Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(7):2003-2016. doi:10.7150/thno.28057 This issue Cite
Research Paper
Tolerizing CTL by Sustained Hepatic PD-L1 Expression Provides a New Therapy Approach in Mouse Sepsis
Andreas von Knethen1,2 ![]() , Anne Schäfer1, Laura Kuchler1, Tilo Knape2, Urs Christen3, Edith Hintermann3, Beate Fißlthaler4, Katrin Schröder5, Ralf P. Brandes5, Berit Genz6, Kerstin Abshagen6, Brigitte M. Pützer7, Lisa K. Sha1, Andreas Weigert1, Shahzad N. Syed1, Martin Schulz1, Ajay M. Shah8, Andreas Ernst2,3, Mateusz Putyrski2, Fabian Finkelmeier9, Marina Pesic9, Florian Greten9, Michael Hogardt10, Volkhard A. J. Kempf10, Sandra Gunne2, Michael J. Parnham2, Bernhard Brüne1,2
, Anne Schäfer1, Laura Kuchler1, Tilo Knape2, Urs Christen3, Edith Hintermann3, Beate Fißlthaler4, Katrin Schröder5, Ralf P. Brandes5, Berit Genz6, Kerstin Abshagen6, Brigitte M. Pützer7, Lisa K. Sha1, Andreas Weigert1, Shahzad N. Syed1, Martin Schulz1, Ajay M. Shah8, Andreas Ernst2,3, Mateusz Putyrski2, Fabian Finkelmeier9, Marina Pesic9, Florian Greten9, Michael Hogardt10, Volkhard A. J. Kempf10, Sandra Gunne2, Michael J. Parnham2, Bernhard Brüne1,2
1. Institute of Biochemistry I, Faculty of Medicine, Goethe-University Frankfurt, Frankfurt, Germany.
2. Fraunhofer Institute for Molecular Biology and Applied Ecology IME, Project Group Translational Medicine & Pharmacology TMP, Frankfurt, Germany.
3. Pharmazentrum/ZAFES Frankfurt, Faculty of Medicine, Goethe-University Frankfurt, Frankfurt, Germany.
4. Institute for Vascular Signalling, Centre for Molecular Medicine, Goethe-University Frankfurt, Frankfurt, Germany.
5. Institute for Cardiovascular Physiology, Faculty of Medicine, Goethe-University Frankfurt, Frankfurt, Germany.
6. Institute for Experimental Surgery, Rostock University Medical Center, Rostock, Germany.
7. Institute of Experimental Gene Therapy and Cancer Research, Rostock University Medical Center, Rostock, Germany.
8. King's College London, British Heart Foundation, Centre of Excellence, The James Black Centre, London, UK.
9. Georg Speyer Haus, Institute for Tumor Biology and Experimental Therapy, Frankfurt, Germany.
10. Institute for Medical Microbiology and Infection Control, University Hospital Frankfurt, Paul-Ehrlich-Str. 40, Frankfurt am Main, Germany.
Received 2018-6-22; Accepted 2019-1-16; Published 2019-3-16
Abstract

Cytotoxic T lymphocyte (CTL) activation contributes to liver damage during sepsis, but the mechanisms involved are largely unknown. Understanding the underlying principle will permit interference with CTL activation and thus, provide a new therapeutic option.
Methods: To elucidate the mechanism leading to CTL activation we used the Hepa1-6 cell line in vitro and the mouse model of in vivo polymicrobial sepsis, following cecal-ligation and -puncture (CLP) in wildtype, myeloid specific NOX-2, global NOX2 and NOX4 knockout mice, and their survival as a final readout. In this in vivo setting, we also determined hepatic mRNA and protein expression as well as clinical parameters of liver damage - aspartate- and alanine amino-transaminases. Hepatocyte specific overexpression of PD-L1 was achieved in vivo by adenoviral infection and transposon-based gene transfer using hydrodynamic injection.
Results: We observed downregulation of PD-L1 on hepatocytes in the murine sepsis model. Adenoviral and transposon-based gene transfer to restore PD-L1 expression, significantly improved survival and reduced the release of liver damage, as PD-L1 is a co-receptor that negatively regulates T cell function. Similar protection was observed during pharmacological intervention using recombinant PD-L1-Fc. N-acetylcysteine blocked the downregulation of PD-L1 suggesting the involvement of reactive oxygen species. This was confirmed in vivo, as we observed significant upregulation of PD-L1 expression in NOX4 knockout mice, following sham operation, whereas its expression in global as well as myeloid lineage NOX2 knockout mice was comparable to that in the wild type animals. PD-L1 expression remained high following CLP only in total NOX2 knockouts, resulting in significantly reduced release of liver damage markers.
Conclusion: These results suggest that, contrary to common assumption, maintaining PD-L1 expression on hepatocytes improves liver damage and survival of mice during sepsis. We conclude that administering recombinant PD-L1 or inhibiting NOX2 activity might offer a new therapeutic option in sepsis.
Keywords: sepsis, cytotoxic T cells, reactive oxygen species, PD-L1, liver
Introduction
Despite intensive research, sepsis remains the third leading cause of mortality in intensive care units [1]. During sepsis progression, there is an initial hyper-inflammatory phase, which provokes the onset of a subsequent hypo-inflammatory stage, and these phases often overlap [2, 3]. Recent therapy approaches focus onto attenuation of the hyper-inflammatory response to confine the release of pro-inflammatory mediators, block their function, or remove them from the circulation. A most promising candidate, shown to significantly improve survival in a rodent sepsis model, was TNFα (tumor necrosis factor α) [4]. Using neutralizing antibodies, this approach was translated into the human situation, but failed to improve sepsis survival [5]. However, with this approach, the hyper-inflammation is reduced, and most patients survive this initial phase. Because blocking the pro-inflammatory immune response reduces the host´s ability to combat and control primary and secondary infections, such therapeutic approaches may even promote or enhance the hypo-inflammatory phase. Immunosuppression often provokes multi- organ-dysfunction syndrome and the death of patients [6]. Treatment approaches to rescue the patient during immune paralysis have also been applied. Based on the findings that monocytes are deactivated during this phase [7], granulocyte- macrophage colony-stimulating factor treatment restored monocyte function during sepsis [8]. In the mouse model, antagonism of the nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ) has been shown to avert T cell depletion [9], one of the hallmarks of immune paralysis associated with the hyper-inflammatory phase, an effect that is correlated with reduced sepsis mortality [10]. Because of the multi-causal origin of sepsis, the various pre-existing co-morbidities, or genetic preconditions of the patients, appropriate patient-specific treatment is still difficult to achieve [11]. Generally, sepsis syndrome severity is already advanced when sepsis is first diagnosed, so we were interested to prevent liver damage, a relatively late event during sepsis progression and an etiological factor in multiple organ failure. Recently, we observed that following cecal ligation and puncture-initiated sepsis in mice, cytotoxic T cells (CTL) are activated towards hepatocytes, thus mounting an autoimmune response [12]. Here we show that hepatic downregulation of the co-inhibitory protein PD-L1 causes autoimmune CTL activation. Maintaining PD-L1 expression or applying recombinant PD-L1-Fc chimera, significantly improves liver damage and animal survival. Mechanistically, PD-L1 was downregulated by reactive oxygen species (ROS). Using NOX2- as well as NOX4-knockout mice revealed that the deletion of non-myeloid NOX2 activity restored PD-L1 expression following CLP, consequently ameliorating liver damage.
Methods
Mice
Mice with a specific NOX2-knockout in the myeloid lineage were generated by crossing C57Bl/6 mice bearing conditional loxP-flanked alleles of NOX2 (NOX2fl/fl) [13], kindly provided by Prof. Shah (King´s College London BHF Centre of Excellence, London, UK) with C57Bl/6N-(Tg) LysM-Cre transgenic mice, in which the Cre recombinase had been knocked in behind the LysM promoter [14]. Global NOX2- and NOX4-knockout mice as well as wild type mice used were also on a C57Bl/6 background. OT-I mice were kindly provided by Prof. Knolle (Technical University Munich, Faculty of Medicine, Institute of Molecular Immunology & Experimental Oncology). Mice housing was temperature controlled. Day and night were 12 h each. Filter-topped cages were used. Mice had access to standard laboratory chow and water ad libitum. PCR using tail DNA verified genotypes of mice.
Animal procedures
For sepsis experiments we used NOX2- and NOX4-global, NOX2-LysM Cre as well as C57Bl/6 WT and albino (B6N-Tyrc-Brd/BrdCrCrl) mice. OT-I mice were used to isolate ovalbumin (OVA)-specific CD8+ T cells (Supplementary Figure 1). The cecal ligation and puncture model (CLP) followed the methodology of Rittirsch et al. [15]. The setups used are shown in Supplementary Figure 2. In detail, ketamine (Ketavet®)/xylazine (Rompun®) 100 mg/ 200 mg per kg body weight was used for anesthesia. A midline laparotomy incision was done. One third of the cecum was ligated with an orientation distal to the ileocecal valve. Importantly, the bowel continuity was not disrupted. Following a double puncture using a 20-gauge needle, the laparotomy was sutured. To avoid dehydration, 1 ml 0.9% NaCl was given to the mice i.p. directly following surgery. Moreover, buprenorphine (Temgesic®) 0.5 mg/kg s. c. was applied directly after surgery and every 6 h for 24 h (Supplementary Figure 2A). When hydrodynamic injection is performed in advance to the CLP operation, mice were allowed to recover and to establish transgene expression for 10 days (Supplementary Figure 2B). To apply recombinant PD-L1-Fc (R&D Systems GmbH, Wiesbaden, Germany), mice were directly following CLP i.v. injected with 12 mg/kg of recombinant PD-1-Fc dissolved in PBS/0.5% BSA (Supplementary Figure 2C). Liver damage was determined 24 h following CLP. Therefore, in some experiments, blood was taken beforehand by heart puncture to isolate serum to determine liver damage markers alanine- and aspartate aminotransferase using a Reflotron Plus hematology analyzer (Roche Diagnostics, Mannheim, Germany). Before liver dissection, the organ is flushed with PBS before. Afterwards, a part of the liver is used to prepare a single cell suspension for FACS analysis and CD8+ T cell enrichment. A second part of the liver is used for immunohistochemistry.
All animal experiments were approved by the State of Hesse animal care and use committee (authorization no. F144/15 and FU/1148).
Cell culture
RPMI1640 (PAA Laboratories) was used to culture Hepa1-6 cells [16] supplemented with 100 U/ml penicillin (PAA Laboratories), 100 µg/ml streptomycin (PAA Laboratories), and 10% heat inactivated fetal calf serum (PAA Laboratories).
Cloning
To overexpress murine PD-L1 in vitro, we amplified PD-L1 from murine mRNA of Hepa1-6 cells by PCR using the following primer pair (NM_021893): forward 5´-CGC CCG GGG GGG ATC ATG AGG ATA TTT GCT GGC ATT ATA TTC ACA-3´; reverse 5´ -TCA AGC TTG CAT GCC TTA CTT GTA CAG CTC GTC CA-3´. The primers were used to clone mPD-L1 into the lentiviral vector pSEW [17] in front of the EGFP encoding sequence, already present in the pSEW vector. Coding sequences of PD-L1 are shown in italics. Following linearization of pSEW with BamHI, the amplified mPD-L1 fragment was inserted with the InFusion system (Takara Bio Europe, Saint-Germain- en-Laye, France). Correct sequence was verified by sequencing. Functionality of the vector was proven in Hepa1-6 cells (Supplementary Figure 3).
For in vivo transduction of PD-L1 into the liver of mice, PD-L1 EGFP in the pSEW vector was subcloned into the pShuttle-CMV vector of the adEasy adenoviral vector system [18]. The following primer pair was used containing flanking sequence appropriate for InFusion cloning into the BglII/EcoRV site of pShuttle-CMV: forward 5´-GAT CCG CTA GAG ATC GCC ACCATG AGG ATA TTT GCT GGC ATT ATA TTC ACA GC-3´, reverse 5´-TCC GGT GGA TCG GAT TTA CTT GTA CAG CTC GTC CAT GCC-3´. Coding sequences of PD-L1 (forward primer) and EGFP (reverse primer) are displayed in italics. Correct sequence was verified by sequencing. As a negative control the pAdTrack vector, only encoding EGFP, was used. To overexpress PD-L1 by hydrodynamic injection, PD-L1 amplified from Hepa1-6 RNA as described above was cloned into the transposon-based vector pCAGGS-IRES-EGFP [19].
Adenovirus preparation
Ad-293 cells were seeded in DMEM (PAA Laboratories) with supplements (100 U/ml penicillin (PAA Laboratories), 100 µg/ml streptomycin (PAA Laboratories), 10% heat inactivated fetal calf serum (PAA Laboratories), HEPES and non-essential amino acids. Following cell expansion after 4 days, cells were detached with 1 ml trypsin. 9 ml culture medium were added, and cells were centrifuged at 500 g for 5 min. Following cell expansion for 11 days, 0.5 ml of virus stock [1x1011 particles/ml] was thawed at RT and resuspended in 15 ml medium. After tightly closing and mixing, 15 ml of diluted adenovirus was added to each flask. After 3 days, most infected cells were detached as clusters. Non-infected cells were still spread-out and attached to the plastic. Cells were detached by tapping the flasks against the hand. Medium was transferred into 10 x 50 ml tubes and spun at 500 g for 5 min at 4ºC. Pellets were suspended in medium and transferred into a cryo-vial, which is snap-frozen in liquid N2 and transferred to a -80ºC freezer. Adenoviral particles were enriched following 4 rapid thaw/freeze cycles and CsCl gradient centrifugation. To determine the colony forming unit capacity, a plaque assay was applied. Evaluation was performed by fluorescence-microscopy due to the EGFP-tag of transduced genes. For mouse transduction 5x1010 infectious particle were administered in 100 µl PBS.
Hydrodynamic injection
Transposon-based gene transfer in mice was achieved with a 5:1 molar ratio of transposon- to transposase encoding vector (30 µg total DNA). The Qiagen EndoFreeMaxi Kit (Qiagen, Hilden, Germany) was used to isolate vector DNA for hydrodynamic tail injection. Plasmids were dissolved in 0.9% NaCl solution to a final volume of 10% of the weight of the animals. 25 μg transposon plasmids and 5 μg transposase were injected into 5-6 weeks old animals within 10 s.
Cytotoxicity assay
OT-I mice derived CD8+ T cells were used as effector cells in allogenic cytotoxicity assay with Hepa 1-6 cells as target cells [12]. In brief, CD8+ T cells derived from the spleen of C57Bl/6N OT-I mice (haplotype H-2b) as effector cells were co-incubated for 24 h with Hepa1-6 cells, originating from the C57L strain (haplotype H-2b) as target cells. Prior to this, Hepa1-6 cells were pulsed for 2 h with 10 nM of the ovalbumin (OVA) peptide 257-264 (AnaSpec, Fremont, USA), or the hepatitis B virus (HBV) peptide ILSPFLPLL derived from the HBsAgas a negative control (IBA, Goettingen, Germany) or remained untreated as control. Following loading with antigen, CellTracker™ Orange (Life Technologies GmbH, Frankfurt, Germany) was used to stain Hepa1-6 cells before CD8+ T cells were added. When indicated, the PD-L1-Fc chimera protein (R&D Systems GmbH, Wiesbaden, Germany) was added with the indicated concentration. Target cells which survived after 24 h were determined by FACS analysis (FACS Fortessa, BD, Heidelberg, Germany).
qPCR
Total RNA from 5x105 CD8+ T cells, Hepa1-6 cells, or primary liver cells were isolated using peqGOLD RNAPure Kit (Peqlab, Erlangen, Germany) following the distributor´s manual. The iScript™ cDNA Synthesis kit (Bio-Rad, Munich, Germany) was used to reverse transcribe 2 µg RNA into complementary DNA (cDNA). The iQ™ SYBR® Green Supermix (Bio-Rad) was used for Quantitative PCR (qPCR). For qPCR analysis and data quantification, the CFX real-time PCR system from Bio-Rad was used. Used primer pairs (Biomers, Ulm, Germany) derived from murine target genes were as follows: PD-L1 (NM_21893) forward: 5´-TGC AGC AGT AAA CGC CTG CG-3´, reverse: 5´-CGC TGC CAA AGG ACC AGC TT-3´; IL-2 (NM_008366) forward: 5´-TGA GCA TCC TGG GGA GTT TC-3´, reverse: 5´-GTG ACC TCA AGT CCT GCA GG-3´; Fas-L (NM_010177) forward: 5'-ACC AAC CAA AGC CTT AAA-3', reverse: 5'-ATA CTT CAC TCC AGA GAT-3'; granzyme B (NM_013542) forward: 5'-CTC CAC GTG CTT TCA CCA AA-3', reverse: 5'-GGA AAA TAG TAC AGA GAG GCA-3'; perforin (NM_011073) forward: 5'-TGC TAC ACT GCC ACT CGG TCA-3', reverse: 5'-TTG GCT ACC TTG GAG TGG GAG-3'. IFNγ (NM_008337) forward: 5`-TTT GCA GCT CTT CCT CAT GG-3´, reverse: 5´-TCG CCT TGC TGT TGC TGA AG-3´. Values were normalized to 18s rRNA.
Flow cytometry and antibodies
Purity of OT-I CD8+ T cells were verified by flowcytometry, using anti-mouse V alpha 2 TCR‑FITC (eBioscience, San Diego, CA, USA) and anti-mouse Vβ 5.1, 5.2 TCR-PE (BD Bioscience, Heidelberg, Germany) antibodies. A CD16/CD32 anti‑mouse antibody, incubated for 15 min, was used to block Fc receptor binding, before the α-CD8α-APC antibody for T cells was added on ice. Surface expression of PD-L1 and PD-L2 on the surface of primary hepatocytes was determined by FACS analysis, using α-PD-L1-PE or α-PD-L2-PE. Immune cells were excluded by CD45-FITC staining, consequently analyzing CD45- cells only.
To identify regulatory T cells (Tregs) in liver single cell preparations, cells were stained for anti-mouse CD45-Vioblue (Miltenyi Biotec, Bergisch-Gladbach, Germany), anti-mouse CD3-APC-Cy7 (BioLegend, Eching, Germany) , anti-mouse CD4-BV711 (BD Horizon, Heidelberg, Germany), anti-mouse CD11b-BV605 (BioLegend), anti-mouse CD25-PE-Cy7 (BD Pharmingen, Heidelberg, Germany), anti-mouse GITR-FITC (BioLegend), and anti-mouse CD44-AF700 (BD Pharmingen) in parallel. CD4+ T cells showing expression of CD25, GITR, combined with CD44 were judged as Tregs. FACS measurements were performed by a FACS Fortessa LSR and data analysis was done with the FlowJo software.
Western analysis
PD-L1, PD-1, Fas, and EGFP expression were analyzed by Western blotting. Briefly, following equivalent numbers of primary hepatocytes or Hepa1-6 cells were washed twice with PBS, lysed in RIPA buffer containing 1x complete Protease Inhibitor Cocktail Tablets (Roche, Basel, Switzerland) and sonicated for 10 impulses, followed by centrifugation for 10 min at 16000 g (4°C). Supernatants were denatured with SDS-PAGE sample buffer (250 mM Tris pH 6.8, 40% glycerol, 10% 2-ME, 8% SDS, 0.02% bromophenol blue) for 10 min at 95°C. Comparable protein concentrations were maintained by measurement with the Lowry method (Bio-Rad). Proteins were separated on 10% SDS-polyacrylamide gels and transferred onto a nitrocellulose membrane by semi-dry blotting. Membranes were blocked with 5% BSA/TTBS followed by incubation with α-PD-L1- (R&D systems), α-PD-L2- (Santa Cruz), α-PD-1- (Santa Cruz), α-Fas-antibody (Santa Cruz) in 5% BSA/TBS at 4°C overnight. Loading was normalized to ß-actin (α-actin, Sigma-Aldrich). Proteins were detected by incubating the membrane with secondary antibodies labelled with IRDye (LI-COR, Bad Homburg, Germany) in 5% BSA/TTBS. Visualization and densitometric analysis were performed with the Odyssey infrared imaging system.
Statistics
All statistical analyses were performed using Prism 5 (GraphPad Software). Experiments were performed at least five times. The unpaired t-test was used for statistical analysis. P-values ≤ 0.05 were considered as significant. Otherwise representative data are shown.
Results
Cytotoxic T cells (CTLs) accumulate in the liver of septic mice
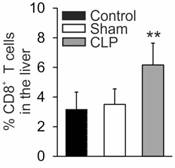
First, we determined the number of CTLs in livers derived from sham operated mice vs. animals subjected to cecal ligation and puncture (CLP). CTL numbers increased in livers derived from septic mice, 24 h following CLP-operation, compared to sham-treated or control mice, respectively (Figure 1). This result suggests activation-induced migration of CTL into the liver tissue.
Enhanced percentage of CD8+ T cells in livers of septic mice. Twenty-four hours following sham- or CLP-operation mice were sacrificed. Livers were removed and single cell suspensions were prepared. Cell subpopulation was determined by FACS analysis. Data from five mice per treatment are shown and represent the mean ± SD (**p<0.01).
Expression of PD-L1 is downregulated in a polymicrobial sepsis model
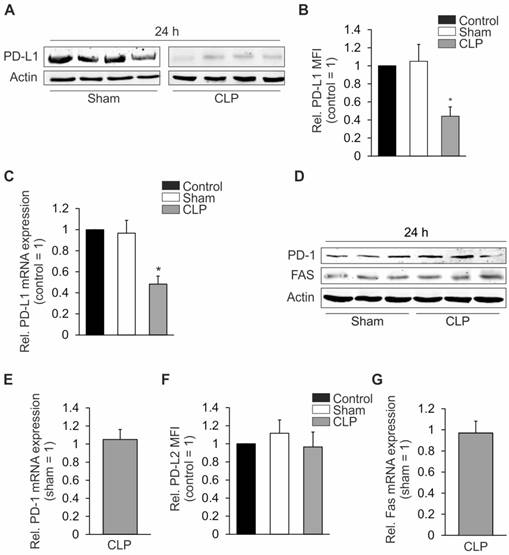
Autoimmune CTL-activation typically is prevented by co-inhibitory proteins such as PD-L1 (CD274 or B7-H1) or PD-L2 (CD273 or B7-DC) [20], expressed on antigen presenting cells (APC) which, in the case of sepsis, also include hepatocytes [21]. We, therefore, determined the expression of these co-inhibitory factors on hepatocytes isolated from mice with CLP sepsis. Expression of PD-L1 was downregulated at the protein level (Figure 2A) and its hepatocyte cell surface expression was diminished (Figure 2B). PD-L1 mRNA expression was also downregulated in livers from CLP-operated compared with sham-operated mice (Figure 2C). In contrast, protein and mRNA expression of PD-1, the PD-L1 receptor, remained unaltered (Figures 2D and 2E). Twenty- four hours after CLP, total hepatic protein expression of PD-L2 was very low (data not shown) but cell surface expression of the protein remained unchanged (Figure 2F). It is well established that Fas ligand/receptor interactions play an important role in regulating immune responses in mice and humans [22]. The hepatic expression of Fas protein or mRNA, however, was not changed 24 h after CLP (Figures 2D and 2G).
Expression of PD-L1, PD-L2, Fas, and PD-1 in the liver. Twenty-four hours following sham- or CLP-operation, mice were sacrificed. Livers were removed to prepare single cell suspensions. Total lysates were prepared in (A) and (D) to analyze expression of PD-L1, PD-1, and Fas. Hepatocyte specific surface expression of (B) PD-L1 and (F) PD-L2 was assessed by FACS analysis. In (C), (E), and (G) mRNA was isolated from total lysates as described in “Methods”. mRNA expression of (C) PD-L1, (E) PD-1, and (G) Fas was determined by quantitative PCR. The untreated control was set as 1 in (C) and the sham mice in (E) and (G). All experiments were performed at least five times. Data represent the means ± SD (*p<0.05) or show representative blots.
Maintaining PD-L1 expression inhibits liver damage after CLP
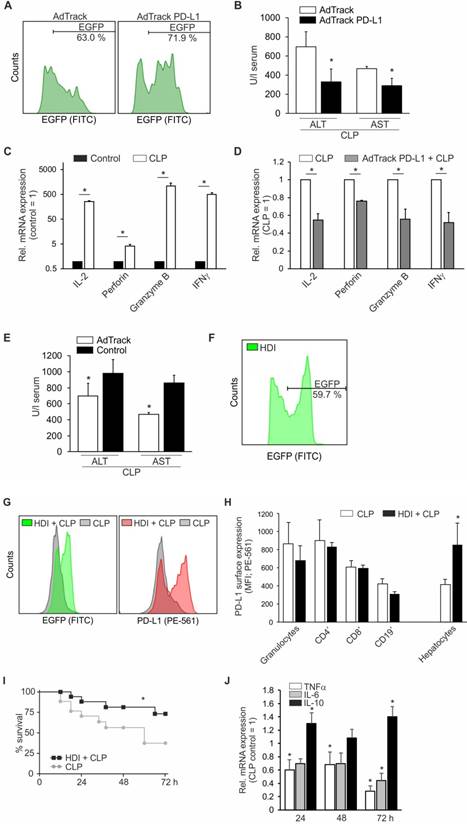
To prove a causal role for PD-L1 in sepsis, we established an adenoviral approach to overexpress PD-L1 in the mouse liver in vivo. Following optimization of the protocol, we achieved a transduction efficiency of roughly 70% in hepatocytes (Figure 3A). Following transduction, which consequently induces an anti-viral immune response, mice were allowed to recover for four days. Afterwards, CLP was initiated, and mice were sacrificed twenty-four hours later to determine disease severity by the liver damage markers ALT and AST. As shown in Figure 3B, adenoviral PD-L1 overexpression attenuated liver damage, i.e. reducing ALT/ AST level significantly. In support of our assumption, cytotoxic T cell expression of IL-2, perforin, granzyme B, and IFNγ, which was induced following CLP-operation (Figure 3C), compared to untreated mice, was significantly reduced in mice which had been subjected to adenoviral transduction to overexpress PD-L1 (Figure 3D) before CLP-operation. However, liver damage in control adenovirus transduced mice was not as high as without adenoviral treatment (Figure 3E). Taking this into consideration, we used a second approach to overexpress PD-L1 in the mice livers. With a sleeping beauty transposon-based vector system, which was applied to the animal by hydrodynamic injection (HDI), we achieved a transduction efficiency of roughly 60% of all hepatocytes (Figure 3F). For liver recovery and establishment of transgene expression, mice were allowed to rest for 10 days. Then, CLP was induced and mouse survival was followed for up to seventy-two hours. As shown in Figures 3G und 3H, in mice surviving seventy-two hours after CLP, PD-L1 expression was still significantly upregulated, i.e. about twofold, in approximately 60% of all hepatocytes with HDI before CLP, compared to only CLP-operated animals. In contrast, PD-L1 expression on granulocytes, CD4+ and CD8+ T cells as well as CD19+ B cells was not altered (Figure 3H). In extension to the data obtained following adenoviral PD-L1 overexpression, survival of CLP-operated mice with a prior HDI, was significantly improved (Figure 3I). Moreover, the induction of the mRNA of pro-inflammatory cytokines such as TNF-α and IL-6 was significantly reduced following HDI compared to only CLP-treated animals, whereas the mRNA amount of anti-inflammatory IL-10 increased (Figure 3J).
Maintaining PD-L1 expression ameliorates liver damage and survival following CLP. Mice were injected intravenously with 5x1010 adenoviral particles encoding EGFP (AdTrack) or PD-L1 EGFP (AdTrack PD-L1). Four days following administration of these particles, mice were subjected to polymicrobial sepsis by CLP operation. After twenty-four hours, mice were sacrificed, livers were removed and blood was collected. (A) Transduction efficiency was determined by FACS analysis of liver single cell suspensions, gated for non-immune cells, i.e. CD45- negative cells. One representative result is shown. (B) Serum was isolated from blood and ALT/AST release determined as described in “Methods”. Results of four mice each are shown as means ± SD (*p<0.05). (C) and (D) cytotoxic T cells were enriched from livers of control and CLP-treated mice without or with (D) AdTrack PD-L1 administration as described in (A). Following mRNA isolation from the purified T cells, mRNA expression of IL-2, perforin, granzyme B, and IFNγ was analyzed by quantitative PCR. T cell expression of control mice was set as 1 in (C) and expression of T cells derived from CLP-operated mice was set as 1 in (D). Results of four mice are shown as means ± SD (*p<0.05). (E) Comparison of ALT/AST release in mice 24 h following CLP operation without (Control) and with adenoviral pretreatment (AdTrack). (F)-(J) Mice were injected via the tail vein with 10% of body weight PBS containing the transposon-based PD-L1 expression cassette in combination with an EGFP cassette separated by an internal ribosomal entry site (IRES), which allows translation of two proteins. After ten days of recovery and transgene establishment (as shown for EGFP in (F)), CLP operation was performed. Mice surviving for seventy-two hours were sacrificed and PD-L1- and EGFP expression were determined by FACS analysis of CD45- ((G), quantification is shown in (H), right panel) vs. CD45+ cells. These leukocytes were further divided into subpopulations of granulocytes, CD4+ and CD8+ T cells as well as CD19+ B cells ((H), left panel). (I) Survival of mice was followed up to 72 h (n=9, *p<0.05). (J) mRNA of hepatocytes was isolated and qPCR for mRNA expression of TNFα, IL-6, and IL-10 was performed. Expression of hepatocytes from control CLP mice was set as 1. Results of four mice are shown as means ± SD (*p<0.05)
Pharmacological mimicry of the effect of PD-L1 expression
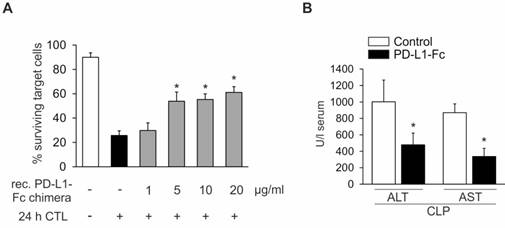
Sustaining PD-L1 expression as a therapeutic approach might also be achieved by exogenously adding PD-L1. To test this hypothesis, we added simultaneously to an in vitro cytotoxicity assay with OVA257-264 pulsed Hepa1-6 target cells both recombinant PD-L1-Fc chimera and allogenic CTL effector cells derived from OT-I mice, thereby mimicking sepsis-dependent CTL activation. As shown in Figure 4A, the PD-L1-Fc chimera dose-dependently inhibited CTL-mediated cytotoxicity. While 1 µg/ml recombinant PD-L1-Fc chimera did not alter target cell killing, 5 µg/ml enhanced target cell survival to approximately 50%. Increasing the concentration of recombinant PD-L1- Fc chimera up to 20 µg/ml did not further enhance survival. Translating this in vitro result to the in vivo situation, we applied recombinant PD-L1-Fc chimera intravenously (i.v.) into the tail vein, directly after the CLP-operation. Twenty-four hours later, blood was collected to prepare serum followed by ALT/AST measurements. Recombinant PD-L1-Fc chimera significantly reduced ALT/AST release, which is indicative of an improved septic outcome in vivo (Figure 4B).
Recombinant PD-L1-Fc chimera prevents CTL-dependent liver damage. (A) Cytotoxic T cell-dependent hepatocyte killing was determined using Hepa1-6 cells as target cells and CD8+ T cells derived from OT-I mice as effector cells. CellTrackerOrange stained Hepa1-6 cells were pulsed for two hours with the OVA257-264 peptide. Afterwards, Hepa1-6 cells were co-cultured with enriched CD8+ T cells derived from the spleen of OT-I mice at a ratio of 5:1 (effector: target cells). In parallel, recombinant PD-L1-Fc chimera was added at the indicated concentrations. The number of surviving target cells was examined by FACS analysis. Data from five independent experiments are provided. Data represent the means ± SD (*p<0.05). (B) Wild type mice were subjected to CLP-operation. Directly afterwards, 12 mg/kg PD-L1-Fc were administered intravenously. PBS alone was administered as a solvent control. Liver damage following CLP-operation was assessed by determining the ALT/AST release into the serum as described in “Methods” (PD-L1-Fc treated vs. control; CLP; n=5/5, *p<0.05).
Cell wall components of gram-positive and gram-negative bacteria downregulate PD-L1 expression in Hepa1-6 cells
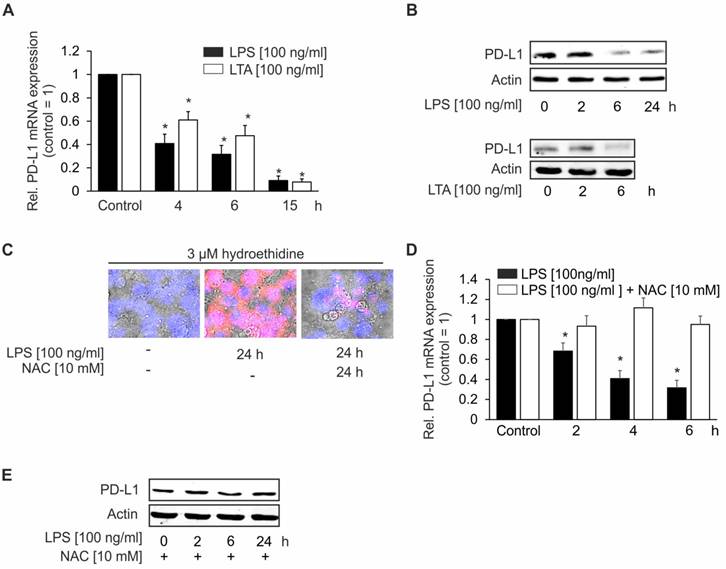
Primary cultures of hepatocytes express the mRNA for all TLRs and respond to TLR2 and TLR4 ligands [23]. Therefore, we hypothesized that bacterial components, which are present in the liver during sepsis, may decrease PD-L1 expression in hepatocytes via TLR-dependent signaling. For mechanistic studies, we used a cell culture model of the murine hepatoma cell line Hepa 1-6 [16]. These cells express TLR2 and -4 [24, 25]. To mimic bacterial infection, we treated Hepa1-6 cells with LPS, a cell wall component of gram-negative bacteria and LTA, a cell wall constituent of gram-positive bacteria. As depicted in Figure 5, LPS and LTA time-dependently downregulated mRNA (Figure 5A) and protein (Figures 5B) of PD-L1. Based on these observations, we inferred that TLR-dependent downregulation of PD-L1 may increase the susceptibility of hepatocytes to CTL. Mechanistically, we observed that reactive oxygen species (ROS) are generated in LPS [100 ng/ml] stimulated Hepa1-6 cells (Figure 5C, middle picture), which was significantly reduced when 10 mM N-acetylcysteine (NAC) were added simultaneously (Figure 5C, right picture). To elucidate whether ROS are important to downregulate PD-L1 expression, we performed a set of experiments using LPS and NAC in combination to stimulate Hepa1-6 cells. Indeed, NAC addition prevented LPS-dependent PD-L1 downregulation in Hepa1-6 cells on mRNA (Figure 5D) as well as on protein level (Figure 5E).
PD-L1 expression in Hepa1-6 cells following LPS- or LTA-stimulation. Hepa1-6 cells were stimulated for the indicated times with 100 ng/ml LPS or 100 ng/ml LTA. Afterwards, cells were harvested and mRNA or proteins were recovered as described in “Methods”. mRNA expression of (A) PD-L1 was analyzed by quantitative PCR. 18s rRNA was used as a house-keeping gene. (B) PD-L1 protein expression following LPS and LTA stimulation was determined by Western analysis. (C) Reactive oxygen species (ROS) formation was determined in Hepa1-6 cells by seeding Hepa1-6 cells on slides in 10 cm petri-dishes. After twenty-four hours, cells were incubated simultaneously with LPS [100 ng/ml] and the redox-sensitive dye hydroethidine [3 µM]. NAC [10 mM] was added in parallel where indicated. ROS generation was assessed twenty-four hours later by fluorescence microscopy. Cells were counterstained with DAPI. For determining PD-L1 (D) mRNA and (E) protein, Hepa1-6 cells were treated for the indicated times with LPS [100 ng/ml] with or without NAC [10 mM]. Afterwards, cells were harvested and mRNA and proteins were prepared as described in “Methods”. (D) PD-L1 mRNA expression was analyzed by qPCR and (E) PD-L1 protein expression was determined by Western analysis. All experiments were performed at least five times. Data represent the means ± SD (*p<0.05) or show a representative blot.
Source of ROS-dependent PD-L1 downregulation
As with hepatocytes, Hepa1-6 cells also express Nox4 [26] and these cells also produce H2O2 constitutively. To determine the contribution of Nox4 to the signaling of these cells, we first incubated Hepa1-6 cells with the Nox4 inhibitor GKT137831 [10 µM] for 24 h without any further treatment [27]. Nox4 inhibition increased PD-L1 surface expression by up to 50% in Hepa1-6 cells (Figure 6A). Similarly, and importantly, also in livers of global NOX4-knockout mice, PD-L1 was upregulated at both the protein and mRNA level, as compared to wild type controls (Figures 6B-D). Densitometric quantification of protein data demonstrated a roughly twofold higher expression of PD-L1 in livers of NOX4-knockout mice (Figure 6C). Using NOX4-knockout mice in the CLP model, we observed sepsis-induced downregulation of PD-L1 expression in both, wild type as well as knockout mice (Figure 6E, right columns). However, expression of PD-L1 in NOX4-deficient cells remained higher compared to wild type mice 24 h following sepsis initiation. Moreover, disease severity was not attenuated in NOX4-knockout mice (Figure 6F). Therefore, blocking Nox4 activity, though modifying PD-L1 expression, is not sufficient to improve sepsis survival.
Source of ROS downregulating PD-L1 expression. (A) Hepa1-6 cells were incubated with the NOX4 inhibitor GKT13781 for 24 h. Afterwards cells were harvested and surface expression of PD-L1 was determined by FACS-analysis. (B) Western blotting. A representative blot is shown. Quantification of data of PD-L1 expression (WT vs. NOX4-KO; n=5/5; *p<0.05) is provided in (C). PD-L1 mRNA expression is shown in (D). PD-L1 expression on hepatocytes following polymicrobial sepsis initiation by cecal ligation and puncture (CLP) was studied in hepatic single cell suspensions by FACS-analysis gating for CD45-, i.e. non-immune cells as described in “Methods”. Data are shown in (E), (sham vs. CLP, WT sham was set as 1; n=5/5/5/5; *p<0.05). (F) Liver damage in global NOX4-knockout mice after CLP operation followed by the release of ALT/AST into the serum as described in “Experimental Procedures” (NOX4-KO; sham vs. CLP; n=5/5, *p<0.05). In (G) and (H) mice with a global NOX2- knockout (NOX2-KO) and a myeloid lineage-specific knockout (LysM-Cre NOX2-KO) were used as well as wild type littermates (WT). Twenty-four hours following CLP- or sham-operation, mice were sacrificed. Blood was collected and the liver was removed. (G) PD-L1 protein expression on hepatocytes was determined in single cell suspensions of livers by FACS-analysis gating for CD45-, i.e. non-immune cells as described in “Methods”. (WT vs. NOX2-KO vs. LysM-Cre NOX2-KO; sham vs. CLP; sham treated WT is set as 1; n=5/5/5; *p<0.05). (H) Serum was isolated from blood and ALT/AST release was assessed as described in “Methods” (LysM-Cre NOX2-KO vs. NOX2-KO; sham vs. CLP; n=5/5/5/5; *p<0.05).
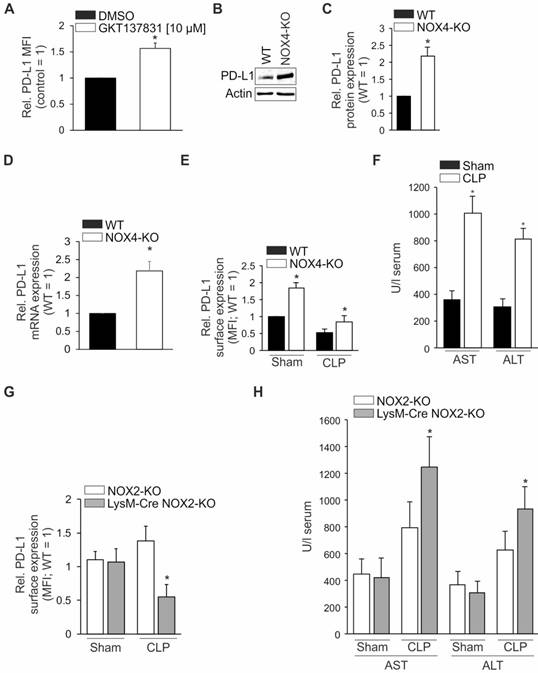
Next, we tested global NOX2-deficient (NOX2-KO), as well as mice with a myeloid lineage (LysM-Cre NOX2-KO) specific NOX2-knockout in our sepsis model. As shown in Figure 6G, the expression of PD-L1 in hepatocytes was similar 24 h following sham operation in both the two genotypes. Interestingly, 24 h after CLP, the expression of PD-L1 was downregulated in mice with a NOX-2 deletion in the myeloid lineage (grey column) similar to wild type mice (Figures 2B vs. 6G). In mice with a global NOX2-knockout (white column), PD-L1 expression remained high. Liver damage markers ALT and AST, released into the serum, revealed a significant increase in mice with a myeloid lineage NOX2-deletion (Figure 6H, grey columns) but activities remained low in global NOX2-knockout mice (Figure 6H, white columns).
Accumulation of CTLs is attenuated in livers of septic mice when PD-L1 expression is retained
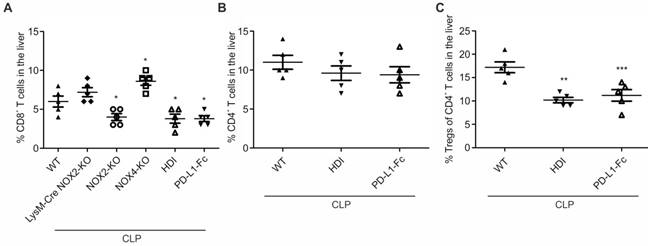
In a final set of experiments, we determined the numbers of CD8+ and regulatory T cells. Returning to our initial observation that the CTL percentage was increased in livers from mice following CLP (Figure 1) compared to sham-treated mice, we were interested to prove our hypothesis that CTLs are involved in liver damage following CLP. 24 h following CLP, CTL numbers increased in livers of LysM-Cre NOX2-KO, and NOX4-KO mice, similarly to wildtype (WT) mice (Figure 7A). In contrast, CTL numbers remained significantly below those in wildtype mice, in global NOX2-KO (NOX2-KO) mice, HDI-treated animals (HDI) as well as mice which received recombinant PD-L1-Fc (PD-L1-Fc) directly following CLP. Interestingly, the percentage of CD4+ T cells was not altered by HDI or PD-L1-Fc treatment (Figure 7B). However, the percentage of regulatory T cells (Tregs) was significantly reduced following these two treatments (Figure 7C).
% of CD8+, CD4+ T cells as well as Tregs in livers of septic mice. Operation was performed (A) in mice with the indicated genetic background (WT, LysM-Cre NOX2-KO, NOX2-KO, NOX-KO). Moreover in (A)-(C), wildtype mice treated with hydrodynamic injection (HDI) as described in “Methods” to overexpress PD-L1 in the liver, ten days before and wildtype mice, which directly after CLP-operation received recombinant 12 mg/kg PD-L1-Fc (PD-L1-Fc) were used. Twenty-four hours following CLP mice were sacrificed. Livers were removed to prepare single cell suspensions. Cell subpopulations were determined by FACS analysis. Data from five mice per treatment are shown and represent the mean ± SD (*p<0.05, **p<0.01, ***p<0.001).
Discussion
Recent clinical trials have provided evidence that the use of PD-1 neutralizing antibodies, such as nivolumab, prevents CD8+ T cell tolerance toward tumor cells, consequently improving the anti-tumor immune response [28-30]. Taking into consideration that during sepsis T cells are often depleted, thus contributing to immune paralysis a hallmark of sepsis progression, a similar approach would seem reasonable to maintain T cell immunity in this condition. This would alleviate the hypo-inflammatory phase of sepsis, most likely improving sepsis outcome. Thus, neutralizing antibodies towards PD-L1, intended to block its binding to PD-1 expressed on T cells, have been used previously in animal sepsis models and rescued T cell depletion [31, 32]. Chang et al. observed that in vitro treatment of cells derived from septic patients with anti-PD-1 or anti-PD-L1 antibodies decreased apoptosis and proposed that this would improve immune cell function in septic patients [33]. Besides monocytes [33, 34] , neutrophils also might be involved in sepsis-induced immunosuppression by PD-L1 upregulation [31]. Our approach described here exclusively focuses on the role of CD8+ T cells at 24 h following sepsis initiation, which have been shown previously, in the polymicrobial mouse sepsis model triggered by CLP, to contribute to liver damage [35] and to be activated in an autoimmune fashion. Blocking this immune response by activating the anti-inflammatory nuclear receptor PPARγ improved septic outcome [12]. This relatively early time point of 24 h after CLP operation might explain differences from the above mentioned studies, in which PD-L1 signaling to PD-1 was inhibited to improve sepsis outcome.
Insights into the mechanism causing CTL activation during sepsis remain obscure. In our setup, we identified downregulation of hepatocyte PD-L1 in response to CLP as a possible mechanism. In analogy to our data, the loss of PD-L1 expression by recipient parenchymal cells leads to expansion of infiltrating donor CD8+ T cells [36]. Zhu et al. showed immunohistochemically, in the CLP model an increase in PD-L1 expression [37]. However, the authors speculated that upregulation of PD-L1 was mainly on KCs and infiltrated lymphocytes. The expression of PD-L2 and the receptor for both of the co-inhibitory proteins PD-L1 and PD-L2, i.e. PD-1, in contrast, remained unaltered following the CLP operation. Moreover, it is well established that Fas is an important mediator of CD8+ T-cell dependent cytotoxicity, including that during sepsis [35] but in our model Fas expression was not changed. This is in line with our recent report. There, we found that in the CLP model, CTL-dependent cytotoxicity was not blocked by a Fas-neutralizing antibody or the SuperFasLigand in an ex vivo cytotoxicity assay [12], ruling out the involvement of the Fas/Fas-L system in our setup.
To translate our in vitro data to an in vivo setting, we used an adenoviral gene transfer approach to overexpress PD-L1 in the liver of mice. Although intravenous application of adenoviral particles has not been developed specifically for liver transduction, the majority of the viral particles are retained in the liver, thus effectively transducing liver tissue [38]. Using these mice transduced with PD-L1 EGFP, we observed in agreement with our hypothesis, that the release, 24 h following CLP, of the hepatic damage markers AST and ALT was significantly attenuated compared with EGFP transduced control mice. Because the anti-viral immune response is activated in mice following intravenous adenovirus application, mice were left untreated for four days after adenovirus injection, to allow the immune system to recover. However, disease severity followed by ALT and AST release into the serum 24 h after CLP operation in the EGFP-transduced mice (AdTrack) was not as high as in mice without gene transfer (Figure 3E). Therefore, minor effects of adenovirus infection, possibly involving desensitization of the immune system, cannot be excluded completely.
To circumvent this problem, we used a second approach to overexpress PD-L1 in the liver. With hydrodynamic injection (HDI), a technique which was originally described in 2005 by Carlson et al. [19], we used a transposon-based sleeping beauty vector system to achieve transduction in hepatocytes. To allow liver recovery and transgene establishment, mice could rest for 10 days. In addition to our demonstration that maintaining PD-L1 expression on hepatocytes prevents liver damage, mouse survival was significantly improved when HDI was performed before CLP-operation. In line with this finding, the cytokine storm [39], which is also indicative for the human situation [40], was significantly reduced compared to that in mice, which suffered only from CLP. Interestingly, liver expression of the pro-inflammatory cytokines TNFα and IL-6 was significantly reduced. Correspondingly, expression of anti-inflammatory cytokines such as IL-10 was significantly elevated. These data support the conclusion that CLP-dependent inflammation in the liver was attenuated by maintaining PD-L1 expression on hepatocytes. Results from other groups also support the notion that abolition of the cytokine storm is a marker of diminished inflammation and associated with improved survival during sepsis [39, 41, 42].
As these two gene transfer approaches cannot be performed in humans, we took advantage of a recombinant PD-L1-Fc chimera [43], which when exogenously added, should saturate PD-1 binding moieties exposed by CD8+ T cells, thus, mimicking PD-L1 expression on antigen presenting cells and consequently inhibit CTL activation. In our in vitro cytotoxicity assay, this treatment significantly reduced CTL-dependent cytotoxicity. Altering PD-L1/PD-1 signaling has been approached mainly to inhibit PD-1, thereby provoking CTL activation to kill tumor cells [44]. For this purpose, PD-1 inhibiting antibodies have entered clinical trials [45], whereas recombinant PD-L1 has not been used so far to prevent CTL-dependent cytotoxicity. Interestingly, recombinant PD-L1-Fc, already at 5 µg/ml, was most effective with no further decrease in CTL-mediated cytotoxicity at higher concentrations. In the CLP mouse model, we administered 12 mg/kg, directly following CLP-operation. With this approach, liver damage, as estimated by ALT/AST release, was significantly reduced. Whereas in the present study, recombinant PD-L1-Fc chimera was only applied once, further studies are necessary to characterize whether sequential administration will improve disease outcome. Because regulatory T cells (Treg) are involved in tolerance induction in CTLs [46], we determined their percentage in liver T cells. Interestingly, our data (Figure 7) support the assumption that during CLP-mediated sepsis the percentage of liver Tregs is increased after 24 h. In contrast, PD-L1-Fc application and functional expression of PD-L1 on hepatocytes following hydrodynamic injection (HDI), both decreased the percentage of Tregs in the liver. This is in line with the report of Cao et al. showing in wildtype mice an increase of Tregs 24 h following CLP [47]. Moreover, neutralization of Tregs has been found to provoke improved survival [48], as in some analogy was observed in our setting following PD-L1-Fc treatment or HDI-dependent PD-L1 overexpression in hepatocytes. It will be interesting to determine putative differences in the expression profile of Tregs isolated from CLP-treated control animals compared to mice with restored PD-L1 expression or following recombinant PD-L1-Fc application. Also a time course following the Treg number, would be interesting taking the recent study of Nascimento et al. into consideration, who demonstratied a role of Treg expansion during sepsis, contributing to long-term immunosuppression [49].
To characterize mechanisms responsible for PD-L1 downregulation, we studied the involvement of ROS formation. First evidence for this possibility came from in vitro experiments with the glutathione precursor N-acetylcysteine (NAC), which restored PD-L1 expression in response to LPS- or LTA-treatment. NAC therapy has already been used to treat sepsis patients [50]. Although protective in animal models [51, 52], NAC did not improve patient survival, while inhibiting the pro-inflammatory transcription factor NF-κB [51]. In none of these previous studies was expression of PD-L1 determined. Further proof for our hypothesis was obtained from in vivo experiments using global NOX4-deficient mice. In these mice, basal PD-L1 expression was significantly higher compared with wild type controls. This suggested a role for Nox4 generated ROS, most likely H2O2 [53], but possibly superoxide (O2‑) as well [26], in fine-tuning PD-L1 expression in the control situation. NOX4-knockout, however, did not protect mice against CLP-mediated sepsis, although hepatocyte NOX4 is involved in liver fibrosis [54]. Therefore, we employed mice deficient in the classical phagocytic NADPH oxidase, NOX2. Nox2 is expressed in cells of myeloid origin, including neutrophils or liver resident macrophages, namely Kupffer cells, but also in other cells, like hepatocytes [55] or endothelial cells [56]. Thus, to distinguish the cellular source of the ROS involved in inhibition of PD-L1 expression, we used global NOX2-knockout (NOX2-KO) mice as well as myeloid-specific NOX2-deficient (LysM-Cre NOX2-KO) mice. Following CLP operation, only global NOX2-knockout mice were protected, whereas Kupffer cell-dependent Nox2 activity was not important. These data are analogous to those in a recent report, published by Spencer et al. [57], claiming that hepatocyte-mediated TNFα production following liver ischemia-reperfusion, is caused, at least in part, by Nox2-dependent ROS formation in hepatocytes, activating NF-κB.
We cannot completely exclude a role of T cell-dependent ROS generation. Although not very likely, in mouse as well as human T cells, Nox-2 activity has been shown during autoimmune diseases, such as systemic sclerosis and type 1 diabetes [58, 59]. Taken together, we suggest that in hepatocytes, Nox2 is activated in response to LPS or LTA via TLR2/4-mediated O2- generation, downregulating PD-L1 expression at the mRNA and protein level. The absence of this co-inhibitory molecule allows activation of CTLs in an autoimmune fashion, provoking liver damage. Blocking hepatocyte ROS generation restores PD-L1 expression, consequently maintaining CTL in a tolerant state. As a more applicable therapeutic regimen, recombinant PD-L1-Fc chimera, administered intravenously during sepsis, blocks CTL PD-1 molecules, thus, inhibiting autoimmune activation of CD8+ cells.
Conclusions
We conclude that maintaining PD-L1 expression in the liver or pharmacologically mimicking PD-L1 using a recombinant protein improves sepsis survival in the CLP-mouse model. Therefore, translating our treatment regime to humans might open a new therapy approach to treat septic patients. Considering that PD-L1 is upregulated on classical antigen presenting cells such as macrophages or dendritic cells during sepsis progression, consequently contributing to immune paralysis by T cell depletion, the time point for modulating PD-L1 expression is an important issue.
Abbreviations
ALT: alanine aminotransferase; AST: aspartate aminotransferase; APC: allophycocyanin; B7-H: B7-homologue; CD: cluster of differentiation; CLP: cecal ligation and puncture; CMV: cytomegaly virus; CTL: cytotoxic T lymphocyte; DMSO: dimethyl sulfoxide; EGFP: enhanced green fluorescence protein; Fc: fragment crystallizable; FCS: fetal calf serum; FITC: fluorescein-isothiocyanate; HBV: hepatitis B virus; HDI: hydrodynamic injection; IL: interleukin; IFN: interferon; IRES: internal ribosomal entry site; KO: knock out; LPS: lipopolysaccharide; LTA: lipoteichoic acid; LysM: lysozyme M; MHC-I: major histocompatibility complex I; NAC: N-acetylcysteine; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B-cells; NOX: NADPH oxidase; OVA: ovalbumin; PD-1: programmed cell death protein 1; PD-L1: PD-1 ligand 1; PE: phycoerythrin; PPARγ: peroxisome proliferator-activated receptor γ; qPCR: quantitative polymerase chain reaction; ROS: reactive oxygen species; RT: room temperature; SD: standard deviation; TCR: T cell receptor; TLR: Toll-like receptor; TNFα: tumor necrosis factor α; Treg: regulatory T cell; WT: wild type.
Acknowledgements
We thank Nadja Wallner for excellent technical assistance. This research was supported by a grant from the Deutsche Forschungsgemeinschaft (KN493/ 13-1 and SFB815 TP3, TP8). The work was supported by the Else Kröner-Fresenius Foundation (EKFS), Research Training Group Translational Research Innovation-Pharma (TRIP) and the Landesoffensive zur Entwicklung wissenschaftlich-ökonomischer Exzellenz (LOEWE) Research Centre for Translational Medicine and Pharmacology.
Author Contributions
AvK, KS, RPB, VAJK, and BB developed the study concept and design with initial contributions from MJP. KS, RPB, and AMS provided expertise in ROS signaling. UC, EH, BF, BG, KA, and BMP provided expertise in adenoviral gene transfer. FF, MP, and FG provided expertise in hydrodynamic injection. AvK, AE, MP, AS, LK, TK, BG, KA, LKS, SNS, MH, SG, and MS performed experiments and analyzed the data. AvK wrote the manuscript and KS, RPB, UC, BMP, AW, AMS, MJP, and BB provided critical review of the manuscript.
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Vincent JL, Marshall JC, Namendys-Silva SA, Francois B, Martin-Loeches I, Lipman J. et al. Assessment of the worldwide burden of critical illness. Lancet Respir Med. 2014;2:380-386
2. Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:840-851
3. Vincent JL, Opal SM, Marshall JC, Tracey KJ. Sepsis definitions. Lancet. 2013;381:774-775
4. Marquez-Velasco R, Bojalil R, Buelna A, Flores-Guzman F, Estevez-Ramirez J, Laguna J. et al. Anti-tumor necrosis factor alpha F(ab')2 antibody fragments protect in murine polymicrobial sepsis. Inflamm Res. 2006;55:378-384
5. Reinhart K, Karzai W. Anti-tumor necrosis factor therapy in sepsis. Crit Care Med. 2001;29:S121-5
6. Otto GP, Sossdorf M, Claus RA, Rödel J, Menge K, Reinhart K. et al. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care. 2011;15:R183
7. Docke WD, Randow F, Syrbe U, Krausch D, Asadullah K, Reinke P. et al. Monocyte deactivation in septic patients. Nat Med. 1997;3:678-681
8. Meisel C, Schefold JC, Pschowski R, Baumann T, Hetzger K, Gregor J. et al. Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression. Am J Respir Crit Care Med. 2009;180:640-648
9. Schmidt MV, Paulus P, Kuhn AM, Weigert A, Morbitzer V, Zacharowski K. et al. Peroxisome proliferator-activated receptor gamma-induced T cell apoptosis reduces survival during polymicrobial sepsis. Am J Respir Crit Care Med. 2011;184:64-74
10. Wesche-Soldato DE, Swan RZ, Chung CS, Ayala A. The apoptotic pathway as a therapeutic target in sepsis. Curr Drug Targets. 2007;8:493-500
11. Hotchkiss RS, Opal S. Immunotherapy for sepsis-a new approach against an ancient foe. N Engl J Med. 2010;363:87-89
12. von Knethen A, Sha L K, Knape T, Kuchler L, Giegerich AK, Schulz M. et al. Activation of the peroxisome proliferator-activated receptor gamma counteracts sepsis-induced T cell cytotoxicity toward alloantigenic target cells. J Mol Med. 2015;93:633-644
13. Weissmann N, Sydykov A, Kalwa H, Storch U, Fuchs B, Mederos y Schnitzler M. et al. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat Commun. 2012;3:649
14. Hume DA. Applications of myeloid-specific promoters in transgenic mice support in vivo imaging and functional genomics but do not support the concept of distinct macrophage and dendritic cell lineages or roles in immunity. J Leukoc Biol. 2011;89:525-538
15. Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31-36
16. Darlington GJ. Liver cell lines. Methods Enzymol. 1987;151:19-38
17. Demaison C, Parsley K, Brouns G, Scherr M, Battmer K, Kinnon C. et al. High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum Gene Ther. 2002;13:803-813
18. Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J. et al. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc. 2007;2:1236-1247
19. Carlson CM, Frandsen JL, Kirchhof N, McIvor RS, Largaespada DA. Somatic integration of an oncogene-harboring sleeping beauty transposon models liver tumor development in the mouse. Proc Natl Acad Sci U S A. 2005;102:17059-17064
20. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239-245
21. Ueki S, Castellaneta A, Yoshida O, Ozaki K, Zhang M, Kimura S. et al. Hepatic B7 homolog 1 expression is essential for controlling cold ischemia/reperfusion injury after mouse liver transplantation. Hepatology. 2011;54:216-228
22. Galle PR, Hofmann WJ, Walczak H, Schaller H, Otto G, Stremmel W. et al. Involvement of the CD95 (APO-1/Fas) receptor and ligand in liver damage. J Exp Med. 1995;182:1223-1230
23. Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease. Hepatology. 2008;48:322-335
24. Matsumura T, Ito A, Takii T, Hayashi H, Onozaki K. Endotoxin and cytokine regulation of toll-like receptor (TLR) 2 and TLR4 gene expression in murine liver and hepatocytes. J Interferon Cytokine Res. 2000;20:915-921
25. Romics L Jr, Dolganiuc A Kodys K, Drechsler Y Oak S, Velayudham A et al. Selective priming to Toll-like receptor 4 (TLR4), not TLR2, ligands by P. acnes involves up-regulation of MD-2 in mice. Hepatology. 2004;40:555-564
26. Boudreau HE, Emerson SU, Korzeniowska A, Jendrysik MA, Leto TL. Hepatitis C virus (HCV) proteins induce NADPH oxidase 4 expression in a transforming growth factor beta-dependent manner. J Virol. 2009;83:12934-12946
27. Jiang JX, Chen X, Serizawa N, Szyndralewiez C, Page P, Schröder K. et al. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med. 2012;53:289-296
28. Larkin J, Lao CD, Urba WJ, McDermott DF, Horak C, Jiang J. et al. Efficacy and Safety of Nivolumab in Patients With BRAF V600 Mutant and BRAF Wild-Type Advanced Melanoma. JAMA Oncol. 2015;1:433-440
29. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD. et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23-34
30. Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B. et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037). Lancet Oncol. 2015;16:375-384
31. Wang JF, Li JB, Zhao YJ, Yi WJ, Bian JJ, Wan XJ. et al. Up-regulation of programmed cell death 1 ligand 1 on neutrophils may be involved in sepsis-induced immunosuppression. Anesthesiology. 2015;122:852-863
32. Zhang Y, Zhou Y, Lou J, Li J, Bo L, Zhu K. et al. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care. 2010;14:R220
33. Chang K, Svabek C, Vazquez-Guillamet C, Sato B, Rasche D, Wilson S. et al. Targeting the programmed cell death 1: programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit Care. 2014;18:R3
34. Zhang Y, Li J, Lou J, Zhou Y, Bo L, Zhu J. et al. Upregulation of programmed death-1 on T cells and programmed death ligand-1 on monocytes in septic shock patients. Crit Care. 2011;15:R70
35. Wesche-Soldato DE, Chung CS, Gregory SH, Salazar-Mather TP, Ayala CA, Ayala A. CD8+ T cells promote inflammation and apoptosis in the liver after sepsis. Am J Pathol. 2007;171:87-96
36. Li X, Deng R, He W, Liu C, Wang M, Young J. et al. Loss of B7-H1 expression by recipient parenchymal cells leads to expansion of infiltrating donor CD8+ T cells and persistence of graft-versus-host disease. J Immunol. 2012;188:724-734
37. Zhu W, Bao R, Fan X, Tao T, Zhu J, Wang J. et al. PD-L1 blockade attenuated sepsis-induced liver injury in a mouse cecal ligation and puncture model. Mediators Inflamm. 2013;2013:361501
38. van Dijk KW, Kypreos KE, Fallaux FJ, Hageman J. Adenovirus-mediated gene transfer. Methods Mol Biol. 2011;693:321-343
39. Huber-Lang M, Barratt-Due A, Pischke SE, Sandanger Ø, Nilsson PH, Nunn MA. et al. Double blockade of CD14 and complement C5 abolishes the cytokine storm and improves morbidity and survival in polymicrobial sepsis in mice. J Immunol. 2014;192:5324-5331
40. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. 2017;39:517-528
41. Danahy DB, Anthony SM, Jensen IJ, Hartwig SM, Shan Q, Xue HH. et al. Polymicrobial sepsis impairs bystander recruitment of effector cells to infected skin despite optimal sensing and alarming function of skin resident memory CD8 T cells. PLoS Pathog. 2017;13:e1006569
42. Wang X, Huang W, Yang Y, Wang Y, Peng T, Chang J. et al. Loss of duplexmiR-223 (5p and 3p) aggravates myocardial depression and mortality in polymicrobial sepsis. Biochim Biophys Acta. 2014;1842:701-711
43. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111-122
44. Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24:207-212
45. Sunshine J, Taube JM. PD-1/PD-L1 inhibitors. Curr Opin Pharmacol. 2015;23:32-38
46. Kalekar LA, Mueller DL. Relationship between CD4 Regulatory T Cells and Anergy In Vivo. J Immunol. 2017;198:2527-2533
47. Cao C, Chai Y, Shou S, Wang J, Huang Y, Ma T. Toll-like receptor 4 deficiency increases resistance in sepsis-induced immune dysfunction. Int Immunopharmacol. 2018;54:169-176
48. Hiraki S, Ono S, Tsujimoto H, Kinoshita M, Takahata R, Miyazaki H. et al. Neutralization of interleukin-10 or transforming growth factor-β decreases the percentages of CD4+ CD25+ Foxp3+ regulatory T cells in septic mice, thereby leading to an improved survival. Surgery. 2012;151:313-322
49. Nascimento DC, Melo PH, Piñeros AR, Ferreira RG, Colón DF, Donate PB. et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat Commun. 2017;8:14919
50. Szakmany T, Hauser B, Radermacher P. N-acetylcysteine for sepsis and systemic inflammatory response in adults. Cochrane Database Syst Rev. 2012;9:CD006616
51. Paterson RL, Galley HF, Webster NR. The effect of N-acetylcysteine on nuclear factor-kappa B activation, interleukin-6, interleukin-8, and intercellular adhesion molecule-1 expression in patients with sepsis. Crit Care Med. 2003;31:2574-2578
52. Ozdulger A, Cinel I, Koksel O, Cinel L, Avlan D, Unlu A. et al. The protective effect of N-acetylcysteine on apoptotic lung injury in cecal ligation and puncture-induced sepsis model. Shock. 2003;19:366-372
53. Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med. 2008;45:1340-1351
54. Bettaieb A, Jiang JX, Sasaki Y, Chao TI, Kiss Z, Chen X. et al. Hepatocyte nicotinamide adenine dinucleotide phosphate reduced oxidase 4 regulates stress signaling, fibrosis, and insulin sensitivity during development of steatohepatitis in mice. Gastroenterology. 2015;149:468-480
55. Diaz-Cruz A, Vilchis-Landeros MM, Guinzberg R, Villalobos-Molina R, Pina E. NOX2 activated by alpha1-adrenoceptors modulates hepatic metabolic routes stimulated by beta-adrenoceptors. Free Radic Res. 2011;45:1366-1378
56. Drummond GR, Sobey CG. Endothelial NADPH oxidases. Trends Endocrinol Metab. 2014;25:452-463
57. Spencer NY, Zhou W, Li Q, Zhang Y, Luo M, Yan Z. et al. Hepatocytes produce TNF-alpha following hypoxia-reoxygenation and liver ischemia-reperfusion in a NADPH oxidase- and c-Src-dependent manner. Am J Physiol Gastrointest Liver Physiol. 2013;305:G84-94
58. Amico D, Spadoni T, Rovinelli M, Serafini M, D'Amico G, Campelli N. et al. Intracellular free radical production by peripheral blood T lymphocytes from patients with systemic sclerosis. Arthritis Res Ther. 2015;17:68
59. Padgett LE, Tse HM. NADPH oxidase-derived superoxide provides a third signal for CD4 T cell effector responses. J Immunol. 2016;197:1733-1742
Author contact
![]() Corresponding author: Andreas von Knethen, Institute of Biochemistry I, Faculty of Medicine, Goethe-University Frankfurt, Theodor-Stern-Kai 7, 60590 Frankfurt/Main. Tel.: +49-69-63016989; Fax: +49-69-63014203; Email: vonknethenuni-frankfurt.de
Corresponding author: Andreas von Knethen, Institute of Biochemistry I, Faculty of Medicine, Goethe-University Frankfurt, Theodor-Stern-Kai 7, 60590 Frankfurt/Main. Tel.: +49-69-63016989; Fax: +49-69-63014203; Email: vonknethenuni-frankfurt.de