Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2019; 9(9):2662-2677. doi:10.7150/thno.32060 This issue Cite
Research Paper
A self-assembled, modular nucleic acid-based nanoscaffold for multivalent theranostic medicine
Veronica Liv Andersen1, Mathias Vinther1, Rajesh Kumar2, Annika Ries2, Jesper Wengel2, Jesper Sejrup Nielsen1, Jørgen Kjems1 ![]()
1. Interdisciplinary Nanoscience Center (iNANO), Aarhus University, Gustav Wieds Vej 14, 8000 Aarhus C, Denmark
2. Biomolecular Nanoscale Engineering Center, Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, 5230 Odense M, Denmark
Received 2018-12-6; Accepted 2019-2-21; Published 2019-4-13
Abstract

Rationale: Within the field of personalized medicine there is an increasing focus on designing flexible, multifunctional drug delivery systems that combine high efficacy with minimal side effects, by tailoring treatment to the individual.
Methods: We synthesized a chemically stabilized ~4 nm nucleic acid nanoscaffold, and characterized its assembly, stability and functional properties in vitro and in vivo. We tested its flexibility towards multifunctionalization by conjugating various biomolecules to the four modules of the system. The pharmacokinetics, targeting capability and bioimaging properties of the structure were investigated in mice. The role of avidity in targeted liver cell internalization was investigated by flow cytometry, confocal microscopy and in vivo by fluorescent scanning of the blood and organs of the animals.
Results: We have developed a nanoscaffold that rapidly and with high efficiency can self-assemble four chemically conjugated functionalities into a stable, in vivo-applicable system with complete control of stoichiometry and site specificity. The circulation time of the nanoscaffold could be tuned by functionalization with various numbers of polyethylene glycol polymers or with albumin-binding fatty acids. Highly effective hepatocyte-specific internalization was achieved with increasing valencies of tri-antennary galactosamine (triGalNAc) in vitro and in vivo.
Conclusion: With its facile functionalization, stoichiometric control, small size and high serum- and thermostability, the nanoscaffold presented here constitutes a novel and flexible platform technology for theranostics.
Keywords: LNA, drug delivery, bioconjugation, nanomedicine, personalized medicine
Introduction
Conventional cancer therapy is typically based on small cytotoxic drugs that preferentially kill fast growing cells throughout the body. Due to their unspecific nature, chemotherapeutics often display a variety of side effects that can be devastating to the patient and, in some cases, even prevent treatment [1,2]. Furthermore, small molecule chemotherapeutics are associated with highly prevalent drug resistance [1,3].
Designing drug delivery systems that can specifically target molecular disease markers is a leading strategy in modern drug development for increasing efficacy and circumventing the adverse effects of cytotoxic drugs [4]. Such targeting can be achieved through the use of ligands with high specificity towards receptors that are overexpressed on diseased cells, which leads to a high local drug concentration in the affected tissue. Targeted drugs, such as T-DM1 (Kadcyla), utilize the specific cell-targeting properties of an antibody (trastuzumab) combined with the potent cytotoxic properties of an attached tubulin inhibitor (DM1) [5]. Presently, several such antibody-drug conjugates (ADCs) have been FDA-approved and are being used in the clinic [6]. However, the development of ADCs is costly and complicated due to the complex nature of monoclonal antibodies and their production.
In addition, there is a need for drug delivery systems that can combine more functionalities than simply a targeting moiety and a drug or imaging agent. Studies from both animals and humans have shown that a synergistic effect of tissue targeting and drug delivery can be achieved by multimerizing targeting ligands with specificity for different sites on the target receptor, and by co-administering or co-delivering multiple drugs with different modes of action [7]. Such combinatorial drug design addresses the issue of tumor heterogeneity and thereby helps to increase the efficacy of the drug while simultaneously reducing the likelihood of induced drug resistance [7]. By further combining delivery of a therapeutic drug with diagnostic imaging, theranostic treatment can be achieved, leading to more precise diagnosis and treatment optimization with better patient outcomes [8]. For these reasons, nanostructures that allow for flexible bioconjugation and can accommodate a combination of drugs, targeting ligands, and imaging agents for theranostic purposes are highly desired.
Nanoparticles have shown promise for co-delivery of drugs and diagnostic imaging agents with the use of multiple targeting ligands. In most cases, a biologically inert polymer is used to condense the desired functionalities into a single noncovalent structure with improved pharmacokinetic (PK) and tissue-targeting properties [9,10]. However, such nanoparticles suffer drawbacks from uncontrolled synthesis causing variations in size, stoichiometry, and surface properties with resulting unpredictable biocompatibility problems [11]. Moreover, their size (usually 50-200 nm) often prevents extravasation into diseased tissue, e.g. a solid tumor. An additional limitation for this type of delivery system is that the molecular core of the nanoparticle, and thereby its principle structural components, is often chosen based on the biophysical properties of its cargo, i.e. charge, size, and hydrophobicity [12]. As a result, many nanoparticle systems are incompatible with the varying nature of different cargo types, and substitution of one type of cargo or targeting moiety with another most often requires extensive redesign, resynthesis and recharacterization.
To circumvent the challenges of low stoichiometric control, several conjugated systems have also been proposed, such as ADCs and small molecule-drug conjugates, that are generally designed following a simple targeting ligand-chemical linker-drug/imaging agent scheme [13]. Such conjugated designs can also include additional functionalities, as in the case of targeted dye-drug conjugates developed for theranostics [14]. However, these types of conjugated systems generally lack modular properties, i.e. the possibility of readily exchanging either the targeting or therapeutic moeities to allow for broader applications and personalization without requiring a resynthesis of the entire structure.
The inherent programmability, and thereby modularity, of nucleic acids have led to the proposal of DNA- and RNA-based nanostructures for theranostic medicine. Such designs take advantage of the sequence-specific self-assembly of nucleic acids into complex structures with interchangable modules containing targeting moieties, as well as bioimaging- and drug molecules [15,16]. With these unique properties, the field of DNA nanotechnology holds the potential to solve many of the challenges of combining multiple functional conjugates with predictable stoichiometry. However, nucleic acid nanostructures proposed for nanomedicine are in many cases large and complex in nature [17] and most often exhibit low in vivo stability and poor PK properties, as well as a potential for high immunogenicity through signaling by pattern recognition receptors of the innate immune system [18]. Even in those instances where DNA nanostructures have shown efficacy, definitive proof of structural integrity in vivo is not provided [19,20].
The implementation of chemical modifications, such as 2'-Fluoro and 2'-O-methyl (2'-OMe) nucleotides into nucleic acid nanostructures allows for a higher degree of nuclease resistance, particularly in the case of larger RNA nanostructures [21], aptamers [22], siRNA [23], and antisense oligonucleotides [24]. The highly stabilizing nucleotide, locked nucleic acid (LNA), has been shown to increase both the serumstability and melting temperature (Tm) of a duplex by approximately 4 degrees per base pair, and can therefore allow for stable assembly of smaller and simpler structures [25,26]. However, while the use of LNA for stabilization is common in antisense oligonucleotides [27] and siRNA [28], these modifications have to our knowledge not been used as central structural components in well-defined self-assembled nucleic acid nanostructures.
Here we describe a defined, modular system that can be used for bioimaging and targeted drug delivery. Four complementary 2'-OMe oligonucleotides, stabilized by LNA, are assembled into a discrete structure resembling a Holliday Junction (HJ). This structure is nuclease resistant and remains stable in biological fluids for several days. We demonstrate that each oligonucleotide can be functionalized with essentially any type of molecule without compromising the self-assembly process. Accordingly, we successfully produced structures containing fluorophores, high molecular weight polyethylene glycol (PEG), carbohydrates, peptides, lipids, and cytotoxic drugs, in up to four copies. Assembly of the four functionalized arms occurs rapidly and requires only room temperature mixing of stoichiometric amounts of the oligonucleotides. In vivo, the non-conjugated HJs are quickly cleared by renal filtration. However, the attachment of one or more PEG chains or fatty acids enabled fine-tuning of the circulation time. Conjugation of one or more targeting ligands resulted in specific and effective cellular uptake both in vitro and in vivo. With these results, the novel LNA-modified HJ presents an unusually robust and simple platform for the controlled and modular assembly of multiple complex functionalities for targeted bioimaging, and with a strong potential for applications within drug delivery and theranostics.
Methods
Synthesis of oligonucleotides. All standard DNA oligonucleotides were purchased from IDT. All modified oligonucleotides were synthesized on an ÄKTA oligopilot plus 10 system in 1.0 μmol scale (polystyrene support) using the phosphoramidite approach and following manufacturer's standard protocols. Oligonucleotides (Q1-Q4) were synthesized according to reference [29]. Amino-modifier C6 (Glen Research) was incorporated into oligonucleotides on the 5' end via manual-coupling using 5-[3,5-bis(trifluoromethyl)phenyl]-H-tetrazole (0.25 M, in anhydrous acetonitrile) as activator and extended coupling time (20 min) [30]. Oligonucleotide (Q3-2xpalmitoyl) was synthesized as described previously [31]. Purification to at least 80% purity of all modified ONs was performed by RP-HPLC or IE-HPLC, and the composition of all synthesized ONs was verified by MALDI-MS analysis recorded using 3-hydroxypicolinic acid as a matrix. The sequences of all oligonucleotides used in this study are shown in Table 1.
Oligonucleotides used in this study.
| Name | Sequence (5'-3')* |
|---|---|
| Q1 | NH2C6 mClCmGlTmCmCmTlGmAlGmCmC |
| Q2 | NH2C6 mCmAlCmAlGmTmGlGmAlCmGmG |
| Q3 | NH2C6 mGlGmClTmCmAmCmClGmAlTmC |
| Q4 | NH2C6 mGmAlTmCIGmGmAmClTmGlTmG |
| Q3-2xPalmitoyl | aT(Pal)aT(Pal)mGlGmClTmCmAmCmClGmAlTmC |
| DNA control | TAGAATTCTAATACGACTCACTATAGGGCTCACCGATCGGGAGGACGATGCGGA |
* Unless otherwise specified all nucleotides are deoxynucleotides. m = 2'-O-methyl, l = LNA, aT(Pal) = 2'-N-palmitoyl-2'-amino-LNA-T.
Bioconjugation. For conjugation to the 5' end of each oligo, commercially available NHS-functionalized molecules were used when possible. For conjugations where an NHS-functionalized variant was not available or was incompatible with the inherent chemistry of the molecule to be conjugated, a two-step reaction was used, in which the oligo was first reacted with an NHS-DBCO heterobifunctional linker, purified, and subsequently reacted with an azide-functionalized biomolecule. All attached molecules and their sources are listed in Table 2.
List of molecules attached to the HJ modules.
| Name | Description | Reference |
|---|---|---|
| NHS-PEG20K | 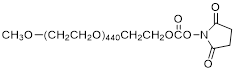 | Nanocs (PG1-SC-20k) |
| NHS-PEG10K | 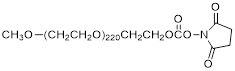 | Nanocs (PG1-SC-10k) |
| NHS-PEG5K | 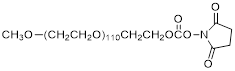 | Nanocs (PG1-SC-5k) |
| NHS-Cy3 | 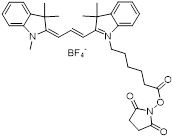 | Lumiprobe (#41020) |
| NHS-Cy5 | 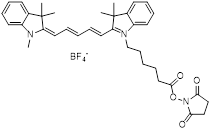 | Lumiprobe (#43020) |
| NHS-Cy5.5 | 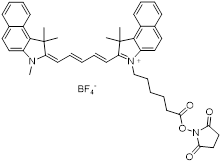 | Lumiprobe (#47020) |
| NHS-DBCO | 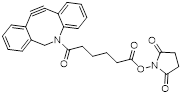 | Lumiprobe (#54720) |
| NHS-C3-Azide | 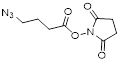 | BaseClick (BCL-014-5) |
| TriGalNAc-Azide | 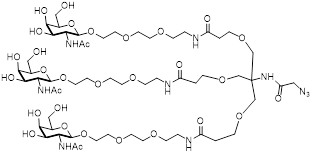 | This study, see suppl. methods |
| N2'-palmitoyl-2'-amino-LNA monomer | 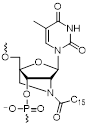 | This study |
| Tfrs | HN-SQETFSDLWKLLPEN-OH | Biosynthan |
| DM1 | 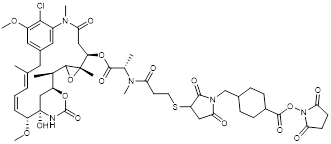 | XDC Explorer |
The 5'-amine oligos were reacted with NHS esters following the general protocol: 1, 10 or 50 nmol oligo was prepared in NHS reaction buffer (100 mM HEPES, pH 8.2, 30% DMSO) at a final concentration of 0.5 mM. To this was added from different stock solutions 5-50-fold molar excess of the appropriate NHS ester. For NHS esters that were insoluble in water (such as NHS-DBCO) the DMSO concentration was adjusted to 50%. Reactions were incubated at RT with 500-700 RPM shaking and stopped after 4 h or after over-night (ON) incubation by ethanol precipitation. The resulting conjugation reactions were dissolved in 100 μL RNase free water and purified by RP-HPLC using a MeCN gradient on a C18 column. Fractions containing the appropriate conjugates were collected and quantified by UV spectroscopy. The purity and integrity of all conjugates was verified by denaturing polyacrylamide gel electrophoresis (PAGE). A representative HPLC chromatogram for each conjugate produced in this study as well as a gel image of the purified product compared to unreacted oligo are shown in Figure S1.
In cases where no NHS ester was available, or the NHS function was incompatible with the function of the molecules, azide-functionalized reagents were used. These were reacted with DBCO-coupled oligos in a 3:1 (azide to oligo) ratio in DBCO reaction buffer (100 mM HEPES, 30% DMSO, pH 7.5) at a concentration of at least 100 μM. The final conjugates were purified by RP-HPLC followed by quantification and freeze drying for long term storage.
Assembly of Holiday Junctions. Equimolar amounts of each HJ oligo (Q1-4) were mixed at a final concentration of 1-100 μM in one of the following buffers: assembly buffer (200 mM KOAc, pH 7.5), phosphate buffered saline (PBS, pH 7.4), Tris (10 mM, pH 7.4), or TAEM (40 mM tris, pH 8.3, 1 mM EDTA, 12 mM MgCl2). The mixtures were incubated at RT for at least 30 min prior to analysis. For animal studies, HJs were assembled and analyzed one day prior to injection and stored at 4 °C ON. HJs for cell studies were all assembled in 200 mM KOAc.
For studies of assembly kinetics, oligos were pre-incubated at 10 μM concentration in water to prevent the formation of intermolecular base pairing before the addition of a buffer. At specific time points, 1 μL of the pre-assembled HJs was transferred to 9 μL of a specific buffer to allow complex formation to initiate. The assembly process was visualized by non-denaturing PAGE gel electrophoresis.
FRET studies. The four HJ oligos were 5'end-labeled with Cy3 or Cy5 by NHS ester-amine reactions. All HJ combinations were assembled with two labeled oligos, one with Cy3 and one with Cy5. These were analyzed on a non-denaturing 10% PAGE gel scanned for Cy3, Cy5 and for Cy5 emission when excited for the Cy3 fluorophore to measure the FRET signal, using the software ImageQuant TL. The Cy5 signal obtained from excitation of Cy3 was normalized to the Cy5 signal from the Cy5-only scan.
Melting curves. HJs were assembled in PBS at 2 μM concentration with or without the following functionalities: 1-3 PEG20K and 1-3 triGalNAc. The HJs were incubated with SYBR Gold (1:10,000) in 20 μL volumes in LightCycler well plates, and the stability was monitored as a decrease in fluorescence as the HJ oligos dissociated. In all cases tested, the melting temperature was approximately 80 °C, which is significantly higher than a corresponding 48 nucleotide double-stranded (ds)DNA (Tm: 65 °C, OligoAnalyzer https://eu.idtdna.com/calc/analyzer) but somewhat lower than a corresponding 48 nucleotide dsDNA containing 16 LNA residues (Tm: 93 °C, OligoAnalyzer).
In vitro stability in serum. HJs were pre-assembled at a final concentration of 2 μM in PBS for one hour. To also study the stability of the fluorophore, a small amount of Q1-Cy3 was spiked in (0.1 μM final concentration). The experiment was conducted over a total period of 72 h. At different time points, 1 μL of the pre-assembled HJ was added to a 10 μL PBS containing 10% fetal bovine serum (FBS) (Gibco) and incubated at 37 °C. A negative control sample was transferred to 10 μL PBS without FBS and stored for 72 h at 37 °C. As a positive control for degradation, a 54 nt single-stranded DNA oligo was included. We also included an RNA control. However, this was degraded almost immediately following incubation with FBS.
Cell culture. Human hepatocarcinoma cells (HepG2) were grown in EMEM medium (ATCC) supplemented with 10% FBS (Gibco) and 1% penicillin/streptomycin (P/S) (full medium), at 37 °C, 5% CO2. Prior to seeding, the cells were washed with PBS (Dulbecco) and released by incubating with 0.05% Trypsin-EDTA (1X, Gibco) for 5-10 min. Cells were centrifuged at 200g for 5 min and resuspended in full medium. Cell number and viability was determined by use of a Via 1-Casette counting chamber (Chemometec) using the software NucleoView NC-200.
Flow cytometry. For uptake studies, cells were seeded in full medium as 150,000 cells per well in a 24-well plate one day prior to treatment. Cells were incubated in three biological replicates with medium for autofluorescence controls and with Cy5-labeled HJs in concentrations of 25 nM, 50 nM, and 100 nM for 45 min. After incubation, the cells were washed three times with PBS and trypsinated for 10 min. Cells were resuspended in full medium and centrifuged for 10 min at 2000 g. The supernatant was removed, and the cells were resuspended in 130 µL PBS and transferred to 96 well plates for analysis. Flow cytometry was performed on a NovoCyte flow cytometer (Acea) and analyzed using NovoExpress. The median fluorescence for each sample was normalized to the average of the three HJ-Cy5 controls.
Albumin binding. HJs carrying a single module (Q3) with two amino LNA-modified palmitoyl groups were assembled at 1 µM concentration in PBS by heating to 75 °C for 5 min, followed by incubation at room temperature for 60 min. 1 pmol assembled HJ-Palmitoyl was transferred to tubes containing 10 μL PBS and various concentrations of human serum albumin (Sigma, A6608, two fold dilution series starting at 16 μM). The samples were incubated for 15 min at RT and analyzed on a 12% denaturing PAGE gel.
Confocal microscopy. HepG2 cells were seeded as 100,000 cells per well in an 8 well microscopy slide in full medium one day prior to treatment. Cells were incubated at 37 °C with either full medium or Cy5-labeled HJs in 100 nM for 45 min. The medium was removed, and the cells were washed three times with PBS. The cells were stained with 200 µL 2.5 µg/mL wheat germ agglutinin (WGA)-Alexa 488 for 15 min at 37 °C. The cells were washed three times with PBS and fixed by adding 200 µL 4% paraformaldehyde (PFA) and incubated at 37 °C for 15 min. Samples were washed with PBS, dried and stained with one drop of DAPI ProLong Gold (Invitrogen) to each position on the slide. After mounting of the cover slide, the samples were incubated at 4 °C ON. Cells were imaged on a confocal laser scanning microscope (Zeiss LSM 700) with a 63X oil objective.
Immunogenicity. Human monocytes and macrophages were isolated as peripheral mononuclear blood cells (PMBCs) from a buffy coat from a healthy donor, using Ficoll-Paque density gradient separation. The isolated collection of leukocytes was diluted in RPMI medium with endotoxin-free FBS (Gibco) and seeded out as 100,000 cells per well in a round-bottom 96 well plate. 4 h after seeding, cells were treated with lipoplexes (0.3 µL Lipofectamine2000 (Invitrogen) per well) containing the different variations of HJs in three replicates and incubated for 18 h at 37 °C. The cells were centrifuged, and the cell-free supernatant was collected and diluted with ELISA diluent buffer. The induction of TNF-α was measured using a human TNF-α ELISA kit (BioLegend) following the manufacturer's instructions. The samples were added to a Maxisorp 96-well plate, pre-coated with capture antibodies, along with a serial titration of recombinant TNF-α standard for the calculation of a standard curve. From this standard curve, the amount of TNF-α measured in the different samples was calculated.
Animal experiments. All animal procedures were approved by the Animal Experiments Inspectorate, under the Danish Veterinary and Food Administration Licence # 2013-15-2934-00901. Studies were performed on 10-week old adult female Balb/c mice. All animals had free access to a standard rodent diet and water. During the experiments, animals were kept in groups of 4-5 mice per cage. For scanning of live animals, the animals were anaesthetized with isoflurane.
Unless otherwise specified, all fluorescence scanning was performed on an IVIS 200 instrument (Xenogen, Caliper Life Sciences, Hopkinton, MA, USA) using Living image 4.3 software (Caliper Life Science). Fluorescent signals were subjected to spectral unmixing using the system's in-build Cy5.5 filter settings and an untreated mouse control.
For PK and biodistribution experiments, 500 pmol of Cy5.5-labeled HJs (with or without a specific set of functionalities) were administered through I.V. tail vein or S.C. injection (n ≥ 3 for each injection). Blood samples were drawn from the tail of each animal at different time points over 24 h using Microvette 300 tubes (Sarstedt). The blood serum was collected by centrifugation of the tubes for 5 min at 10,000 g. A total volume of 5 µL serum was mixed with 50 µl PBS and subsequently scanned in a 96-well black Illumino plate (Thermo Scientific). For experiments involving palmitoylated HJs, blood samples were collected using heparinated 20 µl capillary pipettes (Vitrex). The capillary tubes were sealed with wax and placed in a non-refective placeholder before scanning. In all cases, individual blood samples were normalized to the injected total fluorescence assumed from the 2 min time point. Animals were sacrificed by cervical dislocation after approximately 24 h to ensure a high clearance from blood and urine. The organs were removed and scanned in the IVIS instrument as described above.
For tissue slicing, the relevant organs were fixed in formalin and subsequently embedded in paraffin (FFPE). Sections were prepared by the Department of Pathology, Aarhus University Hospital, and mounted on glass slides. De-paraffinization was done using Tissue Clear Xylene substitute (Tissue-Tek/Sakura Finetek) and sections were rehydrated in decreasing ethanol solutions with concentrations from 100% to 70% and placed under running cold tap water. The slides were stained with DAPI ProLong Gold.
Statistical analysis. T-tests were performed in GraphPad Prism to test significance between each group within one experiment. * P < 0.05, ** P < 0.01, *** P>0.001.
Results
Design and characterization of the HJ scaffold. By exploiting the unique properties of nucleic acids to self-assemble, we designed a small, modular scaffold that can bring together multiple functionalities relevant to drug delivery and/or bioimaging. Ideally, such a scaffold should be as small as possible to reduce production costs, promote tissue penetration, and ensure rapid renal clearance when used for bioimaging. In addition, it should be sufficiently stable to remain intact in biological fluids, and should be amiable to conjugation to a wide range of biomolecules.
After investigating different structural designs in terms of assembly kinetics and stability, a four-way junction resembling a Holiday Junction (HJ) was selected for further experiments. The HJ is a central intermediate of homologous recombination, but has also proven to be remarkably versatile as the underlying structural basis of DNA origami [32]. Importantly, stacking of the terminal nucleotides of a HJ is not required for structural integrity, and bioconjugation at the 5' or 3' positions is therefore unlikely to compromise the overall assembly of the HJ.
To stabilize the scaffold thermodynamically, and to reduce its susceptibility towards nucleases, the structure includes four LNAs and eight 2'-OMe nucleotides in each of the four strands (denoted Q1, Q2, Q3 and Q4). Since the stabilizing effects of LNA extend to neighboring base pairs [33], these were strategically positioned between the 2'-OMe nucleotides to achieve maximum stabilization of the duplex. We considered different variations, including longer arms and alternative positioning of the LNAs, but ultimately decided on the design illustrated in Figure 1A. This structure has a minimal arm length of only six base pairs and showed robust assembly with no apparent unspecific structural interactions (Figure 1B).
Assembly of the HJ. (A) Schematic illustration of the HJ and its assembly. The four strands are denoted Q1-Q4 and each consists of 12 nucleotides and a 5'-NH2 group for bioconjugation. The position of each LNA is indicated by an asterisk (*). The remaining nucleotides are 2'-OMe. (B) Native polyacrylamide gel showing the assembly of the HJ. Lanes 1-4 contain single stranded oligos, lanes 5-8 show the incomplete assemblies of the four dimers, and lane 9 shows the assembly of the complete HJ. The gel was stained with SYBR Gold. (C) Kinetics of HJ assembly in PBS, TAEM, KOAc, 10 mM Tris, and water. The four HJ oligos were mixed in equimolar amounts in H2O and distributed in separate tubes. At the indicated time points, concentrated buffer was added and assembly monitored by gel electrophoresis. (D) Polyacrylamide gel electrophoresis of HJs assembled with various combinations of Cy5 and Cy3 labeling. The image shown is an overlay of three signals: the red Cy3-signal, the green Cy5-signal and the blue 'FRET' signal. The average 'FRET' signal between the various oligo pairs normalized to the Cy5 signal in each well. The data represents averages from three independent gels. * P<0.05, ** P<0.01, *** P>0.001.
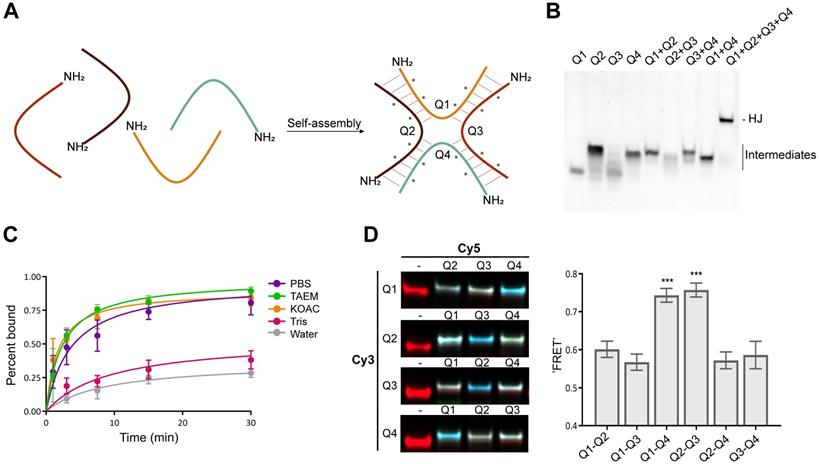
The assembly process was studied using gel electrophoresis under various buffer conditions. In low-salt buffer (10 mM Tris pH 7.4) assembly of the HJ was slow and remained incomplete after 30 min, while in pure water, no assembly could be detected (Figure 1C). In contrast, in high-salt buffers such as PBS, TAEM or KOAc the HJs were assembled efficiently after 15-30 min (Figure 1C).
Functionalization of HJ modules and assembly of multifunctional complexes. Each of the four oligonucleotide modules that make up the HJ were synthesized with 5' terminal amino groups to allow for straightforward bioconjugation. In order to test the flexibility of the system towards functionalization, a variety of different therapeutically relevant molecules including fluorophores, peptides, lipids, high molecular weight PEGs, carbohydrates, and cytotoxic drugs were attached to each of the four HJ arms (Figure S1 and Table 2), and their effect on the assembly process of the HJ complex was assessed (Figure S2). We found that structures containing up to four different classes of functional molecules could be assembled with >90% efficiency. As a consequence of the high purity of the individual functionalized modules and the effective assembly process, post-assembly purification was not required for any of our subsequent studies.
To study the structure of the HJ under various conditions, Förster resonance energy transfer (FRET) between pairs of conjugated fluorophores was measured. In a high Mg2+ buffer (TAEM), combinations of Cy3 and Cy5 FRET pairs on the HJ showed a distinct pattern in which the Q1-Q4 and Q2-Q3 FRET signals were significantly higher than any other combination, suggesting that the 5' ends of those oligos are in closest proximity (Figure 1D). The formation of such coaxially stacked HJs is well established for DNA, which is known to form stacked X-structures in the presence of divalent cations [34]. Accordingly, when similar measurements were carried out in KOAc buffer or PBS, no particular pattern in the FRET data was seen (data not shown), suggesting that the HJ assumes a non-stacked conformation with approximately equal distances between each arm, possibly with a favored conformation determined by the attached bioconjugate.
Stability and immunogenicity of the HJ scaffold. To test the stability of the HJ in serum, a sample of pre-assembled HJ, of which a small fraction was labeled with a Cy3 fluorophore, was incubated for up to 72 h in 10% FBS at 37 °C (Figure 2A). The HJ scaffold, detected by SYBR Gold nucleic acid stain, remained stable for the entire period (72 h; Figure 2A, middle panel), while unmodified DNA was degraded within a few hours (Figure 2A, lower panel). The Cy3-labeled fraction of the HJs also appeared to be partially degraded over time (Figure 2A, upper panel), most likely due to degradation of the Cy3 dye rather than loss of the HJ structure itself.
Stability and immunogenicity of the HJ. (A) Serum stability of the HJ. The HJs were pre-assembled in PBS as described in the methods section. In order to discriminate between degradation of HJ and the fluorescent probes, the HJ sample was spiked with a small amount of HJ-Cy3 and visualized with Cy3 (upper panel) or SYBR Gold imaging (middle panel). SYBR Gold stained 45 nt ssDNA was included as control (lower panel). At specific time points samples were transferred to new tubes containing 10% FBS. (B) Melting curve of HJ scaffold based on SYBR Gold binding. The apparent Tm is 80 °C. (C) Induction of TNF-α in human PBMC culture, measured by ELISA. * P<0.05, ** P<0.01, *** P>0.001 compared to polyI:C control.
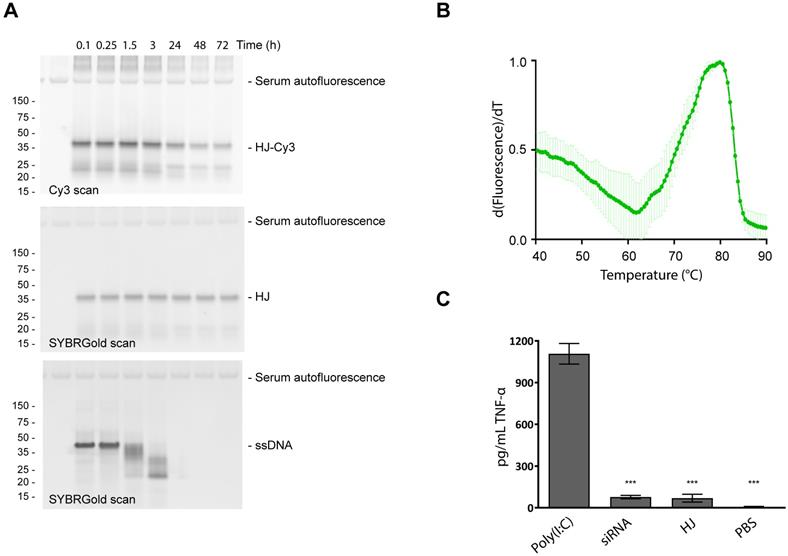
Next, the thermostability of the scaffold was measured using a SYBR Gold binding assay. For non-functionalized HJs, a melting temperature (Tm) of 80 °C was measured (Figure 2B). HJs assembled with 1-3 PEG20K or 1-3 triGalNAcs exhibited similar Tm values (78-81 °C, figure S3). In comparison, an equivalent DNA duplex with six paired bases in each arm was not able to assemble (data not shown), indicating that the LNA residues contribute significantly to the stability of the HJ structure.
An important property for any drug delivery system is high biocompatibility and minimal immunogenicity. To assess this, peripheral mononuclear blood cells (PMBCs) from a healthy donor were incubated with 100 nM of HJ and the resulting induction of TNF-α was measured. The results show that addition of HJs triggered only a small increase in TNF-α levels relative to the PBS control (Figure 2C). This effect was not significantly different from that caused by a similar treatment with a control siRNA. In contrast, the addition of the highly immunogenic polyI:C resulted in a significant induction of TNF-α. We conclude that within the concentration range intended for future use, the HJ by itself is not significantly immunostimulatory which supports its potential for in vivo use.
Pharmacokinetics and biodistribution. In order to investigate the potential of the HJ for drug delivery and theranostics, its half-life and biodistribution profile were studied in mice. We anticipated that the small size of the HJ would ensure fast renal clearance from the blood stream. Indeed, following intravenous (I.V.) injections in mice (Figure 3A), Cy5.5-labeled HJs were quickly cleared from the bloodstream (half-life: ~21 min), and after 24 h the majority of the HJs were observed in the kidney (Figure 3B). When examining slices of the kidneys at higher resolution by 2D fluorescent scanning, the HJs were primarily detected in the renal cortex (Figure 3C), which by confocal microscopy could be identified as proximal tubule cells (Figure 3D). Importantly, the HJs did not appear to accumulate in these cells as the signal decreased over time and became undetectable after 72 h (Figure S4).
Circulation time and biodistribution of unconjugated HJs. (A) Blood circulation time of Cy5.5-conjugated HJ after I.V. or S.C. injections. The lower panel shows a representative image of the collected samples imaged on an IVIS instrument (B) Biodistribution of Cy5.5-labeled HJs after 24 h. * P<0.05, ** P<0.01, *** P>0.001. The lower panel shows a representative image of the collected organs from a mouse. (C) Fluorescent scan of a paraffin-embedded mouse kidney. HJs were labeled with Cy5. The color scale is rainbow (red: high, blue: low). (D) Confocal microscopy images of a paraffin-embedded mouse kidney showing Cy5-labeled HJs (in red) as they pass through the kidney. The slide has been stained with DAPI to indicate nuclei and overlayed with a brightfield image.
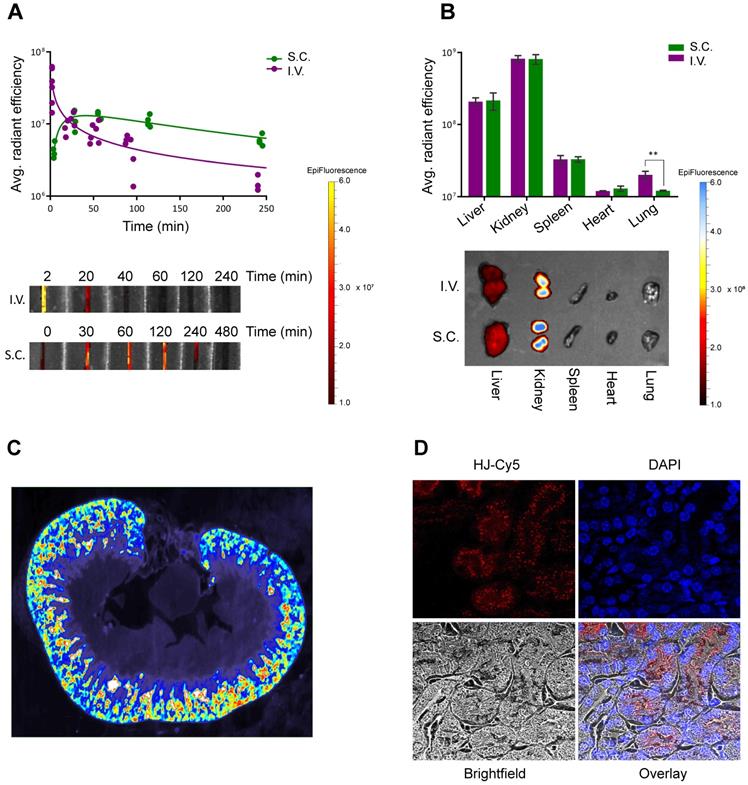
HJs administered subcutaneously (S.C.) showed markedly different blood clearance kinetics (Figure 3A). The HJ blood levels reached a maximum approximately 30-60 min post injection. However, this maximum level corresponded to only about 10% of the maximum level observed from I.V. injections. Nonetheless, the overall biodistribution profile after 24 h was almost identical to that of the I.V. injected samples, with the majority of the HJs observed in the kidney followed by the liver. S.C. injections did result in significantly lower levels in the lungs, which may be attributed to the lower maximum blood levels achieved following S.C. injections (Figure 3B).
Pharmacokinetic modification of HJs with PEG and palmitoyl. The high stoichiometric control of the HJ allowed us to conduct a systematic investigation of the PK properties of the structure with different types and number of conjugates. Initially, HJ oligos were conjugated and assembled with variable number and sizes of PEG polymers, as PEGylation is one of the most widely used approaches for prolonging circulation times [35]. As seen from the agarose gel in Figure 4A, HJs assembled with different numbers and combinations of 5 and 20 kDa PEG chains (PEG5K and PEG20K) all formed well-defined structures.
Pharmacokinetic enhancement of the HJ using PEG and palmitoyl. (A) Agarose gel electrophoresis of HJs assembled with variable number and sizes of PEG chains. HJs were assembled with PEG20K at two different positions to ensure that a flexible placement was allowed. (B) Relative blood levels of HJ without modification (red), with one PEG20K (light blue), two PEG20K (dark blue) and palmitoyl (green) after I.V. injection. On the right side is shown a representative image of the fluorescent scan of the blood samples. (C) Biodistribution of unfunctionalized, PEGylated, and palmitoylated HJs in mice. * P<0.05, ** P<0.01, *** P>0.001 compared to unfunctionalized HJ. (D) In vitro binding between HJ-Palmitoyl conjugates and human serum albumin. HJs carrying two palmitoyl groups on Q3 were preassembled in PBS and subsequently incubated with increasing amounts of albumin.
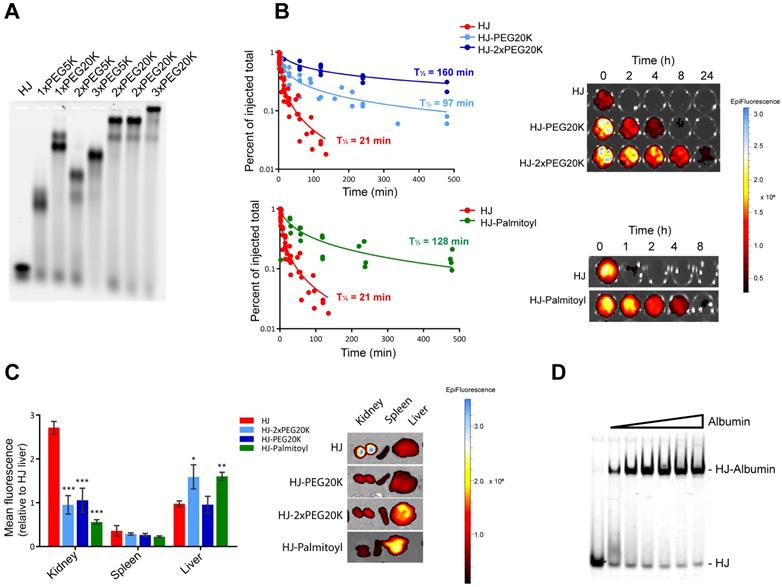
As the renal filtration cut-off is usually observed to be in the 30-50 kDa range, we expected a significant change in circulation time from attaching just a single PEG20K molecule. To test this, we injected Cy5.5-labeled HJs either without any modified modules, or with one, two, or three PEG20K modules into mice and collected blood samples at different time points over 24 h. The results presented in Figure 4B demonstrate that while naked HJs are quickly cleared from the blood (half-life ~21 min), HJs functionalized with a single PEG20K chain continued to circulate with a calculated half-life of ~95 min, while HJs with two PEG20K chains could still be detected in the blood after 24 h (half-life ~160 min). The addition of three PEG20K chains did not significantly increase circulation time any further (Figure S5).
PEGylation of the HJ also had a significant effect on the biodistribution pattern (Figure 4C). Where unfunctionalized HJs were excreted through the kidneys, HJs with one or more PEG20K showed a more peripheral distribution pattern. Imaging of live animals after 24 h showed no peripheral signals above background in mice injected with unfunctionalized HJs, whereas mice injected with HJs carrying one or more PEG20Ks exhibited a widely distributed signal across the body including the extremities, such as paws, tail and nose (Figure S6). These combined results demonstrate that both the half-life and biodistribution of the small structure can be controlled, and that one or two PEG20K moieties is sufficient to extend circulation times in vivo.
A different strategy for half-life extension is to conjugate drugs with biodegradable fatty acids. This approach is utilized in the design of several different FDA-approved drugs, including Levemir and Victoza [36,37], of which the latter contains a C16 palmitoyl group. These drugs exploit the extraordinarily long half-life of circulating human serum albumin (HSA) of ~19 days by non-covalent association with fatty acid binding sites on the protein [38]. To investigate the utility of this approach for the HJ scaffold, one module (Q3) was synthesized with two palmitoyl groups at the 5'-terminus. Although the palmitoylated oligo tended to form aggregates by itself, no aggregation was observed by gel electrophoresis once it was inserted into the HJ structure (Figure 4D). Gel shift assays with purified HSA confirmed that the palmitoylated HJ bound albumin with high binding affinity and fast kinetics (Figure 4D), similar to what has previously been observed for palmitoylated antisense oligos [39].
To analyze the effect of the palmitoylation in vivo, Cy5.5-labeled HJs were injected into mice and blood samples were collected at different time points over a period of 24 h, whereafter the animals were sacrificed, and their organs were scanned for fluorescent signal. As shown in Figure 4B, the palmitoyl groups had a significant effect on the circulation half-life of the HJs, increasing it from approximately 21 min for the unconjugated HJ to more than 2 h for the HJ-Palmitoyl construct. Importantly, after 24 h HJ-Palmitoyl was detected predominantly in the liver with only a small fraction found in the kidney, suggesting that renal clearance had to a large extent been circumvented (Figure 4C). This is consistent with the reported PK properties of previous palmitoyl-conjugated constructs [40].
Targeted delivery in vitro. The asialoglycoprotein receptor (ASGP-R) is conserved in all mammals and is highly expressed on the surface of hepatocytes [41]. The functional entity is a trimer comprised of two proteins that, in the presence of calcium, exhibits high affinity for N-acetyl galactosamine (GalNAc)-terminated oligosaccharides [42]. The primary function of ASGP-R appears to be the clearance of glycoproteins by way of receptor-mediated endocytosis. However, more recently this receptor has also been used for targeted delivery of a variety of drugs, including oligonucleotides, to the liver in mice [43,44].
Using the potential to control the valency of the HJ, we investigated the effect of multimerization of the triGalNAc units on the cellular uptake in hepatocytes. The tri-antennary conformation of GalNAc sugars has been shown to bind with high affinity to the ASGP-R, and we hypothesized that a multivalent display of trivalent sugars would be able to crosslink multiple receptors and therefore further enhance endocytosis.
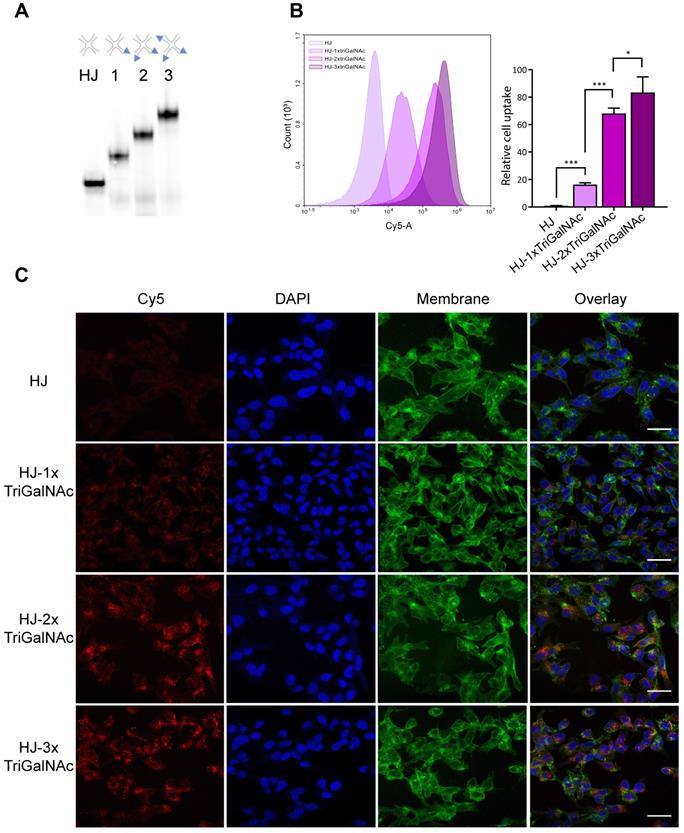
HJs were assembled with one, two, or three triGalNAc moieties and a Cy5 group for detection (Figure 5A). Human hepatocarcinoma cells (HepG2) were incubated with non-targeted HJ-Cy5 or HJ-Cy5 carrying the different numbers of triGalNAc groups. After 45 min, the cells were washed and subsequently analyzed by flow cytometry to compare cellular uptake between the different structures (Figure 5B). Relative to the non-functionalized HJs, scaffolds carrying a single triGalNAc showed ~16-fold increased cell uptake whereas HJs with two and three triGalNAcs showed ~65 and ~80-fold increased uptake, respectively. To ensure that the fluorescence signal detected by flow cytometry did indeed originate from internalized HJs, confocal microscopy was used to image HepG2 cells treated with these same samples (Figure 5C). Here, a clear intracellular signal with a punctuate cytoplasmic distribution was observed, indicative of endosomal localization. Collectively, these data show that the efficient internalization via the ASGP-R receptor can be further potentiated by a multivalent display of tri-antennary ligands on the HJ.
Hepatocyte-specific uptake of triGalNAc-functionalized HJs. (A) Non-denaturing polyacrylamide gel electrophoresis of assembled HJs with variable numbers of triGalNAc residues, scanned for Cy5. (B) Relative cellular uptake of HJs with 1-3 triGalNAcs compared to non-targeted HJs, assessed by flow cytometry. Flow cytometry histograms show the cell-associated fluorescence from one replicate from each group within one experiment, and the bars show the median fluorescence from three biological replicates from two independent experiments, normalized to the HJ-Cy5 control. * P<0.05, ** P<0.01, *** P>0.001. (C) Confocal microscopy images of HepG2 cells treated with the samples shown in B. Cell membranes are shown in green, DAPI nucleus stain in blue, and the Cy5 signal is shown in red. Scalebars: 30 µM.
Targeted delivery in vivo. To test if the multivalency effect from conjugation of multiple triGalNacs observed in the in vitro uptake studies would also result in higher uptake in hepatocytes in vivo, we injected mice with Cy5.5-labeled HJ alone or conjugated with one, two, or three triGalNAcs. Blood samples were collected at different intervals and the organs were harvested 2 h post injection. All injected samples displayed similar blood circulation half-lives (Figure S7) and were essentially undetectable in the blood after 2 h. However, their biodistribution profiles were markedly different. Unfunctionalized HJs were excreted via the kidney as previously observed, while the triGalNAc-functionalized HJs were all found predominately in the liver (Figure 6A). When comparing the three triGalNAc groups, a clear trend was observed that a higher valency of triGalNAcs resulted in stronger liver accumulation, although only the difference between a single and three triGalNAc units was statistically significant (Figure 6A).
Targeting of HJs using triGalNAc in mice. (A) Ex vivo biodistribution of Cy5.5-labeled HJs alone or conjugated to 1-3 triGalNAcs. A representative image of the organs is shown to the right. (B) Fluorescent scan of an entire lobe excised from the liver. The color scale is rainbow (red: high, blue: low). (C) Confocal microscopy images of a paraffin-embedded mouse liver lobe showing Cy5.5-labeled HJs evenly distributed in the liver. The slide has been stained with DAPI to indicate nuclei and overlayed with a bright fieldimage.
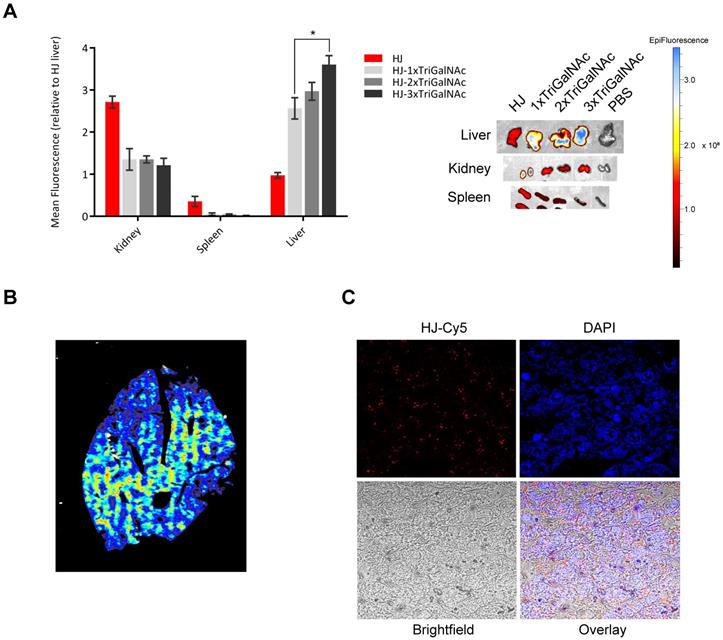
Imaging of tissue slices of the collected livers in a 2D fluorescence scanner showed that the triGalNAc-functionalized HJs were distributed evenly throughout the entire organ (Figure 6B), suggesting that uptake predominantly occurs in hepatocytes. These data were confirmed by confocal microscopy (Figure 6C), which showed that the HJs were not confined to areas near blood vessels or in macrophages, but rather distributed evenly throughout the tissue. Thus, we conclude that the triGalNAc-functionalized HJs are indeed taken up by hepatocytes and that the multivalency of triGalNAc groups contributes to higher uptake.
Discussion
In this study, we have constructed a small (~16 kDa), LNA-based nanoplatform that structurally resembles a HJ. The assembled scaffold appears to either exist in a relaxed form with approximately equal distances between the four arms, or as a stacked, X-shaped structure, depending on the presence of divalent cations. This is consistent with previous observations that Mg2+ induces a stacked conformation of DNA HJs [45]. Each of the four arms can be functionalized and assembled in a modular fashion with, in principle, any biomolecule with an accessible chemical handle for site-specific conjugation. Due to the stabilizing effects of the modified nucleotides, the HJ oligos robustly self-assemble into uniform and stable constructs, even when functionalized with large hydrophilic or charged macromolecules on each of the four arms. This simple covalent coupling approach, in combination with the high thermostability of the duplexes, thus allows us to create structures that are exceptionally stable with a low risk of dissociation under physiological conditions. This represents a significant improvement compared to other nucleic acid-based platforms with a potential use for drug delivery, such as doxorubicin-loaded DNA origami structures [46] that risk dissociation and drug leakage at the physiological salt concentrations and low concentrations in vivo [47]. Due to its small size and highly chemically modified nature, the HJ presented here also separates itself from similar proposed structures, such as a significantly larger DNA HJ consisting of four 50 nt long DNA strands, all modified with fluorophores for high-signal protein labeling [48].
In mice, the small non-functionalized HJ scaffold was shown to be excreted through the kidneys with a half-life of only minutes. This effect can be advantageous when rapid clearance is needed in connection with bioimaging of for instance tumors. However, for drug delivery purposes, a sufficiently long circulatory half-life is vital to ensure maximum exposure of the payload to its target. To accommodate both potential applications, we investigated the possibility of adjusting this circulation time by conjugating various PEG or palmitoyl groups. PEG is widely used in drug delivery and nanotechnology where its main purpose is to increase the hydrodynamic size of small compounds in order to avoid glomerular filtration, or for nanoparticles as a ”stealth” agent to avoid recognition by the immune system [49]. By attaching one or more PEG modules, the circulation time of the HJ was significantly increased, and the HJ remained detectable in the blood after 24 h. However, in spite of its wide use and popularity, there has in recent years been an increased focus on the disadvantages of the use of PEG [50,51]. One of the primary concerns is the presence of antibodies that specifically recognize and bind to PEG, including both pre-existing and treatment-induced antibodies [50,52]. In addition, while PEG prolongs blood circulation and presumably increases bioavailability at the target tissue, heavy PEGylation can lead to shielding of the targeting ligands on the structure. Using PEG for PK enhancement is in this way a balancing act between achieving optimal circulation time and effective target recognition. We therefore also investigated an alternative strategy by attaching biodegradable fatty acids to the HJ. These are known to associate non-covalently with fatty acid binding sites on circulating HSA and take advantage of its extraordinarily long half-life [53]. We synthesized a HJ module carrying two palmitoyl groups in its 5' end. The palmitoyl module was shown to facilitate efficient binding to HSA in vitro and afforded significant PK enhancement by extending the half-life of the HJ from approximately 20 min to more than two hours.
In this way, both the half-life and biodistribution of the HJ in circulation can be tightly controlled by conjugation of PEG or palmitoyl groups, thereby providing a method to fine-tune its PK properties for diagnostic and therapeutic applications. It is likely that such PK optimization could translate into higher drug efficacy and consequently reduce the required drug dose. This could thereby allow for the delivery of cytotoxic drugs with low half-lives and narrow therapeutic windows with a reduced risk of adverse effects. In addition, the modular nature of the HJ provides a unique opportunity to explore different combinations of PK enhancers without the need for extensive resynthesis to identify the best suited configurations for a particular clinical application.
The ability to mediate active and specific tissue targeting is an important property for any drug delivery platform intended for personalized and theranostic medicine. To investigate the potential of the HJ for cell-specific uptake, we functionalized the scaffold with 1-3 triGalNAcs with precise stoichiometry. This led to pronounced hepatocyte-specific uptake of the HJ in HepG2 cells and also fast liver accumulation in vivo. The increase in valency of the triGalNAc targeting ligands also led to an increasing hepatocyte internalization both in vitro and in vivo, demonstrating that although the trivalent GalNAc moiety is an effective targeting ligand by itself, its targeting potential can be further enhanced by a multivalent display of two or more triGalNAcs. This approach could thereby add further potency to sugar-based delivery approaches including the triGalNAc-conjugated siRNA systems developed by Alnylam [54] and the triGalNAc-conjugated antimiR system from Regulus [55]. Furthermore, this indicates a potential of the HJ to improve the effect of receptor-mediated targeting with low-affinity systems. In clinical development of specific targeting ligands, such as peptides derived from phage display, many candidates are discarded from further development due to insufficient affinity for receptors under in vivo conditions. A controlled multivalent display of such candidates could enhance the utility of these ligands and produce effective targeted carriers for imaging modalities or therapeutic drugs.
We have shown that it was possible to assemble the HJ structure with stoichiometric ratios of up to four functionalized modules in >90% pure form. For many applications, this precludes the need for post-assembly purification. This feature of the HJ therefore represents a significant improvement compared to for example nanoparticles or DNA origami where the functionalized substructures do not associate in a 1:1 ratio and must be purified post assembly to avoid off-target effects. Additionally, this unique property opens up the possibility for rapid on-site combination of imaging agents or drugs targeted to selected disease conditions, based on larger libraries of differently functionalized oligonucleotide modules. In this way, the modular nature of the system could enable the creation of personalized medicine for specific diseases in individual patients by combining relevant functional entities in a LEGO® block-like fashion.
The current report focuses on the structural characterization of the HJ scaffold and on establishing the most vital properties required for a safe, stable and versatile delivery platform for imaging agents and drugs. The system was shown to be useful for molecular targeting with an adjustable half-life and could specifically deliver fluorophores for optical imaging to the target tissues. For clinical translation of this, attachment of radioactive compounds for positron emission tomography (PET) imaging or magnetic contrast for magnetic resonance imaging (MRI) is desired, and this work is therefore currently on-going. With the incorporation of such clinically relevant imaging agents, the HJ could provide a flexible platform for highly detailed multi-modal diagnostics of cancer. Furthermore, with the planned production of modules containing different classes of bioactive cargos, including cytotoxic drugs and nucleic acid-based therapeutics, more complex multifunctional HJs could be assembled for applications in cancer, viral- inflammatory- and metabolic diseases. As a consequence of the highly modular nature of the HJ, this system could be envisioned to either function as a combinatorial screening platform in pharmaceutical industries or as a theranostic platform in the clinic, in which modules could be functionalized with PK modifiers, targeting ligands, PET tracers, and/or cytotoxic drugs for a fast and efficient combined therapy and diagnosis of cancer.
Supplementary Material
Supplementary methods and figures.
Acknowledgements
The work was funded, in part, by the Villum Foundation to the Center for Biomolecular Nanoscale Engineering (BIONEC), in part, by the Danish National Research Foundation to the Center for Cellular Signal Patterns (CellPat) and, in part, by the Novo Nordisk Foundation to the Center for Multifunction Biomolecular Drug Design (CEMBID). A. R. was supported by the German Research Foundation (DFG) for a postdoctoral scholarship.
Author contributions
J.K, V.L.A and J.S.N. designed and supervised the project and edited the manuscript. J.S.N. drafted the manuscript. V.L.A. and J.S.N. conducted most of the experimental work. M.V. helped with the mice studies. J.W. supervised part of the oligonucleotide synthesis work and contributed to the molecular designs. R.K. and A.R. contributed with synthesis of oligonucleotides and sugar moeities.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615-627
2. Chabner BA, Roberts Jr TG. Chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5:65
3. Bozic I, Nowak MA. Resisting Resistance. Annu Rev Cancer Biol. 2017;1:203-221
4. de Bono JS, Ashworth A. Translating cancer research into targeted therapeutics. Nature. 2010;467:543-549
5. Diamantis N, Banerji U. Antibody-drug conjugates—an emerging class of cancer treatment. Br J Cancer. 2016;114:362
6. Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16:315-337
7. Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30:679-692
8. Lim E-K, Kim T, Paik S. et al. Nanomaterials for theranostics: recent advances and future challenges. Chem Rev. 2014;115:327-394
9. Kemp JA, Shim MS, Heo CY. “Combo” nanomedicine: Co-delivery of multi-modal therapeutics for efficient, targeted, and safe cancer therapy. Adv Drug Deliv Rev. 2016;98:3-18
10. Janib SM, Moses AS. Imaging and drug delivery using theranostic nanoparticles. Adv Drug Deliv Rev. 2010;62:1052-1063
11. Naahidi S, Jafari M, Edalat F. et al. Biocompatibility of engineered nanoparticles for drug delivery. J Control release. 2013;166:182-194
12. Sun T, Zhang YS, Pang B. et al. Engineered nanoparticles for drug delivery in cancer therapy. Angew Chemie Int Ed. 2014;53:12320-12364
13. Srinivasarao M, Galliford C V, Low PS. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat Rev Drug Discov. 2015;14:203
14. Ji C, Gao Q, Dong X. et al. A Size-Reducible Nanodrug with an Aggregation-Enhanced Photodynamic Effect for Deep Chemo-Photodynamic Therapy. Angew Chemie Int Ed. 2018;57:11384-11388
15. Douglas SM, Bachelet I, Church GM. A logic-gated nanorobot for targeted transport of molecular payloads. Science (80- ). 2012;335:831-834
16. Jiang D, England CG, Cai W. DNA nanomaterials for preclinical imaging and drug delivery. J Control Release. 2016;239:27-38
17. Kumar V, Palazzolo S, Bayda S. et al. DNA nanotechnology for cancer therapy. Theranostics. 2016;6:710
18. Okholm AH, Kjems J. DNA nanovehicles and the biological barriers. Adv Drug Deliv Rev. 2016;106:183-191
19. Halley PD, Lucas CR, McWilliams EM. et al. Daunorubicin-Loaded DNA Origami Nanostructures Circumvent Drug-Resistance Mechanisms in a Leukemia Model. Small. 2016;12:308-20
20. Li S, Jiang Q, Liu S. et al. A DNA nanorobot functions as a cancer therapeutic in response to a molecular trigger in vivo. Nat Biotechnol. 2018;36:258-264
21. Li H, Zhang K, Pi F. et al. Controllable Self-Assembly of RNA Tetrahedrons with Precise Shape and Size for Cancer Targeting. Adv Mater. 2016;28:7501-7507
22. Röthlisberger P, Hollenstein M. Aptamer chemistry. Adv Drug Deliv Rev. 2018;134:3-21
23. Bramsen JB, Kjems J. Development of therapeutic-grade small interfering RNAs by chemical engineering. Front Genet. 2012;3:154
24. Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46-51
25. Wengel J. Synthesis of 3 '-C-and 4 '-C-branched oligodeoxynucleotides and the development of locked nucleic acid (LNA). Acc Chem Res. 1999;32:301-310
26. Vester B, Wengel J. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry. 2004;43:13233-13241
27. Wahlestedt C, Salmi P, Good L. et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc Natl Acad Sci. 2000;97:5633-5638
28. Bramsen JB, Laursen MB, Nielsen AF. et al. A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Res. 2009;37:2867-2881
29. Fontenete S, Guimarães N, Leite M. et al. Hybridization-Based Detection of Helicobacter pylori at Human Body Temperature Using Advanced Locked Nucleic Acid (LNA) Probes. PLoS One. 2013;8:e81230
30. Kvaernø L, Kumar R, Dahl BM, Olsen CE, Wengel J. Synthesis of abasic locked nucleic acid and two seco-LNA derivatives and evaluation of their hybridization properties compared with their more flexible DNA counterparts. J Org Chem. 2000;65:5167-76
31. Johannsen MW, Crispino L, Wamberg MC, Kalra N, Wengel J. Amino acids attached to 2′-amino-LNA: synthesis and excellent duplex stability. Org Biomol Chem. 2011;9:243-252
32. Ho PS. Structure of the Holliday junction: applications beyond recombination. Biochem Soc Trans. 2017;45:1149-1158
33. Petersen M, Wengel J. LNA: a versatile tool for therapeutics and genomics. Trends Biotechnol. 2003;21:74-81
34. Lilley DMJ. Structures of helical junctions in nucleic acids. Q Rev Biophys. 2000;33:109-159
35. Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99:28-51
36. Havelund S, Plum A, Ribel U. et al. The mechanism of protraction of insulin detemir, a long-acting, acylated analog of human insulin. Pharm Res. 2004;21:1498-504
37. Jacobsen L V, Flint A, Olsen AK, Ingwersen SH. Liraglutide in Type 2 Diabetes Mellitus: Clinical Pharmacokinetics and Pharmacodynamics. Clin Pharmacokinet. 2016;55:657-72
38. Chaudhury C, Mehnaz S, Robinson JM. et al. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J Exp Med. 2003;197:315-22
39. Hvam ML, Cai Y, Dagnæs-Hansen F. et al. Fatty Acid-Modified Gapmer Antisense Oligonucleotide and Serum Albumin Constructs for Pharmacokinetic Modulation. Mol Ther. 2017;25:1710-1717
40. Larsen MT, Kuhlmann M, Hvam ML, Howard KA. Albumin-based drug delivery: harnessing nature to cure disease. Mol Cell Ther. 2016;4:3
41. Properties P, Specificity A, Baenziger JU, Maynard Y. Human Hepatic Lectin. J Biol Chem. 1980;255:4607-4613
42. Meier M, Bider MD, Malashkevich VN, Spiess M, Burkhard P. Crystal Structure of the Carbohydrate Recognition Domain of the H1 Subunit of the Asialoglycoprotein Receptor. J Mol Biol. 2000;300:857-865
43. Rensen PCN, van Leeuwen SH, Sliedregt LAJM, van Berkel TJC, Biessen EAL. Design and synthesis of novel N-acetylgalactosamine-terminated glycolipids for targeting of lipoproteins to the hepatic asialoglycoprotein receptor. J Med Chem. 2004;47:5798-5808
44. Prakash TP, Graham MJ, Yu J. et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014;42:8796-8807
45. Duckett DR, Murchie AI, Lilley DM. The role of metal ions in the conformation of the four-way DNA junction. EMBO J. 1990;9:583-90
46. Zhang Q, Jiang Q, Li N. et al. DNA origami as an in vivo drug delivery vehicle for cancer therapy. ACS Nano. 2014;8:6633-6643
47. Hahn J, Wickham SFJ, Shih WM, Perrault SD. Addressing the instability of DNA nanostructures in tissue culture. ACS Nano. 2014;8:8765-8775
48. Li Z, Theile CS, Chen G. et al. Fluorophore-Conjugated Holliday Junctions for Generating Super-Bright Antibodies and Antibody Fragments. Angew Chemie Int Ed. 2015;54:11706-11710
49. Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew Chemie Int Ed. 2010;49:6288-6308
50. Ganson NJ, Povsic TJ, Sullenger BA. et al. Pre-existing anti-polyethylene glycol antibody linked to first-exposure allergic reactions to pegnivacogin, a PEGylated RNA aptamer. J Allergy Clin Immunol. 2016;137:1610-1613
51. Duncan R. Polymer therapeutics at a crossroads? Finding the path for improved translation in the twenty-first century. J Drug Target. 2017;25:759-780
52. Hershfield MS, Ganson NJ, Kelly SJ. et al. Induced and pre-existing anti-polyethylene glycol antibody in a trial of every 3-week dosing of pegloticase for refractory gout, including in organ transplant recipients. Arthritis Res Ther. 2014;16:R63
53. Howard KA. Albumin: the next-generation delivery technology. Ther Deliv. 2015;6:265-268
54. Nair JK, Willoughby JLS, Chan A. et al. Multivalent N -Acetylgalactosamine-Conjugated siRNA Localizes in Hepatocytes and Elicits Robust RNAi-Mediated Gene Silencing. J Am Chem Soc. 2014;136:16958-16961
55. van der Ree MH, de Vree JM, Stelma F. et al. Safety, tolerability, and antiviral effect of RG-101 in patients with chronic hepatitis C: a phase 1B, double-blind, randomised controlled trial. Lancet. 2017;389:709-717
Author contact
![]() Corresponding author: jkau.dk; * Phone: +45 4528992086
Corresponding author: jkau.dk; * Phone: +45 4528992086