supernatants were then removed. The cells were fixed using 80% acetone and subjected to immunofluorescence analysis by labeling infected cells with a mouse monoclonal anti-NP primary antibody and an Alexa488-coupled anti-mouse secondary antibody (Invitrogen, Carlsbad, USA). The plates were further analyzed using an inverted fluorescence microscope.
Red blood cell fusion assay
The RBC fusion assay was performed as follows. Briefly, 100 μL of human red blood cells (2% final red cell concentration) were mixed with 100 μL of B/Florida/4/2006 or B/Brisbane/60/2008 virus (1×107 TCID50) and incubated on ice for 30 min. Antibodies were serially diluted and added to RBC/virus mixtures, then incubated for 30 min at room temperature. The samples were spun down at 3,000 ×g for 5 min and the supernatant discarded. 200 μL of a buffered solution (15 mM citric acid pH 5.0, 150 mM NaCl) was added and the mixture incubated at 37 ºC for 30 min. The samples were spun at 3,000 ×g and the supernatant harvested. Lysis of RBC was measured by the presence of NADPH (absorbance at OD 340 nm) in the supernatant.
Statistical analysis
The bars in this study represent the mean ± standard error of the mean (SEM). To establish significant differences for survival curves, we used the log-rank test in GraphPad Prism 6.0. To determine significant differences for virus titers, inhibition, body weight and body temperature changes, the t-test was used. Statistical analyses were performed with Prism software (GraphPad). Statistical significance is indicated, where tested, as follows: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Accession numbers
Nucleotide sequences for the variable regions of antibodies 7G6-IgM, 3G10-IgM, 11B10-IgG and 10H10-IgG and the HA genes of the viruses used in this study have been deposited in GenBank: MF033139 (7G6-IgM, VH), MF033138 (7G6-IgM, VL), MF033141 (3G10-IgM, VH), MF033140 (3G10-IgM, VL), MF136409 (11B10-IgG, VH), MF136410 (11B10-IgG, VL), MF136411 (10H10-IgG, VH), MF136412 (10H10-IgG, VL), KY848342 (B/Victoria/507/2007-like), KY848345 (B/Taiwan/94/2005-like), KY848346 (B/New York/1093/2006-like), KY848343 (B/Brisbane/3/2007-like), KY848347 (B/Hong Kong/537/2009-like), KY848348 (B/Rhode Island/01/2012-like), KY848349 (B/New York/1352/2012-like), KY848344 (B/Massachusetts/02/2012-like), KY848350 (B/Washington/07/2014-like), MF102256 (B/Singapore/Gp414/2014-like), KY273068 (B/California/11/2016-like), KY273076 (B/California/10/2016-like).
Abbreviations
bnAbs: broadly neutralizing antibodies; ELISA: enzyme-linked immunosorbent assay; HA: hemagglutinin; HI: hemagglutination inhibition; HPLC: high-performance liquid chromatography; IBV: influenza B virus; IC50: half maximal inhibitory concentration; IgG: Immunoglobulin G; IgM: immunoglobulin M; MA: mouse-adapted; MLD50: 50% mouse lethal dose; MN: micro-neutralization; RBC: red blood cell; RBS: receptor binding site; SEM: standard error of the mean; TCID50: median tissue culture infectious dose; TRBC: turkey red blood cell.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Shaojun Ke, Yanbin Wu and Qianyuan Han for their technical support. We are grateful to Dr. Jane Rayner for editing the manuscript. We thank NIAID Biodefense and Emerging Infections Research Resources Repository (BEI Resources) for providing some of the reagents used in this study. This work was funded by grants from the National Natural Science Foundation of China (31670934, 81871651), National Key Research and Development Program of China (2018YFC1200100), and Science and Technology Program of Xiamen, China (3502Z20154006).
Competing interests
YXC, CGS, JYC, GSW, and NSX are inventors on a patent application (PCT/CN2016/084496) submitted by Xiamen University that covers the generation and uses of the anti-Flu B bnAbs 7G6 and 11B10 reported in this paper. All other authors have declared that no competing interest exists.
References
1. Caini S, Huang QS, Ciblak MA, Kusznierz G, Owen R, Wangchuk S. et al. Epidemiological and virological characteristics of influenza B: results of the Global Influenza B Study. Influenza Other Respir Viruses. 2015;9(Suppl 1):3-12
2. Pan Y, Zhang Y, Yang P, Qian H, Shi W, Wu S. et al. Epidemiological and Phylogenetic Characteristics of Influenza B Infection in Severe Acute Respiratory Infection Cases in Beijing, 2014 to 2015. Medicine (Baltimore). 2015;94(52):e2399
3. Thompson WW, Shay DK, Weintraub E, Brammer L, Bridges CB, Cox NJ. et al. Influenza-associated hospitalizations in the United States. Jama. 2004;292(11):1333-40
4. Jennings L, Huang QS, Barr I, Lee PI, Kim WJ, Buchy P. et al. Literature review of the epidemiology of influenza B disease in 15 countries in the Asia-Pacific region. Influenza Other Respir Viruses. 2017
5. Paul Glezen W, Schmier JK, Kuehn CM, Ryan KJ, Oxford J. The burden of influenza B: a structured literature review. Am J Public Health. 2013;103(3):e43-51
6. Vijaykrishna D, Holmes EC, Joseph U, Fourment M, Su YC, Halpin R. et al. The contrasting phylodynamics of human influenza B viruses. Elife. 2015;4:e05055
7. Franco LM, Bucasas KL, Wells JM, Nino D, Wang X, Zapata GE. et al. Integrative genomic analysis of the human immune response to influenza vaccination. Elife. 2013;2:e00299
8. Wu X, Wu X, Sun Q, Zhang C, Yang S, Li L. et al. Progress of small molecular inhibitors in the development of anti-influenza virus agents. Theranostics. 2017;7(4):826-45
9. Harvala H, Smith D, Salvatierra K, Gunson R, von Wissmann B, Reynolds A. et al. Burden of influenza B virus infections in Scotland in 2012/13 and epidemiological investigations between 2000 and 2012. Euro Surveill. 2014:19 (37)
10. Heikkinen T, Ikonen N, Ziegler T. Impact of influenza B lineage-level mismatch between trivalent seasonal influenza vaccines and circulating viruses, 1999-2012. Clin Infect Dis. 2014;59(11):1519-24
11. Su S, Chaves SS, Perez A, D'Mello T, Kirley PD, Yousey-Hindes K. et al. Comparing clinical characteristics between hospitalized adults with laboratory-confirmed influenza A and B virus infection. Clin Infect Dis. 2014;59(2):252-5
12. Subbarao K, Matsuoka Y. The prospects and challenges of universal vaccines for influenza. Trends Microbiol. 2013;21(7):350-8
13. McClellan K, Perry CM. Oseltamivir: a review of its use in influenza. Drugs. 2001;61(2):263-83
14. Stiver G. The treatment of influenza with antiviral drugs. Cmaj. 2003;168(1):49-56
15. Kawai N, Ikematsu H, Iwaki N, Kawashima T, Maeda T, Mitsuoka S. et al. Longer virus shedding in influenza B than in influenza A among outpatients treated with oseltamivir. J Infect. 2007;55(3):267-72
16. Sugaya N, Mitamura K, Yamazaki M, Tamura D, Ichikawa M, Kimura K. et al. Lower clinical effectiveness of oseltamivir against influenza B contrasted with influenza A infection in children. Clin Infect Dis. 2007;44(2):197-202
17. Xu L, He D, Li Z, Zheng J, Yang L, Yu M. et al. Protection against lethal enterovirus 71 challenge in mice by a recombinant vaccine candidate containing a broadly cross-neutralizing epitope within the VP2 EF loop. Theranostics. 2014;4(5):498-513
18. Dreyfus C, Laursen NS, Kwaks T, Zuijdgeest D, Khayat R, Ekiert DC. et al. Highly conserved protective epitopes on influenza B viruses. Science. 2012;337(6100):1343-8
19. Yasugi M, Kubota-Koketsu R, Yamashita A, Kawashita N, Du A, Sasaki T. et al. Human monoclonal antibodies broadly neutralizing against influenza B virus. PLoS Pathog. 2013;9(2):e1003150
20. Chai N, Swem LR, Park S, Nakamura G, Chiang N, Estevez A. et al. A broadly protective therapeutic antibody against influenza B virus with two mechanisms of action. Nat Commun. 2017;8:14234
21. Shen C, Chen J, Li R, Zhang M, Wang G, Stegalkina S. et al. A multimechanistic antibody targeting the receptor binding site potently cross-protects against influenza B viruses. Sci Transl Med. 2017:9 (412)
22. Ochsenbein AF, Fehr T, Lutz C, Suter M, Brombacher F, Hengartner H. et al. Control of early viral and bacterial distribution and disease by natural antibodies. Science. 1999;286(5447):2156-9
23. Brekke OH, Sandlie I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat Rev Drug Discov. 2003;2(1):52-62
24. MacLennan IC, Gulbranson-Judge A, Toellner KM, Casamayor-Palleja M, Chan E, Sze DM. et al. The changing preference of T and B cells for partners as T-dependent antibody responses develop. Immunol Rev. 1997;156:53-66
25. Gui X, Ge P, Wang X, Yang K, Yu H, Zhao Q. et al. Identification of a highly conserved and surface exposed B-cell epitope on the nucleoprotein of influenza A virus. J Med Virol. 2014;86(6):995-1002
26. Ditzel H, Rasmussen JW, Erb K, Borup-Christensen P, Titlestad I, Lassen E. et al. Tumor detection with 131I-labeled human monoclonal antibody COU-1 in patients with suspected colorectal carcinoma. Cancer Res. 1993;53(24):5920-8
27. Gubareva LV, Kaiser L, Hayden FG. Influenza virus neuraminidase inhibitors. Lancet. 2000;355(9206):827-35
28. Velkov T. The specificity of the influenza B virus hemagglutinin receptor binding pocket: what does it bind to? J Mol Recognit. 2013;26(10):439-49
29. Wohlbold TJ, Podolsky KA, Chromikova V, Kirkpatrick E, Falconieri V, Meade P. et al. Broadly protective murine monoclonal antibodies against influenza B virus target highly conserved neuraminidase epitopes. Nat Microbiol. 2017;2(10):1415-24
30. Vigil A, Estelles A, Kauvar LM, Johnson SK, Tripp RA, Wittekind M. Native Human Monoclonal Antibodies with Potent Cross-Lineage Neutralization of Influenza B Viruses. Antimicrob Agents Chemother. 2018:62 (5)
31. Doyle TM, Li C, Bucher DJ, Hashem AM, Van Domselaar G, Wang J. et al. A monoclonal antibody targeting a highly conserved epitope in influenza B neuraminidase provides protection against drug resistant strains. Biochem Biophys Res Commun. 2013;441(1):226-9
32. Rasche L, Duell J, Castro IC, Dubljevic V, Chatterjee M, Knop S. et al. GRP78-directed immunotherapy in relapsed or refractory multiple myeloma - results from a phase 1 trial with the monoclonal immunoglobulin M antibody PAT-SM6. Haematologica. 2015;100(3):377-84
33. Gautam S, Loh KC. Immunoglobulin-M purification-challenges and perspectives. Biotechnol Adv. 2011;29(6):840-9
34. Irie RF, Ollila DW, O'Day S, Morton DL. Phase I pilot clinical trial of human IgM monoclonal antibody to ganglioside GM3 in patients with metastatic melanoma. Cancer Immunol Immunother. 2004;53(2):110-7
35. Mordoh J, Silva C, Albarellos M, Bravo AI, Kairiyama C. Phase I clinical trial in cancer patients of a new monoclonal antibody FC-2.15 reacting with tumor proliferating cells. J Immunother Emphasis Tumor Immunol. 1995;17(3):151-60
36. Macmillan ML, Couriel D, Weisdorf DJ, Schwab G, Havrilla N, Fleming TR. et al. A phase 2/3 multicenter randomized clinical trial of ABX-CBL versus ATG as secondary therapy for steroid-resistant acute graft-versus-host disease. Blood. 2007;109(6):2657-62
37. Alejandria MM, Lansang MA, Dans LF, Mantaring JB. Intravenous immunoglobulin for treating sepsis and septic shock. Cochrane Database Syst Rev. 2002 (1):Cd001090
38. Tchoudakova A, Hensel F, Murillo A, Eng B, Foley M, Smith L. et al. High level expression of functional human IgMs in human PER.C6 cells. MAbs. 2009;1(2):163-71
39. Valasek C, Cole J, Hensel F, Ye P, Conner MA, Ultee ME. Production and purification of a PER. C6-expressed IgM antibody therapeutic. BioProcess Int. 2011;9(11):28-37
40. Rasche L, Duell J, Morgner C, Chatterjee M, Hensel F, Rosenwald A. et al. The natural human IgM antibody PAT-SM6 induces apoptosis in primary human multiple myeloma cells by targeting heat shock protein GRP78. PLoS One. 2013;8(5):e63414
41. Skowronski DM, Hamelin ME, Janjua NZ, De Serres G, Gardy JL, Rheaume C. et al. Cross-lineage influenza B and heterologous influenza A antibody responses in vaccinated mice: immunologic interactions and B/Yamagata dominance. PLoS One. 2012;7(6):e38929
42. Maroof A, Yorgensen YM, Li Y, Evans JT. Intranasal vaccination promotes detrimental Th17-mediated immunity against influenza infection. PLoS Pathog. 2014;10(1):e1003875
43. Rose MA, Zielen S, Baumann U. Mucosal immunity and nasal influenza vaccination. Expert Rev Vaccines. 2012;11(5):595-607
44. Wang TT, Tan GS, Hai R, Pica N, Petersen E, Moran TM. et al. Broadly protective monoclonal antibodies against H3 influenza viruses following sequential immunization with different hemagglutinins. PLoS Pathog. 2010;6(2):e1000796
45. Lin Q, Li T, Chen Y, Lau SY, Wei M, Zhang Y. et al. Structural Basis for the Broad, Antibody-Mediated Neutralization of H5N1 Influenza Virus. Journal of virology. 2018:92 (17)
46. Muramatsu M, Yoshida R, Miyamoto H, Tomabechi D, Kajihara M, Maruyama J. et al. Heterosubtypic antiviral activity of hemagglutinin-specific antibodies induced by intranasal immunization with inactivated influenza viruses in mice. PLoS One. 2013;8(8):e71534
47. Tewawong N, Suwannakarn K, Prachayangprecha S, Korkong S, Vichiwattana P, Vongpunsawad S. et al. Molecular epidemiology and phylogenetic analyses of influenza B virus in Thailand during 2010 to 2014. PLoS One. 2015;10(1):e0116302
48. Zhang TY, Yuan Q, Zhao JH, Zhang YL, Yuan LZ, Lan Y. et al. Prolonged suppression of HBV in mice by a novel antibody that targets a unique epitope on hepatitis B surface antigen. Gut. 2016;65(4):658-71
49. Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R. et al. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc. 2009;4(3):372-84
50. Wang D, Fan F, Li Z, Liu X, Song S, Wei S. et al. Stop codon mutagenesis for homogenous expression of human papillomavirus L1 protein in Escherichia coli. Protein Expr Purif. 2017;133:110-20
51. Li GM, Chiu C, Wrammert J, McCausland M, Andrews SF, Zheng NY. et al. Pandemic H1N1 influenza vaccine induces a recall response in humans that favors broadly cross-reactive memory B cells. Proc Natl Acad Sci U S A. 2012;109(23):9047-52
52. O E, Lee YT, Ko EJ, Kim KH, Lee YN, Song JM. et al. Roles of major histocompatibility complex class II in inducing protective immune responses to influenza vaccination. J Virol. 2014;88(14):7764-75
53. Yang D, Singh A, Wu H, Kroe-Barrett R. Comparison of biosensor platforms in the evaluation of high affinity antibody-antigen binding kinetics. Anal Biochem. 2016;508:78-96
54. Andrews SF, Huang Y, Kaur K, Popova LI, Ho IY, Pauli NT. et al. Immune history profoundly affects broadly protective B cell responses to influenza. Sci Transl Med. 2015;7(316):316ra192
55. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605-12
56. Chen R, Weng Z. A novel shape complementarity scoring function for protein-protein docking. Proteins. 2003;51(3):397-408
57. Li L, Chen R, Weng Z. RDOCK: refinement of rigid-body protein docking predictions. Proteins. 2003;53(3):693-707
Author contact
![]() Corresponding author: Yixin Chen (yxchen2008edu.cn), National Institute of Diagnostics and Vaccine Development in Infectious Diseases, Xiamen University, Xiamen, 361102, China. Fax: +86-592-2181258.
Corresponding author: Yixin Chen (yxchen2008edu.cn), National Institute of Diagnostics and Vaccine Development in Infectious Diseases, Xiamen University, Xiamen, 361102, China. Fax: +86-592-2181258.