Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2019; 9(6):1741-1751. doi:10.7150/thno.30972 This issue Cite
Research Paper
A Novel TNF-α-Targeting Aptamer for TNF-α-Mediated Acute Lung Injury and Acute Liver Failure
Wei-Yun Lai1,2, Jen-Wei Wang1,2, Bo-Tsang Huang2, Emily Pei-Ying Lin3, Pan-Chyr Yang2,4 ![]()
1. Aptamer Core Facility, Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan.
2. Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan.
3. National Center of Excellence for Clinical Trials and Research Center, Department of Medical Research, National Taiwan University Hospital, Taipei, Taiwan
4. Department of Internal Medicine, National Taiwan University Hospital and College of Medicine, Taipei, Taiwan.
Received 2018-10-25; Accepted 2019-1-18; Published 2019-2-28
Abstract

Rationale: The TNF-α pathway plays as a double-edged sword that simultaneously regulates cell apoptosis and proliferation. The dysregulated TNF-α signaling can trigger cytokine storms that lead to profound cell death during the phase of acute tissue injury. On the other hand, an optimal level of TNF-α signaling is required for tissue repair following the acute injury phase. The TNF-α pathway is commonly upregulated in acute lung injury (ALI) and acute liver failure (ALF). Previous studies investigated the feasibility of adopting protein-based TNF-α blockers as disease modifiers in ALI and ALF, but none of these came out with a positive result. One of the potential reasons that resides behind the failure of the trials might be the long half-life of these inhibitors that led to undesired side effects. Developing alternative TNF-α blockers with manageable half-lives remain an unmet need in this regard.
Methods: In the current study, we developed a novel TNF-α-targeting aptamer (aptTNF-α) and its PEG-derivate (aptTNF-α-PEG) with antagonistic functions. We investigated the in vivo antagonistic effects using mouse ALI and ALF models.
Results: Our data showed that aptTNF-α possessed good in vitro binding affinity towards human/mouse TNF-α and successfully targeted TNF-α in vivo. In the mouse ALI model, aptTNF-α/aptTNF-α-PEG treatment attenuated the severity of LPS-induced ALI, as indicated by the improvement of oxygen saturation and lung injury scores, the reduction of protein-rich fluid leakage and neutrophil infiltration in the alveolar spaces, and the suppression of pro-inflammatory cytokines/chemokines expressions in the lung tissues. In the mouse ALF model, we further showed that aptTNF-α/aptTNF-α-PEG treatment not only attenuated the degree of hepatocyte damage upon acute injury but also potentiated early regeneration of the liver tissues.
Conclusion: The results implicated potential roles of aptTNF-α/aptTNF-α-PEG in ALI and ALF. The data also suggested their translational potential as a new category of TNF-α blocking agent.
Keywords: aptamer, acute lung injury, acute liver failure, TNF-α
Introduction
The TNF-α pathway plays a pivotal role in acute inflammatory diseases, including systemic inflammatory response syndrome (SIRS) that comes along with acute organ damages, such as acute lung injury (ALI) and acute liver failure (ALF) [1-3]. At the phase of acute tissue injury, dysregulated TNF-α signaling triggers cell death, releases danger associated- molecular patterns, and activates tissue resident neutrophils and macrophages [3-7]. These activated resident inflammatory cells secrete TNF-α and other pro-inflammatory cytokines/chemokines, such as IL-1β, IL-6, and IL-8, which in turn recruit polymorphonuclear leukocytes (PMNs) into the damaged tissues and aggravate the cascade of cell death signaling. On the other hand, the TNF-α pathway is also the main regulator for tissue repair following acute injury [8, 9]. TNF-α activates p38 MAPK and NF-κB signaling pathways that subsequently induce cell proliferation and differentiation, activate resident progenitor cells, and mobilize circulating stem cells for tissue regeneration [10-12].
ALI is a frequently encountered issue in critical care medicine [13]. ALF is a life-threatening disease that occurs in patients without pre-existing liver diseases [1]. Both diseases are related to the activation of the TNF-α-mediated inflammatory cascade that leads to an excessive cytokine/chemokine release, PMNs infiltration, and cell death. Evidences also showed an inverse correlation between serum TNF-α levels and survival outcomes in these patients [4, 14, 15]. Therefore, previous studies attempted to investigate the feasibility of adopting TNF-α blocking agents in the management of ALI, ALF, and SIRS of various etiologies. However, none of these trials showed a positive result [16-20].
Many reasons might contribute to the failure of these trials, and one of these could be the long half-life of the agents [16-20]. Antibodies or Fc-fusion receptor proteins usually possess long half-lives, ranging from 4 to 20 days [21, 22]. Therefore, use of these agents may disturb tissue repair processes and raise infection probabilities due to sustained suppression of the TNF-α signaling. In addition, all the TNF-α antagonists used in these trials contained Fc region [16-20]. The Fc region recognizes membrane-bound TNF-α molecules located on macrophages, activated T lymphocytes, and PMNs [21]. The binding of the Fc to the membrane-bound TNF-α may trigger antibody- dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) effects, which may in turn lead to drug-induced liver toxicity observed in patients receiving TNF-α blocking agents [21].
Molecular mechanisms underline ALI and ALF suggest that targeting TNF-α pathway could be a direction to go. Past experiences from negative clinical trial results implicate that an appropriate timing for intervention may be the key to success. Nevertheless, how and when should the inhibition be executed during the disease course with an appropriate agent remains an unsolved task.
In terms of precise and timely drug delivery, aptamer can be an attractive therapeutic modality [23, 24]. Without modification, the half-life of DNA aptamers can be as short as one hour [25]. With PEGylation, the half-life can be extended up to 9 days, depending on the molecular size of the PEG used [26, 27]. Therefore, it is possible that a TNF-α-targeting aptamer suppresses the TNF-α-induced cell apoptosis signaling at the early tissue injury phase and then be degraded and excreted without affecting the cell proliferating signaling at the late tissue repair phase. In addition, aptamers do not have the Fc region and are thus devoid of the unwanted ADCC and CDC that may lead to undesired side effects. Taking all these advantages into account, it is worthwhile to develop a TNF-α-targeting aptamer with therapeutic effect. This study investigated the in vivo antagonistic effects of a novel TNF-α-targeting aptamer using the mouse ALI and ALF models.
Materials and methods
Chemicals and oligonucleotides
All chemicals were purchased from Sigma- Aldrich and oligonucleotides were synthesized by Integrated DNA technologies. The sequences of aptTNF-α are 5'-GCGCCACTACAGGGGAGCTGCCATTCGAATAGGTGGGCCGC-3'.
SELEX
Human TNF-α-targeting aptamers were identified by nitrocellulose filter SELEX. The synthetic single-stranded DNA library was composed of 80-nucleotide-long single-stranded DNAs with 40 random sequences flanked by primer sequences, 5'-ACGCTCGGATGCCACTACAG[N]40CTCATGGACGTGCTGGTGAC, N=A, T, G, C. In the first SELEX round, the 1015-molecule ssDNA library was incubated with recombinant human TNF-α proteins (R&D Systems). The ssDNAs that bound to TNF-α proteins were collected by nitrocellulose filter and the unbound ssDNAs were removed through repetitive washing. The TNF-α-bound ssDNAs were then eluted by heating, incubated with albumin for negative selection, and then passed through the nitrocellulose filter. The flow-through was collected and amplified by PCR. The SELEX was repeated for ten rounds. The TNF-α-bound ssDNAs and the albumin-bound ssDNAs were both subjected to next-generation sequencing (Illumina MiSeq System). The output reads were clustered by FASTApatmer [28] and subtracted with the clusters appeared in the albumin-bound group. The representative sequences that had the highest reads in the remaining clusters were then subjected to structure analysis using Mfold. Their truncated derivatives were designed according to the secondary structures predicted.
PEG conjugation
An excess amount of aptTNF-α with primary amine modification at 5' end was incubated with bifunctional N-hydroxylsuccinimide polyethylene glycol (NHS-PEG-NHS, molecular weight 20kDa, Polysciences Inc.) in sodium bicarbonate buffer (pH 8.3) at 37℃ for 18 h. The PEGylated dimeric aptTNF-α (aptTNF-α-PEG) were purified by non-denaturing polyacrylamide gel electrophoresis and the concentration was determined by Nanodrop spectrophotometer (Thermo Scientific).
Binding affinity determination
Human TNF-α proteins (0, 8.75, 17.5, 35, 70, 140 nM, R&D Systems) were incubated with aptTNF-α (50 nM) at 37℃ for 1 h. In addition, mouse TNF-α proteins (140 nM) or BSA (140 nM) were incubated with aptTNF-α (50 nM) or aptTNF-α-PEG (50 nM) as well. The protein-bound aptamers were then collected by nitrocellulose filter and eluted by heating. The amount of the eluted aptamers was quantified by quantitative PCR (LightCycler 480 system, Roche Applied Science). The dissociated constant (Kd) was calculated by GraphPad Prism 5 (GraphPad Software), using the equation Y = Amax × X/(Kd + X). The relative amounts of protein-binding aptamers (human TNF-α proteins and mouse TNF-α proteins) were represented as fold changes, using BSA as the reference (1 fold).
The biodistribution of aptTNF-α
The mice were purchased from the National Laboratory Animal Center. All the animal experiments were done according to the guidance of animal facility at Academia Sinica. Six-week-old Balb/c male mice were administrated with LPS (10 mg/kg, intratracheal) for the induction of ALI. IRDye® 800CW-labeled aptTNF-α or random sequences pool (Integrated DNA technologies) was intravenously injected 1 h after LPS administration. The fluorescent signals emitted from the aptTNF-α or random sequences pool were detected by Xenogen IVIS Imaging System 200 Series (Caliper Life Sciences) at 2, 4, 7, 10 and 24 h post aptamer administration, respectively [12]. In addition, a group of IRDye® 800CW-labeled aptTNF-α-treated mice were sacrificed at 4 h post aptamer administration. Vital organs, including heart, liver, spleen, lung, kidney, and bladder, were collected and the fluorescent signal emitted from the aptTNF-α were detected. The blood was sampled and the levels of LDH, AST, and ALT were measured by Fuji Dri-Chem 4000i (Fujifilm). Besides, a group of mice treated with Alexa Fluor 647-labeled aptTNF-α or random sequences pool (Integrated DNA technologies) were sacrificed at 2 h post drug administration. The mice were perfused with PBS and fixed with 4% paraformaldehyde (Electron Microscopy Sciences). The lung tissues were collected, immersed in 30% sucrose at 4℃ for 24 h, and then frozen-embedded by O.C.T. Compound (Sakura Finetek USA, Inc.) for cryosection. The thickness of tissue sections was 14 μm.
Cell culture and luciferase activity assays
HEK293 cells were cultured in DMEM (Gibco BRL, Life Technologies) with 10% FBS (Gibco) and transfected with NF-κB reporter (pGL4.32, Promega). At 24 h post transfection, cells were treated with hygromycin for the selection of antibiotic-resistant clones. The NF-κB reporter-expressing HEK293 cells were seeded into 96-well plates (2000 cells each) for overnight culture. TNF-α proteins (5 ng) along with aptTNF-α (50, 500 nM), aptTNF-α-PEG (10, 50 nM), or anti-human TNF-α antibody (10, 50 nM, R&D Systems) were added into each independent well. After 4 or 24 h incubation, the luciferase activity was determined by the luciferase assay system (Promega) following the manufacturer's protocol. Data was expressed as relative luciferase activity using the group without treatment as the negative control (0% of activity) and the group with TNF-α treatment as the positive control (100% of activity). To investigate the inhibitory effect of aptTNF-α-PEG on TNF-α-induced ERK signaling, HEK293 cells (3 x 106 cells each) were treated with TNF-α (0, 10, 20, 40 ng/ml) with or without aptTNF-α-PEG (10, 50 nM) for 15 min. The total cell lysates were collected for immunoblot analyses.
Acute lung injury (ALI) animal study
For the induction of ALI, six-week-old Balb/c male mice were intratracheally treated with LPS (10 mg/kg). One hour after LPS treatment, aptTNF-α (1600 μg/kg) or aptTNF-α-PEG (32, 320 μg/kg) were intratracheally or intravenously administrated. Blood oxygen saturation was recorded at 24 h post treatment by MouseMonitorTM S plus pulse oximeter module (Indus Instruments). In one study group, the mice were sacrificed at 24 or 48 h. The weights of lungs were measured and tissues were subjected to RNA and protein extractions as well as immunochemistry staining. In the other, the bronchoalveolar lavage fluid (BALF) was collected for the counting of its total cell number, the measuring of total protein concentration (Nanodrop spectrophotometer), and the determination of the myeloperoxidase (MPO) activity (MPO fluorometric activity assay, BioVision).
Acute liver failure (ALF) animal study
To induce ALF, six-week-old Balb/c male mice were injected with D-galactosamine (D-GalN, 100 mg/kg, intraperitoneal) and human TNF-α (40 μg/kg, intravenous). Treatment with N-acetylcysteine (NAC, 600 mg/kg), aptTNF-α (1600 μg/kg), or aptTNF-α-PEG (3.2, 32, 320 μg/kg) was then given intravenously and blood was sampled 6 h after the treatment [29, 30]. The blood was measured for AST and ALT levels (Fuji Dri-Chem 4000i). The mice were sacrificed 6 or 24 h after treatment. The liver tissues were collected for RNA and protein extractions, as well as immunohistochemistry studies.
Quantitative PCR
RNAs were extracted from mouse tissues with Trizol (Invitrogen) and cDNAs were synthesized using SuperScript III reverse transcriptase (Invitrogen) with random hexamers (Invitrogen). Quantitative PCR was performed on a LightCycler 480 system (Roche Applied Science). Primer sequences against mouse cDNAs used in qPCR were listed as follows: il-1β, 5'-agttgacggaccccaaaag-3' (forward) and 5'-agctggatgctctcatcagg-3' (reverse); il-6, 5'-gctaccaaactggatataatcagga-3' (forward) and 5'-ccaggtagctatggtactccagaa-3' (reverse); cxcl2, 5'-aatcatccaaaagatactgaacaaag-3' (forward) and 5'-ttctctttggttcttccgttg-3' (reverse); and actb, 5'-ctaaggccaaccgtgaaaag-3' (forward) and 5'-accagaggcatacagggaca-3' (reverse).
Western blot and immunohistochemistry staining
Primary antibodies used in the western blotting included anti-GAPDH (Santa Cruz), anti-phospho- ERK (T202/Y204, Cell Signaling Technology), anti- ERK (Santa Cruz), and anti-PCNA (Cell Signaling Technology). The haematoxylin and eosin stain (H&E) was performed by the pathology core at the Institute of Biomedical Sciences, Academia Sinica. The lung injury score was calculated according to the scoring system established by the ALI in Animals Study Group [31]. For immunohistochemistry staining, neutrophils were stained by anti-Ly6G (clone 1A8, Biolegend) antibody at 1:100 dilution and proliferating cells were stained by anti-Ki-67 (Cell signaling) antibody at 1:400 dilution. ImmPRESS™ HRP anti-rat IgG, mouse adsorbed (peroxidase) polymer detection kit (Vectors laboratories), or VECTASTAIN Elite ABC HRP kit (Vectors laboratories) was used for signal amplification and DAB peroxidase (HRP) substrate kit (Vectors laboratories) was used for color development. For frozen tissue staining, anti-TNF-α (Genetex) antibody was used at 1:100 dilution and Alexa Fluor 488-labeled anti-rabbit IgG (Abcam) secondary antibodies were used at 1:400 dilution. Fluoroshield mounting medium with DAPI (Abcam) was used for nuclei staining. Images were captured by LSM 700 laser scanning confocal microscope (Carl Zeiss Microscopy) and analyzed by ZEN imaging software.
Statistics
The results were showed as mean ± standard error of mean and the P value were calculated by Student t-test. The two-tailed P value lower than 0.05 was defined as statistically significant.
Results
AptTNF-α binds to TNF-α with high affinity and targets TNF-α in vivo
A TNF-α-targeting aptamer (aptTNF-α, Figure 1A) was selected by nitrocellulose filter SELEX, analyzed by FASTAptamer, and optimized based on the predicted secondary structure using Mfold. The dissociation constant (Kd) of aptTNF-α and human TNF-α was 8 nM (Figure 1B, left panel). As the data further showed that aptTNF-α also bound to mouse TNF-α (Figure 1B, right panel), we subsequently investigated in vivo binding effects of aptTNF-α using the ALI mouse model.
AptTNF-α binds to human TNF-α with high affinity and serves as a molecular imaging probe for monitoring TNF-α in vivo. (A) The predicted secondary structure of aptTNF-α. (B) The dissociation constant of aptTNF-α and human TNF-α (left panel); aptTNF-α binds to both human and mouse TNF-α (n=3, right panel). (C) In vivo detection of the aptTNF-α or random sequences pool signals in mice with and without ALI (n=3). (D) Biodistribution of aptTNF-α at 4 h post aptamer administration (n=3). (E) The LDH, AST, and ALT level in blood serum at 4 h post aptamer administration (n=5). The data are presented as mean ± standard error of the mean and were analyzed by Student's t-test. Asterisks denote statistical significant differences (P < 0.05).
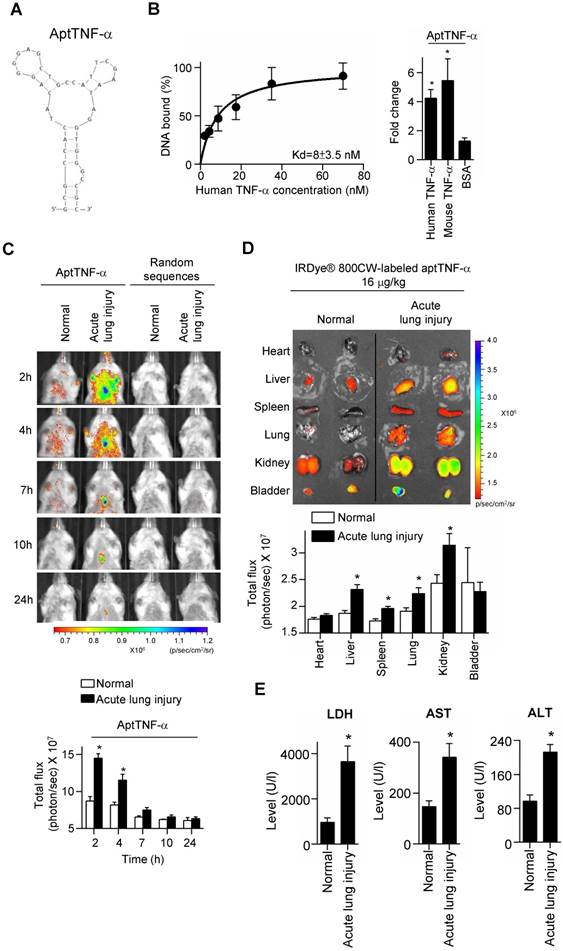
Our data showed that in the ALI group, aptTNF-α signals were clearly observed in the thorax at 2 h after LPS-induced ALI. The signal intensity gradually decreased and were nearly undetectable at 24 h. (Figure 1C). In contrast, no signal in the thorax was detected in the group received random sequences pool injection at all testing time points (Figure 1C). As SIRS can occur following ALI and lead to multiple organ damages, we collected vital organs at 4 h after the administration of aptTNF-α and examined its biodistribution. Our data showed that with identical dosage of aptTNF-α administration, aptTNF-α signals were significantly increased in liver, spleen, lung, and kidney in the ALI group comparing to the control group (Figure 1D). In the control group, aptTNF-α signals were mainly observed in kidney and bladder, the organs involved in aptamer excretion, but with a lower signal intensity than the ALI group. Further blood tests also showed that LDH, AST and ALT levels were significantly increased in the ALI group. The data supported the occurrence of end-organ damages observed in the aptTNF-α biodistribution image studies (Figure 1E).
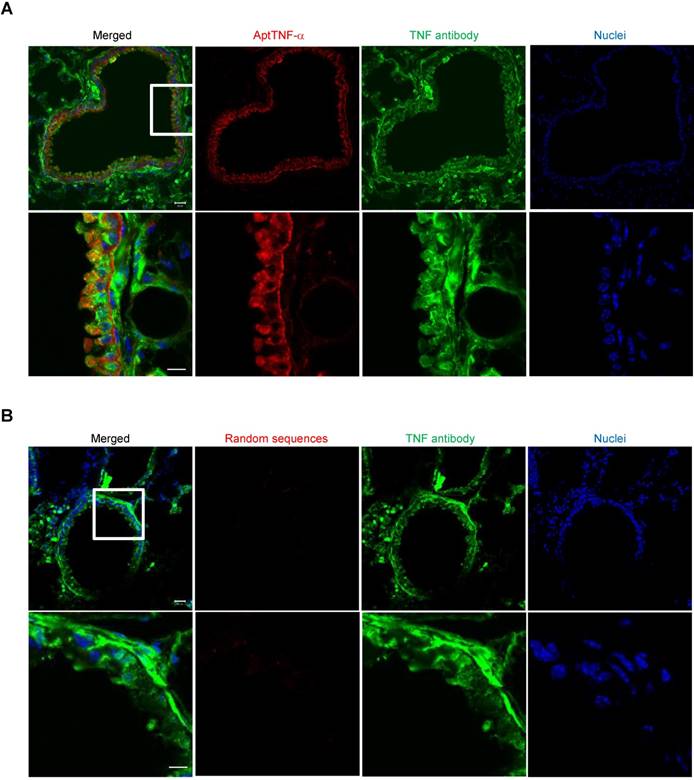
To further verify that the signals detected in the lung of the ALI mice was derived from the binding of aptTNF-α to TNF-α, we collected the lung tissues at 2 h post aptamer administration (Alexa Fluor 647-labeled aptTNF-α or random sequences pool) and TNF-α was stained by anti-TNF-α antibody. The specimens were examined by confocal microscopy. The results showed the colocalization of the aptTNF-α signal (red) and the signals derived from the TNF-α antibody (green), indicating the binding of aptTNF-α to TNF-α in the LPS-induced ALI lung tissues (Figure 2A). On the contrary, no obvious signal colocalization was detected in the random sequences pool group (Figure 2B). Taken together, we showed that aptTNF-α possesses good in vitro binding affinity toward human TNF-α and can target TNF-α in vivo as shown in the mouse ALI model.
AptTNF-α binds to TNF-α in the LPS-induced ALI lung tissues. (A) Confocal microscopy images showed the co-localization of aptTNF-α (red) and TNF-α antibody (green) signals retrieved from the injured lung tissues. (B) Confocal microscopy images of random sequences pool (red) and TNF-α antibody (green) signals retrieved from the injured lung tissues. The magnification is 200 x and the scale bar is 20 μm in the upper panel of (A) and (B). The magnification is 400 x and the scale bar is 10 μm in the lower panel of (A) and (B). The white box in upper panel of (A) or (B) indicated the field in the lower panel of (A) or (B).
AptTNF-α-PEG suppresses TNF-α-mediated signaling
We next investigated whether aptTNF-α or its derivatives efficiently inhibit TNF-α-mediated signaling. As biologically active TNF-α exists in a trimeric form in physiological conditions, we synthesized dimeric aptTNF-α by adding a polyethylene glycol (PEG) linker between two aptTNF-α monomers (aptTNF-α-PEG, Figure 3A) to strengthen the potential antagonistic effect of aptTNF-α. The data showed that the dimeric aptTNF-α-PEG also has specific binding activity towards human and mouse TNF-α proteins (Figure 3B).
AptTNF-α-PEG has a shorter suppression duration on the TNF-α pathway than anti-TNF-α antibody. (A) The design of dimeric aptTNF-α-PEG. (B) aptTNF-α-PEG binds to mouse TNF-α (n=3). (C) Suppressive effects of aptTNF-α, aptTNF-α-PEG, and anti-TNF-α antibody shown by the TNF-α/NF-κB reporter assay after 4 and 24 h post TNF-α treatment (n=3). (D) Suppressive effects of aptTNF-α-PEG on TNF-α-induced ERK signaling. These data are presented as mean ± standard error of the mean and were analyzed by Student's t-test. Asterisks denote statistical significant differences (P < 0.05).
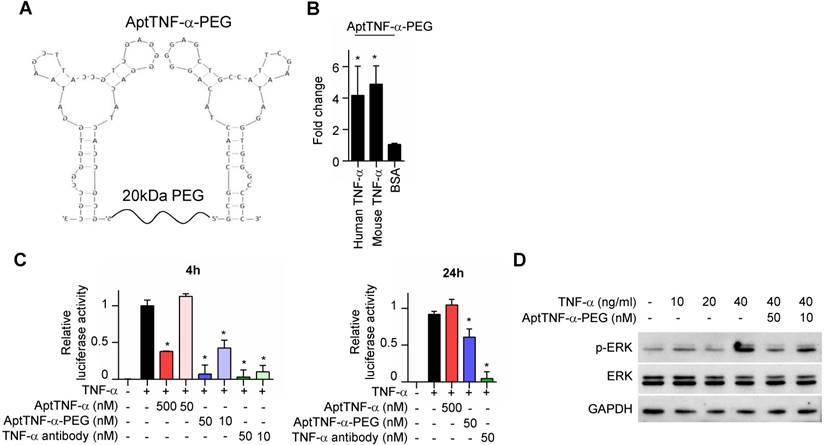
In addition, the reporter assay revealed that while the monomeric aptTNF-α effectively suppressed TNF-α/NF-κB signaling at the concentration of 500 nM, the dimeric aptTNF-α-PEG bore a better potency that inhibited TNF-α/NF-κB signaling at 50 nM as measured at 4 h after TNF-α treatment (Figure 3C, left panel). Moreover, the suppressive effect of the monomeric aptTNF-α subsided at 24 h post TNF-α treatment and that of the dimeric aptTNF-α-PEG reduced to about 40% of the original efficacy. On the contrary, the suppressive effect of the anti-TNF-α antibody remained at 24 h post TNF-α treatment (Figure 3C, right panel). Furthermore, the aptTNF-α- PEG suppressed the TNF-α-induced phosphorylation of ERK signaling (Figure 3D). These data suggested potential roles of aptTNF-α and aptTNF-α-PEG in acute illness with systemic inflammatory response as they only suppress the acute phase TNF-α signaling. This may avoid the interference of basal TNF-α signaling needed in the tissue repair phase as well as the side effects related to sustained suppression of the innate immunity.
AptTNF-α attenuates the severity of acute lung injury
We next investigated in vivo effects of aptTNF-α and aptTNF-α-PEG using the LPS-induced ALI mouse model. Our data showed that LPS treatment induced respiratory distress as indicated by a significant reduction in oxygen saturation (Figure 4A). The increased wet lung weight in the LPS-treated group suggested an enhanced permeability of the alveolar-capillary membrane upon LPS-induced injury (Figure 4B). The histological examinations of the LPS-induced ALI group showed phenomena of alveolar septal thickening and accumulation of red blood cells, neutrophils, and fibrin strands in the alveolar spaces, and was accompanied with an increased lung injury score (Figure 4C-D). Further analyses of the BALF showed increased total protein levels, total cell numbers, an enhanced myeloperoxidase (MPO) activity, and an upregulated expression of pro-inflammatory cytokines/ chemokines (il-1β, il-6, and cxcl2) (Figure 4E-J). These phenotypical and molecular results fitted into the expected cascade of tissue reaction orchestrated by cytokines and chemokines in response to ALI.
AptTNF-α and aptTNF-α-PEG suppress LPS-induced ALI through intratracheal (i.t.) or intravenous (i.v.) delivery. (A) The blood oxygen saturation level, (B) the wet lung weight normalized by body weight, (C) H&E and neutrophil stain of the lung tissues (The magnification is 200x and the scale bar is 100μm), (D) the lung injury scores, (E) total protein in BALF, (F) total cell numbers in BALF, (G) MPO activity in BALF and (H-J) the cytokine/chemokine expression levels in lung tissues at 24 h, (K) the immunohistochemistry staining of Ki-67+ cells in the lung tissues at 48 h after aptamer administration from different treatment group (n=6). The magnification is 400x and the scale bar is 20μm. The treatment doses are represented as μg/kg. These data are presented as mean ± standard error of the mean and were analyzed by Student's t-test. Asterisks denote statistical significant differences (P < 0.05).
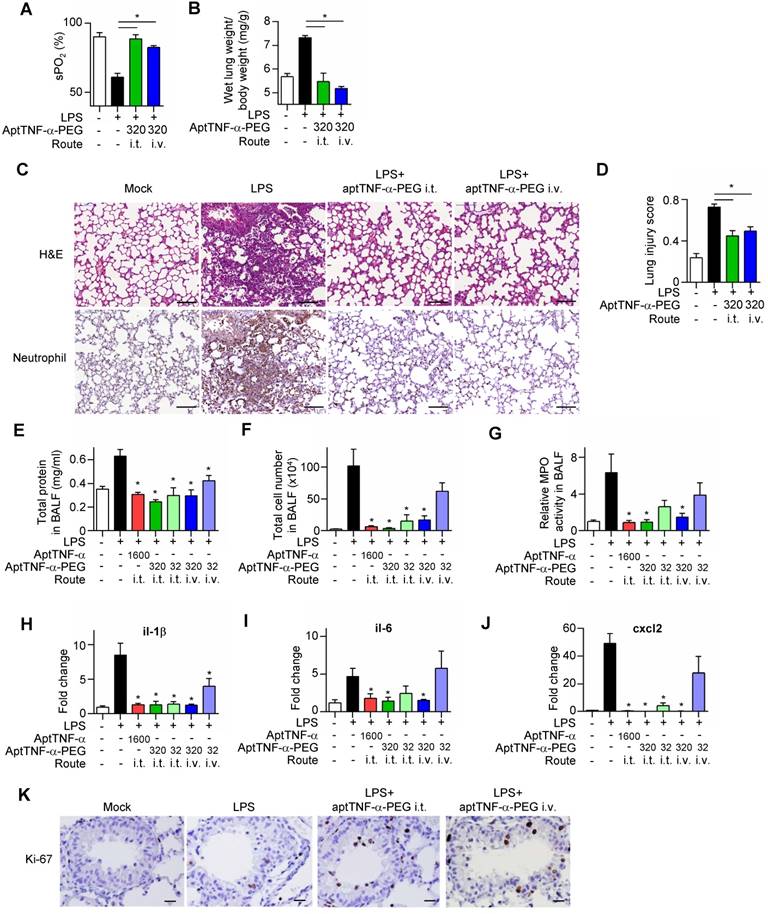
Subsequently, we showed that intratracheal or intravenous administration of aptTNF-α-PEG rescued LPS-induced injury phenotypically, histologically, and molecularly, in a dose-response manner (Figure 4A-J). Furthermore, we also showed that aptTNF-α- PEG treatment increased the number of proliferating cells (Ki-67+ cells) in the repair phase of ALI (Figure 4K).
The data revealed a better efficacy of aptTNF-α- PEG when delivered via the intratracheal route, which might be related to a higher local concentration comparing to systemic delivery. Although intratracheal administration of aptTNF-α also suppressed LPS- induced ALI to some extent, this was only achieved in a relatively high drug concentration (5-fold of aptTNF-α-PEG), indicating a higher potency of aptTNF-α-PEG. Taken together, our data indicated that aptTNF-α-PEG or aptTNF-α could suppress the acute-phase apoptosis signaling mediated by the TNF-α pathway and the subsequent cytokine storm that ultimately results in tissue damage in ALI.
AptNF-α attenuates TNF-α-mediated acute liver failure and potentiates early liver regeneration
For ALF patients with fulminant outcomes, currently available treatment before liver transplantation is systemic infusion of N-acetylcysteine (NAC) [1]. As ALF is TNF-α-mediated, we subsequently investigated the role of aptTNF-α and aptTNF-α-PEG in ALF and compared their effects with NAC using the D-galactosamine (D-GalN)/ TNF-α-induced mouse ALF model.
Our data showed that injection of D-GalN/ TNF-α induced severe hepatocyte death accompanied with tissue hemorrhage and neutrophil infiltration. The observed liver damage was reduced with aptTNF-α-PEG treatment (Figure 5A). Further analyses of serum AST/ALT and tissue pro-inflammatory cytokines/chemokine (il-1β, il-6, and cxcl2) showed superior liver protective effects of either aptTNF-α (1600 μg/kg) or aptTNF-α-PEG (3.2 μg/kg to 320 μg/kg) to NAC (600 mg/kg) (Figure 5B-F). In addition, aptTNF-α-PEG possessed a dose-dependent liver protective effect and was again better than aptTNF-α (Figure 5B-F).
AptTNF-α and aptTNF-α-PEG attenuate the degree of D-GalN/TNF-α-induced acute liver injury and potentiate early liver regeneration. (A) H&E and neutrophil stains of liver tissues at 24 h after treatment. The magnification is 400x and the scale bar is 20μm, (B) AST and (C) ALT levels in blood, (D-F) cytokine/chemokine expression levels, and (G) the PCNA expression at 6 h in liver tissues from different treatment groups (n=6). The treatment doses are showed as μg/kg. These data are presented as mean ± standard error of the mean and were analyzed by Student's t-test. Asterisks denote statistical significant differences (P < 0.05).
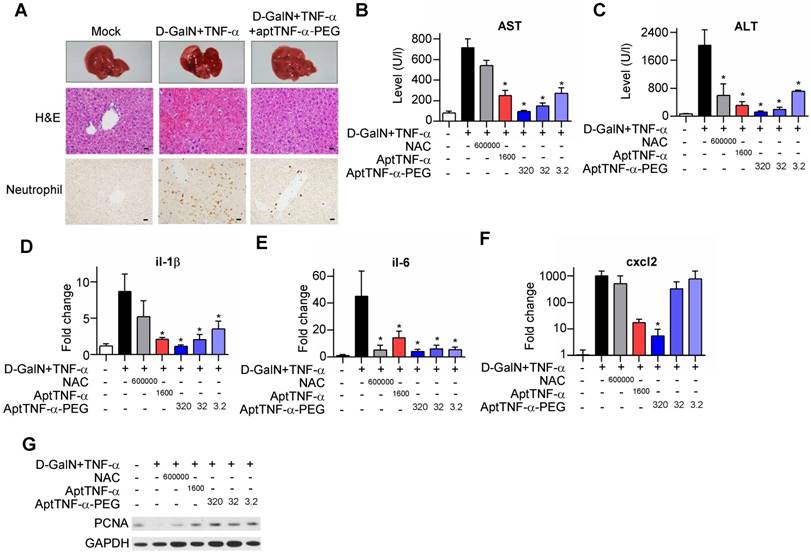
Moreover, as hepatocytes transit from G0 to G1 phase after acute injury phase, upregulation of PCNA expression can be generally observed during liver regeneration. Our data showed that both the aptTNF- α- and the aptTNF-α-PEG-treated groups had higher PCNA expression compared to the NAC-treated groups. The data suggested that hepatocytes enter G1 phase at an earlier time point in the aptTNF-α/ aptTNF-α-PEG-treated group following acute injury (Figure 5G).
Furthermore, we showed that the degree of AST and ALT elevation reversion was identical in the groups received aptTNF-α or aptTNF-α-PEG treatment. Nevertheless, while ALT elevation could be reversed in the NAC-treated group, AST remained high. (Figure 5B-C). ALT is an enzyme mainly expressed in liver. On the contrary, AST is an enzyme expressed not only in liver, but also in heart, muscle, and brain tissues. As liver failure is not merely a single organ disease but can lead to SIRS and multiple organ failures, our data suggested that treatment with aptTNF-α/aptTNF-α-PEG not only rescued acute liver damage but also suppressed the process of SIRS. The data implicate potential systemic protective effects of aptTNF-α/aptTNF-α-PEG in ALF. Taken together, we showed that aptTNF-α/aptTNF-α-PEG possessed good liver protective effects and may also suppressed the process of SIRS so as to prevent multiple organ failures commonly observed in clinical practice.
Discussions
In the current study, we showed that aptTNF-α/ aptTNF-α-PEG has tissue protective effect and systemic anti-inflammatory effect upon acute tissue injury using the mouse ALI and ALF models. We also showed that aptTNF-α/aptTNF-α-PEG possesses less suppressive effect on tissue regeneration in the tissue repair phase. These results suggest potentials for their clinical translation.
The TNF-α pathway plays as a double-edged sword in immune regulation [7]. While excessive TNF-α causes cell apoptosis and triggers cytokine storms, an optimal TNF-α level is essential for innate immunity against bacterial infection and tissue repair. Therefore, timing and duration of intervention are keys to success for TNF-α pathway inhibition in the context of TNF-α mediated acute tissue injury. Comparing to protein-based inhibitors, aptTNF-α/ aptTNF-α-PEG can have shorter and adjustable inhibitory durations. This advantage helps minimize unwanted interference on TNF-α signaling at tissue repair phase as shown in our ALI and ALF models. In addition, as antidote for aptTNF-α/aptTNF-α-PEG, which is the complementary sequences of the aptamer itself, can be easily designed and synthesized, timely administration of the antidote is doable. These characteristics of aptTNF-α/aptTNF-α-PEG facilitate its timely TNF-α pathway inhibition at the acute tissue injury phase while avoiding improper suppression of regeneration at the tissue repair phase, and potentiate quick termination of its antagonistic effects whenever needed.
SIRS is a common phenomenon upon end-organ damages [1, 3, 13]. Cytokine storms that follow organ damages can reshape a single organ disease into a systemic inflammatory disorder that eventually leads to multiple organ failures. As shown in our LPS-induced ALI mouse model, aptTNF-α signals were detected not only in the lungs but also other major vital organs. At the same time, blood tests also showed increased systemic LDH, AST and ALT levels. Aptamers are small in molecular size and possess good tissue penetration efficiency [32-35]. It can be anticipated that with this inborn nature, aptamers may help reduced SIRS-related end-organ damages, such as the SIRS-related encephalopathy commonly encountered in critical care medicine [36-37]. Furthermore, aptamers can be made into different formulations for different applications. Current understandings on drug delivery indicate that topical delivery may increase local effective drug concentration in one hand and reduce systemic side effects in the other. In our ALI model, we showed that intratracheal delivery of aptTNF-α/aptTNF-α-PEG achieved its optimal effects at a lower drug concentration comparing to intravenous delivery. Topic drug delivery is especially useful in inflammatory diseases, with asthma being probably one of the best examples in this regard [38].
In summary, we developed a TNF-α-targeting aptamer with antagonistic effect. Our successful demonstration of its tissue protective effects using the mouse ALI and ALF models suggested its translational potential as a new category of TNF-α blocking agent for acute tissue injury mediated by TNF-α commonly observed in critical care medicine.
Abbreviations
ADCC: antibody-dependent cell-mediated cytotoxicity; ALF: acute liver failure; ALI: acute lung injury; ALT: alanine aminotransferase; AptTNF-α: TNF-α-targeting aptamer; AST: aspartate aminotransferase; BALF: bronchoalveolar lavage fluid; CDC: complement-dependent cytotoxicity; D-GalN: D-galactosamine; H&E: haematoxylin and eosin; i.t.: intratracheal; i.v.: intravenous; LDH: lactic dehydrogenase; MPO: myeloperoxidase; NAC: N-acetylcysteine; PEG: polyethylene glycol; PMNs: polymorphonuclear leukocytes; SELEX: systematic evolution of ligands by exponential enrichment; SIRS: systemic inflammatory response syndrome.
Acknowledgements
This work was supported by Academia Sinica (Nano project), MOST 106-2314-B-002-103-MY2, and collaboration project with Industrial Technology Research Institute. The authors thank the National Center for Genome Medicine for NGS technical and bioinformatics support.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bernal W, Wendon J. Acute liver failure. N Engl J Med. 2013;369:2525-34
2. Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731-40
3. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334-49
4. Spragg RG, Bernard GR, Checkley W, Curtis JR, Gajic O, Guyatt G. et al. Beyond Mortality Future Clinical Research in Acute Lung Injury. Am J Resp Crit Care. 2010;181:1121-7
5. Bantel H, Schulze-Osthoff K. Mechanisms of cell death in acute liver failure. Front Physiol. 2012;3:79-88
6. Krenkel O, Mossanen JC, Tacke F. Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surg Nutr. 2014;3:331-43
7. Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745-56
8. Kotton DN, Morrisey EE. Lung regeneration: mechanisms, applications and emerging stem cell populations. Nat Med. 2014;20:822-32
9. Bohm F, Kohler UA, Speicher T, Werner S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol Med. 2010;2:294-305
10. Crisostomo PR, Wang Y, Markel TA, Wang M, Lahm T, Meldrum DR. Human mesenchymal stem cells stimulated by TNF-alpha, LPS, or hypoxia produce growth factors by an NF kappa B- but not JNK-dependent mechanism. Am J Physiol Cell Physiol. 2008;294:C675-82
11. Broekman W, Amatngalim GD, de Mooij-Eijk Y, Oostendorp J, Roelofs H, Taube C. et al. TNF-alpha and IL-1beta-activated human mesenchymal stromal cells increase airway epithelial wound healing in vitro via activation of the epidermal growth factor receptor. Respir Res. 2016;17:3-14
12. Cakarova L, Marsh LM, Wilhelm J, Mayer K, Grimminger F, Seeger W. et al. Macrophage tumor necrosis factor-alpha induces epithelial expression of granulocyte-macrophage colony-stimulating factor: impact on alveolar epithelial repair. Am J Respir Crit Care Med. 2009;180:521-32
13. Gattinoni L, Quintel M. Why Is Acute Respiratory Distress Syndrome So Important for Critical Care? Am J Resp Crit Care. 2016;194:1051-2
14. Terpstra ML, Aman J, van Nieuw Amerongen GP, Groeneveld AB. Plasma biomarkers for acute respiratory distress syndrome: a systematic review and meta-analysis*. Crit Care Med. 2014;42:691-700
15. Tokushige K, Yamaguchi N, Ikeda I, Hashimoto E, Yamauchi K, Hayashi N. Significance of soluble TNF receptor-I in acute-type fulminant hepatitis. Am J Gastroenterol. 2000;95:2040-6
16. Boetticher NC, Peine CJ, Kwo P, Abrams GA, Patel T, Aqel B. et al. A randomized, double-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology. 2008;135:1953-60
17. Naveau S, Chollet-Martin S, Dharancy S, Mathurin P, Jouet P, Piquet MA. et al. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology. 2004;39:1390-7
18. Abraham E, Anzueto A, Gutierrez G, Tessler S, Pedro GS, Wunderink R. et al. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. The Lancet. 1998;351:929-33
19. Abraham E, Wunderink R, Silverman H, Perl TM, Nasraway S, Levy H. et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAb Sepsis Study Group. JAMA. 1995;273:934-41
20. Fisher CJ Jr, Agosti JM, Opal SM, Lowry SF, Balk RA, Sadoff JC. et al. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N Engl J Med. 1996;334:1697-702
21. French JB, Bonacini M, Ghabril M, Foureau D, Bonkovsky HL. Hepatotoxicity Associated with the Use of Anti-TNF-alpha Agents. Drug Safety. 2016;39:199-208
22. Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol Therapeut. 2008;117:244-79
23. Zhou JH, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nature Reviews Drug Discovery. 2017;16:181-202
24. Lao YH, Phua KK, Leong KW. Aptamer nanomedicine for cancer therapeutics: barriers and potential for translation. ACS Nano. 2015;9:2235-54
25. Sun H, Zhu X, Lu PY, Rosato RR, Tan W, Zu Y. Oligonucleotide aptamers: new tools for targeted cancer therapy. Mol Ther Nucleic Acids. 2014;3:e182
26. Siller-Matula JM, Merhi Y, Tanguay JF, Duerschmied D, Wagner DD, McGinness KE. et al. ARC15105 Is a Potent Antagonist of Von Willebrand Factor Mediated Platelet Activation and Adhesion. Arterioscl Throm Vas. 2012;32:902-U100
27. Gilbert JC, DeFeo-Fraulini T, Hutabarat RM, Horvath CJ, Merlino PG, Marsh HN. et al. First-in-human evaluation of anti-von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation. 2007;116:2678-86
28. Alam KK, Chang JL, Burke DH. FASTAptamer: A Bioinformatic Toolkit for High-throughput Sequence Analysis of Combinatorial Selections. Mol Ther Nucleic Acids. 2015;4:e230
29. Saito C, Zwingmann C, Jaeschke H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology. 2010;51:246-54
30. Sass G, Heinlein S, Agli A, Bang R, Schumann J, Tiegs G. Cytokine expression in three mouse models of experimental hepatitis. Cytokine. 2002;19:115-20
31. Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS. et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44:725-38
32. Pluen A, Boucher Y, Ramanujan S, McKee TD, Gohongi T, di Tomaso E. et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: cranial vs. subcutaneous tumors. Proc Natl Acad Sci U S A. 2001;98:4628-33
33. Cheng C, Chen YH, Lennox KA, Behlke MA, Davidson BL. In vivo SELEX for Identification of Brain-penetrating Aptamers. Mol Ther Nucleic Acids. 2013;2:e67
34. Monaco I, Camorani S, Colecchia D, Locatelli E, Calandro P, Oudin A. et al. Aptamer Functionalization of Nanosystems for Glioblastoma Targeting through the Blood-Brain Barrier. J Med Chem. 2017;60:4510-6
35. Macdonald J, Henri J, Goodman L, Xiang DX, Duan W, Shigdar S. Development of a Bifunctional Aptamer Targeting the Transferrin Receptor and Epithelial Cell Adhesion Molecule (EpCAM) for the Treatment of Brain Cancer Metastases. Acs Chem Neurosci. 2017;8:777-84
36. Tsuge M, Yasui K, Ichiyawa T, Saito Y, Nagaoka Y, Yashiro M. et al. Increase of tumor necrosis factor-alpha in the blood induces early activation of matrix metalloproteinase-9 in the brain. Microbiol Immunol. 2010;54:417-24
37. Lv S, Song HL, Zhou Y, Li LX, Cui W, Wang W. et al. Tumour necrosis factor-alpha affects blood-brain barrier permeability and tight junction-associated occludin in acute liver failure. Liver Int. 2010;30:1198-210
38. Chung KF. Neutrophilic asthma: a distinct target for treatment? Lancet Respir Med. 2016;4:765-7
Author contact
![]() Corresponding authors: Pan-Chyr Yang, MD, PhD, Professor of Internal Medicine, National Taiwan University College of Medicine. Phone: +886-2-33662000; Fax: +886-2-23621877; E-mail: pcyangedu.tw; Address: No. 1, Sec. 4, Roosevelt Rd., Taipei 1061, Taiwan and Emily Pei-Ying Lin, MD, PhD, Attending Physician, National Taiwan University Hospital, Taipei, Taiwan. Phone: +886-2-3356-2905; E-mail: pylinsinica.edu.tw; Address: No. 7, Chung Shan S. Rd., Taipei 1061, Taiwan.
Corresponding authors: Pan-Chyr Yang, MD, PhD, Professor of Internal Medicine, National Taiwan University College of Medicine. Phone: +886-2-33662000; Fax: +886-2-23621877; E-mail: pcyangedu.tw; Address: No. 1, Sec. 4, Roosevelt Rd., Taipei 1061, Taiwan and Emily Pei-Ying Lin, MD, PhD, Attending Physician, National Taiwan University Hospital, Taipei, Taiwan. Phone: +886-2-3356-2905; E-mail: pylinsinica.edu.tw; Address: No. 7, Chung Shan S. Rd., Taipei 1061, Taiwan.