Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(12):3595-3607. doi:10.7150/thno.33100 This issue Cite
Research Paper
Endocytosis-mediated mitochondrial transplantation: Transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity
Chao Sun1,2,3,4*, Xiongxiong Liu1,2,3,4*, Bing Wang6, Zhenhua Wang5, Yang Liu1,2,3,4, Cuixia Di1,2,3,4, Jing Si1,2,3,4, Hongyan Li1,2,3,4, Qingfeng Wu1, Dan Xu1, Ji Li5, Gang Li5, Yupei Wang1,2,3,4, Fang Wang1,2,3,4, Hong Zhang1,2,3,4 ![]()
1. Institute of Modern Physics; Chinese Academy of Sciences; Lanzhou, China
2. Key Laboratory of Heavy Ion Radiation Medicine of Chinese Academy of Sciences; Lanzhou, China
3. Key Laboratory of Heavy Ion Radiation Medicine of Gansu Province; Lanzhou, China
4. University of Chinese Academy of Sciences; Beijing, China
5. Center of Mitochondria and Healthy Ageing; Yantai University; Yantai, China
6. National Institute of Radiological Sciences; National Institutes for Quantum and Radiological Science and Technology; Chiba, Japan.
*These authors contributed equally to this work.
Received 2019-1-13; Accepted 2019-5-6; Published 2019-5-26
Abstract

Emerging evidence indicates that reprogramming of energy metabolism involving disturbances in energy production from a defect in cellular respiration with a shift to glycolysis is a core hallmark of cancer. Alterations in cancer cell energy metabolism are linked to abnormalities in mitochondrial function. Mitochondrial dysfunction of cancer cells includes increased glycolysis, decreased apoptosis, and resistance to radiotherapy. The study was designed for two main points: firstly, to investigate whether exogenous functional mitochondria can transfer into glioma cells and explore the underlying molecular mechanisms from the perspective of endocytosis; secondly, to further verify whether the mitochondrial transplantation is able to rescue aerobic respiration, attenuate the Warburg effect and enhance the radiosensitivity of gliomas.
Methods: Mitochondria were isolated from normal human astrocytes (HA) and immediately co-incubated with starved human glioma cells (U87). Confocal microscopy and gene sequencing were performed to evaluate the ability of isolated mitochondria internalization into U87 cells. The interaction between endocytosis and isolated mitochondria transfer were captured by 3D tomographic microscopy and transmission electron microscopy. NAD+, CD38, cADPR and Ca2+ release were determined by commercial kits, western blot, HLPC-MS and Fluo-3 AM respectively. PCR array expression profiling and Seahorse XF analysis were used to evaluate the effect of mitochondrial transplantation on energy phenotypes of U87 cells. U87 cells and U87 xenografts were both treated with mitochondrial transplantation, radiation, or a combination of mitochondrial transplantation and radiation. Apoptosis in vitro and in vivo were detected by cytochrome C, cleaved caspase 9 and TUNEL staining.
Results: We found that mitochondria from HA could be transferred into starved U87 cells by simple co-incubation. Starvation treatment slowed the rate of glycolysis and decreased the transformation of NAD+ to NADH in U87 cells. A large amount of accumulated NAD+ was released into the extracellular space. CD38 is a member of the NAD+ glycohydrolase family that catalyzes the cyclization of extracellular NAD+ to intracellular cADPR. cADPR triggered release of Ca2+ to promote cytoskeleton remodeling and plasma membrane invagination. Thus, endocytosis involving isolated mitochondria internalization was mediated by NAD+-CD38-cADPR-Ca2+ signaling. Mitochondrial transfer enhanced gene and protein expression related to the tricarboxylic acid (TCA) cycle, increased aerobic respiration, attenuated glycolysis, reactivated the mitochondrial apoptotic pathway, inhibited malignant proliferation of U87 cells. Isolated mitochondria injected into U87 xenograft tumors also entered cells, and inhibited glioma growth in nude mice. Mitochondrial transplantation could enhance the radiosensitivity of gliomas in vitro and in vivo.
Conclusion: These findings suggested that starvation-induced endocytosis via NAD+-CD38-cADPR-Ca2+ signaling could be a new mechanism of mitochondrial transplantation to rescue aerobic respiration and attenuate the Warburg effect. This mechanism could be a promising approach for radiosensitization.
Keywords: mitochondrial transplantation, NAD+-CD38-cADPR-Ca2+ signaling, endocytosis, energy metabolism, radiation therapy, gliomas
Introduction
Mitochondria are powerful and dynamic organelles responsible for essential cell functions, including energy metabolism, maintenance of calcium homeostasis, cell survival and apoptosis. Mitochondrial dysfunction is involved in serious health problems such as neurodegenerative diseases [1], metabolic disorders [2] and cancer [3]. An electron micrographic study showed that mitochondria ultrastructure was abnormal to some degree in 778 patients with breast cancer. Mitochondria were severely reduced in number or were undetectable in tumor tissue from over 80% of patients. The degree of malignancy in breast tumors correlated directly with the degree of mitochondrial structural abnormality [4]. Otto Warburg hypothesized in the 1920s that altered tumor cell metabolism is caused by a defect in cellular respiration with a shift to glycolysis and this is the initiating step in tumorigenesis. Warburg observed in cancer cells a marked increase in glycolysis and lactate production in the presence of oxygen without an increase in oxidative phosphorylation. This phenomenon of aerobic glycolysis is known as the Warburg effect [5]. In 2011, a report indicated that reprogramming of energy metabolism is a core hallmark capability of cancer cells [6]. Mitochondrial defects may be the main reason for the reprogramming of cancer cell energy metabolism. Therefore, malignant tumors may be a type of energy metabolic disease involving respiratory insufficiency caused by mitochondrial dysfunction [3, 7, 8].
Delivery of exogenous functional mitochondria into damaged cells has been proposed as a mechanism for cell transplant and physiological repair of damaged tissue [9]. Several studies found that the intercellular transfer of mitochondrial organelles might contribute to rescuing of energy metabolism [10, 11]. Jeffrey et al. found that mitochondrial DNA (mtDNA)-depleted human lung carcinoma A549 cells (ρ0 cells) had enhanced membrane potentials, increased oxygen consumption, reduced extracellular lactate production and recovered aerobic respiration after intercellular mitochondrial transfer from intact human mesenchymal stem cells [12]. A study reported that isolated, functional mitochondria selectively entered human breast cancer MCF-7 and MDA-MB-231 cells, but not normal breast epithelia MCF-12a cells. Mitochondrial organelle transplantation inhibited aerobic glycolysis and increased the sensitivity of human breast cancer cells to multiple antitumor drugs (doxorubicin, abraxane, and carboplatin) [13]. In addition to chemotherapy, radiotherapy is one of the most important treatment modalities for cancer. Mitochondrial defects increased the radiation resistance of human liver cancer HepG2 cells and the effect was potentially involved in alleviating reactive oxygen species -mediated oxidative damage, blocking the mitochondrial apoptosis pathway [14]. These findings led us to hypothesize that the radiation cytotoxicity of cancer cells is dependent on mitochondria and mitochondrial organelle transplantation might be an effective mode for radiosensitization. To our knowledge, isolated mitochondrial internalization enhancing the radiosensitivity of cancer cells has not been previously demonstrated or reported.
Tunneling membrane nanotubes, macropinocytosis and endocytosis have been suggested as the most likely pathways for mitochondrial transfer between cells [15-17]. However, the physiological nature and specific mechanism of mitochondrial transfer remains uncertain. CD38 is an ADP-ribosyl cyclase that catalyzes the conversion of extracellular oxidized nicotinamide adenine dinucleotide (NAD+) to intracellular cyclic ADP-ribose (cADPR) [18, 19]. FK-506 binding proteins 12.6 (FKBP12.6) can bind to ryanodine receptors (RyRs) of the endoplasmic reticulum (ER), the activity of RyRs (intracellular Ca2+ release channels) is inhibited [20]. cADPR induces the dissociation of FKBP12.6 from RyRs, and results in Ca2+ release from intracellular calcium store [21, 22]. High levels of cytosolic Ca2+ binding to troponin C trigger a conformational change in filamentous actin (F-actin), a cytoskeletal protein [23]. Cytoskeleton remodeling promotes invaginations of the plasma membrane and causes endocytosis. This study focused on the radiosensitization effects induced by mitochondrial transplantation in gliomas, especially the mechanism of free mitochondrial transfer into host cells via an NAD+-CD38-cADPR-Ca2+-endocytosis pathway.
Results
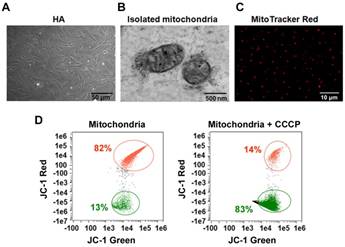
Isolation of intact mitochondria from human astrocytes. Mitochondria were isolated from HA cells (Figure 1A). Transmission electron microscopy (TEM) showed that this isolated fraction contained morphologically intact mitochondria (Figure 1B). MitoTracker Red CMXRos, a red fluorescent dye, was used to stain the mitochondria of living cells, with dye accumulation dependent on membrane potential. MitoTracker labelling suggested that the extracellular mitochondria were functional (Figure 1C). To further determine the integrity and function of isolated mitochondria, we measured the mitochondrial membrane potential (ΔΨm) by flow cytometry. As shown in Figure 1D, isolated mitochondria stained with JC-1 had high red fluorescence signal. Fluorescence decreased with pretreatment with carbonyl cyanide 3-chlorophenylhydrazone (CCCP), a mitochondrial membrane potential disrupter.
The integrity and function of isolated mitochondria. (A) Morphology of cultured HA cells. Scale bar: 50 μm (B) TEM of isolated mitochondria. Scale bar: 500 nm. (C) Isolated mitochondria fluorescence-labeled with MitoTracker Red CMXRos. Scale bar: 10 μm. (D) Flow cytometry analysis of the mitochondrial membrane potential (ΔΨm) of the isolated mitochondria stained with JC-1.
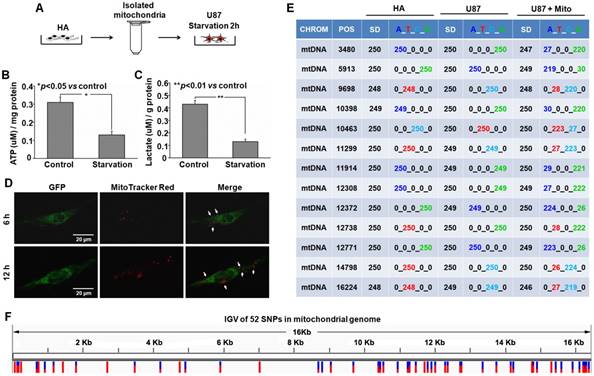
Transfer of isolated mitochondria into the starved U87 cells. Serum-glucose deprivation of U87 cells for 2 h reduced the production of ATP and lactate (Figure 2B-C) and induced cell starvation. About 3×106 intact mitochondria (Figure S1) from HA cells were stained with MitoTracker Red CMXRos and immediately co-incubated with starved U87 cells (Figure 2A). Live fluorescence imaging suggested that MitoTracker Red CMXRos-labelled mitochondria were present within starved U87 cells stably expressing green fluorescent protein (GFP) (Figure 2D). To further investigate the hypothesis that isolated mitochondria can be internalized into U87 cells, mitochondrial genotypes of HA, U87 and U87+Mito (U87 cells co-incubated with isolated mitochondria for 12h) cells were assayed by direct sequencing. 52 single nucleotide polymorphisms (SNPs) that were sites with nucleotides derived from both HA donors and parental U87 cells were detected in the mitochondrial genome of U87+Mito cells. 13 SNPs were randomly selected for detailed presentation in Figure 2E, the distribution of all 52 SNPs in the mitochondrial genome of U87+Mito cells was shown in Figure 2F. This result showed that mtDNA derived from HA cells were present in U87+Mito cells. These results indicated that exogenous mitochondria could be transferred into starved recipient cells by co-incubation. Furthermore, we also found that free mitochondria could not enter U87 cells without starvation treatment (Figure S2).
Transplantation of isolated mitochondria into the starved U87 cells. (A) Experimental schematic for co-incubation studies. After treatment with serum-glucose deprivation, the intracellular ATP (B) and lactate (C) were detected using commercial kits. (D) Live images of GFP-expressing U87 cells containing MitoTracker Red CMXRos-labelled mitochondria. White arrow: transferred mitochondria. Scale bar: 20 μm. (E) Mitochondrial genotypes of HA, U87 and U87+Mito cells were examined by direct sequencing. CHROM: chromosome; POS: position; SD: sequencing depth. (F) Integrative genomics viewer (IGV) of 52 SNPs in the mitochondrial genome of U87+Mito cells.
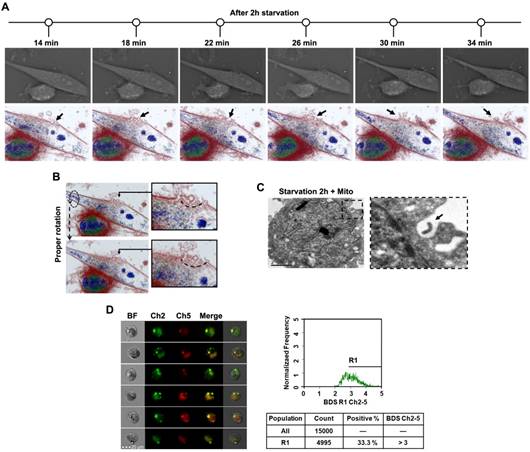
Transfer of mitochondria into U87 cells through endocytosis induced by starvation. The dynamic behavior of endocytosis during starvation treatment was captured by 3D tomographic microscope colored by refractive index, not staining. After 2 h starvation, extracellular substances gradually entered U87 cells by invagination of the plasma membrane (Figure 3A, Video 1). When cells were rotated moderately, endocytosis structures on the cell membrane could be observed more clearly (Figure 3B, Video 2). TEM revealed that invagination of the plasma membrane encapsulated isolated mitochondria (Figure 3C). The fluorescence probe pHrodo, developed for endocytosis, showed that 2 h starvation resulted in colocalization of isolated mitochondria by MitoTracker Red labeling with endosomes by pHrodo (Figure 3D). Cells with a high degree of colocalization were gated with R1 (BDS Ch2-3 > 3). The positive rate of colocalization was 53.3%. These results indicated that isolated mitochondria from HA cells were transferred into starved U87 cells through endocytosis.
Transplantation of isolated mitochondria into U87 cell through endocytosis. (A) After 2h starvation, the whole process of endocytosis was captured with a time lapse 3D tomographic microscope. Cell morphology, colored by the individual refractive indexes of each component, is clearly visible. Black arrow: plasma membrane invagination. (B) Endocytosis was observed after proper rotation of the stereoscopic structure of U87 cells. Arc line: endocytosis structure. (C) TEM of mitochondrial interaction with endocytosis. Black dashed box: endocytic region; black arrow: exogenous mitochondria. Scale bar: 2 μm. (D) The multispectral imaging flow cytometry was used to measure the intracellular colocalization of isolated mitochondria (MitoTracker Red labeling) with the endosomes (stained with pHrodo). Ch2: pHrodo; Ch5: MitoTracker Red; BF: brightfield; BDS Ch2-5: bright detail similarity Ch2-Ch5.
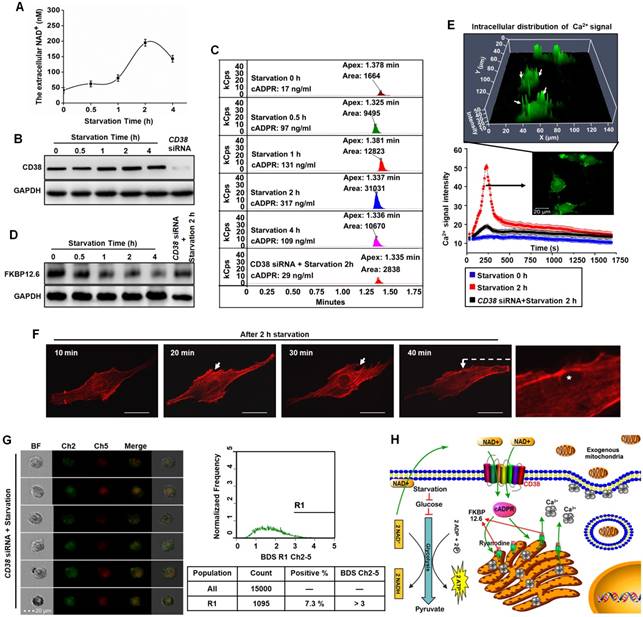
Starvation promoted endocytosis via NAD+-CD38-cADPR-Ca2+ signaling. Human glioma cells exhibit features of Warburg metabolism including low mitochondrial respiration, increased glucose uptake and high glycolysis [24], which may be associated with abnormalities in mitochondrial morphology or function. Starvation treatment with glucose deprivation effectively slowed glycolysis and decreased the transformation of NAD+ to NADH. A large amount of accumulated NAD+ was released into the extracellular space. At 2 h after starvation, NAD+ levels reached their highest levels (Figure 4A). CD38 is a member of NAD+ glycohydrolase family that catalyzes the cyclization of extracellular NAD+ to intracellular cADPR. CD38 protein was measured by western blots and the intracellular contents of cADPR by HLPC-MS. Expression of CD38 increased to a maximum value at 2 h after starvation treatment and cADPR also increased approximately 18.6-fold compared with the control (Figure 4B-C). cADPR is a second messenger in the activation of RyRs by dissociating of FKBP12.6 from ER and mobilization of intracellular Ca2+ [21, 22]. Western blot analysis was performed on lysates of ER from U87 cells, as shown in Figure 4D, the binding of FKBP12.6 in ER was reduced after starvation treatment. Flou-3 fluorescence data indicated that 2 h starvation induced rapid, extensive release of Ca2+ in U87 cells (Figure 4E, Video 3). Released Ca2+ was mainly distributed near the plasma membrane (Figure 4E) to promote endocytosis by F-actin polymerization and cytoskeleton remodeling (Figure 4F). Moreover, as shown in Figure 4B-E, the knockdown of CD38 with siRNA assay followed by 2h starvation treatment would block the NAD+-CD38-cADPR-Ca2+ signaling pathway, simultaneously, the isolated mitochondria were not colocalized with endosomes (Figure 4G) and the positive rate of colocalized cells decreased from 53.3% (Figure 3D) to 7.3% (Figure 4G). These results demonstrated that NAD+-CD38-cADPR-Ca2+ signaling-mediated endocytosis contributed to the exogenous mitochondria transplantation (Figure 4H).
Endocytosis induced by starvation via NAD+-CD38-cADPR-Ca2+ signaling in U87 cells. (A) After starvation treatment for different time, the extracellular NAD+ was detected using commercial kits. (B) The expression of CD38 was measured by western blot. (C) The concentration of cADPR was measured in U87 cells by HLPC-MS. (D) The FKBP12.6 in ER protein extract was measured by western blot. (E) U87 cells were loaded with Fluo-3 AM, the cytosolic Ca2+ release in two spatial dimensions plus time (XYT) were acquired using confocal microscope. White arrow: the enrichment of Ca2+ to cell membrane. (F) The image of cytoskeleton remodeling in mCherry-actin expressing U87 cells was taken by confocal microscope. White arrow: actin-dependent endocytosis. White asterisk: endocytic vesicle. Scale bar: 20 μm. (G) Effects of CD38 siRNA pretreatment followed by starvation on colocalization of isolated mitochondria with the endosomes in U87 cells. Ch2: pHrodo; Ch5: MitoTracker Red; BF: brightfield; BDS Ch2-5: bright detail similarity Ch2-Ch5. (H) Schematic diagram of endocytosis-mediated mitochondrial transplantation via NAD+-CD38-cADPR-Ca2+ signaling.
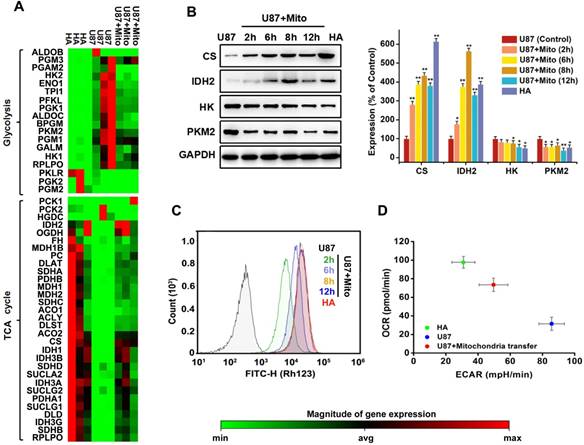
Mitochondrial transplantation changed energy metabolism in cancer cells. Metabolic reprogramming of cells contributes to oncogenesis [6, 25]. A heat map showed that most genes associated with glycolysis were upregulated in U87 cells. Genes associated with the TCA cycle were generally downregulated (Figure 5A). After completing screening, 2 genes (IDH2 and PKLR) with large errors on the heat map were validated by PCR (Figure S3). Basal oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were tested using Seahorse analyser. Comparing overall energy metabolic phenotypes (OCR vs. ECAR) for U87 cells and HA cells showed that U87 cells had relatively lower OCR and higher ECAR (Figure 5D). This result was consistent with the concept of Warburg effect in tumor cells, with increased ECAR indicating higher glycolytic flux. We found that transfer of exogenous functional mitochondria mitochondria into U87 cells for 12 h generally downregulated genes associated with glycolysis (Figure 5A); increased the expression of citrate synthase (CS) and isocitrate dehydrogenase 2 (IDH2), two critical rate-limiting enzymes in TCA cycle (Figure 5B); decreased expression of hexokinase (HK) and pyruvate kinas 2 (PKM2), two critical rate-limiting enzymes in glycolysis (Figure 5B); and restored mitochondrial membrane potentials (Figure 5C). The OCR/ECAR ratio also increased (Figure 5D). The major energy metabolism pathway of U87 cells changed from aerobic glycolysis to oxidative phosphorylation. These results suggested that mitochondrial transplantation rescued aerobic respiration and attenuated the Warburg effect in cancer cells.
Effects of mitochondrial transplantation on energy metabolic phenotype. (A) The expression of 49 key genes involved in glycolysis and TCA cycle was assessed by Human Signal Transduction Pathway Finder PCR Array. Red denotes high expression levels, whereas green denotes low expression levels. (B) Expression levels of critical rate-limiting enzymes in TCA cycle and glycolysis were measured by western blot. AlphaView SA software was used for quantification of western blot. Data were presented as relative protein level normalized to GAPDH, and the ratio of control samples was taken as 100%. *p<0.05, **p<0.01 vs. control group. (C) Effects of mitochondrial transplantation on membrane potential in U87 cells measured by Rhodamine 123. (D) Energy phenotypes of U87, HA and U87+Mito cells.
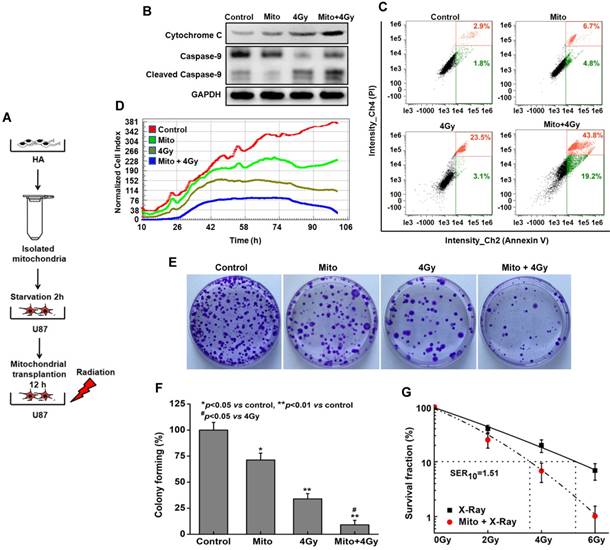
Mitochondrial transplantation enhanced radiosensitivity in vitro. U87 cells were treated with isolated mitochondria or irradiation alone, or pretreated with isolated mitochondria for 12 h followed by irradiation exposure (Figure 6A). Compared with irradiation alone, mitochondrial transplantation combined with X-ray irradiation promoted the release of cytochrome C, increased cleaved-caspase 9, and efficiently activated the mitochondrial apoptotic pathway (Figure 6B). To further investigate the radiosensitization effect of mitochondrial transplantation, apoptosis of U87 cells was examined by annexin V-FITC/PI staining. At 48 h post-irradiation, the apoptotic rate increased to 63.0% (early apoptosis, 43.8 ± 1.4%; late apoptosis, 19.2 ± 1.6%) in the combination group, in contrast to approximately 26.6% (early apoptosis, 23.5 ± 1.3%; late apoptosis, 3.1 ± 1.5%) in the irradiation-alone group (Figure 6C). Besides, mitochondrial transplantation enhanced the inhibition effect of X-ray irradiation on the proliferation of U87 cells (Figure 6D). Colonies in Figure 6E colony-forming ratios are in Figure 6F. For example, when cells were pretreated with mitochondrial transplantation plus 4 Gy X-ray irradiation, the colony forming ratio of U87 cells decreased by 24.8 ± 1.5% compared to cells treated with 4 Gy X-ray irradiation alone. Using mitochondrial transplantation, the sensitizer enhancement ratio at 10% survival level (SER10) was 1.51 for U87 cells (Figure 6G). To further demonstrate the radiobiological effects of mitochondrial transplantation, comet assays, γ-H2AX foci and colony formation assays were performed with ρ0 U87 cells. As shown in Figure S4, mitochondrial transplantation also effectively improved the radiosensitivity of ρ0 cells.
Radiosensitization by mitochondrial transplantation in vitro. (A) Experimental schematic to test radiosensitizing effect of mitochondrial transplantation in U87 cells. (B) Expression levels of key proteins in mitochondrial apoptotic pathway were measured by western blot. (C) Apoptosis was quantified by combined staining of annexin V and PI, and fluorescence was analyzed using flow cytometry. (D) Cell index values were determined every 15 min using a real-time cell electronic sensing (RT-CES) system for up to 106 h. (E) Effects of mitochondrial transplantation followed by X-ray irradiation on clonogenic potential of U87 cells. (F) Statistical analysis of colony formation assay. *p<0.05, **p<0.01 vs. control group; #p<0.05 vs. 4 Gy irradiation alone. (G) Statistical analysis of the sensitizer enhancement ratios at 10% survival level (SER10).
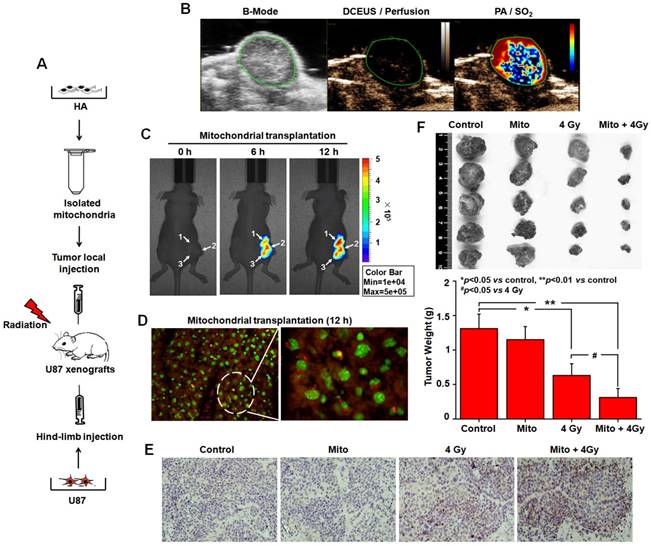
In vivo mitochondrial transplantation and radiosensitization. We also explored the radiosensitization of mitochondrial transplantation in a xenograft tumor model (Figure 7A). Comparison of dynamic contrast-enhanced ultrasound (DCEUS) parametric maps and processed photoacoustic (PA) scans showed the spatial relationship between blood perfusion and oxygen saturation (SO2) in tumors. While the rims of U87 tumors were perfused (golden), their cores contained large, poorly perfused and nonperfused regions (dark) (Figure 7B). Large, resolvable vessels were not detected in any perfusion maps. Areas of high SO2 are bright red, with dark red for low SO2 regions. Black represents no SO2. High SO2 levels were measured throughout tumor rims, with low SO2 in the core (Figure 7B). These data showed large starved regions suitable for mitochondrial transplantation within solid tumors. After 6 h and 12 h injections of MitoTracker Red CMXRos-labelled mitochondria, mitochondrial fluorescence was diffused in U87 tumors (Figure 7C). Fluorescence microscope studies of tissue sections confirmed that functional mitochondria injected into solid tumors entered cells (Figure 7D). To further investigate the radiosensitization effect of mitochondrial transplantation in vivo, in situ cell apoptosis was detected by TUNEL staining. A large number of TUNEL-positive nuclei (brown) were observed. The apoptotic rate was elevated in the group with mitochondrial transplantation combined with X-ray irradiation (Figure 7E). Tumor weight was 1.31 ± 0.21 g in the control group, 1.15 ± 0.19 g in the mitochondrial transplantation alone group, 0.63 ± 0.17 g in the 4 Gy irradiation-alone groups, and 0.31 ± 0.13 g in the combination group (Figure 7F). Compared with the irradiation-alone group, mitochondrial transplantation combined with X-ray irradiation inhibited glioma growth in nude mice (p < 0.05).
Radiosensitization by mitochondrial transplantation in vivo. (A) Experimental schematic to test radiosensitizing effect of mitochondrial transplantation in U87 xenograft tumors. (B) Spatial patterns of blood perfusion and oxygen saturation in xenograft tumors. (C) At 6 h and 12 h after injection, the distribution of isolated mitochondria in xenograft tumors was detected by In-Vivo Xtreme II optical imaging system. White arrow: three-point injection. (D) The localization of isolated mitochondria in xenograft tumors was observed using tissue sections. Red: isolated mitochondria; Green: nuclei. (E) Apoptosis in xenograft tumors was measured by TUNEL-stained histology of tissue sections. Brown: TUNEL-positive cells. (F) Tumors were excised and weighed at the end of the experiment. *p<0.05, **p<0.01 vs. control group; #p<0.05 vs. 4 Gy irradiation alone.
Discussion
The endosymbiotic theory holds that the eukaryotic ancestor was an anaerobic, phagocytic cell that gained energy by glycolysis [26]. Mitochondria have an α-proteobacteria (Alphaproteobacteria) ancestry [27], possessing enzymes required for the TCA cycle and electron transport chain. The mitochondrial ancestor used oxygen to decompose pyruvate, a product of glycolysis, for more energy than glycolysis. Thus, the enrichment of oxygen in the atmosphere created a more suitable survival environment for the mitochondrial ancestor [28]. Starvation induced by inefficient energy metabolism in the eukaryotic ancestor may be a direct cause of engulfing a mitochondrial ancestor and setting up an endocellular symbiotic relationship of anaerobic eukaryotes with aerobic prokaryotes. From the perspective of the endosymbiotic theory, mitochondria might still be transferrable into recipient cells. Solid tumors possess hypoxic regions and insufficient blood supply and most cancer cells use glycolysis as the major energy source [29, 30]. Therefore, the microenvironment of cancer cells is similar to the adverse circumstances of the eukaryotic ancestor. Malignant tumors are the most likely region for transcellular implantation of exogenous mitochondria. Our study showed that starvation treatment with serum-glucose deprivation induced invagination of the plasma membrane and an endocytosis structure in U87 cells (Figure 3A-B, Video 1-2). We found that culturing starved U87 cells with mitochondria isolated from HA cells resulted in mitochondrial transfer into U87 cells through endocytosis (Figure 2D-E, 3C-D). The origin of organelles is a debated step in eukaryotic evolution but contributed to the emergence of cellular complexity that characterizes modern eukaryotes. Although the endosymbiotic theory of the origin of mitochondria was suggested by Lynn Margulis in 1967 [31], doubts continue about the theory because of the lack of direct evidence. Our work on mitochondrial transfer into cancer cells through endocytosis supported the endosymbiotic theory of mitochondrial origin.
In 1982, internalization of isolated mitochondria was reported, by simple co-incubation in vitro, without transfection reagents, medium supplements or other interventions [32]. Cellular uptake of exogeneous mitochondria might be involved in endocytosis. In the following decades, this phenomenon was not widely investigated and little data exists regarding its mechanisms. Katrangi E et al. reported that xenogenic mitochondria isolated from mouse liver were transferred into human colon cancer Colo205 cells, human breast tumor MDA MB435 cells and MCF7 cells via endocytosis [10]. A study with cardiomyocytes found that mitochondrial transfer was unaffected by pharmacological inhibitors of macropinocytosis, clathrin-dependent endocytosis or tunneling nanotube formation but was blocked by inhibition of actin polymerization [17]. The endocytosis involved in internalization of exogenous mitochondria into recipient cells is actin-dependent. However, it is not clear how actin-dependent endocytosis is regulated in the process of mitochondrial transcellular implantation. Our study found that starvation treatment effectively slowed glycolysis (Figure 2B-C) and decreased the transformation of NAD+ to NADH (Figure 4H) in glioma cells. A large amount of accumulated NAD+ was released into the extracellular space (Figure 4A). Extracellular NAD+ that may leak from neighboring cells might function similar to cytokines, as autocrine or paracrine elements inducing receptor-mediated responses in target cells. CD38 is a type-II transmembrane glycoprotein that catalyzes the cyclization of extracellular NAD+ to intracellular cADPR (Figure 4C). cADPR serves as a second messenger to activate RyRs of the endoplasmic reticulum and mobilize intracellular Ca2+. Released Ca2+ was were mainly distributed near the plasma membrane (Figure 4E, Video 3) to promote endocytosis by F-actin polymerization and cytoskeleton remodeling (Figure 4F). Finally, the starvation signal in cancer cells was effectively converted into actin-dependent endocytosis via NAD+-CD38-cADPR-Ca2+ transduction pathways. NAD+-CD38-cADPR-Ca2+ is a signaling pathway that is closely related to mitochondrial transplantation. As a novel mechanism of cell-cell communication, tunneling nanotubes facilitate the exchange of cellular components and signaling molecules between connected cells such as plasma membrane components, calcium ions, and organelles, including mitochondria [15, 33]. We hypothesize that tunneling nanotubes may be able to promote the spread of exogenous mitochondria transplanted into cancer cells between adjacent cells. If endocytosis can be combined with tunneling nanotubes, the efficiency of mitochondrial transplantation will be greatly improved.
Optimal mitochondrial internalization significantly normalizes mitochondrial membrane potential, mitochondrial reactive oxygen species levels and oxygen consumption rate and improves mitochondrial function in mtDNA-depleted ρ0 cells [10, 12]. A 2016 paper reported that functional mitochondria released from astrocytes enter damaged neurons after stroke, increasing ATP levels and recovering neuronal viability [34]. In vivo injection of isolated mitochondria into the ischemic heart of rabbits during early reperfusion significantly decreases myocardial ischemia-reperfusion injury and increases oxygen consumption rates and ATP production, resulting in postischemic functional recovery [35, 36]. These studies established that mitochondria that are transferred into recipient cells remain active and function properly. This phenomenon is similar to organ transplantation such as bone marrow or renal transplantation and is often called mitochondrial transplantation. This type of transplantation has been successfully applied in clinical practice at Boston Children's Hospital. Healthy autologous mitochondria harvested from nonischemic skeletal muscle were injected into the damaged myocardium of five pediatric patients with ischemia-reperfusion-associated myocardial dysfunction after a cardiac surgical procedure to improve ventricular function [37]. Mitochondrial transplantation will be a promising therapeutic strategy for various types of mitochondrial diseases [38]. Many studies show that malignant tumors are probably a type of energy metabolic disease involving respiratory insufficiency caused by mitochondrial dysfunction [3, 7, 8]. Our study showed that functional mitochondrial transplantation rescued aerobic respiration and attenuated the Warburg effect (Figure 5), inhibited malignant proliferation (Figure 6D), reactivated the mitochondrial apoptotic pathway (Figure 6B-C, 7E), and significantly enhanced glioma radiosensitivity (Figure 6E-G, 7F). In addition, we also found that exogenous mitochondria transplanted into U87 cells might successfully undergo fission and proliferation (Figure S5). The biological function of transplanted mitochondria would be gradually amplified in cancer cells. Mitochondrial transplantation might therefore be an effective treatment for malignant tumors.
With low astrocyte viability, and therefore astrocytic mitochondria, especially following transplantation, alternative source of highly viable mitochondria may be required for optimal therapeutic benefits. Along this line, stem cells have been shown to be a very good source of healthy mitochondria [39], which provides us with the idea of optimizing mitochondrial transplantation. We propose that autologous mitochondrial transplantation could be a therapeutic intervention that uses mitochondria from patients who are both the donor and recipient. Since mitochondria are processed outside the body and reintroduced into the same patient, this therapy would be of high specificity and would eliminate post-transplantation immune rejection. We also propose in the near future, every cancer patient will contribute their own mitochondrial biological agents.
Methods
Cell culture. Human glioma cell line U87 was obtained from Shanghai Genechem Co., Ltd, authenticated by short tandem repeat profiling, and cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (Gibco). Normal human astrocytes (HA) were purchased from ScienCellTM, authenticated by immunostaining of glial fibrillary acidic protein, and cultured in astrocyte medium (ScienCellTM).
Mitochondrial isolation and fluorescent labeling. Isolation of mitochondria from HA cells was performed using the Cell Mitochondria Isolation Kit (Beyotime) according to the manufacturer's instructions. Briefly, cells were incubated in mitochondrial lyses buffer for 10 min and homogenized for 30 strokes using a tight pestle on ice. The homogenate was subjected to centrifuging at 600 g for 10 min. Then the supernatant was collected and centrifuged again at 12,000 g for 10 min at 4°C to obtain pure mitochondria. After that, the isolated mitochondria were fluorescently labeled by staining with MitoTracker Red CMXRos (Invitrogen) for 20 min.
Mitochondrial membrane potential (ΔΨm). Assay for the intactness of the isolated mitochondria was performed by measurement of mitochondrial membrane potential (ΔΨm). After incubation with 10 μM JC-1 (Sigma-Aldrich) staining solution at 37°C, mitochondria were washed with JC-1 staining buffer two times and then analyzed using a flow cytometry (FlowSight, Amnis) [40].
Starvation treatment. U87 cells were seeded into a 60 mm culture dish and allowed to attach overnight. The cells were washed once in PBS and cultured with starvation solution composed of serum- and glucose-free DMEM (Gibco). After 2 h challenge, the starvation solution was replaced with maintenance medium and then the cells were used in the mitochondria transfer experiment.
Confocal microscopy. U87 cells stably expressing GFP were generated with a recombinant retrovirus carrying GFP driven by the pMSCV-puro retroviral vector (Wuhan Miaoling Bioscience Technology). MitoTracker Red CMXRos-labelled mitochondria were isolated from HA cells and immediately co-incubated with U87 cells stably expressing GFP to exclude the false positive result. After 6 h or 12 h co-incubation in glass-bottom dishes, confocal images were acquired using a LSM-700 confocal microscope (Zeiss).
Identification of mitochondrial genotypes. After 2 h starvation, U87 cells were co-incubated with isolated mitochondria for 12 h. Then, cells were washed twice for 3 min each with phosphate-buffered saline and harvested by trypsin-EDTA solution to produce a single cell suspension. The cells were pelleted by centrifugation at 600 g for 5 min and washed twice with phosphate-buffered saline to remove extracellular mitochondria. The mitochondrial genotypes of HA, U87 and U87+Mito cells were separately examined through direct sequencing (Hiseq X-ten, Illumina), and the unique mutational sites bearing featured SNPs were chosen to determine the origin of mtDNA in U87+Mito cells [15].
3D tomographic microscopy. U87 cells were seeded into 35 mm glass-bottom dishes and allowed to attach overnight. Dishes were then placed on the 3D tomographic microscope (3D Explorer, NanoLive) and 3D tomographic images (z-stacks) were taken at regular intervals (4 min). The plane with the best focus was picked from every z-stack and used to generate time-lapse sequences [41]. To get a smooth motion movie, the number of frame was increased by a combination of motion interpolation and frame blending.
Transmission electron microscopy (TEM). The ultrastructural analysis of endocytosis was performed with TEM assay as reported previously [42].
Colocalization of endocytosis and isolated mitochondria. To visualize the co-localization of endocytosis and isolated mitochondria in U87 cells, the multispectral imaging flow cytometry (FlowSight, Amnis) was used. pHrodo™ Green Zymosan Bioparticles™ Conjugate (Invitrogen) is a novel fluorogenic reagent developed for quantitative measurements of endocytosis. The starved U87 cells were treated with pHrodo™ Green and the isolated mitochondria were stained with MitoTracker Red. pHrodo™ Green and MitoTracker Red were detected in Channels 2 and 5, and brightfield was collected in Channels 1. The BDS (Bright Detail Similarity Ch2-Ch5) greater than 3 represents the colocalization of fluorescence signals of Channels 2 and 5 [43].
Extracellular NAD+ measurement. The extracellular NAD+ was quantified by a luciferase assay using the NAD/NADH-Glo™ assay kit (Promega). After starvation treatment on U87 cells, 100 μL of culture medium were treated with 0.4 N HCL and heat quenched at 60°C for 15 min in a 96-well plate. An equal volume of NAD/NADH-Glo Detection Reagent was added to each well with cell lysate, incubated at room temperature for 60 min, and luminescence was read on a microplate reader (Tecan Infinite M200, Grödig).
High performance liquid chromatography-tandem mass spectrometry (HLPC-MS) analysis. The intracellular cADPR was detected using an EVOQ Qube LC-TQ system (Bruker). Cells were pulverized and extracted with the mixture of methanol and water (volume ratio 4:1). The chromatographic separation was performed with a Bruker liquid chromatography system, using a ZORBAX Eclipse Plus C18 column (Agilent Technologies). The separation followed gradient elution where solvent A was water containing 0.1% formic acid, and solvent B was methanol also containing 0.1% formic acid [44]. The gradient elution time was 7 min per sample with cADPR showing a retention time close to 1.3 min. Mass spectrometer used electrospray ionization in the positive mode.
Fast XYT imaging of elementary calcium release events with confocal microscopy. U87 cells were loaded with Fluo-3 AM (Beyotime) for intracellular Ca2+ measurements in glass-bottom dishes. Fluo-3 AM is hydrolyzed intracellularly to the green fluorescent calcium indicator Fluo-3 in the presence of Ca2+. Images fast, localized elevations of the cytosolic Ca2+ concentration in two spatial dimensions plus time (XYT) were acquired using a Zeiss LSM-700 confocal microscope. Images were acquired every 2 seconds for a total duration of 25 minutes [45].
PCR array expression profiling. The Human Signal Transduction Pathway Finder™ RT² Profiler™ PCR Array (Qiagen) was used to screen a panel of 49 genes representative of glycolysis and TCA in U87, HA and U87+Mito cells. Microarray data were normalized against the house-keeping genes by calculating the ΔCt for each gene of interest in the plate. Fold changes of gene expression, scatterplot and heatmap were analyzed and generated by using RT2 PCR array data analysis web portal version 3.5.
Cellular metabolic phenotype measurements. Cells were seeded in a Seahorse XFp cell culture miniplate (Seahorse Bioscience) at 12,000 cells per well in complete medium. After overnight attachment, the medium was replaced with Seahorse XF assay medium (Seahorse Bioscience) supplemented with 1 mM sodium pyruvate, 10 mM glucose and 2 mM glutamin. Cells were then incubated in a non-CO2 incubator at 37°C for 60 min. OCR and ECAR were measured with a Seahorse XFp cell energy phenotype test kit (Seahorse Bioscience) and Seahorse XFp analyzer (Seahorse Bioscience) [46].
Observation of cytoskeleton. To investigate the involvement of F-actin at the site of endocytosis induced by cytoskeleton remodeling, the red fluorescent mCherry-actin expression plasmid constructed by Wuhan Miaoling Bioscience Technology Co., Ltd, was used. Cells were transiently transfected with the Lipofectamine 3000 (Invitrogen) according to the supplier's protocol. Fluorescence images were captured every 10 min for up to 40 min using a Zeiss LSM-700 confocal microscope.
Western blot analysis. Cells were lysed in RIPA buffer (Beyotime). Proteins were separated by SDS-PAGE and transferred to a methanol activated PVDF membrane. Antibodies against IDH2, cytochrome C, caspase 9, CD38 and GAPDH were from Abcam. Antibodies against CS, HK and PKM2 were from Cell Signaling Technology.
Isolation of the endoplasmic reticulum (ER) was performed with a Sigma endoplasmic reticulum isolation kit according to the directions of the protocol. Antibodies against FKBP12.6 were from Santa Cruz Biotechnology.
In vitro cell proliferation analysis. The rate of cellular proliferation was analyzed with a RT-CES system (ACEA Bioscience) as described previously [42]. U87 cells were seeded into a 16-well strip at a density of 1×103 cells/well. The sensor devices were placed into the 5% CO2 incubator and cell index values were determined every 15 min automatically by the RT-CES system for up to 106 h.
Detection of apoptosis. Quantification of apoptosis at the single cell level was as described previously by a combination of flow cytometry with classical Annexin V/PI staining using the Annexin V-FITC Apoptosis Detection Kit (Invitrogen) [47].
Colony formation assay. U87 cells were seeded in 100 mm petri dishes and left overnight to attach. After 2 h starvation, the cells were co-incubated with isolated mitochondria for 12 h before exposure to X-Rays. After irradiation, the cells were cultured in fresh medium for 10 days and fixed with ethanol and stained with 0.5% crystal violet [48].
In vivo studies. To form a tumor xenograft, athymic BALB/c nude mice (Beijing HFK Bioscience) were subcutaneously injected at hind-limb with the U87 cell suspension. 13 days later, the ultrasound contrast agent were intravenously injected into mice, the ultrasound and photoacoustic imaging of xenograft tumor was performed using the Vevo LAZR photoacoustic imaging system (VisualSonics). All images including B-Mode imaging for high-resolution anatomical images, DCEUS for functional tissue perfusion and PA imaging for SO2 were generated with the LZ250 transducer at 21 MHz [49].
For mitochondria injection treatment, MitoTracker Red CMXRos-labelled mitochondria were injected into the tumor by trigger point injection and the distribution of mitochondria in the tumor was detected by In-Vivo Xtreme II optical imaging system (Bruker). For animals receiving both mitochondria injection and radiation exposure, the tumors sites were exposed to a single dose of 4 Gy generated by a Faxition 43885D X-ray machine 12 h after mitochondria injection. The xenograft tumors were removed and weighted on the 15th day after radiation. In situ cell apoptosis was evaluated using a terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end-labeling (TUNEL) kit (Roche) following the manufacturer's protocol.
Statistical analysis. Data are presented as means ± SEM from at least three independent experiments and evaluated by analysis of variance (ANOVA) followed by Student Newman-Keuls test. Values of P < 0.05 were considered statistically significant.
Abbreviations
cADPR: cyclic ADP-ribose; CCCP: carbonyl cyanide 3-chlorophenylhydrazone; CS: citrate synthase; DCEUS: dynamic contrast-enhanced ultrasound; ECAR: extracellular acidification rate; ER: endoplasmic reticulum; F-actin: filamentous actin; FKBP12.6: FK-506 binding proteins 12.6; GFP: green fluorescent protein; HA: human astrocytes; HK: hexokinase; HLPC-MS: high performance liquid chromatography-tandem mass spectrometry; IDH2: isocitrate dehydrogenase 2; IGV: integrative genomics viewer; mtDNA: mitochondrial DNA; NAD+: oxidized nicotinamide adenine dinucleotide; OCR: oxygen consumption rate; PA: photoacoustic; PKM2: pyruvate kinas 2; RT-CES: real-time cell electronic sensing; RyRs: ryanodine receptors; SER10: sensitizer enhancement ratio at 10% survival level; SNPs: single nucleotide polymorphisms; SO2: oxygen saturation; TCA cycle: tricarboxylic acid cycle; TEM: transmission electron microscopy.
Supplementary Material
Supplementary figures and tables.
Video 1.
Video 2.
Video 3.
Acknowledgements
This work was supported by grants from the Ministry of Science and Technology National Key R & D Project (SQ2018YFE020524), the National Natural Science Foundation of China (11775280), the CAS “Light of West China” Program (Y866020XB0), the Natural Science Foundation of Gansu Province (18JR3RA391).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther. 2012;342:619-30
2. McInnes J. Mitochondrial-associated metabolic disorders: foundations, pathologies and recent progress. Nutr Metab (Lond). 2013;10:63
3. Seyfried TN, Flores RE, Poff AM, D'Agostino DP. Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis. 2014;35:515-27
4. Elliott R, Barnett B. Ultrastructural observation of mitochondria in human breast carcinoma cells. Microsc Microanal. 2011;17:194-195
5. Warburg O, Wind F, Negelein E. The metabolism of tumours in the body. J Gen Physiol. 1927;8:519-30
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
7. Seyfried TN, Shelton LM. Cancer as a metabolic disease. Nutr Metab (Lond). 2010;27:7
8. Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell. 2008;13:472-482
9. Hayakawa K, Bruzzese M, Chou SH, Ning M, Ji X, Lo EH. Extracellular Mitochondria for Therapy and Diagnosis in Acute Central Nervous System Injury. JAMA Neurol. 2018;75:119-122
10. Katrangi E, D'Souza G, Boddapati SV, Kulawiec M, Singh KK, Bigger B. et al. Xenogenic transfer of isolated murine mitochondria into human rho0 cells can improve respiratory function. Rejuvenation Res. 2007;10:561-70
11. Kim MJ, Hwang JW, Yun CK, Lee Y, Choi YS. Delivery of exogenous mitochondria via centrifugation enhances cellular metabolic function. Sci Rep. 2018;8:3330
12. Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci USA. 2006;103:1283-8
13. Elliott RL, Jiang XP, Head JF. Mitochondria organelle transplantation: introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res Treat. 2012;136:347-54
14. Sun C, Wang Z, Liu Y, Liu Y, Li H, Di C. et al. Carbon ion beams induce hepatoma cell death by NADPH oxidase-mediated mitochondrial damage. J Cell Physiol. 2014;229:100-7
15. Liu K, Ji K, Guo L, Wu W, Lu H, Shan P. et al. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res. 2014;92:10-8
16. Kitani T, Kami D, Matoba S, Gojo S. Internalization of isolated functional mitochondria: involvement of macropinocytosis. J Cell Mol Med. 2014;18:1694-703
17. Pacak CA, Preble JM, Kondo H, Seibel P, Levitsky S, Del Nido PJ. et al. Actin-dependent mitochondrial internalization in cardiomyocytes: evidence for rescue of mitochondrial function. Biol Open. 2015;4:622-6
18. De Flora A, Franco L, Guida L, Bruzzone S, Zocchi E. Ectocellular CD38-catalyzed synthesis and intracellular Ca2+-mobilizing activity of cyclic ADP-ribose. Cell Biochem Biophys. 1998;28:45-62
19. Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RM. et al. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science. 1993;262:1056-9
20. Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasová E, Moschella MC. et al. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513-23
21. Tang WX, Chen YF, Zou AP, Campbell WB, Li PL. Role of FKBP12.6 in cADPR-induced activation of reconstituted ryanodine receptors from arterial smooth muscle. Am J Physiol Heart Circ Physiol. 2002;282:1304-10
22. Lee HC, Aarhus R, Graeff R, Gurnack ME, Walseth TF. Cyclic ADP ribose activation of the ryanodine receptor is mediated by calmodulin. Nature. 1994;370:307-9
23. Rubio Ayala M, Syrovets T, Hafner S, Zablotskii V, Dejneka A, Simmet T. Spatiotemporal magnetic fields enhance cytosolic Ca2+ levels and induce actin polymerization via activation of voltage-gated sodium channels in skeletal muscle cells. Biomaterials. 2018;163:174-184
24. Zhou Y, Zhou Y, Shingu T, Feng L, Chen Z, Ogasawara M. et al. Metabolic alterations in highly tumorigenic glioblastoma cells: preference for hypoxia and high dependency on glycolysis. J Biol Chem. 2011;286:32843-53
25. Liang J, Cao R, Zhang Y, Xia Y, Zheng Y, Li X. et al. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat Commun. 2016;3:12431
26. Embley TM, Martin W. Eukaryotic evolution, changes and challenges. Nature. 2006;440:623-30
27. Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283:1476-81
28. Martin WF, Garg S, Zimorski V. Endosymbiotic theories for eukaryote origin. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140330
29. Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med. 2011;365:537-47
30. Casazza A, Di Conza G, Wenes M, Finisguerra V, Deschoemaeker S, Mazzone M. Tumor stroma: A complexity dictated by the hypoxic tumor microenvironment. Oncogene. 2014;33:743-1754
31. Sagan L. On the origin of mitosing cells. J Theor Biol. 1967;14:255-74
32. Clark MA, Shay JW. Mitochondrial transformation of mammalian cells. Nature. 1982;295:605-7
33. Vallabhaneni KC, Haller H, Dumler I. Vascular smooth muscle cells initiate proliferation of mesenchymal stem cells by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev. 2012;21:3104-13
34. Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C. et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535:551-5
35. McCully JD, Cowan DB, Pacak CA, Toumpoulis IK, Dayalan H, Levitsky S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am J Physiol Heart Circ Physiol. 2009;296:94-105
36. Masuzawa A, Black KM, Pacak CA, Ericsson M, Barnett RJ, Drumm C. et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2013;304:966-82
37. Emani SM, Piekarski BL, Harrild D, Del Nido PJ, McCully JD. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2017;154:286-289
38. McCully JD, Cowan DB, Emani SM, Del Nido PJ. Mitochondrial transplantation: From animal models to clinical use in humans. Mitochondrion. 2017;34:127-134
39. Borlongan CV, Nguyen H, Lippert T, Russo E, Tuazon J, Xu K. et al. May the force be with you: Transfer of healthy mitochondria from stem cells to stroke cells. J Cereb Blood Flow Metab. 2019;39:367-370
40. Paglin S, Lee NY, Nakar C, Fitzgerald M, Plotkin J, Deuel B. et al. Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res. 2005;65:11061-70
41. Ali A, Abouleila Y, Amer S, Furushima R, Emara S, Equis S. et al. Quantitative live single-cell mass spectrometry with spatial evaluation by three-dimensional holographic and tomographic laser microscopy. Anal Sci. 2016;32:125-7
42. Sun C, Liu X, Di C, Wang Z, Mi X, Liu Y. et al. MitoQ regulates autophagy by inducing a pseudo-mitochondrial membrane potential. Autophagy. 2017;13:730-738
43. Beum PV, Lindorfer MA, Hall BE, George TC, Frost K, Morrissey PJ. et al. Quantitative analysis of protein co-localization on B cells opsonized with rituximab and complement using the ImageStream multispectral imaging flow cytometer. J Immunol Methods. 2006;317:90-9
44. da Silva CP, Potter BV, Mayr GW, Guse AH. Quantification of intracellular levels of cyclic ADP-ribose by high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl. 1998;707:43-50
45. Von Wegner F, Both M, Fink RH, Friedrich O. Fast XYT imaging of elementary calcium release events in muscle with multifocal multiphoton microscopy and wavelet denoising and detection. IEEE Trans Med Imaging. 2007;26:925-34
46. Cheng G, Zielonka J, Ouari O, Lopez M, McAllister D, Boyle K. et al. Mitochondria-targeted analogues of metformin exhibit enhanced antiproliferative and radiosensitizing effects in pancreatic cancer cells. Cancer Res. 2016;76:3904-15
47. Sun C, Wang ZH, Liu XX, Yang LN, Wang Y. Disturbance of redox status enhances radiosensitivity of hepatocellular carcinoma. Am J Cancer Res. 2015;5:1368-81
48. Tan Z, Xiao L, Tang M, Bai F, Li J, Li L. et al. Targeting CPT1A-mediated fatty acid oxidation sensitizes nasopharyngeal carcinoma to radiation therapy. Theranostics. 2018;8:2329-2347
49. Bar-Zion A, Yin M, Adam D, Foster FS. Functional flow patterns and static blood pooling in tumors revealed by combined contrast-enhanced ultrasound and photoacoustic imaging. Cancer Res. 2016;76:4320-31
Author contact
![]() Corresponding author: Prof. Hong Zhang; Key Laboratory of Heavy Ion Radiation Medicine of Chinese Academy of Sciences, Institute of Modern Physics, Chinese Academy of Sciences, 509 Nanchang Road, Lanzhou 73000, China. E-mail: zhanghac.cn
Corresponding author: Prof. Hong Zhang; Key Laboratory of Heavy Ion Radiation Medicine of Chinese Academy of Sciences, Institute of Modern Physics, Chinese Academy of Sciences, 509 Nanchang Road, Lanzhou 73000, China. E-mail: zhanghac.cn