Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(7):1952-1964. doi:10.7150/thno.30890 This issue Cite
Research Paper
Repurposing Ponatinib as a Potent Agent against KIT Mutant Melanomas
Yong Han1,2,3*, Ziyue Gu1,2,3*, Jing Wu1,2,3*, Xiaojuan Huang1,2,3, Rong Zhou1,2,3, Chaoji Shi2, Wenjie Tao1,2,3, Lizhen Wang4, Yanan Wang1, Guoyu Zhou1, Jiang Li4, Zhiyuan Zhang1,2,3 ![]() , Shuyang Sun1,2,3
, Shuyang Sun1,2,3 ![]()
1. Department of Oral and Maxillofacial-Head & Neck Oncology, Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200011, P.R. China.
2. National Clinical Research Center for Oral Diseases, Shanghai 200011, P.R. China.
3. Shanghai Key Laboratory of Stomatology & Shanghai Research Institute of Stomatology, Shanghai 200011, P.R. China.
4. Department of Oral Pathology, Ninth People's Hospital, Shanghai Jiao Tong University, School of Medicine, Shanghai 200011, P.R. China.
*These authors contributed equally to this work.
Received 2018-10-23; Accepted 2019-1-22; Published 2019-3-16
Abstract

Rationale: Mutations in KIT, a major cancer driver gene, are now considered as important drug targets for the treatment of melanomas arising from mucosal and acral tissues and from chronically sun-damaged sites. At present, imatinib is the only targeted drug for KIT-mutation-bearing melanomas that is recommended by the National Comprehensive Cancer Network (NCCN) Clinical Practice guidelines. Patients with KIT mutations, however, are either insensitive or rapidly progress to imatinib insensitivity, which restricts its clinical use. Thus, effective inhibitors of KIT-mutation-bearing melanomas are urgently needed.
Methods: A cohort of patient-derived tumor xenograft (PDX) models and corresponding PDX-derived cells (PDCs) from patients with melanomas harboring KIT mutations (KITV560D, KITK642E and KITD816V) were established, characterized, and then used to test the in vitro and, subsequently, in vivo inhibitory effects of a panel of known KIT inhibitors.
Results: Ponatinib was more potent than imatinib against cells bearing KIT mutations. In vivo drug efficacy evaluation experiments showed that ponatinib treatment caused much stronger inhibition of KIT-mutation-bearing melanomas than did imatinib. Mechanistically, molecular dynamics (MD) simulations revealed a plausible atomic-level explanation for the observation that ponatinib has a higher affinity for the KITD816V mutant protein than does imatinib.
Conclusions: Our study of KIT-mutation-and KITWT-bearing melanomas demonstrates that ponatinib is a far more potent inhibitor than is imatinib for KIT-mutation-bearing melanomas and thus underscores that ponatinib should be given priority consideration for the design of precision treatments for melanoma patients triaged to have KIT mutations. Moreover, our work provides a rationale for undertaking clinical trials to examine the repurposing of ponatinib, which is already approved for use in leukemia, for use in treating a large subset of melanoma patients.
Keywords: ponatinib, melanomas, KIT, patient-derived xenograft models
Introduction
Melanomas are tumors that arise from melanocytes located in a large variety of anatomic sites. The incidence of melanoma is rising steadily worldwide; it has increased nearly fourfold in the past 30 years [1]. Fortunately, as recent insights from oncobiological research have identified some of the driver genes for many cancers, melanoma patients now can be clinically stratified based on the molecular characteristics of their tumors [2], and some of these patients have benefited enormously from significant clinical breakthroughs in precision medicine (e.g., vemurafenib treatment has been successfully applied in patients harboring the BRAF V600E/V600K mutation) [3]. Certain melanomas, particularly those arising from mucosal and acral tissues and chronically sun-damaged sites, frequently feature oncogenic mutations in the KIT gene [4]. KIT mutations induce ligand-independent receptor activation, which constitutively activates a series of downstream pathways, including the MAPK and PI3K-mTOR signaling cascades [5]; KIT is thus now viewed as a critically vulnerable target for the treatment of melanomas.
Imatinib, a multikinase inhibitor of KIT, ABL, and PDGFR [6], is currently the only targeted drug for KIT-mutant melanomas recommended in the Clinical Practice Guidelines of the National Comprehensive Cancer Network. However, it is now well documented that the efficacy of imatinib in treating melanomas varies greatly depending on the presence of the particular type of KIT mutation sites in a given tumor, and the therapeutic effect of imatinib is apparently limited to KIT mutations located on exons 11 and 13 but not exon 17 [7]. Specifically, clinical trials have indicated that the average time to disease progression after imatinib treatment for patients bearing KIT-associated (mutation or copy number amplification) tumors is approximately 3-4 months [8-10].
Notably, several alternative KIT inhibitors have been evaluated for use in treating KIT-mutation-bearing melanomas that are resistant to imatinib, including sunitinib [11], dasatinib [12], and nilotinib [13]. However, with each of these three compounds, despite the fact that early disease control was achieved, their efficacy was still modest, and the cancers of most patients in the studies ultimately progressed. Given the limited clinical benefits of these drugs and considering that between 7.6% and 47% of melanomas harbor KIT mutations (varying by subtype) [14-16], new therapies for patients with KIT-mutant melanomas are critically needed.
Patient-derived tumor xenograft (PDX) models are powerful preclinical experimental tools that can retain the histological and genetic characteristic of patient tumors to a large extent [17]. PDX models can faithfully and characteristically respond to targeted therapies and are now popularly used in preclinical drug testing [18]. Indeed, many recent studies have underscored the suitability of using PDX models as precision medicine platforms for drug screening, biomarker discovery, and the development of other personalized medicine strategies [19, 20].
Here, in light of the poor efficacy of imatinib in treating KIT-mutation-bearing melanomas, and with the goal of identifying effective KIT inhibitors to provide alternative therapeutic options for KIT mutant melanoma patients, we developed a series of KIT-mutant PDX models from melanoma patients and used these models to evaluate the antitumor efficacy of a variety of known KIT inhibitor compounds. This examination of drug efficacy, first via screening in cells and then a successive in vivo validation in our genetically characterized PDX models, revealed that ponatinib, an FDA-approved chronic myeloid leukemia (CML) drug, was the most potent inhibitor of tumor growth of against KIT-mutation-bearing melanomas. Furthermore, immunoblotting with phosphorylation-specific antibodies confirmed that, both in vitro and in vivo, ponatinib-induced inhibition of KIT reduced the extent of phosphorylation of its downstream signaling targets. Finally, our molecular dynamics simulations suggested that the significantly improved inhibitory activity of ponatinib compared with imatinib can be attributed to its stronger binding affinity to mutated KIT proteins. Our study demonstrated that ponatinib is a potent agent against KIT-mutation-bearing melanomas and underscored how the repurposing of drugs can help develop effective precision treatments for a substantial proportion of melanomas that are insensitive to the drugs that are currently recommended by NCCN guidelines.
Results
Establishing PDX models of KIT-mutation-bearing melanomas and screening of kinase inhibitors for antitumor efficacy
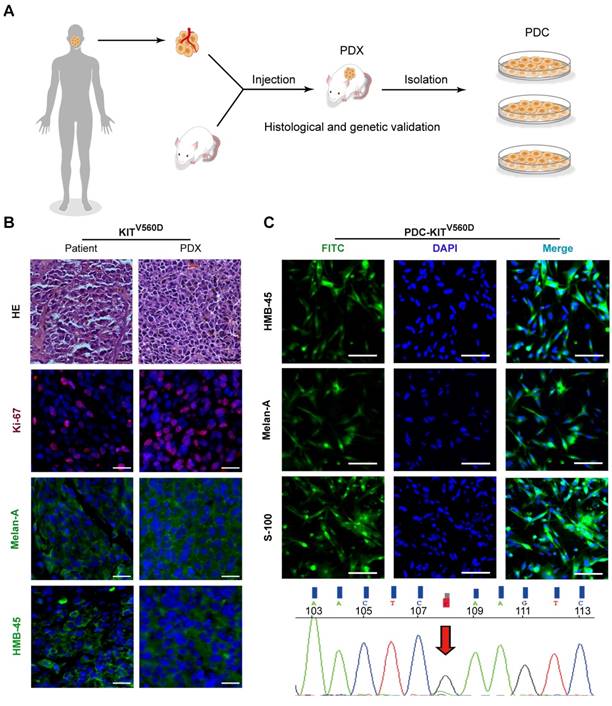
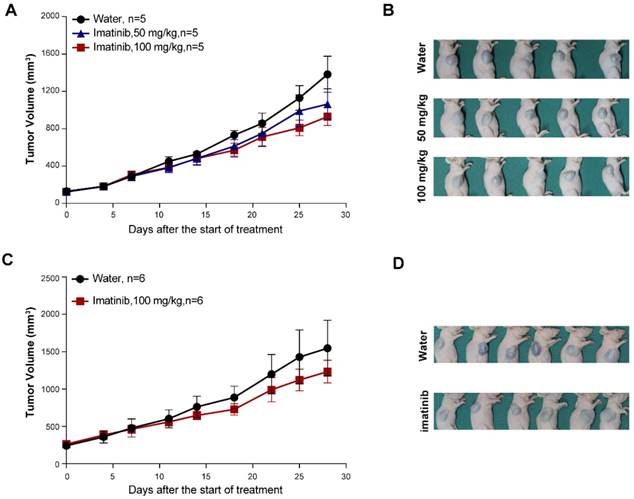
Seeking to establish an experimental platform to explore possible alternative treatments for imatinib-resistant melanomas, we identified mucosal melanoma patients, each with a different KIT mutation (KITV560D, KITK642E, KITD816V), as well as a non-KIT mutation melanoma patient, and generated PDX models from each patient (Figure 1A). The similarity of the established PDX models with the corresponding patient samples was validated using DNA sequencing, HE staining, and immunostaining against the melanoma markers HMB-45 and Melan-A and the cell growth marker Ki-67 (Figure 1B and Figure S1A and Figure S2A). Previous studies have reported that two of these KIT mutations (KITV560D, KITK642E) are highly sensitive to imatinib treatment and that KITD816V is completely insensitive to imatinib [8-10, 21]. Unexpectedly, when we treated the reportedly imatinib-sensitive KITV560D and KITK642E mutations in PDX models with imatinib, we did not find a significant difference in tumor growth inhibition (TGI) between the imatinib (100 mg/kg) and the control groups (Figure 2A-D, PDX-KITV560D, TGI = 35.81%, P > 0.05; PDX-KITK642E, TGI = 25.58%, P > 0.05). This surprising result is not in accord with previous results on the sensitivity of these mutations to imatinib and thus suggests that more research will be needed to develop more potent drugs for these patients.
Establishment and characterization of KITV560D PDX models. (A) Schematic of PDX and PDC establishment. (B) Representative hematoxylin and eosin (H&E) staining and immunofluorescence staining of patient tumors and corresponding PDX model tumors. HMB-45 and Melan-A (both green), Ki-67 (red) and DAPI (blue). The scale bar is 100 μm. (C) Representative immunofluorescence staining of patient-derived tumor cells from KITV560D mutant melanoma PDX models are shown in the upper panel, and the KIT mutation status of the corresponding PDX-derived cells is shown in the bottom panel. HMB-45, Melan-A and S-100 (both green), and DAPI (blue). The scale bar is 100 μm.
In vivo evaluation of imatinib efficacy in KITV560D and KITK642E mutant PDX models. (A-B) Inhibition efficacy of imatinib in PDX models with KITV560D mutation (n= 5 mice per group). Imatinib, 50 mg/kg, tumor growth inhibition, TGI= 25.04%, P>0.05; imatinib, 100 mg/kg, TGI= 35.81%, P >0.05, Student's t-test. (C-D) Inhibition efficacy of imatinib in PDX models with KITK642E mutation (n=6 mice per group). Imatinib, 100 mg/kg, TGI = 35.81%, Student's t-test, P > 0.05.
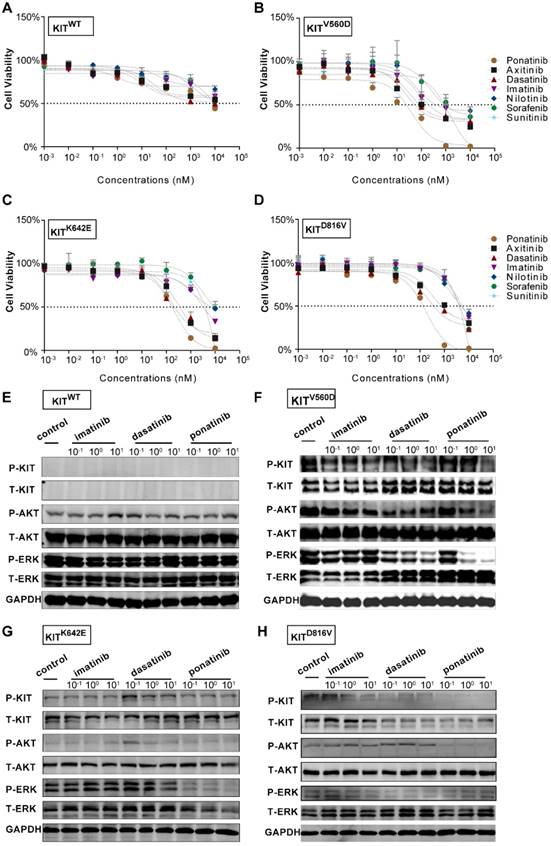
Next, we isolated primary tumor cells from the established KIT mutant PDX models for in vitro inhibition assays (Figure 1C and Figure S1B and Figure S2B); we conducted drug dose-response assays of these cells for a series of clinically evaluated kinase inhibitors with reported activity against KIT, including axitinib, dasatinib, nilotinib, ponatinib, sorafenib and sunitinib. The PDX-KITWT-derived cells had poor responses to all of these inhibitors (Figure 3A). These assays revealed that ponatinib, an FDA-approved drug for the treatment of CML, was more potent than was imatinib against the PDX-derived cell (PDC)-KITV560D, with an IC50 value of 39.05 nM versus 75.66 nM for imatinib (Figure 3B and Table S1). For the PDC-KITV560D, dasatinib and axitinib were also more effective than was imatinib, with IC50 values of 34.59 nM and 23.98 nM, respectively; nilotinib and sorafenib showed poor inhibition effects against PDC-KITV560D (Table S1). Similar results were observed in the PDC with the KITK642E mutation (Figure 3C and Table S1). For the PDC harboring the reportedly imatinib-resistant KITD816V mutation, imatinib induced very little inhibition (IC50 value of 4840 nM), but ponatinib exhibited the highest inhibitory effect (IC50 value of 174.3 nM, Figure 3D and Table S1).
In vitro drug efficacy evaluation using KITWT, KITV560D, KITK642E and KITD816V PDX-derived cells. (A-D) Dose-response curve of imatinib, dasatinib, axitinib, nilotinib, ponatinib, sorafenib, and sunitinib in 72-h proliferation assays with KITWT, KITV560D, KITK642E and KITD816V PDX-derived cells. (E-F) Immunoblot analysis of KIT signaling in KITWT, KITV560D, KITK642E and KITD816V PDX-derived cells treated with imatinib, dasatinib, or ponatinib for 2 hours.
We also compared the inhibitory effect of imatinib and ponatinib on serial KIT-dependent Ba/F3 cells, including KITWT, KITV560D, KITK642E and KITD816V mutations. Consistent with the results in PDCs, ponatinib demonstrated a higher potency for KITV560D, KITK642E and KITD816V in Ba/F3 cells than did imatinib (Figure S3 and Table S2). Notably, all the IC50s of ponatinib in these KIT-dependent Ba/F3 cells were lower than that of dasatinib, indicating ponatinib could also exerted greater inhibitory effect when compared with dasatinib (Figure S3 and Table S2).
To confirm that the observed decreases in cell viability resulted from specific inhibition of KIT signaling, we conducted immunoblotting assays to test the phosphorylation status of KIT and its downstream signaling targets in the PDCs after 2 hours of treatment with imatinib, ponatinib, or dasatinib. Consistent with the cell viability results, ponatinib and dasatinib reduced the phosphorylation levels of KIT, AKT, and ERK in a dose-dependent manner, whereas imatinib did not strongly affect the phosphorylation levels of these downstream KIT target proteins (Figure 3E-H).
In vivo evaluation of ponatinib efficacy
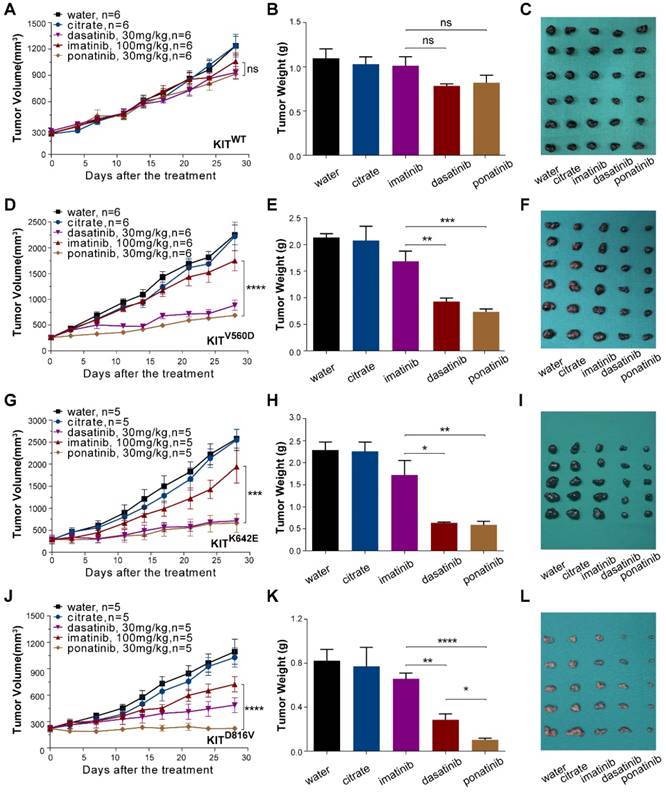
PDX models are now popular in vivo models for drug evaluation. We conducted a preclinical evaluation based on our genetically characterized melanoma PDX models to validate the drug efficacies observed in our in vitro studies. Single-agent treatments were performed with imatinib (100 mg/kg), dasatinib (30 mg/kg) and ponatinib (30 mg/kg) in PDX models. Treatment of PDX-KITWTmodel with imatinib, ponatinib, or dasatinib did not cause significant differences in tumor growth (Figure 4A-C and Table S3). For the PDX-KITV560D, ponatinib (TGI=78.33%) exhibited the greatest tumor growth inhibition effects, and dasatinib (TGI=68.65%) had better inhibition efficacy than did imatinib (TGI=25.25%, Figure 4D-F and Table S3). Similarly, in PDX-KITK642E, both ponatinib (TGI=83.66%) and dasatinib (TGI=81.38%) exhibited more effective inhibition than did imatinib (TGI=27.59%, Figure 4G-I and Table S3). Strikingly, for the PDX-KITD816V, ponatinib (TGI=99.95%) displayed almost complete suppression of tumor growth, while dasatinib (TGI=67.73%) and imatinib (TGI=42.67%) showed much more modest effects (Figure 4J-L and Table S3). In addition, the drug toxicities of these inhibitors were all well tolerated during the preclinical trial (Figure S4).
In vivo drug efficacy evaluation in KITWT, KITV560D, KITK642E and KITD816V mutant PDX models. We administered imatinib (100 mg/kg), dasatinib (30 mg/kg) and ponatinib (30 mg/kg) to PDX mice daily for 28 days. Tumor volume and tumor weight were measured, and the results are summarized as the mean ± SEM (Student's t-test). Representative tumor images of these experiments (taken on day 28). (A-C) PDX-KITWT, treated with imatinib (TGI = 17.96%), dasatinib (TGI = 33.85%), and ponatinib (TGI= 33.26%), n = 6. (D-F) PDX with KITV560D mutation, treated with imatinib (TGI = 25.25%), dasatinib (TGI = 68.65%), and ponatinib (TGI = 78.33%), n = 6. (G-I) PDX with KITK642E mutation, treated with imatinib (TGI = 27.59%), dasatinib (TGI = 81.38%) and ponatinib (TGI = 83.66%), n = 5. (J-L) PDX with KITD816V mutation, treated with imatinib (TGI = 42.67%), dasatinib (TGI = 67.73%) and ponatinib (TGI = 99.95%), n = 5 *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant; n indicates the number of tumors per arm.
To confirm that the observed TGI of the KIT-mutation-bearing melanomas was specifically due to the downregulation of KIT signaling, we measured the phosphorylation levels of KIT, ERK, and AKT in the PDX tumors after 28 days of treatment. Treatment of the PDX-KITWT with ponatinib, dasatinib, and imatinib caused no significant difference in the levels of pKIT, pERK, or pAKT (Figure S5A). In contrast, treatment of the KIT-mutation-bearing PDX-KITV560D, PDX-KITK642E and PDX-KITD816V models with ponatinib and dasatinib significantly reduced the levels of pKIT, pERK, and pAKT, while imatinib showed only modest effects (Figure 5A and Figure S6A and Figure S7A).
Inhibitory efficacy of ponatinib in PDX with KITD816V mutation. (A) Immunoblot analysis of KIT signaling in tumors after 28 days of treatment with imatinib, dasatinib and ponatinib. (B) Scoring of Ki-67 staining is summarized as the mean ± SEM. Student's t-test, **, P < 0.01; ***, P< 0.001. (C) Scoring of TUNEL staining is summarized as the mean ± SEM. Student's t-test, ***, P < 0.001. (D) Representative Ki-67 staining in tumors after 28 days treatment with imatinib, dasatinib and ponatinib. Scale bar, 50μm. (E) Representative TUNEL staining in tumors after 28 days of treatment with imatinib, dasatinib and ponatinib. Scale bar, 50μm.
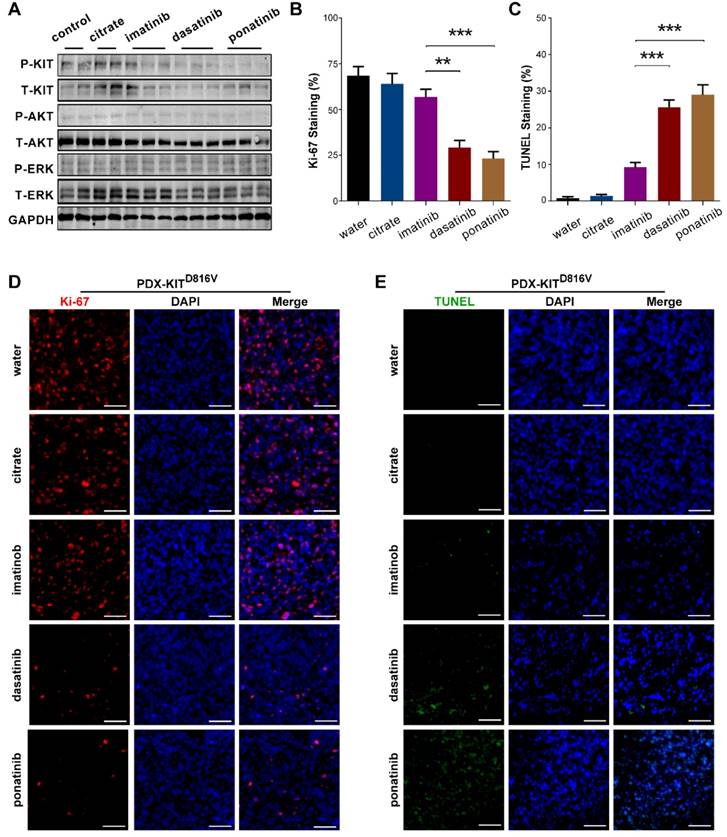
We also used Ki-67 staining and TUNEL-assays to assess the proliferative and apoptotic status of the PDX tumors after 28 days of inhibitor treatment. Ponatinib treatment of PDX-KITV560D, PDX-KITK642E and PDX-KITD816Vmodels resulted in a remarkable reduction in Ki-67-labeled cell proliferation and induced more TUNEL-labeled apoptotic cells than did imatinib, while no significant difference was observed for PDX-KITWT (Figure 5B-E and Figure S5B-E, Figure S6B-E and Figure S7B-E).
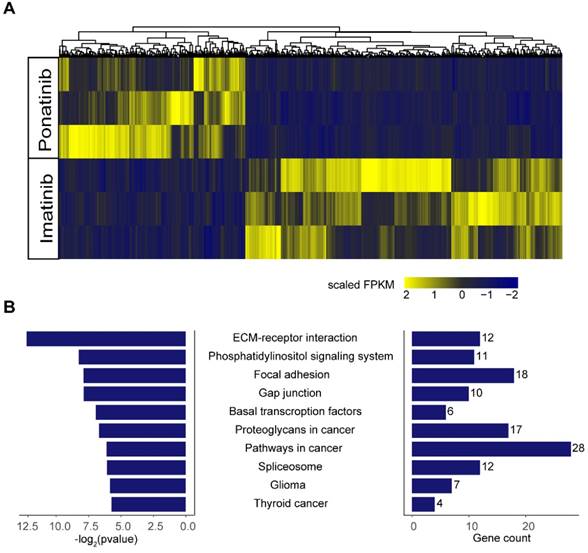
To further explore the potential mechanisms of the inhibitory effects of ponatinib when compared with imatinib, we performed RNA-sequencing in KITK642E PDXs treated with imatinib (n=3) and ponatinib (n=3). A total of 1130 differential expressed genes were found in ponatinib-treated tumors when compared with imatinib-treated tumors, including 712 genes that were down-regulated and 418 genes that were up-regulated (Figure 6A). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was then performed on these differential expressed genes, and demonstrated that the extracellular matrix (ECM)-receptor interaction pathway was the most significantly downregulated pathway in ponatinib treated tumors (Figure 6B). Thus, these data indicated that downregulation of the ECM-receptor interaction pathway was a potential mechanism mediating the robust efficacy of ponatinib in tumor growth inhibition when compared with imatinib.
RNA-sequencing in tumors of PDX-KITK642E after 28 days of treatment with imatinib (n = 3) and ponatinib (n = 3). (A) Heatmap of differential expressed genes between the imatinib-treated group and ponatinib-treated group. (B) KEGG analysis showed that the ECM-receptor interaction pathway was the most significantly downregulated pathway in ponatinib-treated tumors compared with imatinib-treated tumors.
Molecular dynamics simulations showing that ponatinib has a stronger affinity for KITD816V than imatinib
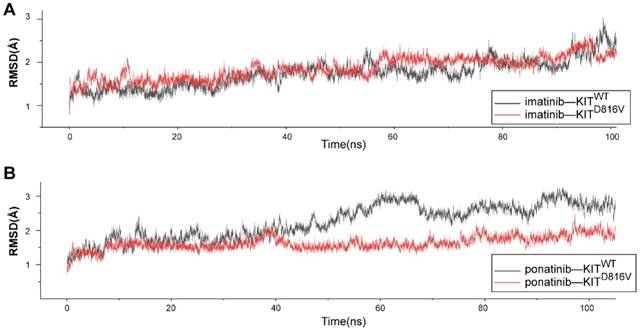
We used MD simulations to investigate the atomic basis of the observed dramatic difference in the inhibition efficacy of ponatinib versus imatinib for melanomas bearing KIT mutations. The crystal structure of ponatinib/imatinib-bound KITWT has been solved [22, 23]; but to date, there have been no studies exploring whether KIT mutations specifically affect the affinity of ponatinib for the KIT protein. Considering that KITD816V is the most common imatinib-resistant mutation in melanomas [21], systemic mastocytosis [24] and several other malignancies [25], we selected KITD816V from among the KIT mutations and compared the binding dynamics using 100-ns MD simulations for four complexes: KITWT-imatinib, KITWT-ponatinib, KITD816V-imatinib, and KITD816V-ponatinib. To check for structural stability and to assess the movements of the four complexes during the course of the simulation, the fluctuations of the KIT-inhibitor complex were analyzed by measuring root mean square deviation (RMSD) for the distance between the backbone atoms of the protein kinase domain and imatinib/ponatinib. After 40 ns, each of the four complexes reached a relatively stable state (Figure 7A-B), which indicated that these protein structures can be stably maintained in all four cases without a significant conformational change under the simulation conditions. Notably, the KITD816V-ponatinib complex had a lower RMSD value than did the KITWT-ponatinib complex, while there were no significant differences between the KITD816V-imatinib and KITWT-imatinib complexes. As imatinib and ponatinib exert their effect by blocking the ATP-binding site of the protein kinase, the result suggests that ponatinib was more constrained in the ATP-binding sites of KITD816V than was imatinib, leading to a relatively stronger affinity for KITD816V than did imatinib. As the mechanism of imatinib resistance to KITD816V has been investigated thoroughly using MD simulations, and the electrostatic interactions were considered to be a decisive factor affecting the binding energy, a significant decrease in electrostatic interactions contributed to imatinib resistance to KITD816V [26]. We calculated the free binding energy in the ponatinib-KITWT and KITD816V complexes. Notably, the electrostatic energy (ΔG elect) sharply increased in the ponatinib-KITD816V complex, while minor changes were observed in other binding energy components (Table S4), suggesting that the change in electrostatic interactions was the principle force contributing to the final binding energy (ΔG bind). The salt bridge D792•••R815 is an important contributor to the electrostatic interaction between imatinib and KIT, and most importantly, it may contribute to the resistance of imatinib to KITD816V [26]. Next, we analyzed the salt bridge D792•••R815 in our inhibitor-KIT complexes. Significantly, divergent salt bridges were shown in the ponatinib-KITD816V complex but absent in the imatinib-KITD816V complex (Figure S8A-B). As D792 and R815 were charged residues located near the ATP-binding site, the salt bridge apparently changed the distribution of the charges of the resides in the region of the ATP-binding site, which was shown in the electrostatic potential (EP) surface of the imatinib-KITD816V-complex and the ponatinib-KITD816V -complex. (Figure S8C-D).
Molecular dynamic simulations of KITWT/ KITD816V-ponatinib interactions. (A) RMSD of KITWT/ KITD816V-imatinib complexes along 100 ns of MD simulations. (B) RMSD of KITWT/ KITD816V-ponatinib complexes along 100 ns of MD simulations.
Discussion
This work successfully established a series of melanoma PDX models and PDCs with KIT mutations and used them for drug efficacy evaluations. Although the in vitro drug assays indicated that ponatinib was not the most effective inhibitor in all PDCs, except in the PDC-KITD816V, ponatinib induced the greatest growth inhibition efficacy against KITD816V-bearing PDX models compared with the other KIT inhibitors that were tested. Ponatinib is an FDA-approved kinase inhibitor for T315I-positive CML and Ph+ ALL patients, known to inhibit the kinase activity of KIT, FGFR, and RET [23, 27]. Previous studies have demonstrated the growth inhibitory efficacy of ponatinib in Ba-F3 cells with KITV560D or KITK642E mutations [23, 28]. Two additional studies indicated that ponatinib exerts growth inhibition and induces apoptosis in neoplastic mast cell lines harboring the KITD816V mutation [24]. However, no study has reported the use of ponatinib in melanoma. Our study thus demonstrated an unexpected opportunity to potentially repurpose ponatinib for use as a potent inhibitor for the treatment of KIT-mutant-bearing melanomas.
Mutations in the cancer driver gene KIT strongly affect the individual responses of patients to targeted therapies. In this vein of work, our study further complicates our knowledge of the interaction of imatinib with melanomas bearing KIT mutations: our finding that the PDX models with the KITV560D or KITK642E mutations did not respond to imatinib treatment does not align with the previously reported findings that melanomas with these mutations are sensitive to treatment with imatinib [8-10]. Thus, the prior conclusion that melanomas with KITV560D or KITK642E mutations should be considered imatinib-sensitive will need to be examined in future work.
As more evidence accumulates about the KIT-mutation-related limits of the clinical benefits of imatinib as a treatment for melanomas, there have been multiple studies examining the use of other kinase inhibitor drugs. For example, dasatinib has shown potent preclinical activity against cells with KITL576P or KITD820Y mutation [29]. However, a phase II clinical trial of dasatinib in locally advanced or stage IV mucosal or acral melanoma patients showed a low response rate of among KIT-mutant or -amplified patients [12]. A notably different result from our findings in the present study, which showed strong TGI effects from dasatinib treatment for all three of the KIT mutation PDX models (Figure 4). Nilotinib can achieve disease control in patients with melanoma-harboring KIT alterations whose disease progressed after imatinib therapy [13]. For the other alternative kinase inhibitors that we tested in this study, the reported responses of melanomas to sunitinib were found to be unrelated to a KIT mutation, and no prolonged responses were observed compared to the response to imatinib. There is almost no KIT mutation-related information for sorafenib, but one patient with a KIT mutation-bearing melanoma was reported to respond well [30]. In addition, there is no study evaluating axitinib for the treatment of KIT-mutated tumor. Some other targeted strategies are also under investigation in KIT-driven melanoma. For example, SEL201, a MNK1/2 inhibitor was reported to suppress clonogenicity, cell migration, and in vivo tumor metastasis in KITL576P and KITD820Y mutant melanoma [29]; the translation of KIT is regulated by LMTK3 in KIT-mutant gastrointestinal stromal tumor (GIST) and melanoma while LMTK3 silencing using a siRNA could reduce the viability of KIT-mutant cancer cells [31].
The application of MD simulations has been increasing in the field of protein kinase drug discovery; these simulations enable mechanistic investigations of biological and chemical events at an atomic level. Multiple MD studies have focused on KIT mutations. Additionally, electrostatic interactions play a significant role in the binding affinity of imatinib to KIT [32], and reduced electrostatic interactions may be responsible for the imatinib resistance to KITD816V [26], which is in accord with our simulation results that imatinib has a relatively lower binding affinity for KITD816V than does ponatinib. Consistently, our experiments with PDCs revealed that ponatinib treatment decreased the phosphorylation of both KIT and its downstream signaling targets, and RNA-seq analysis revealed that ponatinib significantly downregulated the ECM-receptor interaction pathway, which functions in the metastasis, proliferation, and apoptosis of tumor cell [33]. Thus, these data indicated that downregulation of the ECM-receptor interaction pathway was a potential mechanism mediating the robust efficacy of ponatinib in tumor growth inhibition when compared with imatinib. Looking forward, further investigation of ponatinib's molecular mechanisms, in concert with clinical studies about precisely which melanoma patient groups can benefit from ponatinib treatment, will help to further characterize the scientific basis and medicinal impact of the particularly strong efficacy that we observed for ponatinib drug in treating melanomas with KIT mutations.
Materials and Methods
Reagents
Axitinib, imatinib, dasatinib, ponatinib, sorafenib, sunitinib, and nilotinib were purchased from Selleck Chemical Inc (Houston, TX, USA). The following commercially available antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA): KIT (#3074), phospho-KIT (Tyr719, #3391), AKT (#4685), phospho-AKT (Ser474, #4060), ERK1/2 (#4695), phospho-ERK1/2 (Tyr202/Tyr204, #4370), and GAPDH (#2118). Rabbit polyclonal antibody against human Ki67 (ab15580) was purchased from Abcam (Cambridge, UK). Rabbit polyclonal antibody against human S-100A (catalog, GZ031129), mouse polyclonal antibodies against human Melan-A (catalog, GM719629) and HMB-45(catalog, GM063429) were purchased from GeneTech company limuted (shanghai, China). Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) KIT and CCK-8 KIT were purchased from Bimake (Houston, TX, USA).
Establishment of KIT mutant patient-derived tumor xenografts from melanomas
Fresh tumor samples from melanoma patients were collected following informed consent for engraftment in accordance with the ethical guidelines approved for this study by the Institutional Review and the Ethics Committee of Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine. Mice were maintained and all procedures were performed under the approval and supervision of the Institutional Animal Care and Use Committee (IACUC) of Shanghai Jiao Tong University School of Medicine.
Before engraftment, necrotic tissues were carefully removed from the tumor samples using a sterilized scalpel. Approximately 3×3×3-mm3 tissue fragments were implanted subcutaneously into the flank region of nude or NOD-SCID mice (male, 5-6 weeks old). Successfully engrafted tumor models were then passaged and banked using standard methods. The passage directly harboring the patient samples was termed P0; subsequent generations were consecutively named P1, P2, P3, P4, etc. After each passage, harvested tumor samples were divided into several segments for subsequent implantation, for cryopreservation in liquid nitrogen and freezing media, and for histopathological analysis in paraffin embedding.
Generation of patient-derived tumor cells
Tumor tissues from the KIT mutant PDXs were extensively washed with serum-free RPMI1640 (Gibco, Waltham, MA, USA)) and minced using a sterile scalpel and dissociated in RPMI1640 with 1 mg/ml Ⅳ collagenase (Sigma-Aldrich, St. Louis, MO, USA) and 0.5 mg/mL dispase (Gibco) for 2h at 37°C with agitation. Cells were filtered through a 40-μm filter and washed twice in PBS and suspended in RPMI 1640 medium with 10% fetal bovine serum (Gibco) and 1% penicillin and streptomycin. Cells were cultured at 37°C in a humidified atmosphere of 95% air and 5% CO2.
TUNEL assay
The incidence of apoptotic tumor cells was assessed by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining. TUNEL staining was performed according to the manufacturer's protocols. The slides were deparaffinized in xylene and hydrated in graded ethanol. Then, sections were washed with PBS and incubated with proteinase K for 20 min at 37°C. After washing three times with PBS, the sections were treated with 50 µl of TUNEL reaction mix and then washed three times. Nuclei were counterstained with 4, 6-diamidino-2-phenylindole (DAPI) at 37°C for 10 minutes.
Immunofluorescence staining
For tissue sections, after deparaffinization, sections were hydrated and underwent sodium citrate antigen retrieval and then incubated with 3% hydrogen peroxide to block endogenous peroxidase activity. For PDCs, the cells were grown on glass coverslips, washed with PBS, fixed with 4% paraformaldehyde in PBS for 15 minutes, permeabilized with 0.5% Triton X-100 for 10 minutes, and blocked with 5% goat serum in PBS at 37°C for 30 minutes. Sections or cells were incubated with primary antibodies against Ki67 (1:200 dilution), Melan-A (1:100 dilution), and HMB-45 (1:100 dilution) overnight at 4°C, followed by Alexa Fluor 488-conjugated AffiniPure goat anti-mouse IgG (H+L) or Alexa Fluor 594-conjugated goat anti-rabbit IgG (H+L) second antibodies for 60 minutes. Nuclei were counterstained with 4, 6-diamidino-2-phenylindole (DAPI) at 37°C for 10 minutes. In the negative control samples, the primary antibody was replaced with PBS.
Cell viability assay
Cell viability was determined using a cell counting kit-8 (CCK-8) assay. Cells were seeded into 96-well plates at a density of 2,000 cells per well and treated with vehicle (DMSO) control or inhibitors with serial dilution for 72 h. The IC50 values were calculated using GraphPad Prism software.
Generation of Ba-F3 stable cell lines
Full-length cDNAs of KIT containing KITWT, KITV560D, KITK642E and KITD816V were generated by mutagenesis and were cloned into the pLVX-IRES-Puro vector according to the manufacturer's instructions. Ba-F3 cells expressing KITWT, KITV560D, KITK642E and KITD816V mutation constructs were selected in RPMI 1640 supplemented with SCF (20 ng/ml) and puromycin (1 µg/ml). These cells were maintained until IL-3-independent cells emerged. Parental Ba/F3 cells, as a control, were cultured in RPMI 1640 media supplemented with IL-3.
In vivo drug efficacy evaluation
PDX mice were randomized into treatment groups when the average tumor volume reached ~250 mm3. Treatment compounds were administered to mice daily by oral gavage as follows: water and citrate (25 mM, pH=2.75), imatinib mesylate (100 mg/kg, diluted in water), dasatinib (30 mg/kg), and ponatinib (30 mg/kg). Both dasatinib and ponatinib were diluted in citrate buffer (25 mM, pH=2.75). Tumor size was measured with calipers, and mouse weight was recorded twice weekly. Tumor volume (mm3) was calculated as follows: tumor volume = L×W2×0.5, where L represents the largest diameter and W represents the minor tumor axis. When the treatment was terminated, tumor growth inhibition (TGI) was calculated as follows: TGI= 1 - [(TVf, treated - TVi, treated)/ (TVf, control - TVi, control)] ×100%, where TVf is the average tumor volume at the end of the experiment, and TVi is the average tumor volume at the start of treatment.
Immunoblotting
Immunoblotting was performed using standard SDS-PAGE protocols. Briefly, the proteins were separated on SDS-PAGE gels and transferred to PVDF membranes. The membrane was incubated with primary antibodies against KIT (dilution 1:1000), phospho-KIT (dilution 1:1000), AKT (dilution 1:1000), phospho-AKT (dilution 1:1000), ERK1/2 (dilution 1:1000), phospho-ERK1/2 (dilution 1:2000), or GAPDH (dilution 1:1000) overnight at 4°C. After washing and incubation with secondary antibodies, signals were visualized using an Odyssey Infrared Imaging System (Biosciences) according to the product instructions.
RNA-Sequencing and analysis
RNA was isolated from snap-frozen tumors from vehicle-, imatinib-, and ponatinib-treated mice. Total RNA was extracted using RNeasy Mini Kit (Cat# 74106, Qiagen) following the manufacturer's instructions and checked for a RIN number to inspect RNA integrity by an Agilent Bioanalyzer 2100 (Agilent technologies, Santa Clara, CA, US). Qualified total RNA was further purified by RNAClean XP Kit (Cat A63987, Beckman Coulter,Inc.Kraemer Boulevard Brea, CA,USA)and RNase-Free DNase Set (Cat#79254, QIAGEN, GmBH, Germany). The Illumina TruSeq RNA Sample Preparation Kit (Illumina) was used for library preparation. RNA sequencing were performed on the Illumina Hiseq 2000 platform. Reads were aligned with human transcript genome (hg38) using TopHat v2. Gene expression abundance was estimated by Stringtie (version: 1.3.0). Differentially expression analysis was determined using negative binomial model via EdgeR/DESeq2, by comparing imatinib-treated with ponatinib-treated tumors and P-value was calculated using Wald test, multiple test correction was performed using B-H procedure, with a fold change (FC) >2.0 and P < 0.05 as the cut-off criteria. Pathway analysis on differentially expressed genes was performed using KEGG pathway analysis.
Molecular dynamics simulations
The co-structure of KITWT-ponatinib (PDB ID: 4U0I) [23]and KITWT-imatinib (PDB ID: 1T46) [34]were retrieved from the PDB library. Pymol was used for the in silico substitution of the Asp (D) to Val (V) KIT mutation at position 816. Molecular dynamics (MD) simulations of four complexes (KITWT-imatinib, KITWT-ponatinib, KITD816V-imatinib, and KITD816V-ponatinib) were performed using Amber (V14.0) with a previously reported approach [35]. Briefly, the energy of the systems was minimized, relaxed and then heated to reach equilibration. The four complexes underwent 100-ns MD simulations in isothermal and isobaric conditions. The trajectories were recorded every 20 ps. The root-mean-square deviation (RMSD) of the trajectories was calculated to present the dynamic conformational changes throughout the MD simulations. Superposition of structural conformations was taken at 100 ns from the MD simulations. The binding free energy(△G binding) of KITD816V-ponatinib was calculated using Molecular Mechanics Generalized Born Surface Area (MM-GBSA) analysis. The electrostatic potential surface was computed with the APBS software through the PDB2PQR webserver (http://nbcr-222.ucsd.edu/pdb2pqr/). The conformations of the KITD816V-imatinib and KITD816V-ponatinib complexes after 100 ns MD simulations were used as input data. Plots were generated using APBS tools plugin available with Pymol software[36].
Statistics
Analyses were conducted with GraphPad Prism version 6.0 for Windows (GraphPad Software, La Jolla, CA. USA). The results are represented as the mean ± SEM of at least three replicate samples for each group, unless noted otherwise. Unpaired Student's t-tests were performed to compare differences between the two groups. P< 0.05 was considered statistically significant.
Abbreviations
ECM: Extracellular matrix; FC: Fold change; IC50: half maximal inhibitory concentration; KEGG: Kyoto Encyclopedia of Genes and Genomes; NCCN: National Comprehensive Cancer Network; MD: molecular dynamics; PDX: patient-derived tumor xenograft; PDC: PDX-derived tumor cells; TGI: tumor growth inhibition.
Acknowledgements
The authors thank Professor Qiancheng Shen, Department of Pathophysiology, Key Laboratory of Cell Differentiation and Apoptosis of Ministry of Education, Shanghai Jiao Tong University School of Medicine, for helpful direction regarding the MD simulations.
Funding
This work was funded by The National Key Research and Development Program of China (2017YFC0908500) and also funded by grants from the National Natural Science Foundation of China (81572656, 8187219, 81202131).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ossio R, Roldán-Marín R, Martínez-Said H, Adams DJ, Robles-Espinoza CD. Melanoma: a global perspective. Nat Rev Cancer. 2017;17:393
2. Genomic Classification of Cutaneous Melanoma. Cell. 2015; 161: 1681-96.
3. McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R. et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-32
4. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24:4340-6
5. Todd JR, Scurr LL, Becker TM, Kefford RF, Rizos H. The MAPK pathway functions as a redundant survival signal that reinforces the PI3K cascade in c-Kit mutant melanoma. Oncogene. 2014;33:236-45
6. Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000;96:925-32
7. Carvajal RD. Another option in our KIT of effective therapies for advanced melanoma. J Clin Oncol. 2013;31:3173-5
8. Hodi FS, Corless CL, Giobbie-Hurder A, Fletcher JA, Zhu M, Marino-Enriquez A. et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. 2013;31:3182-90
9. Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J. et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305:2327-34
10. Guo J, Si L, Kong Y, Flaherty KT, Xu X, Zhu Y. et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29:2904-9
11. Minor DR, Kashani-Sabet M, Garrido M, O'Day SJ, Hamid O, Bastian BC. Sunitinib therapy for melanoma patients with KIT mutations. Clin Cancer Res. 2012;18:1457-63
12. Kalinsky K, Lee S, Rubin KM, Lawrence DP, Iafrarte AJ, Borger DR. et al. A phase 2 trial of dasatinib in patients with locally advanced or stage IV mucosal, acral, or vulvovaginal melanoma: A trial of the ECOG-ACRIN Cancer Research Group (E2607). Cancer. 2017;123:2688-97
13. Guo J, Carvajal RD, Dummer R, Hauschild A, Daud A, Bastian BC. et al. Efficacy and safety of nilotinib in patients with KIT-mutated metastatic or inoperable melanoma: final results from the global, single-arm, phase II TEAM trial. Ann Oncol. 2017;28:1380-7
14. Yun J, Lee J, Jang J, Lee EJ, Jang KT, Kim JH. et al. KIT amplification and gene mutations in acral/mucosal melanoma in Korea. APMIS. 2011;119:330-5
15. Hintzsche JD, Gorden NT, Amato CM, Kim J, Wuensch KE, Robinson SE. et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res. 2017;27:189-99
16. Kong Y, Si L, Zhu Y, Xu X, Corless CL, Flaherty KT. et al. Large-scale analysis of KIT aberrations in Chinese patients with melanoma. Clin Cancer Res. 2011;17:1684-91
17. Bruna A, Rueda OM, Greenwood W, Batra AS, Callari M, Batra RN. et al. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell. 2016;167:260-74.e22
18. Izumchenko E, Paz K, Ciznadija D, Sloma I, Katz A, Vasquez-Dunddel D. et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann Oncol. 2017;28:2595-605
19. Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C. et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998-1013
20. Aparicio S, Hidalgo M, Kung AL. Examining the utility of patient-derived xenograft mouse models. Nat Rev Cancer. 2015;15:311-6
21. Phung B, Kazi JU, Lundby A, Bergsteinsdottir K, Sun J, Goding CR. et al. KIT(D816V) Induces SRC-Mediated Tyrosine Phosphorylation of MITF and Altered Transcription Program in Melanoma. Mol Cancer Res. 2017;15:1265-74
22. Mol CD, Lim KB, Sridhar V, Zou H, Chien EY, Sang BC. et al. Structure of a c-kit product complex reveals the basis for kinase transactivation. J Biol Chem. 2003;278:31461-4
23. Garner AP, Gozgit JM, Anjum R, Vodala S, Schrock A, Zhou T. et al. Ponatinib inhibits polyclonal drug-resistant KIT oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients. Clin Cancer Res. 2014;20:5745-55
24. Jin B, Ding K, Pan J. Ponatinib induces apoptosis in imatinib-resistant human mast cells by dephosphorylating mutant D816V KIT and silencing beta-catenin signaling. Mol Cancer Ther. 2014;13:1217-30
25. Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H. et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342-9
26. Da Silva Figueiredo Celestino Gomes P, Chauvot De Beauchene I, Panel N, Lopez S, De Sepulveda P, Geraldo Pascutti P. et al. Insight on Mutation-Induced Resistance from Molecular Dynamics Simulations of the Native and Mutated CSF-1R and KIT. PloS one. 2016;11:e0160165
27. O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F. et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401-12
28. Lierman E, Smits S, Cools J, Dewaele B, Debiec-Rychter M, Vandenberghe P. Ponatinib is active against imatinib-resistant mutants of FIP1L1-PDGFRA and KIT, and against FGFR1-derived fusion kinases. Leukemia. 2012;26:1693-5
29. Zhan Y, Guo J, Yang W, Goncalves C, Rzymski T, Dreas A. et al. MNK1/2 inhibition limits oncogenicity and metastasis of KIT-mutant melanoma. J Clin Invest. 2017;127:4179-92
30. Quintas-Cardama A, Lazar AJ, Woodman SE, Kim K, Ross M, Hwu P. Complete response of stage IV anal mucosal melanoma expressing KIT Val560Asp to the multikinase inhibitor sorafenib. Nat Clin Pract Oncol. 2008;5:737-40
31. Klug LR, Bannon AE, Javidi-Sharifi N, Town A, Fleming WH, VanSlyke JK. et al. LMTK3 is essential for oncogenic KIT expression in KIT-mutant GIST and melanoma. Oncogene. 2018
32. Lin YL, Roux B. Computational analysis of the binding specificity of Gleevec to Abl, c-Kit, Lck, and c-Src tyrosine kinases. J Am Chem Soc. 2013;135:14741-53
33. Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer. 2014;14:430-9
34. Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN. et al. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 2004;279:31655-63
35. Case DA, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, Merz KM Jr. et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668-88
36. Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665-7
Author contact
![]() Corresponding authors: shuyangsedu.cn and zhzhyedu.cn
Corresponding authors: shuyangsedu.cn and zhzhyedu.cn