Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(8):2315-2324. doi:10.7150/thno.30254 This issue Cite
Research Paper
Human LAP+GARP+FOXP3+ regulatory T cells attenuate xenogeneic graft versus host disease
Huihui Wang1,2 ![]() , Hyo Song2, Anha V. Pham2, Laurence J. Cooper3, Janika J. Schulze4, Sven Olek4, Dat Q. Tran2
, Hyo Song2, Anha V. Pham2, Laurence J. Cooper3, Janika J. Schulze4, Sven Olek4, Dat Q. Tran2 ![]()
1. School of Public Health, China Medical University, Shenyang, China
2. Pediatrics, UTHealth Medical School, Houston, TX, USA
3. Pediatrics, Children's Cancer Hospital, University of Texas MD Anderson Cancer Center, Houston, TX, USA
4. Ivana Türbachova Laboratory for Epigenetics, Epiontis GmbH, Precision for Medicine Group, Rudower Chaussee 29, 12489 Berlin, Germany
Received 2018-9-28; Accepted 2019-2-26; Published 2019-4-12
Abstract

Adoptive transfer of regulatory T cells (FOXP3+ Tregs) has been developed as a potential curative immune therapy to prevent and treat autoimmune and graft-versus-host diseases (GVHD). A major limitation that has hindered the use of Treg immunotherapy in humans is the difficulty of consistently isolating and obtaining highly purified Tregs after ex vivo expansion.
Methods: We isolated bona fide Tregs from expansion cultures based on their selective surface expression of latency-associated peptide (LAP). The TCR Vβ diversity and intracellular cytokine production of Tregs were determined by flow cytometer. The TSDR methylation was determined by epigenetic human FOXP3 qPCR Assay. Their in vitro and in vivo potency was confirmed with suppression assay and humanized xenogeneic GVHD (xGVHD) murine model, respectively.
Results: LAP+ repurification results in >90% LAP+FOXP3+ Tregs, leaving behind FOXP3- and FOXP3+ nonTregs within the LAP- population. After 4-week expansion, the LAP+ Tregs were >1 billion cells, highly suppressive and anergic in vitro, >90% demethylated in the TSDR and able to maintain TCR Vβ diversity. In the xGVHD model, exogenous CD25-PBMC administered alone results in a median survival of 32 days. The co-transfer of LAP+ Tregs increased median survival to 47 days, while the LAP parent (CD25+) and LAP- nonTregs had median survival of 39 and 31 days, respectively.
Conclusions: These preclinical data together provide evidence that LAP+ Tregs are highly purified with fully suppressive function for cell therapy. This population results in a more effective and safer product for immunotherapy to treat GVHD and provides the necessary preclinical data for transition into a clinical trial with LAP+ Tregs to prevent or treat GVHD and other autoimmune diseases.
Keywords: regulatory T cells (Tregs), latency-associated peptide (LAP), Treg-specific demethylated region (TSDR), T cell receptor (TCR) repertoire, Graft-versus-host disease (GVHD)
Introduction
FOXP3+ regulatory T cells (Tregs), a subset of CD4+ T cells, maintain immune homeostasis and prevent autoimmunity by suppressing auto-reactivity and regulating immune responses to foreign and self-antigens [1]. A hallmark of Tregs is their expression of the transcription factor, FOXP3, and the high level of CD25, the IL2 receptor alpha chain [2]. Due to their role in maintaining immune homeostasis and tolerance, Tregs have been considered as potentially immunotherapy to prevent a variety of immunological diseases in human. Among potential diseases that may be tackled with such immune-modulators are most graft versus host disease (GVHD), a frequent and often severe complication following allogeneic hematopoietic cell transplantation [3]. Murine studies have demonstrated convincing evidence of the power of Tregs as a cell therapy to prevent and treat various immunological conditions, such as multiple sclerosis, GVHD, inflammatory bowel disease and lupus [4-10]. In translating these studies to human therapeutic applications, one major limitation is the expansion of Tregs ex vivo to achieve consistently sufficient number and purity of bona fide Tregs. In the peripheral blood of healthy adults, there are ~5-10% FOXP3+ Tregs within total CD4+ T cells. Since FOXP3 is an intracellular transcription factor, only the CD4+ T cells with low CD127 and high CD25 expression, i.e., no more than 2-4% can be sorted to functionally isolate >90% FOXP3 purity. However, this population is not homogeneously Tregs, particularly in diseases where these markers are less correlative due to activated T cells. It has been established that there are two subsets of Tregs: those developed in the thymus (tTregs) and those generated in the periphery (pTregs). Moreover, naïve CD4+ T cells can be induced in vitro with TCR stimulation in the presence of TGFβ1 and IL2 to express FOXP3 (iTregs) [11]. The functional patterns for these different subgroups of Tregs are widely varying during the expansion of Tregs over a 3-4 week period. In addition, the purity and composition of Tregs frequencies with respect to tTregs, iTregs, pTregs and CD4+FOXP3- nonTregs are also varying, resulting in an impure Treg product.
We have demonstrated previously that iTregs lack the cardinal features of tTregs based on anergy and suppressive function [11]. While >90% FOXP3+ cells can be isolated by FACS-sorting, frequently the percentage of FOXP3+ T cells decreases to 75% after two weeks and 50% after three weeks of expansion [12-14]. Due to these limitations that have hindered the advancement of Treg analysis and therapy, we have developed an innovative technique to repurify Tregs from expanded cultures contaminated with activated nonTregs. By targeting the selective expression of latency-associated peptide (LAP), this novel method allows for expansion and consistent re-purification of sufficient quantity of highly purified, bona fide Tregs (>90% FOXP3), even from an initial limited blood volume of 5-10 ml [15]. Without this technique, it has been difficult to analyze and assess definitively the function of Tregs due to variable contaminants of nonTregs. Moreover, this method has the potential to optimize Treg therapy by permitting the manufacturing of a Treg product that is consistently >90% bona fide Tregs regardless of methods of expansion or donor's variability. Using the LAP marker, we can selectively isolate LAP+ Tregs from any time point during expansion that are typically contaminated by >25% FOXP3+ and FOXP3- nonTregs. This purity and selectivity will allow for separation of different subsets and definitive determination of Tregs function in GVHD. Another important factor is the cost effectiveness for production of a Treg product. In this study, we use CD25+ cells isolated from buffy coats with anti-CD25 microbeads. These CD25+ cells are expanded with OKT3-loaded KT64/86 artificial APC (aAPC) expressing CD86 and CD64 and re-purified based on LAP expression. We perform an analysis of the TCR repertoire, function, and phenotype within CD25+ cells and the subsets of LAP+ Tregs and LAP- nonTregs isolated from expansion cultures. We demonstrate that LAP+ Tregs after 4-week ex vivo expansion still maintain diverse TCR Vβ repertoire, are highly demethylated in the TSDR region, and possess suppressive function. In contrast, the LAP- nonTregs possess no suppressive function, while the CD25+ parent population had variable functionality depending on the level of expanded, contaminated nonTregs.
Our study demonstrates a simple single-step CD25+ isolation, robust expansion on aAPCs, and LAP+ repurification to obtain highly purified, bona fide FOXP3+ Tregs. Ultimately, clinical investigators can apply our techniques to further define Tregs in patients with other immunologic conditions and generate highly purified Treg products for therapeutic use. A highly purified Treg product after ex vivo expansion would not only reduce any potential adverse reactions but would also enhance the efficacy and interpretability of multi-center clinical trials on Treg immunotherapy.
Materials and Methods
Flow cytometry and Antibodies
Various fluorescent conjugated antibodies to human CD4 (RPA-T4), CD8 (RPA-T8), CD16 (3G8), CD19 (HIB19), CD25 (BC96), LAP (TW4-2F8) and GARP (7B11) were from BioLegend. Cells were surface stained first using appropriate combinations of mAbs, and either used directly for flow cytometric analyses or further processed for intracellular staining of cytokines, FOXP3 and HELIOS. The cells were fixed and permeabilized with a Fixation/Permeabilization kit (eBioscience) and stained with anti-FOXP3 (259D) and/or anti-HELIOS (22F6) mAbs (BioLegend). FACSCalibur and Attune flow cytometers were used for data acquisition, and the data were analyzed with FlowJo software (TreeStar).
Reagents
Anti-CD4, anti-biotin, anti-CD25 and anti-CD14 magnetic microbeads were from Miltenyi Biotec. All cells were cultured in complete media consisting of RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin (100 U/mL), streptomycin (100 μg/mL), 1×GlutaMAX (Gibco) and 1% MEM non-essential amino acids, 1% sodium pyruvate (Hyclone).
Treg isolation and expansion
Peripheral blood mononuclear cells (PBMC) were isolated from adult peripheral buffy coats (Gulf Coast Regional Blood Center) by Ficoll gradient centrifugation. Tregs were purified from PBMC by positive selection using directly conjugated anti-CD25 magnetic microbeads with AutoMACS (Posseld2 program). Bead-purified CD4+CD25high cells (~5-10×106) were stimulated with OKT3-loaded KT64/86 artificial APC (aAPC) expressing CD86 and CD64 at 1:1 ratio (kindly provided by Dr. Carl H. June and Dr. James L. Riley). KT64/86 cells were loaded with anti-CD3 mAb (OKT3, BioLegend). CD4+CD25high cells were cultured in complete media supplemented with 100 U/mL IL2 (PeproTech) in the presence of 25 nM rapamycin (Sigma-Aldrich). The cells were cultured in 25-cm2 culture flasks (ThermoFisher Scientific). On day 5, the cells were transferred to 75-cm2 culture flasks with additional fresh complete media containing 100 U/mL IL2. No additional rapamycin was added to the cultures except for the first day. On day 8, the cells were transferred to 175-cm2 culture flasks with fresh IL2 media. The cultures were maintained at cell concentration of 106/mL and split every 3 days with additional fresh IL2 media. On day 10, the cells were spun down and given fresh IL2 media before re-stimulation with additional OKT3 loaded KT64/86 at 6:1 of cell: aAPC. After 48-hour stimulation, the cells were analyzed and purified based on the expression of LAP. For LAP+ cells isolation, biotinylated anti-hLAP mAbs (TW4-2F8, BioLegend) followed by secondary incubation with anti-biotin magnetic microbeads (Miltenyi Biotecs) were used. LAP+ Tregs were purified from expanded CD25+ cells by positive selection with AutoMACS (Posseld2 program). The CD25+, LAP+ and LAP- populations were cultured in fresh IL2 media and maintained at cell concentration of 106/mL, splitting every 3 days with additional fresh IL2 media. On day 20, these populations got the 3rd stimulation with OKT3-loaded KT64/86 cells at 6:1 ratio.
In vitro suppression assay
CD4+CD25-and CD14+ cells were isolated from PBMC of healthy donor buffy coats using the AutoMACS and related Miltenyi Biotecs kits. Fresh CD4+CD25- T cells (50,000) were labeled with 2 μM CFSE (Invitrogen) and stimulated with non-irradiated CD14+ APC (12,500) loaded with 0.25 μg/mL OKT3 alone or with 2:1, 4:1, 8:1 and 16:1 responder to suppressor cells (CD25+, LAP+ Tregs or LAP- cells). The cells were cultured for 4 days in 96-well flat-bottom plates (Corning), and CFSE dilution was analyzed by FACS.
Intracellular cytokine staining
For analysis of intracellular cytokine production, the cells were stimulated for 5 hours with 50 ng/mL PMA, 1 μg/mL ionomycin and 3 μg/mL brefeldin A (Sigma-Aldrich). Afterward, the cells were fixed and permeabilized with eBioscience FOXP3 kit and stained with mAbs against FOXP3 (259D), IL2 (MQ1-17H12), IL4 (8D4-8), IL10 (JES3-9D7), IL13 (JES10-5A2), IL17A (BL168) and IFNγ (4S.B3) (BioLegend) for FACS analysis.
Xenogeneic GVHD model
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice were obtained from Jackson Laboratory and maintained in specific pathogen free conditions at the CLAMC (University of Texas Health Science Center at Houston). In this protocol, the mice were not irradiated. On day 0, mice were injected with human CD25-PBMC (30×106) with or without expanded CD25+, LAP+ Tregs and LAP‾ nonTregs (30×106). Following injection of cells, animals were monitored twice a week for weight loss and other signs of xGVHD, e.g. hunched back, ruffled fur and reduced mobility [16, 17]. Mice were euthanized if they experienced >15% weight loss or showed signs of compromised health and were considered to have human graft-related disease if an expanded population of human cells was detected simultaneously in blood. Mice were evaluated for human cells in blood by flow cytometer weekly until day 70 or until they were euthanized. To document CD25-PBMC-associated cell expansion, the animals were bled (10-40 μl) and red blood cells were lysed with ACK Lysing Buffer (Quality Biological, Inc.). All animal protocols were approved by CLAMC at the University of Texas Health Science Center at Houston.
TSDR Methylation status
To determine the methylation status of the Foxp3 gene, DNA was purified from frozen aliquots of expanded CD25+, LAP+ Tregs and LAP‒ nonTregs. Coded samples were sent to Epiontis (Berlin, Germany) for bisulfite modification and quantification of TSDR methylation by epigenetic human FOXP3 qPCR Assay.
TCR repertoire status
T cell receptor (TCR) Vβ repertoire was analyzed using flow cytometry with the IOTest Beta Mark TCR Repertoire Kit (Beckman Coulter), which consists of fluorochrome conjugated monoclonal antibodies that identify 24 TCR Vβ subfamilies, covering about 70% of the normal human Vβ T cell repertoire.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA). Data were analyzed by analysis of variance (ANOVA) or Student's t test. All group results are expressed as mean plus or minus SEM, if not stated otherwise. The paired Student's t test was used for the comparison of group values and discriminatory parameters, where appropriate. Survival data was analyzed with SPSS software (version 16.0; SPSS Inc., Chicago, IL, USA) by using the Kaplan-Meier method and compared using a Log-rank Test. p values less than 0.05 were considered significant.
Results
Expansion and purification of LAP+ Tregs
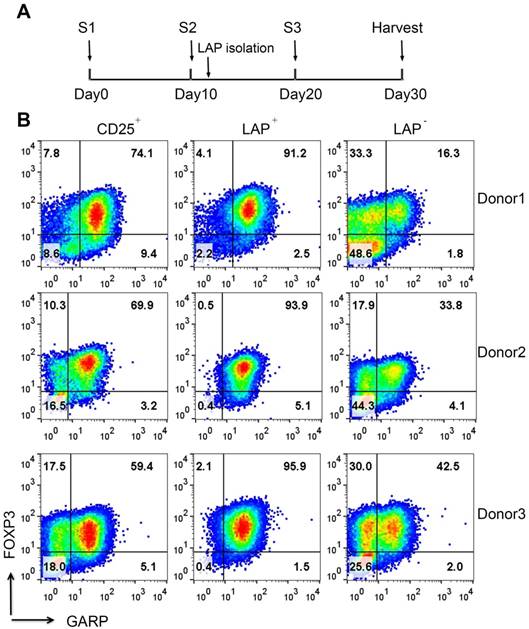
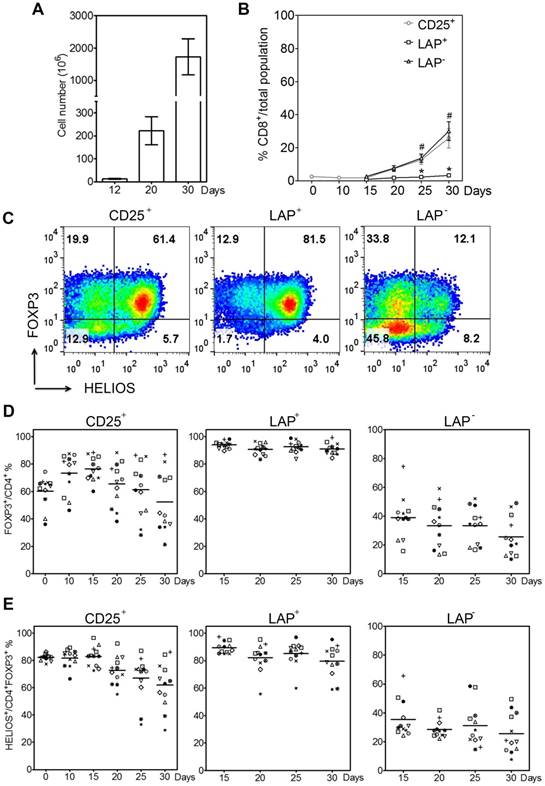
Previously, we have demonstrated that it is feasible and economical to use an initial one-step method with anti-CD25 bead to purify and expand Tregs with anti-CD3/CD28 beads [15]. This time we use aAPCs, because they are more cost-effective at large scale production and do not require a removal process. Figure 1A shows the expansion and re-isolation protocol. Instead of previously isolating for LAP+ Tregs at day 21 (3rd round of stimulation), this time we isolate LAP+ Tregs on the 2nd round of stimulation at day 12. We choose this approach to reduce the amount of anti-LAP mAbs, since the cell numbers in the cultures are less. With this method, the CD25+ population contains an average of 72% FOXP3+ cells within CD4+ cells (range, 60%-84%), while the percentages of FOXP3 purity post LAP purification (LAP+) are typically ≥90%. The LAP‒ population still has some FOXP3+ Tregs left. Furthermore, the LAP+ cells are mainly GARP+ cells (Figure 1B). Starting with 4-25×106 LAP+ Tregs at day 12, we are able to achieve >1 billion LAP+ Tregs by day 30 of expansion (Figure 2A). The starting CD25+ population at day 0 contains mainly CD4+FOXP3+ Tregs with the remainder being CD8+ T, CD19+ B and CD16+ NK cells (Figure 2B and S1). During the expansion, these contaminating non-T cells disappeared by day 10. Furthermore, the CD8+ T cell population is mainly found in the LAP‒ population after repurification (Figure 2B). There is a progressive reduction of FOXP3 purity in CD25+ population during the expansion from an average of 60% (range, 36%-74%) at the starting point to a mean of 73% (range, 46%-88%) on day 10 and 52% (range, 21%-87%) on day 30. After LAP repurification, the means of CD4+FOXP3+ T cells maintain higher than 90% in LAP+ Tregs and lower than 40% in LAP‒ population during expansion (Figure 2C and D). The mean percentages of FOXP3+HELIOS+ T cells maintain higher than 80% in LAP+ Tregs and lower than 40% in LAP‒ population during expansion (Figure 2C and E).
Expansion and purification of LAP+ Tregs. (A) Schema showing time course of experiment, stimulation (S1=one stimulation, etc.) and LAP isolation. (B) After LAP purification, LAP+ Tregs are mainly FOXP3+GARP+ Tregs and leave the FOXP3- and FOXP3+ nonTregs contaminants in LAP- population. Representative data come from 3 donors. The number in each quadrant represents the percentage within CD4+ population.
Purification of LAP+ Tregs over 30-day expansion. (A) Fold expansion of magnetic bead-purified LAP+ Tregs during expansion. (B) CD8+ T cells percentage in LAP parents (CD25+), LAP+ Tregs and LAP- population. *, p < 0.05 vs. CD25+ cells at same timepoint. #, p < 0.05 vs. LAP+ Tregs at same timepoint. (C) Representative data for FOXP3+HELIOS+ cells percentage of CD4+ population in CD25+ cells, LAP+ Tregs and LAP- population. (D) FOXP3+ cells percentage out of CD4+ cells in CD25+ cells, LAP+ Tregs and LAP- population. (E) HELIOS+ cells percentage out of CD4+FOXP3+ cells in CD25+ cells, LAP+ Tregs and LAP- population. Horizontal lines represent the mean of each group. Data come from 12 donors.
TCR Vβ diversity in expanded LAP+ Tregs
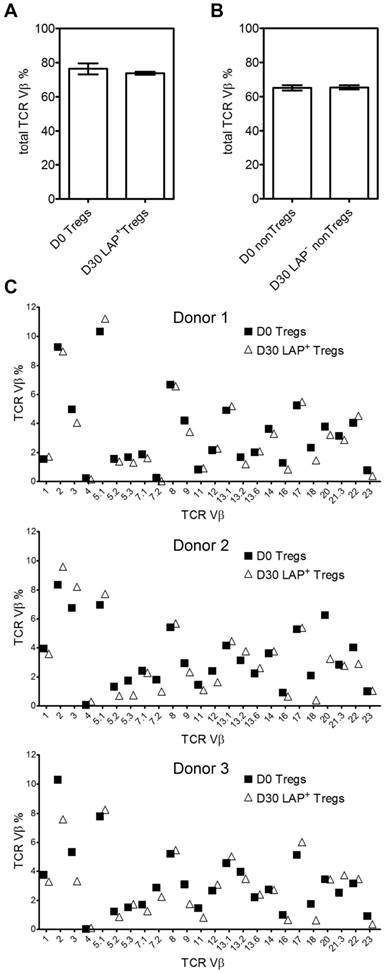
In order to investigate the changes in TCR Vβ repertoire during expansion, we next examine TCR repertoire usage of the Tregs (CD4+FOXP3+) before (day 0) and after (day 30) expansion. After three rounds of stimulation and LAP purification, the total Treg TCR Vβ usages are essentially unchanged between Day 0 and Day 30 (Figure 3A and B). Figure 3C shows data from three representative donors for TCR Vβ distribution before and after expansion.
TCR Vβ diversity during expansion. (A) Total %TCR Vβ in Tregs and (B) nonTregs before expanded (Day 0) and after a total of three stimulations (Day 30). (C) Three representative donors' %TCR Vβ changes between D0 Tregs and D30 LAP+ Tregs. Data come from 3 donors.
Cytokine profile in LAP+ Tregs
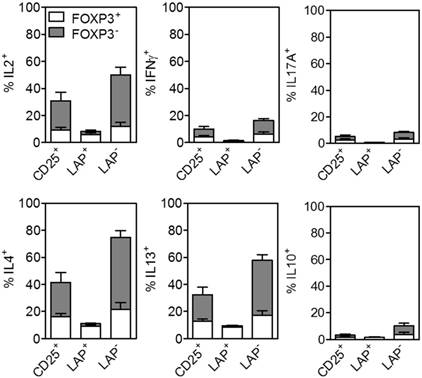
Effector cells that secrete cytokines can exacerbate GVHD [18]. A cardinal feature of Tregs is their anergic state. The CD25+, LAP+ Tregs and LAP‒ nonTregs are stimulated to measure the levels of cytokine production at day 30 end of culture. There are substantially lower percentages of IL2+, IFNγ+, IL17A+, IL4+, IL13+ and IL10+ producing cells out of CD4+ cells detected in the LAP+ Tregs as compared to the other two populations (Figure 4 and S2).
Cytokine production by FOXP3+ and FOXP3- cells in CD25+ cells, LAP+ Tregs and LAP- populations. Percent of cells secreting IL2, IFNγ, IL17A, IL4, IL13 and IL10. Means are for 8 independent experiments. The percent cytokines for CD4+FOXP3+ and CD4+FOXP3‾ are gated from CD4+ T cells within CD25+, LAP+ and LAP- populations.
LAP+ Tregs are highly demethylated in TSDR
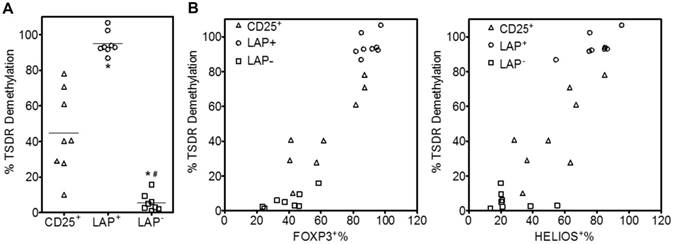
The level of methylation at the promoter region upstream of FOXP3 termed Treg-specific demethylated region (TSDR) has provided specific differentiation between activated T effector cells and bona fide Tregs [19, 20]. Several studies have demonstrated that FOXP3- cells are fully methylated at this site, while iTregs are intermediate and tTregs are fully demethylated [21, 22]. The level of demethylation at the TSDR region is critical for establishment of a stable Treg lineage and correlation with suppressive function. In this study, we demonstrate that after 30 days expansion the purified LAP+ Tregs are highly demethylated with an average of 95% demethylation in the TSDR region, while the CD25+ cells with mean of 45% demethylation (Figure 5A). The LAP‒ nonTregs are highly methylated, with less than 20% demethylation even when the population contains close to 60% FOXP3+ and HELIOS+ cells (Figure 5B and C). It should be noted that the LAP‒ populations still contain LAP+ cells ranging from 10-50% which represent most of the demethylated TSDR and HELIOS signals (Figure 1 and 2).
LAP+ Tregs are highly demethylated in TSDR. (A) TSDR demethylation percentage of CD25+, LAP+ Tregs and LAP- population. Mean percent of cells demethylation are for 8 independent experiments. Horizontal lines represent the mean of each group. *, p < 0.05 vs. CD25+ cells. #, p < 0.05 vs. LAP+ Tregs. (B) The correlation between %FOXP3 or %HELIOS and TSDR demethylation in CD25+ cells, LAP+ Tregs and LAP- population.
LAP+ Tregs are anergic and suppressive in vitro
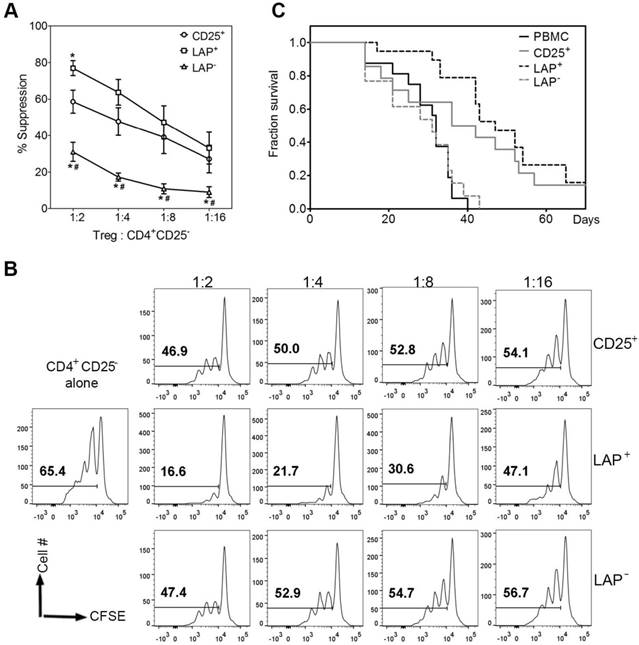
Previously we have demonstrated that LAP+ Tregs isolated from CD3/CD28 Dynabead stimulated expansion cultures maintained suppressive function. Since these LAP+ Tregs are obtained from aAPC stimulated expansion cultures, it is important to confirm their suppressive function. We evaluate the capacity of purified LAP+ Tregs to suppress the proliferative responses of CD4+25- T cells in CFSE dilution assays. As expected, LAP+ Tregs are the most potent suppressors among these three populations (Figure 6A and B).
LAP+ Tregs are suppressive in vitro and in vivo. (A) CD25+ cells, LAP+ Tregs and LAP- population percent suppression for different ratio of Tregs: CD4+CD25-. *, p < 0.05 vs. CD25+ cells suppression. #, p < 0.05 vs. LAP+ Tregs suppression. (B) Representative data for CD4+CD25- cells proliferation w/wo CD25+ cells, LAP+ Tregs or LAP- population. (C) Tregs are co-transferred at a 1:1 ratio with allogeneic 25- PBMC (30 × 106 each) into NSG recipients. Kaplan-Meier survival curves depict the percentage of mice with >15% weight loss. Log-rank Test was used to analyze the statistic different. Data come from 5 independent experiments (5 donors). There were 2-4 mice in each group for one donor.
LAP+ Tregs ameliorate disease in a xenogeneic murine model of GVHD
To determine whether expanded LAP+ Tregs are effective in suppressing human CD25-depleted PBMC-induced xenogeneic GVHD, we used a xenogeneic model of GVHD in which Tregs are co-transferred at a 1:1 ratio with allogeneic CD25-depleted PBMC (30 × 106 each) into NSG recipients to compare the potency and safety of in vitro-expanded LAP+ Tregs and LAP- cells (Figure 6C). CD25-PBMC administered alone results in a median survival of 32 days. The co-transfer of LAP parent (CD25+) or LAP+ Tregs increased median survival to 39 (p < 0.05) and 47 days (p < 0.001), respectively. The LAP- nonTregs showed similar median survival (31 days) with PBMC group. The survival of LAP+ group was significantly higher than LAP- group (p < 0.001). Compared with CD25+ group, LAP+ Tregs increased median survival from 39 to 47 days. However, no statistic different was found, since some of the CD25+ parent populations have less contaminating nonTregs and therefore higher level of FOXP3+ Tregs (Figure 2D, 5B). Average body weight (% of initial weight) for mice surviving on given day for different groups was shown in Figure S3.
Discussion
Adoptive transfer of FOXP3+ Tregs to prevent or cure multiple autoimmune diseases or GVHD has proven to be highly efficacious in animal models and human [23-25]. But a major obstacle to overcome is to achieve a consistent and highly purified FOXP3+ Treg product after ex vivo expansion in a cost-effective manner. In this study, we use LAP as a unique cell surface marker that distinguishes bona fide Tregs from activated FOXP3+ and FOXP3- nonTregs and show that it is feasible to sort expanded FOXP3+ Tregs from nonTregs based on expression of LAP. We obtain a post-sort FOXP3 purity of greater than 90% which would be ideal for cellular immunotherapy. Furthermore, we find that ex vivo expanded LAP+ Tregs with aAPCs maintain TCR Vβ repertoire diversity, instead of clonal expansion due to selective cell death. Similar data has been detected in expanded cord blood (CB) Tregs: the process of ex vivo expansion did not skew the CB Tregs TCR Vβ repertoire [26].
Although it is possible to obtain a highly purified FOXP3+ population after re-stimulation of the expanded CD25hi cells, it has also been shown that the effector T cells can transiently possess this feature, particularly after repeated in vitro stimulation [27]. Moreover, several studies have pointed out the potential plasticity of Tregs and caution that this instability could result in the generation of pathogenic memory T cells in vivo [28]. In our study, a quantitative assay to assess the anergic state of Tregs is used to detect the cytokine secretion. Only minimal percentages of IL2, IFNγ, IL17, IL4, IL13 and IL10 are detected in the purified LAP+ Tregs as compared to CD25+ and LAP‾ population. This method offers the opportunity to reduce most of the cytokine secreting cells in our culture compared with rapamycin culture system, which still has >50% of cells secreting IL4 (both FOXP3+ and FOXP3- cells) after the fourth re-stimulation [4]. In this study, only minimal percentages of LAP+ Treg cells secrete IL-10. While the LAP+ cells are mainly GARP+ cells, the immunosuppressive role of LAP+ might be through GARP/LAP complexes. Glycoprotein A repetitions predominant (GARP), also known as leucine rich repeat containing 32 (LRRC32), plays important role in immunosuppressive function of Tregs by inhibiting T effector cell activity [29]. Targeting GARP by siRNA or monoclonal antibodies specific for GARP inhibit immunosuppressive activity of Treg in vitro and in vivo [30, 31]. However, the role of LAP/GARP on Tregs remains unclear and controversial. Deletion of GARP on mouse Tregs is not sufficient to inhibit the growth of transplanted tumors. They found the suppressive function of Tregs was not affected by knockout of garp in Tregs [32]. Further studies are needed to reveal the mechanisms responsible for suppressive function of LAP+ Tregs.
Tregs represent a heterogeneous population comprised of a committed lineage and an uncommitted subpopulation with developmental plasticity [33]. Therefore, it is crucial to certify that the final Treg products consist of a committed Treg lineage and possess a stable phenotype with the highest purity as possible. A reliable method is the quantification of demethylation in the TSDR, which correlates positively with Treg phenotype and function. This epigenetic marker is highly specific for Tregs and can discriminate them from activated FOXP3+ conventional T cells [34, 35]. When compared with LAP+ Tregs, significant levels of FOXP3 TSDR methylation are demonstrated in the LAP- nonTregs. Even after 30 days expansion, the purified LAP+ Tregs are highly demethylated in the TSDR region. Some of the LAP- populations contain over 40% FOXP3+ cells, yet they have <20% TSDR demethylation, supporting that these FOXP3+ cells are not bona fide Tregs.
Adoptive transfer of LAP+ Tregs delayed the development of xenogeneic GVHD. Our finding supports a prior observation showing that GARP+ Treg protect from xGVHD. Dr. Lucas group found that the administration of antibody blocking GARP or GARP/TGF-β1 abrogate the protection of Tregs in xGVHD [31]. While we are able to demonstrate the suppressive function of LAP+ Tregs, these assays have limitations. Although the LAP+ Tregs demonstrate suppressive function by delaying the development of xenogeneic GVHD, we are unable to detect their existence in peripheral blood and spleens of the NSG mice by day 14 post adoptive transfer. Similar to our finding, Parmar et al. have previously shown that ex vivo-expanded cord blood Tregs in NSG xenogeneic model did not persist beyond day 3 [26]. One possible explanation might be due to cytokine deprivation, particularly IL2. Enhancing the stability of LAP+ Tregs is critical for the development of an efficacious cell therapy. At this point, we are unclear whether their lack of persistence is intrinsic to the human Tregs or extrinsic due to the absence of critical growth factors compatible with human Tregs in the NSG mice. Further studies are needed to answer these two issues.
In the absence of a repurification process, the final expanded Treg product would have highly variable content of bona fide Tregs and contaminated, expanded nonTregs (Figure 2D). To address this issue, other protocols have implemented rapamycin which has improved the percentage of Tregs, but variability still exists [15, 36]. Consistent with other studies, some of our expanded CD25+ populations maintained >75% FOXP3+ and >60% TSDR demethylation (Figure 5B). These CD25+ populations did possess sufficient suppressive function similar to the repurified LAP+ Tregs (Figure 6A, C). We were unable to demonstrate a statistical difference between CD25+ populations and LAP+ Tregs due to the high variability in Treg percentage within some of the CD25+ populations. Therefore, it would be prudent to manufacture highly purified Treg immunotherapy, similar to the standard required for conventional drug manufacturing. Regardless of the variability in bona fide Tregs within the final product, implementing LAP repurification would normalize and produce a highly purified and more homogenous Treg product.
Overall, our study provides greater characterizations of LAP+ Tregs to support their therapeutic application for GVHD and other autoimmune conditions. Regardless of the methods and strategies used to expand Tregs, co-expansion of other cells, particularly FOXP3+ nonTregs, is almost inevitable. With the application of LAP or GARP repurification, a highly purified, TSDR demethylated Treg population can be achieved which maintains diversity and functionality. The next step is to demonstrate their safety, stability and functionality in a human clinical trial.
Abbreviations
APC: antigen presenting cell; CB: cord blood; FBS: fetal bovine serum; GARP: glycoprotein A repetitions predominant; GVHD: graft-versus-host diseases; LAP: latency-associated peptide; PBMC: peripheral blood mononuclear cells; TCR: T cell receptor; Treg: regulatory T cell; TSDR: Treg-specific demethylated region.
Supplementary Material
Supplementary figures.
Acknowledgements
This research was supported by the National Institutes of Health Grant R01HL113304 (DQT) and the National Natural Science Foundation of China 81502841 (HW). The content is solely the responsibility of the authors. All authors have agreed to its content. The authors thank Dr. Carl H. June and Dr. James L. Riley for providing artificial APC (aAPC) expressing CD86 and CD64.
Contributions
Huihui Wang conducted all the experiments and wrote the manuscript. Hyo Song and Anha V. Pham assisted in the experiments. Dat Q. Tran supervised the project and edited the manuscript. Janika J. Schulze and Sven Olek conducted and interpreted the epigenetic TSDR analyses. Laurence J. Cooper provided guidance on clinical-grade methodologies.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z. et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8-27
2. Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell. 2000;101:455-8
3. Trzonkowski P, Dukat-Mazurek A, Bieniaszewska M, Marek-Trzonkowska N, Dobyszuk A, Juscinska J. et al. Treatment of graft-versus-host disease with naturally occurring T regulatory cells. BioDrugs. 2013;27:605-14
4. Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM. et al. Massive ex vivo expansion of human natural regulatory T cells (T(regs)) with minimal loss of in vivo functional activity. Sci Transl Med. 2011;3:83ra41
5. Horwitz DA. Regulatory T cells in systemic lupus erythematosus: past, present and future. Arthritis Res Ther. 2008;10:227
6. Mayne CG, Williams CB. Induced and natural regulatory T cells in the development of inflammatory bowel disease. Inflamm Bowel Dis. 19. 1772 -88
7. Ogino H, Nakamura K, Iwasa T, Ihara E, Akiho H, Motomura Y. et al. Regulatory T cells expanded by rapamycin in vitro suppress colitis in an experimental mouse model. J Gastroenterol. 2012;47:366-76
8. Hao L, Gao L, Chen XH, Zou ZM, Zhang X, Kong PY. et al. Human umbilical cord blood-derived stromal cells prevent graft-versus-host disease in mice following haplo-identical stem cell transplantation. Cytotherapy. 2011;13:83-91
9. Trandem K, Anghelina D, Zhao J, Perlman S. Regulatory T cells inhibit T cell proliferation and decrease demyelination in mice chronically infected with a coronavirus. J Immunol. 2010;184:4391-400
10. Luth S, Huber S, Schramm C, Buch T, Zander S, Stadelmann C. et al. Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen-specific Tregs. J Clin Invest. 2008;118:3403-10
11. Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. 2007;110:2983-90
12. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y. et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010;184:3433-41
13. Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O'Shea JJ. et al. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med. 2008;205:1975-81
14. Krenger W, Hollander GA. The thymus in GVHD pathophysiology. Best Pract Res Clin Haematol. 2008;21:119-28
15. Tran DQ, Andersson J, Hardwick D, Bebris L, Illei GG, Shevach EM. Selective expression of latency-associated peptide (LAP) and IL-1 receptor type I/II (CD121a/CD121b) on activated human FOXP3+ regulatory T cells allows for their purification from expansion cultures. Blood. 2009;113:5125-33
16. Ali N, Flutter B, Sanchez Rodriguez R, Sharif-Paghaleh E, Barber LD, Lombardi G. et al. Xenogeneic graft-versus-host-disease in NOD-scid IL-2Rgammanull mice display a T-effector memory phenotype. PLoS One. 2012;7:e44219
17. Deaglio S, Morra M, Mallone R, Ausiello CM, Prager E, Garbarino G. et al. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J Immunol. 1998;160:395-402
18. Hippen KL, Merkel SC, Schirm DK, Nelson C, Tennis NC, Riley JL. et al. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am J Transplant. 2011;11:1148-57
19. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U. et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol. 2008;38:1654-63
20. MacDonald KG, Orban PC, Levings MK. T regulatory cell therapy in transplantation: stability, localization and functional specialization. Curr Opin Organ Transplant. 2012;17:343-8
21. Fazilleau N, Bachelez H, Gougeon ML, Viguier M. Cutting edge: size and diversity of CD4+CD25high Foxp3+ regulatory T cell repertoire in humans: evidence for similarities and partial overlapping with CD4+CD25- T cells. J Immunol. 2007;179:3412-6
22. Scheinberg P, Melenhorst JJ, Hill BJ, Keyvanfar K, Barrett AJ, Price DA. et al. The clonal composition of human CD4+CD25+Foxp3+ cells determined by a comprehensive DNA-based multiplex PCR for TCRB gene rearrangements. J Immunol Methods. 2007;321:107-20
23. Shevach EM, DiPaolo RA, Andersson J, Zhao DM, Stephens GL, Thornton AM. The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol Rev. 2006;212:60-73
24. Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med. 2002;196:389-99
25. Taylor PA, Lees CJ, Blazar BR. The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood. 2002;99:3493-9
26. Parmar S, Liu X, Tung SS, Robinson SN, Rodriguez G, Cooper LJ. et al. Third-party umbilical cord blood-derived regulatory T cells prevent xenogenic graft-versus-host disease. Cytotherapy. 2014;16:90-100
27. Baron U, Floess S, Wieczorek G, Baumann K, Grutzkau A, Dong J. et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol. 2007;37:2378-89
28. Mercer F, Kozhaya L, Unutmaz D. Expression and function of TNF and IL-1 receptors on human regulatory T cells. PLoS One. 2010;5:e8639
29. Hahn SA, Stahl HF, Becker C, Correll A, Schneider FJ, Tuettenberg A. et al. Soluble GARP has potent antiinflammatory and immunomodulatory impact on human CD4(+) T cells. Blood. 2013;122:1182-91
30. Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A. 2009;106:13445-50
31. Cuende J, Lienart S, Dedobbeleer O, van der Woning B, De Boeck G, Stockis J. et al. Monoclonal antibodies against GARP/TGF-beta1 complexes inhibit the immunosuppressive activity of human regulatory T cells in vivo. Sci Transl Med. 2015;7:284ra56
32. Vermeersch E, Lienart S, Collignon A, Lucas S, Gallimore A, Gysemans C. et al. Deletion of GARP on mouse regulatory T cells is not sufficient to inhibit the growth of transplanted tumors. Cell Immunol. 2018;332:129-33
33. Brinster C, Shevach EM. Costimulatory effects of IL-1 on the expansion/differentiation of CD4+CD25+Foxp3+ and CD4+CD25+Foxp3- T cells. J Leukoc Biol. 2008;84:480-7
34. Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS. et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576-87
35. Li L, Kim J, Boussiotis VA. IL-1beta-mediated signals preferentially drive conversion of regulatory T cells but not conventional T cells into IL-17-producing cells. J Immunol. 2010;185:4148-53
36. Basu S, Golovina T, Mikheeva T, June CH, Riley JL. Cutting edge: Foxp3-mediated induction of pim 2 allows human T regulatory cells to preferentially expand in rapamycin. J Immunol. 2008;180:5794-8
Author contact
![]() Corresponding authors: Huihui Wang, hhwangedu.cn and Dat Q. Tran, Dat.Q.Trantmc.edu
Corresponding authors: Huihui Wang, hhwangedu.cn and Dat Q. Tran, Dat.Q.Trantmc.edu