Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(6):1634-1650. doi:10.7150/thno.30206 This issue Cite
Research Paper
Pseurotin A Inhibits Osteoclastogenesis and Prevents Ovariectomized-Induced Bone Loss by Suppressing Reactive Oxygen Species
Kai Chen1*, Pengcheng Qiu2,3*, Yu Yuan1,4, Lin Zheng2,3, Jianbo He1,5, Chao Wang1, Qiang Guo1,6, Jacob Kenny1, Qian Liu1,7,8,9, Jinmin Zhao7,8,9, Junhao Chen1, Jennifer Tickner1, Shunwu Fan2,3, Xianfeng Lin2,3 ![]() , Jiake Xu1,7
, Jiake Xu1,7 ![]()
1. School of Biomedical Sciences, University of Western Australia, Perth, WA 6009, Australia.
2. Department of Orthopedic Surgery, Sir Run Run Shaw Hospital, Medical College of Zhejiang University, Hangzhou, Zhejiang 310000, China
3. Key Laboratory of Musculoskeletal System Degeneration and Regeneration Translational Research of Zhejiang Province, Hangzhou, Zhejiang 310000, China
4. School of Physical Education and Sports Science, South China Normal University, Guangzhou, Guangdong 510631, China
5. Guangzhou University of Chinese Medicine, Guangzhou, Guangdong 510000, China
6. Department of Spine Surgery, Xiangya Hospital, Central South University, Changsha, Hunan 410008, China
7. Research Centre for Regenerative Medicine, Guangxi Medical University, Nanning, Guangxi 530021, China
8. Guangxi Key Laboratory of Regenerative Medicine, Guangxi Medical University, Nanning, Guangxi 530021, China
9. International Joint Laboratory on Regeneration of Bone and Soft Tissues, Guangxi Medical University, Nanning, Guangxi 530021, China
*Kai Chen and Pengcheng Qiu contributed equally to this work.
Received 2018-9-21; Accepted 2019-1-1; Published 2019-2-28
Abstract

Rationale: Growing evidence indicates that intracellular reactive oxygen species (ROS) accumulation is a critical factor in the development of osteoporosis by triggering osteoclast formation and function. Pseurotin A (Pse) is a secondary metabolite isolated from Aspergillus fumigatus with antioxidant properties, recently shown to exhibit a wide range of potential therapeutic applications. However, its effects on osteoporosis remain unknown. This study aimed to explore whether Pse, by suppressing ROS level, is able to inhibit osteoclastogenesis and prevent the bone loss induced by estrogen-deficiency in ovariectomized (OVX) mice.
Methods: The effects of Pse on receptor activator of nuclear factor-κB (NF-κB) ligand (RANKL)-induced osteoclastogenesis and bone resorptive function were examined by tartrate resistant acid phosphatase (TRAcP) staining and hydroxyapatite resorption assay. 2',7'-dichlorodihydrofluorescein diacetate (H2DCFDA) was used to detect intracellular ROS production in vitro. Western blot assay was used to identify proteins associated with ROS generation and scavenging as well as ROS-mediated signaling cascades including mitogen-activated protein kinases (MAPKs), NF-κB pathways, and nuclear factor of activated T cells 1 (NFATc1) signaling. The expression of osteoclast-specific genes was assessed by qPCR. The in vivo potential of Pse was determined using an OVX mouse model administered with Pse or vehicle for 6 weeks. In vivo ROS production was assessed by intravenous injection of dihydroethidium (DHE) into OVX mice 24h prior to killing. After sacrifice, the bone samples were analyzed using micro-CT and histomorphometry to determine bone volume, osteoclast activity, and ROS level ex vivo.
Results: Pse was demonstrated to inhibit osteoclastogenesis and bone resorptive function in vitro, as well as the downregulation of osteoclast-specific genes including Acp5 (encoding TRAcP), Ctsk (encoding cathepsin K), and Mmp9 (encoding matrix metalloproteinase 9). Mechanistically, Pse suppressed intracellular ROS level by inhibiting RANKL-induced ROS production and enhancing ROS scavenging enzymes, subsequently suppressing MAPK pathway (ERK, P38, and JNK) and NF-κB pathways, leading to the inhibition of NFATc1 signaling. Micro-CT and histological data indicated that OVX procedure resulted in a significant bone loss, with dramatically increased the number of osteoclasts on the bone surface as well as increased ROS level in the bone marrow microenvironment; whereas Pse supplementation was capable of effectively preventing these OVX-induced changes.
Conclusion: Pse was demonstrated for the first time as a novel alternative therapy for osteoclast-related bone diseases such as osteoporosis through suppressing ROS level.
Keywords: Pseurotin A, reactive oxygen species, osteoclast, osteoporosis
Introduction
The maintenance of bone homeostasis is achieved by the delicate balance between osteoblastic bone formation and osteoclastic bone resorption. Uncoordinated activities of osteoblasts and osteoclasts can result in various perturbations in skeletal structure and function thus leading to bone diseases such as Paget's disease of bone and osteoporosis [1, 2]. Estrogen deficiency-induced bone loss, mainly due to enhanced osteoclast activity, plays an essential role in osteoporosis in postmenopausal women, leading to fragility fractures that are often associated with life- threatening mortality and morbidity, as well as massive economic cost to both individuals and society [3].
Osteoclasts are large multinucleated cells which are derived from hematopoietic progenitors. The formation of mature osteoclasts is dependent on several crucial factors, including macrophage-colony stimulating factor (M-CSF) and receptor activator of nuclear factor kappa B (NF-κB) ligand (RANKL). Osteoclast precursor cells undergo survival and proliferation in response to the binding of M-CSF to its receptor c-Fms [4]. RANKL is an indispensable and sufficient factor for osteoclastogenesis, via its action on RANK receptor [5]. After stimulation with RANKL, recruitment of TNF receptor-associated factor 6 (TRAF6) is induced to activate multiple downstream targets including mitogen-activated protein kinases (MAPKs) and NF-κB pathways, leading to activation of c-Fos and nuclear factor of activated T cells 1 (NFATc1) [6-8]. These signaling cascades enable the expression of genes that typify the osteoclast lineage including Acp5 (encoding tartrate-resistant acid phosphatase [TRAcP]), Ctsk (encoding cathepsin K), and Mmp9 (encoding matrix metalloproteinase 9), thus eventually leading to the formation of mature osteoclasts [6].
Growing evidence also indicates that intracellular reactive oxygen species (ROS) play a crucial role during osteoclast formation and bone resorption [9-11]. ROS are endogenously produced in osteoclast precursors following stimulation with RANKL, via a signaling cascade involving TRAF6, Rac1, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 1 (Nox1)[9]. Application of oxidant scavenger like N-acetylcysteine (NAC) or Nox inhibitor such as diphenylene iodonium (DPI), was found to inhibit osteoclastogenesis by suppressing RANKL-mediated ROS production [9], indicating ROS are required for osteoclast differentiation. Cellular protective mechanisms against oxidative stressors also include a variety of cytoprotective or antioxidant enzymes, such as heme oxygenase-1 (HO-1), catalase, glutathione- disulfide reductase (GSR), NAD(P)H: quinone reductase (NQO1), and γ-glutamylcysteine synthetase (GCS) [12, 13]. Antioxidants were shown to attenuate osteoclast formation and bone resorption by enhancing expression of the cytoprotective enzymes [14, 15]. The downstream targets of ROS in RANKL-mediated signaling still remain unclear; however, a higher level of oxidative stress was suggested to promote osteoclast formation and function through the activation of NF-κB and MAPKs [13, 16]. In addition, ROS production is highly involved in bone remodeling and bone homeostasis by promoting bone resorption [16, 17]. Estrogen deficiency-induced osteoporosis is associated with a higher level of oxidative stress and can be prevented by increasing antioxidant defenses [18-20]. Therefore, these findings might provide a rationale for suppressing ROS as a potential strategy for the treatment of osteoporosis.
Pseurotin A (Pse) is a bioactive secondary metabolite originally isolated from Pseudoeurotium ovalis [21] and later also from several species of Aspergillus [22-24]. A single hybrid polyketide synthase/non-ribosomal peptide synthetase (PKS/ NRPS) [25] gene and the rtfA gene [26] are associated with the biosynthesis of Pse in Aspergillus fumigatus. The expression of Pse is also induced in response to hypoxia [27]. So far, Pse has demonstrated potential therapeutic applications due to its immunosuppressive activity [28], antibacterial activity [29], nematicidal activity [30], antiparasitic as well as anticancer activity [22]. Furthermore, Pse was found to have antioxidant and radical-scavenging activity, as demonstrated by its ability to scavenge the DPPH (2, 2-diphenyl-1-picrylhydrazyl) radical [31].
Given the significant role of ROS on osteoclast formation and function, the potential antioxidant activity as well as other various potential therapeutic applications of Pse, we hypothesized that Pse might inhibit osteoclasts and thus prevent osteoclast-related osteoporosis. In the present study, we assessed the therapeutic effects of Pse on RANKL-induced osteoclastogenesis in vitro and ovariectomized (OVX)- induced osteoporosis mouse models in vivo, with a focus on determining the antioxidant capacity of Pse as well as elucidating its underlying mechanisms.
Materials and Methods
Materials and reagents
Pse, obtained from SHANGHAI ZZBIO CO., LTD (Shanghai, China), was dissolved in Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, Sydney, NSW, Australia) at a concentration of 100 mM and further diluted to working concentrations with culture medium. DMSO of the same dilution was used as vehicle control. Alpha modified Minimal Essential Medium (a-MEM) and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific (Scoresby, VIC, Australia). MTS assay kit and luciferase analysis system were purchased from Promega (Sydney, NSW, Australia). Primary antibodies for TRAF6, IκB-α, β-actin, phospho-ERK, ERK, phospho-P38, P38, NFATc1, Integrin αV, cathepsin K, and V-ATPase-d2 were purchased from Santa Cruz Biotechnology (Dallas, CA, USA). Primary antibody for GSR was purchased from Bioss Inc (Woburn, MA, USA). Primary antibodies for HO-1, catalase, phospho-JNK, JNK, and Active Rac1 Detection Kit were purchased from Cell Signaling Technology (Danvers, MA, USA). Primary antibody for vinculin was purchased from Sigma-Aldrich (Sydney, NSW, Australia). Recombinant M-CSF was purchased from R&D Systems (Minneapolis, MN, USA). Recombinant Glutathione S-transferase (GST)-rRANKL protein was expressed and purified as previously described [32]. Rhodamine Phalloidin, ProLong Gold Antifade Mountant, and 2',7'-dichlorodihydrofluorescein diacetate (H2DCFDA) were obtained from Thermo Fisher Scientific (Scoresby, VIC, Australia) and 4,6-diamidino-2-phenylindole (DAPI) was purchased from Santa Cruz Biotechnology (Santa Cruz, Dallas, CA, USA). Dihydroethidium (DHE) was purchased from ApexBio (Boston, MA, USA).
In vitro osteoclastogenesis assay
Fresh bone marrow macrophages (BMMs) from C57BL/6J mice were isolated using methods approved by the University of Western Australia Animal Ethics Committee (RA/3/100/1244) as described [16]. In brief, bone marrow was flushed from the femur and tibia and then cultured in α-MEM/10% FBS/Penicillin/streptomycin (complete αMEM). To obtain pure BMMs, non-adherent cells were then collected and cultured in complete αMEM containing M-CSF (50 ng/mL). After a further 3 days in culture, the attached cells were used for experimental purposes. BMMs were plated in 96-well plates at a density of 6 × 103 cells/well overnight. The following day, cells were stimulated with M-CSF and GST-rRANKL (50 ng/mL) in the presence or absence of increasing concentrations of Pse (2.5, 5, 7.5, 10 μM). Medium was replaced every 2 days until osteoclasts formed. The cells were then fixed with 2.5% glutaraldehyde solution for 10 min and stained for tartrate resistant acid phosphatase (TRAcP) enzymatic activity using a leucocyte acid phosphatase staining kit (Sigma-Aldrich, Sydney, NSW, Australia). Cells with more than 3 nuclei were counted as osteoclasts.
Cytotoxicity assay
BMMs were seeded in 96-well plates at a density of 6 × 103 cells/well and incubated overnight to allow adherence. Cells were then treated with different concentrations of Pse (0, 2.5, 5, 7.5, 10 μM) for 48 h. At the end of the experiment, MTS solution (20 μL/well) was added and the plate was incubated for an additional 2 h. The optical density (OD) was measured by spectrophotometric absorbance at 490nm using a microplate reader (BMG LABTECH, Ortenberg, Germany).
Staining for podosome belts
To visualize podosome belts, BMMs were seeded onto FBS-coated coverslips (5-mm diameter) in 96-well plates and were induced to form osteoclasts, as described above, in the presence of Pse (0, 5, 10 μM). Osteoclasts were then fixed 4% paraformaldehyde (PFA) for 10 min, permeabilized with 0.1% (v/v) Triton X-100 for 10 min and blocked with 3% BSA in PBS for 1h. Cells were then incubated with mouse monoclonal anti-vinculin (1:200) at 4 °C overnight, followed by washing in PBS and incubation with a fluorescent secondary anti-mouse antibody (Alexa Fluor 488, green). F-actin was stained with Rhodamine Phalloidin in the dark for 1 h. After that, cells were washed with PBS, stained with DAPI for 10 min, washed again, and mounted in ProLong Gold Antifade Mountant prior to visualization using a NIKON A1Si confocal microscope (Nikon Corporation, Minato, Tokyo, Japan).
Quantitative real-time PCR
BMMs were cultured in 6-well plates (1 × 105 cells/well) and stimulated with GST- rRANKL and M-CSF in the presence of Pse (0, 5, 10 μM) until osteoclasts formed. Total RNA was isolated from the cells using Trizol reagent (Life Technologies, Sydney, Australia) and single stranded cDNA was prepared from 1 μg of total RNA template using moloney murine leukemia virus (MMLV) reverse transcriptase with oligo-dT primer (Promega, Sydney, NSW, Australia). For relative quantitative real-time PCR (qPCR), SYBR Green PCR MasterMix (Thermo Fisher Scientific, Scoresby, VIC, Australia) was used. The cycling parameters for PCR were set as follows: 94 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 60 s. The specific primers used are as following: Ctsk (forward: 5'-GGGAGAAAAACCTGA AGC-3'; reverse: 5' -ATTCTGGGGACTCAGAGC-3'), Acp5 (forward: 5'-TGTGGCCATCTTTATG CT-3'; reverse: 5'-GTCATTTCTTTGGGGCTT-3'), Atp6v0d2 (Forward: 5′-GTGAGACCTTGGAA GACCTGAA-3′; Reverse: 5′-GAGAAATGTGCTCAGGGGCT-3′), Mmp9 (Forward: 5′-CGTGTCTG GAGATTCGACTTGA-3′; Reverse: 5′-TTGGAAACTCACACGCCAGA-3′), Nfatc1 (Forward: 5′- CA ACGCCCTGACCACCGATAG-3′; Reverse: 5′-GGCTGCCTTCCGTCTCATAGT- 3′), C-fos (Forward: 5′- GCGAGCAACTGAGAAGAC-3′; Reverse: 5′- TTGAAACCCGAGAACATC- 3′), Hmbs (Forward: 5′- AAGGGCTTTTCTGAGGCACC -3′; Reverse: 5′-AGTTGCCCATCTTTCATCACTG- 3′), Hprt1 (Forward: 5′- TCAGTCAACGGGGGACATAAA-3′; Reverse: 5′-GGGGCTGTACTGCTTAACCAG - 3′). The qPCR procedure was performed on a ViiA 7 Real-time PCR machine (Applied Biosystems, Warrington, Cheshire, UK). Target gene expression levels were normalized to average expression of Hprt1 and Hmbs.
Hydroxyapatite resorption assay
To determine whether Pse affects osteoclast function, hydroxyapatite resorption assay was performed as described previously [15, 33, 34]. BMMs were plated in 6-well collagen-coated plates (Corning Inc., Corning, NY, USA) in complete αMEM with M-CSF at a density of 1 x 105 cells/well. The following day cells were stimulated with 50 ng/mL GST-RANKL until osteoclasts began to form. Cells were then gently detached using cell dissociation solution (Sigma-Aldrich, Sydney, NSW, Australia) and an equal number of cells were seeded into each well of hydroxyapatite-coated plate (Corning Inc., Corning, NY, USA). Cells were treated with Pse at increasing concentrations (0, 5, 10 μM) in complete αMEM containing 50 ng/mL GST-rRANKL and M-CSF. After 48 h incubation, half of the wells for each group were washed with 10% bleach solution to remove cells and the images of hydroxyapatite resorption areas were captured using a using a Nikon microscope (Nikon Corporation, Minato, Tokyo, Japan) and quantified using Image J software (NIH, Bethesda, Maryland, USA). The remaining wells were fixed and stained for TRAcP activity as above to count the number of osteoclasts. The resorbed area per well and the percentage of resorbed area per osteoclast were used to quantify the osteoclast activity.
Detection of intracellular ROS production
The intracellular production of ROS was measured as described previously [9]. Briefly, after stimulation with GST-rRANKL (50 ng/mL) in the absence or presence of Pse at stated concentrations (5, 10 µM), BMMs were then incubated in Hank's balanced salt solution containing 5 mM H2DCFDA for 1 h. Upon oxidation, the nonfluorescent H2DCFDA was converted to the highly fluorescent 2',7'-dichlorofluorescein (DCF). The fluorescence of DCF was measured at an excitation wavelength of 488 nm and an emission wavelength of 515-540 nm using a NIKON A1Si confocal microscope. Cells images were also captured by digital interference contrast (DIC). The mean fluorescence intensity for each cell and the number of ROS-positive cells per field were analyzed using Image J software.
Luciferase reporter assays of NF-κB or NFATc1
RAW264.7 cells (ATCC, Manassas, Virginia, USA), stably transfected with an NF-κB responsive luciferase construct [35] and an NFATc1 responsive luciferase reporter construct [36], were seeded in 48-well plates at a density of 1.5 × 105 cells/well or 5 × 104 cells/ well overnight, respectively. Cells were then pre-treated with Pse for 1 h and stimulated with GST-rRANKL (50 ng/mL) for 6 h and 24 h respectively. At the end of the time points, cells were lysed, and luciferase activity was measured using the luciferase reporter assay kit (Promega, Sydney, NSW, Australia) and a luminescence reader (BMG LABTECH, Ortenberg, Germany).
Western blot Assay
BMM cells were seeded in 6-well plates (1 × 105 cells/well) and stimulated with GST- rRANKL and M-CSF in the presence or absence of Pse of indicated concentrations for the stated times. Cells were lysed in radioimmunoprecipitation (RIPA) lysis buffer to harvest protein. For GTP-Rac1 detection, cell lysates were collected and incubated with GST-Human PAK1-PBD fusion protein that binds to GTP-bound Rac1 using Active Rac1 Detection Kit (Cell Signaling Technology, Danvers, MA, USA). Protein was separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane (GE Healthcare, Silverwater, Australia). Non-specific binding was blocked by incubation in 5% skim milk powder and the membrane was then incubated with primary antibodies (1:1000) with gentle shaking overnight at 4 ℃. The next day, membranes were washed and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 1 h. Antibodies were detected with enhanced chemiluminescence substrate (PerkinElmer, Waltham, MA, USA). Images were acquired on an Image-quant LAS 4000 (GE Healthcare, Silverwater, NSW, Australia) and analyzed by ImageJ software.
Ovariectomized (OVX)-induced osteoporosis mouse model
All in vivo experimental procedures were approved by the Institutional Animal Ethics Committee of Sir Run Run Shaw Hospital. Eighteen C57BL/6J mice (females; 18.6 ± 1.4 g, 11 weeks old) were supplied by Animal Experiment Center of Sir Run Run Shaw Hospital (Hangzhou, Zhejiang, China). All mice were randomly divided into three groups: sham group (n=6), OVX group (n=6), and OVX+Pse (5 mg/kg) group (n=6). Bilateral ovariectomy was carried out to induce osteoporosis under chloral hydrate anesthesia for the mice in OVX group and OVX+Pse group, a sham procedure in which the ovaries were only exteriorized but not resected was performed for the mice in sham group. All mice had a 1-week-recovery after operations, then the mice in the OVX +Pse group were administrated an intraperitoneal injection of Pse at 5 mg/kg every 2 days for 6 weeks. The mice in the sham and OVX groups were intraperitoneally injected with vehicle (1% DMSO in PBS) as a control. ROS production in vivo was determined using DHE following previously described protocols with modifications [37, 38]. In brief, 24h before killing, each mouse received a 200-μL intravenous injection of DHE at 25 mg/kg.
Micro-CT scanning and analysis
After sacrifice, the femurs were collected and fixed in 10% neutral buffered formalin for 24 h and the excess soft tissue was removed. Left femurs were scanned using a Skyscan 1176 micro-CT instrument (Bruker microCT, Kontich, Belgium) using the following settings: source voltage, 50 kV; source current, 500 μA; AI 0.5 mm filter; pixel size 9 μm; rotation step, 0.4 degree. The images were then reconstructed with NRecon software (Bruker microCT, Kontich, Belgium) using the following settings: ring artefact correction, 7; smoothing, 2; beam hardening correction, 40%. A refined volume of interest was generated 0.5 mm above the growth plate of the distal femur and 1 mm in height. The trabecular bone region of interest (ROI) within this volume was manually defined and bone parameters within this ROI were all determined using a constant threshold (50 ~ 255) for binarization of the trabecular bone. The following parameters, including bone volume per tissue volume (BV/TV), trabecular number (Tb.N), connectivity density (Conn.Dn), and trabecular thickness (Tb.Th), were analyzed by the program CTAn (Bruker microCT, Kontich, Belgium). Cortical bone analysis was performed in the mid shaft (4 mm below the growth plate with a height of 1 mm) and a constant threshold (100~ 255) for binarization was used.
Bone histomorphometry analysis
Following micro-CT analysis, femurs were decalcified in 14% EDTA (Sigma-Aldrich, Sydney, NSW, Australia) at 37 ℃ for 7 days, and then embedded into paraffin for sectioning. Hematoxylin and eosin (H&E) staining and TRAcP activity staining were performed. Section images were acquired using Aperio Scanscope (Mt Waverley, VIC, Australia), and bone histomorphometric analyses were performed using BIOQUANT OSTEO software (Bioquant Image Analysis Corporation, Nashville, TN, USA).
For in vivo ROS fluorescence detection, cryosections of bone tissues were prepared as previously described [39]. Fresh bone tissues were dissected, cleaned and immediately fixed in 4% paraformaldehyde (Sigma-Aldrich, Sydney, NSW, Australia) solution at 4 ℃ for 4 h. Next, decalcification was carried out with 0.5 M EDTA overnight at 4 ℃ under constant rotation, after which the EDTA was exchanged for cryoprotective solution composed of 20% sucrose and 2% polyvinylpyrrolidone (PVP) (Sigma-Aldrich, Sydney, NSW, Australia) for 24 h. Finally, the resultant tissues were embedded and frozen in 8% gelatin in the presence of 20% sucrose and 2% PVP. 5 μm sections were generated and air-dried at room temperature prior to permeabilization for 10 min in 0.3% Triton X-100 (Sigma-Aldrich, Sydney, NSW, Australia). Nuclei were stained with DAPI for 30 min. After washing with PBS, sections were mounted, and coverslips were sealed with nail polish. A NIKON A1Si confocal microscope (Nikon Corporation, Minato, Tokyo, Japan) was used for imaging. The fluorescence intensity of 6 random areas of each group were quantified and analyzed using NIS-Elements software (Nikon Corporation, Minato, Tokyo, Japan).
Statistical analysis
Each experiment was repeated at least three times. All quantitative data are presented as mean ± standard deviation (SD). Statistical significance was determined by Student's t test. A p-value of less than 0.05 was considered to be significant.
Results
Pse suppresses RANKL-induced osteoclastogenesis in vitro
The chemical structure and formula of Pse are shown in Figure 1A. To identify the effect of Pse on osteoclastogenesis, BMMs were seeded in 96-well plates and treated with both M-CSF and RANKL in the presence or absence of various concentrations of Pse as indicated. The purity of isolated precursor BMMs was approximately 99%, as assessed by flow cytometry for CD11b (Figure S1). Numerous TRAcP-positive multinucleated osteoclasts formed in the RANKL-induced control group (without Pse), whereas increasing concentrations of Pse exerted a dose-dependent inhibition on osteoclastogenesis (Figure 1B and C). To determine any potential cytotoxicity of Pse on BMMs, MTS assay was performed to assess cell viability. Pse was found to have no effect on BMM proliferation over the concentrations used in this study (Figure 1D). To examine which stage of osteoclastogenesis was affected, cells were treated with 10 μM Pse at indicated time phases (Figure 1E). Pse was found to predominantly exert its suppressive effect during mid-late stage (Day 3-6) of osteoclast differentiation, rather than early stage (Day 1-3) (Figure 1F).
Pse suppresses RANKL-induced osteoclastogenesis in vitro. (A) The chemical structure and formula of Pse. (B) Representative images of TRAcP staining showing that Pse inhibited osteoclastogenesis dose-dependently. BMMs were stimulated with RANKL for 5 days in the absence or presence of indicated concentrations of Pse. (C) Quantification of TRAcP-positive multinucleated cells (nuclei >3) (n=3 per group). (D) Effects of Pse on BMM viability after 48 h treatment as measured by MTS assay (n=3 per group). (E) Representative images of TRAcP staining showing BMMs treated with Pse for the indicated days during osteoclastogenesis. (F) Quantification of TRAcP-positive multinucleated cells treated with Pse in different time periods (n=3 per group). All bar graphs are presented as mean ± SD. *p<0.05, **p<0.01 compared with control group (without Pse treatment). Scale bar = 200 µM. BMMs, bone marrow macrophages; OD, optical density; Pse, Pseurotin A; RANKL, receptor activator of nuclear factor‐κB ligand; TRAcP, tartrate-resistant acid phosphatase
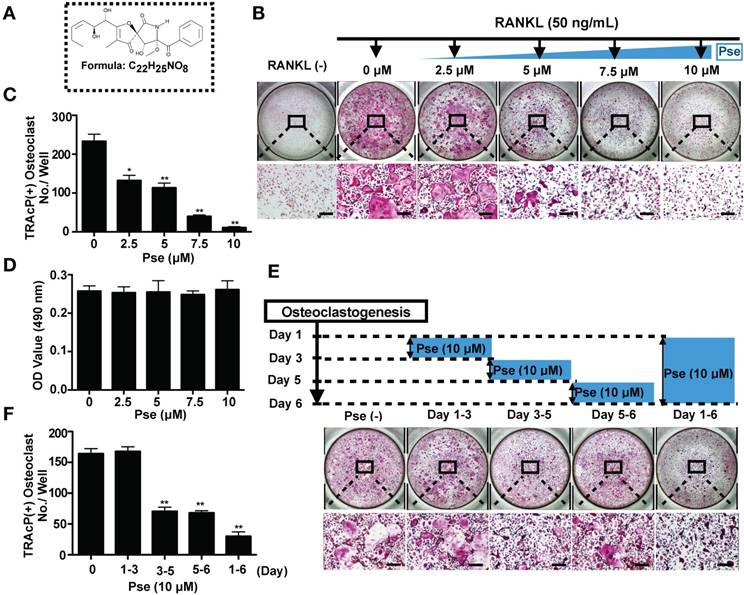
Pse affects podosome belt formation and inhibits osteoclast-specific genes expression
Cells were co-stained with rhodamine-phalloidin and anti-vinculin to visualize podosome belt formation and morphological changes in cells treated with or without Pse. As shown in Figure 2A, after RANKL stimulation, well-defined podosome belts with intact nuclei formed in mature osteoclasts. In contrast, smaller osteoclasts with fewer nuclei were observed after Pse treatment (5, 10 μM) (Figure 2B and C).
Pse affects podosome belts formation and inhibits osteoclast-specific genes expression. (A) Representative images showing the impaired podosome belts formation in osteoclasts treated with Pse. Vinculin (green), F-actin (red) and nuclei (blue) staining of osteoclasts on glass coverslip and observed by confocal microscopy. Overlapping staining of vinculin and F-actin is shown in yellow. (B) Quantification of the nuclei number per osteoclast (n=16 per group). (C) Quantification of the relative area of osteoclast (n=16 per group). (D-I) qPCR analysis of osteoclast-specific genes expression of c-fos, Nfatc1, Ctsk, Acp5, Atp6v0d2, and Mmp9 relative to Hprt1 and Hmbs in BMMs stimulated with RANKL for 5 days in the presence of Pse (n=3 per group). All bar graphs are presented as mean ± SD. **p<0.01 compared with the control group (treated with RANKL but without Pse). Scale bar = 200 µM. Acp5, acid phosphatase 5, tartrate resistant; Atp6v0d2, ATPase H+ Transporting V0 Subunit D2; BMMs, bone marrow macrophages; c-fos, Proto-oncogene C-Fos; Ctsk, cathepsin K; Mmp9, matrix metallopeptidase 9; Nfatc1, nuclear factor of activated T cells 1; PCR, polymerase chain reaction; Pse, Pseurotin A; RANKL, receptor activator of nuclear factor‐κB ligand; TRAcP, tartrate-resistant acid phosphatase
Several osteoclast-specific genes, including c-fos, Nfatc1, Ctsk, Acp5, Atp6v0d2, and Mmp9 are upregulated in BMMs when osteoclast differentiation is induced. We examined these genes at mRNA level using quantitative PCR to observe how Pse affected osteoclast-specific genes expression and found that their expression was inhibited during RANKL-induced osteoclastogenesis following the addition of Pse (Figure 2D-I). Collectively, these results further confirmed that Pse suppressed the expression of osteoclast-specific genes and thus osteoclastogenesis in vitro.
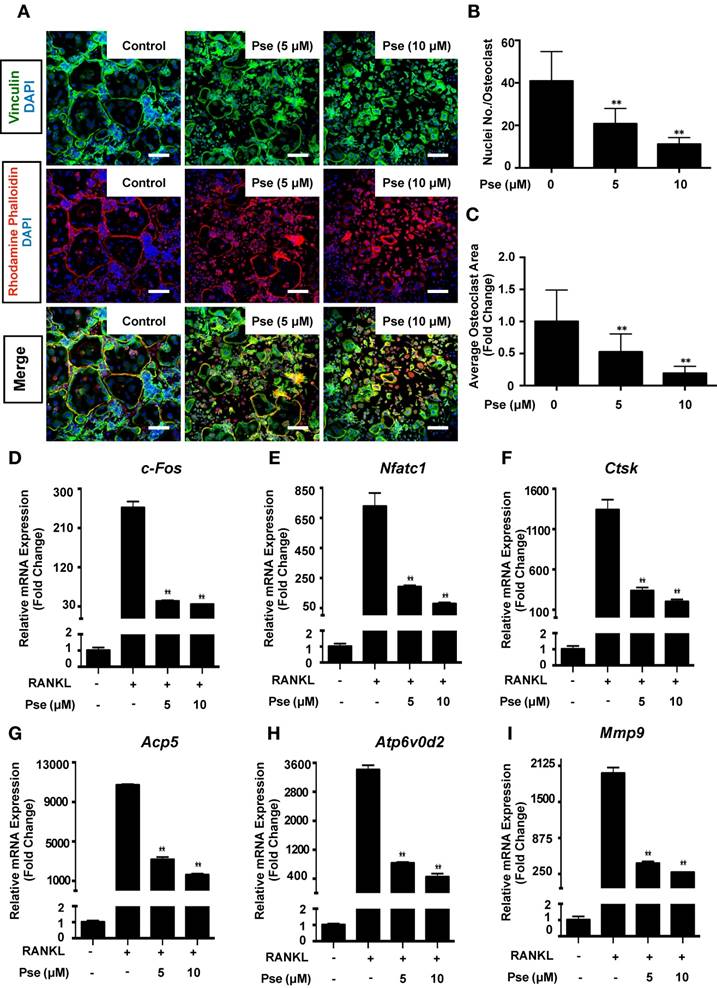
Pse inhibits osteoclast resorptive function
We next determined whether Pse has an effect on osteoclast resorptive activity using hydroxyapatite-coated plates. A reduction in the number of osteoclasts and the resorption area were observed at both Pse concentrations (5, 10 μM) (Figure 3A and B). In addition, following normalization of resorption area to the number of osteoclasts, a dose-dependent inhibitory effect on osteoclast resorptive function was observed as compared to the control group (Figure 3C).
Pse inhibits osteoclast resorptive function. (A) Representative images showing the osteoclastogenesis and hydroxyapatite resorption in each group. Osteoclasts were seeded in hydroxyapatite-coated plates and treated by RANKL with or without Pse. Half of the wells for each group were stained by TRAcP or washed with 10% bleach solution. (B-C) Quantification of resorbed hydroxyapatite area per well and resorbed area per osteoclast in each group (n=3 per group). All bar graphs are presented as mean ± SD. *p<0.05, compared with control. Scale bar = 200 µM. Pse, Pseurotin A; RANKL, receptor activator of nuclear factor‐κB ligand; TRAcP, tartrate-resistant acid phosphatase
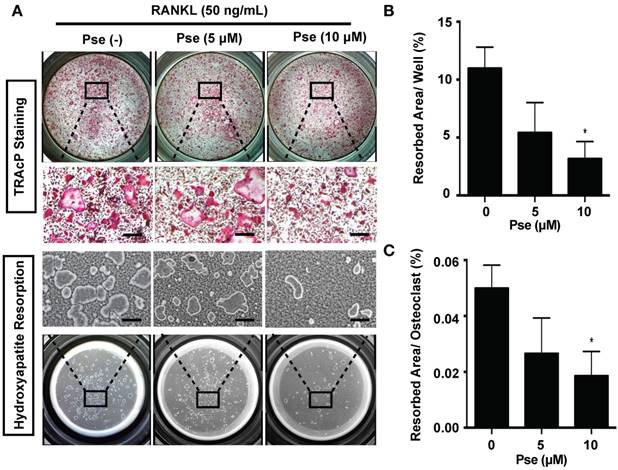
Pse reduces RANKL-induced intracellular ROS level in BMMs
To investigate whether RANKL-induced ROS production during osteoclast differentiation was reduced by Pse, ROS level was visualized using a cell-permeable, oxidation-sensitive dye H2DCFDA. ROS oxidation of this dye was detected as a fluorescent signal derived from the oxidation product DCF using confocal microscopy. The intensity of DCF fluorescence was significantly decreased in a dose-dependent manner in BMMs when stimulated with M-CSF and RANKL in the presence of Pse (5, 10 μM) (Figure 4 A-C). Therefore, our results indicated that Pse inhibited osteoclast formation and activity via suppressing ROS.
Pse attenuates RANKL-induced ROS generation in vitro. (A) Representative confocal images of RANKL-induced ROS generation in BMMs with or without pre-treatment of Pse. Intracellular ROS generation reacted with the cell permeant, oxidation-sensitive dye H2DCFHDA and was detected in the form of highly fluorescent DCF. The lower panel is a merge of DCF fluorescence and confocal digital interference contrast (DIC) images. (B) Quantification of DCF fluorescence intensity averaged on cells of each well (n=3 per group). (C) Quantification of the number of ROS-positive cells per field (n=3). (D) Representative Western Blot images of the effects of Pse on TRAF6 and Nox1 expression. BMMs were stimulated with RANKL (50 ng/mL) in the absence or presence of Pse (2.5, 5, 10 μM) for 2 days before protein collection for Western Blot. (E and F) Quantification of the ratios of band intensity of TRAF6 and Nox1 relative to β-actin (n=3 per group). (G) Representative Western Blot images of the effects of Pse on GTP-Rac1. BMMs were pretreated with Pse at indicated concentrations and were then stimulated with RANKL for 0, 5, and 15 min. Cell lysates were collected and incubated with GST-Human PAK1-PBD fusion protein that binds to GTP-bound Rac1. Rac1 activation levels were then detected with anti-Rac1 antibody. (H) Quantification of the ratios of band intensity of GTP-Rac1 relative to Rac1 (n=3 per group). (I) Representative Western Blot images of the effects of Pse on expression of antioxidant enzymes, including HO-1, Catalase, and GSR. (J-L) Quantification of the ratios of band intensity of HO-1, Catalase, and GSR relative to β-actin (n=3 per group). All bar graphs are presented as mean ± SD. *p<0.01, **p<0.01 relative to RANKL-induced control group. Scale bar = 200 μm. BMMs, bone marrow macrophages; DCF, 2',7'-dichlorofluorescein; GSR, glutathione-disulfide reductase; GTP, Guanosine-5'-triphosphate; H2DCFDA, 2',7'-dichlorodihydrofluorescein diacetate; HO-1, heme oxygenase-1; NOX, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase; Pse, Pseurotin A; Rac1, Ras-related C3 botulinum toxin substrate 1; RANKL, receptor activator of nuclear factor‐κB ligand; ROS, reactive oxygen species; TRAF6, TNF receptor-associated factor 6.
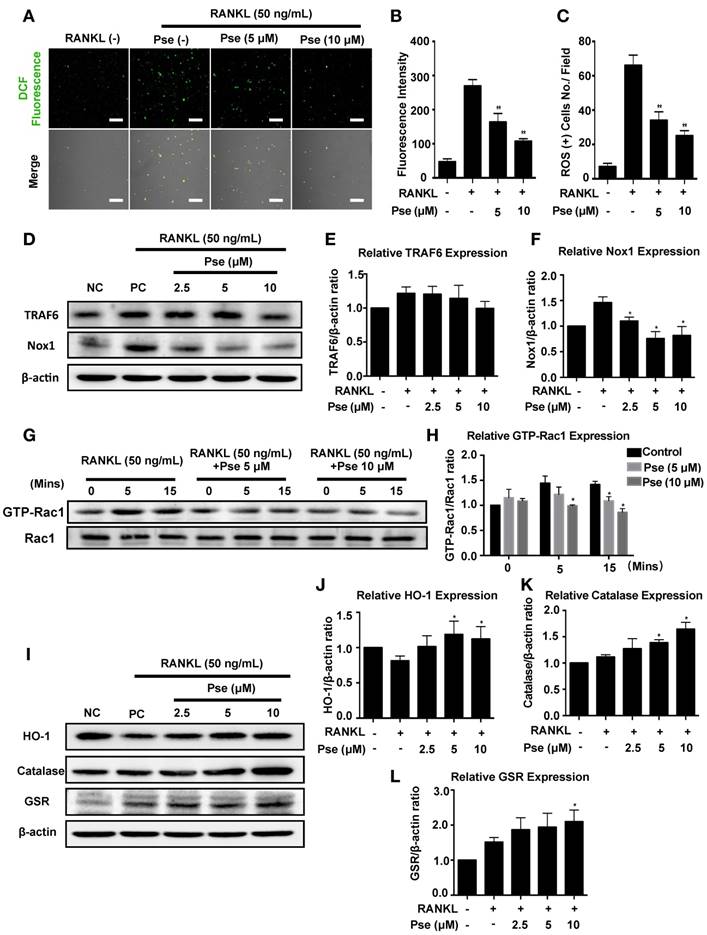
Pse suppresses intracellular ROS level by down-regulating the TRAF6/Rac1/Nox1 signaling cascade as well as enhancing expression of antioxidant enzymes
To determine whether Pse affects ROS generation, the activation level of Nox1, known as a major contributing factor in ROS generation [9, 40], was investigated using western blot. BMMs were incubated with RANKL for 2 days in the absence or presence of Pse at different concentrations as indicated. Nox1 expression was significantly up-regulated by RANKL, but dose-dependently inhibited by Pse at the concentrations of 2.5-10 μM (Figure 4 D and F). TRAF6 and GTP-Rac1 are required for the Nox1 activation, we therefore examined whether Pse inhibited Nox1 by attenuating TRAF6 and GTP-Rac1. Adaptor protein TRAF6 expression was augmented by RANKL and showed a trend of down-regulation at high concentrations (10 μM), which was not significant (Figure 4 D and E). GTP-bound Rac1 is a cytosolic component of Nox1 and is responsible for Nox1 activation. As shown in Figure 1G and H, GTP-Rac1 activation was significantly enhanced after 5 min and 15 min of RANKL stimulation but was attenuated by Pse treatment in a dose-dependent manner. Next, to examine whether Pse is able to scavenge ROS by up-regulating antioxidant enzymes, the expression of antioxidant enzymes including HO-1, catalase, and GSR were examined. As shown in Figure I and J, the expression of HO-1 was reduced by RANKL stimulation but was recovered and enhanced dose- dependently by Pse treatment. Similarly, Pse also enhanced the expression levels of catalase (Figure 4 I and K) and GSR (Figure 4 I and L). Taken together, these data mechanistically suggest that Pse suppresses RANKL-induced intracellular ROS level via inhibition of ROS generation as well as the enhancement of ROS scavenging.
Pse interferes with RANKL-induced activation of NF-κB and MAPK pathways
NF-κB and MAPK pathways are regarded as the main signaling pathways activated during osteoclastogenesis [6], and RANKL-induced ROS generation may trigger these signaling events. In order to further explore whether Pse blocked ROS activated osteoclastogenesis via attenuating NF-κB and MAPK signaling, we investigated these two pathways using Western Blot and luciferase assay. For the NF-κB pathway, we analyzed the expression of IκB-α. NF-κB transcription factors are bound to IκB-α and retained in an inactive state, these are activated and released when IκB-α is degraded after stimulation with RANKL [41]. Within 60 minutes of stimulation with RANKL, IκB-α degradation was significantly inhibited by Pse (Figure 5A and B), thus indicating failure to activate NF-κB signaling. Consistent with inhibition of IκB-α degradation, the results of luciferase assay showed the activation of NF-κB was significantly inhibited by treatment with Pse (Figure 5C). For the MAPK signaling pathway, as shown in Figure 5D, phosphorylation of ERK (Figure 5E), p38 (Figure 5F), and JNK (Figure 5G) relative to total ERK, total p38, and total JNK was suppressed significantly by Pse treatment in BMMs. Collectively, these data suggested Pse had an inhibitory effect on both NF-κB and MAPK activation pathways mediated by RANKL.
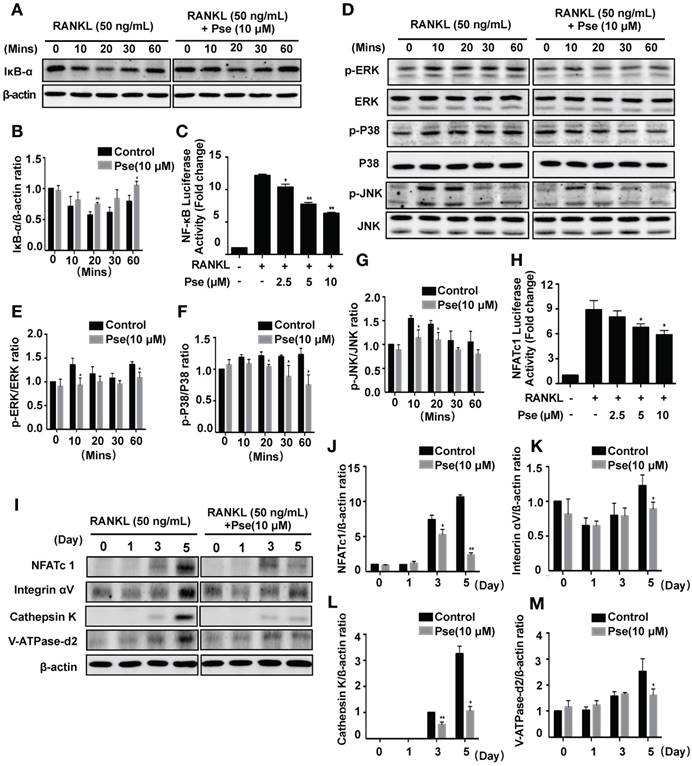
Pse suppresses osteoclast differentiation by inhibiting the activation of NF-κB pathway, MAPK pathway, and NFATc1 signaling. (A) Representative Western Blot images of the effects of Pse on IκBα degradation induced by RANKL. (B) Quantification of the ratios of band intensity of IκBα relative to β-actin (n=3 per group). (C) NF-κB luciferase assay showing that Pse inhibited NF-κB transcriptional activity dose-dependently (n=3 per group). (D) Representative Western Blot images of the effects of Pse on MAPKs pathway, including p-ERK, p-P38, and p-JNK. BMMs were pretreated with 10 μM Pse for 2 h prior to the addition of RANKL at the indicated time points and the indicated proteins were determined. (E-G) Quantification of the ratios of band intensity relative to ERK, P38, and JNK were analyzed (n=3 per group). (H) NFATc1 luciferase assay showing Pse significantly inhibited NFATc1 transcriptional activity (n=3 per group). (I) Representative Western Blot images of the expression levels of NFATc1 and osteoclast-related proteins including Integrin αV, Cathepsin K, and V-ATPase-d2 during osteoclastogenesis. BMMs were stimulated with RANKL in the absence or presence of 10 µM Pse for 0, 1, 3, and 5 days before protein collection for Western Blot. (J-M) Quantification of the ratios of band intensity of NFATc1, Integrin αV, Cathepsin K, and V-ATPase-d2 relative to β-actin (n=3 per group). All bar graphs are presented as mean ± SD. *P<0.05, **P<0.01 relative to RANKL-induced control group at the same time point. BMMs, bone marrow macrophages, MAPKs, mitogen-activated protein kinases; NFATc1, nuclear factor of activated T cells 1; NF-κB, nuclear factor-κB; Pse, Pseurotin A
Pse attenuates NFATc1 activity and downstream factors
NFATc1 acts as the master transcriptional regulator of osteoclastogenesis [7]. In this study, RAW264.7 cells stably expressing an NFATc1-driven luciferase reporter gene were used to measure the NFATc1 transcriptional activity. After stimulation with RANKL, NFATc1 activity was very highly elevated (Figure 5H). In contrast, NFATc1 activity was abrogated significantly in a dose-dependent manner after pre-treatment with Pse (2.5, 5, 10 μM) (Figure 5H). NFATc1 expression was also found to be suppressed over the course of osteoclast differentiation using Western Blot assay (Figure 5I and J). In addition, our results showed that Pse abrogates the elevation of integrin αV (Figure 5K), cathepsin K (Figure 5L), and V-ATPase-d2 (Figure 5M), which are all downstream proteins needed for osteoclast formation and function. Consequently, Pse treatment strongly inhibits NFATc1 activity, thus affecting downstream protein expression.
Pse prevents OVX-induced bone loss
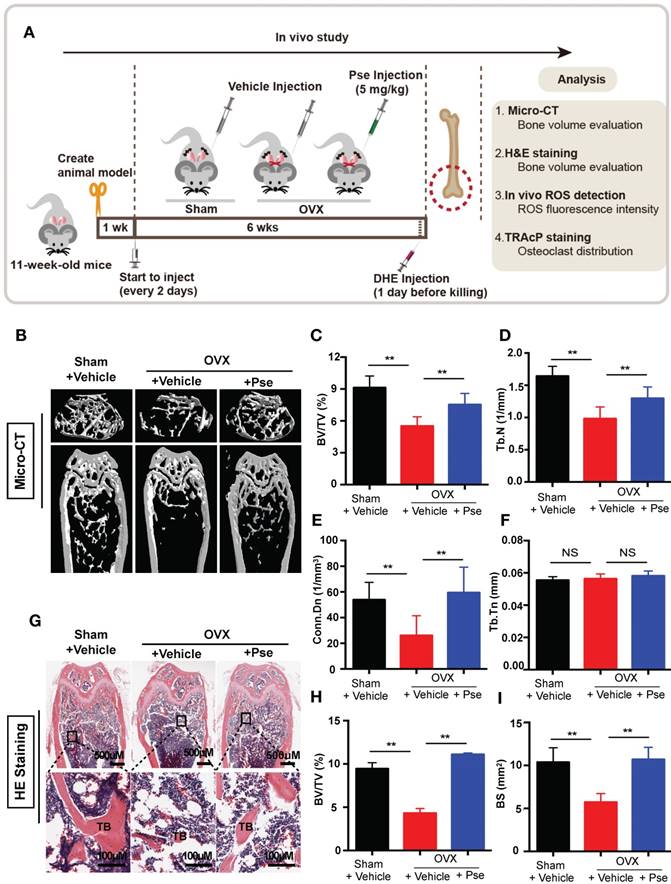
Our results established that Pse has inhibitory effects on osteoclast formation and function in vitro. The potential of Pse to act as a prophylactic agent to protect against osteoclast-related bone disease in vivo was further explored using an OVX-induced osteoporosis mice model. Mice were either OVX or sham operated and were then intraperitoneally injected with either Pse at a concentration of 5 mg/kg every 2 days, or vehicle only for 6 weeks post-surgery (Figure 6A). No adverse events or fatalities were recorded after the OVX procedure and Pse administration. Furthermore, there were no observable effects of Pse on body weight in treated mice relative to non-treated mice (Figure S5A). Micro-CT confirmed that Pse prevented the extensive bone loss induced by the OVX procedure in mice femurs (Figure 6B). Quantitative analysis of bone parameters also confirmed increased BV/TV, Tb.N and Conn.Dn in Pse treated group relative to the OVX mice without Pse treatment (Figure 6C-E). However, Tb.Th remained unchanged in this study (Figure 6F). Consistently, histological examination showed that Pse reduced the extent of bone loss induced by OVX (Figure 6G). Quantification of H&E staining demonstrated that the bone volume and bone surface were both well maintained in Pse treated group compared with non-Pse treated OVX mice (Figure 6H and I). Cortical bone related parameters, including cortical thickness (Ct.Th), total cross-sectional area (Tt.Ar), cortical bone area (Ct.Ar), and cortical area fraction (Ct.Ar/Tt.Ar), were also analyzed (Figure S5B-F). Pse showed no effect on cortical bone.
Pse treatment prevents ovariectomized (OVX)-induced bone loss in vivo. (A) Schematic illustration of the establishment of OVX mouse model and the experimental design to evaluate Pse's therapeutic effects. (B) Representative μCT images showing that the bone loss was prevented by Pse administration. (C-F) Quantitative analyses of parameters regarding bone microstructure, including BV/TV, Tb.N, Conn.Dn, and Tb.Th (N=6 per group). (G) Representative images of HE staining of decalcified bone sections. (H-I) Quantitative analyses of BV/TV and BS in tissue sections (n=4 per group). All bar graphs are presented as mean ± SD. **P<0.01 relative to the OVX group. BS, bone surface; BV/TV, bone volume per tissue volume; Conn.Dn, connectivity density; HE, hematoxylin and eosin; NS, non-significant; Pse, Pseurotin A; TB, trabecular bone; Tb.N, trabecular number; Tb.Th, trabecular thickness
Pse reduces osteoclasts and ROS production in the OVX mouse model
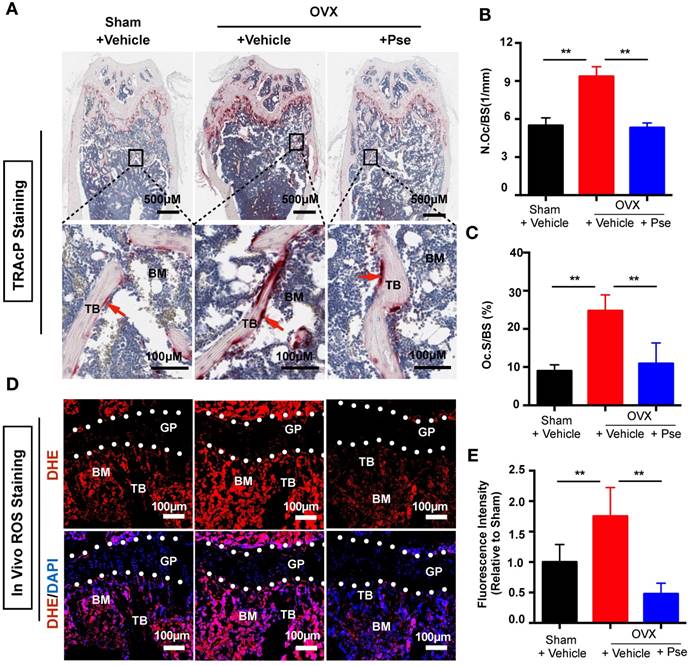
Histological staining using TRAcP was performed on femur sections to examine the number of osteoclasts in vivo. As shown in Figure 7A, the OVX procedure led to an increased number of TRAcP- positive osteoclasts, whereas Pse treatment prevented the increase of osteoclast number in vivo. This was supported by quantification of osteoclast parameters, demonstrating a reduction in the number and surface area of osteoclasts on the bone surface in Pse treated mice (Figure 7B and C). In addition, as Pse exhibited in vitro anti-oxidant activity, in vivo ROS level was also assessed in cryosections of bone tissue using DHE, a probe that detects ROS. Consistent with previous publications [42, 43], ROS fluorescence intensity was highly elevated due to OVX procedure and Pse dramatically reversed the ROS production within the bone marrow microenvironment (Figure 7 D and E). Taken together, these data indicated that Pse prevented OVX-induced bone loss in vivo by scavenging ROS thus inhibiting osteoclast activity.
Pse treatment reduces osteoclasts and ROS production in OVX mice. (A) Representative images of TRAcP staining of decalcified bone sections. (B-C) Quantitative analyses of N.Oc/BS and Oc.S/B (n=4 per group). (D) Representative images of bone cryosections showing ROS fluorescence in different groups. (E) Quantitative analyses of ROS fluorescence intensity relative to sham group (n=5 per group). All bar graphs are presented as mean ± SD. **P<0.01 relative to the OVX+Vehicle group. BM, bone marrow; DAPI, 4,6-diamidino-2-phenylindole; DHE, dihydroethidium; GP, growth plate; ROS, reactive oxygen species; N.Oc/BS, osteoclast number/bone surface; Oc.S/BS, osteoclast surface/bone surface; TB, trabecular bone; TRAcP, tartrate resistant acid phosphatase
Discussion
Osteoporosis is a skeletal disease which is characterized by low bone mass, leading to reduced bone strength and susceptibility to fracture. It is more frequently diagnosed in the postmenopausal population majorly due to excessive osteoclast activity. Therefore, osteoclasts remain as the major target in dealing with osteoporosis. Current clinically available therapies for osteoporosis such as estrogen replacement, bisphosphonates, and denosumab are effective but have limitations and side-effects including increased risk of breast cancer, osteonecrosis of the jaw, and atypical femur fracture [44, 45]. A search for novel alternative drugs is always required for the improved treatment of osteoporosis. In the present study, we elucidated for the first time that Pse inhibited osteoclastogenesis in vitro and prevented the development of OVX-induced osteoporosis in vivo via suppressing ROS level.
Firstly, osteoclastogenesis assay and hydroxyapatite resorption assay in vitro, showed Pse as a potentially novel therapy for osteoclast-related bone disease. Further, we investigated the mechanisms underlying this inhibitory effect on osteoclast formation and function, especially regarding the intracellular ROS level, NF-κB pathway, MAPK pathway, and NFATc1.
Upon stimulation with RANKL, intracellular ROS level was hugely suppressed by Pse treatment, which was accompanied by the reduced activation of NF-κB and MAPKs, leading to the subsequent attenuation of NFATc1. The level of intracellular ROS depends on the balance between the rate of production and the rate of scavenging. ROS are generated during osteoclastogenesis through a signaling cascade including TRAF6, Rac1, and Nox1 [9]. Nox1-mediated ROS production was found to regulate RANKL-induced signaling and is required for osteoclastogenesis [9, 40]. Therefore, we hypothesized that the down-regulated ROS signaling might be partly due to the inhibition of Nox1. Indeed, Pse effectively attenuated Nox1 expression via the suppression of GTP-Rac1. To scavenge oxidative stress and maintain redox homeostasis, a wide range of antioxidant enzymes are induced. Several ROS scavengers were determined to be upregulated by Pse in this study, such as HO-1, catalase, and GSR. HO-1 induction may act as a defense mechanism to catalyze heme liberated by oxidants [46]. Catalase plays a key role in converting hydrogen peroxide into water and oxygen [47]. GSR converts glutathione disulphide back to glutathione in an NADPH-consuming process and GSR inhibitor was also found to induce the activation of the NF-κB pathway [48]. The mechanisms behind the upregulation of antioxidant enzymes still remain unclear. Nuclear factor- erythroid 2 related factor 2 (Nrf2) is regarded as an important redox-sensitive transcription factor that positively regulates antioxidant enzymes [12, 13]. RANKL stimulation attenuated the gene expression of Nrf2 to favour ROS signalling, whereas Pse treatment recovered and upregulated Nrf2 expression dose dependently (Figure S4), suggesting that Pse may upregulate antioxidant enzymes at least in part via augmenting Nrf2. Taken together, Pse attenuates the ROS level by suppressing ROS production as well as enhancing ROS scavenging.
Accumulating evidence suggests that RANKL- induced ROS signaling regulates MAPK and NF-κB activation [9, 18, 49-51]. NF-κB signaling serves a crucial role and appears to be the first event in early osteoclast development from precursors, followed by c-Fos and NFATc1 activation [7]. Loss of NF-κB signaling in mice leads to an osteopetrotic bone phenotype due to a defect of osteoclastogenesis [52]. Tumor necrosis factor α (TNFα)-induced NF-κB activation is redox-dependently regulated through the dynein light chain LC8 [49]. ROS is able to oxidize LC8 to a homodimer linked by a disulphide bond between the Cys2 residues of each subunit, which promotes its dissociation from IκBα and thereby allows IκBα's phosphorylation and degradation by IκB kinase (IKK), thus releasing NF-κB dimers to translocate to nucleus [49]. In our study, Pse treatment effectively suppressed RANKL-induced IκB-α degradation, as demonstrated by a higher expression level of IκB-α in the Pse treated group, suggesting the involvement of IκB-dependent inactivation of NF-κB in the inhibitory effect of Pse on osteoclast formation. Furthermore, luciferase assay results consistently supported the inhibitory effect of Pse on NF-κB transcriptional activation.
The MAPK pathways, including three major MAPK family members (ERKs, JNKs, and p38), are also important signaling events involved in osteoclast differentiation stimulated by RANKL [6]. ERK is crucial for the survival of osteoclasts [53, 54]. Similarly, JNK and p38 are also phosphorylated during osteoclastogenesis in response to RANKL stimulation [54, 55]. MAPKs are activated by MAPK kinases through phosphorylation of tyrosine and threonine and inactivated by MAPK phosphatases (MKPs) through dephosphorylation [56]. ROS may act as a physiologic second messenger and oxidize tyrosine phosphatases, thus inhibiting MKPs and subsequently activating MAPKs [9, 57]. In the present study, the Western Blot results showed that Pse comprehensively attenuated the phosphorylation of ERK, JNK, and p38 thus achieving its inhibitory effects. Thus, ROS is suggested to incorporate into the activation of NF-κB and MAPKs during osteoclastogenesis.
NFATc1 is well-known as a master transcriptional regulator of terminal osteoclast differentiation, and its initiation is regulated in an auto-amplification loop to maintain robust expression by binding to its own promoter [8]. NFATc1 modulates the expression of osteoclast downstream gene expression, and NFATc1- deficiency results in inhibition of osteoclastogenesis in vitro and osteopetrosis in vivo [58]. Here, our data implicated that the expression level and transcriptional activity of NFATc1 were reduced by Pse following RANKL stimulation. The mechanism behind the reduction of NFATc1 still remains unclear. It could result from the suppression of upstream NF-κB and MAPK signaling or a direct intervention of NFATc1 activation. We further demonstrated that Pse suppressed the expression of osteoclast-specific genes, including Ctsk, Acp5, Atp6v0d2, and Mmp9, which are regulated by NFATc1 directly or indirectly [59, 60], and participate in osteoclast formation and their bone resorptive function. C-Fos, which cooperates with NFATc1 in the context of activator protein 1 (AP-1) and contributes to osteoclast differentiation [61], was also found to be inhibited by Pse treatment at mRNA expression level.
Based on these convincing in vitro results, an OVX mouse model was further used to evaluate whether Pse has therapeutic effects in vivo through its antioxidant activity. It is interesting to note that oxidative stress status has a negative correlation with osteoporotic status. OVX rats were found to have higher oxidative stress in the femurs accompanied by a decreased activity of antioxidant systems compared to sham-operated control rats [42, 62]. Furthermore, Altindag et al. found that the oxidative stress index values and total plasma oxidant status in osteoporotic postmenopausal women was higher than that in the healthy group [43]. The development of osteoporosis is at least partly due to the imbalance between antioxidant defenses and oxidative stress. This compelling evidence provides a new insight into a potential approach for the treatment of postmenopausal osteoporosis via suppressing oxidative stress.
Our results indicated that Pse exhibited a significant protective effect on OVX-induced bone loss in mice by scavenging ROS. A previous study on ROS detection in bone used direct staining on histological sections to assess ROS level [16]. However, the stability of ROS level is likely to be highly variable using this method due to the lengthy tissue fixation and processing times. In this study, a ROS probe was intravenously injected 24h prior to euthanasia, which allowed DHE to be distributed to cells and tissues via the circulation. DHE is a probe that is particularly sensitive to superoxide anion levels and it was previously reported as an in vivo marker for ROS [37]. The imaging of bone tissue remains challenging because of its calcified nature. Herein, a protocol of mild fixation, decalcification, and cryo-sectioning was used to process the bone samples, which was able to effectively preserve cellular morphology and tissue architecture [39]. To our knowledge, this is the first time that DHE was used as a ROS probe to detect ROS in vivo using histological sections in an OVX model. Our data showed that ROS level within bone marrow were dramatically suppressed in the Pse treated group, which was accompanied by a reduced number of TRAcP-positive osteoclasts. The prevention of bone loss by Pse was thought to be due to its suppression of oxidative stress and subsequent compromised osteoclast formation and function. However, this probe is not cell or tissue-specific, this data only indicated the total ROS level in bone marrow microenvironment, osteoclast-specific ROS in this study still remains to be further investigated.
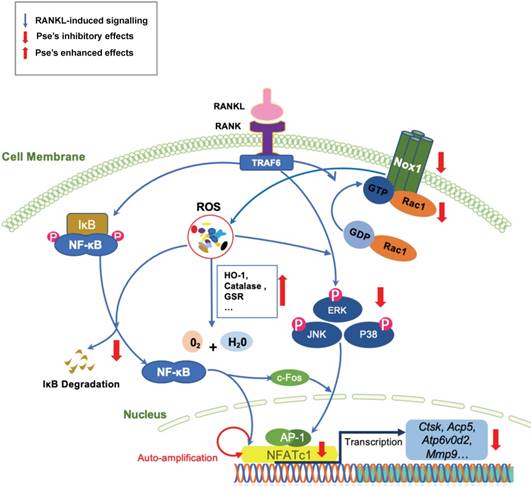
In summary, this study has demonstrated for the first time that Pse can suppress intracellular ROS level by inhibiting RANKL-induced ROS production and enhancing expression of antioxidant enzymes, which attenuates the activation of MAPK and NF-κB pathways, subsequently leading to attenuation of NFATc1 as well as its downstream proteins (Figure 8). These signaling events contribute to decreased osteoclast formation and bone resorptive function in vitro. Additionally, Pse was also found to prevent estrogen deficiency-induced osteoporosis in vivo via suppressing oxidative stress in the bone marrow microenvironment. Therefore, Pse may serve as a novel candidate or an alternative therapeutic treatment for osteoclast-related bone disease such as osteoporosis.
A proposed scheme for the inhibition of Pse on osteoclastogenesis. Upon RANKL binding to RANK, both NF-κB and MAPKs pathways are activated, leading to the amplification of NFATc1. Several osteoclast-specific genes such as Ctsk, Acp5, Atp6v0d2, and Mmp9 are upregulated as a result. These signaling events are mediated by RANKL-induced ROS signaling. Our results demonstrated for the first time that Pse inhibits osteoclastogenesis via suppressing ROS level by inhibiting RANKL-induced ROS production and enhancing expression of antioxidant enzymes. Acp5, acid phosphatase 5, tartrate resistant; AP-1, activator protein 1; Atp6v0d2, ATPase H+ Transporting V0 Subunit D2; NFATc1, nuclear factor of activated T cells 1; c-fos, Proto-oncogene C-Fos; Ctsk, cathepsin K; Mmp9, matrix metallopeptidase 9; NOX, nicotinamide adenine dinucleotide phosphate oxidase; Pse, Pseurotin A; Rac1, Ras-related C3 botulinum toxin substrate 1; RANKL, receptor activator of nuclear factor‐κB (NF-κB) ligand; ROS, reactive oxygen species
Abbreviations
Acp5: acid phosphatase 5, tartrate resistant; Atp6v0d2: ATPase H+ Transporting V0 Subunit D2; BMMs: bone marrow macrophages; BV/TV: bone volume per tissue volume; c-fos: Proto-oncogene C-Fos; Ctsk: cathepsin K; Conn.Dn: connectivity density; DAPI: 4,6-diamidino-2-phenylindole; DCF: 2',7'-dichlorofluorescein; DHE: dihydroethidium; FBS: fetal bovine serum; GCS: γ-glutamylcysteine synthetase; GSR: glutathione-disulfide reductase; GTP: Guanosine-5'-triphosphate; H2DCFDA: 2',7'-dichlorodihydrofluorescein diacetate; HO-1: heme oxygenase-1; IKK: IκB kinase; MAPKs: mitogen-activated protein kinases; M-CSF: macrophage-colony stimulating factor; Mmp9: matrix metallopeptidase 9; NADPH: nicotinamide adenine dinucleotide phosphate; NAD(P)H: quinone reductase (NQO1); NFATc1: nuclear factor of activated T cells 1; NF-κB: nuclear factor-κB; NOX: NADPH oxidase; PBS: phosphate buffered saline; PCR: polymerase chain reaction; Pse: Pseurotin A; PVP: polyvinylpyrrolidone; OVX: ovariectomized; RANKL: receptor activator of nuclear factor‐κB ligand; Rac1: Ras- related C3 botulinum toxin substrate 1; ROS: reactive oxygen species; Tb.N: trabecular number; TNF: tumor necrosis factor; Tb.Th: trabecular thickness; TRAcP: tartrate resistant acid phosphatase; TRAF6: TNF receptor-associated factor 6.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was supported by grants from the Australian Health and Medical Research Council (NHMRC, No. 1107828, 1127156, 1163933). This study was also supported by the National Nature Science Fund of China (81702143). Kai Chen is supported by Australian Government Research Training Program Scholarship. We acknowledge the facilities and technical assistance of the Centre for Microscopy, Characterization & Analysis, the University of Western Australia.
Contributions
Kai Chen, Yu Yuan, Jianbo He, Chao Wang, and Qiang Guo performed cell culture, qPCR and signaling studies; Pengcheng Qiu, Lin Zheng, and Xianfeng Lin performed animal experiments; Kai Chen, Jacob Kenny, and Qian Liu performed histological analyses; Kai Chen, Junhao Chen, and Jianbo He prepared the figures; Kai Chen, Junhao Chen, Jennifer Tickner wrote the manuscript; Jinmin Zhao designed animal experiments; Kai Chen, Jiake Xu, and Jianbo He revised the manuscript; Jiake Xu, Xianfeng Lin, and Shunwu Fan designed and supervised the overall study and revised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science. 2000;289:1508-14
2. Kular J, Tickner J, Chim SM, Xu J. An overview of the regulation of bone remodelling at the cellular level. Clin Biochem. 2012;45:863-73
3. Harvey N, Dennison E, Cooper C. Osteoporosis: impact on health and economics. Nat Rev Rheumatol. 2010;6:99-105
4. Gingery A, Bradley E, Shaw A, Oursler MJ. Phosphatidylinositol 3-kinase coordinately activates the MEK/ERK and AKT/NFkappaB pathways to maintain osteoclast survival. J Cell Biochem. 2003;89:165-79
5. Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Yano K. et al. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem Biophys Res Commun. 1998;253:395-400
6. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337-42
7. Yamashita T, Yao Z, Li F, Zhang Q, Badell IR, Schwarz EM. et al. NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. J Biol Chem. 2007;282:18245-53
8. Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H. et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202:1261-9
9. Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS. et al. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106:852-9
10. Kim H, Lee YD, Kim HJ, Lee ZH, Kim HH. SOD2 and Sirt3 Control Osteoclastogenesis by Regulating Mitochondrial ROS. J Bone Miner Res. 2017;32:397-406
11. Yip KH, Zheng MH, Steer JH, Giardina TM, Han R, Lo SZ. et al. Thapsigargin modulates osteoclastogenesis through the regulation of RANKL-induced signaling pathways and reactive oxygen species production. J Bone Miner Res. 2005;20:1462-71
12. Kanzaki H, Shinohara F, Kajiya M, Kodama T. The Keap1/Nrf2 protein axis plays a role in osteoclast differentiation by regulating intracellular reactive oxygen species signaling. J Biol Chem. 2013;288:23009-20
13. Hyeon S, Lee H, Yang Y, Jeong W. Nrf2 deficiency induces oxidative stress and promotes RANKL-induced osteoclast differentiation. Free Radic Biol Med. 2013;65:789-99
14. Yamaguchi Y, Kanzaki H, Katsumata Y, Itohiya K, Fukaya S, Miyamoto Y. et al. Dimethyl fumarate inhibits osteoclasts via attenuation of reactive oxygen species signalling by augmented antioxidation. J Cell Mol Med. 2017
15. Song D, Cao Z, Liu Z, Tickner J, Qiu H, Wang C. et al. Cistanche deserticola polysaccharide attenuates osteoclastogenesis and bone resorption via inhibiting RANKL signaling and reactive oxygen species production. J Cell Physiol. 2018;233:9674-84
16. Kim HS, Nam ST, Mun SH, Lee SK, Kim HW, Park YH. et al. DJ-1 controls bone homeostasis through the regulation of osteoclast differentiation. Nat Commun. 2017;8:1519
17. Li J, Wang Q, Yang R, Zhang J, Li X, Zhou X. et al. BMI-1 Mediates Estrogen-Deficiency-Induced Bone Loss by Inhibiting Reactive Oxygen Species Accumulation and T Cell Activation. J Bone Miner Res. 2017;32:962-73
18. Lean JM, Davies JT, Fuller K, Jagger CJ, Kirstein B, Partington GA. et al. A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J Clin Invest. 2003;112:915-23
19. Wauquier F, Leotoing L, Coxam V, Guicheux J, Wittrant Y. Oxidative stress in bone remodelling and disease. Trends Mol Med. 2009;15:468-77
20. Sendur OF, Turan Y, Tastaban E, Serter M. Antioxidant status in patients with osteoporosis: a controlled study. Joint Bone Spine. 2009;76:514-8
21. Bloch P, Tamm C, Bollinger P, Petcher TJ, Weber HP. Pseurotin, a new metabolite of Pseudeurotium ovalis Stolk having an unusual hetero-spirocyclic system. Helv Chim Acta. 1976;59:133-7
22. Martinez-Luis S, Cherigo L, Arnold E, Spadafora C, Gerwick WH, Cubilla-Rios L. Antiparasitic and anticancer constituents of the endophytic fungus Aspergillus sp. strain F1544. Nat Prod Commun. 2012;7:165-8
23. Igarashi Y, Yabuta Y, Sekine A, Fujii K, Harada K, Oikawa T. et al. Directed biosynthesis of fluorinated pseurotin A, synerazol and gliotoxin. J Antibiot (Tokyo). 2004;57:748-54
24. Shaaban M, El-Metwally MM, Nasr H. A new diketopiperazine alkaloid from Aspergillus oryzae. Nat Prod Res. 2014;28:86-94
25. Maiya S, Grundmann A, Li X, Li SM, Turner G. Identification of a hybrid PKS/NRPS required for pseurotin A biosynthesis in the human pathogen Aspergillus fumigatus. Chembiochem. 2007;8:1736-43
26. Myers RR, Smith TD, Elsawa SF, Puel O, Tadrist S, Calvo AM. rtfA controls development, secondary metabolism, and virulence in Aspergillus fumigatus. PLoS One. 2017;12:e0176702
27. Vodisch M, Scherlach K, Winkler R, Hertweck C, Braun HP, Roth M. et al. Analysis of the Aspergillus fumigatus proteome reveals metabolic changes and the activation of the pseurotin A biosynthesis gene cluster in response to hypoxia. J Proteome Res. 2011;10:2508-24
28. Ishikawa M, Ninomiya T, Akabane H, Kushida N, Tsujiuchi G, Ohyama M. et al. Pseurotin A and its analogues as inhibitors of immunoglobulin E [correction of immunoglobuline E] production. Bioorg Med Chem Lett. 2009;19:1457-60
29. Pinheiro EA, Carvalho JM, dos Santos DC, Feitosa Ade O, Marinho PS, Guilhon GM. et al. Antibacterial activity of alkaloids produced by endophytic fungus Aspergillus sp. EJC08 isolated from medical plant Bauhinia guianensis. Nat Prod Res. 2013;27:1633-8
30. Hayashi A, Fujioka S, Nukina M, Kawano T, Shimada A, Kimura Y. Fumiquinones A and B, nematicidal quinones produced by Aspergillus fumigatus. Biosci Biotechnol Biochem. 2007;71:1697-702
31. Lu QQ, Tian JM, Wei J, Gao JM. Bioactive metabolites from the mycelia of the basidiomycete Hericium erinaceum. Nat Prod Res. 2014;28:1288-92
32. Xu J, Tan JW, Huang L, Gao XH, Laird R, Liu D. et al. Cloning, sequencing, and functional characterization of the rat homologue of receptor activator of NF-kappaB ligand. J Bone Miner Res. 2000;15:2178-86
33. Zhou L, Liu Q, Yang M, Wang T, Yao J, Cheng J. et al. Dihydroartemisinin, an Anti-Malaria Drug, Suppresses Estrogen Deficiency-Induced Osteoporosis, Osteoclast Formation, and RANKL-Induced Signaling Pathways. J Bone Miner Res. 2016;31:964-74
34. Chen K, Yuan Y, Wang Z, Song D, Zhao J, Cao Z. et al. Helvolic acid attenuates osteoclast formation and function via suppressing RANKL-induced NFATc1 activation. J Cell Physiol. 2019;234:6477-88
35. Wang C, Steer JH, Joyce DA, Yip KH, Zheng MH, Xu J. 12-O-tetradecanoylphorbol-13-acetate (TPA) inhibits osteoclastogenesis by suppressing RANKL-induced NF-kappaB activation. J Bone Miner Res. 2003;18:2159-68
36. van der Kraan AG, Chai RC, Singh PP, Lang BJ, Xu J, Gillespie MT. et al. HSP90 inhibitors enhance differentiation and MITF (microphthalmia transcription factor) activity in osteoclast progenitors. Biochem J. 2013;451:235-44
37. Rodriguez-Muela N, Germain F, Marino G, Fitze PS, Boya P. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ. 2012;19:162-9
38. Kim GW, Kondo T, Noshita N, Chan PH. Manganese superoxide dismutase deficiency exacerbates cerebral infarction after focal cerebral ischemia/reperfusion in mice: implications for the production and role of superoxide radicals. Stroke. 2002;33:809-15
39. Kusumbe AP, Ramasamy SK, Starsichova A, Adams RH. Sample preparation for high-resolution 3D confocal imaging of mouse skeletal tissue. Nat Protoc. 2015;10:1904-14
40. Sasaki H, Yamamoto H, Tominaga K, Masuda K, Kawai T, Teshima-Kondo S. et al. NADPH oxidase-derived reactive oxygen species are essential for differentiation of a mouse macrophage cell line (RAW264.7) into osteoclasts. J Med Invest. 2009;56:33-41
41. Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IkappaB kinase. Cell. 1997;90:373-83
42. Almeida M, Han L, Martin-Millan M, Plotkin LI, Stewart SA, Roberson PK. et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. 2007;282:27285-97
43. Altindag O, Erel O, Soran N, Celik H, Selek S. Total oxidative/anti-oxidative status and relation to bone mineral density in osteoporosis. Rheumatol Int. 2008;28:317-21
44. Black DM, Bauer DC, Schwartz AV, Cummings SR, Rosen CJ. Continuing bisphosphonate treatment for osteoporosis-for whom and for how long? N Engl J Med. 2012;366:2051-3
45. Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet. 2011;377:1276-87
46. Hayashi S, Takamiya R, Yamaguchi T, Matsumoto K, Tojo SJ, Tamatani T. et al. Induction of heme oxygenase-1 suppresses venular leukocyte adhesion elicited by oxidative stress: role of bilirubin generated by the enzyme. Circ Res. 1999;85:663-71
47. Glorieux C, Calderon PB. Catalase, a remarkable enzyme: targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol Chem. 2017;398:1095-108
48. Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47-95
49. Jung Y, Kim H, Min SH, Rhee SG, Jeong W. Dynein light chain LC8 negatively regulates NF-kappaB through the redox-dependent interaction with IkappaBalpha. J Biol Chem. 2008;283:23863-71
50. Bax BE, Alam AS, Banerji B, Bax CM, Bevis PJ, Stevens CR. et al. Stimulation of osteoclastic bone resorption by hydrogen peroxide. Biochem Biophys Res Commun. 1992;183:1153-8
51. Kim HJ, Chang EJ, Kim HM, Lee SB, Kim HD, Su Kim G. et al. Antioxidant alpha-lipoic acid inhibits osteoclast differentiation by reducing nuclear factor-kappaB DNA binding and prevents in vivo bone resorption induced by receptor activator of nuclear factor-kappaB ligand and tumor necrosis factor-alpha. Free Radic Biol Med. 2006;40:1483-93
52. Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med. 1997;3:1285-9
53. Lee SE, Chung WJ, Kwak HB, Chung CH, Kwack KB, Lee ZH. et al. Tumor necrosis factor-alpha supports the survival of osteoclasts through the activation of Akt and ERK. J Biol Chem. 2001;276:49343-9
54. Lee SE, Woo KM, Kim SY, Kim HM, Kwack K, Lee ZH. et al. The phosphatidylinositol 3-kinase, p38, and extracellular signal-regulated kinase pathways are involved in osteoclast differentiation. Bone. 2002;30:71-7
55. David JP, Sabapathy K, Hoffmann O, Idarraga MH, Wagner EF. JNK1 modulates osteoclastogenesis through both c-Jun phosphorylation-dependent and -independent mechanisms. J Cell Sci. 2002;115:4317-25
56. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239-52
57. Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649-61
58. Aliprantis AO, Ueki Y, Sulyanto R, Park A, Sigrist KS, Sharma SM. et al. NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J Clin Invest. 2008;118:3775-89
59. Song I, Kim JH, Kim K, Jin HM, Youn BU, Kim N. Regulatory mechanism of NFATc1 in RANKL-induced osteoclast activation. FEBS Lett. 2009;583:2435-40
60. Feng H, Cheng T, Steer JH, Joyce DA, Pavlos NJ, Leong C. et al. Myocyte enhancer factor 2 and microphthalmia-associated transcription factor cooperate with NFATc1 to transactivate the V-ATPase d2 promoter during RANKL-induced osteoclastogenesis. J Biol Chem. 2009;284:14667-76
61. Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H. et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889-901
62. Muthusami S, Ramachandran I, Muthusamy B, Vasudevan G, Prabhu V, Subramaniam V. et al. Ovariectomy induces oxidative stress and impairs bone antioxidant system in adult rats. Clin Chim Acta. 2005;360:81-6
Author contact
![]() Corresponding authors: Jiake Xu, Email: jiake.xuedu.au; Xianfeng Lin, Email: xianfeng_linedu.cn
Corresponding authors: Jiake Xu, Email: jiake.xuedu.au; Xianfeng Lin, Email: xianfeng_linedu.cn