Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(3):661-675. doi:10.7150/thno.27794 This issue Cite
Research Paper
VEGFC acts as a double-edged sword in renal cell carcinoma aggressiveness
Papa Diogop Ndiaye1, Maeva Dufies2, Sandy Giuliano2, Laetitia Douguet1, Renaud Grépin2, Jérôme Durivault2, Philippe Lenormand1, Natacha Glisse1, Janita Mintcheva1, Valérie Vouret-Craviari1, Baharia Mograbi1, Maud Wurmser3, Damien Ambrosetti4, Nathalie Rioux-Leclercq5, Pascal Maire3, Gilles Pagès1,2 ![]()
1. University of Nice Sophia Antipolis, Institute for research on cancer and aging of Nice, CNRS UMR 7284; INSERM U1081, Centre Antoine Lacassagne, France
2. Centre Scientifique de Monaco, Biomedical Department, 8 Quai Antoine Ier, MC-98000 Monaco, Principality of Monaco.
3. Université Paris Descartes, Sorbonne Paris Cité, INSERM U1016, Institut Cochin, Paris, 75014 France; CNRS UMR 8104, Paris, 75014 France, Paris, 75014 France.
4. Nice University Hospital, Central laboratory of Pathology.
5. Rennes University, Rennes University Hospital, Department of Pathology, Rennes, France.
Received 2018-6-11; Accepted 2018-7-30; Published 2019-1-21
Abstract

Hypoxic zones are common features of metastatic tumors. Due to inactivation of the von Hippel-Lindau gene (VHL), renal cell carcinomas (RCC) show constitutive stabilization of the alpha subunit of the hypoxia-inducible factor (HIF). Thus, RCC represents a model of chronic hypoxia. Development of the lymphatic network is dependent on vascular endothelial growth factor C (VEGFC) and lies at the front line of metastatic spreading. Here, we addressed the role of VEGFC in RCC aggressiveness and the regulation of its expression in hypoxia.
Methods: Transcriptional and post transcriptional regulation of VEGFC expression was evaluated by qPCR and with reporter genes. The involvement of HIF was evaluated using a siRNA approach. Experimental RCC were performed with immuno-competent/deficient mice using human and mouse cells knocked-out for the VEGFC gene by a CRISPR/Cas9 method. The VEGFC axis was analyzed with an online available data base (TCGA) and using an independent cohort of patients.
Results: Hypoxia induced VEGFC protein expression but down-regulated VEGFC gene transcription and mRNA stability. Increased proliferation, migration, over-activation of the AKT signaling pathway and enhanced expression of mesenchymal markers characterized VEGFC-/- cells. VEGFC-/- cells did not form tumors in immuno-deficient mice but developed aggressive tumors in immuno-competent mice. These tumors showed down-regulation of markers of activated lymphocytes and M1 macrophages, and up-regulation of M2 macrophages markers and programmed death ligand 1 (PDL1). Over-expression of lymphangiogenic genes including VEGFC was linked to increased disease-free and overall survival in patients with non-metastatic tumors, whereas its over-expression correlated with decreased progression-free and overall survival of metastatic patients.
Conclusion: Our study revisited the admitted dogma linking VEGFC to tumor aggressiveness. We conclude that targeting VEGFC for therapy must be considered with caution.
Keywords: VEGFC, lymphangiogenesis, sunitinib, angiogenesis, renal cell carcinoma, metastasis, CRISPR/Cas9
Introduction
The incidence of renal cell carcinoma (RCC) has increased during the last decade. Surgical resection of small and non-invasive tumors detected by chance is curative most of the time. However, metastatic RCC is intrinsically resistant to radio- and chemo-therapy, which worsens the prognosis. Mutation/inactivation of the von Hippel-Lindau (VHL) gene, an E3 ubiquitin ligase that regulates the stability of the hypoxia-inducible factor-1 or -2α (HIF-1α, HIF-2α), occurs in almost eighty percent of RCC. The subsequent stabilization of HIF-1α leads to over-expression of VEGF and exacerbated vascularization. Therefore, RCC represents a paradigm for the use of anti-angiogenic (AAG) therapies. These treatments together with previous treatments with interleukin 2 or interferon have changed the survival rate of patients with metastatic disease from a few months to several years in the most favorable cases [1]. However, in the case of relapse, death generally occurs within a few months. The first-line reference treatment is sunitinib, an inhibitor of tyrosine kinase receptors (TKI) including the VEGF (VEGFR1/2/3), PDGF, CSF1 receptors and c-Kit [2]. At relapse on sunitinib, other TKIs or mTOR inhibitors are available for second- or third-line treatments including axitinib [3], pazopanib [4], cabozantinib [5], lenvatinib [6] and everolimus [7]. Immuno-therapies have also shown promising results in first-line treatment or after relapse on sunitinib [8, 9]. However, these treatments are not curative. Therefore, understanding the mechanism of metastatic propagation from an indolent disease in non-metastatic patients (M0) to a more aggressive pathology in metastatic patients (M1) and of relapse on treatment may bring to light new therapeutic strategies.
The lymphatic network has long been considered as an inert system for the return of interstitial fluids to the bloodstream and drainage of leukocytes and antigens to the lymph nodes [10]. It transports the tumor cells to the lymph nodes where they are eliminated by immune cells. This system therefore constitutes a natural barrier to metastatic dissemination. However, tumor cells migrating to the lymph nodes produce VEGFC, a growth factor for lymphatic endothelial cells [11]. The neo-formed lymphatic vessels bypass the lymph node and accelerate the propagation of the tumor cells to the next lymph node and then to other organs [12]. Therefore, the lymphatic network represents an important route of dissemination of tumor cells. The number of invaded lymph nodes correlated with the severity of the disease. Therefore, detection of the sentinel lymph node has become a routine practice in hospitals. At this stage, clinicians are faced with a serious situation limiting the therapeutic options. This observation highlights two antagonistic roles of the lymphatic vessels: a beneficial effect in the initial phase of tumor development and a detrimental effect when the lymphatic network is tumor bearing [13].
This antagonism has not been addressed in depth and deserves to be investigated. We recently reported that the different TKIs used for the treatment of metastatic RCC, including sunitinib, stimulate the development of lymphatic vessels in experimental tumors and in tumors from patients treated in a neo-adjuvant setting [14]. These results suggested that TKIs reduce the tumor burden but favor metastatic dissemination. Our study reconciles in part the results of preclinical studies showing that AAG may elicit metastatic cell phenotypes compromising tumor-reducing benefits [15, 16]. However, these results were not confirmed by clinical trials showing that TKI do not stimulate tumor growth in patients with metastatic RCC [17].
Understanding the different roles of the lymphatic network and studying the molecular actors involved in its development represent major therapeutic issues. The major growth factor of lymphatic endothelial cells is VEGFC. However, only the regulation of its mRNA has been addressed in response to growth factors and oncoproteins [18]. Only the sine oculis (SIX1) gene and its co-activator eye absent homolog 2 (EYA2) were reported to up-regulate VEGFC transcription and to favor metastatic dissemination of breast cancer cells via the lymphatics [19, 20]. A close correlation exists between reduced survival, presence of hypoxic zones and high levels of VEGFC in these areas [21]. Conventional or targeted radio- and chemo-therapy induce intra-tumor hypoxia [22] and production of VEGFC [14, 23]. Hypoxia is a pathophysiological condition for the selection of aggressive tumor cells and is dependent on HIF-1 and/or -2. HIF-1 has tumor suppressor characteristics whereas HIF-2 has oncogenic properties in RCC [24]. Testing the role of hypoxia in RCC cells and the involvement of HIF-1 or -2 appears inappropriate since HIF-1 and/or HIF-2 are constitutively present because of VHL inactivation in 80% of cases. However, a small fraction of tumors present an active form of VHL and these tumors have the poorest prognosis [25]. Therefore, these fast growing tumors may present hypoxic zones with subsequent induction of HIF-1, 2.
The presence of lymphatic vessels and the metastatic potential of tumors have been studied extensively but these investigations have mainly been performed on advanced tumors. The role of lymphatic vessels on non-metastatic (M0)/metastatic (M1) tumor aggressiveness has not been investigated. In addition, knowledge of the molecular mechanisms responsible for the expression of VEGFC at diagnosis and in response to treatments is a major research issue. Controlling VEGFC's action on lymphatic vessel development would improve the effectiveness of current treatments.
Lymphatic metastasis is the main dissemination pathway in many solid tumors. We recently discovered that the formation of new lymphatic vessels in AAG-resistant RCC is primarily induced by VEGFC [14]. However, little is known about the regulation of VEGFC expression and its direct roles in RCC development and metastasis.
We show here that the basal expression of VEGFC depended on HIF-2 in VHL-deficient RCC cell lines. Hypoxia, a common feature of metastatic tumors, further stimulated VEGFC protein expression at both transcriptional and post-transcriptional levels, in which NF kappa B (NFκB) was involved. Whereas tumors developed rapidly and metastasized in immuno-competent mice, their growth was greatly inhibited in immuno-deficient mice. Our findings suggest that VEGFC regulation by hypoxia is subtle and depends on hypoxia in a HIF-2-dependent manner. VEGFC appears to be beneficial or detrimental for tumor growth. Thus, targeting VEGFC should be considered with caution for the treatment of RCC patients.
Methods
Reagents and antibodies
Sunitinib was purchased from Selleckchem (Houston, USA). Anti-ARD1 antibodies were home-made and previously described [26]. Anti-Twist and anti-P65 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-Slug and anti-phospho P65 antibodies were from Cell Signaling Technology (Beverly, MA, USA). Anti-HIF-2α antibodies were from Novus Biologicals (Littleton, CO, USA).
Cell culture
786-0 (786), RCC4 (R4) and RENCA RCC cell lines were purchased from the American Tissue Culture Collection. RCC10 (R10) cells were a kind gift from Dr. W.H. Kaelin (Dana-Farber Cancer Institute, Boston, MA) and derived in the laboratory of Dr KH Plate [27]. These cells present a difference in sensitivity to HIF-2α antagonists [28]. RENCA cells express a wild-type form of VHL, whereas the VHL gene is inactivated in R4, R10 and 786 cells. RENCA cells are mouse cells syngenic of Balb-C mice. R4, R10 and 786 are of human origin.
Immunoblotting
Cells were lysed in buffer containing 3% SDS, 10% glycerol and 0.825 mM Na2HPO4. 30 to 50 μg of proteins were separated on 10% SDS-PAGE, transferred onto a PVDF membrane (Immobilon, Millipore, France) and then exposed to the appropriate antibodies. Proteins were visualized with the ECL system using horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies.
Quantitative real-time PCR (qPCR) experiments
One microgram of total RNA was used for reverse transcription using the QuantiTect Reverse Transcription kit (QIAGEN, Hilden, Germany), with a blend of oligo (dT) and random primers to prime first-strand synthesis. SYBR master mix plus (Eurogentec, Liege, Belgium) was used for qPCR. The mRNA level was normalized to 36B4 mRNA. Oligo sequences of the VEGFC mRNA have already been described [14]. A list of the primers used in the manuscript is given in Table S1.
Genomic disruption of VEGFC using CRISPR-Cas9
786-O or RENCA cells were transfected with PX458 plasmids containing CRISPR-Cas9 targeting regions of the first exon of the VEGFC gene using JetPRIME (Polyplus). Control cells were obtained by transfecting the empty plasmid. The pSpCas9 (BB)-2A-GFP (PX458) plasmid was a gift from Dr. Feng Zhang (Addgene plasmid # 48138) [29]. The sgRNA sequence that we cloned into the vector to target the VEGFC gene was: 5'-GAGTCATGAGTTCATCTACAC-3' for the human gene and 5'- GACCGTCGCCGCCTTCGAGTC -3' for the mouse gene. As the PX458 plasmid contains GFP, cells were first sorted using flow cytometry to obtain cells containing the CRISPR-Cas9 and followed by clonal selection and screening. Sequencing of human genomic DNA to confirm the mutations leading to VEGFC invalidation was performed using the following primers: forward, 5'- TTGTGTTAGGGAACGGAGCAT-3'; reverse, 5'-AGAACCAGGCTGGCAACTTC3'.
Tumor xenograft experiments
Ectopic model of RCC: Five million 786-O or 105 RENCA cells were injected subcutaneously into the flank of 5-week-old nude (nu/nu) or Balb-C mice (Janvier, France) respectively. Three-month-old six1flox/flox; RosaCreERt2 mice (six1-/-) were given intraperitoneal injection of tamoxifen (1 mg per mouse per day; Sigma) for five consecutive days. Two weeks later, injection of 105 RENCA cells was performed. Days three and four after cells injection, mice received again an injection of tamoxifen [20]. eya2 knock-out mice (eya2-/-) [30] and corresponding littermates were injected with an equivalent number of RENCA cells. The tumor volume was determined with a caliper (V = L × W2 × 0.5). This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals. Our experiments were approved by the ''Comité national institutionnel d'éthique pour l'animal de laboratoire (CIEPAL)'' (reference: NCE/2013-97).
Measurement of cytokines
After stimulation, the cell supernatant was recovered for VEGFC measurement using the Human DuoSet ELISA kit (R&D Systems, MN, USA). A specific ELISA kit was used to evaluate mouse VEGFC (LifeSpan Biosciences, WA, USA).
Luciferase assays
Transient transfections were performed using 2 µL of lipofectamine (GIBCO BRL) and 0.5 µg of total plasmid DNA-renilla luciferase in a 500 µL final volume. The firefly control plasmid was co-transfected with the test plasmids to control for the transfection efficiency. 24 h after transfection, cell lysates were tested for renilla and firefly luciferase. All transfections were repeated four times using different plasmid preparations. LightSwitch™ Promoter Reporter VEGFC (S710378) and LightSwitch™ 3´UTR reporter VEGFC (S803537) were purchased from Active motif (CA, USA). The short and long forms of the VEGFC promoter were a kind gift of Dr. Heide L Ford and Kari Alitalo [19]. Mutation of the NFκB binding site in the VEGFC promoter was obtained as previously described (wild-type sequence of the NFκB site: 5'-GGGAAACGGGGAGCT-3'; mutated: 5'-GGGAAACAAGGAGCT-3' [31]).
Chromatin immuno-precipitation (ChIP)
These experiments were performed as previously described [32]. Briefly, cells were grown in normoxia or hypoxia for 24 h (5-10×106 cells were used per condition). Cells were then fixed with 1% (v/v) formaldehyde (final concentration) for 10 min at 37 °C and the action of the formaldehyde was then stopped by the addition of 125 mM glycine (final concentration). Next, cells were washed in cold PBS containing a protease inhibitor cocktail (Roche, Basel, Switzerland), scraped into the same buffer and centrifuged. The pellets were re-suspended in lysis buffer, incubated on ice for 10 min, and sonicated to shear the DNA into fragments of between 200 and 1,000 base pairs. Insoluble material was removed by centrifugation and the supernatant was diluted 10-fold by addition of ChIP dilution buffer and pre-cleared by addition of salmon sperm DNA/protein A agarose 50% slurry for 1 h at 4 °C. About 5% of the diluted samples was stored and constituted the input material. Immuno-precipitation was then performed by addition of anti-HIF-2α or anti-tubulin as IgG control antibodies for 24 h at 4 °C. Immuno-complexes were recovered by adding 50% of salmon sperm DNA/protein A agarose and washed sequentially with low salt buffer, high salt buffer, LiCl buffer and TE. DNA complexes were extracted in elution buffer, and cross-linking was reversed by incubating overnight at 65 °C in the presence of 200 mM NaCl (final concentration). Proteins were removed by incubating for 2 h at 42 °C with proteinase K and the DNA was extracted with phenol/chloroform and precipitated with ethanol. Immuno-precipitated DNA was amplified by PCR with the following primers. HIF primers first couple of primers: promoter region, -458/-344; forward primer, 5'-GGACAAGAACTCGGGAGTGG-3'; reverse primer, 5'-ACCGGCTTTAGAGGTGATGC. HIF primers second couple of primers: promoter region, -457/-342; forward primer, 5'-GACAAGAACTCGGGAGTGGC-3'; reverse primer, 5'-GGACCGGCTTTAGAGGTGAT-3'. NFκB primers: promoter region, -364/-105; forward primer, 5'-GCATCACCTCTAAAGCCGGT-3'; reverse primer, 5'-TGCCTGCGCTTATGTGAGAG-3'.
siRNA assay
siRNA transfection was performed using Lipofectamine RNAiMAX (Invitrogen). Cells were transfected with either 50 nM of validated siRNA against HIF-1α [33] or HIF-2α [34]; si-HIF-1α-sense: 5′-CCTATATCCCAATGGATGATG-3′; si-HIF-1α-antisense: 5′-CUTCATCCATTGGGATATAGG-3′; siHIF-2α-sense: 5'-CAGCAUCUUUGAUAGCAGU-3'; siHIF-2α-antisense: 5'-ACUGCUAUCAAAGAUGCUG-3; si-Control (Ambion, 4390843). Two days later, qPCR was performed, as described above.
Single cell suspension procedures
Samples of spleen were mechanically dissociated, homogenized, and passed through a 100 µM cell strainer in PBS with 5% FCS and 0.5% EDTA. Tumors were mechanically dissociated and digested with 1 mg/mL collagenase A (Roche) and 0.1 mg/mL DNase I (Roche) for 30 min at 37 °C.
Cell staining and flow cytometry
Surface staining was performed by incubating cells on ice for 20 min, with saturating concentrations of labeled Abs in PBS, 5% FCS and 0.5% EDTA. Mouse cell-staining reactions were preceded by a 15-min incubation with purified anti-CD16/32 Abs (2.4G2). The following anti-mouse antibodies were used: FITC-conjugated anti-B220 (RA3-632), PE-conjugated anti-δTCR (GL3), APC-conjugated anti-CD11b (M1/70), PerCP-Cy5.5-conjugated anti-CD3 (145-2C11), V450-conjugated anti-Ly6C (AL-21), PE-Cy7-conjugated anti-CD11c (HL3), AF700-conjugated anti-Ly6G (1A8), BV786-conjugated anti-CD45.2 (104), BV711-conjugated anti-CD4 (RM4-5), and BV650-conjugated anti-CD8 (53-6.7). Antibodies were purchased from BD Biosciences except anti-CD11b and anti-B220 from eBioscience. Data files were acquired on Aria II and analyzed using Diva software (BD Biosciences).
Gene expression microarray analysis
Normalized RNA sequencing (RNA-Seq) data produced by The Cancer Genome Atlas (TCGA) were downloaded from cBiopotal (www.cbioportal.org, TCGA Provisional; RNA-Seq V2) and were analyzed as previously described [14]. The results published here are in whole or in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/ [35, 36].
Patients and association studies
Primary tumor samples of M0 RCC patients were obtained from the Rennes University hospital [37]. The disease-free survival (DFS) and overall survival (OS) were calculated from patient subgroups with VEGFC mRNA levels that were less or greater than the third quartile value. DFS was defined as the time from surgery to the appearance of metastasis. Progression-free survival (PFS) was defined as the time between surgery and subsequent blood sampling and progression, or death from any cause, censoring live patients and progression-free at last follow-up. OS was defined as the time from blood sample collection to the date of death from any cause, censoring those alive at last follow-up. The Kaplan Meier method was used to produce survival curves and analyses of censored data were performed using Cox models.
Statistical analysis
For the analysis of in vitro and mice experiments: All data are expressed as the mean ± the standard error (SEM). Statistical significance and p values were determined by the two-tailed Student's t-test or Mann-Whitney test. One-way ANOVA was used for statistical comparisons. Data were analyzed with Prism 5.0b (GraphPad Software) by one-way ANOVA with Bonferroni post hoc.
For patients: The Student's t-test was used to compare continuous variables and the chi-square test, or Fisher's exact test (when the conditions for use of the χ2-test were not fulfilled), were used for categorical variables. DFS was defined as the time from surgery to the appearance of metastasis. PFS was defined as the time between surgery and progression, or death from any cause, censoring live patients and progression-free at last follow-up. Overall survival (OS) was defined as the time between surgery and the date of death from any cause, censoring those alive at the last follow-up. The Kaplan-Meier method was used to produce survival curves and analyses of censored data were performed using Cox models. All analyses were performed using R software, version 3.2.2 (Vienna, Austria, https://www.r-project.org/).
Results
VEGFC expression depends on HIF-2 in RCC
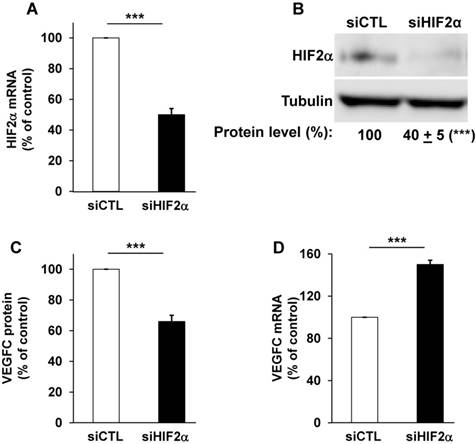
The relative presence of HIF-1α and -2α have already been investigated in different RCC cell lines and we confirmed that certain RCC cells inactivated for vhl express the two HIFα isoforms (RCC4), or only HIF-2α for the most aggressive (RCC10 and 786-O, Figure S1A, [38]) cells. Therefore, we focused on the most aggressive cells that only express HIF-2α [39]. We already demonstrated that VEGFC expression correlated to the relative aggressiveness of RCC cells with respect to their ability to form tumors in nude mice [14]. VEGFC expression is induced by hypoxia in breast, pancreatic cancers and melanoma cells but such induction is independent of HIF-1 [21]. However, the role of HIF-2α, the “oncogenic form of HIFα for RCC”, in VEGFC expression has not been investigated. Therefore, we hypothesized that HIF-2α may be one of the drivers of VEGFC expression in aggressive cells that mimic chronic hypoxia through VHL inactivation. Consistent with this, HIF-2α silenced with siRNA (Figure 1A-B and Figure S1B) decreased VEGFC protein levels in two independent cell lines expressing only HIF-2α (Figure 1C, 786-O cells and Figure S1C, RCC10 cells). However, HIF-2α silencing resulted in an increase in mRNA levels in these two independent cell lines (Figure 1D and Figure S1D). Moreover, HIF-1α and -2α silenced in cells expressing both proteins (RCC4) also showed an increase in VEGFC mRNA levels (Figure S1E-G). These results suggest that HIF-1α and -2α are involved in negative control of VEGFC mRNA expression probably via inhibition of transcription and/or mRNA stability.
HIF-2α induced a high basal level of VEGFC mRNA in 786-O cells. (A) HIF-2α expression was down-regulated by siRNA. HIF-2α mRNA levels were evaluated by qPCR in siRNA control (siCTL) and HIF-2α-directed siRNA (siHIF-2α). (B) HIF-2α protein levels were evaluated by immuno-blotting; tubulin is shown as a loading control. A quantification of protein levels (mean ± SEM) is shown. HIF-2α in the siCTL condition was considered as the reference value (100%); *** p <0.001. (C) VEGFC protein expression was assessed by ELISA for the siRNA control (siCTL) and HIF-2α-directed siRNA (siHIF-2α). (D) VEGFC mRNA levels were evaluated by qPCR in siRNA control (siCTL) and HIF-2α-directed siRNA (siHIF-2α), Results are represented as the mean of three independent experiments ± SEM. *** p<0.001.
Opposite effects of hypoxia on VEGFC gene transcription, mRNA half-life and protein expression
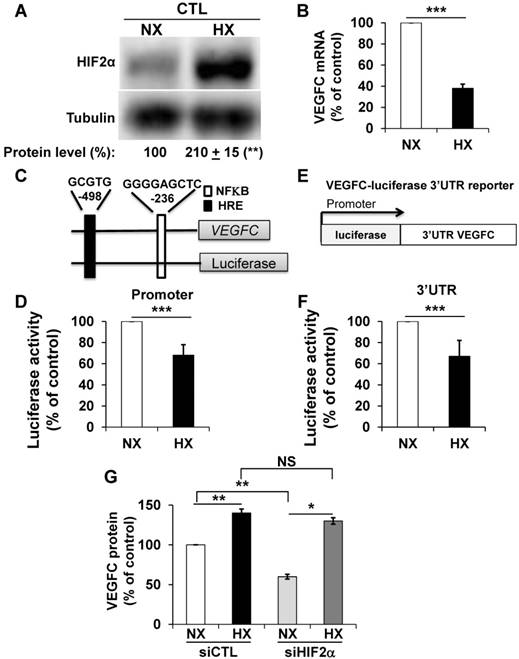
Morfoisse and co-workers have elegantly shown that hypoxia induced down-regulation of the amount of VEGFC mRNA but stimulated VEGFC protein production through intra ribosomal entry sites present in the 5'UTR of the VEGFC mRNA [21]. Therefore, we focused on the hypoxia-dependent down-regulation of the VEGFC mRNA. Considering the negative role of HIF-2α in the control of the amount of mRNA but its positive role in VEGFC protein expression (see above), we hypothesized that an experimental condition resulting in an increase in the HIF-2 level would down-regulate the VEGFC mRNA and up-regulate the protein. Although 786-O cells constitutively stabilize HIF-2α because of VHL inactivation, hypoxia (24 h 1% oxygen; equivalent conditions for the experiments described below) further induced HIF-2α levels (Figure 2A). Consistent with the fact that HIF-2α acts as a negative regulator on the level of mRNA (Figure 1D), the amount of VEGFC mRNA decreased in hypoxia (Figure 2B). To understand the molecular mechanism associated with this down-regulation, we investigated the VEGFC promoter activity as a readout of its transcriptional control. A schema of this promoter containing functional hypoxia (HRE [40]) and NFκB response elements [41], is shown in Figure 2C. The luciferase activity representing the VEGFC promoter activity was down-regulated by hypoxia, which strongly suggests that the decrease in mRNA levels in hypoxia concerns inhibition of transcription (Figure 2D). To address the effect of hypoxia on the mRNA half-life, which mainly relies on its 3'UTR, we used a reporter vector in which the luciferase mRNA half-life/activity is controlled by the VEGFC mRNA 3'UTR (Figure 2E) [14]. The luciferase activity, readout of the 3'UTR-dependent mRNA half-life, was inhibited by hypoxia (Figure 2F). The decrease in the amount of mRNA (Figure S2A), inhibition of transcription (Figure S2C), mRNA stability (Figure S2D) and increase in the amount of protein (Figure S2B) were confirmed in an independent cell line (R10). Whereas VEGFC protein production was dependent on HIF-2α in normoxia, HIF-2α down-regulation by siRNA did not affect VEGFC production in hypoxia (Figure 2G). Equivalent results were obtained with an independent cell line (Figure S2E). These results suggest that chronic hypoxia mediated by VHL inactivation and acute hypoxia induced by incubation of cells in low oxygen concentrations regulate VEGFC expression through independent mechanisms.
Hypoxia induced down-regulation of VEGFC mRNA and increased the level of VEGFC protein. (A) HIF-2α protein expression was stimulated by hypoxic (Hx) as compared to normoxic conditions (Nx) in 786-O cells as shown by immuno-blotting; tubulin is shown as a loading control. Quantification of protein levels (mean ± SEM) is shown. HIF-2α in normoxic condition was considered as the reference value (100%); ** p <0.01. (B) VEGFC mRNA levels in 786-O cells were evaluated by qPCR in normoxia (NX) and hypoxia (HX). (C) Schematic representation of the VEGFC promoter showing the hypoxia response element (HRE) and the NFκB binding site together with their respective localization according to the transcription initiation start site. (D) 786-O cells were transfected with a renilla luciferase reporter gene under the control of the VEGFC promoter and incubated in normoxia (NX) or hypoxia (HX) for 48 h. The renilla luciferase activity normalized to the firefly luciferase (control vector) was the readout of the VEGFC promoter activity. (E) Schematic representation of the VEGFC mRNA 3'UTR down-stream of the renilla luciferase reporter gene under the control of the cytomegalo virus promoter (CMV). (F) 786-O cells were transfected with the above-mentioned reporter gene and incubated in normoxia (NX) or hypoxia (HX) for 48 h. The renilla luciferase activity normalized to the firefly luciferase (control vector) was the readout of the VEGFC mRNA half-life. (G) VEGFC protein expression was assessed by ELISA in siRNA control (CTL) and HIF-2α-directed siRNA (HIF-2) in normoxia (NX) or hypoxia (HX). Results are represented as the mean of at least three independent experiments ± SEM. * p<0.05, ** p<0.01, *** p<0.001, NS, non-significant.
Inhibitory effects of hypoxia on VEGFC gene transcription depend on cross talk between HIF-2 and NFκB
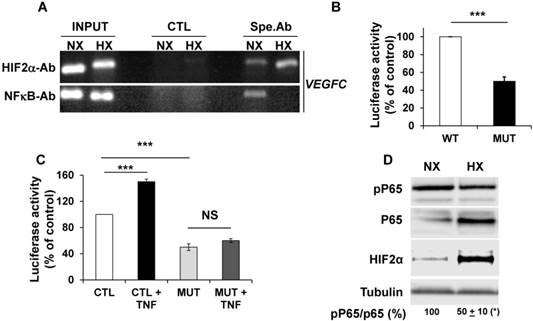
The NFκB-dependent transcriptional regulation of VEGFC by tumor necrosis factor alpha (TNFα) has already been described [41]. Considering that NFκB and HIF-2 conversely control each other's transcription [42], we analyzed their respective expression and effect on VEGFC transcription in hypoxia. ChIP experiments clearly demonstrated that NFκB and HIF-2α, bound to the VEGFC promoter (Figure 3A). Moreover, Guo et al. showed that HIF-1 stimulated VEGFC expression via its direct binding to HRE present in the VEGFC promoter region [40]. We demonstrated that HIF-2α also binds to the same domain of the VEGFC promoter. HIF-2α recruitment increased and NFκB binding decreased in hypoxia (Figure 3A). These results and the ones presented in the previous figures strongly suggest that the inhibition in VEGFC transcription observed in hypoxia involves an inhibitory mechanism mediated by enhanced HIF-2α binding and decreased NFκB binding to the promoter. The basal but not the hypoxia-driven VEGFC promoter activity depends on the integrity of the NFκB binding site (Figure 3B and Figure S3). Moreover, VEGFC promoter activity is stimulated by the inflammatory cytokine TNFα and this induction is dependent on the NFκB binding site, as demonstrated in gallbladder carcinoma, thereby confirming the functionality of NFκB in RCC cells (Figure 3C, [41]). These results strongly suggest that the inhibition in VEGFC transcription in hypoxia involves dissociation of NFκB from its binding domain. Since the NFκB activity is dependent on phosphorylation, we investigated its post-translational modification in hypoxia. Although the amount of NFκB/P65 was increased by hypoxia, probably reflecting the HIF-dependent transcriptional up-regulation of NFκB, the phosphorylated active form of NFκB was down-regulated. In the same experimental conditions, HIF-2α expression was up-regulated by hypoxia (Figure 3D). Therefore, these results strongly suggest that hypoxia inhibits the activity/phosphorylation of NFκB limiting its affinity for the VEGFC promoter, thereby participating in the down-regulation of VEGFC transcription.
NFκB plays a key role in the control of VEGFC transcription in normoxia and hypoxia. (A) Chromatin immuno-precipitation of cells incubated in normoxia (NX) or hypoxia (HX) was performed using control (CTL) or specific antibodies (Spe.Ab) for HIF-2α or NFκB. The specific DNA domains containing the HIF-2α or NFκB binding sites in the VEGFC promoter amplified by PCR are shown. (B) 786-O cells were transfected with a renilla luciferase reporter gene under the control of the VEGFC promoter that was wild-type (WT) or mutated for the NFκB binding site. The luciferase activity was measured 48 h post transfection. (C) The same transfections were performed but the cells were stimulated or not with TNFα (150 ng/mL) for 48 h. (D) 786-O cells were incubated in normoxia (NX) or hypoxia (HX, 24 h 1% oxygen). Total (P65) and phosphorylated (pP65) HIF-2α were detected by immuno-blotting; tubulin is shown as a loading control. Quantification of the blot is indicated. Quantification of pP65/P65 (mean ± SEM) is shown. This ratio in normoxia was considered as the reference value (100%); * p <0.05. When indicated, results are represented as the mean of at least three independent experiments ± SEM. *** p<0.001, NS, non-significant.
Knock-out of VEGFC resulted in increased proliferation, migration and mesenchymal marker expression
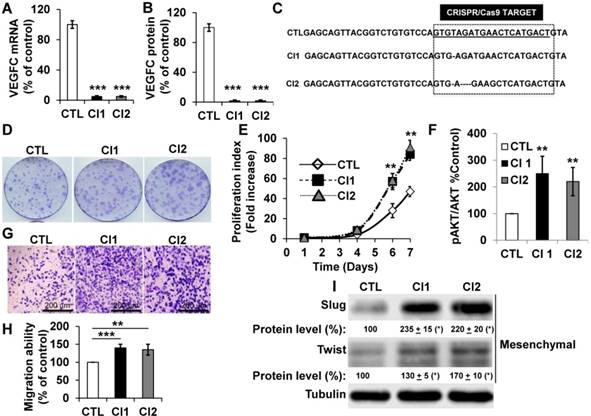
To further address the role of VEGFC in the hypoxic environment throughout tumor development, we knocked out the gene in human (786-O) and mouse (RENCA) RCC cells. RENCA cells express low whereas 786-O cells express high VEGFC levels (Figure S4A). RENCA cells have an active form of VHL and we observed induction of HIF-2α in hypoxia (Figure S4B). Consistently, we observed the same hypoxia-dependent induction of VEGFC in these cells (Figure S4C). Two independent knock-out clones of 786-O (Figure 4A-C), and RENCA cells were obtained and characterized (Figure S4D). A clone heterozygous for vegfc knock-out was obtained for RENCA cells (Figure S4D). VEGFC mRNA and protein were not detected in 786-O (Figure 4A-B) and RENCA clones (Figure S4D). Half the normal level of VEGFC was obtained for the vegfc -/+ clone of RENCA cells (Figure S4D). The VEGFC genomic locus was sequenced and showed small deletions in 786-O cells (Figure 4C), demonstrating the specificity of the CRISPR/Cas9 knock-out. 786-O knock-out clones (Cl1, Cl2) presented more proliferation when assessed with clonogenic assays (bigger size of the colonies, Figure 4D) and through measurement of live cell counts after six and seven days (Figure 4E). Increased proliferation was associated with an enhanced AKT activity (Figure 4F). VEGFC-knock-out 786-O clones were also more invasive (Figure 4G-H), which was consistent with an increase in the mesenchymal markers Slug and Twist (Figure 4I) [43]. The vegfc-/- cells presented more proliferation when assessed with clonogenic assays (more colonies with a bigger size, Figure S4E) and after measurement of the cell number (Figure S4F). The proliferation of vegfc-/+ cells resembled that of wild-type cells (Figure S4E-F). Only vegfc-/- cells presented a fibroblast-like morphology (Figure S4G), which was consistent with increasing Slug and Twist expression (Figure S4H). These results suggest that VEGFC lowers the intrinsic aggressiveness of RCC cells.
786-O cells knocked-out for VEGFC show increased proliferation and migration. (A) VEGFC mRNA levels were tested by qPCR in control (CTL) and two independent clones (Cl1, Cl2) knocked-out for VEGFC. (B) VEGFC protein levels were evaluated by ELISA in the supernatant of control (CTL) and two independent clones (Cl1, Cl2) knocked-out for VEGFC. (C) The locus of VEGFC was sequenced in control (CTL) and two independent clones (Cl1, Cl2) knocked-out for VEGFC; single or multi-base deletions were detected in the VEGFC locus. (D-E) The proliferation of control (CTL) or knock-out (Cl1, Cl2) cells were determined using clonogenic assays (D) or by quantification of the number of live cells (E). (F) Increased proliferation correlated with enhanced AKT activity evaluated by measuring the ratio of amount of phosphorylated to total AKT (pAKT/AKT). (G-H) VEGFC knock-out cells showed increased migration in Boyden chambers (G). (H) Quantification of the results shown in (G). (I) VEGFC knock-out cells showed increased levels of mesenchymal markers (Slug, Twist) detected by immuno-blotting; tubulin is shown as a loading control. Quantification of protein levels (mean ± SEM) is shown. Slug and Twist expressions in control cells (CTL) were considered reference values (100%); * p <0.05. When indicated, results are represented as the mean of at least three independent experiments ± SEM. ** p<0.01, *** p<0.001.
Knock-out VEGFC cells did not form tumors in immuno-deficient mice whereas they formed very fast-growing and invasive tumors in immuno-competent mice
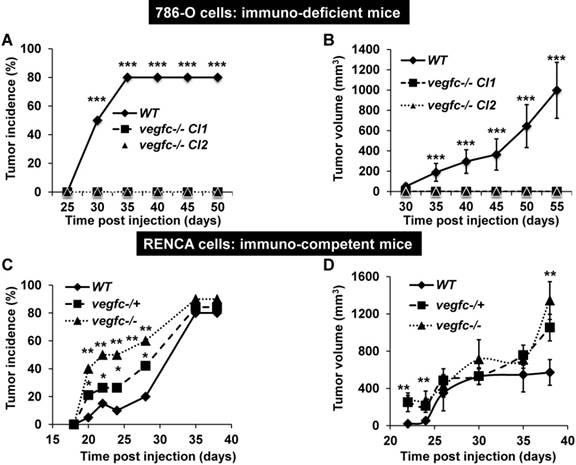
To further address the role of VEGFC in the microenvironment and on tumor aggressiveness, we subcutaneously implanted the knock-out cells in different mouse models: i) immuno-deficient nude mice were implanted with human cells; ii) immuno-competent Balb-C mice were implanted with mouse cells. Control 786-O cells formed tumors in 80% of the inoculated mice after a delay of 35 days and then grew rapidly, whereas VEGFC-/- cells did not form tumors in nude mice (Figure 5A-B). This result suggests that VEGFC is a driver of aggressiveness when the immune system is absent, a situation mimicking the immune tolerance observed in advanced tumors. In contrast, the tumor incidence (presence of a visible tumor or abdominal metastasis, see below) with RENCA cells was dependent on time and vegfc down-regulation/knock-out, vegfc-/- RENCA cells formed tumors with the highest incidence more rapidly (Figure 5C). The size of the tumors generated with vegfc-/+ or vegfc-/- cells was bigger than those formed with control cells, reaching a plateau 30 days post injection (Figure 5D). However, tumors generated with vegfc-/- cells were bigger than control tumors when mice were euthanized for ethical reasons 40 days after cell injection (Figure 5D). Moreover, 15% (3 mice out of 20) and 20% (4 mice out of 20) of mice injected with vegfc-/+ or vegfc-/- cells, respectively, presented an early ascite-like formation, which corresponded to peritoneal metastatic dissemination (Figure S5A). These results in immuno-competent mice suggest that the VEGFC produced by tumor cells educated anti-tumor immune cells during initiation of tumor development, a situation mimicking low-grade tumors developing in a context of an efficient immune system. Strikingly, immuno-deficient mice developed tumors of the same size and with an equivalent incidence to the control or heterozygous RENCA cells. Although, the incidence and the size of the tumors generated with knock-out RENCA cells were lower (Figure S5B-C), the differences were less spectacular than for 786-O cells, reflecting the higher level of aggressiveness of the RENCA model probably linked to the presence of an active VHL [44] (tumors developed 15 days after injection for RENCA as compared to 35 days for 786-O cells). This result strongly suggests that VEGFC exerts a potent anti-tumor effect via the immune system.
Knock-out of VEGFC in tumor cells has opposite effects on the formation of tumors in immuno-deficient or immuno-competent mice. (A) Experimental tumors in nude mice (20 mice per condition) were obtained after subcutaneous injection of 5×106 wild-type (WT) or VEGFC-knock-out 786-O cells. Two independent VEGFC-/- clones were injected (VEGFC-/- Cl1 and Cl2). Tumor incidence (percentage of mice with tumors) is presented. (B) Evaluation of tumor size for the experiment described in (A). (C) Experimental tumors in immuno-competent Balb-C mice (10 mice per condition) were obtained after subcutaneous injection of 105 wild-type (WT), heterozygous (vegf -/+) or knock-out RENCA cells (vegfc -/-). The tumor incidence (percentage of mice with a tumor) is shown. (D) Evaluation of the tumor size for the experiment described in (C). When indicated, results are represented as the mean ± SEM. ** p<0.01, *** p<0.001.
The microenvironment favors aggressiveness in vegfc-/- tumors
We first analyzed the composition of the immune cells infiltrate within control vegfc-/+ and vegfc-/- tumors. Recruitment of immune cells was equivalent in the three different tumor types. No modulation in the lymphoid compartment was observed (CD4+T, CD8+T, γδT and B cells). While recruitment of total dendritic cells (CD11c+) and monocytic myeloid-derived suppressor cells (M-MDSC) was not affected by the presence or absence of VEGFC, we found a significant decrease in polymorphonuclear myeloid derived suppressor cells (PMN-MDSC) in tumors obtained with vegfc-/- cells (Figure 6A). This observation suggests that the VEGFC produced by tumor cells is responsible for PMN-MDSC recruitment. This result may be in contradiction with the tumor aggressiveness obtained with vegfc-/- RENCA cells since PMN-MDSC are generally associated with poor prognosis.
Shaping of the microenvironment by VEGFC. (A) Detection by flow cytometry of PMN-MDSC cells in tumors generated with wild-type (WT), heterozygous (vegfc -/+) and knock-out RENCA cells (vegfc -/-). (B) Detection by flow cytometry of tumor-associated macrophages (TAM) in tumors generated with wild-type (WT), heterozygous (vegfc -/+) or knock-out RENCA cells (vegfc -/-). (C) The levels of indicated mRNA were determined by qPCR.
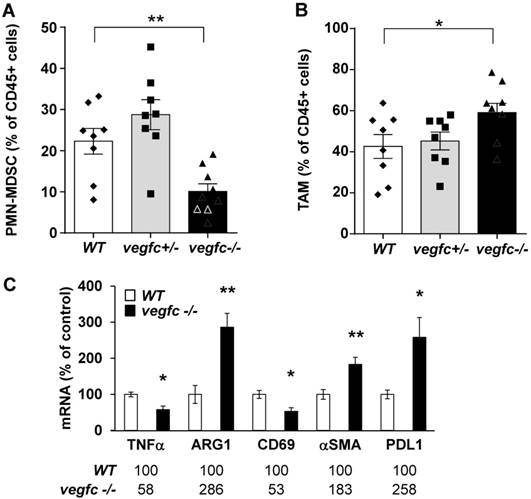
However, we also observed an increase in tumor-associated macrophages (TAM) in vegfc-/- tumors (Figure 6B), which may compensate for the decreased infiltration of PMN-MDSC in driving tumor growth. These results (initial aggressiveness, decrease and increase of different immune cells synonymous of poor prognosis) illustrate the ambivalence of the VEGFC activity during the initiation of tumor development and during the phase of accelerated growth and dissemination. During tumor growth, activation/proliferation of T lymphocytes plays a key role in driving the anti-tumor immunity. Moreover, the nature of macrophages infiltrating the tumors is an important parameter since M1 macrophages are associated with good prognosis whereas M2 macrophages are synonymous with poor prognosis. We further assessed the presence of activated lymphocytes and the nature of infiltrated macrophages by testing specific markers using qPCR. We observed a decrease in a marker of M1 macrophages (tumor necrosis factor α) and an increase in a marker of M2 macrophages (arginase 1) suggesting polarization of macrophages towards the M2 phenotype (Figure 6C). We also observed a decrease in CD69, a marker of lymphocyte activation. We also observed an increase in the marker of tumor-associated fibroblasts, α smooth muscle actin (αSMA) and up-regulation in the immune checkpoint, program death ligand 1 (PDL1). These different markers are related to tumor aggressiveness, which is consistent with the accelerated tumor growth and metastasis observed in experiments into tumorigenesis.
To further address the role of VEGFC produced by cells of the microenvironment and because vegfc gene knock-out is lethal [45], we injected wild-type RENCA cells into mice knocked out for genes coding for the two major regulators of vegfc transcription, SIX1 and its co-activator EYA2. Tumors generated with RENCA cells developed faster and were bigger in the six1-/- mice (Figure S6A). The incidence of tumor formation was equivalent in wild-type and six1-/- mice (almost 70% of mice developed a tumor). In eya2-/- mice, bigger tumors appeared rapidly within fifteen days after injection but reached the same incidence and volume as in wild-type and knock-out mice at the end of the experiment (Figure S6B-C). These results suggest that the VEGFC produced by tumor cells (above) or cells of the microenvironment slow down tumor initiation.
Genes associated with the VEGFC pathway have an opposite prognostic role in non- metastatic and metastatic patients
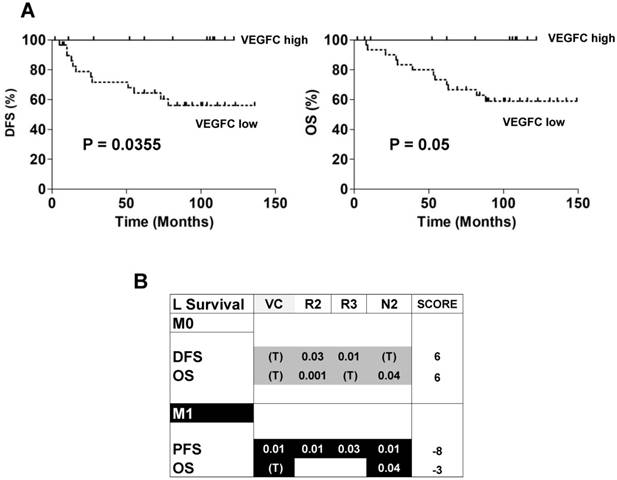
The ambivalence observed for the role of VEGFC in animal models prompted us to analyze the prognostic impact of lymphangiogenic genes on a cohort of M0 patients from the Rennes University Hospital (see Table 1 for the patients' characteristics). 34 patients out of 38 did not present signs of metastatic dissemination. Only 4 out of 38 presented with invaded lymph nodes. High levels of VEGFC mRNA in the tumors of M0 patients (third quartile cut-off, n=25/38) correlated with a longer DFS (p = 0.0355, Figure 7A left) and OS (p = 0.05, Figure 7A right). These results suggest that VEGFC reduced the aggressiveness of non-invasive tumors and were in agreement with the results of mice tumors. Then, 535 patients of The Cancer Genome Atlas (TCGA) were analyzed for mRNA levels of the VEGFC (VC) gene and its receptors (VEGFR2 (R2); VEGFR3 (R3))/co-receptors (NEUROPILIN 2 (N2)) and subsequent follow-up in terms of DFS, PFS and OS. High VEGFC level (> third quartile) was associated with longer DFS and OS, especially for grade I/II M0 patients (298 patients out of 425 (70.1%)), whereas it was associated with shorter PFS and OS in M1 patients (111 patients). The same trend was observed for high VEGFR2, VEGFR3 and N2 gene levels (> third quartile) but without discriminating the less aggressive grade I/II M0 patients but including the grade III (123 patients out of 425 (28.9%)) and grade IV (3 patients out of 425 (0.7%)) patients. Based on this result, we established a prognostic score according to the VC/R2/R3/N2 levels. The relative weight that we attributed to each gene, depending on a trend or a significant correlation with DFS, PFS or OS, highlighted the high levels of VC/R2/R3/N2 genes associated with a longer DFS/OS in M0 patients (respective score 6 and 6) whereas it was exactly the opposite for M1 patients (respective scores, -8, -3, Figure 7B). To reconcile the patient and experimental results (correspondence between mRNA and protein levels) and because it was almost impossible to determine the tumor hypoxic status, we hypothesized that if VEGFC mRNA and protein levels correlated, VEGFC would stimulate the formation of lymphatic vessels. Therefore, tumors with high VEGFC mRNA/protein should express highly lymphatic vessel markers. This is exactly what we observed in the cohort of our Institute and in the TCGA cohort (Figure S7A-B). The correlation between VEGFC levels was kept for R3 but not for N2 (Figure S7C). Hence, it appears that a strong correspondence exists between VEGFC mRNA and protein levels in these specific cases.
Characteristics of the patients included in the study.
| GROUP | |
|---|---|
| Number of patients | 38 |
| ccRCC | 38 (100%) |
| Sex | |
| - Woman | 14 (36.8%) |
| - Man | 24 (63.2%) |
| Age (year) | 62 (42-82) |
| Furhman grade | |
| - 2 | 17 (44.7%) |
| - 3 | 16 (42.1%) |
| - 4 | 5 (13.2%) |
| Metastatic status pM | |
| - M0 | 38 (100%) |
| Lymph node status pN | |
| N0 | 34 (89.5%) |
| N1 | 4 (10.5%) |
| Size status pT | |
| - 1 | 17 (44.7%) |
| - 2 | 6 (15.8%) |
| - 3 | 15 (39.5%) |
Patient outcome depended on the expression of genes of the VEGFC pathway. (A) Kaplan-Meier analysis of DFS and OS of M0 patients. DFS and OS were calculated from patient subgroups with VEGFC mRNA levels that were less or greater than the third quartile. Statistical significance (p-value) is indicated. (B) Correlation between genes of the VEGFC pathway and survival (DFS/PFS/OS) in M0 and M1 patients. The tested genes of the VEGFC pathway were the following: VEGFC (VC), VEGFR2 (R2), VEGFR3 (R3) and NEUROPILIN 2 (N2). The p-values of genes associated with shorter DFS/PFS/OS appear white on a black background; the p-values of genes associated with a longer DFS/PFS/OS appear black on a gray background. Significant p-values are given; a trend to significance is indicated by a “T” in black on a gray background. A score was established as follows: a positive point was given for a gene with a trend to good prognosis; two positive points for a gene associated with good prognosis and with a significant p-value; two negative points were given for a gene associated with poor prognosis and with a significant p-value. Positive scores were found for DFS and OS for M0 patients. The scores were negative for PFS and OS for M1 patients.
Discussion
The comparison of the levels of VEGFC in normal and tumor tissues and the regulation of its expression have been poorly investigated [18]. We showed that VEGFC levels are elevated in primary cells derived from the healthy portion of the kidney. The levels of VEGFC produced by tumor cells were highly variable; only the cells derived from metastatic tumors reached levels comparable to those of healthy cells [14]. This situation evokes the beneficial effect of VEGFC on the early stages of tumor development, while high levels become detrimental during metastatic dissemination. This ambiguity was addressed by implanting tumor cells in immuno-deficient mice, mimicking a pathological situation in which the immune system is bypassed and no longer exerts control on the tumor. In this situation, VEGFC promoted tumor initiation and tumor growth. Therefore, inhibiting its expression or activity may lower tumor growth. This hypothesis was confirmed by our experiments. However, the main role of VEGFC is to induce lymphatic vessel expansion in primary tumors. Dendritic cells present in the primary tumor capture tumor antigens and migrate to the draining lymph nodes where they activate specific lymphocytes [46]. Hence, an efficient lymphatic network participates in control of the tumor by the immune system [47]. Considering this early primary event, the VEGFC activity may contribute to keeping tumor expansion in check and therefore appears to be beneficial. So, limiting VEGFC production would probably result in local tumor growth and selection of tumor cells with high proliferative and invasive abilities. VEGFC expressed by tumor cells migrated to the sentinel lymph nodes acts directly on preexisting lymphatic vessels to induce lymphangiogenesis [13] and tumor dissemination. The local inflammation that results from this rapid growth educates inflammatory cells for local production of VEGFC and tumor cell spreading via the newly formed lymphatic network [48]. Moreover, VEGFC was shown to induce immune tolerance and to protect against preexisting anti-tumor immunity, which is consistent with the presence of PMN-MDSC observed in our study [49]. The presence of tumor-associated lymphoid structures is generally associated with good prognosis because they educate intra-tumor CD8+ T cells. CD8+ T cells are beneficial for most tumors. However, for RCC, two types of CD8+ T cells are linked to good or poor prognosis and the absence of fully functional mature dendritic cells has been described [50]. It is possible that these two populations were selected according to the relative levels of VEGFC present in the microenvironment. This second step was addressed in immuno-competent mice by reducing the production of VEGFC in tumor cells or in cells of the microenvironment. These two ambivalent roles of VEGFC probably explain the discrepant results in the literature showing that VEGFC may have or not have prognostic significance for the detrimental evolution of different cancers [51-53]. Therefore, the presence of lymphatic vessels within a tumor specimen or the presence of VEGFC in the plasma as a surrogate marker of lymphangiogenesis and metastasis may highlight two opposite situations. Thus, correlating tumor or plasmatic VEGFC to prognosis may show opposite results and puzzle the investigator. Hence, considering that VEGFC participates in the dissemination of tumor cells, immediate targeting of VEGFC in metastatic patients should be considered with caution. The role of VEGFC as a predictive marker of anti-angiogenic drugs may also represent an interesting tool for patient follow-up when considering the development of lymphatic vessels when the patient is on treatment [14]. Lower baseline levels of VEGFC were associated with longer PFS and OS for patients treated with sunitinib and the drug induced down-regulation of plasmatic levels in a cohort of sixty-one patients [54]. However, in a recent independent cohort of patients, we were unable to demonstrate a correlation between plasmatic VEGFC and PFS or OS in patients treated with sunitinib [55]. These contradictory results also highlight our substantial knowledge on how VEGFC expression is controlled. The hypoxia-HIF-dependent regulation of VEGFC expression is currently under debate and probably involves different partners, as we demonstrated in this study. We showed that HIF-2 is probably the major regulator of VEGFC transcription in a pathological situation mimicking chronic hypoxia in RCC cells knocked-out for VHL. However, the HIF-2 binding site on the VEGFC promoter in the tumor cells described in this study (proximal to the transcription initiation site) is different from that described for HIF-1 in inflamed macrophages [40]. HIF-1 may play a role in the induction of VEGFC expression in hypoxia in normal cells but not in tumor cells as was shown recently [21]. Moreover, the partners involved in VEGFC transcription may also depend on the acute or chronic hypoxia in tumor cells. The decrease in VEGFC mRNA levels and the concomitant up-regulation in protein levels described in our study were consistent with previous published results [21]. However, it was really surprising to observe induction of HIF-2α protein expression in cells mutated for VHL. The decrease in the transcriptional activity observed in acute hypoxia is mainly due to a decrease in NFκB binding to the VEGFC promoter although HIF-2α is up-regulated. The decrease in mRNA stability depended on the VEGFC mRNA 3'UTR. We demonstrated recently the role of an equilibrated balance in tristetraprolin and HuR in the enhanced VEGFC mRNA half-life in response to anti-angiogenic drugs [14]. We tested both proteins in hypoxia but their levels and phosphorylation were not modified. Additional work is needed to discover mRNA binding factors involved in the decreased mRNA half-life observed in hypoxia.
The detrimental role of hypoxia in several tumors has been well documented. However, it was counterintuitive to believe that in RCC hypoxia may play an important role because of VHL inactivation. Further induction of VEGFC in hypoxic zones in a VHL inactivation context may represent a missing link to understand the mechanisms related to RCC aggressiveness depending on the presence of an efficient or tolerant immune system. The increase in proliferation and migration of knock-out cells also suggests that VEGFC participates in an autocrine loop. However, we and others did not detect VEGF receptors on the different RCC cell lines [56]. VEGFC signaling in RCC depends on the VEGFR3 co-receptor neuropilin 2 [57], and this autocrine loop probably maintains stemness as already described in skin cancers, and consequently, a low ability to proliferate [58]. α9β1 integrin was described as a VEGFC receptor mediating the migration of breast cancer cells [59, 60]. Moreover, the prostaglandin E2 receptor EP4 mediates autocrine and paracrine VEGFC production by tumor cells or macrophages [61, 62]. Integrin α9 and β1 and EP4 expression is equivalent in tumor and normal tissues, as revealed by analysis of the TCGA database. However, levels of expression of α9 and β1 integrin and EP4 above the median value are synonymous with longer OS (p = 0.0006, p = 0.008 and p = 0.017 respectively). The same beneficial role of α9, β1 and EP4 was observed if M0 and M1 patients were considered separately. These results suggest that the VEGFC pathway may play a different role depending on the cancer type. In conclusion, regulation of VEGFC involves a concert of transcription factors and mRNA binding proteins. Depending on whether hypoxia is chronic or acute, they may act differently on VEGFC expression. Deciphering this subtle regulation may allow better understanding of the sequence of events leading to a beneficial or detrimental role of lymphangiogenesis.
Abbreviations
AAG: anti-angiogenic therapy; ChIP: chromatin immune-precipitation; CRISPR: Clustered Regularly Interspaced Short Palindromic Repeats; CSF: colony-stimulating factor; DFS: disease-free survival; EYA2; eye absent 2; HIF: hypoxia-inducible factor; M0; non metastatic patients; M1: metastatic patients; mTOR: mammalian target of rapamycin; NKκB: nuclear factor kappa B; OS: overall survival; PDGF; platelet-derived growth factor; PDL1: programmed death ligand 1; PFS: progression-free survival; PMN-MDSC: poly morphonuclear-myeloid derived suppressor cell; qPCR: quantitative polymerase chain reaction; RCC: renal cell carcinoma; SIX1: sine oculis 1; SMA: smooth muscle actin; TAM: tumor associated macrophage; TCGA; tumor cancer genome atlas; TKI; tyrosine kinase inhibitor; UTR: unstranslated region; VEGFC: vascular endothelial growth factor C; VEGFR: VEGFR receptor; VHL: von Hippel Lindau.
Acknowledgements
We thank Dr. Heide L. Ford and Dr. Kari Alitalo for the kind gift of VEGFC promoter reporter genes. We thank O. Brégerie and M. Mihoc (UMS 28, Paris) for six1 and eya2 animal care.
Grant Support
This work was supported by the French Association for Cancer Research (ARC; to G. Pagès), the French National Institute for Cancer Research (INCA; to G. Pagès), the Fondation de France (S. Giuliano and M. Dufies), The “Ligue Nationale contre le cancer (Equipe labellisée 2019), the Fondation d'entreprise Groupe Pasteur Mutualité (M. Dufies), the Framework Program 7 of the European Commission -Marie Curie Intra-European grant "VELYMPH", Fondation François Xavier Mora (S Giuliano), the association “Le Cancer du Sein, Parlons-en” and IRIS Pharma (P. Diogop NDiaye).
Authors' Contributions
Conception and design: P.Diogop Ndiaye, M.Dufies, S. Giuliano, K.S., L.Douguet, R.Grépin, G. Pagès. Development of methodology: P.Diogop Ndiaye, G. Pagès. Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): P.Diogop Ndiaye, M.Dufies, N.Glisse, J.Mintcheva, L.Douguet, M. Wurmser, G. Pagès. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): P.Diogop Ndiaye, M.Dufies, N.Glisse, J.Mintcheva, G. Pagès. Writing, review, and/or revision of the manuscript: G. Pagès. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): V.Vouret-Craviari, B. Mograbi, G. Pagès. Study supervision: G. Pagès.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no conflict of interest exists.
References
1. Gore ME, Szczylik C, Porta C, Bracarda S, Bjarnason GA, Oudard S. et al. Final results from the large sunitinib global expanded-access trial in metastatic renal cell carcinoma. Br J Cancer. 2015;113:12-9
2. Eisen T, Sternberg CN, Robert C, Mulders P, Pyle L, Zbinden S. et al. Targeted therapies for renal cell carcinoma: review of adverse event management strategies. J Natl Cancer Inst. 2012;104:93-113
3. Motzer RJ, Escudier B, Tomczak P, Hutson TE, Michaelson MD, Negrier S. et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013;14:552-62
4. Motzer RJ, Hutson TE, Cella D, Reeves J, Hawkins R, Guo J. et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med. 2013;369:722-31
5. Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F. et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373:1814-23
6. Motzer RJ, Hutson TE, Ren M, Dutcus C, Larkin J. Independent assessment of lenvatinib plus everolimus in patients with metastatic renal cell carcinoma. Lancet Oncol. 2016;17:e4-5
7. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S. et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449-56
8. Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK. et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med. 2018;378:1277-90
9. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S. et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373:1803-13
10. Aebischer D, Iolyeva M, Halin C. The inflammatory response of lymphatic endothelium. Angiogenesis. 2014;17:383-93
11. Hirakawa S, Brown LF, Kodama S, Paavonen K, Alitalo K, Detmar M. VEGF-C-induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood. 2007;109:1010-7
12. Alitalo A, Detmar M. Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene. 2012;31:4499-508
13. Karaman S, Detmar M. Mechanisms of lymphatic metastasis. J Clin Invest. 2014;124:922-8
14. Dufies M, Giuliano S, Ambrosetti D, Claren A, Ndiaye PD, Mastri M. et al. Sunitinib Stimulates Expression of VEGFC by Tumor Cells and Promotes Lymphangiogenesis in Clear Cell Renal Cell Carcinomas. Cancer Res. 2017;77:1212-26
15. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F. et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220-31
16. Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232-9
17. Blagoev KB, Wilkerson J, Stein WD, Motzer RJ, Bates SE, Fojo AT. Sunitinib does not accelerate tumor growth in patients with metastatic renal cell carcinoma. Cell Rep. 2013;3:277-81
18. Enholm B, Paavonen K, Ristimäki A, Kumar V, Gunji Y, Klefstrom J. et al. Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene. 1997;14:2475-83
19. Wang CA, Jedlicka P, Patrick AN, Micalizzi DS, Lemmer KC, Deitsch E. et al. SIX1 induces lymphangiogenesis and metastasis via upregulation of VEGF-C in mouse models of breast cancer. J Clin Invest. 2012;122:1895-906
20. Le Grand F, Grifone R, Mourikis P, Houbron C, Gigaud C, Pujol J. et al. Six1 regulates stem cell repair potential and self-renewal during skeletal muscle regeneration. J Cell Biol. 2012;198:815-32
21. Morfoisse F, Kuchnio A, Frainay C, Gomez-Brouchet A, Delisle MB, Marzi S. et al. Hypoxia induces VEGF-C expression in metastatic tumor cells via a HIF-1alpha-independent translation-mediated mechanism. Cell Rep. 2014;6:155-67
22. Varna M, Gapihan G, Feugeas JP, Ratajczak P, Tan S, Ferreira I. et al. Stem cells increase in numbers in perinecrotic areas in human renal cancer. Clin Cancer Res. 2015;21:916-24
23. Lupu-Plesu M, Claren A, Martial S, N'Diaye PD, Lebrigand K, Pons N. et al. Effects of proton versus photon irradiation on (lymph)angiogenic, inflammatory, proliferative and anti-tumor immune responses in head and neck squamous cell carcinoma. Oncogenesis. 2017;6:e354
24. Shen C, Kaelin WG Jr. The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol. 2013;23:18-25
25. Kammerer-Jacquet SF, Crouzet L, Brunot A, Dagher J, Pladys A, Edeline J. et al. Independent association of PD-L1 expression with noninactivated VHL clear cell renal cell carcinoma-A finding with therapeutic potential. Int J Cancer. 2017;140:142-8
26. Bilton R, Mazure N, Trottier E, Hattab M, Dery MA, Richard DE. et al. Arrest-defective-1 protein, an acetyltransferase, does not alter stability of hypoxia-inducible factor (HIF)-1alpha and is not induced by hypoxia or HIF. J Biol Chem. 2005;280:31132-40
27. Krieg M, Haas R, Brauch H, Acker T, Flamme I, Plate KH. Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene. 2000;19:5435-43
28. Cho H, Du X, Rizzi JP, Liberzon E, Chakraborty AA, Gao W. et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature. 2016;539:107-11
29. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281-308
30. Grifone R, Demignon J, Giordani J, Niro C, Souil E, Bertin F. et al. Eya1 and Eya2 proteins are required for hypaxial somitic myogenesis in the mouse embryo. Dev Biol. 2007;302:602-16
31. Lidke DS, Huang F, Post JN, Rieger B, Wilsbacher J, Thomas JL. et al. ERK nuclear translocation is dimerization-independent but controlled by the rate of phosphorylation. J Biol Chem. 2010;285:3092-102
32. Simon MP, Tournaire R, Pouyssegur J. The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J Cell Physiol. 2008;217:809-18
33. Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J. et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570-81
34. Turchi L, Aberdam E, Mazure N, Pouyssegur J, Deckert M, Kitajima S. et al. Hif-2alpha mediates UV-induced apoptosis through a novel ATF3-dependent death pathway. Cell Death Differ. 2008;15:1472-80
35. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
36. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
37. Kammerer-Jacquet SF, Brunot A, Pladys A, Bouzille G, Dagher J, Medane S. et al. Synchronous Metastatic Clear-Cell Renal Cell Carcinoma: A Distinct Morphologic, Immunohistochemical, and Molecular Phenotype. Clin Genitourin Cancer. 2017;15:e1-e7
38. Shinojima T, Oya M, Takayanagi A, Mizuno R, Shimizu N, Murai M. Renal cancer cells lacking hypoxia inducible factor (HIF)-1alpha expression maintain vascular endothelial growth factor expression through HIF-2alpha. Carcinogenesis. 2007;28:529-36
39. Schodel J, Grampp S, Maher ER, Moch H, Ratcliffe PJ, Russo P. et al. Hypoxia, Hypoxia-inducible Transcription Factors, and Renal Cancer. Eur Urol. 2016;69:646-57
40. Guo YC, Zhang M, Wang FX, Pei GC, Sun F, Zhang Y. et al. Macrophages Regulate Unilateral Ureteral Obstruction-Induced Renal Lymphangiogenesis through C-C Motif Chemokine Receptor 2-Dependent Phosphatidylinositol 3-Kinase-AKT-Mechanistic Target of Rapamycin Signaling and Hypoxia-Inducible Factor-1alpha/Vascular Endothelial Growth Factor-C Expression. Am J Pathol. 2017;187:1736-49
41. Du Q, Jiang L, Wang X, Wang M, She F, Chen Y. Tumor necrosis factor-alpha promotes the lymphangiogenesis of gallbladder carcinoma through nuclear factor-kappaB-mediated upregulation of vascular endothelial growth factor-C. Cancer Sci. 2014;105:1261-71
42. Szade A, Grochot-Przeczek A, Florczyk U, Jozkowicz A, Dulak J. Cellular and molecular mechanisms of inflammation-induced angiogenesis. IUBMB Life. 2015;67:145-59
43. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178-96
44. Dagher J, Kammerer-Jacquet SF, Brunot A, Pladys A, Patard JJ, Bensalah K. et al. Wild-type VHL Clear Cell Renal Cell Carcinomas Are a Distinct Clinical and Histologic Entity: A 10-Year Follow-up. Eur Urol Focus. 2016;1:284-90
45. Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV. et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol. 2004;5:74-80
46. Dieu-Nosjean MC, Giraldo NA, Kaplon H, Germain C, Fridman WH, Sautes-Fridman C. Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol Rev. 2016;271:260-75
47. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480-9
48. Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol. 2014;5:75
49. Lund AW, Duraes FV, Hirosue S, Raghavan VR, Nembrini C, Thomas SN. et al. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep. 2012;1:191-9
50. Giraldo NA, Becht E, Pages F, Skliris G, Verkarre V, Vano Y. et al. Orchestration and Prognostic Significance of Immune Checkpoints in the Microenvironment of Primary and Metastatic Renal Cell Cancer. Clin Cancer Res. 2015;21:3031-40
51. Liang B, Li Y. Prognostic Significance of VEGF-C Expression in Patients with Breast Cancer: A Meta-Analysis. Iran J Public Health. 2014;43:128-35
52. Bierer S, Herrmann E, Kopke T, Neumann J, Eltze E, Hertle L. et al. Lymphangiogenesis in kidney cancer: expression of VEGF-C, VEGF-D and VEGFR-3 in clear cell and papillary renal cell carcinoma. Oncol Rep. 2008;20:721-5
53. Kyzas PA, Cunha IW, Ioannidis JP. Prognostic significance of vascular endothelial growth factor immunohistochemical expression in head and neck squamous cell carcinoma: a meta-analysis. Clin Cancer Res. 2005;11:1434-40
54. Rini BI, Michaelson MD, Rosenberg JE, Bukowski RM, Sosman JA, Stadler WM. et al. Antitumor activity and biomarker analysis of sunitinib in patients with bevacizumab-refractory metastatic renal cell carcinoma. J Clin Oncol. 2008;26:3743-8
55. Dufies M, Giuliano S, Viotti J, Borchiellini D, Cooley LS, Ambrosetti D. et al. CXCL7 is a predictive marker of sunitinib efficacy in clear cell renal cell carcinomas. Br J Cancer. 2017;117:947-53
56. Cao Y, Wang L, Nandy D, Zhang Y, Basu A, Radisky D. et al. Neuropilin-1 upholds dedifferentiation and propagation phenotypes of renal cell carcinoma cells by activating Akt and sonic hedgehog axes. Cancer Res. 2008;68:8667-72
57. Cao Y, Hoeppner LH, Bach S, E G, Guo Y, Wang E. et al. Neuropilin-2 promotes extravasation and metastasis by interacting with endothelial alpha5 integrin. Cancer Res. 2013;73:4579-90
58. Yeh YW, Cheng CC, Yang ST, Tseng CF, Chang TY, Tsai SY. et al. Targeting the VEGF-C/VEGFR3 axis suppresses Slug-mediated cancer metastasis and stemness via inhibition of KRAS/YAP1 signaling. Oncotarget. 2017;8:5603-18
59. Timoshenko AV, Rastogi S, Lala PK. Migration-promoting role of VEGF-C and VEGF-C binding receptors in human breast cancer cells. Br J Cancer. 2007;97:1090-8
60. Majumder M, Tutunea-Fatan E, Xin X, Rodriguez-Torres M, Torres-Garcia J, Wiebe R. et al. Co-expression of alpha9beta1 integrin and VEGF-D confers lymphatic metastatic ability to a human breast cancer cell line MDA-MB-468LN. PLoS One. 2012;7:e35094
61. Majumder M, Xin X, Liu L, Girish GV, Lala PK. Prostaglandin E2 receptor EP4 as the common target on cancer cells and macrophages to abolish angiogenesis, lymphangiogenesis, metastasis, and stem-like cell functions. Cancer Sci. 2014;105:1142-51
62. Timoshenko AV, Chakraborty C, Wagner GF, Lala PK. COX-2-mediated stimulation of the lymphangiogenic factor VEGF-C in human breast cancer. Br J Cancer. 2006;94:1154-63
Author contact
![]() Corresponding author: Gilles Pagès: gpagesfr
Corresponding author: Gilles Pagès: gpagesfr