Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(2):436-448. doi:10.7150/thno.27576 This issue Cite
Research Paper
MSC-Delivered Soluble TRAIL and Paclitaxel as Novel Combinatory Treatment for Pancreatic Adenocarcinoma
Filippo Rossignoli1, Carlotta Spano1,2, Giulia Grisendi1,2, Elisabetta Manuela Foppiani1,2, Giulia Golinelli1, Ilenia Mastrolia1, Marco Bestagno3, Olivia Candini1,2, Tiziana Petrachi4, Alessandra Recchia5, Francesca Miselli5, Giulia Rovesti1, Giulia Orsi1, Elena Veronesi1,4, Gregorio Medici7, Benedetta Petocchi7, Massimo Pinelli8, Edwin M. Horwitz9,10, Pierfranco Conte11,12, Massimo Dominici1,2 ![]()
1. Division of Oncology, Department of Medical and Surgical Sciences for Children & Adults, University-Hospital of Modena and Reggio Emilia, Modena, Italy;
2. Rigenerand srl, Medolla, Modena, Italy;
3. International Centre for Genetic Engineering and Biotechnology, Trieste, Italy;
4. Technopole of Mirandola TPM, Mirandola, Modena;
5. Department of Life Sciences, University of Modena and Reggio Emilia, Modena, Italy;
6. Department of Diagnostic and Clinical Medicine and of Public Health, Institute of Pathology, University of Modena and Reggio Emilia, Modena, Italy;
7. Pharmacy Unit, University-Hospital of Modena and Reggio Emilia, Modena, Italy;
8. Division of Plastic and Reconstructive Surgery, University-Hospital of Modena and Reggio Emilia, Modena, Italy;
9. Aflac Cancer and Blood Disorders Center, Children's Healthcare of Atlanta, Atlanta, GA, USA;
10. Department of Pediatrics, Emory University, Atlanta, GA, USA;
11. Department of Surgery, Oncology and Gastroenterology, University of Padua, Padua, Italy;
12. Medical Oncology 2, Veneto Institute of Oncology IRCCS, Padua, Italy.
Received 2018-1-27; Accepted 2018-12-9; Published 2019-1-1
Abstract

Pancreatic cancer is the fourth leading cause of cancer death in western countries with more than 100,000 new cases per year in Europe and a mortality rate higher than 90%. In this scenario, advanced therapies based on gene therapies are emerging, thanks to a better understanding of tumour architecture and cancer cell alterations. We have demonstrated the efficacy of an innovative approach for pancreatic cancer based on mesenchymal stromal cells (MSC) genetically engineered to produce TNF-related Apoptosis Inducing Ligand (TRAIL). Here we investigated the combination of this MSC-based approach with the administration of a paclitaxel (PTX)-based chemotherapy to improve the potential of the treatment, also accounting for a possible resistance onset.
Methods: Starting from the BXPC3 cell line, we generated and profiled a TRAIL-resistant model of pancreatic cancer, testing the impact of the combined treatment in vitro with specific cytotoxicity and metabolic assays. We then challenged the rationale in a subcutaneous mouse model of pancreatic cancer, assessing its effect on tumour size accounting stromal and parenchymal organization.
Results: PTX was able to restore pancreatic cancer sensitivity to MSC-delivered TRAIL by reverting its pro-survival gene expression profile. The two compounds cooperate both in vitro and in vivo and the combined treatment resulted in an improved cytotoxicity on tumour cells.
Conclusion: In summary, this study uncovers the potential of a combinatory approach between MSC-delivered TRAIL and PTX, supporting the combination of cell-based products and conventional chemotherapeutics as a tool to improve the efficacy of the treatments, also addressing possible mechanisms of resistance.
Keywords: pancreatic cancer, combinatory, synergy, TRAIL, paclitaxel
Introduction
Pancreatic cancer is the fourth leading cause of cancer death in western countries characterized by more than 100,000 new cases per year in Europe and a mortality rate higher than 90% [1]. Although there has been some progress in the use of improved diagnostic methods and development of novel targeted therapies, the overall survival rate has not improved over the last decade with a 5-year survival rate settled around 8% [2,3]. Among the most important factors contributing to this inauspicious outcome, a pivotal role is played by late stage diagnosis, the abundant stromal component and a frequent relapse even after resection [4]. Moreover, intrinsic or acquired drug resistance often characterizes pancreatic cancer and its treatment remains a major challenge demanding novel, more effective therapies also accounting for the robust desmoplastic reaction that reduces drug bioavailability inside pancreatic cancer [5].
In the settings of locally advanced or metastatic pancreatic cancer, even in the presence of modest improvements in survival, newer standard treatments have become available, such as systemic chemotherapy with FOLFIRINOX or gemcitabine alone or in combination with the promising Nab-Paclitaxel [6,7]. In particular, Paclitaxel (PTX) is a microtubule- stabilizing agent belonging to the family of Taxanes. The potent suppression of microtubule dynamics ultimately leads to cell death [8,9]. Currently, PTX is administered in a Nanoparticle Albumin Bound (Nab) formulation, improving its solubility in water and also its bioavailability, thanks to the albumin-specific receptors on the cell membrane binding Nab-PTX particles and transporting them into the cell by transcytosis. Another key mechanism resulting in drug accumulation is the enhanced perfusion of Nab-PTX through endothelial gaps in tumour vasculature and its retention inside the stroma as key element to be additionally considered for more effective treatments [10,11].
Stromal cells in cancer are increasingly considered as targets of pre-clinical and clinical research. In this scenario, cell and gene therapies are emerging as tools to manipulate tumour microenvironment, thanks to a better understanding of the cross-talks between cancer cells and stromal elements [12]. Among non-immune gene-modified effectors, mesenchymal stromal/stem cells (MSC) have been studied for their possible use as tumour-specific delivery vehicles for therapeutic compounds, suicide genes and oncolytic viruses. Their tropism for tumours and metastases has been exploited to transform MSC by genetic manipulation, in bullets capable of delivering biological agents to the tumour site [13]. MSC have been engineered for the production of several compounds such as interleukins, interferons, pro-drug converting enzymes, oncolytic viruses, anti-miR, anti-angiogenic agents and pro-apoptotic molecules such as TNF-related Apoptosis Inducing Ligand (TRAIL) with pre-clinical and clinical promising results [13-16].
Our group sought to introduce MSC as delivery approach to selectively direct TRAIL variants against the tumour, empowering its anti-cancer activity and overcoming its poor bioavailability [17-19]. TRAIL-expressing gene-modified MSC can reach and engraft into tumour burden effectively delivering the anti-cancer agent. Their efficacy has also been demonstrated by others in in vivo models of lung, colon-rectal, breast, cervical, brain tumours with pro-apoptotic effects [20-24]. While this strategy has potential, there are several aspects that will need to be optimized, such as MSC homing, persistence and the possible resistance to MSC delivering a single drug, especially for cancers that are not particularly chemo-sensitive.
Recent findings suggest that successful treatments for pancreatic cancer may proceed through the combination of conventional chemo- and radio- therapy with cell and gene approaches, originating novel therapeutic concepts in order to achieve a greater efficacy and deal with resistance mechanisms [25,26]. In this perspective, we previously demonstrated in vitro that a combinatory treatment with MSC expressing TRAIL and bortezomib resulted in the sensitization of a TRAIL-refractory breast cancer cell line [27]. Moreover, other drugs such as valproic acid, anisomycin, doxorubicin, trifluorothymidine and decitabin revealed TRAIL-sensitizing properties in several tumour models [28]. However, there is little consensus on how different chemotherapeutics synergize with TRAIL signalling to mediate enhanced tumour cell death, with various studies showing upregulation of TRAIL receptors, downregulation of anti-apoptotic genes and enhanced grouping of TRAIL receptors on the membrane as being important for the combination effect. Moreover, sustained activation of the NFκB pathway can suppress TRAIL-mediated apoptosis through the upregulation of cFLIP, IAP and Bcl2 family proteins. Accordingly, agents that directly or indirectly suppress NFκB, cFLIP and IAP activity have been shown to strongly synergize with activators of the TRAIL pathway in vitro and in vivo [29]. Even if PTX mechanism of action has not been completely elucidated, it involves the downregulation of NFkB and Bcl2 [30,31] and has been successfully employed to sensitize gastric cancer cells to TRAIL [32]. For these reasons and accounting its role as a standard of care in pancreatic cancer, it can be considered as a possible combinatory player with TRAIL-delivering strategies.
Starting from an early study demonstrating the capacity of a MSC membrane bound form of TRAIL to target pancreatic cancer [19], relying on adipose-derived MSC delivering a novel soluble TRAIL variant [17,33] and additionally accounting the risk of TRAIL-resistant cancer, we originally combined a cell-based approach with the administration of PTX-based schedule. To explore the possible TRAIL-sensitizing role of Nab-PTX, the efficacy of the combined treatment was assessed in a laboratory-generated model of TRAIL-resistant pancreatic cancer both in vitro and in vivo.
Materials and Methods
Tissue culture
AD-MSC were obtained as previously described from lipoaspirate specimens of individuals undergoing liposuction for aesthetic purposes after approval by local Ethical Committee [27]. Cells were cultured in αMem (Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA) supplemented with 2.5% human platelet lysate (Modena Blood Bank, Policlinic of Modena, Modena, Italy), 1% L-glutamine (BioWhittaker, Lonza, Verviers, Belgium), 0.5% ciprofloxacin (Fresenius Kabi Italia S.r.l., Verona, Italy) and 1 IU/mL heparin (Sigma Aldrich, St. Louis, MO, USA). BXPC3 cells (Interlab Cell Line Collection, ICLC, Genova, Italy), instead, were maintained in RPMI (Gibco) integrated with 10% FBS, 1% L-glutamine and 1% penicillin/ streptomycin (all from Carlo Erba Reagents Srl). Authentication of BXPC3 cells was performed last year by Leibniz-Institut DSMZ GmbH (Braunschweig, Germany).
Establishment of sTRAIL-resistant BXPC3 cell lines
Supernatants conditioned by AD-MSC secreting soluble TRAIL (AD-MSC sTRAIL) were produced as previously reported [17,33]. Briefly, when AD-MSC sTRAIL reached almost 80% confluence, culture medium was replaced with BXPC3 maintenance medium. After 48 hours, conditioned supernatant was collected, filtered through 0.45 µm PES low binding protein filter (Corning Incorporated, NY, USA) and stored at -80°C until use. Conditioned medium from AD-MSC infected with an empty vector (AD-MSC EMPTY) was collected in the same way and used as control.
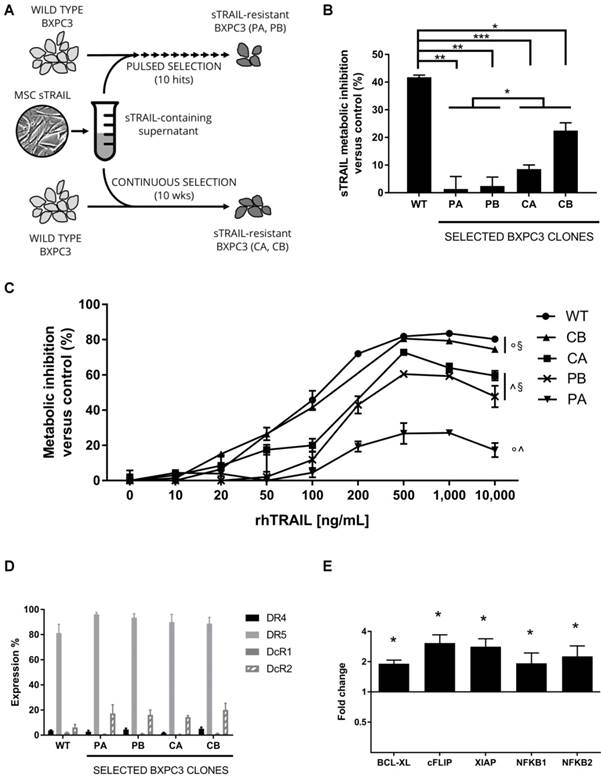
Development of sTRAIL-resistant BXPC3 cell lines has been pursued by two distinct approaches (Figure 1A). A pulsed strategy, resembling the chemotherapy cycles, consisted in exposing cultured BXPC3 to sTRAIL-containing supernatant for 24 h once weekly with surviving cells allowed to recover in complete medium until the subsequent hit. In an additional strategy, a continuous treatment was introduced and cells were constantly maintained in sTRAIL-containing supernatant. To quantify the resistance gain, cells were seeded at the established time points in 12-well multiwell plates at 6000 cells/cm2 and incubated on the following day with sTRAIL-containing supernatant for 24 h. Death rate was assessed by FACS following Propidium Iodide (PI) staining. Supernatant deriving from AD-MSC EMPTY was used as control. Wild type (WT) BXPC3 were evaluated for comparison. After selection, BXPC3 clones were characterized for resistance to a range of recombinant human TRAIL (rhTRAIL) concentrations by MTS, FACS, microarray analysis and qRT-PCR.
Generation of BXPC3 clones resistant to sTRAIL and rhTRAIL. (A) Representation of the selection strategy. Wild type BXPC3 were treated with sTRAIL- containing supernatant once a week for 10 weeks (pulsed selection), obtaining PA and PB clones, or constantly for 10 weeks (continuous selection), obtaining CA and CB clones. (B) Wild type BXPC3 and selected clones were tested for susceptibility to sTRAIL treatment by MTS assay. Each bar represents metabolic inhibition respect to untreated control. (C) rhTRAIL dose-response assay for wild type and selected BXPC3 clones in MTS. Cells were treated with a range of concentrations of rhTRAIL for 24 h. (D) Expression of TRAIL functional (DR4, DR5) and decoy (DcR1, DcR2) receptors by flow cytometry on wild type and selected BXPC3 clones. (E) qPCR analysis on PA clone for the expression of key pro-survival genes respect to BXPC3 wild type cells. Data are represented as mean±SD. All data are represented as the mean of two independent experiments performed in triplicate. WT: Wild type BXPC3; CA, CB, PA, PB: selected BXPC3 clones. * p<0.01, ** p<0.001, *** p<0.0001 with Student's T-test. § p<0.05, ^ p<0.0001, ° p<0.0001 by ANOVA and multiple comparison test.
ELISA
sTRAIL levels from AD-MSC supernatants were measured by Quantikine Human TRAIL/TNFSF10 kit (R&D Systems, Inc, Minneapolis, MN, USA) according to manufacturing instructions. At least 4 supernatants from different cell cultures were analysed in duplicate.
MTS metabolic assay
In the MTS assay (CellTiter 96 AQueous One Solution Cell Proliferation Assay, Promega Corporation, Madison, WI, USA), for each tested sample and condition, 5000 cells were seeded in a 96-wells plate. The following day, medium was replaced with sTRAIL-containing supernatant or AD-MSC EMPTY conditioned medium containing the appropriate concentration of rhTRAIL/Apo2 Ligand (rhTRAIL, Peprotech Inc., Rocky Hill, NJ, USA) (10, 20, 50, 100, 200, 500, 1000 and 10,000 ng/mL). After 24 h, the plate was analysed with Multiskan FC Microplate Photometer (Thermo Fisher Scientific Inc.) according to the manufacturer's recommendations.
FACS analyses
The expression of TRAIL functional receptors (DR4 and DR5) and decoy receptors (DcR1 and DcR2) was assessed by flow cytometry and compared with wild type BXPC3 cells. Briefly, cells were aliquoted in FACS analyses polypropylene tubes (VWR International, Radnor, PA, USA) and incubated in 100 μL blocking buffer composed of Dulbecco's Modified Eagle Medium (Gibco), 10% FBS, 0.1 M Sodium Azide and human immunoglobulin G (both from Sigma Aldrich) for 20 min on ice. After a washing step, cells were resuspended in PBS with 0.5% Bovine Serum Albumin (BSA, Sigma Aldrich) and stained on ice and in the dark for 30 min with the following monoclonal antibodies: PE-anti-DR4, APC-anti-DR5, PE-anti-DcR1 (all from BioLegend, San Diego, CA, USA), APC-anti-DcR2 (R&D Systems, Minneapolis, MN, USA). In all the experiments, the corresponding isotype-matched antibodies were used as negative controls (provided by BD Biosciences, San Diego CA, USA and BioLegend).
PI staining was done at the final concentration of 50 µg/mL (stock solution 1 mg/mL in PBS, Sigma Aldrich) and incubation proceeded at 37°C in the dark for 30 min. Cells and supernatants from each well were centrifuged for 10 min at 1400 rpm and resuspended in 100 µL of 1x Annexin V Buffer Solution containing APC-Annexin V antibody (both from BD Biosciences). Data were collected using a FACS Aria III flow cytometer (BD Biosciences) and analysed on FACS Diva software (BD Biosciences).
Microarray and qRT-PCR analyses
A whole transcriptome microarray analysis was performed by the facility of Consorzio Futuro in Ricerca (University of Ferrara, Ferrara, Italy) on the most TRAIL-resistant clone (defined as BXPC3 PA) in comparison with parental BXPC3 WT cells (Agilent SurePrint G3 Human Gene Expression v3 8x60K Microarray Kit #G4851C, Agilent Technologies, Palo Alto, CA). A subset of differentially expressed key genes involved in death ligand-induced apoptosis resistance was identified and included BCL-XL, cFLIP, XIAP, NFKB1 and NFKB2 genes. Differential expression of these genes was then analysed by qRT-PCR before and after PTX treatment (50 ng/mL for 24 h, Paclitaxel, Accord Healthcare Italia S.r.l., Milan, Italy). In detail, total cellular RNA was isolated from wild type and BXPC3 PA, using Trizol reagent (Invitrogen Corporation, Carlsbad, CA, USA) and quantified by spectrophotometry using a DU730 UV/VIS Spectrophotometer (Beckman Coulter, Brea, CA, USA). A 2 µg of template was reverse-transcribed into cDNA by the RevertAid H minus first-strand cDNA synthesis kit (Fermentas, Waltham, MA, USA) using Veriti 96-Well Thermal Cycler (Applied Biosystems, Foster City, CA, USA).
qPCR was performed with Fast SYBR Green Master Mix (Applied Biosystems) using Step One Real-Time PCR System Thermal Cycling Block (Applied Biosystems). The program ended with melt curve analysis to check specificity of amplification. Levels of mRNA for tested genes were compared using ∆∆CT method with β-Actin and GAPDH as reference genes. All primers were purchased from Integrated DNA Technologies (Coralville, IA, USA) and the sequences are shown in Table 1. Data have been analysed using StepOne software (version 2.3, Life Technologies Corporation, Carlsbad, CA, USA).
Primers sequences for qPCR
| Gene | Primers sequences |
|---|---|
| BCL-XL | FW: GTG GAA AGC GTA GAC AAG GAG |
| RV: CTG CAT TGT TCC CAT AGA GTT C | |
| cFLIP | FW: CCT CAC CTT GTT TCG GAC TAT AG |
| RV: TCC TTG CTT ATC TTG CCT CG | |
| XIAP | FW: CTT GGA CCG AGC CGA TC |
| RV: TGA TGT CTG CAG GTA CAC AAG | |
| NFKB1 | FW: GTG ACA GGA GAC GTG AAG ATG |
| RV: CAA GTT GAG AAT GAA GGT GGA TG | |
| NFKB2 | FW: GAA GCC AGT CAT CTC CCA G |
| RV: CAT CTT TCT GCA CCT TGT CAC |
PTX cytotoxicity and combinatory approach in vitro
Wild type BXPC3 and selected BXPC3 clones were tested in a dose-response assay for sensitivity to PTX treatment using MTS metabolic assay with the same protocol reported above. Tested concentrations were 0.1, 0.5, 1, 5, 10, 50, 100 and 500 ng/mL.
The BXPC3 PA clone was challenged in a combinatory treatment by both sTRAIL-containing supernatants and Paclitaxel. In detail, BXPC3 PA cells were seeded in a 12-wells multiwell plate at the density of 15000 cells/cm2 in complete medium. The next day, the medium was replaced with AD-MSC EMPTY-derived supernatant either with or without the addition of 5 ng/mL PTX. This concentration was chosen based on dose-response result which showed 50% metabolic inhibition at this dose. After 24 h of pre-treatment, wells were washed with PBS and the medium was replaced with sTRAIL-containing supernatant or AD-MSC EMPTY-derived supernatant, also adding PTX if present during pre-treatment. Four experimental conditions were thus obtained: AD-MSC EMPTY-derived conditioned medium (“EMPTY CM”), sTRAIL-containing conditioned medium (“sTRAIL CM”), AD-MSC EMPTY-derived conditioned medium plus PTX (“EMPTY CM+PTX”), sTRAIL-containing conditioned medium plus PTX (“sTRAIL CM+PTX”). After another 24 h incubation, death rate was assessed by PI and Annexin V double staining as reported above. In an alternative experimental setting without pre-treatment, cells were directly treated as in the four conditions stated above for 24 h and then stained with PI and Annexin V to assess mortality.
NFkB-inhibition mediated cytotoxicity and sTRAIL combinatory approach
BXPC3 PA clone was tested in a dose-response assay for sensitivity to a known NFkB-inhibitor, such as dehydroxymethylepoxyquinomicin (DHMEQ, MedchemExpress, NJ, USA). Treatment using MTS metabolic assay was performed with the same protocol reported above. Tested concentrations were 1, 5, 10, 15, 20, 25, 40, 50 µg/mL for 48 h. Moreover, BXPC3 PA cells were also challenged in a combinatory treatment by both sTRAIL-containing supernatants and 20 µg/mL DHMEQ with the same protocol described above for the combination with PTX.
In vivo studies
To further strengthen the rationale of the combined approach, an ectopic animal model of pancreatic cancer was established in nude mice. 28 female 6 weeks-old CD-1 nude mice from Charles River (Charles River Laboratories Italia SRL, Lecco, Italy) were housed at 12 h light, 12 h dark cycle with no restrictions on food and water supply. The animals were implanted subcutaneously with 2 million BXPC3 PA cells. Absence of mycoplasma contamination was tested on BXPC3 cells before in vivo use using the MycoAlert Mycoplasma Detection Kit (Lonza, Verviers, Belgium). One week after cell implantation, tumour mass was palpable sub cutis and the animals were randomly divided into 4 groups for treatment: the control group received no treatment; the “MSC sTRAIL” group received weekly peri-tumoral injection of 1 million AD-MSC sTRAIL in 200 μl PBS; the “Nab-PTX” group received weekly intra-peritoneal injection of 20 mg/kg PTX (corresponding to 200 mg/kg of Abraxane, Celgene Italia, Milan, Italy) in 200 μl PBS; the “MSC sTRAIL+Nab-PTX” group received Nab-PTX treatment followed by AD-MSC sTRAIL inoculum on the following day. Starting from day 1 post-injection, the health of the animals was monitored regularly, and weight and tumour size were measured weekly. Tumour volume was estimated according to the formula: Volume = (length · width2) / 2. Treatments were repeated four times, the animals were then euthanized, and tumour masses removed for histologic evaluation. All procedures were conducted in accordance with the institutional and national guidelines and under approved protocols by the Local Ethical Committee on Animal Experimentation and by the Italian Ministry of Health.
Histology
Formalin-fixed, paraffin-embedded tumour sections were evaluated by hematoxylin-and-eosin staining (Sigma Aldrich). For immunohistochemistry, sections were retrieved in citrate buffer (pH 6) for 15 min and incubated overnight at 4 °C with anti-cytokeratin 7 (CK-7) SP52 antibody (Ventana, Tucson, AZ, USA) or Cleaved Caspase-3 (Asp175) (5A1) antibody (Cell Signaling Technology, Danvers, MA, USA). Slides were then incubated with a biotinylated goat anti-rabbit IgG (H+L) (1:200; Vector Laboratories, Burlingame, CA) for 1h at room temperature. Negative controls were run simultaneously omitting primary antibody while incubating with buffer. Staining was performed and visualized by 3,3'-diaminobenzidine (DAB, Vector Laboratories). All slides were counterstained with Harris hematoxylin (Bio Optica, Milan, Italy).
Statistical analyses
Data have been analysed using Microsoft Excel 2010 (Microsoft Corporation, Redmond, WA, USA) and are expressed as mean values ±SEM unless otherwise noted. Unpaired 2-tailed Student's T-test was used considering p≤0.05 as statistically signifycant. Anova test was performed using Prism software (GraphPad Software Inc., La Jolla, CA, USA) considering p≤0.05 as statistically significant.
Results
Selected BXPC3 cell lines demonstrate a remarkable tolerance to TRAIL-mediated killing
To generate TRAIL resistant clones, parental BXBC3 WT cells were treated by sTRAIL-containing supernatants either continuously or intermittently for several weeks, as represented in Figure 1A. Supernatants, obtained from gene modified AD-MSC, contained an average of 407±97 pg/mL sTRAIL. After 10 hits of pulsed selection or 10 weeks of continuous treatment, selected BXPC3 clones (n=4: CA and CB for continuous selection, PA and PB for pulsed selection) were tested for resistance to sTRAIL in MTS assay compared with wild type BXPC3. All selected BXPC3 clones demonstrated between 2 and 30-fold increase in sTRAIL tolerance compared to wild type BXPC3. These differences were significantly marked in the pulsed selection group with the BXPC3 PA clone as the most resistant. Particularly, inhibition rate for wild type BXPC3 was 41.8±0.8% while for resistant clone PA it was 1.3±4.5%, 2.4±3.2% for PB, 8.6±1.5% for CA and 22.5±2.9% for CB (Figure 1B). Moreover, we performed a dose-response assay to verify whether the resistant BXPC3 cells could be additionnally cross-resistant to the recombinant form of TRAIL. All tested clones revealed an inhibition rate that reached a plateau at 500 ng/mL rhTRAIL (Figure 1C). Among resistant clones, again the BXPC3 PA clone showed the highest tolerance, with a maximum inhibition of 27.1±1.6%. On the contrary, CB clone had a maximum inhibition rate of 80.6±0.7%, very close to the 83.5±0.7% of wild type BXPC3. CA and PB clones showed an intermediate resistance with a growth inhibition of 72.8±1.0% and 60.6±1.5%, respectively. These data indicated the BXPC3 PA clone as the most resistant to either sTRAIL-containing supernatant or rhTRAIL, thereby prompting its use for further analyses.
BXPC3 resistance to TRAIL is led by overexpression of pro-survival genes
To assess whether the lower response to TRAIL on selected BXPC3 clones was due to an impaired expression of TRAIL surface receptors, cells were analysed by FACS (Figure 1D). Expression of TRAIL functional receptors was statistically similar between all tested samples, with low DR4 expression ranging from 1.8±0.6% in the CA clone, to 5.1±1.3% in the CB clone and 3.8±0.5% for wild type BXPC3. On the contrary, DR5 levels were consistently higher in all samples with 81.3±7.0% in WT, 96.2±1.4% in PA, 93.5±3.3% in PB, 90.0±6.2% in CA and 88.8±4.9% in CB. A modest although not statistically significant increase in DcR2 decoy receptor was observed in selected clones (14.5±1.2% in CA, 16.2±3.9% in PB, 17.3±6.9% in PA and 20.2±5.3% in CB) compared with wild type BXPC3 (6.2±2.3%). Finally, positivity of decoy receptor DcR1 was very low in all tested samples, ranging from 1% to 2% (Figure 1D). Having demonstrated that the resistance to TRAIL was not due to the selection of clones with down-regulated receptors expression or an increase in decoy levels, we began to address whether the resistance could be due to the regulation of downstream factors. TRAIL resistance is reported in relationship to a series of key pro-survival genes [28], therefore the most resistant clone BXPC3-PA was compared with WT cells by microarray analysis to identify candidate transcripts to be further investigated (Supplementary Table S1). Differential expression of selected genes involved in TRAIL apoptotic pathway [29] is summarized in Table 2. Subsequent qPCR on a subset of differentially-expressed pro-survival genes showed a significant 2- to 4-fold increase in expression of BCL-XL, cFLIP, XIAP, NFKB1 and NFKB2 compared to WT cells (Figure 1E). These data depicted a pro-survival pattern that justifies the outlined BXPC3 resistance, suggesting how combinatory strategy based on agents targeting one or more of these pro-survival genes could be effective in rescuing the BXPC3 sensitivity to TRAIL.
Summarized differential expression of selected TRAIL pathway-related genes from microarray analysis (from Supplementary Table S1) of the TRAIL resistant BXPC3 PA versus BXPC3 WT parental cells
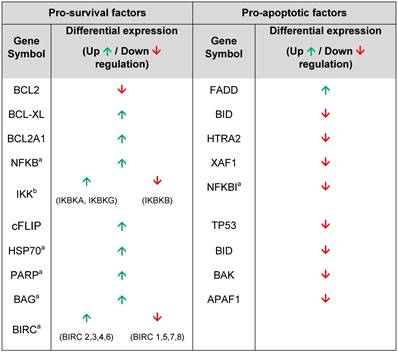
afamily of genes; bcomponents of a molecular complex
In vitro Paclitaxel treatment induces cytotoxicity in wild type and resistant BXPC3 clones counteracting sTRAIL resistance
Considering the role of PTX in hindering TRAIL-related pro-survival pathways [30-32], we sought to challenge all resistant BXPC3 clones with PTX in comparison with wild type cells. Dose-response assay by MTS showed a similar sensitivity of both wild type and resistant BXPC3 clones after 72 h PTX treatment (Figure 2A). In particular, 50% inhibition was obtained at PTX dose of 5 ng/mL and a plateau was reached after 50 ng/mL in all samples, indicating that the TRAIL resistant pattern was not cross-influencing with a PTX apoptosis induction, further justifying its role as sensitizer agent for sTRAIL.
Paclitaxel treatment sensitizes resistant BXPC3 to sTRAIL by reversing survival pathways (A) PTX dose-response assay for wild type (WT) and selected BXPC3 clones by MTS. Cells were treated with a range of concentrations of PTX for 72 h. (B) In vitro combined treatment of sTRAIL- containing supernatant with PTX. PA clone cells were treated for 24 h with AD-MSC EMPTY derived conditioned medium (EMPTY CM) or AD-MSC sTRAIL derived conditioned medium (sTRAIL CM) or the same with 5 ng/mL PTX (EMPTY CM+PTX and sTRAIL CM+PTX). Death rate was evaluated by PI and Annexin V staining in flow cytometry. (C) Pre-treatment strategy included a 24 h incubation with 5 ng/mL PTX for EMPTY CM+PTX and sTRAIL CM+PTX groups. The subsequent treatment was performed as in B for a further 24 h. (D) Expression of main pro-survival genes by qPCR assay on BXPC3 PA after 24 h treatment with 50 ng/mL PTX (BXPC3 PA-PTX) compared with untreated cells (BXPC3 PA) and BXPC3 WT as baseline. Data are represented as mean±SD. All data are represented as the mean of two independent experiments performed in triplicate. * p<0.01; ** p<0.05 with Student's T-test.
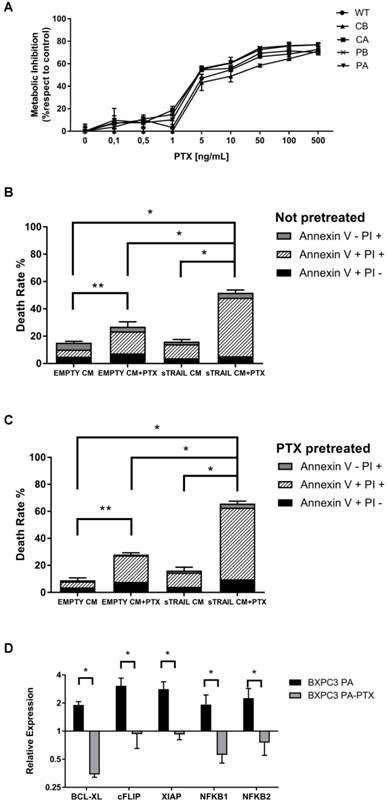
Therefore, a combined treatment of sTRAIL-containing supernatant with 5 ng/mL PTX for 24 h was introduced, generating a significant increase in apoptosis of BXPC3 PA clone (51.8±2.0%) compared either with sTRAIL-containing supernatant alone (16.0±1.5%) or PTX alone (27.0±3.6%) and suggesting a synergy between the two agents (Figure 2B).
A pre-treatment strategy was additionally developed by incubating BXPC3 cells with PTX 24 h prior to the treatment with sTRAIL-containing supernatant. This setting again confirmed the sensitizing effect of PTX which was found to be even increased with 65.8±1.8% death rate in combination with sTRAIL-containing supernatant, 28.1±1.2% with PTX only and 16.1±2.5% with AD-MSC sTRAIL supernatant only (Figure 2C). Similar in vitro results have been obtained by Nab-PTX that was then introduced in the in vivo model for its significantly better toxicity and delivery profile versus PTX [34] (Supplementary Figure S1).
Having accounted for the combinatory impact of sTRAIL and PTX against artificially-induced TRAIL resistance, we wanted to additionally challenge that in spontaneously TRAIL-resistant PDAC cell lines expressing TRAIL receptors. Therefore, CFPAC-1 and PaTu 8988t (PaTu) cell lines were selected and treated with sTRAIL and PTX alone or in combination (Supplementary Figure S3). Collectively these data on additional cell lines reinforce the rationale of a combined therapy with Nab-PTX as a sensitizing agent for TRAIL-resistant tumours while also highlighting the high variability of PC tumours which showed variegated responses and demand for a deeper investigation on effective doses.
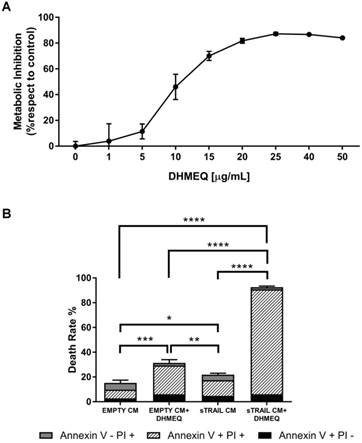
Considering the previously mentioned genes involved in sTRAIL resistance (Figure 1E), we began to identify the molecular mechanisms that may be involved in the PTX sensitization observed in the BXPC3 clone. Interestingly, the expression of pro-survival genes (BCL-XL, cFLIP, XIAP, NFKB1 and NFKB2) significantly dropped after PTX treatment to levels close to or lower than wild type cells, suggesting a restoring action by PTX in sTRAIL resistant cells (Figure 2D). To further strengthen this observation, we combined sTRAIL treatment with pharmacological inhibition of NFkB by incubation with DHMEQ. Cells were treated with DHMEQ alone in a dose response assay, showing a progressive inhibition of tumour cell metabolism reaching a plateau from the dose of 25 µg/mL (Figure 3A). We then treated sTRAIL-resistant cells with 20 µg/mL of DHMEQ along with sTRAIL CM in a combined treatment (Figure 3B). We observed a 31.4±2.7% cell death when cells were incubated with DHMEQ alone which dramatically rose to 92.6±0.8% in the combined setting, thus confirming the role of NFkB in the TRAIL resistance pathway. Control death rate was 15.2±2.2% while sTRAIL conditioned medium alone led to 21.9±1.2% cell death.
Inhibition of NFkB confirms its main role in sTRAIL resistance of selected BXPC3. (A) DHMEQ dose-response assay for wild type (WT) and selected BXPC3 clones by MTS. Cells were treated with a range of concentrations of DHMEQ for 48 h. (B) In vitro combined treatment of sTRAIL-containing supernatant with DHMEQ. PA clone cells were treated for 24 h with AD-MSC EMPTY derived conditioned medium (EMPTY CM) or AD-MSC sTRAIL derived conditioned medium (sTRAIL CM) or the same with 20 µg/mL DHMEQ (EMPTY CM+DHMEQ and sTRAIL CM+DHMEQ). Death rate was evaluated by PI and Annexin V staining in flow cytometry. All data are represented as the mean of two independent experiments performed in triplicate. * p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001 with Student's T-test
AD-MSC sTRAIL and Nab-PTX combined treatment has a potent anti-tumour effect on tumour size and vitality
Based on in vitro findings, a xenotransplant model was established to challenge the potential of AD-MSC sTRAIL and Nab-PTX combined treatment. Mice bearing BXPC3 PA clones were treated weekly by 20 mg/kg PTX i.p. and/or peri-tumoral injection of 1 million AD-MSC sTRAIL. Tumour volume monitoring confirmed that BXPC3 PA clone maintains sTRAIL resistance in vivo and, after 35 days from implantation, tumours from the group treated by AD-MSC sTRAIL were similar to untreated control mice (Figure 4A and 4B).
In vivo impact of combined treatment with MSC sTRAIL and Nab-PTX. (A) Monitoring of tumour volumes during treatment. Nab-PTX was administered i.p. with a PTX final concentration of 20 mg/kg once weekly for four weeks (grey arrowheads). AD-MSC sTRAIL were inoculated peri-tumoral the day following Nab-PTX treatment (black arrowheads). Tumour volume was calculated from the measurement of tumour diameters. (B) Pictures of representative tumours from each experimental group. (C) Representative images of hematoxylin and eosin staining of tumours from the different treatment groups. Stars indicate the stromal bundles, while black arrows point to areas of pancreatic cancer tissue damage. Magnification 100x, scale bar 100 μm. (D) Representative images of the IHC staining for the human CK-7 (brown DAB). CK-7 positive areas represent human PDAC islets. Tumour stroma (black stars), of murine origin, do not show any positivity for CK7 in any group. Again, large areas of empty cancer tissues were evident in the combinatory treatment group (black arrows). Magnification 100x, scale bar 100 μm. (E) Representative images of IHC staining for cleaved Caspase-3 (brown DAB). Brown elements, indicated by white arrows, represent cells bearing cleaved Caspase-3. Positive cells are more frequent in Nab-PTX and MSC sTRAIL+Nab-PTX groups and, in particular, around PC tissue damaged areas (white dotted region). Magnification 100x, scale bar 100μm. ^ p<0.0001, * p<0.05, § p<0.05 by ANOVA coupled with multiple comparison test.
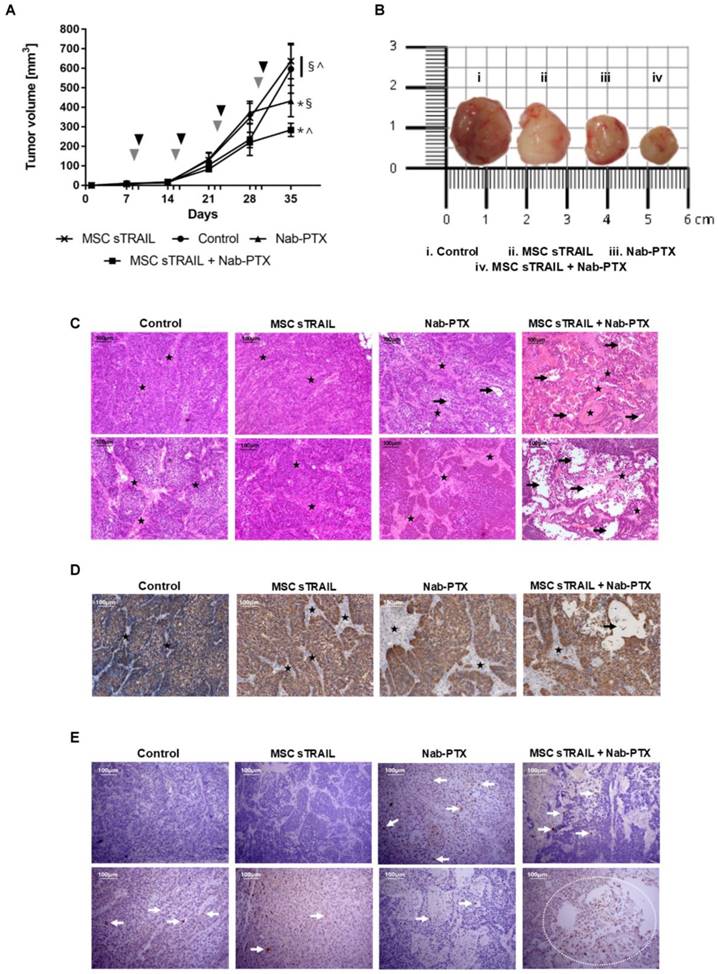
Nab-PTX treatment was effective in counteracting pancreatic cancer development and, most interestingly, its combination with AD-MSC-delivered sTRAIL demonstrated a robust anti-cancer effect compared to all other conditions. BXPC3 PA tumour size in the combinatory group was 2 times smaller versus both AD-MSC sTRAIL alone and the untreated controls, while a 1.5 fold decrease was observed against the Nab-PTX only group (Figure 4A and 4B).
Histological studies confirmed the potential of the strategy with a hematoxylin and eosin staining showing a compact structure in both untreated control and AD-MSC sTRAIL groups with islets of small pancreatic cancer cells surrounded by a dense stromal network (Figure 4C upper and lower rows). Nab-PTX group showed a looser and less organized architecture with areas characterized by an increase in the stromal compartment as against the pancreatic cancer areas. It is notable how the MSC sTRAIL+Nab-PTX treated xenotransplants showed a dramatic reduction in pancreatic cancer component with large holes in the histological preparation together with an apparent increase in stromal fraction. Moreover, an immunohistochemical staining for human Cytokeratin-7 confirmed the morphological features described (Figure 4D), in particular the relationship between pancreatic cancer and stroma in the group treated by the combinatory strategy. Finally, the staining for cleaved Caspase-3 (Figure 4E) showed areas of positive cells particularly evident around compromised regions, thus highlighting the role of apoptosis in the architecture modifications described. Collectively, these findings confirmed the in vitro data providing further evidence of a taxane-based combinatory approach capable of restoring sTRAIL sensitivity within a relevant pre-clinical pancreatic cancer model.
Discussion
Pancreatic cancer is a highly lethal disease with yet limited therapeutic options. The low survival rate is connected with a late diagnosis, a high metastatic potential and the lack of game changing therapies. In this scenario, innovative strategies also based on cell and gene therapy are needed. In particular, a deeper characterization of the individual patient's tumour coupled with the action of targeted therapies could lead to more effective and personalized treatments [35,36]. Among these strategies, the employment of biological vectors such as cells delivering targeted therapeutic agents may allow a greater precision, augmenting local compound bioavailability and reducing off-target toxicity.
MSC may represent this paradigm by virtue of their immunological characteristics and innate migration capacity to inflamed tissues, such as tumours [12]. Several studies have shown the advantages and efficacy of MSC-based gene therapy, and our research focuses on cancer treatment by gene modified MSC producing TRAIL variants, showing an effect both in vitro and in vivo [17,27]. In these settings, cellular vehicles can deliver a single compound with pre-clinical evidence of efficacy and safety. However, it is well known that a single agent delivery, even by innovative tools, may have limitations in oncology, and several groups are now identifying combinatory schedules in which conventional therapies can be combined with new therapeutic tools [25]. This is generally done to counteract cancer cell resistance prompting the combination of different ways of delivery and action to improve long-term efficacy.
We previously demonstrated that TRAIL variants delivered by MSC could be effective in targeting human pancreatic cancer [17,19,33]. Being aware of the possibility related to TRAIL resistance and with the purpose of empowering our gene delivery, here we originally propose to combine Taxanes as known agents against pancreatic cancer [37], with AD-MSC delivering sTRAIL in a challenging TRAIL-resistant cancer.
To generate a TRAIL-resistant pancreatic cancer model, we incubated BXPC3 cells with sTRAIL-containing supernatants for up to 3 months with different schedules to select resistant populations mimicking the situation of an induced resistance. The clones obtained showed different levels of sTRAIL resistance, resulting in a mortality rate 2 to 30 times lower than unselected BXPC3 control, with a sTRAIL resistance that was more evident in cells obtained through discontinuous selection versus samples maintained in constant selection. Another possibility would have been to work with an intrinsically resistant clone or to clonally dilute a BXPC3 cell suspension and test individual colonies for resistance to sTRAIL [38]. In particular, we focused only on the first aspect demonstrating that even naturally TRAIL resistant lines (PaTu and CFPAC-1) can be in principle sensitized by PTX. However, we have been more interested in the induced resistance model that is closer to a possible clinical scenario. In particular, pulsed selection resembles repeated doses of chemotherapy as a clinically relevant schedule and, very interestingly, this was the approach capable of generating the most sTRAIL resistant pancreatic cancer lines.
Characterization of selected clones included also a dose-response assay to determine the resistance against the recombinant human form of TRAIL, which led to the same results obtained with sTRAIL-containing supernatants. In particular, maximum mortality rate for non-selected BXPC3 was more than 80% versus less than 30% for the most resistant clone PA, similarly to that reported for pancreatic cancer lines constitutively resistant to TRAIL [39].
We describe a plateau effect from rhTRAIL concentration of 500 ng/mL onwards in all the tested samples. On the one hand, this result confirms an induced resistance in selected cells, while on the other hand, it shows the limits of a rhTRAIL monotherapy in vitro, unable to fully eradicate tumour cells and suggesting a combination with another compound for a better therapeutic profile.
Reported mechanisms for rhTRAIL resistance in vitro included down-regulation of active receptors, up-regulation of decoy receptors and desensitization of the cell to the apoptotic stimulus [29]. FACS analyses revealed that the expression of active receptors was similar between all the selected clones and wild type cells, collectively characterized by low DR4 and high DR5 expression, similarly to other pancreatic cancer cell lines [39]. On the contrary, we observed an increase in DcR2 receptor that, although not statistically significant respect to control, may indicate a trend due to a TRAIL counteracting action.
The most convincing contribution to TRAIL resistance comes from the analyses of key pro-survival genes with a relevant increase in BCL-XL, cFLIP, XIAP and NFKB1 and 2 versus unselected cells. BCL-XL is part of the BCL-2 family of molecules, involved in the mitochondrial pathway of apoptosis and frequently up-regulated in many cancers. It has been shown that its inhibition can improve TRAIL sensitivity in several intrinsically resistant pancreatic cancer cell lines and here we also confirmed its role in a chemotherapy-induced model [39]. cFLIP is also a well-known master anti-apoptotic regulator frequently overexpressed in malignant cells that suppresses TNF-α, Fas-L, and TRAIL-induced apoptosis by creating an apoptosis inhibitory complex which blocks caspase cascade [40]. XIAP belongs to a family of apoptotic suppressor proteins and inhibits several members of the caspase family including Caspase-3, 7 and 9. Its connection with TRAIL resistance has been demonstrated in several tumours such as melanoma, nasopharyngeal carcinoma and pancreatic cancer [41-43]. Moreover, a down-regulation of XIAP has been shown to sensitize pancreatic cancer cells to MSC-delivered sTRAIL [44]. Finally, NFkB effectively prevents TRAIL-mediated killing in a range of different tumours [45,46] also by having a role in the regulation of other pro-survival factors such as cFLIP, IAP and Bcl2 family proteins [29]. Starting from these considerations, we further confirm our observations and those of others about the role of NFkB in sTRAIL resistance by showing that its inhibition with the potent synthetic agent DHMEQ [47] allows a remarkable recovery of sTRAIL sensitivity. This early data additionally indicates how a more targeted manipulation of that specific pathway could empower TRAIL action; future investigations will be implemented to further clarify this combinatory strategy. Taken together, all these data support the notion of a resistance due to the development of a pro-survival cellular state involving master anti-apoptotic regulators. These findings have also been reported by others [29], with the involvement of these same genes in rhTRAIL resistance and support the relevance of our resistance induction even in the presence of sTRAIL released by AD-MSC.
To overcome the established or native resistance, TRAIL refractory cells were treated with PTX. This choice was based on its clinical relevance against pancreatic cancer and because of its distinct mechanism of action with respect to TRAIL minimizing the possibility of a cross-resistance. To test the most effective dose of PTX alone, we performed a dose-response assay with concentrations ranging from 0.1 to 500 ng/mL, demonstrating an effect starting from 5 ng/mL onwards with no difference between wild type and TRAIL-resistant BXPC3 cells. A plateau effect was reached with 50 ng/mL PTX corresponding to 70% of cell inhibition. Beside the identification of an effective dose, the assay confirmed our hypothesis that mechanisms providing protection from TRAIL-induced apoptosis could not protect from the PTX action. This is also indirectly confirmed by data on the synergy between rhTRAIL and using different formulations of the two compounds against multiple tumours, including pancreatic cancer [32,48,49]. However, to our knowledge, this is the first example of a combination of a human cell-based TRAIL delivery and PTX in the setting of a TRAIL-resistant tumour.
In vitro combined treatments showed a remarkable improvement in cytotoxicity when the two compounds were used together either simultaneously or in sequential mode. In the latter case, the PTX pretreatment before incubation with sTRAIL-containing supernatants generated the highest cytotoxicity compared with the two agents given simultaneously. Interestingly, the mortality due to combined protocols has always been higher than the sum of the death rate obtainable after single treatments, suggesting a synergy in the anti-cancer action of the two compounds.
To verify how PTX could restore BXPC3 sensitivity to TRAIL, we checked gene expression after PTX treatment, and noticed a clear turnaround in levels of the master anti-apoptotic genes overexpressed in the resistant clones. Their expression returned to levels similar to wild type BXPC3 or even less in the case of BCL-XL and NFKB1 genes. These findings could justify the rescue in sTRAIL sensitivity by PTX as also described previously using rhTRAIL [32,48, 49]. However, this could depict only a part of the entire picture and a deeper understanding will be needed, with special focus on the synergistic effect of the combination.
The effect of PTX alone or in combination with sTRAIL-containing supernatant was investigated also on AD-MSC and we were able to confirm previous reports of a reduction of AD-MSC metabolic activity and proliferation with little impact on viability [50] even in the combined approach (Supplementary Figure S2).
The in vivo xenotransplant of pancreatic cancer further tested the working hypothesis in a more clinical- relevant model introducing PTX in its albumin-bound formulation (Nab-PTX). This was mainly due to practical reasons; Nab-PTX is easier to deliver in virtue of its solubility in saline solution. In contrast, formulation with ethanol and castor oil for PTX injection has a high viscosity that makes the injection difficult, also being dangerous for mice [51]. Moreover, Nab formulation better fits with the current standard in chemotherapy [4].
The peri-tumoral inoculum of MSC sTRAIL was always performed the day after Nab-PTX, following the in vitro data of a better BXPC3 PA sensitization to TRAIL when the drug was delivered 24 h before the supernatant incubation and also to minimize the impact of PTX on effector cells. Weekly schedule for the combined treatment was decided based on our previous studies with sTRAIL-delivering MSC and on other studies employing PTX in vivo [17,52]. The group treated by AD-MSC sTRAIL only developed tumour size similar to untreated controls, thus confirming that BXPC3 maintain resistance to sTRAIL in vivo and indirectly suggesting that, at least in this model, MSC action alone is incapable of counteracting tumour growth. Cousin et al. originally reported that WT adipose MSC could inhibit pancreatic cancer cell lines proliferation [53]; however, it may be that the sTRAIL resistance of our clones is associated with an up-regulation of pro-survival genes that could, in turn, also act against the apoptosis induction due to MSC themselves. As expected, Nab-PTX treatment alone caused a tumour volume reduction associated with a decrease of the pancreatic cancer component with no change in the desmoplastic reaction that apparently seemed even increased, as in vivo confirmation of PTX efficacy on pancreatic cancer, regardless of TRAIL-resistance.
The combined treatment showed the highest reduction in tumour volume in line with the histological analyses, clearly indicating the impairment of tumour tissue in treated samples impacting both the pancreatic cancer cells and their stromal component.
Translating these findings to a future clinical scenario, this study favourably supports the combination of cell mediated TRAIL therapy with PTX either in up-front treatment or in case of an established TRAIL resistance. To conclude, given the complexity of adenocarcinomas and their ability to adapt and relapse, synergy between different strategies including standard treatments, targeted therapy and immunotherapy should be pursued to improve disease outcome, in particular in still deadly conditions, such as pancreatic adenocarcinoma.
Abbreviations
CK-7: cytokeratin 7; CM: conditioned medium; DHMEQ: dehydroxymethylepoxyquinomicin; MSC: mesenchymal stromal/stem cell; Nab: nanoparticle albumin-bound; PI: propidium iodide; PTX: paclitaxel; TRAIL: TNF-related apoptosis-inducing ligand; sTRAIL: soluble TRAIL; WT: wild type; rh: recombinant human.
Acknowledgements
This work was supported in parts by: Associazione Italiana Ricerca Cancro (AIRC) IG 2012 Grant #12755 (M.D.; C.S.; G. Grisendi); AIRC IG 2015 Grant# 17326 (M.D., C.S., F.R., G. Grisendi) Ministero Italiano Istruzione Università e Ricerca PRIN 2008WECX78 (M.D.), Project “Dipartimenti Eccellenti MIUR 2017” (M.D.) and the Associazione ASEOP (M.D.).
Supplementary Material
Supplementary figures and tables.
Competing Interests
M.D. and P.C. are co-founders of Rigenerand srl, a University start-up company developing gene therapy approaches for cancer. M.D. is also a member of the Board of Directors of Rigenerand srl. The interests of M.D. and P.C. are managed by their Universities (Modena - Reggio Emilia and Padua) in accordance with their conflict of interest policies. C.S., G.Grisendi, O.C., E.M.F. are currently employed by Rigenerand srl. The other authors do not declare any competing interests.
References
1. Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JWW, Comber H. et al. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374-403
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30
3. Pancreatic Cancer - Cancer Stat Facts [Internet]. [cited 2018 Jan 10]. Available from: https://seer.cancer.gov/statfacts/html/pancreas.html.
4. Garrido-Laguna I, Hidalgo M. Pancreatic cancer: from state-of-the-art treatments to promising novel therapies. Nat Rev Clin Oncol. 2015;12:319-34
5. Long J, Zhang Y, Yu X, Yang J, LeBrun D, Chen C. et al. Overcoming Drug Resistance in Pancreatic Cancer. Expert Opin Ther Targets. 2011;15:817-28
6. Javle M, Golan T, Maitra A. Changing the course of pancreatic cancer-Focus on recent translational advances. Cancer Treat Rev. 2016;44:17-25
7. Suker M, Beumer BR, Sadot E, Marthey L, Faris JE, Mellon EA. et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol. 2016;17:801-10
8. Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9:790-803
9. Lemstrova R, Melichar B, Mohelnikova-Duchonova B. Therapeutic potential of taxanes in the treatment of metastatic pancreatic cancer. Cancer Chemother Pharmacol. 2016;78:1101-11
10. Gradishar WJ. Albumin-bound paclitaxel: a next-generation taxane. Expert Opin Pharmacother. 2006;7:1041-53
11. Muniraj T, Jamidar PA, Aslanian HR. Pancreatic cancer: a comprehensive review and update. Dis-Mon DM. 2013;59:368-402
12. Grisendi G, Spano C, Rossignoli F, D Souza N, Golinelli G, Fiori A. et al. Tumor Stroma Manipulation By MSC. Curr Drug Targets. 2016;17:1111-26
13. Dwyer RM, Khan S, Barry FP, O'Brien T, Kerin MJ. Advances in mesenchymal stem cell-mediated gene therapy for cancer. Stem Cell Res Ther. 2010;1:25
14. Mohr A, Zwacka R. The future of mesenchymal stem cell-based therapeutic approaches for cancer - From cells to ghosts. Cancer Lett. 2018;414:239-49
15. Moniri MR, Sun X-Y, Rayat J, Dai D, Ao Z, He Z. et al. TRAIL-engineered pancreas-derived mesenchymal stem cells: characterization and cytotoxic effects on pancreatic cancer cells. Cancer Gene Ther. 2012;19:652-8
16. Hammer K, Kazcorowski A, Liu L, Behr M, Schemmer P, Herr I. et al. Engineered adenoviruses combine enhanced oncolysis with improved virus production by mesenchymal stromal carrier cells. Int J Cancer. 2015;137:978-90
17. Spano C, Grisendi G, Candini O, Golinelli G, Petrachi T, Rossignoli F. et al. Soluble TRAIL-Armed Human AD-MSC as Novel Cell Therapy. Cytotherapy. 2016;18:S24-5
18. Grisendi G, Spano C, D'souza N, Rasini V, Veronesi E, Prapa M. et al. Mesenchymal Progenitors Expressing TRAIL Induce Apoptosis in Sarcomas. Stem Cells. 2015;33:859-69
19. Golinelli G, Grisendi G, Spano C, Dominici M. Surrounding Pancreatic Adenocarcinoma by Killer Mesenchymal Stromal/Stem Cells. Hum Gene Ther. 2014;25:406-7
20. Loebinger MR, Eddaoudi A, Davies D, Janes SM. Mesenchymal stem cell delivery of TRAIL can eliminate metastatic cancer. Cancer Res. 2009;69:4134-42
21. Mohr A, Lyons M, Deedigan L, Harte T, Shaw G, Howard L. et al. Mesenchymal stem cells expressing TRAIL lead to tumour growth inhibition in an experimental lung cancer model. J Cell Mol Med. 2008;12:2628-43
22. Sasportas LS, Kasmieh R, Wakimoto H, Hingtgen S, van de Water JAJM, Mohapatra G. et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Natl Acad Sci U S A. 2009;106:4822-7
23. Mueller LP, Luetzkendorf J, Widder M, Nerger K, Caysa H, Mueller T. TRAIL-transduced multipotent mesenchymal stromal cells (TRAIL-MSC) overcome TRAIL resistance in selected CRC cell lines in vitro and in vivo. Cancer Gene Ther. 2011;18:229-39
24. Wang XG, Jandl T, Dadachova E, Revskaya E. Effect of naive and radiolabeled rhTRAIL on the cervical cancer xenografts in mice. Ther Deliv. 2014;5:139-47
25. Singh HM, Ungerechts G, Tsimberidou AM. Gene and cell therapy for pancreatic cancer. Expert Opin Biol Ther. 2015;15:505-16
26. Yu R, Deedigan L, Albarenque SM, Mohr A, Zwacka RM. Delivery of sTRAIL variants by MSCs in combination with cytotoxic drug treatment leads to p53-independent enhanced antitumor effects. Cell Death Dis. 2013;4:e503
27. Grisendi G, Bussolari R, Cafarelli L, Petak I, Rasini V, Veronesi E. et al. Adipose-Derived Mesenchymal Stem Cells as Stable Source of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Delivery for Cancer Therapy. Cancer Res. 2010;70:3718-29
28. Trivedi R, Mishra DP. Trailing TRAIL Resistance: Novel Targets for TRAIL Sensitization in Cancer Cells. Front Oncol. 2015;5:69
29. Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer. 2008;8:782-98
30. Blagosklonny MV, Schulte T, Nguyen P, Trepel J, Neckers LM. Taxol-induced apoptosis and phosphorylation of Bcl-2 protein involves c-Raf-1 and represents a novel c-Raf-1 signal transduction pathway. Cancer Res. 1996;56:1851-4
31. Huang Y, Johnson KR, Norris JS, Fan W. Nuclear factor-kappaB/IkappaB signaling pathway may contribute to the mediation of paclitaxel-induced apoptosis in solid tumor cells. Cancer Res. 2000;60:4426-32
32. Li L, Wen X-Z, Bu Z-D, Cheng X-J, Xing X-F, Wang X-H. et al. Paclitaxel enhances tumoricidal potential of TRAIL via inhibition of MAPK in resistant gastric cancer cells. Oncol Rep. 2016;35:3009-17
33. Spano C, Grisendi G, Golinelli G, Rossignoli F, Prapa M, Bestagno M. et al. Soluble TRAIL Armed Human MSC As Gene Therapy For Pancreatic Cancer. Sci Rep.
34. Yardley DA. nab-Paclitaxel mechanisms of action and delivery. J Controlled Release. 2013;170:365-72
35. Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388:73-85
36. Liu S-X, Xia Z-S, Zhong Y-Q. Gene therapy in pancreatic cancer. World J Gastroenterol WJG. 2014;20:13343-68
37. Goldstein D, El-Maraghi RH, Hammel P, Heinemann V, Kunzmann V, Sastre J. et al. nab-Paclitaxel Plus Gemcitabine for Metastatic Pancreatic Cancer: Long-Term Survival From a Phase III Trial. JNCI J Natl Cancer Inst. 2015;107:dju413-dju413
38. McDermott M, Eustace AJ, Busschots S, Breen L, Crown J, Clynes M. et al. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front Oncol. 2014;4:40
39. Hari Y, Harashima N, Tajima Y, Harada M. Bcl-xL inhibition by molecular-targeting drugs sensitizes human pancreatic cancer cells to TRAIL. Oncotarget. 2015;6:41902-15
40. Safa AR. c-FLIP, a master anti-apoptotic regulator. Exp Oncol. 2012;34:176-84
41. Hörnle M, Peters N, Thayaparasingham B, Vörsmann H, Kashkar H, Kulms D. Caspase-3 cleaves XIAP in a positive feedback loop to sensitize melanoma cells to TRAIL-induced apoptosis. Oncogene. 2011;30:575-87
42. Yang S, Li S-S, Yang X-M, Yin D-H, Wang L. Embelin prevents LMP1-induced TRAIL resistance via inhibition of XIAP in nasopharyngeal carcinoma cells. Oncol Lett. 2016;11:4167-76
43. Vogler M, Dürr K, Jovanovic M, Debatin K-M, Fulda S. Regulation of TRAIL-induced apoptosis by XIAP in pancreatic carcinoma cells. Oncogene. 2007;26:248-57
44. Mohr A, Albarenque SM, Deedigan L, Yu R, Reidy M, Fulda S. et al. Targeting of XIAP Combined with Systemic Mesenchymal Stem Cell-Mediated Delivery of sTRAIL Ligand Inhibits Metastatic Growth of Pancreatic Carcinoma Cells. Stem Cells. 2010;28:2109-2120
45. de Miguel D, Lemke J, Anel A, Walczak H, Martinez-Lostao L. Onto better TRAILs for cancer treatment. Cell Death Differ. 2016;23:733-47
46. Kim Y-S, Schwabe RF, Qian T, Lemasters JJ, Brenner DA. TRAIL-mediated apoptosis requires NF-kappaB inhibition and the mitochondrial permeability transition in human hepatoma cells. Hepatology. 2002;36:1498-508
47. Matsumoto N, Ariga A, To-e S, Nakamura H, Agata N, Hirano S. et al. Synthesis of NF-κB activation inhibitors derived from epoxyquinomicin C. Bioorg Med Chem Lett. 2000;10:865-9
48. Min SY, Byeon HJ, Lee C, Seo J, Lee ES, Shin BS. et al. Facile one-pot formulation of TRAIL-embedded paclitaxel-bound albumin nanoparticles for the treatment of pancreatic cancer. Int J Pharm. 2015;494:506-15
49. Dorsey JF, Mintz A, Tian X, Dowling ML, Plastaras JP, Dicker DT. et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and paclitaxel have cooperative in vivo effects against glioblastoma multiforme cells. Mol Cancer Ther. 2009;8:3285-95
50. Bonomi A, Coccè V, Cavicchini L, Sisto F, Dossena M, Balzarini P. et al. Adipose tissue-derived stromal cells primed in vitro with paclitaxel acquire anti-tumor activity. Int J Immunopathol Pharmacol. 2013;26:33-41
51. Sparreboom A, van Tellingen O, Nooijen WJ, Beijnen JH. Nonlinear pharmacokinetics of paclitaxel in mice results from the pharmaceutical vehicle Cremophor EL. Cancer Res. 1996;56:2112-2115
52. Gonzalez-Villasana V, Rodriguez-Aguayo C, Arumugam T, Cruz-Monserrate Z, Fuentes-Mattei E, Deng D. et al. Bisphosphonates inhibit stellate cell activity and enhance antitumor effects of nanoparticle albumin-bound paclitaxel in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2014;13:2583-94
53. Cousin B, Ravet E, Poglio S, De Toni F, Bertuzzi M, Lulka H. et al. Adult stromal cells derived from human adipose tissue provoke pancreatic cancer cell death both in vitro and in vivo. PloS One. 2009;4:e6278
Author contact
![]() Corresponding author: Massimo Dominici, MD, Division of Oncology, Department of Medical and Surgical Sciences of Children & Adults, University Hospital of Modena and Reggio Emilia, Via del Pozzo, 71, ZIP: 41100, Modena, Italy. Ph Office 0039-059-422-2858 / 4019; Ph Lab: 0039-059-422-3307 / 2718; Fax Lab: 0039-059-422-3341; massimo.dominiciit
Corresponding author: Massimo Dominici, MD, Division of Oncology, Department of Medical and Surgical Sciences of Children & Adults, University Hospital of Modena and Reggio Emilia, Via del Pozzo, 71, ZIP: 41100, Modena, Italy. Ph Office 0039-059-422-2858 / 4019; Ph Lab: 0039-059-422-3307 / 2718; Fax Lab: 0039-059-422-3341; massimo.dominiciit