Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(7):1878-1892. doi:10.7150/thno.29682 This issue Cite
Research Paper
Dioscin Alleviates Crystalline Silica-Induced Pulmonary Inflammation and Fibrosis through Promoting Alveolar Macrophage Autophagy
Sitong Du1*, Chao Li1*, Yiping Lu1, Xue Lei1, Yiting Zhang1, Siyi Li1, Fangwei Liu1, Ying Chen1, Dong Weng1,2, Jie Chen1 ![]()
1. Division of Pneumoconiosis, School of Public Health, China Medical University, Shenyang, PR China.
2. Department of Respiratory Medicine, Shanghai Pulmonary Hospital, Tongji University School of Medicine, Shanghai, PR China.
*Equal contribution to this study
Received 2018-9-3; Accepted 2019-2-1; Published 2019-3-7
Abstract

Occupational exposure to crystalline silica (CS) particles leads to silicosis, which is characterized by chronic inflammation and abnormal tissue repair. Alveolar macrophages (AMs) play a crucial role in the process of silicosis. Previously, we demonstrated positive effect of dioscin on silicosis through modulating macrophage-elicited innate immune response. However, the concrete molecular mechanism remains to be discovered.
Methods: We established experimental model of silicosis with wildtype and Atg5flox/floxDppa3Cre/+ mice and oral administrated dioscin daily to explore the effects of dioscin on macrophages and pulmonary fibrosis. AM cell line MH-S with Atg5 silence was used to explore specific function of dioscin on macrophage-derived inflammation and the underlying molecular mechanism.
Results: Dioscin could promote autophagy in macrophages. Dioscin-triggered AMs autophagy limited mitochondrial reactive oxygen species (mtROS) mass stimulated by CS, reduced mitochondria-dependent apoptosis pathway activation and facilitated cell survival. Relieved oxidative stress resulted in decreased secretion of inflammatory factors and chemokines. Dioscin treatment alleviated macrophage-derived inflammation and subsequent abnormal collagen repair. All the dioscin's protective effects were diminished in Atg5flox/floxDppa3Cre/+ mice.
Conclusion: Dioscin promoting autophagy leads to reduced CS-induced mitochondria-dependent apoptosis and cytokine production in AMs, which may provide concrete molecular mechanism for the therapy of silicosis.
Keywords: crystalline silica, dioscin, macrophage, autophagy, mitochondrial dysfunction
Introduction
Crystalline silica (CS) exposure, a significant occupational hazard worldwide, seriously impairs the health of exposed workers. Prolonged inhalation of CS leads to silicosis, which is a pulmonary disease characterized progressive fibrosis [1]. Since CS particles can't be removed from lung, it is difficult to diminish pulmonary inflammation, even the workers leave the working environment. The inflammatory response begins with alveolar macrophages (AMs) engulfing particles, further leading to infiltration of diverse immune cells and production of pro-inflammatory cytokines and chemokines [2]. Persistent inflammation results in activation of adaptive immunity, tissue damage, fibroblast proliferation and migration, abnormal collagen deposition, eventually lost lung functions and disability even death [3].
AMs are tissue-resident cells that play a primary role in maintaining immunological homoeostasis and host defense in lung [4]. AMs identify CS particles through scavenger receptors and start phagocytosis [5]. The invaded CS leads to ROS production in AMs, which results in activation of inflammatory pathways and cytokine secretions [6]. CS-induced ROS also leads to mitochondrial dysfunction, forcing AMs to undergo mitochondrial apoptosis. Massive release of ROS and inflammatory factors from apoptotic AMs cause excessive tissue injury [7, 8]. The apoptotic AMs also release the engulfed CS into lung parenchyma recruiting more AMs and perpetuating the damage cycle [9]. Hence, alleviating CS-mediated AMs' apoptosis could offer a relief for pulmonary inflammation and/or fibrosis in silicosis.
Autophagy is an evolutionarily conserved cellular self-protection mechanism, which is tightly linked with apoptosis [10]. It is a dynamic process involved in many cellular processes. The double- membrane autophagosomes fusing with lysosomes sequester dysfunctional macromolecules and injured organelles, which is useful to maintain homeostasis in cell [11]. Increased mitochondrial injury is observed in macrophages in response to CS [7], while autophagy can be a valid way to remove dysfunctional mitochondria and reduce cellular stress [12, 13]. Defects in autophagy lead to enhanced mtROS, which causes oxidative and inflammatory damage to lung tissue during the progression of fibrosis [13, 14]. Also, autophagy plays a crucial role in maintaining cell function and preventing apoptosis, since there is a crosstalk between the autophagy and apoptosis pathways, which is governed by BECN1-caspases/ Bcl-2 family proteins network [10]. BECN1 positively regulates autophagic cascades that may represent a novel anti-apoptotic mechanism [15].
Researches about macrophage autophagy showed opposite effects on inflammatory and fibrotic diseases. On positive side, macrophage autophagy regulated IL-1 production during liver injury and lessened IL-6 secretion in LPS-stimulated macrophages [16-19]. The AM-derived cytokines could recruit fibroblast and promote its proliferation leading to excessive collagen deposition [9, 20]. Our previous study showed autophagosomes increased in AMs from silicosis patients' bronchoalveolar lavage fluid [21]. Besides, another study indicated autophagy led apoptosis resistance in macrophages of IPF patients [22], while autophagy is dysfunction during pulmonary fibrosis [23]. On negative side, study showed that BBC3 and MCPIP1 promote pulmonary fibrosis through inducing AMs autophagy, which might trigger macrophage apoptosis [24, 25]. The controversial issue should be noted. And the precise mechanism of CS-inducing macrophage autophagy and its role in silicosis deserve further exploration.
Our previous study showed that a steroidal saponin, dioscin, exerted anti-fibrosis effects in CS-induced pulmonary fibrosis [26]. It could effectively mitigate CS-induced epithelia injury, smooth lung inflammation response through blocking ASK-p38/JNK pathway. Notably, it plays an important function in regulating cytokine secretion in macrophages. However, the concrete molecular mechanism remains to be undiscovered. Based on this, we further did bioinformatics analysis making a prediction that autophagy might be a biological process triggered by dioscin, which may provide a clue to dioscin's molecular target. In this study, we explored whether dioscin's anti-inflammation and anti-fibrosis effects correlated with its function on autophagy. And it may provide a specific molecular target and mechanism to postpone CS-induced fibrosis.
Materials and Methods
Mice and treatment
Female C57BL/6 mice (6-8 weeks) were purchased from SLAC Laboratory Animal Co. Ltd. (Shanghai, China) and the Atg5flox/floxDppa3cre/+ mice were purchased from Shanghai Model Organisms Center (Shanghai, China), on a C57BL/6 background. Animals were bred under specific pathogen-free conditions. All animal experiments were performed in accordance with the protocols permitted by the Animal Care and Use Committee of China Medical University.
C57BL/6 mice were randomly divided into 4 groups (Saline group, Saline + dioscin group, Crystalline silica group, Crystalline silica + dioscin group; 10 mice per group) by body weight. Atg5flox/floxDppa 3cre/+ mice and their littermates Atg5flox/floxDppa3+/+ were divided into 4 groups (Atg5flox/floxDppa3cre/+ + Crystalline silica group, Atg5flox/floxDppa3cre/+ + Crystalline silica + dioscin group, WT + Crystalline silica group, WT + Crystalline silica + dioscin group; 7 mice per group). The experimental silicosis model was established as described previously [26]. 50 μL CS suspension was introduced to mice by intratracheal injection after anesthetization. 50 μL sterile saline was used as a control. The injections of CS suspension or saline were performed only once. Dioscin or vehicle (0.5%CMC-Na) was administered orally each day to the corresponding groups. The mice were sacrificed at day 7 and day 56 under anesthesia (Fig. S1). Lung tissues were extracted carefully for further study.
Isolation and treatment of primary AMs
Bronchoalveolar lavage fluid (BALF) was obtained by perfusing 1 mL of pre-cooled sterile saline into the lung tissue repeatedly. The BALF was centrifuged to isolate cells. These cells were re-suspended using RPMI medium (BI, Israel) supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, and 1% penicillin- streptomycin (complete RPMI medium) and cultured in an incubator at 37°C, 5% CO2 for 2 h. Cells in suspension were then removed to obtain pure primary AMs for further experiments. The cells were counted and aliquoted to 1×106 cells per well in the culture plate. Fresh medium with 50 ng/mL LPS was added to each well. The media was collected 6 h later for the ELISA assay. The adherent AMs were collected for qPCR and immunofluorescence analysis.
Cell culture and treatment
The mouse-derived AM MH-S cell line, which is used for CS experiments in the literature [27], was purchased from the National Infrastructure of Cell Line Resource (Beijing, China). The cells were cultured in complete RPMI medium with 1 mM sodium pyruvate and 10 mM HEPES at 37°C, humidified 95% air, and 5% CO2. Cells were treated with media containing CS (50 μg/cm2) and/or gradient-dose dioscin (200 nM, 400 nM, 800 nM), and were then evaluated by the MTT assay (Fig. S2A-D). Saline was added as a negative control and rapamycin was added as a positive control to induce autophagy. We collected the cells and supernatants for further analysis. The cells were pre-treated with 25 ng/mL LPS for 3 h before treatment if the concentration of IL-1β was being measured [28].
MTT assay
The MTT assay was performed following the manufacturer's instructions. Briefly, MH-S cells were cultured in 96-well plates (3×103 cells per well). After gradient-dose dioscin, rapamycin, 3MA or chloroquine treatments, 10 µL MTT (0.5 mg/mL) was added and the cells were incubated for 4 h. The media was then removed and 150 μL DMSO was added to dissolve the crystal adequately. The absorbance was measured at 570 nm by a plate reader. The results of MTT were showed in Figure S2.
Apoptosis detection
The Apoptosis Detection Kit (BD Pharmingen, USA) was used to detect apoptosis according to the manufacturer's instructions. Briefly, the cells were incubated with 5 μL Annexin V-FITC and 5 μL PI for 15 min at room temperature avoiding direct light. Fluorescence-activated cell sorting (FACS) analysis was performed to analyze the apoptotic cells. Both Annexin V+/PI- and Annexin V+/PI+ cells were considered apoptotic.
TUNEL assay was performed using the one step TUNEL Apoptosis Assay Kit (Beyotime, China). After paraformaldehyde fixation, serum blocking, and 0.2% Triton X-100 permeabilization, cells were incubated with 50 μL mixed TUNEL working solution at 37°C for an hour away from direct light. A fluorescence microscope was used for observation. Nuclei were marked by DAPI. Apoptotic cells were characterized by co-localization of the TUNEL label and DAPI.
Caspase-glo 3/7 assay
Caspase 3/7 activity was detected using a caspase-glo 3/7 assay Kit (Promega, USA) following manufacturer's instructions. Cells were treated as previously mentioned. Thereafter, the substrate provided with the kit was added in a 1:1 dilution and incubated for 30 min at room temperature. The chemiluminescence signal was read with a multi-mode microplate reader.
Transfection
MH-S cells were transfected with lentivirus containing the siRNA sequence to knockdown Atg5 and BECN1, while a meaningless sequence was used as a negative control (Hanbio Biotechnology, China). The siRNA sequences were as follows: control, 5'- TTCTCCGAACGTGTCACGTAA -3'; Atg5, 5'- CCATCAACCGGAAACTCATGGAATA -3'. BECN1, 5'- CCAATAAGATGGGTCTGAAGTTTCA-3'. Western blot analysis was used to verify the transfection efficiency (Fig. S3, Fig. S4).
mRFP-GFP-LC3 adenovirus vectors were purchased from Hanbio Biotechnology Co. Ltd. MH-S cells were transfected with the double-fluorescent adenovirus according to the manufacturer's protocol. Microscopy was used to observe the fluorescence positive cells.
Immunoblot analysis
Lung tissues or cells were homogenized in RIPA buffer with protease inhibitors (Beyotime) and phosphatase inhibitors (Roche, Suisse) on ice to obtain total protein. The Cell Mitochondria Isolation Kit (Beyotime) was used to obtain the mitochondrial proteins. The lysates were measured by the Pierce BCA Protein Assay Kit (Beyotime) and quantified to 3 μg/μL. Proteins were loaded on an 8%-12% SDS-polyacrylamide gel which was electrophoretically transferred to PVDF membranes (Millipore, Germany) after electrophoresis. The membranes were incubated at 4°C overnight after blocking. The following primary antibodies were used in this experiment: anti-LC3, anti- BECN1, anti-ATG5, anti-phospho-mTOR, anti-AKT, anti-Parkin, anti-β-Actin (CST, 1:1000), anti-MMP9, anti-MMP12, anti-sequestosome1/P62 (Abcam, 1: 2000), anti-PINK1 (NOVUS, 1:1000), anti-phospho- AKT (Merck, 1:1000). The membranes were probed with a horseradish peroxidase-conjugated secondary antibody (CST, 1:5000) at room temperature for an hour. A chemiluminescence detection system was used to detect protein bands.
Quantitative PCR (qPCR) analysis
Total RNA was isolated from cells or lung tissues using TRIZOL reagent (Life Technologies, USA), following the manufacturer's instructions. Extracted RNA was reverse transcribed into cDNA with the PrimeScript RT kit (Takara, Japan). The mRNA levels of Atg5, Atg7, BECN1, Ulk1, P62, Mmp9, Mmp12, Il-1β, Il-6, and Mcp-1 were quantified with the SYBR Green Master Mix Kit (Takara). Gapdh was used as an internal control. The sequences of the primer pairs are described in Table S1.
Immunofluorescence
Treated cells were fixed with 4% paraformaldehyde (if cells were incubated with mitotracker, methanol was used), then blocked by 5% BSA containing 0.2% Triton X-100 for 30 min. The cells were incubated with primary antibodies LC3 (CST, 1:100), P62 (Abcam, 1:100) or Cyt-c (Abcam, 1:100) overnight at 4°C. Alexa Fluor 594-conjugated secondary antibodies (1:200) were incubated at room temperature for an hour in dark. The results were observed under a confocal microscope. Nuclei were marked by DAPI.
Mitochondrial Membrane Potential (MMP) assay
Mitochondrial membrane potential changes were measured by JC-1, TMRE and Rhodamine123 staining. The transfected cells were tagged with 5 mM JC-1 (Beyotime), 20 nM TMRM (Solarbio) or 1μM Rhodamine123 (Beyotime) at 37°C for 30 min, respectively. This step was protected from light. Cells were detected by FACS or fluorescence microscopy within 1 h.
Mitochondrial ROS (mtROS) analysis
The cells were incubated with 5 μM MitoSOXTM reagent working solution (Invitrogen, USA) at 37°C for 10 min to detect mtROS. This step was protected from light. Cells were detected by FACS and fluorescence microscopy.
ATP production analysis
ATP Assay Kit (Beyotime) was used to determine the content of ATP following the manufacturer's instructions. The chemiluminescence signal was read with a multi-mode microplate reader. Protein concentration were measured by the Pierce BCA Protein Assay Kit (Beyotime). ATP concentration was converted into the nmol/mg protein form.
Enzyme-linked immunosorbent assay (ELISA)
Supernatants from primary AMs or MH-S cells and BALF were collected. Levels of IL-1β, IL-6, and MCP-1 were analyzed with ELISA kits according to the manufacturer's instructions (R&D Systems, USA). The absorbance was detected at 450 nm and 570 nm.
Histological analysis and immunohistochemistry
Paraffin-embedded tissues were cut into 5 μm slices and mounted on slides. Hematoxylin & eosin (HE) staining and immunohistochemistry were performed to evaluate inflammation. Sirius red dye was used to measure collagen. For immunohistochemistry, the method of microwave antigen retrieval was used. The sections were covered with F4/80 antibody (CST, 1:100) overnight at 4°C and incubated with horseradish peroxidase polymer secondary antibodies (Santa Cruz, 1:200) for 30 min at room temperature. Positive staining was visualized with DAB. For Sirius red staining, sections were dyed with Sirius red solution for an hour, followed by Mayer hematoxylin for 10 min. HE staining and immunohistochemistry were observed under a microscope. Sirius red staining was observed under a polarizing microscope.
Statistical analysis
SPSS 19.0 was used for statistical analysis. One-way analysis of variance (ANOVA) followed by a Student-Newman-Keuls test was performed to analyze the difference between multiple groups. P<0.05 was considered to be statistically significant. All the data are shown as mean ± standard error of the mean (SEM).
Results
Autophagy is involved in CS-induced pulmonary fibrogenesis
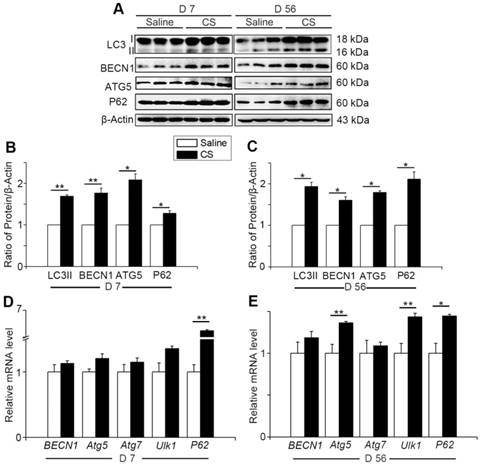
Autophagy activity increases when cells suffer from adverse cellular stress and plays a crucial role in many physiological and pathological conditions [11]. To investigate the changes of autophagy during the process of silicosis, we first measured a series of proteins associated with autophagy in the lung tissue of experimental silicosis mice. Compared to saline controls, the expression of LC3II, P62, ATG5, and BECN1 were all upregulated after CS stimulation at both day 7 (inflammation phase) and day 56 (fibrosis phase) in CS-treated mice (Fig. 1A-C). Next, qPCR analysis was performed to evaluate the level of autophagy-related genes in the mouse lungs. We found that CS stimulation led to increased expression of BECN1, Atg5, Atg7, Ulk1, and P62. Notably, the level of P62 was significantly higher at day 7, and slightly increased at day 56, compared to controls. Atg5 and Ulk1 were also significantly increased at day 56 (Fig. 1D-E). The variations of these genes were consistent with matching protein expressions. The combined results indicate that CS exposure triggers pulmonary autophagy activity.
Autophagy is activated in CS-injured mouse lung tissue. (A) Immunoblot analysis of proteins associated with autophagy in lung tissue (n=3 per group). (B-C) Quantification of LC3II, P62, BECN1, and ATG5 levels at day 7 and day 56 post CS-stimulation. (D-E) qPCR analysis of autophagy related genes in lung tissue at day 7 and day 56 (n=5). (B-E) *, P<0.05; **, P<0.01. Error bars indicate mean ± SEM. All experiments were repeated three times with similar results.
Autophagy plays a protective role against CS-induced AMs apoptosis
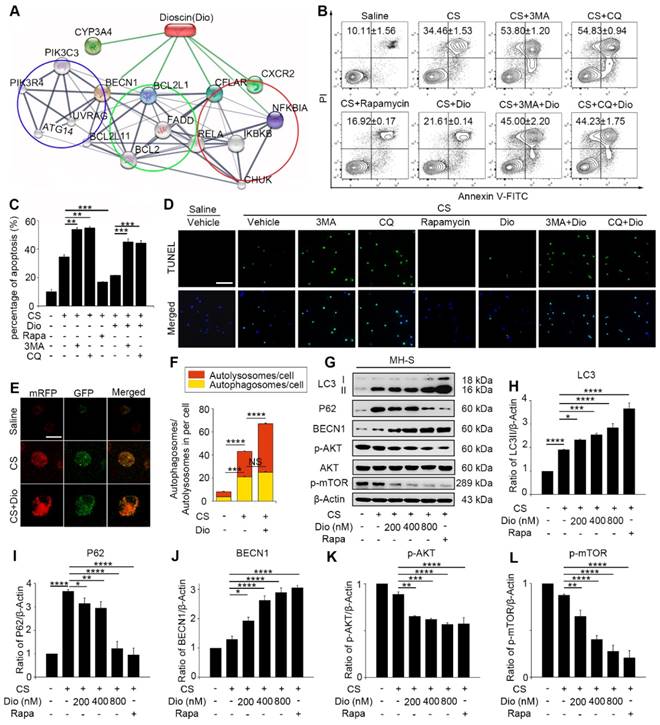
AMs, which are the first line defense to the invaded body, engulf silica and play an important role in silicosis. Our previous study showed that CS particles enhanced autophagosomes formation in the AMs of silicosis patients [21]. CS leads to macrophage apoptosis, which is a fundamental event during the development of silicosis [29, 30]. Therefore, we further focused on AMs and explored the specific effects of autophagy in CS-injured AMs. To achieve this goal, we used an AM cell line — MH-S and treated the cells with an autophagy inducer (rapamycin), inhibitor (3-Methyladenine, 3MA or chloroquine, CQ), and a traditional Chinese medicine (TCM) agent (dioscin) together with CS. Bioinformatic analysis provided clues that dioscin may function as a natural autophagy mediator (Fig. 2A). We first used flow cytometry (FCM) to explore the relationship between autophagy and apoptosis in CS-injured MH-S cells. The doses of chemicals we used had no effect on cell apoptosis (Figure S2). Results showed that CS stimulation gave rise to MH-S cells apoptosis and that autophagy induction (rapamycin treated) could reduce the number of apoptotic cells, yet autophagy inhibition (3MA or CQ treated) increased cell mortality (Fig. 2B-C). TUNEL analysis was performed to confirm these results. The results were similar to those of FCM (Fig. 2D). Intriguingly, dioscin treatment had a positive effect on CS-injured AMs, which was analogous to the effect of rapamycin and could be counteracted by 3MA or CQ. These results suggest that autophagy may play a protective role to AMs and dioscin exerts protective effects on CS-exposed AMs through inducing autophagy.
Dioscin helps AMs resist to CS-induced apoptosis through inducing autophagy. (A) STITCH predicted interaction between dioscin and related proteins. The network contains three associated biological processes: autophagy (blue cycle), apoptosis (green cycle) and inflammatory response (red cycle). (B-C) MH-S cell apoptosis assay by flow cytometry. (D) TUNEL assay (green) and DAPI staining (blue), the scale bar indicates 50 μm (n=3). (E) MH-S cells were transfected with mRFP-GFP-LC3 adenovirus. LC3 puncta were observed by confocal microscopy, the scale bar indicates 10 μm (n=3). (F) Quantification of autophagosome and autolysosome. (G) Immunoblot analysis of autophagy-associated proteins in MH-S cell lysates (n=3). (H-L) Quantification of LC3II, P62, BECN1, P-AKT, P-mTOR levels. *, P<0.05; **, P<0.01; ***, P<0.001, ****, P<0.0001; NS, No significance. Error bars indicate mean ± SEM. All experiments were repeated three times with similar results.
Dioscin promotes autophagy and alleviates AMs apoptosis in vitro and in vivo
It was reported that dioscin may modulate autophagy process [31]. We also demonstrated that dioscin's effects on CS-exposed AMs were similar to those of rapamycin. To explore the autophagy regulatory effects of dioscin, we used mRFP-GFP-LC3 adenovirus-infected MH-S cells as a tool to monitor autophagy flux. Due to GFP's property of acid dissolution, the transformation of the autophagosome to the autolysosome can be displayed as fluorescence conversion in real time. Under normal conditions, fluorescence signals were observed diffusely in cytoplasm with less punctate dots arising. The counts of both dual-fluorescent dots and mRFP-fluorescent dots increased significantly after CS treatment. Dioscin treatment further increased the quantity of both the dual- fluorescent and mRFP-positive dots (Fig. 2E-F). Immunoblot analysis confirmed this result: LC3II expression levels in the dioscin-treated group were upregulated, whereas the P62 levels were downregulated compared to CS group. Saline group and rapamycin group served as negative and positive controls, respectively (Fig. 2G-I). The levels of BECN1 expression increased with the rising dosage of dioscin (Fig. 2J). To further explore the effect of dioscin on autophagy, we also examined the autophagy-related signaling pathways. The results showed that AKT-mTOR signaling was blocked by dioscin treatment (Fig. 2K-L), which suggested that dioscin's autophagy-inducing effect might be related to this pathway. The combination of these results reinforces the conclusion that dioscin activates autophagy in vitro.
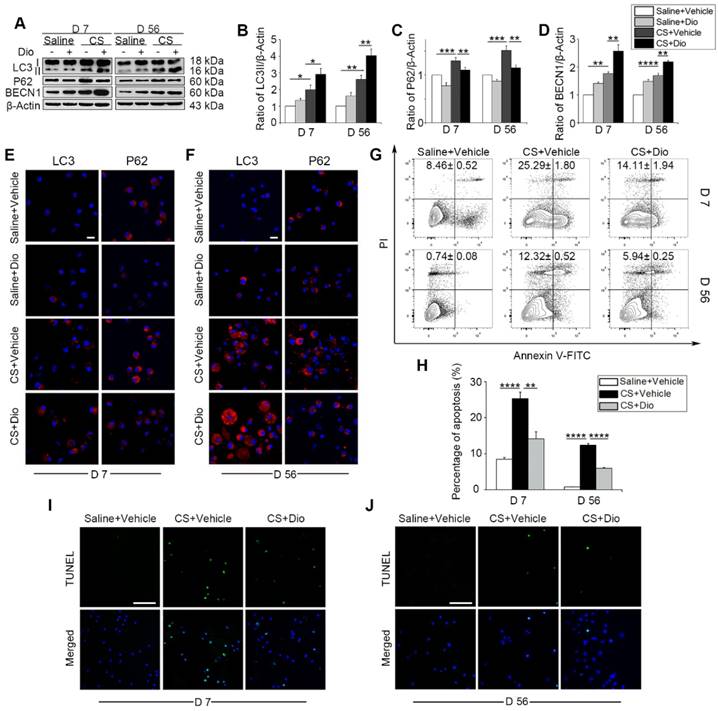
Next, CS-injured experimental animals, who were treated with dioscin orally, were used to test the efficacy of dioscin in vivo. We first examined the levels of autophagy-related proteins in the mice lung tissues. As we expected, up-regulation of LC3II and BECN1, and down-regulation of P62 was observed, which indicates greater autophagy activity in the dioscin- treated mice compared to the CS-treated mice at both day 7 and day 56 (Fig. 3A-D). To clarify dioscin's effects on AMs in mice, we measured autophagy activity in primary AMs with LC3 and P62 fluorescent tags. Consistent with the immunoblotting results in vitro, the number of LC3-positive punctate dots increased in the CS group at day 7, dioscin treated groups showed increased numbers of LC3-postive dots compared to CS group. The expression of P62 was reduced in the dioscin-treated groups compared to the other groups. The results at day 56 were consistent with those at day 7 (Fig. 3E-F). Based on our previous in vitro result, we further demonstrated that dioscin treatment relieved CS-induced AMs apoptosis at both day 7 and day 56 in vivo (Fig. 3G-J). Together, these data suggest that dioscin could promote autophagy and alleviate AMs apoptosis.
Dioscin treatment induces autophagy in mouse lung and attenuates CS-induced AMs apoptosis. (A) Immunoblot analysis of autophagy-associated proteins in lung tissue lysates (n=4). (B-D) Quantification of LC3II, P62, and BECN1. (E-F) Immunofluorescence analysis of LC3 and P62 in primary AMs from bronchoalveolar lavage fluid, the scale bar indicates 10 μm (n=3). (G-H) Primary AMs apoptosis assay by flow cytometry. (I-J) TUNEL assay (green) and DAPI staining (blue) of primary AMs, the scale bar indicates 50 μm (n=4). (B-D and H) *, P<0.05; **, P<0.01; ***, P<0.001, ****, P<0.0001. Error bars indicate mean ± SEM. All experiments were repeated three times with similar results.
Dioscin-induced autophagy activation protects AMs from mitochondria-dependent apoptosis
To explore detailed molecular mechanism of dioscin's effects on autophagy and apoptosis, we introduced Atg5 knockdown MH-S cells. We proved Atg5 interference could offset most autophagy- induced effect of dioscin by testing expressions of LC3II, BECN1 and P62 (Fig. 4A and Fig. S5A-C). The main function of autophagy is scavenging damaged organelles for reuse [11]. CS cause mitochondrial injury which were selectively eliminated through autophagy (also referred as mitophagy) [12, 32]. Immunofluorescence with mitochondria marker Mitotracker and autophagosome maker LC3 revealed that cells treated with dioscin showed more Mitotracker/LC3-colocalized points than the CS group (Fig. 4B), indicating that dioscin may trigger mitophagy. We also determined two predominant proteins, Pink1 and Parkin, increased dramatically in the dioscin group, which were paralleled with expression of autophagy-related protein (Fig. 4C and Fig. S5D-E). Using in vitro model above, we further explored the relationship between mitophagy/ autophagy and apoptosis. Oxidative stress is the start of mitochondria-dependent apoptosis [33]. MtROS were detected by mitoSOX analysis. CS treatment augmented mtROS significantly, while dioscin could downregulate its production. All the reductions were reversed when autophagy was inhibited, regardless dioscin presence or not (Fig. 4D-F). Mitochondrial membrane potential (MMP) were analyzed by JC-1 analysis, as an indicator of mitochondria function. Dioscin protected mitochondrial from depolarization after CS stimulation, while this effect could be reversed after Atg5 knockdown (Fig.4G-H). Moreover, the significant decreases of TMRE and Rhodamine123 fluorescence in the Atg5 knockdown group cells reflected the effect of dioscin-induced autophagy/mitophagy on maintaining Δψm (Fig. 4I-J). We also tested the direct indicator of mitochondria function, ATP production, and found dioscin treatment protected ATP level after CS stimulation while inhibiting autophagy neutralized the effect (Fig. 4K). This phenomenon suggests dioscin reduces CS-induced mtROS production and mitochondrial dysfunction at least partially through inducing autophagy.
Dioscin-induced autophagy protects AMs from mitochondria-dependent apoptosis. (A) Immunoblot analysis of autophagy-associated proteins LC3II, P62, and BECN1 (n=3). (B) Immunofluorescence analysis of LC3 (red) and mitochondria (Mitotracker, green) in MH-S cells, the scale bar indicates 10 μm (n=3). (C) Immunoblot analysis of PINK1 and Parkin in mitochondria isolated from MH-S cells (n=3). (D-F) MitoSOX was detected by fluorescence microscopy and flow cytometry (n=3), the scale bar indicates 10μm (n=3). (G-H) Mitochondrial membrane potential (MMP) was detected by flow cytometry and fluorescence microscopy, the scale bar indicates 50 μm (n=3). (I) Fluorescence image of cells labeled with TMRE and (J) Rhodamine 123, the scale bar indicates 50 μm (n=3). (K) ATP production assay. (L) Immunofluorescence analysis of Cyt-c in MH-S cells, the scale bar indicates 50 μm (n=3). (M) The activities of caspase 3 and 7 by Caspase-Glo 3/7 assay (n=3). (E, K and M) *, P<0.05; **, P<0.01; ***, P<0.001, ****, P<0.0001. Error bars indicate mean ± SEM. All experiments were repeated three times with similar results.
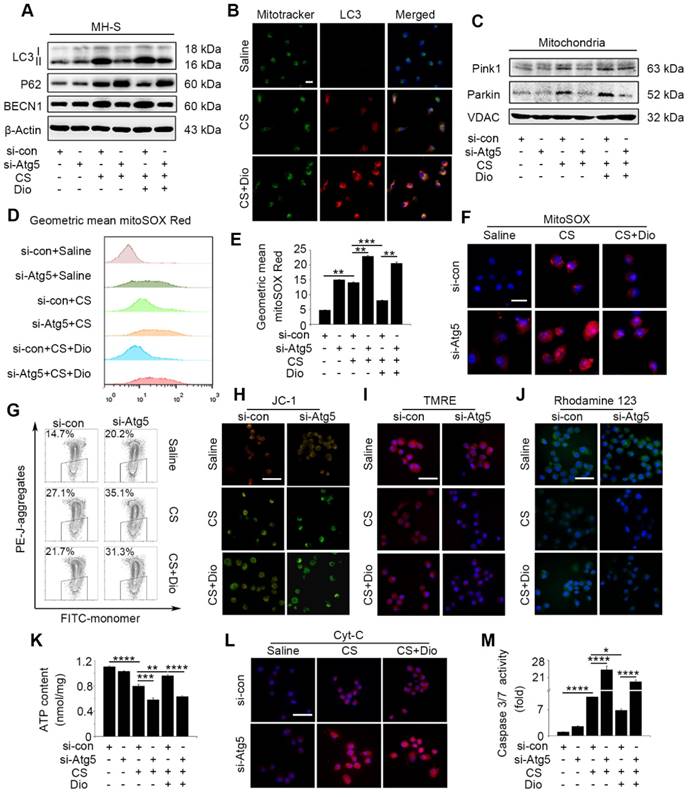
Loss of mitochondrial function activates the cytochrome c (Cyt-c) release and subsequent caspase cascade reaction [34]. We observed Cyt-c distribution by immunofluorescence staining. Compared to cells treated with saline, Cyt-c protein was increased in the cytoplasmic fractions from CS-stimulated cells, while dioscin treatment released this condition. Cyt-c protein were increased greatly in cytoplasm of Atg5 knockdown cells whether treated dioscin or not (Fig. 4L). We also tested caspase 3/7 activity which activated by Cyt-c and found analogous change as Cyt-c (Fig. 4M). These results combined suggest that dioscin-induced AMs autophagy could eliminate injured mitochondria, maintain mitochondria function, and provide a potential molecular mechanism to autophagy's anti-apoptosis effects.
Dioscin suppresses the generation of inflammatory factors in AMs through inducing autophagy
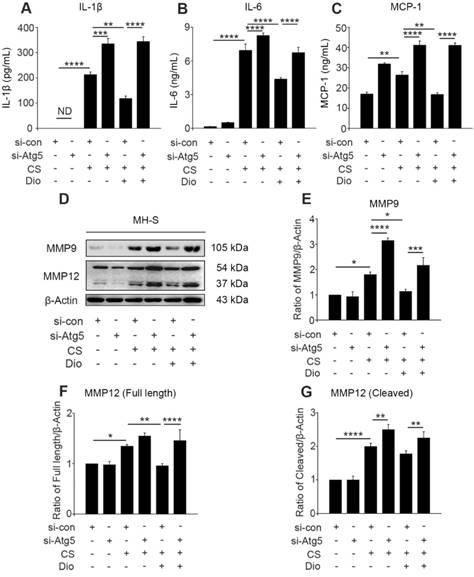
The continuous CS stimulation leads to excessive mtROS, which is the cause and result of mitochondria-depend cell death [7, 14, 34]. Excessive mtROS cause increased cellular oxidative stress, and one of the adverse outcomes is the excessive release of inflammatory factors and cytokines [35]. We found dioscin decreased the amount of IL-1β and IL-6 in cultured supernatants after CS injury. However, autophagy inhibition by Atg5 knockdown always increased the amount of these inflammatory factors whether stimulated by CS alone or combined with dioscin. Moreover, the quantity of monocyte chemotactic protein-1 (MCP-1) was increased after autophagy inhibition and decreased after dioscin treatment (Fig. 5A-C). The combined results illustrate that dioscin reduced the secretion of macrophage inflammatory factors, which is dependent on autophagy. ROS production is also associated with secretion of collagenase MMP9 and macrophage elastase MMP12, which promote inflammatory cell infiltration [36, 37]. We found that MMP9 expression was increased in the CS group compared to the control group. The expression level was further increased after Atg5 gene silencing. Similarly, the secretion and cleavage of MMP12 were also impacted by CS stimulation. These findings show that dioscin treatment can reduce the production of MMP9 and full length of MMP12 in macrophages, but these effects can be reversed by Atg5 knockdown. Dioscin treatment did not affect the cleavage of MMP12, however, autophagy depletion increased its cleavage (Fig. 5D-G).
Dioscin reduces CS-stimulated secretion of inflammatory factors through autophagy induction. (A-C) ELISA analysis of IL-1β, IL-6, and MCP-1 in MH-S cell supernatant (n=3). (D) Immunoblot analysis of MMP9 and MMP12 in lysates from stimulated MH-S cells (n=3). (F-G) Quantification of MMP9 and MMP12 proteins. (A-C and E-G). *, P<0.05; **, P<0.01; ***, P<0.001; ****, P<0.0001; ND, Not detected. Error bars indicate mean ± SEM. All experiments were repeated three times with similar results.
Dioscin treatment alleviates macrophage-induced inflammation in vivo
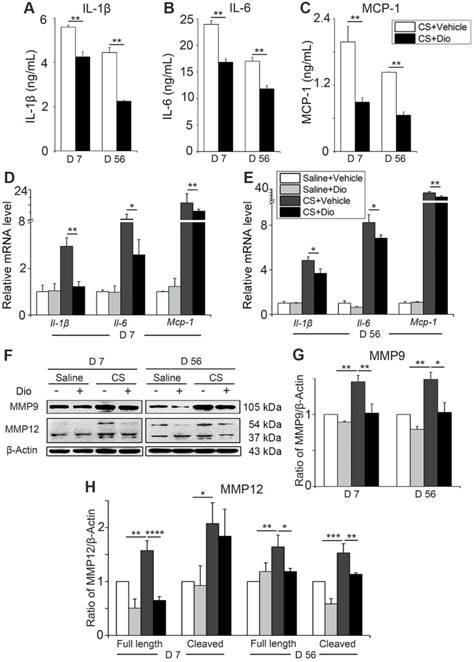
To verify whether dioscin exerts the same anti-inflammatory effect in vivo, we collected primary AMs from different treated mice and cultured them (all the stimuli and treatment were applied to the animals, not to the cells directly). Culture medium and cells were collected. The levels of IL-1β, IL-6, and MCP-1 were reduced significantly after dioscin treatment at both day 7 and day 56 (Fig. 6A-C). Transcription levels of Il-1β, Il-6, and Mcp-1 in the collected cells were downregulated by dioscin treatment as well (Fig. 6D-E). The expression of MMP9 and the two forms of MMP12 decreased at both translational level (Fig. 6F-H) and transcriptional level after dioscin treatment (Fig. S6). Overall, the results of the in vitro cell experiments can be extended to the animal model, suggesting that dioscin treatment could improve the AM-triggered inflammatory microenvironment in lung tissue through autophagy.
Dioscin treatment reduces the secretion of inflammatory cytokines in mice AMs. (A-C) ELISA analysis of IL-1β, IL-6, and MCP-1 in primary AMs culture supernatants (n=3). (D-E) mRNA levels of Il-1β, Il-6, and Mcp-1 in primary AMs (n=5). (F) Immunoblot analysis of MMP9 and MMP12 in lung tissue (n=3-4). (G-H) Quantification of MMP9 and MMP12 proteins. (A-E, G, H). *, P<0.05; **, P<0.01; ***, P<0.001, ****, P<0.0001. Error bars indicate mean ± SEM. All experiments were repeated three times with similar results.
Dioscin protects against pulmonary inflammation and fibrosis caused by CS
AMs play a critical role in orchestrating innate and adaptive immune cells to regulate pulmonary inflammatory response [38]. Paraffin sections taken at day 7 were stained with anti-F4/80 to mark macrophages in the lung tissue. Few macrophages could be seen in the groups treated with saline in presence of dioscin or not. In contrast, a large number of macrophages accumulated in the CS-damaged lung tissue, especially around the terminal bronchus, while dioscin lessened the macrophage accumulation (Fig. 7A). HE staining revealed inflammatory cell infiltration. The CS-treated group showed masses of cells aggregated around the pulmonary airway and damaged alveolar walls. This infiltration of cells was alleviated by dioscin treatment (Fig. 7B). All the data indicate that dioscin-induced autophagy could be an effective therapy for CS-induced pulmonary inflammation.
Dioscin treatment alleviates pulmonary inflammation and fibrosis in mice. (A) Immunohistochemical staining of F4/80 applied to paraffin sections from mice at day 7 post treatment, upper scale bar indicates 100 μm and lower scale bar indicates 50 μm (n=4). (B) HE staining of mouse lungs at day 7, the scale bar indicates 50 μm (n=4). (C) mRNA level of Col1a1 and Col3a1 in the lungs of mice at day 56 (n=4-5). (D) Sirius red staining of mouse lungs at day 56 (upper panel natural light; lower panel polarized light), the scale bar indicates 100 μm (n=4). (C). *, P<0.05; **, P<0.01. Error bars indicate mean ± SEM. All experiments were repeated three times with similar results.
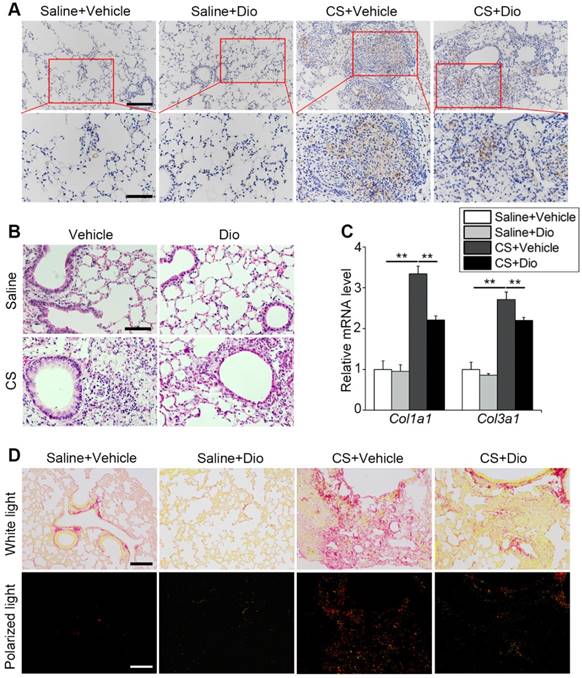
Macrophages participate in collagen repair processes as well [39]. qPCR analysis showed that dioscin treatment reduced collagen I and collagen III expression induced by CS stimulation during the fibrosis phase (Fig. 7C). Sirius red staining was also carried out to measure the degree of fibrosis. Similar to the variations at the gene level, we observed that the distribution of red, yellow (representing collagen I), and green (representing collagen III) lessened after dioscin treatment. In addition, the two types of collagen were concentrated in the nodules near the airway, which is consistent with the location of the early inflammatory injury induced by CS (Fig. 7D). These data suggest that dioscin could decrease collagen deposition, thus reducing the degree of pulmonary fibrosis.
Autophagy inhibition reverses the protective effects of dioscin in vivo
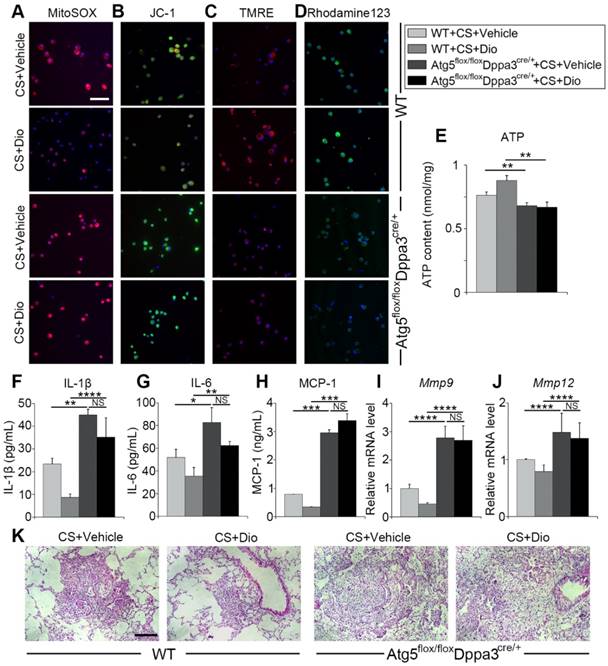
To further verify dioscin inducing autophagy could protect AMs from mitochondrial dysfunction and relieve inflammatory response in the inflammatory stage of experiment silicosis, we introduced Atg5 flox/floxDppa3cre/+ mice, who lacked autophagy function. First, macrophage mitochondrial function was detected. MitoSOX analysis revealed dioscin reduced mtROS production through inducing autophagy (Fig. 8A). Mitochondrial membrane potential was measured by JC-1, TMRE and Rhodamin123 staining, showing that autophagy inhibition reversed dioscin's maintaining mitochondrial membrane potential effects (Fig. 8B-D). ATP analysis further supported the view (Fig. 8E). Next, we evaluated pulmonary inflammation through ELISA, qPCR analysis and lung section HE staining, found that autophagy inhibiting abrogated dioscin's anti-inflammatory effects, even aggravated CS-induced pulmonary inflammation (Figure 8F-K). All the data combined suggest dioscin protects AMs from mitochondrial dysfunction and alleviates inflammatory response through inducing autophagy.
Autophagy inhibition reverses the protective effects of dioscin. (A) Fluorescence image of cells labeled with MitoSOX, (B) JC-1 (C) TMRE and (D) Rhodamine 123, the scale bar indicates 50 μm (n=3). (E) ATP production assay. (F-H) ELISA analysis of IL-1β, IL-6, and MCP-1 in BALF (n=3). (I-J) mRNA levels of Mmp9 and Mmp12 in lung tissue (n=4-5). (K) HE staining of mouse lung sections, the scale bar indicates 50 μm (n=4). *, P<0.05; **, P<0.01, ***, P<0.001, ****, P<0.0001; NS, No significance. Error bars indicate mean ± SEM. All experiments were repeated three times with similar results.
Discussion
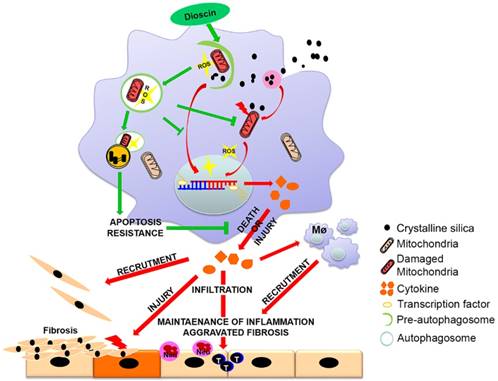
Innate immunity is indispensable for the progression of silicosis [40]. Our preliminary work has demonstrated the anti-fibrotic effects of dioscin in connection with regulation of AMs [26]. In this study, we demonstrated that AMs autophagy exerted protective role in CS-induced apoptosis and reduced inflammatory cytokine release. Dioscin-triggered autophagy mitigated CS-induced excessive mtROS release, mitochondrial dysfunction and AMs apoptosis, resulting in reduced production of inflammatory factors both in vitro and in vivo, consequently decreasing inflammatory infiltration and collagen deposition. We provide a potential molecular mechanism of dioscin's anti-CS-induced pulmonary fibrosis effect through up-regulating AMs autophagy (Fig. 9).
Mechanism of dioscin-induced autophagy in macrophages relieves CS-induced lung inflammation and fibrosis. Dioscin-induced autophagy promotes the removal of damaged mitochondria leading to apoptosis resistance and ROS reduction. Decreased secretion of inflammatory factors and reduced recruitment of immune cells result in alleviation of tissue injury and abnormal collagen repair.
Whether autophagy has a positive effect on human disease remains controversial, however, several studies have shown that macrophage autophagy may play a protective role in fibrogenic diseases [16-18]. In addition, our previous study showed that autophagosomes accumulated in AMs of silicosis patients [21]. Autophagy is closely related to apoptosis. In this study, we showed that autophagy activation reduced AM apoptosis significantly (Fig. 2B-D). Consistent with our findings, macrophages deficient in Atg5 are more susceptible to apoptosis caused by silica exposure [40]. Recent study about pulmonary fibrosis also showed that AMs autophagy activation led to apoptosis resistance, which further supported our results [22]. However, there are still opposite results claiming that macrophage autophagy exerts negative effects in progression of CS-induced pulmonary fibrosis. It was reported that BBC3 and MCPIP1, the two pro-fibrogenic factors, may lead to macrophage apoptosis under CS circumstances through inducing autophagy [24, 25]. The contrary views of macrophage autophagy on silicosis indicated that it is worth being deeply explored, which may further be developed as a potential target for silicosis treatment.
Invaded CS particles lead to AM apoptosis due to not only its cytotoxicity but also the overload of apoptotic cell engulfment. Engulfed particles would damage organelles within the cytoplasm, such as mitochondria. Autophagy may be induced to reuse and eliminate the injured mitochondria. The probable effects of autophagy in AMs can be related to the improved phagocytosis function. The autophagic pathway converges with the phagocytic pathway at amphisomes to enable lysosomal degradation of the debris, thus lightening the phagocytosis load of AMs [41, 42]. Excessive engulfment would trigger mitochondrial apoptosis by increasing intracellular ROS [8]. MtROS accounts for most of the cellular ROS, mitochondria are the most significant source of intracellular ROS in CS-exposed macrophages [7]. Mitochondria dysfunction leads to redistribution of Cyt-c and subsequent caspase cascades activation [14]. We proved CS-induced oxidative stress and mitochondria-dependent apoptosis activation could be reduced by autophagy activation in vitro and in vivo. Dioscin exerts its positive effects, decreasing ROS mass, maintaining MMP and reducing mitochondrial apoptosis pathway activation, all of which were disappeared after autophagy related gene silenced (Fig. 4D-M and Fig. 8A-E). All the findings indicate that dioscin exerts function through regulating macrophage autophagy rather than directly acting on ROS or mitochondria.
Clearance of mitochondria by selective autophagy (mitophagy) is an essential mechanism of mitochondrial quality control [43]. Study on IPF patients showed that increased mitophagy do reduce cell apoptosis in AMs [22]. The increasing overlap of mitotracker and the autophagosome marker LC3 suggested that mitophagy was activated by dioscin (Fig. 4B). PINK1 and Parkin are characterized by expressing on damaged mitochondria and trafficking to phagosomes, which is regulated by BECN1 [15]. In our study, we showed that decreased mitochondrial dysfunction correlated with increased levels of PINK1 and Parkin proteins after dioscin treatment (Fig. 4C and Fig. S5D-E). Meanwhile, the expression of BECN1 was upregulated. Research about BECN1 uncovers its dual functions that it coordinately controls and balances mitophagy and general autophagy [15]. BECN1 promotes Parkin translocation to mitochondria, while BECN1 inhibition results in accumulation of damaged mitochondria, facilitating pulmonary fibrosis [36, 44]. We observed that BECN1 expression was partly reduced after autophagy inhibition, so as PINK1 and Parkin expressions (Fig. 4A and C). Notably, BECN1 is a predicted molecule that dioscin directly acts with. However, due to knockdown efficiency was quite low (only 40%, Fig. S4A), we chose Atg5, the important molecule in autophagy process, as an alternative, since we regarded autophagy process (blue circle) as dioscin's target (Fig. 2A) and found that dioscin influence inflammatory response through autophagy activation. Moreover, we examined IL-1β, IL-6 and MCP-1 in supernatant of BECN1-knockdown MH-S cells. The variation tendencies of inflammatory factors were similar to those of Atg5-knockdown cells (Fig. S4B-D), suggesting it is a good alternative candidate. Our results suggest that increased mitophagy should be a consequence of dioscin-induced autophagy.
CS-induced mtROS production in macrophages is responsible for cell death and tissue injury [14]. MtROS is tightly linked with inflammatory response as it could regulate NLRP3 inflammasome formation and mitogen activated protein kinase (MAPK) signaling activation, which are associated with cytokine secretion [45]. It provides extensive molecular mechanism to our previous study that dioscin could block ASK-p38/JNK signaling pathway by reducing mtROS in macrophages, leading to reduced cytokine productions in BALF [26]. Besides, mtROS mediates its pro-inflammatory and pro-fibrotic effects by increasing IL-1β production in liver fibrosis [18]. Both pro-IL-1β sythesis and bioactive IL-1β secretion depend on mtROS [46]. Additionally, autophagy degrades inflammasome complexes directly may further prevent pro-IL-1β from being cleaved into biologically active forms [47]. The potent chemokine MCP-1, which was increased in serum of silicosis patients [48], possesses the ability recruiting multiple inflammatory cells to damaged areas, in which macrophage and circulating fibrocyte are closely linked with pulmonary fibrosis [49]. Our previous finding showed recruitment of the two cells to lung were clearly decreased by dioscin treatment [26]. The data illustrate that reduced mtROS-mediated inflammatory factor secretion is a key link connecting dioscin-induced autophagy activation and the inflammation relief effects.
Matrix metalloproteinases (MMPs) play vital roles in both the inflammatory stage and tissue repair stage in silicosis [50]. They could degrade basement membrane, which result in inflammatory cells infiltration into damaged area. Dioscin-treated mice showed a decreased accumulation of immune cells including T and B cells in the pulmonary tissue and shrunken injury areas caused by CS during the inflammatory stage of silicosis [26], which are ascribed to autophagy activation on lessening MMP9 and MMP12 production. Dioscin treatment also led to decreased macrophage infiltration. Aggregated macrophages in lung tissue are not just resident AMs but also monocytes from the circulation [4]. Research indicated that monocyte-derived macrophages express higher levels of inflammatory and pro-fibrotic genes than resident macrophages [51], which may lead to more severe inflammation and accelerate fibrogenesis. Besides, MMP9 exerts its pro-fibrotic effect through the cleavage and release of bio-active TGF-β which adds to the deposition of collagen and leads to abnormal tissue repair [52, 53]. All of these data illustrate that dioscin promoting autophagy lead to reduced chemokines and pro- inflammatory cytokines secretion further decreasing inflammatory cell infiltration and collagen deposition.
It also should be noted that AM apoptosis is tightly linked with the progression of silicosis [29, 54]. Apoptotic AMs release ingested CS particles, accompanied by more cell debris and ROS, which lead to next round of phagocytosis and macrophage apoptosis. The iterative phagocytosis and apoptosis cycle further exacerbate the inflammatory cascading, resulting in aggravated pulmonary parenchyma damage. It could provide another explanation that the reduced AMs aggregation in damaged lung tissue treated with dioscin. In addition, the released ROS can further aggravate pulmonary injury and cause alveolar epithelial damage, which may provide a theoretical basis for our previous experimental results that dioscin can protect alveolar epithelium from damage of CS particles [26, 55]. Moreover, our study demonstrates that dioscin reduces the secretion and cleavage of MMPs in AMs by inducing autophagy, thereby decreases a variety of inflammatory cells infiltration protecting basement membrane lead to milder injury.
In summary, our results suggest that dioscin exerts anti-inflammatory and anti-fibrotic effects through the induction of autophagy in AMs. Autophagy protects AMs from CS-induced apoptosis through enhancing mitophagy, eliminating mitochondrial apoptosis pathway activation and lessening excessive mtROS, further reducing the secretion of inflammatory factors and eventual fibrosis induced by CS particles.
Abbreviations
Atg5: Autophagy-related protein 5; Atg7: Autophagy-related protein 7; AKT/PKB: Protein Kinase B; BECN1: Beclin 1; IL-1β: Interleukin 1 beta; IL-6: Interleukin 6; LC3/MAP1LC3: Microtubule-Associated Protein 1 Light Chain 3; MMP9: Matrix metalloprotein 9; MMP12: Matrix metalloprotein 12; m-TOR: Mammalian target of rapamycin; MCP-1: Monocyte chemoattractant protein-1; P62/SQSTM1: Sequestosome 1; PTEN: Phosphatase and tensin homolog deleted on chromosome ten; PINK1: PTEN induced putative kinase 1; ROS: Reactive oxygen species; TGF-β: Transforming growth factor beta; ULK1: Unc-51 like autophagy activating kinase.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81872592) and the Program for Liaoning Innovative Research Team in University (LT2015028).
Author Contributions
SD, CL and JC were responsible for the conception and design of the study. SD, CL, YL, XL, YZ and SL performed experiments. SD and CL analyzed results and interpreted data. JC supervised the study. SD and CL drafted the manuscript. FL, YC, DW and JC helped revise the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Steenland K, Ward E. Silica: a lung carcinogen. CA Cancer J Clin. 2014;64:63-9
2. Li C, Du S, Lu Y, Lu X, Liu F, Chen Y. et al. Blocking the 4-1BB Pathway Ameliorates Crystalline Silica-induced Lung Inflammation and Fibrosis in Mice. Theranostics. 2016;6:2052-67
3. Pollard KM. Silica, Silicosis, and Autoimmunity. Front Immunol. 2016;7:97
4. Byrne AJ, Mathie SA, Gregory LG, Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. 2015;70:1189-96
5. Murthy S, Larson-Casey JL, Ryan AJ, He C, Kobzik L, Carter AB. Alternative activation of macrophages and pulmonary fibrosis are modulated by scavenger receptor, macrophage receptor with collagenous structure. FASEB J. 2015;29:3527-36
6. Hamilton RF Jr, Thakur SA, Holian A. Silica binding and toxicity in alveolar macrophages. Free Radic Biol Med. 2008;44:1246-58
7. Fazzi F, Njah J, Di Giuseppe M, Winnica DE, Go K, Sala E. et al. TNFR1/phox interaction and TNFR1 mitochondrial translocation Thwart silica-induced pulmonary fibrosis. J Immunol. 2014;192:3837-46
8. Zhang L, He YL, Li QZ, Hao XH, Zhang ZF, Yuan JX. et al. N-acetylcysteine alleviated silica-induced lung fibrosis in rats by down-regulation of ROS and mitochondrial apoptosis signaling. Toxicol Mech Methods. 2014;24:212-9
9. Lopes-Pacheco M, Bandeira E, Morales MM. Cell-Based Therapy for Silicosis. Stem Cells Int. 2016;2016:5091838
10. Booth LA, Tavallai S, Hamed HA, Cruickshanks N, Dent P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell Signal. 2014;26:549-55
11. Kim KH, Lee MS. Autophagy-a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014;10:322-37
12. Palikaras K, Lionaki E, Tavernarakis N. Mitophagy: In sickness and in health. Mol Cell Oncol. 2016;3:e1056332
13. Zank DC, Bueno M, Mora AL, Rojas M. Idiopathic Pulmonary Fibrosis: Aging, Mitochondrial Dysfunction, and Cellular Bioenergetics. Front Med (Lausanne). 2018;5:10
14. Liu X, Chen Z. The pathophysiological role of mitochondrial oxidative stress in lung diseases. J Transl Med. 2017;15:207
15. Choubey V, Cagalinec M, Liiv J, Safiulina D, Hickey MA, Kuum M. et al. BECN1 is involved in the initiation of mitophagy: it facilitates PARK2 translocation to mitochondria. Autophagy. 2014;10:1105-19
16. Lodder J, Denaes T, Chobert MN, Wan J, El-Benna J, Pawlotsky JM. et al. Macrophage autophagy protects against liver fibrosis in mice. Autophagy. 2015;11:1280-92
17. Ilyas G, Zhao E, Liu K, Lin Y, Tesfa L, Tanaka KE. et al. Macrophage autophagy limits acute toxic liver injury in mice through down regulation of interleukin-1beta. J Hepatol. 2016;64:118-27
18. Sun K, Xu L, Jing Y, Han Z, Chen X, Cai C. et al. Autophagy-deficient Kupffer cells promote tumorigenesis by enhancing mtROS-NF-kappaB-IL1alpha/beta-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis. Cancer Lett. 2017;388:198-207
19. Lee J, Foote A, Fan H, Peral de Castro C, Lang T, Jones S. et al. Loss of autophagy enhances MIF/macrophage migration inhibitory factor release by macrophages. Autophagy. 2016;12:907-16
20. Le TT, Karmouty-Quintana H, Melicoff E, Le TT, Weng T, Chen NY. et al. Blockade of IL-6 Trans signaling attenuates pulmonary fibrosis. J Immunol. 2014;193:3755-68
21. Chen S, Yuan J, Yao S, Jin Y, Chen G, Tian W. et al. Lipopolysaccharides may aggravate apoptosis through accumulation of autophagosomes in alveolar macrophages of human silicosis. Autophagy. 2015;11:2346-57
22. Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity. 2016;44:582-96
23. Sosulski ML, Gongora R, Danchuk S, Dong C, Luo F, Sanchez CG. Deregulation of selective autophagy during aging and pulmonary fibrosis: the role of TGFbeta1. Aging cell. 2015;14:774-83
24. Liu H, Cheng Y, Yang J, Wang W, Fang S, Zhang W. et al. BBC3 in macrophages promoted pulmonary fibrosis development through inducing autophagy during silicosis. Cell Death Dis. 2017;8:e2657
25. Liu H, Fang S, Wang W, Cheng Y, Zhang Y, Liao H. et al. Macrophage-derived MCPIP1 mediates silica-induced pulmonary fibrosis via autophagy. Part Fibre Toxicol. 2016;13:55
26. Li C, Lu Y, Du S, Li S, Zhang Y, Liu F. et al. Dioscin Exerts Protective Effects Against Crystalline Silica-induced Pulmonary Fibrosis in Mice. Theranostics. 2017;7:4255-75
27. Joshi GN, Knecht DA. Silica phagocytosis causes apoptosis and necrosis by different temporal and molecular pathways in alveolar macrophages. Apoptosis. 2013;18:271-85
28. Lu Y, Li C, Du S, Chen X, Zeng X, Liu F. et al. 4-1BB Signaling Promotes Alveolar Macrophages-Mediated Pro-Fibrotic Responses and Crystalline Silica-Induced Pulmonary Fibrosis in Mice. Front Immunol. 2018;9:1848
29. Yao SQ, Rojanasakul LW, Chen ZY, Xu YJ, Bai YP, Chen G. et al. Fas/FasL pathway-mediated alveolar macrophage apoptosis involved in human silicosis. Apoptosis. 2011;16:1195-204
30. Wang L, Antonini JM, Rojanasakul Y, Castranova V, Scabilloni JF, Mercer RR. Potential role of apoptotic macrophages in pulmonary inflammation and fibrosis. J Cell Physiology. 2003;194:215-24
31. Hsieh MJ, Tsai TL, Hsieh YS, Wang CJ, Chiou HL. Dioscin-induced autophagy mitigates cell apoptosis through modulation of PI3K/Akt and ERK and JNK signaling pathways in human lung cancer cell lines. Arch Toxicol. 2013;87:1927-37
32. Hu S, Zhao H, Al-Humadi NH, Yin XJ, Ma JK. Silica-induced apoptosis in alveolar macrophages: evidence of in vivo thiol depletion and the activation of mitochondrial pathway. J Toxicol Environ Health A. 2006;69:1261-84
33. Kang P, Zhang W, Chen X, Yi X, Song P, Chang Y. et al. TRPM2 mediates mitochondria-dependent apoptosis of melanocytes under oxidative stress. Free Radic Biol Med. 2018;126:259-68
34. Han YQ, Ming SL, Wu HT, Zeng L, Ba G, Li J. et al. Myostatin knockout induces apoptosis in human cervical cancer cells via elevated reactive oxygen species generation. Redox Biol. 2018;19:412-28
35. Harris J, Deen N, Zamani S, Hasnat MA. Mitophagy and the release of inflammatory cytokines. Mitochondrion. 2018;41:2-8
36. Chen L, You Q, Hu L, Gao J, Meng Q, Liu W. et al. The Antioxidant Procyanidin Reduces Reactive Oxygen Species Signaling in Macrophages and Ameliorates Experimental Colitis in Mice. Front Immunol. 2017;8:1910
37. Yun SP, Lee SJ, Oh SY, Jung YH, Ryu JM, Suh HN. et al. Reactive oxygen species induce MMP12-dependent degradation of collagen 5 and fibronectin to promote the motility of human umbilical cord-derived mesenchymal stem cells. Br J Pharmacol. 2014;171:3283-97
38. Zhai R, Ge X, Li H, Tang Z, Liao R, Kleinjans J. Differences in cellular and inflammatory cytokine profiles in the bronchoalveolar lavage fluid in bagassosis and silicosis. Am J Ind Med. 2004;46:338-44
39. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245-57
40. Jessop F, Hamilton RF, Rhoderick JF, Shaw PK, Holian A. Autophagy deficiency in macrophages enhances NLRP3 inflammasome activity and chronic lung disease following silica exposure. Toxicol Appl Pharmacol. 2016;309:101-10
41. Hu B, Sonstein J, Christensen PJ, Punturieri A, Curtis JL. Deficient in vitro and in vivo phagocytosis of apoptotic T cells by resident murine alveolar macrophages. J Immunol. 2000;165:2124-33
42. Zhou P, Tan YZ, Wang HJ, Li T, He T, Yu Y. et al. Cytoprotective effect of autophagy on phagocytosis of apoptotic cells by macrophages. Exp Cell Res. 2016;348:165-76
43. Yoo SM, Jung YK. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol Cells. 2018;41:18-26
44. van der Vliet A, Janssen-Heininger YMW, Anathy V. Oxidative stress in chronic lung disease: From mitochondrial dysfunction to dysregulated redox signaling. Mol Aspects Med. 2018;63:59-69
45. Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126-67
46. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J. et al. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell. 2016;164:896-910
47. Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy. 2016;12:245-60
48. Chen Y, Li C, Lu Y, Zhuang H, Gu W, Liu B. et al. IL-10-Producing CD1d(hi)CD5(+) Regulatory B Cells May Play a Critical Role in Modulating Immune Homeostasis in Silicosis Patients. Front Immunol. 2017;8:110
49. van Deventer HW, Palmieri DA, Wu QP, McCook EC, Serody JS. Circulating fibrocytes prepare the lung for cancer metastasis by recruiting Ly-6C+ monocytes via CCL2. J Immunol. 2013;190:4861-7
50. Scabilloni JF, Wang L, Antonini JM, Roberts JR, Castranova V, Mercer RR. Matrix metalloproteinase induction in fibrosis and fibrotic nodule formation due to silica inhalation. Am J Physiol Lung Cell Mol Physiol. 2005;288:L709-17
51. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC. et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. 2017;214:2387-404
52. Wilson TJ, Nannuru KC, Singh RK. Cathepsin G-mediated activation of pro-matrix metalloproteinase 9 at the tumor-bone interface promotes transforming growth factor-beta signaling and bone destruction. Mol Cancer Res. 2009;7:1224-33
53. Jolly L, Stavrou A, Vanderstoken G, Meliopoulos VA, Habgood A, Tatler AL. et al. Influenza promotes collagen deposition via alphavbeta6 integrin-mediated transforming growth factor beta activation. J Biol Chem. 2014;289:35246-63
54. Guidi P, Nigro M, Bernardeschi M, Lucchesi P, Scarcelli V, Frenzilli G. Does the crystal habit modulate the genotoxic potential of silica particles? A cytogenetic evaluation in human and murine cell lines. Mutat Res Genet Toxicol Environ Mutagen. 2015;792:46-52
55. Bernard O, Jeny F, Uzunhan Y, Dondi E, Terfous R, Label R. et al. Mesenchymal stem cells reduce hypoxia-induced apoptosis in alveolar epithelial cells by modulating HIF and ROS hypoxic signaling. Am J Physiol Lung Cell Mol Physiol. 2018;314:L360-l71
Author contact
![]() Corresponding author: Jie Chen e-mail: jchenedu.cn, Tel: 86 24 31939079
Corresponding author: Jie Chen e-mail: jchenedu.cn, Tel: 86 24 31939079