Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(3):853-867. doi:10.7150/thno.29553 This issue Cite
Research Paper
Monoclonal Antibody against CXCL1 (HL2401) as a Novel Agent in Suppressing IL6 Expression and Tumoral Growth
Makito Miyake1*, Hideki Furuya2,3 *, Sayuri Onishi1, Kanani Hokutan2,3, Satoshi Anai1, Owen Chan2, Sixiang Shi4, Kiyohide Fujimoto1, Steve Goodison5, Weibo Cai4 ![]() , Charles J. Rosser2,3,5
, Charles J. Rosser2,3,5 ![]()
1. Nara Medical University, Department of Urology, Nara, Japan,
2. University of Hawaii Cancer Center, Clinical and Translational Research, Honolulu, Hawaii,
3. Department of Molecular Biosciences and Bioengineering, University of Hawaii at Manoa, Honolulu, HI USA
4. Department of Radiology, University of Wisconsin - Madison, Madison, Wisconsin, USA
5. Nonagen Bioscience Corporation, Jacksonville, Florida
*co-first authors
Received 2018-8-28; Accepted 2018-12-22; Published 2019-1-25
Abstract

Rationale: The expression of the chemokine (C-X-C motif) ligand 1 (CXCL1), an inflammatory protein, has been reported to be up-regulated in many human cancers. The mechanisms through which aberrant cellular CXCL1 levels promote specific steps in tumor growth and progression are unknown.
Methods: We described the anticancer effects and mechanism of action of HL2401, a monoclonal antibody directed at CXCL1 with in vitro and in vivo data on bladder and prostate cancers.
Results: HL2401 inhibited proliferation and invasion of bladder and prostate cells along with disrupting endothelial sprouting in vitro. Furthermore, novel mechanistic investigations revealed that CXCL1 expression stimulated interleukin 6 (IL6) expression and repressed tissue inhibitor of metalloproteinase 4 (TIMP4). Systemic administration of HL2401 in mice bearing bladder and prostate xenograft tumors retarded tumor growth through the inhibition of cellular proliferation and angiogenesis along with an induction of apoptosis. Our findings reveal a previously undocumented relationship between CXCL1, IL6 and TIMP4 in solid tumor biology.
Principal conclusions: Taken together, our results argue that CXCL1 plays an important role in sustaining the growth of bladder and prostate tumors via up-regulation of IL6 and down-regulation of TIMP4. Targeting these critical interactions with a CXCL1 monoclonal antibody offers a novel strategy to therapeutically manage bladder and prostate cancers.
Keywords: bladder cancer, CXCL1, IL6, prostate cancer, TIMP4
Introduction
Chemokines are known to be critical mediators of the inflammatory response by regulating recruitment of immune cells from both the innate and adaptive immune systems to diseased tissues. Dysregulated expression and activity of certain chemokines have been implicated in the initiation and progression of several cancers [1]. Specifically, the chronic exposure of cells to a chemokine-laced milieu is associated with macrophage and T cell accumulation, chronic activation of macrophages, abnormal angiogenesis and DNA damage due to the presence of reactive oxygen species [2, 3]. Furthermore, chemokines have been known to regulate multiple processes during tumor progression including primary tumor growth, tumor angiogenesis and development of metastatic disease [4, 5]. Chemokines have also been known to enhance epithelial-stromal interactions facilitating tumor growth and invasion [6].
Chemokine (C-X-C motif) ligand 1 (CXCL1), also known as growth-regulated oncogene-alpha or melanoma growth stimulatory activity, alpha, is a secreted growth factor that interacts with the G-protein coupled receptor CXCR2, also known as interleukin-8 receptor type beta (IL8RB) [7, 8], that plays a key role in inflammation and as a chemoattractant for neutrophils [9, 10]. It has been reported to be overexpressed in many cancers (Fig. S1) [11-13]. On the other hand, its presence has also been negatively associated with cancers [14], attesting to its complex role in human tumorigenesis.
Little attention has been given to CXCL1 as a therapeutic target, but CXCL1 has been proposed as a potential diagnostic biomarker. We have shown that CXCL1 protein expression in malignant tissue is overexpressed in both bladder and prostate tumors when compared to benign tissue. Specifically in prostate tissue, increased CXCL1 protein levels were associated with higher-grade tumors [15]. Surprisingly, additional studies assessing CXCL1 in human prostate cancer are lacking. In bladder cancer, it was demonstrated that CXCL1 protein expression was present in cancerous tissue but was entirely absent in benign tissue. A combined CXCL1 immunostaining score was significantly higher in high-grade tumors relative to low-grade tumors as well as in high stage tumors (T2-T4) compared to low stage tumors (Ta-T1). An increase in CXCL1 immunostaining was also associated with reduced disease-specific survival [16]. Kawanishi et al. showed that urinary CXCL1 levels were significantly higher in patients with invasive bladder cancer (pT1-4) than those with noninvasive pTa tumors and normal control and thus may provide an independent factor for predicting an invasive phenotype [17]. Additionally, other human tumor types have reported overexpression of CXCL1, thus possibly broadening the implication of CXCL1 as a potential therapeutic target.
Here, we report that the manipulation of CXCL1 expression influences the rates of proliferation, migration and invasion of human bladder and prostate cancer cells. We show that this influence involves a previously unrecognized interplay between CXCL1, interleukin 6 (IL6) and tissue inhibitor of metalloproteinase 4 (TIMP4). Hence, pharmacologic perturbation of CXCL1 might be an attractive therapeutic strategy to restrict cancer development. In order to test this hypothesis, we derived an anti-CXCL1 monoclonal antibody (mAb) HL2401 for in vitro and in vivo testing. We showed that treatment of tumor cells with the HL2401 mAb mimicked the effects of manipulating CXCL1 mRNA in vitro. Systemic administration of HL2401 restricted both bladder and prostate xenograft growth through the inhibition of cellular proliferation and angiogenesis and a concomitant induction of apoptosis, without causing observable toxicity. Collectively, the study provides new information on the role of CXCL1 in tumor growth regulation and reports the derivation of mAb that effectively interferes with CXCL1 activity. This reagent may open new opportunities to study both the biology of CXCL1 and the therapeutic potential of targeting this tumor-associated cytokine.
Results
CXCL1 plays a role in the invasion of cancer cells
Given the upregulation of CXCL1 in bladder and prostate cancers [15-17], we investigated how CXCL1 influences key tumor cell processes. Evaluation of the human cell lines T24, DU145 and PC3 demonstrated a range of CXCL1 expression levels, with the bladder cancer cell line T24 and the prostate cancer cell line PC3 expressing the highest levels of CXCL1 (Fig. 1A). Similarly, CXCR2, the receptor for CXCL1, was expressed in all cells assessed (Fig. 1B). To test the influence of CXCL1 on cellular functions, we created stably transfected CXCL1-overexpressing DU145 cells (DU145-CXCL1-OE3&8), and stably knocked down CXCL1 expression via shRNA targeting vectors in T24 and PC3 cells. CXCL1 expression in these cells at the mRNA level (Fig. S2A) and protein level (Fig. 1C) was confirmed. In addition, secreted CXCL1 was monitored in the cell culture media using a commercial ELISA assay. As expected, secreted CXCL1 correlated with cellular CXCL1 levels (Fig. 1D).
CXCL1 knockdown in cancer cell lines inhibits invasion and tube formation. A, Western blot and real-time RT-PCR analyses of CXCL1 in human cancer cell lines T24 (bladder), DU145 and PC3 (prostate). B, Western blot and real-time RT-PCR analyses of CXCR2 in human cancer cell lines T24, DU145 and PC3, and HEK293T (human embryonic kidney cells). Parental HEK293T cells and HEK293T cells overexpressing CXCR2 were used as negative and positive controls, respectively. C, T24 and PC3 cells were stably knockdown by shRNA targeting CXCL1 (T24-CXCL1-KD4&8 and PC3-CXCL1KD7) or scrambled control (T24-shSCR and PC3-shSCR), while DU145 cells were stably transfected with empty vector (DU145-Empty) or CXCL1 expression plasmid (DU145-CXCL1OE3&8). Western blot analysis demonstrated altered CXCL1 expression in the two selected T24-CXCL1 clones 4 and 8, the two selected DU145 clones 3 and 8 and the one PC3 clone 7. D, CXCL1 ELISA assay confirmed altered secretion of CXCL1 in line with the above western blot analysis. E, T24 cells (T24-shSCR, T24-CXCL1-KD4&8), DU145 cells (DU145-Empty, DU145-CXCL1-OE3&8, PC3 cells (PC3-shSCR, PC3-CXCL1-KD7) were subjected to in vitro migration and invasion assays. Data are average percent of control and SDs of three independent experiments conducted in triplicate. Overexpression of CXCL1 in DU145 cells was noted to correspond with an increase in invasive potential, while silencing of CXCL1 in T24 and PC3 cells resulted in a reduced invasive potential. Data from one representative experiment are presented as mean ± SD, *, p < 0.05; **, p < 0.01 and ***, p < 0.001. All western blot experiments were performed in triplicate, representative experiment was shown.
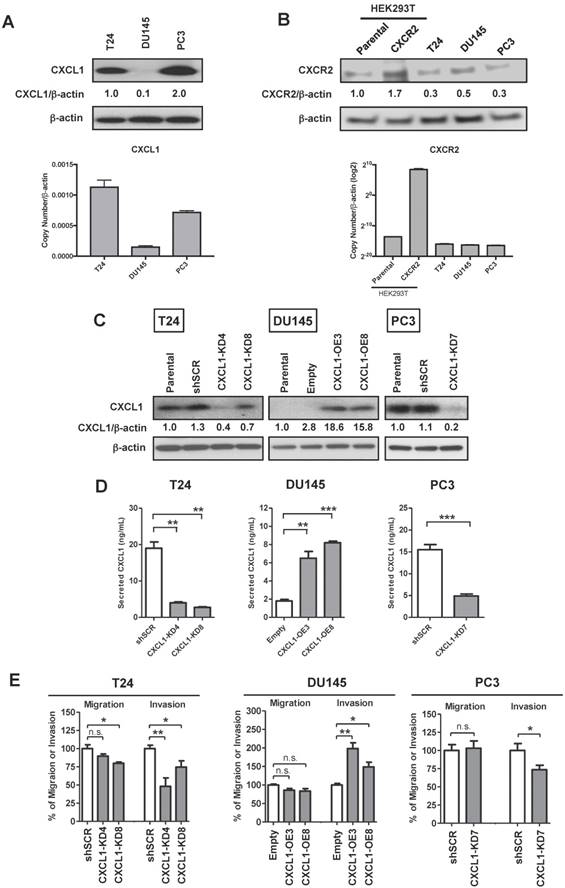
In an in vitro proliferation assay at 72 hours, manipulation of CXCL1 expression did not effect tumor cell proliferation (Fig. S2B). In an in vitro migration and invasion assay (Fig. 1E), the migratory potential of DU145-CXCL1-OE3&8 clones was not enhanced compared to the DU145-Empty control, however the invasive potential of DU145-CXCL1- OE3 & OE8 clones was significantly enhanced by at least 50% compared to DU145-Empty cells (p < 0.01). Similarly, the migratory potential of PC3-CXCL1-KD7 cells was not reduced compared to PC3-shSCR, but the invasive potential was significantly reduced by 27% in PC3-CXCL1-KD7 compared to PC3-shSCR (p < 0.01). The same phenomenon was also observed in the migration and invasion assays of T24 clones. Specifically, T24-CXCL1-KD8 cells (p < 0.01) but not T24-CXCL1-KD4 cells showed an inhibition in cell migration, however both T24-CXCL1-KD4&8 clones demonstrated a significant inhibition (at least 25%) of invasive potential compared to T24-shSCR (p < 0.01). These results suggest that CXCL1 directly stimulates the invasion of human cancer cells.
CXCL1 plays a critical role in the proliferation and sprouting of endothelial cells
We previously found that exogenous CXCL1 induced endothelial cell proliferation and blocked induction of apoptosis of endothelial cells [18]. Here, we tested whether exogenous CXCL1 might influence endothelial cell sprouting by subjecting HUVEC cultures to conditioned media from the above manipulated cell lines. In a tube-formation assay, the total length of structures formed by HUVECs on growth factor-reduced Matrigel was significantly enhanced (~60%) when treated with media from DU145-CXCL1-OE3&8 clones (Fig. S2C). Accordingly, the total length of tube-like structures was significantly reduced when treated with conditioned media from CXCL1-knockdown T24 and PC3 cells (~50% and ~28%, respectively). These results confirm our previous results [18] that CXCL1 plays a critical role in developing and sustaining angiogenesis and tumor vasculature.
CXCL1 effects are mediated by IL6 and TIMP4
Based on the findings that CXCL1 can influence invasion of cancer cells as well as influence HUVEC tube formation, we performed a limited gene expression profiling of our cell line panel in order to identify changes in tumor metastasis and angiogenic factor expression. An Angiogenesis PCR array containing 84 targets was queried from two independent experiments and significant fold change deviations were recorded (Table S1). Among the genes that were noted to consistently correlate with CXCL1 expression were IL6, Jagged 1 protein (JAG1) and Chondromodulin-1 (LECT1) (Fig. 2A). A Tumor Metastasis PCR array containing an additional 84 targets was also queried from two independent experiments and significant fold change deviations (p < 0.05) were recorded (Table S2). Among the genes that were noted to consistently correlate with CXCL1 expression were Insulin-like growth factor 1 (IGF1), Matrix metalloproteinase 2 (MMP2) and TIMP4 (Fig. 2A).
CXCL1 stimulates IL6 and inhibits TIMP4 via EGF and ERK1/2. A, Angiogenesis and Metastasis RT2 Profiler PCR Arrays evaluated by quantitative RT-PCR assessed the expression of 84 gene transcripts related to angiogenesis and 84 genes related to metastasis. Gene expression levels were normalized to the combination of five housekeeping genes (β-actin, B2M, GAPDH, HPRT and RPLP). Changes in expression of IL6, JAG1 and LECT (angiogenesis) and IGF1, MMP2 and TIMP4 (Metastasis) were consistently noted in the three cell lines when CXCL1 expression was manipulated (see other changes listed in Table S1). B, Comparison of proteins related to angiogenesis and matrix metalloproteinase by an antibody array. C, Immunoblots of protein extracts with antibodies for IL6, TIMP4 in T24-shSCR, T24-CXCL1-KD4&8, DU145-Empty, DU145-CXCL1-OE3&8, and PC3-shSCR, PC3-CXCL1-KD7. β-actin served as loading control.
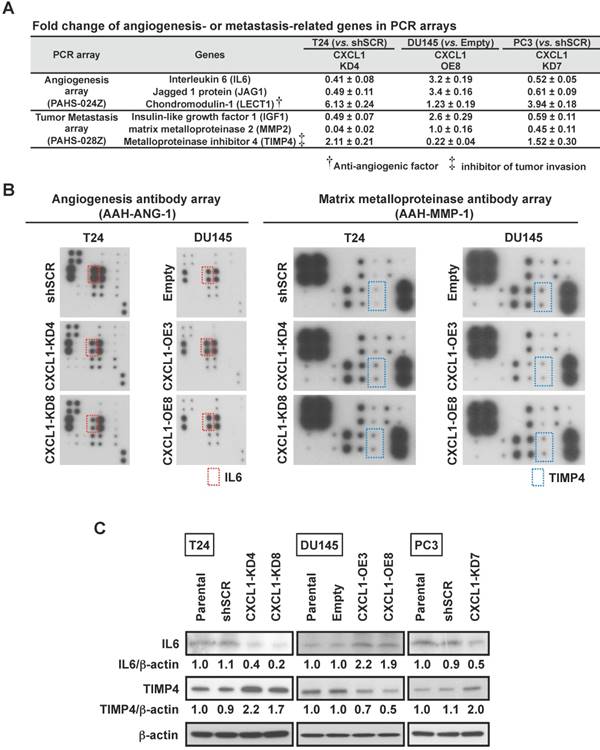
Using an Angiogenesis antibody array and a Matrix Metalloproteinase antibody array, we set out to corroborate the above noted gene expression associations at the protein level. T24-CXCL1-KD4&8 clones had a reduction in IL6 protein levels, while the DU145-CXCL1-OE3&8 had an increase in IL6 protein levels (Fig. 2B). Changes in JAG1 and LECT1 could not be confirmed at the protein level. We also confirmed that TIMP4 protein levels were elevated in T24-CXCL1-KD4&8 clones, while TIMP4 protein levels were reduced in DU145-CXCL1-OE3&8 (Fig. 2B). PC3-CXCL1-KD7 was noted to harbor more TIMP4 and less IL6 compared to PC3-shSCR (data not shown). Changes in IGF1 and MMP2 could not be confirmed at the protein level. We also confirmed by Western blot analysis that knockdown of CXCL1 by shRNA resulted in a reduction of IL6 and an increase in TIMP4 in both T24 and PC3 cell lines, while overexpression of CXCL1 resulted in an increase in IL6 and a reduction of TIMP4 in DU145 cell line (Fig. 2C). Quantitative densitometric analysis was performed to compare the expression level of IL6 and TIMP4 between control cells and CXCL1-manupulated cells (Fig. S3A). Lastly, we confirmed TIMP4 knockdown by siRNA in T24-CXCL1-KD4&8 clones rescued invasion reduced by CXCL1 downregulation, while IL6 knockdown by siRNA in DU145-CXCL1- OE3&8 clones abolished tube formation induced by forced expression of CXCL1 (Fig. S3B). Taken together these results revealed a previously unrecognized interplay between CXCL1, IL6 and TIMP4 in human cancer cells.
Selection of monoclonal antibody against CXCL1 (HL2401)
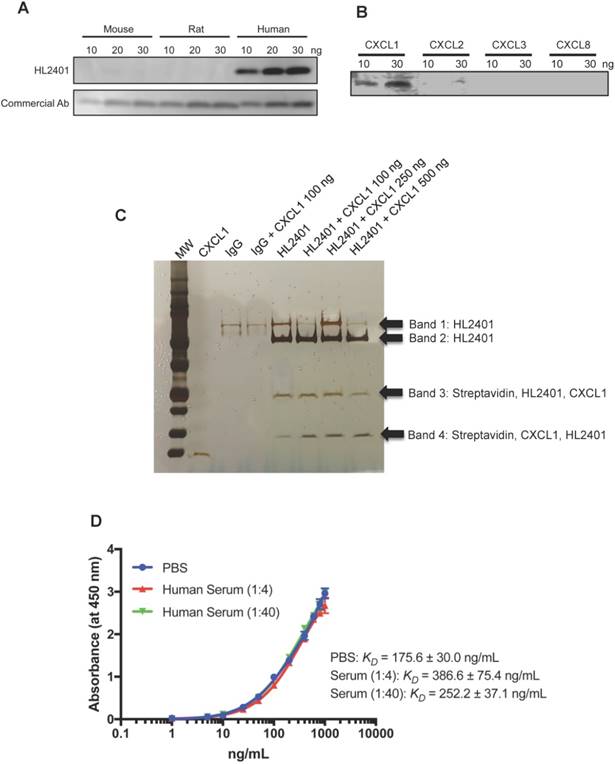
On the basis that CXCL1 may enhance human tumor growth, we generated an anti-CXCL1 neutralizing monoclonal mouse antibody (HL2401) for preclinical anti-tumor testing. HL2401 (IgG1) binds to recombinant human CXCL1, but not mouse and rat CXCL1 (Fig. 3A). Since CXCL2, CXCL3 and CXCL8 could also bind to CXCR2, and their amino acid sequences are similar to CXCL1 (Identities of CXCL2: 90%, CXCL3: 87% and CXCL8: 48%), we tested whether HL2401 binds to recombinant human CXCL2, CXCL3 and CXCL8. We found that HL2401 specifically binds to CXCL1, although HL2401 slightly binds to CXCL2 (Fig. 3B). In addition, immunoprecipitation followed by LC/MS-MS analysis confirmed that HL2401 binds specifically to CXCL1 (Figure 3C and Table S3). The apparent affinity-binding constant (KD) value for CXCL1 in PBS and serum (1:4 dilution and 1:40 dilution) were 175.6 ± 30.0 ng/mL, 386.6 ± 75.4 ng/ and 252.2 ± 37.1 ng/mL, respectively (Fig. 3D).
CXCL1 antibody binding specificity and affinity. A, CXCL1 antibody specifically recognizes human CXCL1, but not mouse and rat. B, CXCL1 antibody specifically recognizes CXCL1, but not CXCL2, CXCL3 and CXCL8. C, Specificity of CXCL1 Antibody. The following precipitate samples (Lane 1 - recombinant CXCL1, Lane 2 - serum precipitated with IgG, Lane 3 - 100 ng CXCL1 spiked serum precipitated with IgG, Lane 4 - serum precipitated with HL2401, Lane 5 - 100 ng CXCL1 spiked serum precipitated with HL2401, Lane 6 - 250 ng CXCL1 spiked serum precipitated with HL2401 and Lane 7 - 500 ng CXCL1 spiked serum precipitated with HL2401) were run on a NuPAGE Novex 4-12% Bis-Tris Gel and then stained with the SilverQuest Kit. Arrows indicate CXCL1. C, saturation curve of CXCL1 antibody binding to CXCL1 in PBS and serum by ELISA shows KD of 175.6 ± 30.0 ng/mL in PBS, 386.6 ± 75.4 ng/mL in serum (1:4 dilution), and 252.2 ± 37.1 ng/mL in serum (1:40 dilution).
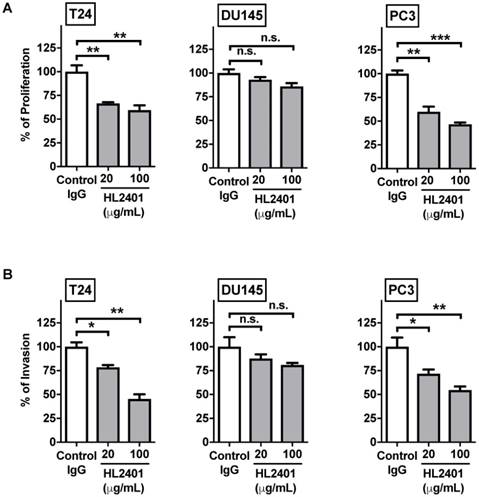
HL2401 decreases both cellular proliferation and invasion of cancer cells overexpressing CXCL1
In an in vitro proliferation assay at 72 hours, proliferation of T24, PC3 and HUVEC cell lines, but not DU145 cells, were significantly inhibited by HL2401 (20 and 100 μg/mL, Fig. 4A and Fig. S4A). In an in vitro invasion assay, the invasive potential (Fig. 4B) of T24 and PC3 was significantly reduced with the addition of HL2401 (20 μg/mL) (p < 0.01). Perhaps since parental DU145 lacks CXCL1, invasive potential was unchanged by the addition of HL2401 (Fig. 4B). Migration evaluation was not performed since it was minimally altered in the CXCL1-manipulated clones. Moreover, we noted that HL2401 in a dose response manner significantly reduced the total length of structures formed by HUVECs in an endothelial tube formation assay (p < 0.01), (Fig. S4B).
Neutralizing monoclonal antibody to CXCL1 (HL2401) suppresses cancer cell proliferation and invasion. A, Cellular proliferation of T24, DU145 and PC3 parental cells was measured by MTT assay at 72 hours after treatment with increasing concentrations of HL2401 (0, 20 and 100 μg/mL). Based on the growth curves, IC50 values were calculated from three independent experiments for each cell line. B, T24, DU145 and PC3 parental cells were subjected to in vitro invasion assays after treatment with HL2401 (20 μg/mL). Data are average values and SDs of three independent experiments conducted in triplicate. Targeting CXCL1 resulted in a reduction in cell invasion in T24 and PC3 cells.
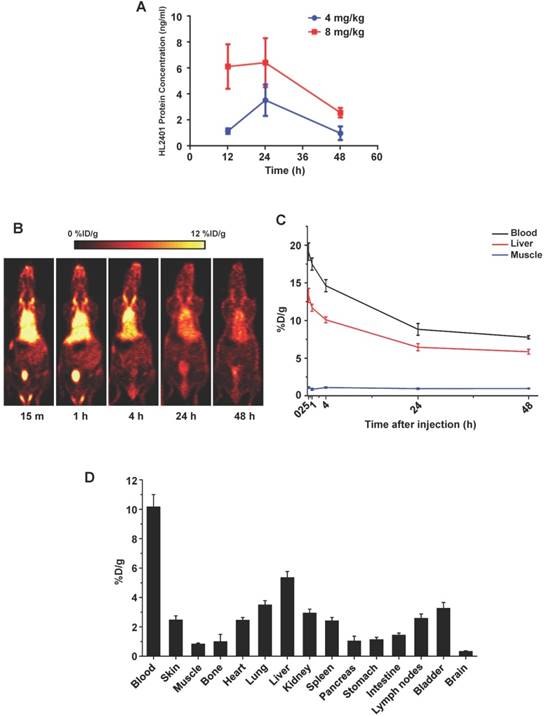
Pharmacokinetic studies and biodistribution of HL2401
Following intraperitoneal administration of HL2401 in C57BL/6 mice, we performed pharmacokinetics study. After administration, the plasma concentration of HL2401 declined rapidly, due to rapid distribution to peripheral components (Fig. 5A). Limitations of assay sensitivity prevented characterization of terminal elimination (i.e., excretion). Concentration time analysis of HL2401 in plasma after a single dose of 4 mg/kg or 8 mg/kg was 22.89 ng/g and 46.71 ng/g (Cmax), 2.49 hours and 2.71 hours (t1/2) and 0.046 units and 0.044 units (clearance), respectively. Similarly after a single intravenous injection of 0.5 mg/kg in CF-1 mice, the radiolabeled antibody was rapidly distributed, remaining above the limits of detection for over 48 hours on PET imaging (Cmax = 19.15 %D/g at 15 min, t1/2α ≈ 3.5 min, t1/2β ≈ 44.0 hours) (Fig. 5B and C). The ex vivo bio-distribution data well matched with the imaging results, confirming the accuracy of PET imaging (Fig. 5D).
Pharmacokinetic and Quantitative analysis of the PET data. A, Pharmacokinetic profile of CXCL1 antibody in plasma from mice receiving a single intraperitoneal dose. In all cases, plasma samples collected at 48 hours post-injection were above lower levels of quantification. B, Representative serial coronal PET images at 15 min, 1, 4, 24 h and 48 h post-injection of 64Cu-NOTA-CXCL1 antibody in ICR mice. C, Time-activity curves of blood, liver, and muscle after intravenous injection of 64Cu-NOTA-CXCL1 antibody (n = 4). D, Biodistribution of 64Cu-NOTA-CXCL1 antibody in ICR mice at 48 h post-injection, after the last PET scans.
HL2401 inhibits tumor growth, reduces proliferation, induces apoptosis, impairs tumor vasculature, and influences IL6 expression in tumor xenografts
Having shown the effects of HL2401 in vitro, we investigated whether targeting CXCL1 with a monoclonal antibody could inhibit xenograft tumor growth. Once again, no observable toxicity (i.e., no weight change or activity change) was evident with the reported doses of HL2401. At the end of 5 wk endpoint of an in vivo study, control T24 xenografts reached an average of 388 mm3 in size. T24 xenografts treated twice weekly with 8 mg/kg of HL2401 reached 224 mm3 (p < 0.05) (Fig. 6A). Similarly, PC3 tumors in mice treated with 8 mg/kg of HL2401 were reduced (p < 0.05) in size at the experimental endpoint (Fig S5A). We noted that long-term treatment of HL2401 did not effect mouse body weight (Fig S6), suggesting that HL2401 has no observable toxicity or adverse effects in the mice.
Effect of targeting CXCL1 on T24 bladder xenograft tumor growth. Tumor growth was established by subcutaneous injection of parental T24 cells into athymic mice (nu/nu). HL2401 (8 mg/kg), a neutralizing monoclonal antibody to CXCL1, was injected intraperitoneally as described in Materials and Methods. Sterile PBS injection served as control. A, Tumor size, recorded over 5 weeks, was plotted as mean ± SD for three treatment groups per cell line (n=10/group). Statistical significance is represented by *, p < 0.05. Treatment with HL2401 (8 mg/kg) was associated with a reduction in tumor burden. B, H&E staining, CXCL1 and CXCR2, immunostaining of tumors are shown (original magnification 200 ×). A reduction in CXCL1 was noted in tumors treated with HL2401. C, Apoptotic index (AI) was quantified based on caspase-3 immunostaining. MVD was quantified based on PECAM-1 immunostaining. Proliferative index (PI) was quantified based on Ki-67 immunostaining. Data are presented as mean ± SD, *, p < 0.05. D, Immunostaining of tumors corresponding to AI, MVD and PI (original magnification 200 ×). E, IL6 and TIMP4 immunostaining of tumors are shown (original magnification 200 ×). A reduction in IL6, while an increase in TIMP4 was noted in tumors treated with HL2401. F, Immunofluorescent staining of xenograft tumors. CXCL1 and PECAM-1 immunofluorescent staining of tumors treatment with HL2401 (200 μg) are shown (original magnification 1000x), demonstrating CXCL1 expression with tumor epithelia as well as in tumor vasculature.
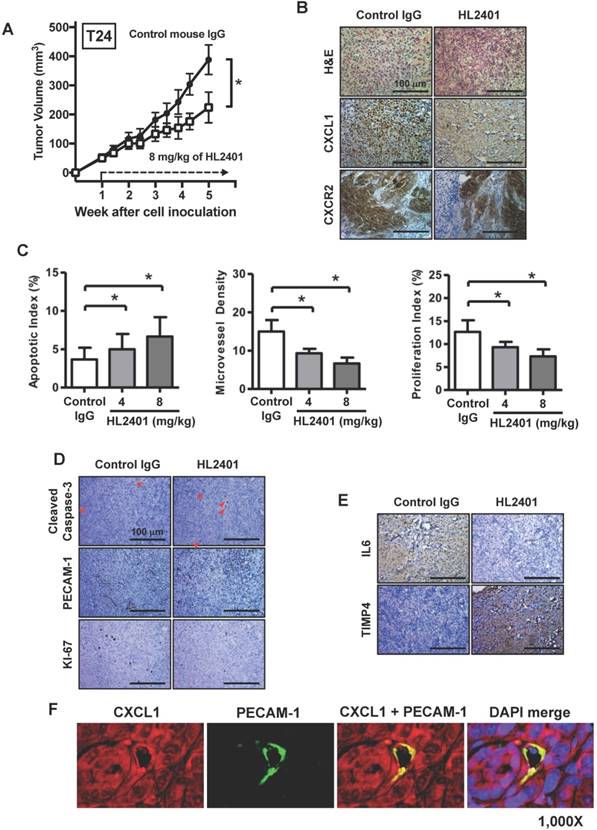
IHC analysis of excised xenografts revealed a reduction in CXCL1 expression when treated with 8 mg/kg of HL2401. It is possible the difference seen between T24 and PC3 xenografts response to H2401 is due to the fact that CXCR2 expression was less prevalent in PC3 xenografts compared to T24 xenografts (Fig. 6B and Fig. S6B). Next, the apoptotic index in xenografts was evaluated using cleaved caspase -3 immunostaining. Analyses revealed a significant increase in cleaved caspase-3 (indication of apoptosis) in T24 xenografts treated with 8 mg/kg of HL2401. The apoptotic index was increased in T24 by 35% (p < 0.05) in tumors from animals treated with 8 mg/kg of HL2401 (Fig. 6C). Apoptotic index in PC3 xenograft tumors was increased by 42% (p < 0.05) in tumors treated with 8 mg/kg of HL2401 (Fig. S5C and S5D). To monitor associated angiogenic index in these xenografts, microvessel density (MVD) was evaluated using PECAM-1 immunostaining. Analyses revealed a significant reduction of MVD (angiogenesis) in T24 xenografts treated with 8 mg/kg of HL2401. MVD was reduced in T24 xenografts by 52% (p < 0.05) treated with 8 mg/kg of HL2401 (Fig. 6C and 6D). Similar reduction in MVD was noted in PC3 xenografts treated with HL2401 (Figure S5C and S5D). To monitor associated proliferative capability in xenografts, proliferation index was evaluated using Ki-67 immunostaining. In line with the observed reduction in MVD, a reduction in proliferation index was evident in T24 xenografts treated with 8 mg/kg of HL2401 (Fig. 6C and 6D). Proliferative index was reduced in T24 by 50% (p < 0.05) in tumors from animals treated with 8 mg/kg of HL2401. Similarly, PC3 xenografts noted a reduction in proliferative index of 39% (p < 0.05) (Figure S5C and S5D). Furthermore, we confirmed a reduction of IL6 and an increase in TIMP4 in both T24 and PC3 tumors from animals treated with 8 mg/kg of HL2401 (Fig. 6E). Lastly, we performed immunofluorescent staining on the T24 and PC3 xenograft tumors for CXCL1 and PECAM-1 to note the location of CXCL1. In both T24 and PC3 xenografts, CXCL1 was expressed in the tumor cells in addition to the tumor-associated endothelial cells (Fig 6F and Fig. S5F). These in vivo observations corroborate the in vitro findings and confirm a role for CXCL1 regulation of tumor growth associated with an increase in IL6 expression and a reduction in TIMP4 expression and support a role for CXCL1 as a viable therapeutic target.
Discussion
CXCL1 is a multifunctional chemokine and a potential target for cancer therapy. Analysis of TCGA data demonstrates that CXCL1 is frequently overexpressed in numerous cancers (Fig. S1), thus it is a relevant cancer target. Herein, we demonstrated that CXCL1 expression in human cancer epithelial cells stimulates invasion, and secreted CXCL1 released by these cells stimulate the sprouting of endothelial cells. In addition, we revealed that an anti-CXCL1 neutralizing monoclonal antibody (HL2401) can: a) inhibit cancer cell proliferation, b) inhibit cancer cell invasion, c) inhibit endothelial sprouting and d) inhibit subcutaneous xenograft tumors expressing CXCL1 via reduction in both proliferation and angiogenesis, and the induction of apoptosis. Subsequently, we demonstrated that CXCL1 expression is positively correlated with the expression of IL6 and inversely correlated with TIMP4 expression.
TIMP4 is a member of the tissue inhibitors of metalloproteinases (MMPs) family, which is comprised of four members (TIMP1-4) with high sequence homology and structural identity, but with different tissue expression, regulation and inhibitory characteristics. The TIMPs regulate such diverse processes as extracellular matrix (ECM) remodeling, and growth factors and their receptors' activities through the inhibition of MMPs. Numerous tumors, including bladder and prostate, have been noted to have lower levels of TIMP4 [19]. IL6 is a multifunctional pro-inflammatory cytokine that functions in inflammation and the maturation of B cells. IL6 expression and function are altered in inflammatory-associated disease states (e.g., arthritis) as well as in several human cancers, including prostate [20] and bladder cancer [21]. Binding of IL6 to its membrane receptor is followed by initiation of signal transduction through one of several pathways: JAK/STAT, MAPK and/or PI3K pathways. In addition to regulation through its membrane receptor, IL6 also acts through trans-signaling in regulation of proliferation, migration, and invasion [22]. Our study supports the idea that CXCL1 influences tumor growth through a) the induction of IL6, which leads to enhancement of cellular proliferation, migration and invasion and b) the inhibition of TIMP4, which may facilitate the activation of MMPs enabling cellular growth and motility. Therapeutically targeting CXCL1 disrupts this molecular axis and halts these processes.
In addition to CXCL1's role within the tumor epithelia to stimulate and sustain tumor growth, recent studies reported that CXCL1 plays a major role in angiogenesis by stimulating human endothelial cells and increasing rates of angiogenesis. Angiogenesis is necessary for the growth and metastasis of tumors and thus is an important factor in the progression of cancer [23-25]. For tumors to expand beyond 1-2 mm3, new blood vessels must sprout from existing blood vessels in order to deliver nutrients and growth factors to the growing tissue mass [26, 27]. Angiogenesis is regulated by both activator and inhibitor molecules released by tumor-associated epithelial cells, endothelial cells, mesothelial cells, and leukocytes [7, 8, 24, 28]. The balance between these regulating molecules determines the extent of tissue angiogenesis [29]. The major endogenous inhibitors of angiogenesis include thrombospondin and interferons [30-32], while the major endogenous activators of angiogenesis include fibroblast growth factor (FGF) [33], vascular endothelial growth factor (VEGF) [34, 35], interleukin 8 (IL8, also known as CXCL8) [36, 37], platelet-derived endothelial cell growth factor [38] and members of the MMP family (MMP2, MMP9) [39]. In quiescent normal tissues, factors that inhibit angiogenesis predominate, whereas in rapidly dividing tissues (e.g., tumors), the balance of angiogenic molecules favors the stimulation of angiogenesis and tumor growth [7, 25].
Blockade of CXCL1 resulted in the reduction of human endothelial cell viability and negated angiogenesis induction. In addition, it was noted that CXCL1's effects require interaction with its receptor CXCR2, involves the ERK1/2 signaling pathway, and leads to the expression and secretion of EGF, a potent stimulator of angiogenesis Furthermore, an in vivo Matrigel plug angiogenesis assay confirmed that interference of CXCL1 function resulted in the inhibition of angiogenesis [18]. Collectively, the apparent significance of CXCL1 in both the tumor and its associated stroma supports the rationale for CXCL1-targeting therapeutics. Here, we demonstrate an important role for CXCL1 in human solid tumor survival and progression through its interactions with IL6 and TIMP4. Reduction in xenograft growth through treatment with the HL2401 antibody was accompanied with desirable changes in apoptosis, cellular proliferation rates and angiogenesis. Together, our data provide evidence to support the potential value of this therapeutic approach and CXCL1 as a novel drug target of human cancers.
Methods
Cells and reagents
Human embryonic kidney cells 293T (HEK293T), human bladder cancer cell line T24 and the human prostate cancer cell lines DU145 and PC3 were purchased from American Type Culture Collection (ATCC, Manassas, VA). Cell lines were maintained in DMEM or RPMI 1640 media as previously described [40, 41]. Primary human umbilical vein endothelial cells (HUVEC, Cambrex Bio Science, Walkersville, MD, USA) were cultured in EBM-2 basal media supplemented with EGM-2 MV Kit (Lonza, Walkersville, MD) containing 2% FBS. HUVEC cells of passage 6 to 8 were used. The above cell lines were authenticated by Genetic Resources Core Facility at Johns Hopkins (Baltimore, MD). All cell lines were confirmed to be mycoplasma free.
Immunoblotting
Cell lysate and immunoblotting were performed using standard protocols as previously described [41, 42]. Total protein from cultured cells was extracted using a RIPA buffer with Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific). Twenty micrograms of total protein (assessed using BCA protein assay) were subjected to SDS-PAGE using Any kD polyacrylamide gels (Bio-Rad Laboratories). Proteins were transferred to Immun-Blot PVDF Membrane (Bio-Rad Laboratories) and stained using a goat anti-human CXCL1 antibody (sc-1374, dilution 1/250; Santa Cruz Biotechnology), mouse anti-human CXCR2 antibody (ab24963, dilution 1:500; Abcam), rabbit anti-human IL6 antibody (sc-1265, dilution1:200, Santa Cruz Biotechnology), goat anti-human TIMP4 antibody (sc-9375, dilution 1:200, Santa Cruz Biotechnology) and mouse anti-human β-actin antibody (AC-15, dilution 1:10,000, Sigma-Aldrich). Stained proteins were detected using the SuperSignal West Pico Chemiluminescent Substrate (Pierce). Equal loading was confirmed by β-actin staining.
Stable transfection of cell lines
DU145 cells were stably transfected with human CXCL1 cDNA cloned within pCMV6-Entry vector and plasmid with vector alone (Origene Technologies). T24 and PC3 cells were stably transfected with CXCL1 short hairpin RNA (shRNA) and scramble (Scr) non-effective shRNA construct (Origene). Plasmids with sequence verified human CXCL1 cDNA cloned within pCMV6-Entry vector and plasmid with vector alone (Origene Technologies) were transfected into DU145 cells using Fugene HD transfection reagent (Roche Diagnostics) to create DU145-CXCL1 and DU145-Empty. Similarly, CXCL1 short hairpin RNA (shRNA) cloned within pRS vector was transfected into T24 and PC3 cells as well as CXCL1 plasmid scramble (Scr) non-effective shRNA construct within pRS vector (Origene) using Fugene HD. Stable transfectants were selected with 1,200 µg/ml of G418 (Life Technologies, Inc., Carlsbad, CA) for DU145 clones and 0.25 µg/ml of puromycin (Life Technologies) for T24 and PC3 clones for 14 days and subcloned by limiting dilution in 96-well plates. Integration of the transfected gene into the genome was confirmed by RT-PCR. Stable cell lines were maintained in media containing 500 µg/ml of G418 for DU145 clones and in media containing 0.25 µg/ml of puromycin for T24 and PC3 clones.
Quantitative reverse transcriptase-PCR
RNA was extracted from cells using RNeasy mini kit (Qiagen, Valencia, CA) as per manufacturer's instructions. Conversion to cDNA was achieved through High Capacity cDNA Reverse Transcription kit (Life Technologies). Quantitative reverse transcriptase (RT)-PCR was carried out using ABI 7300 Real-Time PCR System (Life Technologies) in a 20 μl reaction volume containing 1 μl of the first-strand cDNA, 1 μl of gene-specific TaqMan primer and probe mix. Gene-specific TaqMan primer and probe sets used in this study were Hs00236937_m1 for CXCL1 and Hs01060665_g1 for β -actin. Relative fold changes in mRNA levels were calculated after normalization to β-actin using the comparative Ct method [43].
Measurement of secreted CXCL1 by ELISA
Cells were plated onto 6-well plate at a density of 2 × 105 cells/well. After 48 hours, the conditioned media was collected and centrifuged to remove any dead or floating cells. Conditioned media was analyzed by ELISA for CXCL1 (Human CXCL1/GRO alpha Quantikine ELISA Kit; R&D Systems) using a FLUOstar OPTIMA microplate reader (BMG Labtech, Cary, NC).
Cell migration and invasion assays
Migration assays were performed in 6 well two-tier invasion chambers (Collaborative Biomedical Products, Bedford, MA, USA) [44]. Polycarbonate membranes were coated with 4 mg/mL growth factor reduced Matrigel (BD Biosciences, San Jose, CA) as described for invasion assays, control inserts (migration only) contained no coating. Two separate experimental designs were tested. First, DU145- CXCL1-OE3&8, DU145-Empty, T24-CXCL1-KD4&8, T24-shSCR, PC3-CXCL1-KD7 and PC3-shSCR cells were added to each insert at a density of 105 cells/ml/well in RPMI media. The lower chamber contained RPMI media with 10% FBS as a chemoattractant. The cells were maintained in a humidified incubator in 5% CO2 at 37°C for 24 hours. After the designated time, the cells on the top of the polycarbonate membrane were removed. The cells attached to the bottom of the membrane were stained for 1 hour with cell viability indicator Calcein AM Fluorescent Dye (BD Biosciences, Franklin Lakes, NJ) and quantified using the FLUOstar OPTIMA at 495mm excitation and 515nm emission (BMG LABTECH Inc., Cary, NC).
In addition, T24, DU145 and PC3 cells (105 cells/mL/well) were exposed to 0-200 μg/ml of CXCL1 monoclonal antibody (HL2401) in RPMI media. The lower chamber contained RPMI media with 10% FBS as chemoattractant. After 24 hours, the T24, DU145 and PC3 cells on the top of the polycarbonate membrane were removed, while T24, DU145 and PC3 cells attached to the bottom of the membrane were stained for 1 hour with cell viability indicator Calcein AM Fluorscent Dye and quantified using the FLUOstar OPTIMA. For the migration and invasion assays, at least three independent experiments consisting of each condition tested in triplicate wells was used to calculate mean ± SD values.
Capillary tube formation assays
The capillary tube formation assay was conducted as described previously [18, 41]. Matrigel (BD Biosciences) was added to 96-well plates (50 µl per well) and allowed to solidify for 30 min at 370C. HUVEC cells were incubated in serum- and growth factor-free EBM2 basal media containing 0.1% delipidated BSA for 5 hrs. HUVECs were seeded on top of Matrigel in triplicates at a density of 104 cells per well in conditioned media and incubated for 6 hrs. Similarly, HUVEC cells were resuspended in EBM-2 basal media with 20 μg/ml or 100 μg/ml of CXCL1 monoclonal antibody (HL2401), or control IgG (100 μg/mL) prior to seeding on top of Matrigel and incubated for 6 hrs. Images were acquired with a Nikon ECLIPS E400 microscope (Nikon, Melville, NY). The total length of tube-like structures in at least 4 viewed fields per well was measured using ImageJ. At least three independent experiments consisting of each condition tested in triplicate wells was used to calculate mean ± SD values.
Cell proliferation and soft agar colony formation assay
Cell proliferation was determined by incorporation of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) into T24, DU145 and PC3 treated with control IgG (100 μg/mL) and HL2401 (20 and 100 μg/mL) as previously described [18]. Briefly, 103 cells (T24, DU145 and PC3) per well were plated in 96-microwell plates and incubated for 72 hrs with the indicated concentration of HL2401. Each condition was tested in triplicate wells. At least three independent experiments were performed in triplicate.
RT2 Profiler PCR Arrays for Angiogenesis and Metastasis
Cellular RNA from DU145-Empty, DU145- CXCL1-OE8, T24-shSCR, T24-CXCL1-KD4, PC3- shSCR and PC3-CXCL1-KD7 recovered and converted to cDNA as described above. RT2 Profiler 'human angiogenesis' PCR arrays, (Catalog # PAHS-024ZA; SABiosciences Corporation, Frederick, MD, USA) and RT2 Profiler 'human metastasis' PCR arrays, (Catalog # PAHS-028ZA; SABiosciences) were analyzed in duplicate according to the manufacturer's instructions (www.sabiosciences.com/pcrarraydataanalysis.php) by quantitative reverse transcriptase (RT)-PCR carried out using ABI 7300 Real-Time PCR system (Life Technologies, Carlsbad, CA). The specificity of the SYBR Green assay was confirmed by melting curve analysis. Relative fold changes in mRNA levels were calculated after normalization to housekeeping control gene targets using the comparative Ct method.
Antibody protein array
DU145-CXCL1-OE3&8, DU145-Empty, T24- CXCL1-KD4&8, T24-shSCR, PC3-CXCL1-KD7 and PC3-shSCR were harvested from three individual experiments and lyzed with 2X Cell Lysis Buffer (RayBiotech, Norcross, GA). Protein levels were quantified using a bicinchoninic acid protein assay kit (Applygen Technologies Inc., Beijing, China). Lysates were then analyzed for angiogenesis-related cytokines (60 cytokines total) and Matrix metalloproteinase-related proteins (10 proteins total) using a RayBio® C-Series Human Angiogenesis Antibody Array 1000 Kit (RayBiotech) and RayBio® C-Series Human Matrix Metalloproteinase Antibody Array 1 Kit (RayBiotech), respectively. The assay was performed according to the manufacturer's instruction. Briefly, 100 µl blocking buffer was added into each well and incubated at room temperature for 60 min to block slides. After removing the blocking buffer, 30 µg of samples or serial diluted standards were added to each well containing 70 µl sample diluent and incubated overnight at 4°C. The samples were decanted and wash three times with wash buffer I at room temperature with gentle shaking. Next, the array was washed twice with wash buffer II, then 70 µl diluted detection antibody was added to each well and incubate at room temperature for 2 h, followed by three washes with wash buffer I at room temperature. Subsequently, 80 µl diluted Cy3 equivalent dye-conjugated streptavidin was added to each well. The array was incubated in dark room at room temperature for 1 h and washed as above.
Transient transfection of small interfering RNA (siRNA)
IL6 targeting siRNA, TIMP4 targeting siRNA and Scr-siRNA were purchased from Life Technologies. For siRNA transfection, cells were seeded at a density of 10,000 cells/well in 6-well plates and transfected with 10 nM siRNA using Lipofectamine™ RNAiMAX Transfection Reagent (Life Technologies) according to the manufacturer's instruction. The transfection was performed by the reverse transfection method, in which cells were transfected and plated simultaneously. In order to avoid cytotoxicity of the transfection reagent, the medium was routinely replaced with fresh medium at 24 h after siRNA transfection. After incubation of 48 hours in the fresh medium, the conditioned media was collected for in vitro tube formation assay using HUVEC or invasion assay using Fluoroblok insert system (BD Biosciences, San Jose, CA).
Monoclonal Anti-CXCL1 Antibody Production
A mouse monoclonal antibody against CXCL1 was commercially produced using a standard protocol of the Hybridoma and Protein Core Laboratories, University of Florida Interdisciplinary Center for Biotechnology Research (ICBR) [45]. A mouse monoclonal antibody against CXCL1 was commercially produced using a standard protocol of the Hybridoma and Protein Core Laboratories, University of Florida Interdisciplinary Center for Biotechnology Research (ICBR) [46]. Briefly, two female Balb/cByJ mice were immunized with approximately 100 μg of native CXCL1 protein diluted in sterile physiologic phosphate buffered saline (PBS) and emulsified in Ribi MPL +TDM adjuvant. The immunogen was administered on day 1, 21, 44, and 192. The test bleeds were collected 11 to 14 days after the second and third immunizations. The presence of anti-CXCL1 antibodies in the post-immunized serum was determined by western blots and ELISA. Six days after the fourth immunization, mouse #1 was euthanized and the splenic lymphocytes were collected and fused with mouse myeloma cells to form hybridoma cells [4]. The cultured media of the growing hybridoma mass cultures (n = 30) were collected and screened for anti-CXCL1 antibody production by ELISA. The mass cultures that tested positive by ELISA were subsequently tested for biologic effect in a proliferation assay utilizing HUVEC cells. The cultures (n = 3) that showed reactivity to CXCL1 in ELISA and exhibited anti-proliferative effects were grown out, and further cloned by limiting dilution. The cultured media collected from each clone were tested again by ELISA. The monoclonal antibodies were isotyped by ELISA and IsoStrip tested following manufacturer's protocol. The cultured medium of the final selected hybridoma clone (HL2401) was harvested and purified through a protein G column (GE Healthcare Protein G Sepharose 4 Fast Flow). The concentration of the purified monoclonal anti-CXCL1 antibody (HL2401) was determined by Bradford Protein Assay and stored at 4°C for future validation. A gel clot LAL assay from Lonza (Basel, Switzerland) ensured the antibody was free of endotoxins.
Customized indirect competitive immunoassay (ELISA)
Recombinant human CXCL1 was diluted to a final concentration of 10 μg/mL in 0.01 M phosphate- buffered saline pH 7.2 (PBS). One hundred microliters of the solution were used for coating each well of the ELISA plates (Maxisorp; Nunc, Roskilde, Denmark) at 4°C overnight. The plates were thereafter washed three times with 350 μl of washing solution (PBS). The plates were blocked at room temperature for 1 h with 200 μl of 3% (w/v) albumin from bovine serum (BSA) in PBS and then washed 3 times. Then 6.25 μg of HL2401 (control) or 6.25 μg of serum proteins were added to wells in triplicate. The plate was incubated at room temperature for 1 h. After removal of unbound material (4 washes with 0.05% Tween 20-PBS), 50 μl of IgG-HRP as the secondary antibody; diluted in PBS-T 1:1000 dilution Mouse IgG-HRP (Santa Cruz Biotech) was added to each well and incubated for 30 min at room temperature. After 4 repeated PBS-T washes, 100 μl of TMB Abcam high sensitivity was added to each well for 15 min at room temperature. The enzyme-substrate reaction was stopped by adding 100 μl of 2N sulfuric acid to each well. The optical density (OD450) was measured at 450 nm using a microtitre plate reader (Bio-Rad, Hercules, California, USA).
Radiolabeling of CXCL1 Antibody
64Cu was produced with an onsite cyclotron (GE PETrace). 64CuCl2 (74 MBq) was diluted in 300 μL of 0.1 M sodium acetate buffer (pH 5.5) and mixed with 200 μL of NOTA-CXCL1 antibody (0.5 mg/mL). The reaction was conducted at 37 °C for 45 min with constant shaking. The resulting 64Cu-NOTA-CXCL1 antibody was purified by PD-10 size exclusion column chromatography, using PBS as the mobile phase. The radioactive fraction containing 64Cu-NOTA-CXCL1 antibody was collected for in vivo studies.
PET imaging and bio-distribution study
At different time points post-injection (p.i.) of 5-10 MBq of 64Cu-NOTA-CXCL1 antibody via tail vein, PET scans of ICR mice (Envigo, Indianapolis, IN; n = 4) were carried out using a microPET/microCT Inveon rodent model scanner (Siemens Medical Solutions USA, Inc.). Data acquisition, image reconstruction, and region-of-interest (ROI) analysis of the PET data were performed. Briefly, the images were acquired by 40 million-count static PET scans and reconstructed using maximum a posteriori (MAP) algorithm, without attenuation or scatter correction. ROI analysis of each PET scan was carried out using software (Inveon Research Workplace, IRW) based on decay-corrected whole-body images, calculated with the injected dose measured by a dose calibrator (Capintec, Inc., Ramsey, NJ). Quantitative PET data of the tumor and major organs was presented in the format of percentage injected dose per gram of tissue (%ID/g). After the last scan at 48 h p.i., biodistribution studies were performed to corroborate PET data. Mice were euthanized, and blood and major organs/tissues were collected and wet-weighed. The radioactivity in the tissue was measured using a γ counter (Perkin-Elmer, Norwalk, CT) and presented as %ID/g (mean ± SD).
Pharmacokinetic studies
Pharmacokinetics studies were performed in female C57BL/6 mice to determine plasma exposure to CXCL1 antibody after single administration. Plasma samples at the following time points post-injection were taken: time zero (no treatment), 12, 24 and 48 hours. Plasma was derived from the whole blood by centrifugation at 3,000 rpm at 4°C in plasma separator tubes for 10 minutes. All samples were stored at -80°C until subsequent analysis. Samples were analyzed for CXCL1 antibody using an indirect ELISA. The lower limit of quantifications was 0.94 ng/mL in plasma. Pharamcokinetics parameters were calculated using noncompartmental analysis in WinNonLin v 5.0.3.
In vivo administration of CXCL1 antibody HL2401
The importance of CXCL1 expression for tumorigenicity and angiogenesis was assessed in vivo using bladder cancer and prostate cancer mouse xenograft models. Animal care was in compliance with the recommendations of The Guide for Care and Use of Laboratory Animals (National Research Council) and approved by University of Hawaii local IACUC. The subcutaneous tumorigenicity assay was performed in athymic BALB/c nu/nu male mice (6 to 8 weeks old) purchased from Envigo by inoculating 2 x 106 parental T24 cells and 2 x 106 parental PC3 cells as described previously [47, 48]. After one week, mice bearing subcutaneously xenograft tumors were divided randomly into three groups (Control or 8 mg/kg) of HL2401 and treatment was initiated. Each group was comprised of at least 10 mice. No toxicity or weight loss was noted in any of the treatment groups (Figure S6). HL2401 (100 µl diluted in sterile PBS) was administered via intraperitoneal injection twice weekly for four weeks. Control mice received IgG alone on the same schedule. Tumor volumes were measured weekly with digital calipers and calculated by V (mm3) = length x (width)2 x 0.5236. After five weeks of cell inoculation, the mice were sacrificed, tumors and key organs were resected and analyzed histologically and by immunohistochemical staining.
Immunohistochemical analysis of xenograft tumors
Immunohistochemistry (IHC) was conducted as previously described [15, 16, 18, 41, 42]. Paraffin embedded tumor sectioned were deparaffinized in xylene, rehydrated using graded percentages of ethanol. Slides were treated with 1% hydrogen peroxide in methanol to block endogenous peroxidase activity. Staining was conducted using goat anti-human CXCL1 antibody (sc-1374, dilution 1:200, Santa Cruz Biotechnology), mouse anti-human CXCR2 antibody (ab24963, dilution 1:200; Abcam), human IL6 antibody (sc-1265, dilution1:200, Santa Cruz Biotechnology) human TIMP4 antibody (sc-9375, dilution 1:200, Santa Cruz Biotechnology), cleaved caspase-3 antibody (5A1E, dilution 1:1,500, Cell Signaling Technology), mouse anti-human PECAM-1 (sc-1506-R, dilution 1/1,000, Santa Cruz Biotechnology) and mouse anti-human Ki-67 (MIB-1, dilution, 1:100, Dako). Biotin-labeled horse anti-mouse IgG, rabbit IgG or goat IgG (2 µg/ml in blocking buffer) was used as secondary antibody. Immunoreactive signals were amplified by formation of avidin-biotin peroxidase complexes and visualized using 3, 3'- diaminobenzidine (DAB). Nuclear counterstaining was conducted with hematoxylin. Apoptotic Index (AI), Microvessel density (MVD) and Proliferative Index (PI) analyses were determined as previously described [48-50].
Statistical analyses
All experimental data were expressed as mean with standard deviation. All statistical analyses were conducted using Student's t-test, Mann-Whitney non-parametric U-test, or one-way ANOVA and compared to the control(s). A p < 0.05 was considered significant. All statistical analyses and figures were carried out using GraphPad Prism software 7.0 (GraphPad Software, Inc., La Jolla, CA).
Abbreviations
Chemokine (C-X-C motif) ligand 1: CXCL1; interleukin-8 receptor type beta: IL8RB; interleukin 6: IL6; tissue inhibitor of metalloproteinase 4: TIMP4; Jagged 1 protein: JAG1; Chondromodulin-1: LECT1; Insulin-like growth factor 1: IGF1; Matrix metalloproteinase 2: MMP2; affinity-binding constant: KD; maximum concentration: Cmax; half-life: t1/2; extracellular matrix: ECM; fibroblast growth factor: FGF; vascular endothelial growth factor: VEGF; interleukin 8: IL8; American Type Culture Collection: ATCC; human umbilical vein endothelial cells: HUVEC.
Supplementary Material
Supplementary materials and methods, figures.
Acknowledgements
For their assistance in the animal work: Shreya Goel and Justin J. Jeffrey.
Financial support
The study was funded in part by the Weinman Foundation Fund (CJR), NIH/NCI R01 CA198887 (CJR), NCI/NIH R44 CA173921 (SG).
Competing Interests
Drs. Goodison and Rosser are officers in Nonagen Bioscience Corp. The remaining authors have declared no competing interests.
References
1. Chow MT, Luster AD. Chemokines in cancer. Cancer Immunol Res. 2014;2:1125-31 doi: 10.1158/2326-6066.CIR-14-0160
2. Moll NM, Cossoy MB, Fisher E, Staugaitis SM, Tucky BH, Rietsch AM. et al. Imaging correlates of leukocyte accumulation and CXCR4/CXCL12 in multiple sclerosis. Arch Neurol. 2009;66:44-53 doi: 10.1001/archneurol. 2008.512
3. Gillitzer R, Goebeler M. Chemokines in cutaneous wound healing. J Leukoc Biol. 2001;69:513-21 doi
4. Mukaida N, Sasaki S, Baba T. Chemokines in cancer development and progression and their potential as targeting molecules for cancer treatment. Mediators Inflamm. 2014;2014:170381. doi: 10.1155/2014/170381
5. Sarvaiya PJ, Guo D, Ulasov I, Gabikian P, Lesniak MS. Chemokines in tumor progression and metastasis. Oncotarget. 2013;4:2171-85 doi: 10.18632/ oncotarget.1426
6. Hembruff SL, Cheng N. Chemokine signaling in cancer: Implications on the tumor microenvironment and therapeutic targeting. Cancer Ther. 2009;7:254-67 doi
7. Bochner BH, Cote RJ, Weidner N, Groshen S, Chen SC, Skinner DG, Nichols PW. Angiogenesis in bladder cancer: relationship between microvessel density and tumor prognosis. J Natl Cancer Inst. 1995;87:1603-12 doi
8. Klagsbrun M, D'Amore PA. Regulators of angiogenesis. Annu Rev Physiol. 1991;53:217-39 doi: 10.1146/annurev.ph.53.030191.001245
9. Raghuwanshi SK, Su Y, Singh V, Haynes K, Richmond A, Richardson RM. The chemokine receptors CXCR1 and CXCR2 couple to distinct G protein-coupled receptor kinases to mediate and regulate leukocyte functions. J Immunol. 2012;189:2824-32 doi: 10.4049/jimmunol.1201114
10. Su Y, Raghuwanshi SK, Yu Y, Nanney LB, Richardson RM, Richmond A. Altered CXCR2 signaling in beta-arrestin-2-deficient mouse models. J Immunol. 2005;175:5396-402 doi
11. Cheng WL, Wang CS, Huang YH, Tsai MM, Liang Y, Lin KH. Overexpression of CXCL1 and its receptor CXCR2 promote tumor invasion in gastric cancer. Ann Oncol. 2011;22:2267-76 doi: 10.1093/annonc/mdq739
12. Bieche I, Chavey C, Andrieu C, Busson M, Vacher S, Le Corre L. et al. CXC chemokines located in the 4q21 region are up-regulated in breast cancer. Endocr Relat Cancer. 2007;14:1039-52 doi: 10.1677/erc.1.01301
13. Middleman BR, Friedman M, Lawson DH, DeRose PB, Cohen C. Melanoma growth stimulatory activity in primary malignant melanoma: prognostic significance. Mod Pathol. 2002;15:532-7 doi: 10.1038/modpathol.3880559
14. Baird AM, Gray SG, O'Byrne KJ. Epigenetics underpinning the regulation of the CXC (ELR+) chemokines in non-small cell lung cancer. PLoS One. 2011;6:e14593. doi: 10.1371/journal.pone.0014593
15. Miyake M, Lawton A, Goodison S, Urquidi V, Rosser CJ. Chemokine (C-X-C motif) ligand 1 (CXCL1) protein expression is increased in high-grade prostate cancer. Pathol Res Pract. 2014;210:74-8 doi: 10.1016/j.prp.2013.08.013
16. Miyake M, Lawton A, Goodison S, Urquidi V, Gomes-Giacoia E, Zhang G. et al. Chemokine (C-X-C) ligand 1 (CXCL1) protein expression is increased in aggressive bladder cancers. BMC Cancer. 2013;13:322. doi: 10.1186/1471-2407-13-322
17. Kawanishi H, Matsui Y, Ito M, Watanabe J, Takahashi T, Nishizawa K. et al. Secreted CXCL1 is a potential mediator and marker of the tumor invasion of bladder cancer. Clin Cancer Res. 2008;14:2579-87 doi: 10.1158/1078-0432. CCR-07-1922
18. Miyake M, Goodison S, Urquidi V, Gomes Giacoia E, Rosser CJ. Expression of CXCL1 in human endothelial cells induces angiogenesis through the CXCR2 receptor and the ERK1/2 and EGF pathways. Lab Invest. 2013;93:768-78 doi: 10.1038/labinvest.2013.71
19. Melendez-Zajgla J, Del Pozo L, Ceballos G, Maldonado V. Tissue inhibitor of metalloproteinases-4. The road less traveled. Mol Cancer. 2008;7:85. doi: 10.1186/1476-4598-7-85
20. Culig Z. Proinflammatory cytokine interleukin-6 in prostate carcinogenesis. Am J Clin Exp Urol. 2014;2:231-8 doi
21. Chen MF, Lin PY, Wu CF, Chen WC, Wu CT. IL-6 expression regulates tumorigenicity and correlates with prognosis in bladder cancer. PLoS One. 2013;8:e61901. doi: 10.1371/journal.pone.0061901
22. Santer FR, Malinowska K, Culig Z, Cavarretta IT. Interleukin-6 trans-signalling differentially regulates proliferation, migration, adhesion and maspin expression in human prostate cancer cells. Endocr Relat Cancer. 2010;17:241-53 doi: 10.1677/ERC-09-0200
23. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27-31 doi
24. Folkman J, Klagsbrun M. Angiogenic factors. Science. 1987;235:442-7 doi
25. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353-64 doi
26. Liotta LA, Kleinerman J, Saidel GM. Quantitative relationships of intravascular tumor cells, tumor vessels, and pulmonary metastases following tumor implantation. Cancer Res. 1974;34:997-1004 doi
27. Weidner N, Folkman J, Pozza F, Bevilacqua P, Allred EN, Moore DH. et al. Tumor angiogenesis: a new significant and independent prognostic indicator in early-stage breast carcinoma. J Natl Cancer Inst. 1992;84:1875-87 doi
28. Liotta LA, Steeg PS, Stetler-Stevenson WG. Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell. 1991;64:327-36 doi
29. Blood CH, Zetter BR. Tumor interactions with the vasculature: angiogenesis and tumor metastasis. Biochim Biophys Acta. 1990;1032:89-118 doi
30. Grossfeld GD, Ginsberg DA, Stein JP, Bochner BH, Esrig D, Groshen S. et al. Thrombospondin-1 expression in bladder cancer: association with p53 alterations, tumor angiogenesis, and tumor progression. J Natl Cancer Inst. 1997;89:219-27 doi
31. Folkman J, Ingber D. Inhibition of angiogenesis. Semin Cancer Biol. 1992;3:89-96 doi
32. Sidky YA, Borden EC. Inhibition of angiogenesis by interferons: effects on tumor- and lymphocyte-induced vascular responses. Cancer Res. 1987;47:5155-61 doi
33. O'Brien T, Cranston D, Fuggle S, Bicknell R, Harris AL. Two mechanisms of basic fibroblast growth factor-induced angiogenesis in bladder cancer. Cancer Res. 1997;57:136-40 doi
34. Kollermann J, Helpap B. Expression of vascular endothelial growth factor (VEGF) and VEGF receptor Flk-1 in benign, premalignant, and malignant prostate tissue. Am J Clin Pathol. 2001;116:115-21 doi: 10.1309/1LBM-6X32-JH6W-ENUD
35. Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4-25 doi: 10.1210/edrv.18.1.0287
36. Inoue K, Slaton JW, Eve BY, Kim SJ, Perrotte P, Balbay MD. et al. Interleukin 8 expression regulates tumorigenicity and metastases in androgen-independent prostate cancer. Clin Cancer Res. 2000;6:2104-19 doi
37. Uehara H, Troncoso P, Johnston D, Bucana CD, Dinney C, Dong Z. et al. Expression of interleukin-8 gene in radical prostatectomy specimens is associated with advanced pathologic stage. Prostate. 2005;64:40-9 doi: 10.1002/pros.20223
38. O'Brien TS, Fox SB, Dickinson AJ, Turley H, Westwood M, Moghaddam A. et al. Expression of the angiogenic factor thymidine phosphorylase/platelet- derived endothelial cell growth factor in primary bladder cancers. Cancer Res. 1996;56:4799-804 doi
39. Miyake H, Yoshimura K, Hara I, Eto H, Arakawa S, Kamidono S. Basic fibroblast growth factor regulates matrix metalloproteinases production and in vitro invasiveness in human bladder cancer cell lines. J Urol. 1997;157:2351-5 doi
40. Sakai Y, Goodison S, Cao W, Urquidi V, Namiki K, Porvasnik S. et al. VEGF induces expression of Bcl-2 and multiple signaling factors in microvascular endothelial cells in a prostate cancer model. World J Urol. 2009;27:659-66 doi: 10.1007/s00345-009-0422-0
41. Miyake M, Goodison S, Lawton A, Gomes-Giacoia E, Rosser CJ. Angiogenin promotes tumoral growth and angiogenesis by regulating matrix metallopeptidase-2 expression via the ERK1/2 pathway. Oncogene. 2015;34:890-901 doi: 10.1038/onc.2014.2
42. Gomes Giacoia E, Miyake M, Lawton A, Goodison S, Rosser CJ. PAI-1 Leads to G1-phase Cell Cycle Progression through Cyclin D3/CDK4/6 Up-regulation. Mol Cancer Res. 2014 doi: 10.1158/1541-7786.MCR-13-0543
43. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45
44. Gomes Giacoia E, Miyake M, Lawton A, Goodison S, Rosser CJ. PAI-1 Leads to G1-phase Cell Cycle Progression through Cyclin D3/CDK4/6 Up-regulation. Mol Cancer Res. 2014
45. Chang LW, Fu A, Wozniak E, Chow M, Duke DG, Green L. et al. Immunohistochemical detection of a unique protein within cells of snakes having inclusion body disease, a world-wide disease seen in members of the families Boidae and Pythonidae. PLoS One. 2013;8:e82916. doi: 10.1371/journal.pone.0082916
46. Chang LW, Fu A, Wozniak E, Chow M, Duke DG, Green L. et al. Immunohistochemical detection of a unique protein within cells of snakes having inclusion body disease, a world-wide disease seen in members of the families Boidae and Pythonidae. PLoS One. 2013;8:e82916
47. Uehara H, Troncoso P, Johnston D, Bucana CD, Dinney C, Dong Z. et al. Expression of interleukin-8 gene in radical prostatectomy specimens is associated with advanced pathologic stage. Prostate. 2005;64:40-9
48. Sakai Y, Goodison S, Kusmartsev S, Fletcher B, Eruslanov E, Cao W. et al. Bcl-2 mediated modulation of vascularization in prostate cancer xenografts. Prostate. 2009;69:459-70
49. Anai S, Goodison S, Shiverick K, Hirao Y, Brown BD, Rosser CJ. Knock-down of Bcl-2 by antisense oligodeoxynucleotides induces radiosensitization and inhibition of angiogenesis in human PC-3 prostate tumor xenografts. Mol Cancer Ther. 2007;6:101-11
50. Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis-correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1-8
Author contact
![]() Corresponding authors: Charles J. Rosser, MD, MBA, FACS, Professor, Cedars-Sinai Medical Center, Samuel Oschin Comprehensive Cancer Center. Tel 310-423-7600; Email deacdoccom and Weibo Cai, PhD, Departments of Radiology and Medical Physics, University of Wisconsin - Madison, Madison, WI 53705. Tel: 608-262-1749; Fax: 608-265-0614; Email: wcaiorg
Corresponding authors: Charles J. Rosser, MD, MBA, FACS, Professor, Cedars-Sinai Medical Center, Samuel Oschin Comprehensive Cancer Center. Tel 310-423-7600; Email deacdoccom and Weibo Cai, PhD, Departments of Radiology and Medical Physics, University of Wisconsin - Madison, Madison, WI 53705. Tel: 608-262-1749; Fax: 608-265-0614; Email: wcaiorg