Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(7):1909-1922. doi:10.7150/thno.30578 This issue Cite
Research Paper
HMGB1-Induced p62 Overexpression Promotes Snail-Mediated Epithelial-Mesenchymal Transition in Glioblastoma Cells via the Degradation of GSK-3β
Hong Li1*, Junjie Li1*, Guozhong Zhang1,2,3*, Qian Da4*, Lei Chen1, Shishi Yu5, Qiang Zhou1, Zhijian Weng1, Zong Xin1, Linyong Shi1, Liyi Ma1, Annie Huang6, Songtao Qi1,2,3 ![]() , Yuntao Lu1,2,3
, Yuntao Lu1,2,3 ![]()
1. Department of Neurosurgery, Nanfang Hospital, Southern Medical University, Guangzhou, China.
2. Nanfang Neurology Research Institution, Nanfang Hospital, Southern Medical University, Guangzhou, China.
3. Nanfang Glioma Center, Guangzhou, China
4. Department of Pathology, Ruijing Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China.
5. Editorial Department of the Journal of Southern Medical University, Guangzhou, China.
6. Brain Tumor Research Center, SickKids Hospital, Toronto, Canada.
*These authors contributed equally to this work.
Received 2018-10-11; Accepted 2019-2-5; Published 2019-3-16
Abstract

Rationale: Glioblastoma (GBM) is the most common and aggressive brain tumor, characterized by its propensity to invade the surrounding brain parenchyma. The effect of extracellular high-mobility group box 1 (HMGB1) protein on glioblastoma (GBM) progression is still controversial. p62 is overexpressed in glioma cells, and has been associated with the malignant features and poor prognosis of GBM patients. Hence, this study aimed to clarify the role of p62 in HMGB1-induced epithelial-mesenchymal transition (EMT) of GBM both in vitro and in vivo.
Methods: Immunoblotting, immunofluorescence and qRT-PCR were performed to evaluate EMT progression in both human GBM cell line and primary GBM cells. Transwell and wound healing assays were used to assess the invasion and migration of GBM cells. shRNA technique was used to investigate the role of p62 in HMGB1-induced EMT both in vitro and in vivo orthotopic tumor model. Co-immunoprecipitation assay was used to reveal the interaction between p62 and GSK-3β (glycogen synthase kinase 3 beta). Immunohistochemistry was performed to detect the expression levels of proteins in human GBM tissues.
Results: In this study, GBM cells treated with recombinant human HMGB1 (rhHMGB1) underwent spontaneous EMT through GSK-3β/Snail signaling pathway. In addition, our study revealed that rhHMGB1-induced EMT of GBM cells was accompanied by p62 overexpression, which was mediated by the activation of TLR4-p38-Nrf2 signaling pathway. Moreover, the results demonstrated that p62 knockdown impaired rhHMGB1-induced EMT both in vitro and in vivo. Subsequent mechanistic investigations showed that p62 served as a shuttling factor for the interaction of GSK-3β with proteasome, and ultimately activated GSK-3β/Snail signaling pathway by augmenting the degradation of GSK-3β. Furthermore, immunohistochemistry analysis revealed a significant inverse correlation between p62 and GSK-3β, and a combination of the both might serve as a more powerful predictor of poor survival in GBM patients.
Conclusions: This study suggests that p62 is an effector for HMGB1-induced EMT, and may represent a novel therapeutic target in GBM.
Keywords: HMGB1, p62, glioblastoma, GSK-3β, epithelial-to-mesenchymal transition (EMT)
Introduction
Glioblastoma (GBM) is the most common and aggressive brain tumor, characterized by its propensity to invade the surrounding brain parenchyma [1]. Thus, the prognosis of GBM still remains poor, and dissecting the mechanisms underlying the highly invasive nature of GBM is urgently needed to improve the efficacy of GBM treatment. High-mobility group box 1 (HMGB1) is initially discovered as a highly conserved chromatin-binding nuclear protein [2], which can be passively secreted from dying tumor cells in response to radiation and/or temozolomide [3-5], or actively secreted by inflammatory cells, such as tumor-associated macrophages [6, 7]. HMGB1 can play an important role in tumor infiltration. Although it has been demonstrated that extracellular HMGB1 activates toll-like receptors, such as TLR2, TLR4, and TLR9, to promote tumor invasion [8, 9], other downstream effectors regulating tumor invasiveness are required to be explored.
Epithelial-mesenchymal transition (EMT), a cellular process characterized by the loss of the apical-basal polarity of epithelial cells toward a mesenchymal state, is frequently observed in invasive human tumors [10, 11]. The mesenchymal transition of GBM cells is associated with a more aggressive and treatment-resistant phenotype, leading to rapid progression and poor prognosis in patients with glioblastomas [12, 13]. It has recently been suggested that HMGB1 may promote cancer cell invasion via autocrine and paracrine means, which is associated with enhanced EMT in cancer cells [14-16]. However, the effects of HMGB1 on the invasion and EMT of GBM cells remain unclear. Zinc-finger transcription factor Snail, a transcriptional regulator for EMT, represses the expression of epithelial markers (occludin, claudins and cytokeratin) and increases the expression of mesenchymal markers (fibronectin and vimentin) [17, 18]. Snail has been emerged as a master regulator of cell migration and invasion, as well as irradiation-induced EMT in GBM cells [19]. Additionally, Snail is negatively regulated by glycogen synthase kinase-3β (GSK-3β)-mediated serial phosphorylation and subsequent proteasomal degradation [20]. More recently, it has been reported that the activation of GSK-3β/Snail signaling pathway is associated with microenvironment-induced EMT in epithelial cancers [21], including neuroepithelial GBM [19, 22].
The signaling adaptor sequestosome-1 (SQSTM1, hereafter referred to as p62) can shuttles ubiquitinated targets for proteasomal or autophagic degradation [23, 24]. A recent study has shown that p62 can regulate EMT process by degrading some transcription factors, such as TWIST1 [25]. Moreover, accumulation of p62 is associated with poor prognosis in patients with glioma [26]. These findings support an oncogenic role of p62 in tumor growth and progression. However, the detailed mechanisms behind are still poorly understood and await further investigation, particularly on p62-mediated EMT and invasion of GBM cells.
In this study, we characterized the effect of p62 on the HMGB1-induced EMT in GBM cells and investigated the underlying molecular mechanisms. We observed that p62 was accumulated during HMGB1-induced EMT in GBM cells, and this accumulation could be upregulated by the activation of TLR4-p38-Nrf2 signaling pathway. In addition, p62 might serve as a shuttling factor for the interaction of GSK-3β with proteasome, and ultimately activated GSK-3β/Snail signaling pathway by augmenting the degradation of GSK-3β in GBM cells.
Materials and Methods
An expanded methods section is available in Supplementary Material.
Cell culture and treatment
T98G cells [American Type Culture Collection (ATCC), Manassas, VA, USA] were cultured in DMEM supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA), 100 U/mL penicillin and 100 mg/mL streptomycin (Gibco, Carlsbad, CA, USA). The cells were maintained at 37 °C in a humidified incubator with 5% CO2. HEK 293T cells were cultured as previously described [2]. The methods for culturing patient-derived GBM cell line (G141119) were described previously [19].
Plasmids
p62 shRNAs (#1, #2) and control shRNA (SHC 002) were kindly gifted by Dr. Dong-Fan Xu (Department of Hematology, the Third Affiliated Hospital, Sun Yat-sen University, Guangzhou, China). T98G and G141119 cells were transfected with p62 shRNA and control vector by using Lipofectamine transfection reagent according to the manufacturer's protocol. Flag-p62 pcDNA3.1 and Flag-p62-ΔN or Flag-p62-ΔUBA were kindly provided by Dr. Huan-Sheng Wu (Key Laboratory of Animal Virology of Ministry of Agriculture, Zhejiang University, Hangzhou, China). HA-GSK-3β-pcDNA3 plasmid encoding C-terminal HA-tagged wild-type proteins was purchased from Addgene (Cambridge, MA, #14753). SNAIL shRNA plasmid was obtained from Santa Cruz Biotechnology (sc-38398SH; Santa Cruz, CA). Flag Snail WT was purchased from Addgene (Cambridge, MA, #16218).
Immunofluorescence assays
Cells were grown on glass coverslip in six-well plates and were treated accordingly. Subsequently, the cells were fixed with 4% paraformaldehyde in PBS for 30 min, and 0.2% of Triton solution was added to penetrate the cytomembrane. After overnight incubation with primary antibodies at 4 °C, fluorescence-dye conjugated secondary antibodies and DAPI staining kit were used to detect the binding of primary antibody and the nuclei of fixed cells, respectively, in the dark. Visualization of fixed cells was carried out using a confocal laser scanning microscope (Carl Zeiss, LSM 880). Quantitative analysis of punctate localization of p62, GSK-3β and Rpt1 was performed by ImageJ software (version 1.52i, NIH). Pearson's correlation coefficient (PCC) and Mander's overlap coefficient (MOC) were estimated.
Statistical analysis
All statistical analyses were performed using SPSS version 20.0 for Windows (International Business Machines Corp.). Spearman's rank correlation analysis was used to determine the correlation of protein expression levels in human clinical GBM specimens, while Kaplan-Meier survival analysis was conducted by log-rank test. In addition, Student's t-test and ANOVA were carried out, and the levels of significance were expressed as *P<0.05, **P<0.01 and NS (not significant).
Results
HMGB1 induced EMT in GBM cells
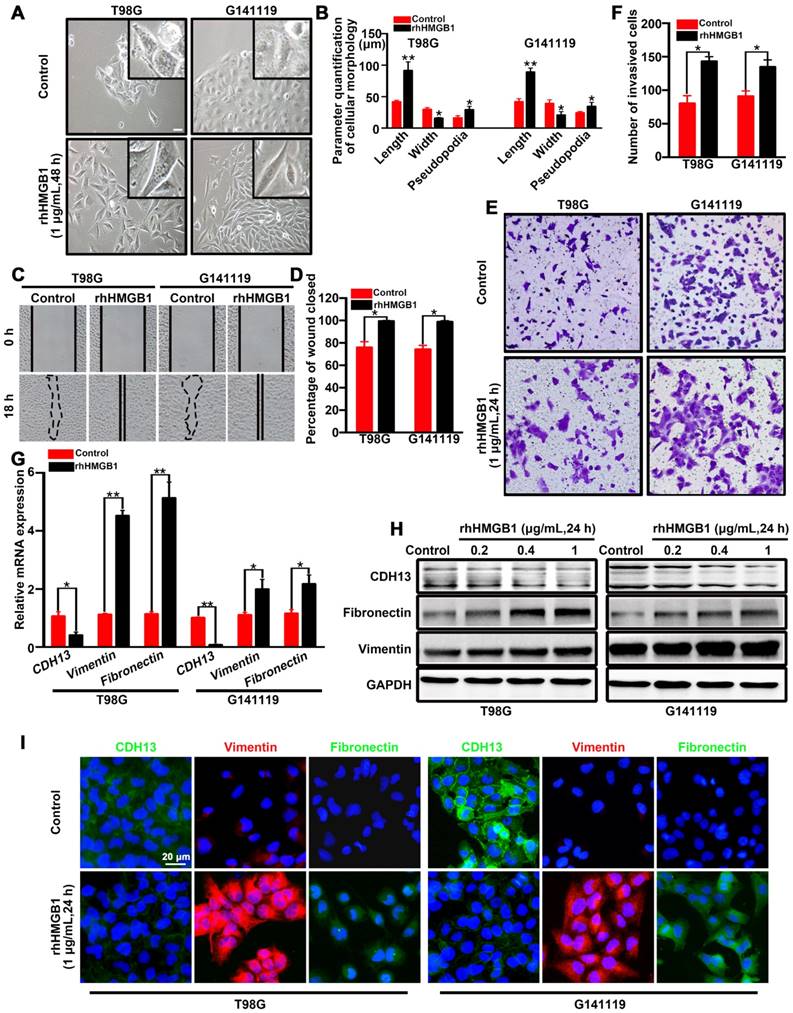
In order to identify the role of HMGB1 in promoting EMT, T98G cells (classical subtype) and patient-derived G141119 primary cells (classical subtype) were used to examine the effects of rhHMGB1 treatment [19]. Under inverted microscopy, we observed that the morphology of rhHMGB1-stimulated cells displayed a more stretched or elongated spindle-shape, which represents a typical feature of EMT [27]. Cell parameters were quantified using ImageJ software. The results showed that the lengths or cellular projections (pseudopodia) of cell bodies were greater after rhHMGB1 treatment compared to those cultured in normal medium (Figure 1A-B). Concomitantly, the migration and invasion capabilities of GBM cells were significantly enhanced following treatment with rhHMGB1 (Figure 1C-F). Moreover, the mRNA level of epithelial marker (CDH13) was decreased in T98G and G141119 cells treated with rhHMGB1, while the mRNA levels of mesenchymal markers (fibronectin and vimentin) were increased (Figure 1G). Since HMGB1 is a danger signal protein that highly expressed in GMB, it is necessary to verify the role of endogenous HMGB1 in EMT induction in addition to exogenous HMGB1 treatment. First, the culture supernatants of GBM cells were at extremely low amounts (Figure S1A). Besides, rhHMGB1 slightly increased the expression levels of endogenous HMGB1, but the difference was not statistically significant (Figure S1B). Moreover, neutralizing anti-HMGB1 antibody caused no significant changes of EMT in GBM cells, indicating that endogenous HMGB1 exerts no effect on EMT induction (Figure S1C-E). In addition, GBM cells were treated with different concentrations of rhHMGB1 (0.2-1 μg/mL) and the protein expression levels of CDH13, fibronectin and vimentin were examined. rhHMGB1 treatment enhanced the expression of mesenchymal markers (fibronectin and vimentin), whereas reduced the expression of epithelial marker (CDH13) in a concentration-dependent manner (Figure 1H and Figure S2A-B). Immunofluorescence assay revealed that rhHMGB1 promoted the expression of mesenchymal markers (fibronectin and vimentin), but suppressed the expression of CDH13 (Figure 1I). Furthermore, 10 mg/mL of anti-HMGB1 antibody was found to completely inhibit rhHMGB1-induced EMT in GBM cells (Figure S1F). These data suggested extracellular rhHMGB1 induces EMT in GBM cells.
HMGB1 induces EMT in GBM cells. (A) Inverted microscope images showing the morphology of GBM cells treated with or without rhHMGB1 (1 μg/mL) for 48 h. Scale bar: 40 μm. (B) The related parameters (length, width, and length of pseudopodia) were measured by NIH ImageJ software. (C-D) In vitro wound healing assay. The migration of T98G and G141119 cells in response to rhHMGB1 was determined by wound healing assay. Cells were monitored for 18 h to evaluate the rate of migration into the scratched area (magnification, × 40). (E-F) In vitro invasion assay. GBM cells were serum starved for 12 h, and then placed in the upper wells of transwell system. After 24 h of incubation, invasive cells were counted. qRT-PCR (G) and Western blot (H) analysis of EMT markers (CDH13, vimentin and fibronectin) in T98G and G141119 cells treated with rhHMGB1. (I) The expression changes of CDH13, vimentin and fibronectin in T98G and G141119 cells were assessed through immunofluorescence staining, as shown in the representative merged figures (scale bar: 20 μm). Unpaired t-test was used for the statistical analysis. **P < 0.01, *P < 0.05. The results are shown as mean ± SEM.
HMGB1 activates GSK-3β/Snail signaling pathway to induce EMT in GBM cells via GSK-3β degradation
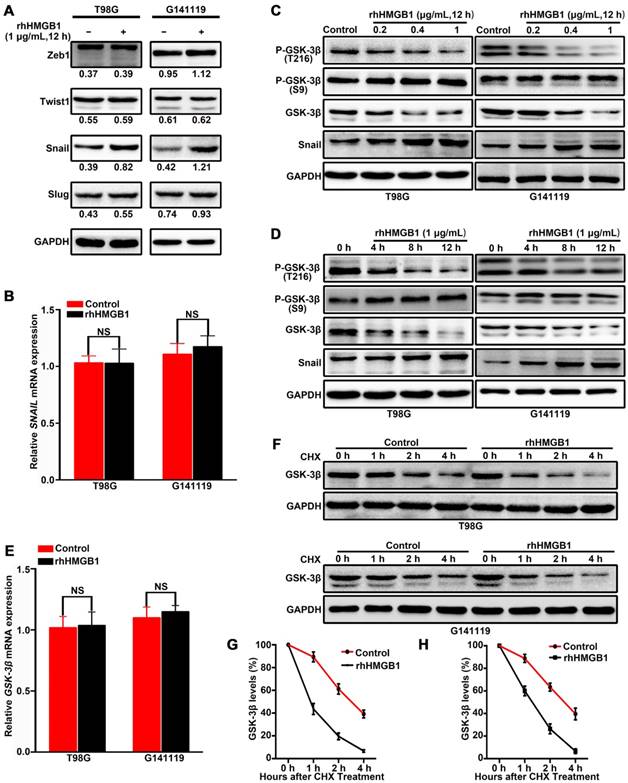
To elucidate the molecular mechanisms underlying HMGB1-induced EMT in GBM cells, the expression levels of EMT regulators (Snail, Slug, Zeb1 and Twist1) were analyzed in GBM cells treated with rhHMGB1. Compared with other regulators, only Snail protein was highly upregulated following rhHMGB1 treatment (Figure 2A). However, rhHMGB1 exerted no significant effect on the mRNA expression level of SNAIL (Figure 2B). To determine the influence of rhHMGB1 on Snail stability, Flag-tagged Snail vector was transfected and the Snail protein abundance was chased by cycloheximide treatment. It was noted that the half-life of Snail protein was significantly prolonged by rhHMGB1 treatment (Figure S3A-B). To further verify the role of Snail during the process of rhHMGB1-induced EMT, the overexpression of Snail was knockdown by small hairpin RNA (shRNA). rhHMGB1 specifically induced the EMT phenotype of cells compared to rhHMGB1-untreated cells, but not SNAIL-knockdown cells (Figure S4A-B). The invasion potential of SNAIL-knockdown cells was significantly decreased under rhHMGB1 treatment (Figure S4C-D). These results indicate that Snail plays a crucial role in rhHMGB1-induced EMT, which is post-translationally regulated by rhHMGB1. Snail is known to be phosphorylated by GSK-3β and subsequent proteasomal degradation [20], thus, the expression levels of total GSK-3β and p-GSK-3β (Ser 9 and Tyr 216) were examined. Western blot analysis revealed that rhHMGB1 significantly decreased the protein expression of p-GSK-3β Tyr 216 (active form of GSK-3β phosphorylation) in GBM cells, in a dose-dependent and time-dependent manner (Figure 2C-D). Similarly, total GSK-3β protein level was decreased, but the level of p-GSK-3β Ser 9 (inactive form of GSK-3β phosphorylation) remained unchanged (Figure 2C-D). Furthermore, the mRNA level of GSK-3β was measured by qRT-PCR after rhHMGB1 treatment, and we found that rhHMGB1 did not affect GSK-3β mRNA expression (Figure 2E). These results prompted us to examine whether GSK-3β is regulated by rhHMGB1 at the post-translational level. Cycloheximide pulse-chase experiments showed that endogenous GSK-3β level was significantly decreased in GBM cells treated with rhHMGB1 (Figure 2F-H). These findings suggested that GSK-3β/snail pathway may contribute to rhHMGB1-induced EMT in GBM cells.
HMGB1 activates GSK-3β/Snail signaling to induce EMT in GBM cells via GSK-3β degradation. Cells were treated with 1 μg/mL of rhHMGB1, total RNA and proteins were extracted within 12 h after treatment. The expression levels of Zeb1,Twist1, Snail and Slug proteins were detected by immunoblotting (A), while SNAIL mRNA expression level was measured by qRT-PCR (B). T98G and G141119 cells were treated with different doses of rhHMGB1 for 12 h (C) or 1 μg/mL of rhHMGB1 at different time points (D), followed by Western blot analysis. (E) GSK-3β mRNA expression level was measured by qRT-PCR. (F) Downregulation of GSK-3β protein expression in a time-dependent manner. After rhHMGB1 treatment, the time-course of GSK-3β protein abundance changes in T98G and G141119 cells were determined by immunoblot analysis. GAPDH was used as the loading control. Quantitative analysis of GSK-3β expression in T98G and G141119 cells were respectively shown in (G) and (H). **P < 0.01, *P < 0.05 by a two-tailed Student's t test. The results are shown as mean ± SEM.
p62 is required for HMGB1-stimulated EMT and invasion of GBM cells in vitro
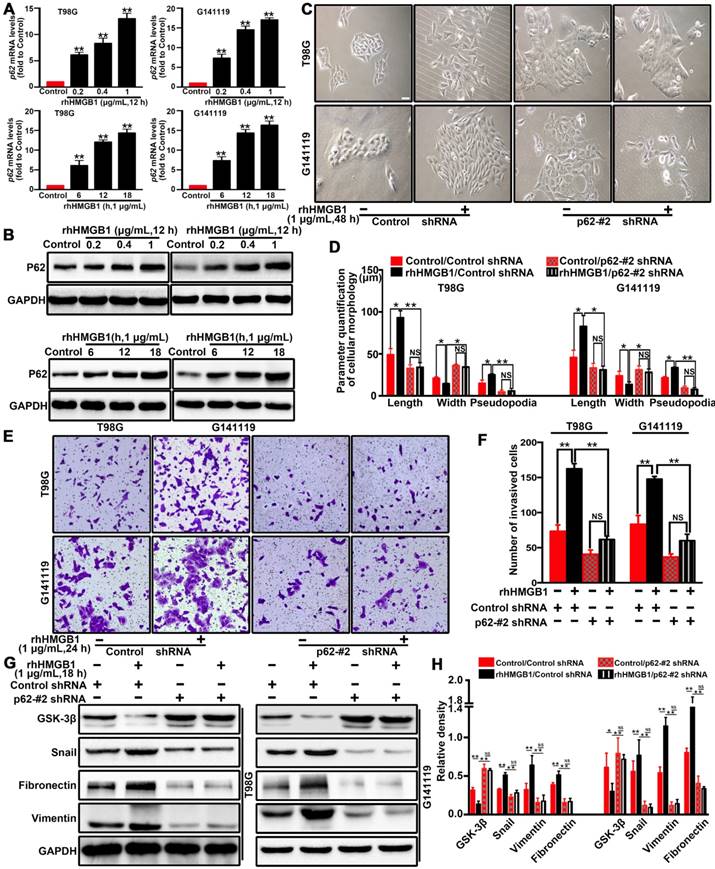
The scaffold protein p62 has recently been reported to be associated with the degradation of proteins [28, 29]. p62, as a multifunctional signal adapter protein, is involved in multiple signaling pathways, including EMT-related pathways [25, 30, 31]. Therefore, we investigated the role of p62 in HMGB1-induced EMT of GBM cells. RT-qPCR (Figure 3A) and Western blot (Figure 3B) results demonstrated that rhHMGB1 significantly increased the expression of p62 in GBM cells. To further investigate whether p62 can contribute to HMGB1-induced EMT, p62 expression was suppressed by two different shRNAs (shp62-#1 and shp62-#2). The effectiveness of knockdown was determined by Western blot and RT-qPCR analysis (Figure S5). Knockdown of p62 significantly attenuated the morphological changes (Figure 3C-D) and cellular invasion (Figure 3E-F) of GBM cells treated with rhHMGB1. In line with this, p62 knockdown cells prevented the induction of EMT marker in cells treated rhHMGB1 compared to those transfected with control shRNA (Figure 3G-H). These findings suggested that p62 is required for HMGB1-induced EMT of GBM cells in vitro.
p62 is required for HMGB1-stimulated GBM invasion and EMT both in vitro. The mRNA (A) and protein (B) expression levels of p62 in T98G and G141119 cells stimulated with rhHMGB1(**P<0.01 compared to Control). (C) Effects of rhHMGB1 stimulation and/or p62 knockdown with p62-#2 shRNA on the morphology of T98G and G141119 cells. Scale bar: 40 μm. (D) The related cellular parameters (length, width, and length of pseudopodia) were measured by NIH ImageJ software. (E-F) Representative images of cell invasion for rhHMGB1 stimulation and/or p62 knockdown in T98G and G141119 cells (magnification, × 40). (G-H) Expression levels of GSK-3β, Snail, fibronectin and vimentin in the indicated GBM cell lines. Statistical analysis used one-way ANOVA.**P<0.01, *P<0.05. The results are shown as mean ± SEM.
p62 is required for HMGB1-stimulated EMT and invasion of GBM cells in vivo
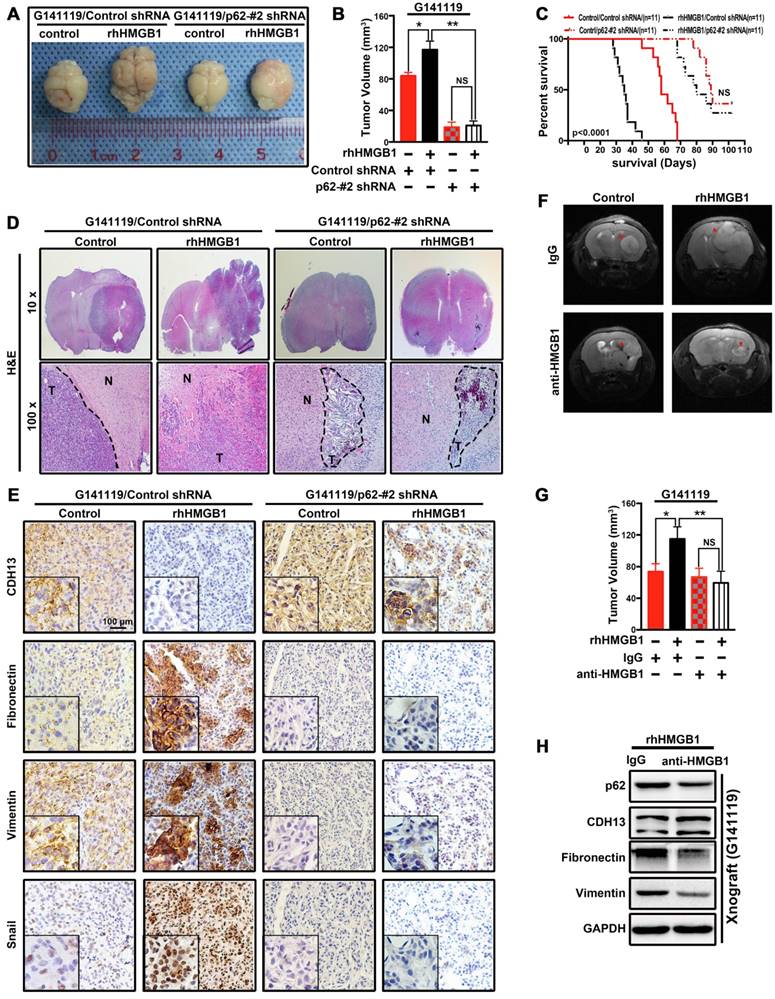
Since p62 is involved in cell proliferation and apoptosis, MTT and clonogenic assays demonstrated p62 increased colony formation and promoted cell growth (Figure S6). Next, we examined whether p62 downregulation can affect HMGB1-induced tumor cell invasion and growth in vivo. Transfected cells were stimulated with rhHMGB1 for 48 h and then implanted into the brain of nude mice. Tumors in the untreated group were weakly proliferative and expanded with well-defined borders. Following rhHMGB1 treatment, knockdown of p62 markedly suppressed rhHMGB1-stimulated GBM tumor proliferation (Figure 4A-B) and led to an increased survival rate (Figure 4C) in G141119/rhHMGB1/ p62-#2 shRNA group. In addition, the tumor boundaries became irregular, and strong invasiveness was observed in G141119/rhHMGB1/Control shRNA group (Figure 4D). Immunohistochemical analysis revealed that rhHMGB1 increased the levels of fibronectin, vimentin and Snail, while decreased CDH13 level in xenograft tumors (Figure 4E). Furthermore, by pre-treating G141119 cells with either anti-HMGB1 Ab or control IgG before implanting them into mice, we also demonstrated HMGB1-Ab diminish the extracellular function of HMGB1 in vivo (Figure 4F-H). These findings further indicated HMGB1 induce EMT and invasion of GBM cells in vivo and p62 is also required for this process.
p62 is required for HMGB1-stimulated EMT and invasion in GBM cells in vivo. (A) Photographs of the excised mouse brain bearing G141119 glioma xenografts. (B) The tumor volume was statistically analyzed and presented as a histogram. (C) Kaplan-Meier survival curves of mice bearing G141119/Control/Control shRNA,G141119/rhHMGB1/Control shRNA,G141119/control/p62-#2 shRNA, G141119/rhHMGB1/p62-#2 shRNA tumor xenografts(p < 0.0001, NS, no statistical significance by the Mantel-Cox test. n = number of animals per cohort). (D) Representative hematoxylin and eosin (H&E)-stained images of intracranial tumors derived from G141119/control/Control shRNA, G141119/rhHMGB1/Control shRNA, G141119/control/p62-#2 shRNA and G141119/rhHMGB1/p62-#2 shRNA groups. (N:normal brain; T:tumor) (E) Immunohistochemical analysis of CDH13, vimentin, fibronectin and Snail in orthotopic tumors. (F) Coronal T2-weighted MRI of tumors acquired from G141119 cells (red arrow) in brain samples on day 28 after implantation. (G) The tumor volume was statistically analyzed by MRI images. (H) Western blot analysis of CDH13, vimentin, fibronectin and p62 in orthotopic tumors. **P < 0.01, *P < 0.05 , NS, no statistical significance(unless otherwise stated, a one-way ANOVA was applied). The results are shown as means ± SEM.
HMGB1 induced p62 overexpression in human GBM cells via TLR4-p38-Nrf2 activation
A previous study has reported that lipopolysaccharides can activate p38-Nrf2 axis through TLR4 signaling to upregulate the expression of p62 [32] and we further confirmed in GBM cells (Figure S7). HMGB1 is also an endogenous TLR4 agonist [33, 34]. Our data indicated that rhHMGB1 treatment increased p62 expression at both mRNA and protein levels (Figure 3A and B). Therefore, we investigated whether HMGB1-induced p62 expression is mediated via TLR4-p38-Nrf2 activation. In addition, nuclear translocation of Nrf2 is an important mechanism for the activation of p62 [35]. To assess the role of Nrf2 in rhHMGB1-stimulated p62 expression, T98G cells were treated with rhHMGB1 at different times and concentrations. As a result, rhHMGB1 significantly increased the mRNA and protein levels of Nrf2 in a time- and dose-dependent manner (Figure 5A and B). Cytosolic and nuclear protein were prepared and analyzed for Nrf2 expression by Western blotting. Figure 5C demonstrated that Nrf2 was expressed in the cytosol of untreated T98G cells, but the expression was decreased over time. In contrast, a low expression of Nrf2 was found in the nucleus of untreated cells, but gradually increased following rhHMGB1 treatment. Furthermore, siRNA of Nrf2 and TLR4 suppressed rhHMGB1-stimulated p62 overexpression at both mRNA and protein levels (Figure 5D). These results indicated the role of Nrf2 in rhHMGB1-induced p62 expression in GBM cells.
HMGB1 induces p62 expression in human GBM cells via TLR4-p38-Nrf2 activation. The mRNA and protein levels of p62 in T98G cells treated with 1 μg/mL of rhHMGB1 at the indicated time points (A) or with different concentrations of rhHMGB1 at 240 min (B). (C) Western blot analysis of cytoplasmic and nuclear fractions from T98G cells. Nuclear segregation was assayed by total Lamin B, while cytoplasmic segregation was assayed by GAPDH. (D) Cells were transfected with siRNAs against indicated genes followed by stimulation with 1 μg/mL of rhHMGB1 for 18 h (**p < 0.01, NS, no statistical significance by a one-way ANOVA). (E) Phosphorylation levels of Akt,p38 MAPK, ERK1/2 and JNK1/2 were measured by Western blotting using phosphorylation-specific antibodies. (F) T98G cells were incubated with or without the indicated inhibitors followed by rhHMGB1 treatment. The mRNA expression levels of p62 are presented. Asterisks indicate statistical significance compared with control (**P < 0.01, NS, no statistical significance). The results are shown as means ± SEM.
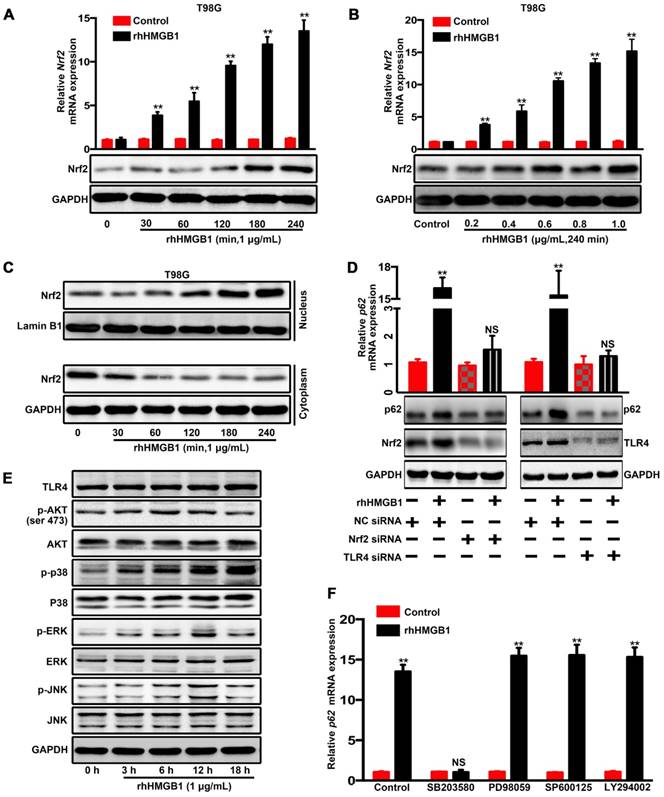
Next, we sought to elucidate the signaling pathways involved in rhHMGB1-activated Nrf2 expression. Western blotting was used to measure the phosphorylation kinetics of signaling kinase mediators, including Akt, p38, ERK1/2 and JNK1/2 (Figure 5E). Subsequent densitometrical analysis indicated that the phosphorylation of Akt, JNK and ERK was transiently upregulated, peaked at 6-12 h, and then gradually declined to the basal levels at 18 h after rhHMGB1 treatment. The phosphorylation level of p38 was steadily increased over 18 h, and possibly higher thereafter (Figure S8). Given these differential signaling results, we sought to further elucidate the signaling pathway responsible for rhHMGB1-stimulated p62 expression. Treatment with SB203580, a p38 inhibitor, substantially reduced HMGB1-induced p62 mRNA expression, while treatment with other inhibitors, such as SB202474 and JNK inhibitor II (JNK inh. II), exhibited little effect on p62 expression (Figure 4F and Figure S9). As reported, HMGB1 combined with TLR4 can be signaled via Wnt/β-catenin, TGF-β and NF-kB pathways, which ultimately induce EMT in different types of cells [36]. Western blotting was performed to determine the expression profiles of active forms of nodes in these pathways. Compared to untreated cells, the expression levels of these proteins were transiently increased by HMGB1, and then gradually declined to basal concentrations (Figure S10). Taken together, these results suggested the contribution of p38-Nrf2 axis to TLR4-mediated p62 overexpression.
p62 serves as a shuttling factor for the interaction of GSK-3β with proteasome
GSK-3β is post-transcriptionally downregulated through rhHMGB1 stimulation (Figure 2F-H), and possibly degraded via the proteasomal pathway [37]. In order to verify whether rhHMGB1-induced GSK-3β downregulation is mediated through the proteasome pathway, the cells were treated with rhHMGB1 and/or MG132, an ubiquitination-proteasome inhibitor. In the absence of MG132, rhHMGB1 accelerated the degradation of GSK-3β, while proteasome inhibition by MG132 reversed this (Figure 6A). In addition, we found that endogenous GSK-3β immunoprecipitated with Rpt1 proteasomal subunit, which was enhanced by rhHMGB1 treatment (Figure 6B). Further, rhHMGB1 promoted the colocalization between GSK-3β and Rpt1 in T98G cells (Figure 6C-D). These results indicated that GSK-3β can be degraded via the proteasome-mediated pathway.
p62 serves as a shuttling factor for the interaction of GSK-3β with proteasome. (A) rhHMGB1 triggers proteasome-dependent degradation of GSK-3β. T98G and G141119 cells were treated with proteasome inhibitor MG132 (1 μM) or lysosome inhibitor CQ (50 μM) in the absence or presence of rhHMGB1 (1 μg/mL) for 6 h, followed by Western blot analysis of GSK-3β protein expression. GAPDH was used as the loading control. (B) Immunoprecipitation of endogenous GSK-3β in T98G cells. IgG was used as a negative control. (C) rhHMGB1-treated T98G cells were stained with Rpt1 (green) and GSK-3β (red), and counterstained with DAPI to visualize nuclei (blue) by confocal microscopy. Scale bar, 10 μm. (D) Calculation of colocalization ratio (*P < 0.05 verse Control). (E) p62 overexpression enhanced the degradation of GSK-3β. Lysates from G141119 cells expressing Myc-p62 were generated after a specific period of cycloheximide treatment. The endogenous GSK-3β levels were detected by immunoblot analysis. (F) Co-localization of p62, Rpt1 and GSK-3β (appears as white in the merged image) in rhHMGB1-treated T98G cells was visualized by immunofluorescence staining using anti-Rpt1 (green), anti-GSK-3β (red) and anti-p62 (purple). Scale bar, 10 μm. (G) Knockdown of p62 expression attenuates GSK-3β-Rpt1 interaction. shRNA, short hairpin RNA. (H) p62 knockdown slows the degradation of GSK-3β after rhHMGB1 treatment. (I) HA-tagged GSK-3β along with Flag-tagged WT p62, N-term or UBA were expressed in HEK 293T cells followed by rhHMGB1 stimulation for 48 h. In order to examine the association of GSK-3β with p62 and Rpt1, cells were lysed in Triton lysis buffer and immunoprecipitated with anti-GSK-3β antibody, followed by Western blotting using anti-HA, anti-Flag and anti-Rpt1 antibodies. (J) Likewise, the same transfected HEK 293T lysates were immunoprecipitated with anti-Flag and Western blotting was performed using anti-HA, anti-Flag and anti-Rpt1 antibodies.
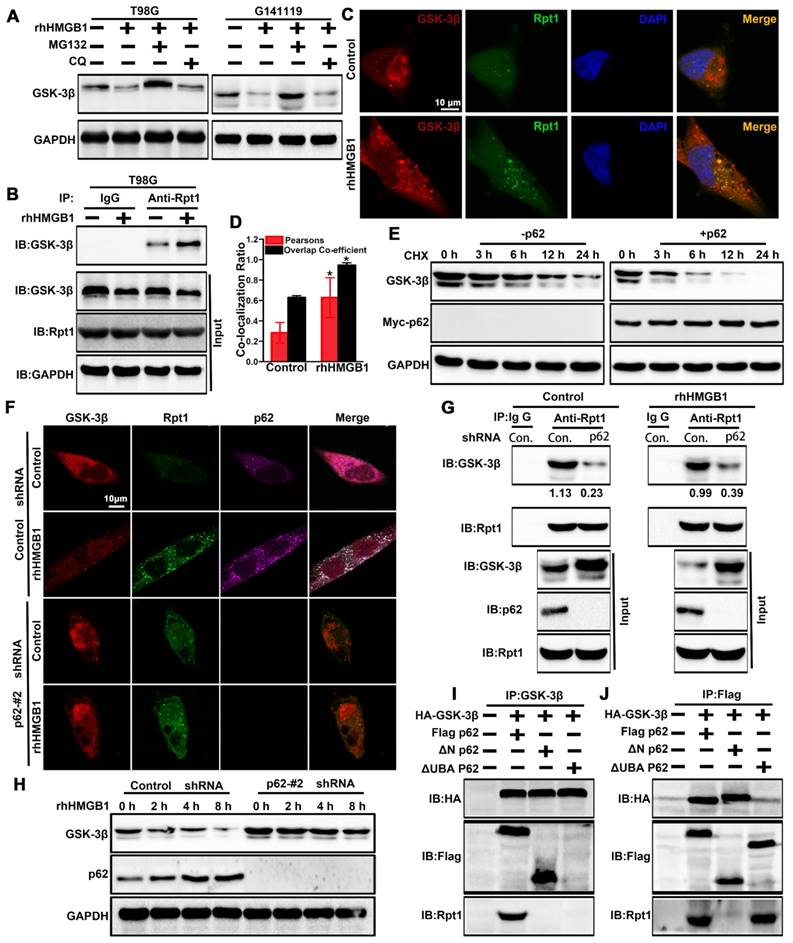
p62 is a UBA/UBL domain protein that interacts directly with the proteasome to facilitate the degradation of target proteins. Since Rpt1 has been implicated as a docking site for interaction with some putative shuttling factors, such as p62, we sought to determine whether p62 can mediate the interaction between GSK-3β and Rpt1. Cycloheximide pulse-chase experiments showed that the endogenous level of GSK-3β was degraded slightly in G141119 cells, and its level was reduced more significantly after transfected with Myc-p62 (Figure 6E). Co-localization of p62 and Rpt1 with GSK-3β was observed in the T98G cells after rhHMGB1 treatment, and p62 knockdown apparently diminished this co-localization (Control shRNA+rhHMGB1 group: p62/GSK-3β:PCC=0.575359, MOC=0.962117; p62/Rpt1:PCC=0.577902, MOC=0.941687; Figure 6F). The endogenous interaction between GSK-3β and Rpt1 might be facilitated by p62, owing to the fact that p62 knockdown apparently attenuated this interaction (Figure 6G). Furthermore, p62 knockdown slowed rhHMGB1-induced GSK-3β degradation (Figure 6H). Based on these findings, we seted out to identify the p62 domain involved in mediating the interaction between GSK-3β and Rpt1. HEK 293T cells were co-transfected with HA-GSK-3β, Flag-p62, and either N-terminal or UBA mutants of p62. Immunoprecipitation analysis revealed that the UBA domain was required for the interaction of GSK-3β with Rpt1 (Figure 6I-J). Taken together, our findings demonstrated that GSK-3β is proteasomally degraded in a p62-dependent manner.
p62 expression inversely correlates with GSK-3β level in human GBM tissues, and their combination exhibits increased prognostic accuracy for GBM
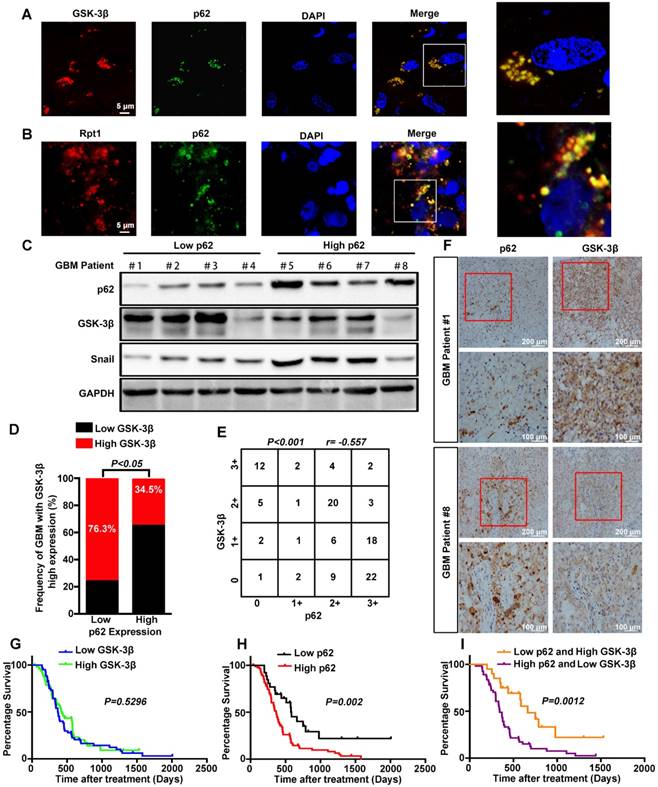
To determine the clinical relevance of both in vitro and in vitro findings, the expression levels of p62 and GSK-3β were evaluated in 110 human primary GBM specimens. First, co-localization of p62 and GSK-3β or Rpt1 was observed mainly in the cytoplasm of GBM biopsies (Figure 7A-B). Next, the expression level of GSK-3β was assessed in p62low and p62high GBM tissues. Immunoblotting analysis revealed a strong reverse correlation between p62 expression and GSK-3β/Snail signaling in tumor tissues (Figure 7C). The results of immunohistochemical staining demonstrated that the percentage of GBM patients (34.5%) with elevated GSK-3β levels in p62high group was lower than that (76.3%) in p62low group (Figure 7D), suggesting an inverse association between p62 and GSK-3β expression levels (Figure 7E-F). Furthermore, we investigated whether p62, GSK-3β and their co-expression are associated with patient survival. There was no significant correlation between GSK-3β expression and GBM survival, although high GSK-3β expression was found to improve the overall survival of GBM patients (Figure 7G). In contrast, the patients with high p62 expression had shorter survival than those with low p62 expression (p = 0.002) (Figure 7H), the combination of high expression of p62 and low expression of GSK-3β may be a powerful predictor of poor prognosis in GBM patients (p = 0.0012) (Figure 7I).
Correlation between the expression levels of p62 and GSK-3β in human GBM specimens. Frozen sections derived from GBM patients were subjected to immunofluorescence analysis for the detection of p62 and GSK-3β (A) or Rpt1 (B) co-localization. (C) Western blot analysis was used to determine the expression levels of p62 and GSK-3β/Snail in p62low or p62high GBM frozen biopsies samples (n=8), with GAPDH as the loading control. (D and E) Correlation between the expression levels of p62 and GSK-3β was assessed in GBM tumor tissues (n=110)(Chi-squared test and Spearman rank correlation test was used respectively). (F) Representative image of the immunostaining of p62 and GSK-3β was denoted by two patient samples. Kaplan-Meier analyses for overall survival were performed on GSK-3β (G) and p62 (H) expression levels, as well as their combination (I);Log-rank test. The results are shown as means ± SEM. **P < 0.01, *P < 0.05.
Discussion
Over the past decade, most GBM research has focused on the cancer cells themselves, without taking the unique but complex tumor microenvironment into account. HMGB1, an autocrine/paracrine stimulus in the tumor microenvironment, plays a prominent role in driving cell invasion [8, 9]. In the present study, we investigated the underlying mechanisms of HMGB1-induced EMT. First, rhHMGB1 induced EMT via enhancing GSK-3β/Snail signaling. Moreover, rhHMGB1-induced p62 expression was dependent on p38-Nrf2 activation. Furthermore, p62 was critically involved in HMGB1-induced EMT by shuttling GSK-3β for proteasomal degradation.
For the invasive-predominant GBM, local invasion can result in a less-defined tumor margin, and thus reduces the success rate of surgical eradication or radiation [38]. Increasing evidence has demonstrated that EMT plays a crucial role in the invasion of GBM cells [39]. Here, we investigated the mRNA/protein expression levels of EMT-related markers and the acquisition of EMT-like phenotype in GBM cells after rhHMGB1 treatment. Compared with Twist1, Slug, and Zeb1, only Snail protein was highly upregulated following rhHMGB1 treatment (Figure 2A). However, rhHMGB1 exerted no significant effect on the mRNA level of SNAIL, suggesting that Snail is regulated post-translationally. Besides, GSK-3β can phosphorylate Snail and lead to ubiquitin-dependent degradation, which is crucial for Snail stabilization [20]. In this study, treatment with rhHMGB1 resulted in a decrease of GSK-3β in GBM cells, which prolonged the Snail protein half-life. Accumulation of Snail following HMGB1 treatment could sustain up without any observable expression of SNAIL mRNA. These findings indicate that that GSK-3β/Snail signaling may play a pivotal role in HMGB1-stimulated EMT in GBM cells.
Signaling adapter p62 (also known as SQSTM1) plays an important role in the degradation of ubiquitinated protein, by binding to the proteasome and ubiquitinated substrates via UBL domain and UBA domain, respectively [23, 24]. Recent studies have implied an oncogenic role of p62 in different types of cancer [40-43], including GBM [31]. Similarly, our findings indicated the oncogenic role of p62 in GBM samples, in which p62 expression inversely correlated with GSK-3β level in human GBM tissues, and the combination of these two markers demonstrated a more accurate prognostic value for GBM. Previous studies have reported that p62 can be transcriptionnally upregulated during inflammation [32, 44]. Likewise, our study revealed that HMGB1-induced p62 expression in human GBM cells was mediated through TLR4-p38-Nrf2 inflammatory pathway, while the activation of other pathways such as Wnt/β-catenin and NF-kB pathways do not seem implicated in HMGB1-mediated p62 upregulation and invasion of GBM cells. Besides, p62 can bind to the oncogenic transcription factor TWIST1 and inhibit degradation of TWIST1 [25]. In addition to the stabilizing effect, p62 has been reported to target β-catenin for autophagic clearance in autolysosomes upon autophagy induction [45]. In the present study, we found that p62 targeted GSK-3β degradation to promote EMT of GBM cells following rhHMGB1 treatment.
GSK-3β is a serine-threonine kinase with over 50 known protein substrates, including Snail [21]. GSK-3β plays a central role in multiple signaling pathways, such as the Wnt/β-catenin, Hedgehog, Notch and insulin signaling pathways [46-50]. GSK-3β activity depends on the balance of phosphorylation levels between Tyr216 activation site and Ser9 inhibition site. Indeed, the level of p-GSK-3β (Ser9), but not total GSK-3β, is significantly upregulated in glioma tissues compared to normal tissues. Forced expression of GSK-3β in glioma cells significantly inhibits both tumor growth and angiogenesis in vivo [51]. Here, we demonstrated that rhHMGB1 decreased the levels of p-GSK-3β (T216) and total GSK-3β. Previous reports have shown that the GSK-3β protein is mostly degraded by proteasome in certain types of cells, but the underlying mechanisms are far from being elucidated. FBXO17 has been identified as an SCF E3 ligase, which can target GSK-3β for proteasomal degradation [52]. In addition, REGγ-proteasome can control the steady state level of GSK-3β protein [53]. Our experimental data revealed that GSK-3β could interact with proteasome subunit Rpt1, and p62 promoted GSK-3β-Rpt1 interaction and subsequent GSK-3β degradation following rhHMGB1 treatment.
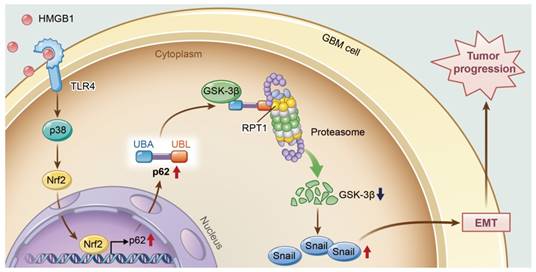
In conclusion, this study provides new insights into the role of p62 in HMGB1-induced EMT of GBM cells (Figure 8). Our findings indicate that HMGB1 induces EMT of GBM cells through upregulation of p62, which leads to the stabilization of Snail via shuttling GSK-3β for proteasome degradation. Therefore, targeting p62 may represent a promising strategy for treating human GBM in the future.
A model illustrating the role of p62 during HMGB1-induced EMT in GBM cells. HMGB1 induces mesenchymal transition of GBM via modulation of p62 and GSK-3β/Snail signaling. rhHMGB1 promotes p62 expression through TLR4-p38-Nrf2 activation, and p62 enhances GSK-3β degradation via the proteasome.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 81772656), Natural Science Foundation of Guangdong Province (Grant no. 2016A030313563), and the National Key Clinical Specialist Construction Program of China.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA. et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. New Engl J. Med. 2014;370:699-708
2. Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L. et al. HMGB1 in health and disease. Mol Aspects Med. 2014;40:1-116
3. Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K. et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. Plos Med. 2009;6:e10
4. Pasi F, Paolini A, Nano R, Di Liberto R, Capelli E. Effects of single or combined treatments with radiation and chemotherapy on survival and danger signals expression in glioblastoma cell lines. Biomed Res. Int. 2014;2014:453-97
5. Candolfi M, Yagiz K, Foulad D, Alzadeh GE, Tesarfreund M, Muhammad AK. et al. Release of HMGB1 in response to proapoptotic glioma killing strategies: efficacy and neurotoxicity. Clin Cancer Res. 2009;15:4401-14
6. Yang H, Wang H, Chavan SS, Andersson U. High Mobility Group Box Protein 1 (HMGB1): The Prototypical Endogenous Danger Molecule. Mol Med. 2015;21:S6-12
7. Wang X, Zhou S, Fu X, Zhang Y, Liang B, Shou J. et al. Clinical and prognostic significance of high-mobility group box-1 in human gliomas. Exp. Ther. Med. 2015;9:513-8
8. Deng S, Zhu S, Qiao Y, Liu Y, Chen W, Zhao G. et al. Recent advances in the role of toll-like receptors and TLR agonists in immunotherapy for human glioma. Protein Cell. 2014;5:899-911
9. Vinnakota K, Hu F, Ku M, Georgieva PB, Szulzewsky F, Pohlmann A. et al. Toll-like receptor 2 mediates microglia/brain macrophage MT1-MMP expression and glioma expansion. Neuro-Oncology. 2013;15:1457-68
10. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014;7:e8
11. Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY. et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318-68
12. Phillips HS, Kharbanda S, Chen RH, Forrest WF, Soriano RH, Wu TD. et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157-73
13. Tso C, Shintaku P, Chen J, Liu Q, Liu J, Chen Z. et al. Primary glioblastomas express mesenchymal stem-like properties. Mol Cancer Res. 2006;4:607-19
14. Li LC, Li DL, Xu L, Mo XT, Cui WH, Zhao P. et al. High-Mobility Group Box 1 Mediates Epithelial-to-Mesenchymal Transition in Pulmonary Fibrosis Involving Transforming Growth Factor-beta1/Smad2/3 Signaling. J. Pharmacol Exp Ther. 2015;354:302-9
15. Bassi R, Giussani P, Anelli V, Colleoni T, Pedrazzi M, Patrone M. et al. HMGB1 as an autocrine stimulus in human T98G glioblastoma cells: role in cell growth and migration. J Neurooncol. 2008;87:23-33
16. Chen YC, Statt S, Wu R, Chang HT, Liao JW, Wang CN. et al. High mobility group box 1-induced epithelial mesenchymal transition in human airway epithelial cells. Sci Rep. 2016;6:18815
17. Han SP, Kim JH, Han ME, Sim HE, Kim KS, Yoon S. et al. SNAI1 is involved in the proliferation and migration of glioblastoma cells. Cell Mol Neurobiol. 2011;31:489-96
18. Ma YS, Wu ZJ, Bai RZ, Dong H, Xie BX, Wu XH. et al. DRR1 promotes glioblastoma cell invasion and epithelial-mesenchymal transition via regulating AKT activation. Cancer Lett. 2018;423:86-94
19. Mahabir R, Tanino M, Elmansuri A, Wang L, Kimura T, Itoh T. et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol. 2014;16:671-85
20. Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH. et al. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8:1398-406
21. Lee DG, Kim HS, Lee YS, Kim S, Cha SY, Ota I. et al. Helicobacter pylori CagA promotes Snail-mediated epithelial-mesenchymal transition by reducing GSK-3 activity. Nat. Commun. 2014;5:4423
22. Fei Y, Xiong Y, Shen X, Zhao Y, Zhu Y, Wang L. et al. Cathepsin L promotes ionizing radiation-induced U251 glioma cell migration and invasion through regulating the GSK-3beta/CUX1 pathway. Cell Signal. 2018;44:62-71
23. Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P. et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011;71:5976-86
24. Cohen-Kaplan V, Livneh I, Avni N, Fabre B, Ziv T, Kwon YT. et al. p62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc Natl Acad Sci U S A. 2016;113:E7490-9
25. Qiang L, Zhao B, Ming M, Wang N, He TC, Hwang S. et al. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci U S A. 2014;111:9241-6
26. Zhao M, Xu H, Zhang B, Hong B, Yan W, Zhang J. Impact of nuclear factor erythroid-derived 2-like 2 and p62/sequestosome expression on prognosis of patients with gliomas. Hum Pathol. 2015;46:843-9
27. Lu Y, Xiao L, Liu Y, Wang H, Li H, Zhou Q. et al. MIR517C inhibits autophagy and the epithelial-to-mesenchymal (-like) transition phenotype in human glioblastoma through KPNA2-dependent disruption of TP53 nuclear translocation. Autophagy. 2015;11:2213-32
28. Geetha T, Seibenhener ML, Chen L, Madura K, Wooten MW. p62 serves as a shuttling factor for TrkA interaction with the proteasome. Biochem Biophys Res Commun. 2008;374:33-7
29. Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055-68
30. Bertrand M, Petit V, Jain A, Amsellem R, Johansen T, Larue L. et al. SQSTM1/p62 regulates the expression of junctional proteins through epithelial-mesenchymal transition factors. Cell Cycle. 2015;14:364-74
31. Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A. et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene. 2013;32:699-712
32. Fujita K, Maeda D, Xiao Q, Srinivasula SM. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A. 2011;108:1427-32
33. Di Candia L, Gomez E, Venereau E, Chachi L, Kaur D, Bianchi ME. et al. HMGB1 is upregulated in the airways in asthma and potentiates airway smooth muscle contraction via TLR4. J Allergy Clin Immunol. 2017;140:584-7
34. Zong M, Bruton JD, Grundtman C, Yang H, Li JH, Alexanderson H. et al. TLR4 as receptor for HMGB1 induced muscle dysfunction in myositis. Ann Rheum Dis. 2013;72:1390-9
35. Ngo H, Kim DH, Cha YN, Na HK, Surh YJ. Nrf2 Mutagenic Activation Drives Hepatocarcinogenesis. Cancer Res. 2017;77:4797-808
36. van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis. 2008;11:91-9
37. Richet E, Pooler AM, Rodriguez T, Novoselov SS, Schmidtke G, Groettrup M. et al. NUB1 modulation of GSK3beta reduces tau aggregation. Hum Mol Genet. 2012;21:5254-67
38. Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, Sarkisian MR. et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. Embo Mol. Med. 2013;5:1196-212
39. Kievit FM, Florczyk SJ, Leung MC, Wang K, Wu JD, Silber JR. et al. Proliferation and enrichment of CD133(+) glioblastoma cancer stem cells on 3D chitosan-alginate scaffolds. Biomaterials. 2014;35:9137-43
40. Kitamura H, Torigoe T, Asanuma H, Hisasue SI, Suzuki K, Tsukamoto T. et al. Cytosolic overexpression of p62 sequestosome 1 in neoplastic prostate tissue. Histopathology. 2006;48:157-61
41. Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z. et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105-20
42. Inoue D, Suzuki T, Mitsuishi Y, Miki Y, Suzuki S, Sugawara S. et al. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 2012;103:760-6
43. Li L, Shen C, Nakamura E, Ando K, Signoretti S, Beroukhim R. et al. SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell. 2013;24:738-50
44. Fujita K, Srinivasula SM. TLR4-mediated autophagy in macrophages is a p62-dependent type of selective autophagy of aggresome-like induced structures (ALIS). Autophagy. 2011;7:552-4
45. Petherick KJ, Williams AC, Lane JD, Ordonez-Moran P, Huelsken J, Collard TJ. et al. Autolysosomal beta-catenin degradation regulates Wnt-autophagy-p62 crosstalk. Embo J. 2013;32:1903-16
46. Farago M, Dominguez I, Landesman-Bollag E, Xu X, Rosner A, Cardiff RD. et al. Kinase-inactive glycogen synthase kinase 3beta promotes Wnt signaling and mammary tumorigenesis. Cancer Res. 2005;65:5792-801
47. Voskas D, Ling LS, Woodgett JR. Does GSK-3 provide a shortcut for PI3K activation of Wnt signalling? F1000 Biol Rep. 2010;2:82
48. Denham M, Bye C, Leung J, Conley BJ, Thompson LH, Dottori M. Glycogen synthase kinase 3beta and activin/nodal inhibition in human embryonic stem cells induces a pre-neuroepithelial state that is required for specification to a floor plate cell lineage. Stem Cells. 2012;30:2400-11
49. Kwon C, Cheng P, King IN, Andersen P, Shenje L, Nigam V. et al. Notch post-translationally regulates beta-catenin protein in stem and progenitor cells. Nat Cell Biol. 2011;13:1244-51
50. Patel S, Doble BW, MacAulay K, Sinclair EM, Drucker DJ, Woodgett JR. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol. 2008;28:6314-28
51. Zhao P, Li Q, Shi Z, Li C, Wang L, Liu X. et al. GSK-3beta regulates tumor growth and angiogenesis in human glioma cells. Oncotarget. 2015;6:31901-15
52. Suber T, Wei J, Jacko AM, Nikolli I, Zhao Y, Zhao J. et al. SCF(FBXO17) E3 ligase modulates inflammation by regulating proteasomal degradation of glycogen synthase kinase-3beta in lung epithelia. J. Biol Chem. 2017;292:7452-61
53. Lv Y, Meng B, Dong H, Jing T, Wu N, Yang Y. et al. Upregulation of GSK3beta Contributes to Brain Disorders in Elderly REGgamma-knockout Mice. Neuropsychopharmacol. 2016;41:1340-9
Author contact
![]() Corresponding authors: Yun-tao Lu, Department of Neurosurgery, Nanfang Hospital, Southern Medical University, 1838 North Guangzhou Avenue, Guangzhou, China, 510515. Phone:+86-20-61641806; Fax:+86-20-61641806; Email: lllu2000edu.cn and Song-tao Qi, Department of Neurosurgery, Nanfang Hospital, Southern Medical University, 1838 North Guangzhou Avenue, Guangzhou, China, 510515. Phone:+86-20-61641806; Fax:+86-20-61641806; Email: sjwk_songtaocn
Corresponding authors: Yun-tao Lu, Department of Neurosurgery, Nanfang Hospital, Southern Medical University, 1838 North Guangzhou Avenue, Guangzhou, China, 510515. Phone:+86-20-61641806; Fax:+86-20-61641806; Email: lllu2000edu.cn and Song-tao Qi, Department of Neurosurgery, Nanfang Hospital, Southern Medical University, 1838 North Guangzhou Avenue, Guangzhou, China, 510515. Phone:+86-20-61641806; Fax:+86-20-61641806; Email: sjwk_songtaocn